
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conventional and emerging strategies for the fabrication and functionalization of PDMS-based microfluidic devices
Amid
Shakeri
 a,
Shadman
Khan
b and
Tohid F.
Didar
*ab
a,
Shadman
Khan
b and
Tohid F.
Didar
*ab
aDepartment of Mechanical Engineering, McMaster University, 1280 Main Street West, Hamilton, ON L8S 4L7, Canada. E-mail: didar@mcmaster.ca
bSchool of Biomedical Engineering, McMaster University, 1280 Main Street West, Hamilton, ON L8S 4L8, Canada
First published on 12th July 2021
Abstract
Microfluidics is an emerging and multidisciplinary field that is of great interest to manufacturers in medicine, biotechnology, and chemistry, as it provides unique tools for the development of point-of-care diagnostics, organs-on-chip systems, and biosensors. Polymeric microfluidics, unlike glass and silicon, offer several advantages such as low-cost mass manufacturing and a wide range of beneficial material properties, which make them the material of choice for commercial applications and high-throughput systems. Among polymers used for the fabrication of microfluidic devices, polydimethylsiloxane (PDMS) still remains the most widely used material in academia due to its advantageous properties, such as excellent transparency and biocompatibility. However, commercialization of PDMS has been a challenge mostly due to the high cost of the current fabrication strategies. Moreover, specific surface modification and functionalization steps are required to tailor the surface chemistry of PDMS channels (e.g. biomolecule immobilization, surface hydrophobicity and antifouling properties) with respect to the desired application. While significant research has been reported in the field of PDMS microfluidics, functionalization of PDMS surfaces remains a critical step in the fabrication process that is difficult to navigate. This review first offers a thorough illustration of existing fabrication methods for PDMS-based microfluidic devices, providing several recent advancements in this field with the aim of reducing the cost and time for mass production of these devices. Next, various conventional and emerging approaches for engineering the surface chemistry of PDMS are discussed in detail. We provide a wide range of functionalization techniques rendering PDMS microchannels highly biocompatible for physical or covalent immobilization of various biological entities while preventing non-specific interactions.
Introduction
The biotechnology market has been vastly expanding due to the high demand for fast analytical procedures, such as high-throughput screening, pharmacogenomics, and gene expression analytics; technologies that could greatly benefit from microfluidic platforms. As a result, microfluidics has garnered significant interest in both academia and industry. Microfluidic channels can be made of different materials such as glass, silicon, polymers, and/or combinations of these materials. Polymeric microfluidics offer several advantages such as flexibility, low environmental impact, biocompatibility and versatile functionalization chemistries for bulk production, making them the most promising materials for microsystem technology.1 Polymeric microfluidics are mainly fabricated from polydimethylsiloxane (PDMS)2,3 or thermoplastic polymers such as poly(methyl methacrylate) (PMMA)4 and cyclic olefin copolymer (COC).5 Undoubtedly, PDMS is the most widely used mineral–organic polymer for fabrication of microfluidic channels by academic researchers. It presents several advantages, which include high optical transparency, low auto fluorescence, low toxicity in its solid form, easy mouldability, biocompatibility and gas permeability making it an ideal choice for research and rapid prototyping.6,7From an industrial perspective however, PDMS is relatively expensive, making low-cost, mass production of PDMS microfluidic devices a challenge. Thus, it has only been adapted in a few commercial products such as the ones developed by Fluidigm and Emulate. In fact, manufacturers prefer thermoplastic-based microfluidics over PDMS-based microfluidics as well as glass and silicon due to their reduced cost and suitable fabrication procedures for scaling up such as injection moulding and hot embossing.8,9
From a performance standpoint, one major drawback for polymers such as PDMS is their intrinsic hydrophobic nature, which leads to biofouling and denaturation, depletion of reagents, changes in local concentrations and bubble formation.10,11 PDMS can physically absorb hydrophobic biomolecules as well as hydrophobic solvents such as toluene, causing the polymer to swell.12 Hydrophobicity can also reduce the sensitivity and specificity of PDMS-based microfluidic devices used for biosensing applications, due to the non-specific attachment of target analytes and background noise generation.11,13 Notably, it has been shown that hydrophobic molecules with low molecular weights are more likely to diffuse into a PDMS matrix compared to molecules with high molecular weights.3,14
However, by using different wet and dry chemical modifications such as plasma activation,15,16 ultra violet (UV) irradiation,17 piranha etching,18 silanization,19 graft polymerization,20 chemical vapor deposition (CVD),21 sol–gel chemistry,22 and 3D scaffold formation (e.g. hydrogels),23 one can effectively alter the wettability and tailor the surface chemistry of the polymeric microchannels to suit specific biotechnological requirements.24,25 In addition to tuning surface energy, conjugation of target biomolecules onto microfluidic channels is a key step in designing target applications. Functionalization of the microchannels with different functional groups such as hydroxyl and thiol groups, epoxy rings, NHS-esters, amine groups, carboxylic and acrylic groups, aldehyde groups, carbodiimide chemistry, maleimide groups, or isocyanate and isothiocyanate groups can result in covalent immobilization of biological entities on the polymeric channels.26–30 Inducing the functional groups after fabrication of the device is usually more favourable as it does not interfere with the device bonding process.
Here, we first discuss various fabrication methods of PDMS microfluidic channels. Next, we provide a thorough run down of the different techniques used for PDMS bonding, followed by various surface modification and functionalization strategies including physical adsorption, silanization, grafting and hydrogel formation. As it will be discussed throughout this review paper, some of the provided approaches for fabrication and functionalization of PDMS-based microfluidics can potentially be incorporated in mass production lines due to the simplicity and cost efficiency of the approach.
Fabrication
Forming the microchannel geometry
The most common approach to fabricate PDMS with microfluidic channel cavities is soft lithography, which provides microfluidic pattern resolution ranging from nanometer to micrometer precision depending on the resolution of the master mould.31 The master mould is usually fabricated via a conventional photolithography technique where the negative photoresist of SU-8 is photopatterned on a silicon wafer to create the protrusions of a microchannel network.6,32 Although photolithography provides master moulds with superior resolution and allows for rapid prototyping of PDMS-chips in the next steps, it requires expensive clean-room facilities hindering its applicability for low-cost mast production of PDMS-microfluidics, especially in resource-limited and developing countries. Therefore, there have been several attempts to reduce the cost of photolithography technique such as modification of the UV lightening as well as the type of substrate, mask, and photoresist used in photolithography. In a study done by Li et al., a portable light emitting diode (LED) as an alternative to high-voltage mercury lamps was suggested to generate UV light for photoetching.33 Their LED was able to emit a ∼365 nm monochromatic light which directly falls in the maximal UV absorption rang of SU-8 (350–400 nm) without any need for further filtering. Unlike the commercial lithographic machines, their approach does not require high-level clean rooms and high-voltage mercury lamps with safety issues. Nguyen et al. utilized low-cost nail polish meth(acrylates) (MA) gels in photolithography as a negative photoresist instead of expensive SU-8.34 In the proposed protocol, the photomask was made via an office laser printer and offset printing using a toner aided spray to increase the contrast. LED-UV light was also emitted to cross link the MA patterns on a glass substrate. Moreover, rather than spin coating, they used vinyl stickers as a spacer on the glass slides to control the thickness of the MA layer. By eliminating the need for all the expensive clean-room facilities, they succeeded in reducing the cost of the setup needed for microfluidic fabrication from $30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–65000 to about $6000. Bubendorfer et al. have also demonstrated the possibility of producing SU-8 on PMMA substrates – a much cheaper substitute for silicon wafers, as a master mould for PDMS soft lithography.35
000–65000 to about $6000. Bubendorfer et al. have also demonstrated the possibility of producing SU-8 on PMMA substrates – a much cheaper substitute for silicon wafers, as a master mould for PDMS soft lithography.35
Photolithography techniques can also be employed to fabricate 3D microfluidic devices with multiple channel layers. Compared to conventional one-layer microfluidics, 3D chips offer several advantages including repeated mixing and sorting and higher efficiency for cell cultures and biological studies.36–38 3D microfluidics could be made through an add-on process where a number of complementary master moulds each of which contains multi-layers of photoresist should first be fabricated via photolithography and after casting PDMS in the moulds, the resulted 2D PDMS layers are stacked together to form the 3D channels.37,39 This makes the whole process very complicated and time-consuming thereby limiting the commercialization of 3D microfluidics. Zhang et al. introduced a novel protocol to make the fabrication process of 3D microfluidics simpler.37 In their design, a PDMS slab layer was made and treated with trichloro(1H,1H,2H,2H-perfluorooctyl)silane. This PDMS slab was then pressed on a master mould containing SU-8 protrusions together with PDMS liquid in a way that the liquid PDMS was sandwiched in between the master mould and the top PDMS slab. After proper curing, the PDMS slab was peeled off together with the resulted PDMS microarrays underneath and placed on another micropatterned PDMS layer. After plasma bonding the layers, the top PDMS slab could then be easily detached (as it was previously treated with fluorosilane) leaving only the micro features on the first layer. This process was repeated to form multilayer PDMS microfluidic. Backside 3D diffuser lithography is another strategy to generate 3D microfluidics.40,41 This method was utilized in Fenech et al.'s work to precisely form blood vessels-mimicking microchannels with a rounded cross section and varying geometries in one-step lithography process.41 Photolithography using grayscale masks also enables to fabricate 3D microfluidics in one step.42 However, these masks are generally very costly to fabricate. Lai et al. employed backside diffused light lithography using pseudo-grayscale masks as a cost-effective and simple approach to fabricate 3D microfluidic in a single UV step.43
Other lithography techniques such as reactive-ion etching, electron-beam lithography, wet etching, multiphoton lithography, direct laser writing, focused ion beam, and stereolithography have also been used to fabricate the master moulds.44–48 These methods allow for high-resolution fabrication and some do not require masks (e.g. direct laser writing, electron-beam, and focused-ion beam all of which are controlled by a software instead of a physical mask) which can potentially reduce the costs. Nevertheless, they are usually low-speed restricting the path to commercialization.48,49 For example, two photon polymerization as a promising stereolithography approach allows for fabrication of master moulds with extremely high resolution.50 However, it takes around 3.5 h to fabricate a 25000 μm3 cube using this technique.49,51 In order to reduce the time, Lin et al. combined this technique with photolithography to only produce the specific parts of the microchannels that need high resolution with two photon polymerization, while the majority of the mould was fabricated using photolithography.49 As another mask-less lithography technique, Rammohan et al. used an array of micro-mirrors controlled by software to fabricate master moulds. This approach could be utilized for batch production of 3D microfluidics where short exposure times of UV light could result in SU-8 patterns in a grayscale fashion.52 Mukherjee et al. demonstrated a new way of prototyping master moulds through the lamination of ADEX dry films, which showcases simplicity, promptness, and non-toxicity as key advantages.53 As this process does not need clean-room environments, it could reduce the cost of master mould fabrication.
