
Autonomous lab-on-paper for multiplexed, CRISPR-based diagnostics of SARS-CoV-2†

Abstract
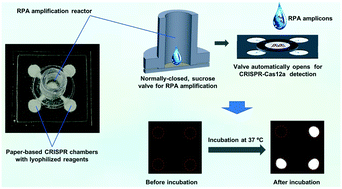
The COVID-19 pandemic, caused by severe acute respiratory coronavirus 2 (SARS-CoV-2), has become a public health emergency and widely spread around the world. Rapid, accurate and early diagnosis of COVID-19 infection plays a crucial role in breaking this pandemic. However, the detection accuracy is limited for current single-gene diagnosis of SARS-CoV-2. Herein, we develop an autonomous lab-on-paper platform for multiplex gene diagnosis of SARS-CoV-2 by combining reverse transcription recombinase polymerase amplification (RT-RPA) and CRISPR–Cas12a detection. The autonomous lab-on-paper is capable of simultaneously detecting nucleoprotein (N) gene and spike (S) gene of SARS-CoV-2 virus as well as human housekeeping RNAse P gene (an internal control) in a single clinical sample. With the developed platform, 102 copies viral RNA per test can be detected within one hour. Also, the lab-on-paper platform has been used to detect 21 swab clinical samples and obtains a comparable performance to the conventional RT-PCR method. Thus, the developed lab-on-paper platform holds great potential for rapid, sensitive, reliable, multiple molecular diagnostics of COVID-19 and other infectious diseases in resource-limited settings.
- This article is part of the themed collections: Coronavirus articles - free to access collection, Lab on a Chip HOT Articles 2021 and Coronavirus collection – Analytical
 Please wait while we load your content...
Please wait while we load your content...