Fabrication of the master moulds is not limited to the lithography methods and can be conducted through other techniques such micromachining of PMMA, brass, glass, and steel as well as 3D printing.54–57 While these processes are usually cheaper than lithography methods, the resulted resolution is not as high as lithography hindering their applicability for both commercialization and rsearch. Yan et al. used laser ablation to engrave microchannels in a PMMA substrate with good resolution.38 To generate a male master mould from the engraved PMMA substrate (female mould), they UV cured Norland Optical Adhesive (NOA) on the PMMA substrate. The NOA layer was then peeled off and used for replicating PDMS microchannels. The whole process could be performed outside of the clean room which could pave the way for low-cost rapid prototyping of microfluidics for commercialization.
Considering the recent advances in three-dimensional (3D) printing technology, master moulds with relatively good resolution can be directly printed by a 3D printer (with feature sizes usually larger than 50 μm).58,59 Villegas et al. demonstrated a mould modification technique, which makes it possible to cast smooth PDMS microchannels using rough 3D printed moulds produced using low-resolution, inexpensive 3D printers.57 In their protocol, rough 3D printed negative moulds are fluorosilanized through CVD of trichloro(1H,1H,2H,2H-perfluorooctyl)silane and cured at 60 °C overnight. Then, perflourorperhydrophenanthrene (PFPP) or perfluorodecalin (PFD) lubricant is added onto the moulds to create omniphobic-lubricant-infused moulds resulting in 10-fold reduction in the surface roughness of the PDMS microchannels produced by these moulds compared to unmodified moulds.
In general, following the fabrication of the master mould with any of the aforementioned methods, it is often fluorosilanized (e.g. with trichloro(1H,1H,2H,2H-perfluorooctyl)silane) to become hydrophobic, which facilitates the peeling off of PDMS after the casting process. PDMS casting involves mixing the silicon elastomer and curing agent with (most commonly at a mass ratio of 10:1), degassing the PDMS in a vacuum desiccator to remove the air bubbles, and curing the PDMS inside the master mould at a temperature higher than 70 °C for 15 min to 6 h (depending on the temperature), which in turn solidifies the polymer and transfers the positive mould's pattern to the PDMS layer.16,60 Spin coating of PDMS on a fluorosilanized flat substrate can also be employed to create thin PDMS membranes used in organ-on-chip applications.55,61–63 In a study done by Kim et al., PDMS was casted in a silicon mould containing micropillars to form a porous membrane with 10 μm pore-size.17
An alternative way to directly create microchannel arrays in a cured PDMS layer is to use a laser cutting machine.63,64 Engeland et al., for instance, used an excimer laser to create pores of 10 μm on a PDMS membrane.63 Li et al. also used laser ablation technique to create microfeatures on a thin PDMS layer.65 Similar to Zhang et al.'s study37 (discussed before), they first created a PDMS transferring slab layer, but they coated the layer with Teflon instead of fluorosilanization. Afterwards, a thin PDMS layer was spin coated and cured on top the Teflon layer and subsequently cut via a CO2 laser plotter to form the microchannels. Then, the microchannels were transferred to a substrate that was initially patterned with electrodes. After peeling off the PDMS transferring slab, another flat substrate with patterns of electrode was placed on top sandwiching the microchannel features in between. As another example, low-energy electron beam (LE-EB) irradiation was employed in Oyama et al.'s work to directly form microarrays in PDMS layers without using master moulds.66 As these methods do not need initial fabrication of master moulds in clean rooms, they could be considered simpler and more cost-effective approaches for commercialization purposes.
PDMS bonding
Subsequent to casting the PDMS layer with the microchannels' geometrical features, inlets and outlets are punched to provide access to the channel. This can also be done by positioning silicon tubing in the precured PDMS during the casting process. The PDMS microchannel is then bonded to another PDMS substrate to form the device (Fig. 1a).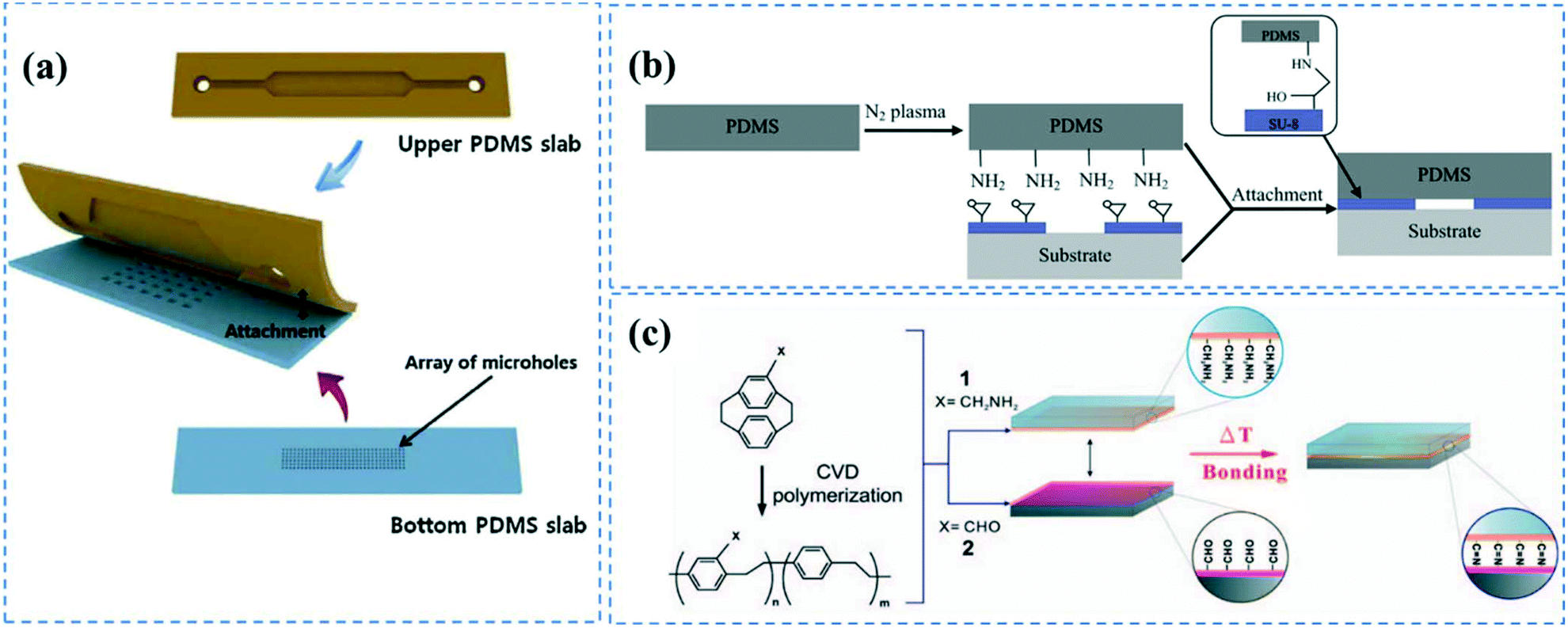 | ||
| Fig. 1 PDMS bonding techniques; (a) PDMS layers containing microchannel cavity and micro holes were aligned and bonded after oxygen plasma treatment. Reprinted from ref. 72 with permission from Elsevier. (b) Nitrogen plasma induced amine groups of PDMS were chemically bonded with epoxy rings of SU-8 substrate. Reprinted from ref. 82 with permission from Elsevier. (c) Chemical bonding of microchannel layers achieved by CVD polymerization of (2.2) paracyclophanes onto the substrates. Reprinted from ref. 84 with permission from Elsevier. | ||
Irreversible PDMS bonding is usually performed though hydroxylation67 of the PDMS surface via different methods such as plasma treatment,57,68 UV/ozone treatment,62 piranha etching,18 or the corona discharge technique.69 The most common technique to bond PDMS layers is oxygen or air plasma activation.20,57,68,70–72 Under plasma conditions (pressures <1 Torr and frequencies >1 MHz), electrons possess high kinetic energy, thus breaking and ionizing the incorporated gas molecules. The reactive species and photons produced by gas discharge bombard the surface causing chemical reactions on the substrate.73 This process does not induce structural changes and maintains the bulk properties of the polymer. The composition of the gas, alongside the pressure, power and exposure time determine the efficiency of the plasma treatment process.74 The silanol groups of the plasma activated PDMS layers can covalently bind to each other creating siloxane bonds once the PDMS layers are brought into contact. This is usually followed by a heat treatment step to promote bonding strength.14,17,55 PDMS–PDMS bonding strength and fracture energy is thoroughly investigated by Chen et al.75,76 Notably, oxygen plasma treatment is usually more favourable for bonding since it can trigger the formation of more hydroxyl groups on PDMS compared to air resulting in PDMS to PDMS bonding to be as strong as 715 kPa.77,78 In addition to the silanol groups, oxygen plasma treatment can induce alcoholic hydroxyl groups (C–OH) and carboxylic groups (COOH) on the PDMS surface.79 It is important to note that the plasma activated silanol groups on the PDMS surface are thermodynamically unstable, and hydrophobicity can be fully recovered in less than an hour. The activated PDMS tends to return to its equilibrium state through the diffusion of oxidized low molecular weight chains at the surface into the bulk substrate, thereby reducing the surface energy.80,81 This is attributed to the low glass transition temperature of PDMS (Tg ≲ −120 °C).1 Consequently, PDMS–PDMS bonding should be performed immediately after the plasma treatment.
This plasma treatment method can also be employed to bond PDMS to other types of polymers such as polystyrene and polyethylene.2,35 Furthermore, it has been shown that plasma activation of PDMS with nitrogen gas allows for PDMS bonding to SU-8 substrates through the covalent reactions between the plasma induced amine groups on the PDMS surface and the epoxy rings present on a SU-8 substrate (Fig. 1b).82 Plasma treatment is a fast and straightforward technique that can also provide hydroxyl groups inside the enclosed microchannels. Nevertheless, it requires advanced plasma equipment and the repeatability of the process is questionable.78
Ultraviolet/ozone (UVO) is another method utilized to hydroxylate PDMS surfaces. UV light at wavelengths of ∼160 to 240 nm (usually 185 nm is used) can dissociate the oxygen molecules and create ozone.83 The generated ozone molecules are again broken apart with a UV light at higher wavelengths (∼254 nm) producing O2 and reactive atomic oxygen molecules capable of oxidizing the substrate and eliminating hydrocarbon-containing organic moieties (the UVO treatment time is usually 30 min).62,84 UVO is performed in air and does not require a vacuuming system. Furthermore, Berdichevsky et al. have demonstrated that the hydrophilic recovery of UVO treated PDMS is much slower than plasma treated samples and the contact angles of UVO treated surfaces remain small months after treatment.62
The third way to hydroxylate PDMS surfaces is via the usage of piranha, an extremely strong oxidizer made of hydrogen peroxide and usually sulfuric acid with a weight ratio of 1:3. The transiently atomic oxygen produced by the reaction between sulfuric acid and hydrogen peroxide can breakdown the Si–CH3 bonds on the PDMS surface and substitute the methyl groups with silanol groups, while producing methane as a by-product.18 Piranha etching does not require any sophisticated facility, nor does it pose any size limitations. However, as it is highly corrosive, explosive, and toxic, application of piranha is always accompanied with safety and healthcare issues.85 Maji et al. have shown that piranha treatment using a solution with a H2O2:H2SO4 ratio of 2:3 followed by a dip in KOH solution for 15 min results in a contact angle of 27° compared to 110° for pristine PDMS.86 They also obtained lower hydrophobic saturation levels after the recovery of PDMS compared to the plasma treatment technique.
The fourth technique that has been employed to induce silanol groups on PDMS surfaces for bonding purposes is the corona discharge technique. Handheld corona discharge apparatus is capable of ionizing the air through the high voltage potential generated across its electrodes creating localized corona discharge and generates functional OH groups at room temperature.17,69,78,87
The main drawback of these hydroxylation techniques is that the layers are irreversibly bonded together as soon as they are brought into contact and will not let any slight readjustment if needed. Thus, precise alignment is required on the first trial. Another approach to irreversibly bond PDMS microchannels to a flat substrate, which can be made of either PDMS or other types of polymers, is use of uncured or partially cured PDMS as an adhesive between the two layers.27,64,88–91 In a protocol described by Villa-Diaz et al., a PDMS microchannel was bonded to a tissue culture Petri dish using a PDMS adhesive.90 This was done by spin-coating a mixture of PDMS prepolymer and toluene onto a glass slide. Then, a PDMS microchannel was placed onto the prepolymer layer, with only the sides adjacent to the channel cavity in contact with the prepolymer, thus keeping the channel cavity dry. Further, the PDMS platform was placed on the Petri dish and cured to induce bonding. The primary drawbacks of this strategy are the possibility of PDMS diffusion inside the channels as well as slight inconsistency in the channel height.89 Furthermore, changing the PDMS components (i.e., silicon elastomer and curing agent) ratios from standard 10:1 ratio could also result in strong bonding between PDMS to PDMS.78 In the “off-ratio” bonding technique, the bottom and top PDMS layers are first cured in a way that one layer contains an excess of one of the PDMS components, for instance the curing agent, while the other layer lacks that component. Once the layers are brought together and heated, due to the presence of concentration gradient, PDMS components could diffuse across the interface resulting in irreversible bonding. Unlike the hydroxylating strategies where the bonding occurs instantaneously, here it is possible to accurately align the channels positions before the heat treatment and irreversible bonding. However, changing the PDMS ratio could adversely affect the material properties and biocompatibility of PDMS, especially for cell studies.93 Lai et al. compared the bonding strength of “off-ratio” PDMS method with partially cured PDMS bonding, and achieved robust attachment when partially cured Sylgard 182 PDMS was bonded to fully cured Sylgard 184 PDMS with the normal rations.92
Other less commonly used methods for PDMS bonding include the utilization of UV-curable glue and adhesive films.78,94,95 Chu et al. used a mixture of silicone-based soft skin adhesive and PDMS liquid to form the adhesive polymer for bonding.91 Commercialized pressure sensitive adhesive (PSA) tapes (e.g. 3 M, Research Adhesive, ABI Tape, Coroplast) have also been cut via laser cutters or other cutting machines (e.g. vinyl cutters or knife plotters) to produce microchannels cavities and then used as a spacer to bind PDMS to different substrates such as PMMA, glass, and silicone so as to form enclosed microfluidic channels.96,97 The channel's height is determined by the thickness of the tape which could be problematic if thin microchannels (<50 μm in height) are required. Unfortunately, this method is not often employed for PDMS–PDMS bonding due to the weak attachment of the tape to PDMS. In order to overcome this issue, a PDMS/tape composite system was suggested, where the tape is embedded in PDMS by adding a thin layer of PDMS on top of the tape after attaching the tape to a PDMS slab and curing it,98 or by just baking the attached tape.99 In general, in comparison to the hydroxylation techniques for PDMS device bonding, the use of adhesive films and uncured/partially cured PDMS are more affordable and simpler in many cases making them good candidates for large-scale production purposes.
Ghaemi et al. introduced a low-cost flame-treatment approach to irreversibly bond PDMS.100 They used a piezo-jet torch placed at ∼1.5 cm above the PDMS layers to treat a substrate in different sequential patterns. The layers were then brought into contact and placed at 85 °C for 24 h to complete the bonding process. The bonding process could be related to the formation of OH groups although it was not confirmed.101 Using this technique, it is possible to bond PDMS to other substrates including SU-8, polystyrene, and even metallic substrates such as aluminium. Furthermore, recently, Oyama et al. introduced a new bonding technique with capability for commercialization and mass production. In his technique, several stacked PDMS layers can be bonded together through irradiation of an electron beam or 60Co γ-rays.36 The proposed method also provides long-lasting hydrophilization as well as sterilization of the 3D PDMS devices, simultaneously.
Chen et al. introduced a solventless adhesive bonding method allowing for irreversibly bonding between PDMS and PDMS or other substrates such as poly(tetrafluoroethylene) (PTFE), stainless steel, silicon, glass, and gold materials.84 In the proposed protocol, the substrates were CVD polymerized by 4-aminomethyl[2.2]paracyclophane and 4-formyl[2,2] paracyclophane to synthesize poly(4-aminomethyl-p-xylylene)-co-(p-xylylene) (polymer 1) and poly(4-formyl-p-xylylene-co-p-xylylene) (polymer 2) on the substrates, respectively (Fig. 1c). CVD polymerization was carried out through subliming the precursors in a vacuum chamber followed by condensation on the substrates, which were maintained at 15 °C. This led to spontaneous polymerization of the chemicals. The substrates were then contacted, and heat treated at 140 °C for 3 h, resulting in covalent bonding between the amine groups of polymer 1 and aldehyde groups of polymer 2. They also showed that the free functional chemical groups remaining inside the channel can be used for covalent immobilization of biomolecules. The aldehyde groups of the polymer 2 can directly bind to the amine, hydrazines, or hydrazides groups of biomolecules such as biotin-hydrazide. For polymer 1, they first activated the amine groups of the polymer via an NHS ester to be able to bind to the carboxylic groups of proteins and form covalent amide bonds.
Reversible sealing of PDMS to PDMS can also be obtained by simply pressing fully cured PDMS layers against each other due to van der Waals forces and hydrophobic contact.102,103 The elastomeric property of PDMS makes it possible to reversibly seal PDMS layers against nonpolar surfaces as well. However, the produced van der Waal forces can only withstand up to 5 psi.2,7 Clamping multiple PDMS layers between two metallic or polymeric slabs using screws,104,105 use of magnetic force to hold together PDMS layers,106 and coating PDMS with a chelating agent93 are other reversible sealing methods used in microfluidic fabrication.
Surface modification and functionalization
Physical adsorption
As mentioned earlier, the hydrophobic nature of PDMS microfluidics can be problematic in biological applications as it brings about non-specific adsorption of biomolecules. Hydrophobic PDMS can adsorb different biological components in cell culture media or biofluids thereby altering local concentrations and reducing the device's efficacy when it is implemented for cell culture applications and bioassays.3 Pre-saturation of PDMS microfluidic devices in cell culture media may be a way to diminish protein adsorption. Although the hydrophobicity in PDMS is not favourable for cell adhesion,80 it can be leveraged for the physical adsorption of extra cellular matrix (ECM) proteins, which effectively promote cell adhesion and growth. For instance, Engeland et al.63 coated a porous PDMS membrane with fibronectin after the fabrication of microchannels. Their microfluidic design was comprised of two PDMS microchannel layers and a middle PDMS membrane with 10 μm pores and was used to mimic artery systems on a chip. They coated the membrane with 100 μg ml−1 fibronectin diluted in PBS solution for 30 minutes at 37 °C. The protein was physically adsorbed onto the PDMS, inducing biocompatibility for cell culture. They first seeded the bottom channel with human aortic smooth muscle cells and then the top channel with human aortic vein endothelial cells, which allowed them to study cell–cell interactions and signalling under hemodynamic conditions.The adsorption of proteins onto PDMS can be derived by the hydrophobic interactions between the unfolded hydrophobic core and/or hydrophobic domains of the protein and the hydrophobic methyl groups of a PDMS surface.107,108 Since the flow regime in microfluidic channels is usually laminar with a parabolic velocity profile and no-slip boundary condition at the walls (these conditions could change depending on the design and application), the proteins close to the channel walls have more time to interact with the hydrophobic surface and diffuse into the polymer.69
Giridharan et al.61 also used hydrophobic adsorption of a fibronectin coating to make a PDMS microfluidic chamber for cardiac cell culture. A PDMS layer containing the cell culture chamber was bonded to a thin PDMS membrane as a substrate for cell adhesion, and another PDMS layer. These two layers were sandwiched between two polycarbonate fixtures. The cell culture chamber was incubated with fibronectin for 24 hours at 37 °C followed by a washing step and seeding with embryonic cardio myoblast line (H9c2). The special design of the device allowed it to replicate the hemodynamics of a normal heart, heart failure, hypertension, hypotension tachycardia, and bradycardia, as the associated strains and stretches could be transferred to the cells through the middle PDMS membrane. Fibronectin was also used in Song et al.'s device to functionalize PDMS microchannels for cell culture.55 Their device was pressed against a Braille display layer which had a grid of piezoelectric pins to control the fluid injection. Before cell culture, the channels were incubated with fibronectin for 30 minutes at room temperature and then human dermal microvascular endothelial cells were injected into the channels. The device was capable of inducing pulsatile shear stress to mimic the natural blood flow in the body. It was also able to provide multiple culture loops whose flow rate could be independent of one another. Adiraj Iyer et al. used the hydrophobic property of PDMS for gradual drug release applications.69 They loaded PDMS microchannels with rhodamine B as a model for hydrophobic molecules and demonstrated the gradual release of the biomolecule via the perfusion of ultrapure water in the microchannels, which generated a concentration gradient.
It is very plausible that hydrophobic adsorption causes dehydration and denaturation of the adsorbed proteins.71,107 Additionally, it is extremely challenging to fill the hydrophobic microchannels with aqueous solutions as the hydrophobicity can increase the chance of air bubble formation inside the device due to the lack of capillary forces. Using the hydroxylation techniques mentioned in the PDMS bonding section, it is possible to render the microchannels hydrophilic. The hydrophilicity after hydroxylation is generated by virtue of cleavage of nonpolar methyl groups, oxidation of the cleaved sites and formation of polar hydrophilic silanol groups, thereby increasing the surface energy.77,86 The benefit with piranha and UVO hydroxylation is that they can be performed after device bonding as well.109 With the plasma method, it is very challenging to uniformly treat complex channels after device bonding. If the plasma bonding is not followed by a heat treatment step, it is still possible to benefit from the remaining plasma induced OH groups inside the channel right after device bonding. Priest et al. demonstrated a method for rapid plasma treatment of PDMS enclosed channels whereby they injected molten gallium electrode pads into adjacent channels, which resulted in the generation of regions of high field-strength along the main microchannel.15 Using this technique, it is possible to pattern plasma activated areas inside a bonded microfluidic device. As discussed earlier, the hydroxyl groups induced by plasma or other described hydroxylation methods are not stable on PDMS due to its hydrophobic recovery. Eddington et al. have shown that thermal aging of PDMS in an oven for several days can elongate the hydrophobic recovery time of PDMS after plasma treatment from 15 min for unaged PDMS to 14 days for aged PDMS samples.80 They also noticed that the thinner a PDMS layer was, the quicker it recovered, which was attributed to the shorter diffusion time of oxidized low molecular weight chains into the thin samples. Moreover, covering plasma treated PDMS with aluminum deposition has been proven to be effective in the stabilization of hydroxyl groups.10 In this case, the hydroxylated PDMS could be stored for several months without losing its hydroxyl groups. The aluminium coating should be etched off before further surface modification. 55 kV LE-EB irradiation has also been proven to be an effective way to create long-lasting hydrophilicity (more than 5 months) due to the high thickness (∼40 μm) of the produced Si–Ox-rich layer.66 However, undesired cracking and warping in the substrate is plausible in this technique because of dissimilar mechanical properties throughout the PDMS thickness.
It is worth mentioning that plasma treatment with oxygen gas can render the surface electronegatively charged owing to the induced silanol and carboxylic groups, which have negative charges. Allylamine (NH2) plasma treatment of the PDMS surface, on the other hand, can induce positive charges on the surface through the production of amine groups on the PDMS surface.110 Charged surfaces are able to electrostatistically adsorb biomolecules with pH-dependent net electrical charges – such as negatively charged BSA and positively charged avidin, in PBS at a pH of 7.4.107,110
In a work done by Kim et al., an ECM consisting of rat type I collagen and Matrigel was physically bonded onto a PDMS membrane.17 They UVO treated the device to decontaminate the microchannels and make the device more hydrophilic before introducing the ECM. Further, Caco-2BBE human intestinal epithelial cells were cultured in the channels. The device was capable of generating the peristaltic motions of the intestine through the vacuum channels. Under both mechanical distortion and continuous fluid flow, the Caco-2 cells were able to grow into a 3D intestinal crypt-villus microstructure to create a human-gut-on-a-chip platform. Choi et al. layer-by-layer deposited polyelectrolytes of poly(allylamine hydrochloride) (PAH) with positive charge and poly(acrylic acid) (PAA) with negative charge to pattern hydrophilicity in the microchannel due to the high polarity of the PAA layer.39 The coating was stable under different organic and aprotic solvents.111 The device was able to generate monodisperse double- and triple- emulsion droplets in a microfluidic device with 3D nonplanar geometry. It is worth noting that the carboxylic groups in the PAA layer as well as amine groups in the PAH layer could potentially be utilized for covalent immobilization of biomolecules as well.
Overall, even though physical attachment of biomolecules to PDMS channels is one the most straightforward ways of PDMS functionalization, this method is usually undesirable as the non-covalent attachment of biomolecules increases the chance of biomolecule and cell dissociation under high shear stresses.19
Surface amination by silanization
Silanization of PDMS microfluidic channels with silane coupling agents such as (3-aminopropyl)triethoxysilane (APTES) and (3-aminopropyl)trimethoxysilane (APTMS) is the most frequently used method to generate amine functional groups on the channels' surfaces and enhance the immobilization of biomolecules.16,68,112–117 The basic concepts of this method is discussed elsewhere in depth.13,118 The process usually starts with hydroxylation of the substrate followed by the introduction of silane coupling agents to the channels. These agents covalently bind to the PDMS silanol groups forming siloxane bonds. A baking step is usually required after the silanization process to promote polymerization and condensation reactions and to create a self-assembled monolayer (SAM) of silane coupling agents by a lateral siloxane (Si–O–Si) network. Amine treatment can facilitate cell adhesion by providing biocompatibility as well as optimized hydrophilicity on PDMS surfaces (contact angles of ∼66° and ∼74° for APTES19 and APTMS54 treatment, respectively).119 It must be underlined that excessive hydrophobicity and hydrophilicity can reduce cell adhesion.54 Moreover, amine functional groups render the surface positively charged,120,121 which can be used for the electrostatic adsorption of negatively charged biomolecules as well as to facilitate cell attachment and proliferation, given the net negative change on plasma membranes.119 Beal et al. indicated that the positive charge density increases in acidic conditions due to the protonation of aminopropyl groups.103 In their work, the untreated PDMS microchannel (without hydroxylation) was immersed in a very concentrated APTES solution diluted in ethanol (APTES:ethanol 1:2 v/v) for 5 min in order for APTES to diffuse into the PDMS surface. Longer incubation times using their technique can cause swelling and irreversible distortion of PDMS. They introduce acidic or basic aqueous catalyst solutions to catalyze the hydrolysis reactions. It has been shown that further plasma treatment of the APTES-modified PDMS for irreversible bonding to another PDMS layer does not deteriorate the functionality of amine groups in the channels for covalent immobilization of the biomolecules.
Siddique et al. have shown that APTES treatment of PDMS microchannels can bring about improved collagen immobilization and supports cell stability and proliferation even at high shear stresses compared to only plasma treated PDMS devices.19 Immediately after bonding the plasma treated layers, they introduced 2% APTES solution in acetone to the channel, incubated it for 15 min at room temperature and then washed with PBS. Afterwards, the channels were incubated with 50 g mL−1 collagen-I at 37 °C. In a similar manner, Jain et al. functionalized oxygen plasma treated PDMS microchannels with 1% APTMS in ethanol for 10 min.68 After flushing the channel with 70% ethanol and 100% ethanol, they heat treated the device at 80 °C for 2 h to form a SAM. The device was coated with 100 g mL−1 collagen-I and subsequently all four sides of the rectangular channel were cultured with endothelial cells for mimicking thrombosis on a chip using whole blood.
Wu et al. compared the effect of (3-cyanopropyl)triethoxysilane (CPTES) silane (containing nitrile group) treatment with APTMS in terms of hydrophilicity and epithelial cell growth.54 They coated oxygen plasma treated PDMS substrates with 1% APTMS or 1% CPTES diluted in acetone for different incubation times (45 min to 20 h). They concluded that CPTES treated PDMS had higher hydrophilicity and lower hydrophobic recovery. Moreover, HeLa cells (human cervical cancer cell line) showed better cell attachment and growth on APTMS treated surface, while MDCK cells (Madin–Darby canine kidney normal cell line) had better growth on CPTES-treated surfaces.
Biomolecules can covalently bind to amine treated surfaces through the activation of their carboxylic groups via carbodiimide chemistry, which is discussed elsewhere in detail.118 The most commonly used carbodiimide include 1-ethyl-3-(-3-dimethylaminopropyl) carbodiimide hydrochloride (EDC or EDAC) soluble in aqueous buffers as well as N′,N′-dicyclohexyl carbodiimide (DCC) and N,N′-diisopropyl carbodiimide (DIC) soluble in organic buffers. Carbodiimides react with carboxylic groups and produce an O-acylisourea intermediate which can rapidly react with the amine groups on the surface, thus producing peptide bonds.16N-Hydroxysuccinimide (NHS) or sulfo-NHS usually accompany the EDC reaction to replace O-acylisourea intermediates with more stable NHS esters capable of reacting with the amine groups of the biomolecules.14,118 Li et al.16 applied carbodiimide chemistry to covalently bind anti-human IgG to APTES treated PDMS microchannels. They demonstrated effective use of double spiral microfluidic channels for chemiluminescent detection of human immunoglobin G (IgG), via the integration of a signal amplification strategy. The plasma treated channel was filled with 2% APTES and incubated for 1 hour and washed with PBS. Further, capture anti-human IgG antibodies were incubated on the surface via EDC (5.0 mM) – NHS (2.5 mM) for 15 min and Au nanoparticle-conjugated IgG detection antibodies and horseradish peroxidase (HRP) were added, allowing for chemiluminescence detection (Fig. 2a). Zhang et al.14 also presented an in situ bio-functionalization method using EDC–NHS to promote cell adhesion of murine cancer cells to fibronectin modified microchannels via covalent bonding. Immediately after the channel bonding through oxygen plasma activation, they perfused 2% APTES in acetone to generate a SAM with amino groups on the PDMS surface after 2 h incubation. The anti-fibronectin IgG antibody could then covalently bind to the amino group using EDC (20 mM) – NHS (10 mM) chemistry. The bio-affinity between the antibody and the antigen is strong and capable of maintaining bio-functionality, ensuring reliability and efficient cell adhesion. In another study, PDMS channels were hydroxylated through passing H2O/H2O2/HCl (in a volume ratio of 5:1:1) into the channels for 5 min followed by APTMS treatment.27 The amine groups were then converted to isothiocyanate groups using a thiophosgene solution so as to be able to bind to amine-terminated biomolecules.
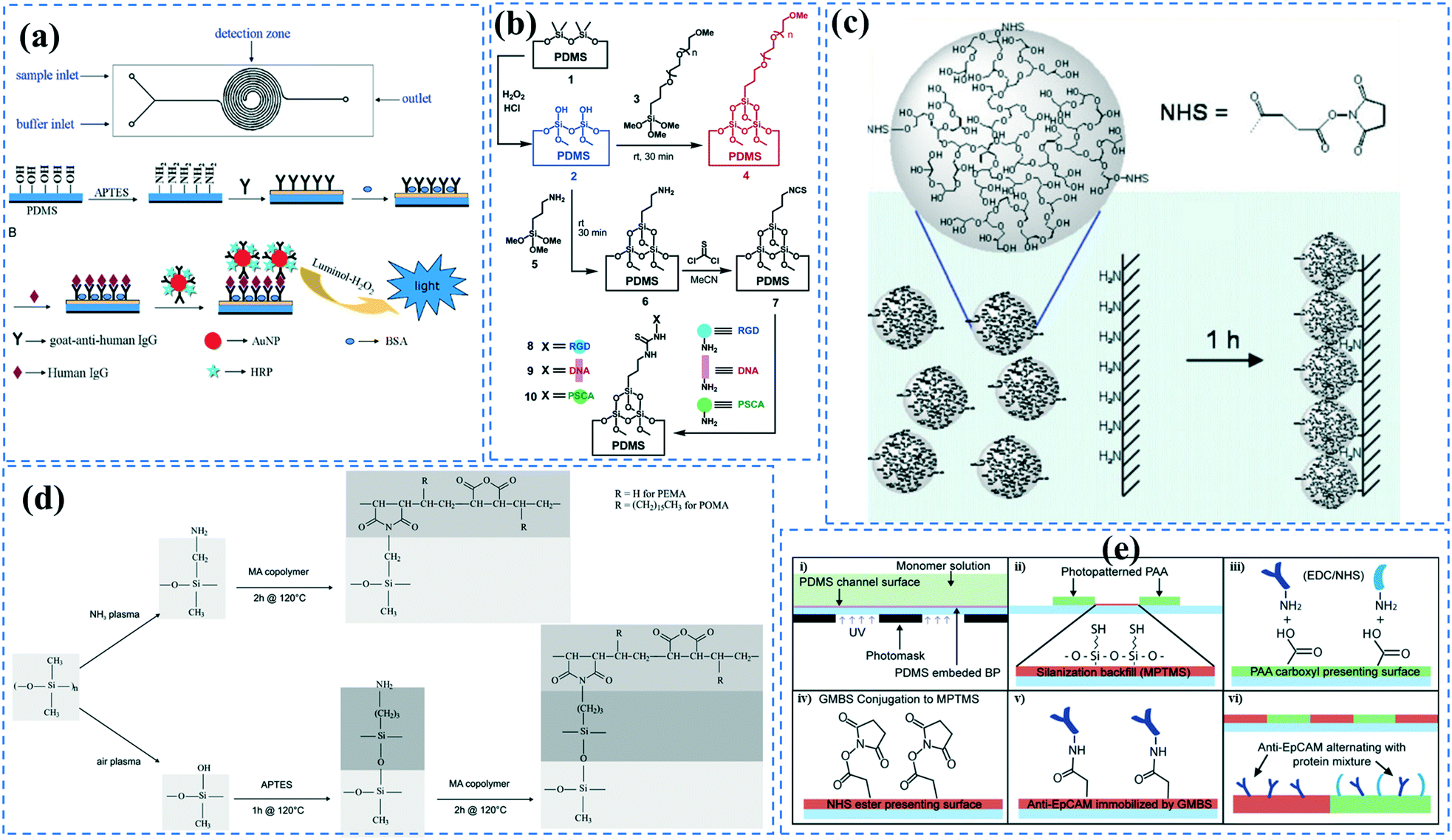 | ||
| Fig. 2 Functionalization of PDMS microchannels; (a) Double spiral microchannels functionalized with (APTES) followed by immobilization of goat anti-human IgG to capture human IgG through chemiluminescence immunoassay. Reprinted from ref. 16 with permission from Elsevier. (b) Microchannels were grafted with either PEG to prevent non-specific adhesion, or isothiocyanate for covalent biomolecule immobilization. Reprinted from ref. 27 with permission from ACS publications. (c) Microchannels grafted with functionalized HPG containing NHS esters. Reprinted from ref. 110 with permission from Springer. (d) Maleic anhydride was grafted on the amine-functionalized surface obtained via either ammonia plasma treatment or APTES treatment. Reprinted from ref. 125 with permission from Springer. (e) (i–vi) The experimental steps performed to create patterns of PAA-grafted and GMBS-grafted regions in microchannels. Reprinted from ref. 122 with permission from ACS publications. | ||
Generally, PDMS amination with silanization methods provides sustainable functional groups allowing for robust immobilization of different biomolecules. That being said, the process requires several steps making it complicated and time consuming. Additionally, crosslinkers such as EDC–NHS can bring about aggregation of the proteins due to the crosslinking of amine groups and carboxyl groups of adjacent biomolecules.
Grafting
Grafting the PDMS microchannels with different polymers is another popular technique for surface modification. Polymer grafting can be performed using one of two mechanisms: “grafting from” or “grafting to”. The “grafting from” approach utilizes the induced radical groups at the PDMS surface to initiate polymerization of the desired polymer. Application of a photoinitiator followed by UV polymerization is a common way to graft polymers from the surface.122 Benzophenone (BP) diluted in acetone is usually used as the photoinitiator in this technique.29,71,122 Swelling the PDMS via acetone allows the photoinitiator to diffuse through the PDMS. Moreover, surface-initiated atom transfer radical polymerization by means of UVO pre-treatment of PDMS surfaces123 or chloromethyl groups124 is another way for initiation of polymerization in the “grafting from” technique. “Grafting to” is referred to covalent attachment of the polymer to the functional tethering groups such as amine groups and thiol (SH) groups induced on the PDMS surface.110,125Polymer grafting is usually used for tailoring the wettability and anti-biofouling properties of PDMS, which is much more effective than physical adsorption of conventionally used blocking agents such as BSA, fat milk, serum, gelatin, or non-grafted PEG.16,125,126 Grafting PEG is proven to be a powerful way for altering the wettability of the PDMS channels and blocking biomolecules adsorption. For instance, Kovach et al. grafted 50% 2-[methoxy(polyethyleneoxy)propyl]trimethoxysilane (PEG-silane diluted in acetone) into assembled microchannels via oxygen plasma activation.20 The PEG-silane covalently bonded to the silanol groups and reduced fibrinogen adsorption and platelet adhesion, thereby suppressing the coagulation response. In another study, 100% PEG-silane was grafted to hydroxylated PDMS microchannels for 30 min (Fig. 2b).27 Using neat PEG-silane is claimed to be necessary for obtaining long-lasting PDMS passivation properties.27 PEG-diacrylate (PEG-DA) is another well-known PEG-based chemical that can be grafted into PDMS microchannels via a UV-mediated grafting technique, using BP as the photoinitiator. The resulting substrate exhibits a contact angle of 30° and non-fouling properties.71,127 As UV-mediated grafting could be performed through a mask, patterning hydrophilic/hydrophobic areas are also possible with this method. The main disadvantage of PEG-silane and PEG-DA is difficulty in immobilization of capture antibodies and biomolecules on such a repellent PEG treated surface for applications such as biosensing. To address this issue, a combination of biotin–PEG and methoxy-PEG (mPEG) is shown to not only prevent non-specific adhesion, but induce capability to capture streptavidin-conjugated biomolecules.128,129 However, streptavidin conjugation is a prerequisite for biomolecule immobilization in this method. NHS-PEG-acrylate is another PEG-based graft that can be UV polymerized and accompanied with PEG-DA to reduce non-specific adsorption and provide functional NHS esters for biomolecules immobilization.130 Using this method, it is possible to covalently pattern many different types of biomolecules on PDMS while the non-specific adsorption is prevented via PED. Nonetheless, since NHS is very reactive and unstable, the biomolecules should quickly be incubated with the surface for covalent attachment. UV-mediated grafting of poly(N-isopropylacrylamide) (PNIPAAm) can provide a thermo-responsive polymeric layer with switchable hydrophilicity at a lower critical solution temperature (LCST; 32 °C).71,127 The hydrophilic anti-fouling PNIPAAm grafted PDMS channels can become hydrophobic once the temperature is raised above the LCST. PNIPAAm coated microchannels can also be used for reversible attachment and release of PNIPAAm coated-grafted nanobeads, cells, and proteins above and below the LCST, respectively.71,127,131,132 Ebara et al. grafted channels with smart copolymers of P[NIPAAm-co-acrylic acid (AAc)] via UV irradiation of the monomer mixture.71 In this case, the wettability of the copolymers was tuneable in response to both pH and temperature. In Sung et al.'s study, oxygen plasma treated PDMS channels were first coated with multiple layers of polyethyleneimine (PEI) and poly(acrylic acid) (PAA), followed by the crosslinking of COOH groups in PAA and amine groups in PEI through EDC–NHS. The remaining NHS esters were blocked with ethylene amine.29 Next, NHS-PEG-acrylate solution mixed with protein G solution was copolymerized inside the channel for antibody immobilization. PEI and PAA multilayers can increase hydrophilicity and suppress non-specific attachment in PDMS. Integration of the polyelectrolyte multilayers with PEG can make PDMS greatly adsorption resistant. Utilization of protein G in the system also helps with the orientation of the antibodies that are further immobilized on the surface thereby enhancing the functionality of the antibodies for capturing the target biomolecules.
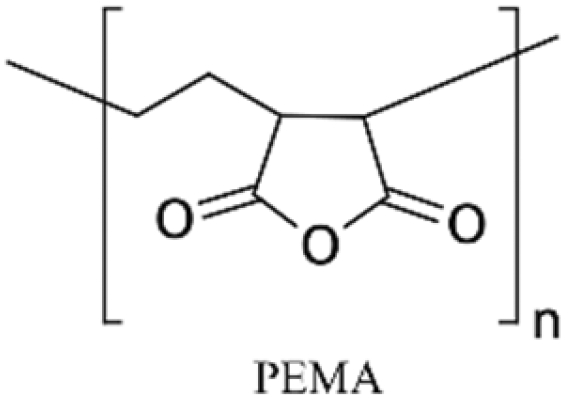
Multilayers of poly(diallyldimethylammonium chloride) (PDADMAC) with positive charge and PAA with negative charge were grafted on PDMS by Demming et al.133 They incubated the already closed microfluidic channel with HCl to induce negatively charged anchoring groups on the PDMS surface. Afterwards, through electrostatic adsorption, multilayers of the aforementioned polymers were alternatively deposited into the channel to obtain 7 polyelectrolyte double layers ending with PAA. The grafted polyelectrolyte provides hydrophilic properties on the surface, which were used for the cultivation of the model microorganism Saccharomyces cerevisiae CCOS 538. Cordeiro et al. grafted the maleic anhydride copolymer of poly(ethylene-alt-maleic acid) (PEMA) and poly(octadecene-alt-maleic anhydride) (POMA) on the PDMS channels to provide anti-fouling properties.125 The anhydride copolymer can covalently bind to amine treated PDMS channels through either ammonia (NH3) plasma treatment or APTES treatment. They concluded that ammonia plasma treatment results in a smoother and more homogeneous PEMA copolymer film (Fig. 2d). The main problem of the multilayer deposition methods is the complexity of the process which makes the functionalization step more time-consuming. Yeh et al. fabricated hyperbranched polyglycerol (HPG) grafts into microfluidic channels.110 They first plasma treated the already bonded PDMS channels using argon gas followed by allylamine (NH2) plasma activation to achieve amine groups. Then, functionalized HPG was perfused into the channel. The hydroxyl groups of HPG reacted with succinic anhydride to create succinimidyl ester groups, which were subsequently hydrolyzed in water to form NHS esters and carboxylic acids at HPG. Functional HPG can covalently bind to the amine functionalized PDMS channel through the NHS ester. The carboxylic groups also provided negative surface charge and could be used for the immobilization of biomolecules containing amine groups through carbodiimide chemistry. The graft can also reduce the non-specific adsorption of biomolecules. They utilized the negatively charged COOH groups in the graft in a special microfluidic design with pillar arrays to separate proteins with positive (e.g. avidin) and negative (e.g. BSA) charges (Fig. 2c).
Yang et al. grafted polyperfluoro-butenylvinylether (CYTOP) through spin coating on APTES treated PDMS surfaces to create a chemically inert layer.134 In order to bond the treated layers, they used thermal fusion bonding, as CYTOP is a thermoplastic polymer. Trantidou et al. presented a quick and low-cost method to graft a polyvinyl alcohol (PVA) polymer inside PDMS microfluidic channels in order to increase hydrophilicity, which is required for the generation of oil-in-water droplets.79 To do so, immediately after bonding the device by oxygen plasma treatment, the microchannels were incubated with 1% PVA in water for 10 min, blow dried, and heated at 110 °C for 15 min. It was also possible to pattern hydrophilic areas inside the channels by passing air through the channels that should remain hydrophobic during the PVA treatment. Even though this process is very simple as well as cost and time efficient, it does not provide functional groups for biomolecules immobilization.
To generate functional groups in PDMS channels for the covalent immobilization of biomolecules, Vu et al. grafted amino-terminated polyacrylamide chains (Am-PAM) on PDMS surfaces (and other surfaces such as SU-8 and silicon) through the radical polymerization of acrylamide using aminoethanthiol hydrochloride as the chain transfer agent and potassium persulfate as the initiator.26 The polyacrylamide (PAM) brush can render the surface non-fouling and hydrophilic. The main advantage of Am-PAM grafting is the presence of amine terminal groups that can covalently bind to isothiocyanate or carboxylic groups using carbodiimide chemistry. In another study, PAA was grafted into PDMS microfluidic channels through a UV polymerization technique.122 This technique was employed for patterning multi- component proteins, using a photomask alternating region of plain PDMS and PAA. The carboxylic groups of the PAA graft were used for the covalent attachment of E-selectin proteins via EDC–NHS. The remaining PDMS regions were silanized with 3-mercaptopropyl trimethoxysilane (MPTMS), which was then attached to amine-to-sulfhydryl cross-linker γ-maleimidobutyryloxy succinimide (GMBS). GMBS could then bind to antiepithelial cell adhesion molecules (anti-EpCAM). This device was used for immunoaffinity-based circulating tumour cell (CTC) capture (Fig. 2e).
Hydrogels
Hydrogels offer adequate porosity for cellular organization, biocompatibility and stiffness. Using hydrogels as a matrix improves the functionality and viability of PDMS microfluidic chips as it allows for spatial and temporal control of cell growth, monitoring of dynamic cellular responses and the application of mechanical and chemical stimuli. Hydrogels can be fabricated in 2D or 3D shapes out of a variety of materials including natural materials such as collagen, alginate, and chitosan as well as synthetic material, such as PEG, PAM, and PAA.135–143 Shin et al. have presented a thorough protocol to integrate microfluidic devices with different hydrogels including polymers such as poly-D-lysine (PDL) and poly-L-lysine (PLL), or extracellular matrix proteins such as fibronectin, collagen, and Matrigel.144 However, inducing adhesion between hydrogels and PDMS is very challenging due to the distinct differences between their chemical properties.Cha et al. incorporated alginate coatings on PDMS microchannels leading to an increase in the hydrophilicity and adhesion properties of PDMS.109 Additionally, the carboxylic functional groups of alginate provide an avenue for the covalent attachment of hydrogels. In their protocol, the alginate solution was covalently bound to APTES treated channels using EDC–sulfo NHS. To fabricate the hydrogel, one method involved perfusing alginate together with calcium for ionic cross linking to the alginate coated PDMS channel. The other method was perfusing alginate together with adipic acid dihydrazide (AAD) to covalently bind the alginate coated PDMS channel using carbodiimide chemistry (Fig. 3a). Polyelectrolyte multilayers of PEI and PAA can also be incorporated into PDMS channels to increase the hydrophilicity for hydrogel attachment. Sung et al. used this technique to UV graft a gel precursor containing acrylamide, bis-acrylamide, glycerol, and acrylate-PEG–protein G, using BP photoinitiator.29 The device was used for capturing estrogen receptors.
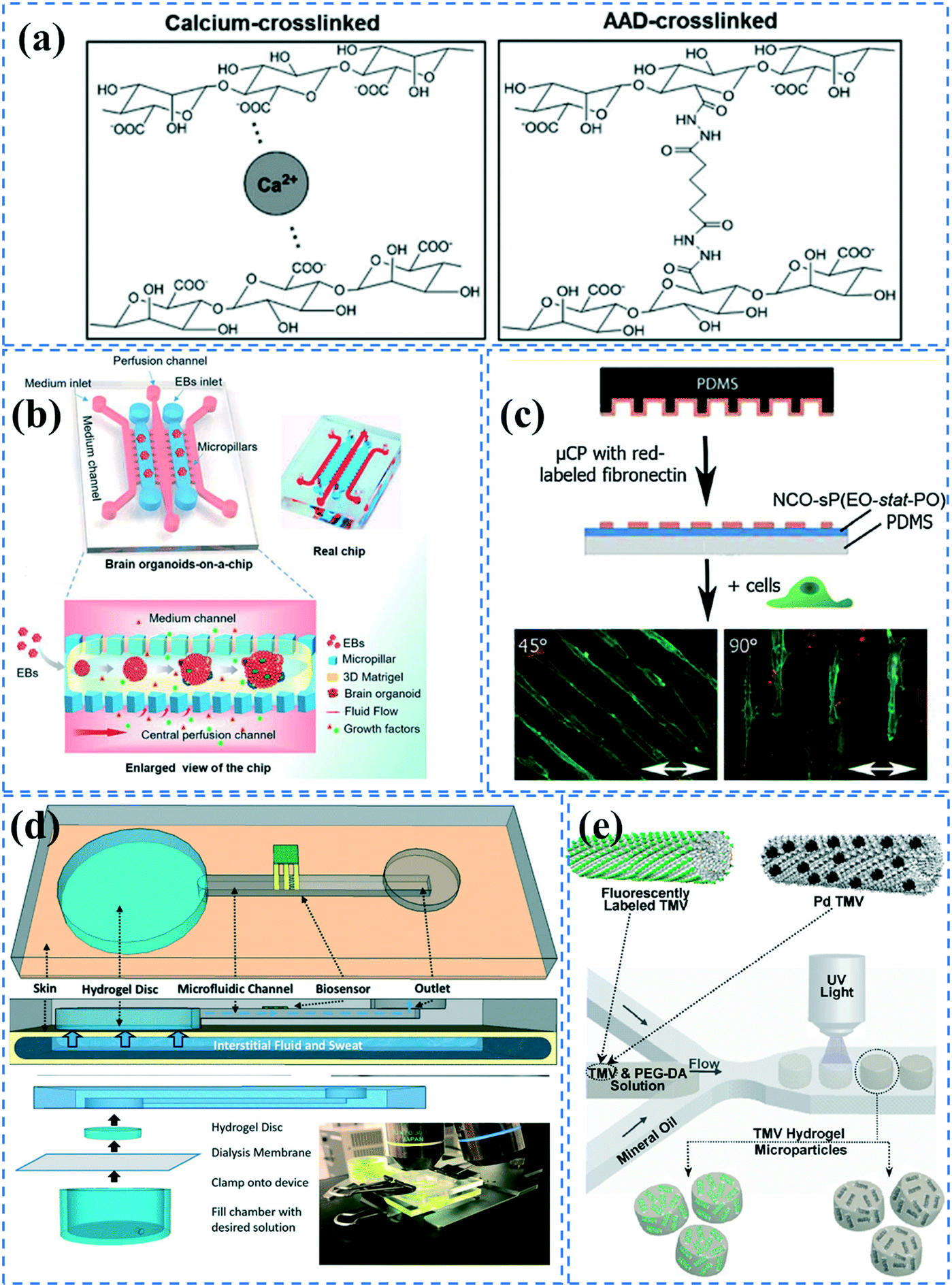 | ||
| Fig. 3 Hydrogel formation in PDMS microchannels; (a) Alginate hydrogels were formed in alginate coated microchannels through either ionic crosslinking or covalent binding using ADD. Reprinted from ref. 109 with permission from Wiley. (b) An organ-on-a-chip device was coated with Matrigel to culture brain organoids. Reprinted from ref. 145 with permission from Royal Society of Chemistry. (c) NCO-sP(EO-stat-PO) coated surfaces were microcontact printed with fibronectin for cell patterning. Reprinted from ref. 28 with permission from Wiley. (d) A microchannel embedded with a hydrogel disk which can transfer sweat samples to microfluidic device. Reprinted from ref. 23 with permission from Royal Society of Chemistry. (e) Hydrogel microparticles embedded with fluorescently labelled or palladium conjugated TMV were fabricated by a droplet-based microfluidic device. Reprinted from ref. 88 with permission from ACS publications. | ||
Wang et al.145 incorporated Matrigel in an organ-on-a-chip system to culture 3D brain organoids. The PDMS device consisted of medium channels and culture chambers separated by trapezoid pillar arrays. They produced embryoid bodies (EBs) suspended within ice-cold Matrigel. The EB-Matrigel was added to the culture channels of the chip and incubated at 37 °C for Matrigel gelation. Subsequently, neural differentiation medium (NDM) was perfused in the middle medium channel (Fig. 3b). Villa-Diaz et al.90 also prepared a PDMS microfluidic device with a Matrigel coating to culture individual colonies of human embryonic stem cell lines. The plasma treated channels were coated with Matrigel solution and incubated at room temperature for 2 hours. Annabi et al. cultured cardiomyocytes in a PDMS-based microfluidic embedded with methacrylated gelatin and methacrylated tropoelastin hydrogels.146 Given that gelatin is achieved by denaturing collagen, it possesses cell-binding sites due to the presence of arginine–glycine–aspartic acid (RGD) peptides that allow it to adhere to cells and facilitate their growth. Tropoelastin is also a natural protein present in elastic human tissues and has resilient characteristics. Use of methacrylate allows for crosslinking under UV light and mouldability to any desired form. The cells showed a higher spontaneous beating rate in the tropoelastin-based hydrogel compared to the gelatin-based one.
Greiner et al. coated ammonia plasma treated PDMS microfluidic channels with star shaped poly(ethylene oxide-stat-propylene oxide) (NCO-sP(EO-stat-PO)) via dip and spin coating methods.28 Given the cell-repellent properties of NCO-sP(EO-stat-PO), they could pattern fibroblasts cells by microcontact printing fibronectin onto the hydrogel.147 Isocyanate (NCO) groups can remain stable for 1 h, which allows for covalent bonding of the amine groups of fibronectin during contact printing. The mechanical stability of the coating was also investigated under long term uniaxial cyclic tensile strain (Fig. 3c). Furthermore, it is possible to embed RGD peptides in the hydrogel during polymerization, for cell adhesion purposes.147 However, this functionalization process is quite complicated for commercialization, and short-lasting functionality of isocyanate groups could be problematic for biomolecule immobilization. Piao et al. coated plasma treated PDMS channels with 3-(trimethoxysilyl) propyl methacrylate to be able to attach PEG-DA hydrogel.148 They UV polymerized PEG-DA mixed with glucose oxidase (GOx) enzyme, using 2-hydroxy-2-methylpropiophenone (HMPP) as the photoinitiator. The device was used for in situ glucose monitoring. Shay el al. have presented an interesting skin-inspired hydrogel interface that can pump up fluid and transfer it to an adjacent microfluidic device via osmotic driving force.23 Their hydrogel is made by UV polymerizing a mixture of acrylamide, n,n′-methylenebisacrylamide crosslinker, and a photoinitiator. The system is doped with NaCl or glycerol at a higher concentration than in the body to be able to create osmotic driving force. The device could thus take sweat samples from the skin and analyze them via sensors embedded in the microfluidic device (Fig. 3d).
PDMS microfluidics have also been employed for the generation of functionalized hydrogel particles. For instance, Lewis et al. fabricated functionalized viral-synthetic hybrid hydrogel particles embedded with genetically modified tobacco mosaic virus (TMV).88 Using a droplet-based microfluidic device, they created droplets of TMV-PEG-DA solution containing HMPP photoinitiator, which were then separately UV polymerized in the channel (Fig. 3e). Functionalized hydrogel microparticles being embedded in a virus minimizes aggregation and stability issues that are usually introduced in microfluidics. In a similar manner, Sheikhi et al. UV polymerized hydrogel beads made of gelatin modified with methacrylic anhydride.149 By decreasing the temperature in the microchannels, physical binding of the beads occurred, which was followed by UV chemical binding outside of the chip. Guo et al. also made core (poly(PEG-DA))–shell (alginate) structured hydrogel particles via droplet-based microfluidics by UV irradiation of aqueous droplets containing PEG-DA, alginate, EDTA–Ca, and hydrophobic corticosteroid-loaded poly(lactic-co-glycolic acid) (PLGA) nanoparticles.150 During the polymerization, drug-loaded PLGA nanoparticles spontaneously diffused into the poly(PEG-DA) core because of the analogous hydrophilicity. The hydrogels could be used for sustainable release of the drug for corticosteroid treatment.
Other surface modification methods
Sol–gel is one of the most widely used techniques to induce anti-biofouling properties on PDMS microchannels. Sol–gel coating of PDMS channels using 2.8% ethylamine after swelling the PDMS by tetraethyl orthosilicate (TEOS), can form nanometer-sized SiO2 particles, generating hydrophilicity and anti-biofouling properties.151 Sol–gel is also used to form metal oxide barriers of titania (CA: 61°), zirconia (CA: 90°), or vanadia (CA: 19°) for preventing biomolecule diffusion in PDMS.152 The metal oxide particles can be further functionalized via oligoethylene oxide, amine, perfluoro, or mercapto groups through silane chemistry.152 Furthermore, tetraethyl orthosilicate (TEOS) in combination with either trimethoxyboroxine153 or methyltriethoxysilane,154 has been used in an in situ sol–gel chemistry manner to create protective and chemically inert glass coatings on the PDMS microchannels that can be further functionalized for biomolecule immobilization. Li et al. have combined vanadia or titania sol–gel coatings and additional dynamic coatings with sodium dodecyl sulfate (SDS) to more effectively inhibit biomolecule adsorption issues.22 They concluded that compared to titania, vanadia coatings can better suppress the adsorption of carboxylate-modified polystyrene nanoparticles (PS NPs), and a combination of either vanadia or titania with a SDS coating can result in completely blocking of PS NPs adsorption. The device was utilized for the separation of PS NPs based on diffusion-based recovery, using a two-layer crossing microdevice (Fig. 4a). Sol–gel methods could provide very uniform coatings on PDMS surfaces and allows for in situ coating the channels after device bonding. However, the process could sometimes be laborious and expensive.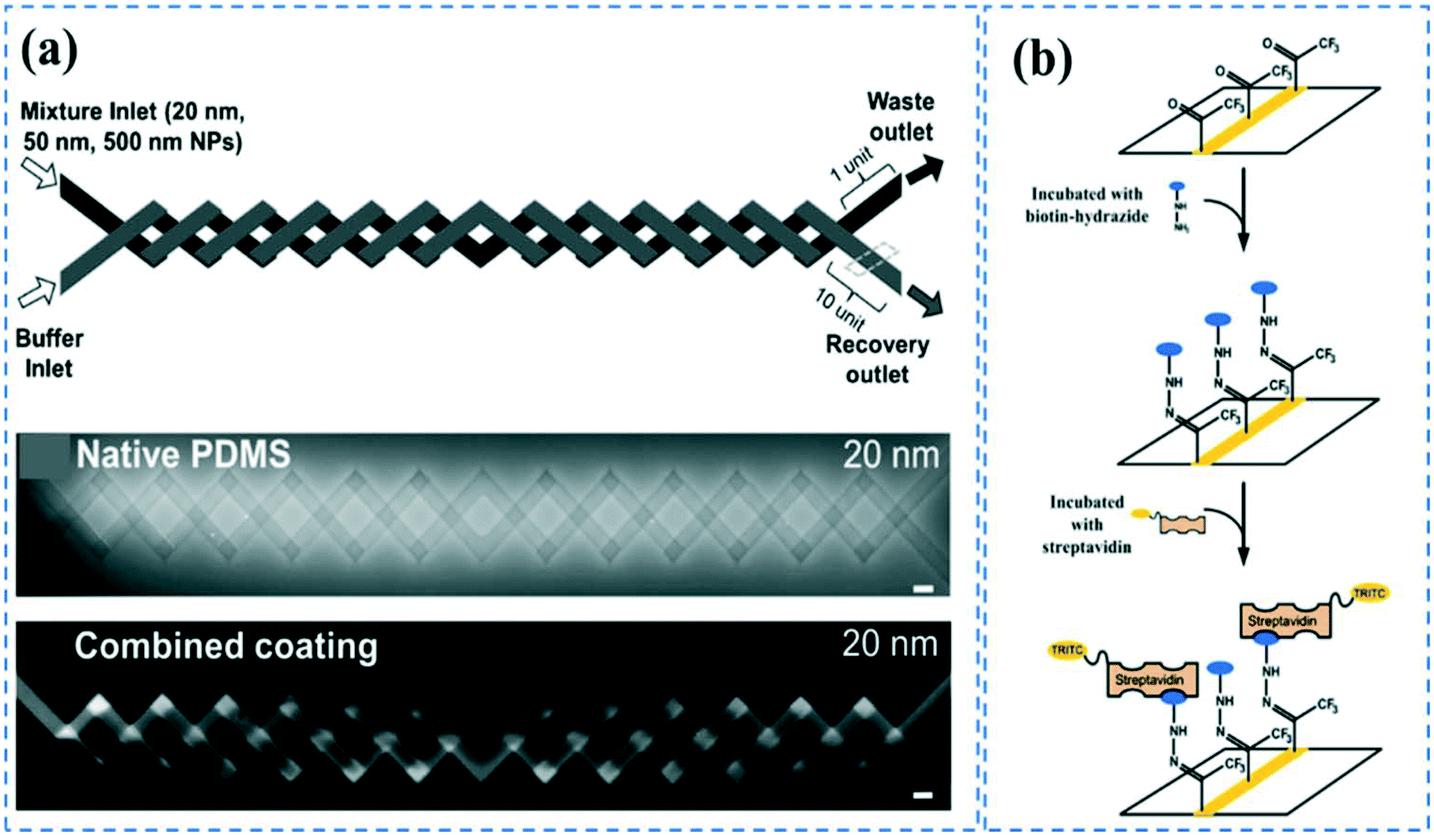 | ||
| Fig. 4 PDMS surface modification; (a) Microchannels coated with titania and SDS could effectively prevent bulk absorption of 20 nm carboxylate-modified polystyrene nanoparticles. Reprinted from ref. 22 with permission from Springer. (b) CVD polymerization of functionalized poly(p-xylylenes) could contribute to the covalent immobilization of biotin-hydrazide. Reprinted from ref. 155 with permission from ACS publications. | ||
In a different approach, Sasaki et al. employed CVD polymerization of p-xylylene derivatives (parylenes) to generate a protective layer on PDMS microchannels to prevent the adsorption of biomolecules.21 After bonding the device, they vaporized the parylenes at different temperatures so that it can diffuse inside a straight channel through the inlets. The uniformity of the coating layer depends on the length and geometry of the channels. In a different study, it was shown that CVD polymerization of functionalized poly-(p-xylylenes) through sublimation of functionalized (2.2) paracyclophanes, followed by pyrolysis and polymerization steps can contribute to the covalent immobilization of the biomolecules.155 The amine functional parylene can covalently bind to the activated carboxylic groups of proteins or pentafluorophenyl esters (as in biotin-pentafluorophenyl ester), while the keto functional parylene can react with hydrazines or hydrazides groups (such as in biotin-hydrazide) (Fig. 4b). The primary disadvantage of these methods is the advanced CVD equipment required for the coating, which makes the process difficult for commercialization.
Alzahid et al. used oxygen plasma treatment without any further modification to covalently bind mineral particles to PDMS microchannels. They immobilized quartz, kaolinite, and calcite from their water suspensions through reactions between the OH groups of the mineral particles and the silanol groups of PDMS.77 Their device could potentially be employed for the visualization of rock-fluid interactions in applications such as hydrocarbon recovery and CO2 sequestration. Entrapment of functional beads in PDMS microfluidic channels has also been utilized to functionalize channels. Han et al. used polystyrene microbeads functionalized with poly(allylamine hydrochloride) and PAA.72 The carboxylic groups of PAA provide a pathway for covalent antibody attachment via EDC–NHS. These beads were then immobilized onto a PDMS substrate containing an array of microholes – each capable of housing one bead, via gravity. Wang et al. used vancomycin-coated magnetic beads – capable of readily binding to bacteria, which were prepared via mixing of commercially available carboxylic modified magnetic beads with vancomycin and incubating with EDC for 18 h.70 Ethanolamine was also used to block the non-specific binding sites on the beads. The modified magnetic beads were then mixed with bacterial samples within the chip and immobilized in the microchambers via the use of an external magnet under the chip.
Another common technique is dopamine coating, a mussel-inspired surface modification for PDMS microchannels. Under alkaline conditions, it is spontaneously polymerized and self-organized on PDMS substrates. Poly dopamine increases the hydrophilicity of the channels and promotes tissue development.156 In a study done by Chuah et al., PDMS channels were first incubated in dopamine prepared in 10 mM Tris-HCl (pH 8.5) and then enclosed via plasma treatment.156 They coated collagen on dopamine treated channels and showed improved cell adhesion and proliferation for the long-term culture of bone marrow stromal cell. In another study, Priest et al. directly cultured different cells including human fibrosarcoma, mouse preosteoblast, and mouse fibroblast on poly dopamine coated channels and showed its efficiency.104 Vu et al. also incorporated dopamine in an amino-terminated PAM graft coating. They used ammonium persulfate for rapid oxidation of dopamine which is required to obtain thin and uniform dopamine layers.26Table 1 summarizes different functionalization methods used for PDMS microfluidics along with the advantages and disadvantages of each strategy.
| Chemical modification | Terminal groups | Applications | Crosslinker | Advantages and disadvantages | Schematica |
|---|---|---|---|---|---|
| a All included schematics use antibodies to depict the immobilization chemistry as an example. All included strategies can be implemented in the immobilization of other biomolecules as well (i.e. nucleotides, proteins, peptides etc.). | |||||
| No modification55,61,63,69 | NA | • Hydrophobic adsorption on naturally hydrophobic polymers such as PDMS, PMMA, COC, PET, and polyester• Gradual release of absorbed biomolecules for drug release systems69 | NA | ✓ Simplicity✓ Time efficiency ✗ High chance of biomolecule dissociation, dehydration, and denaturation71,107 ✗ High chance of air bubble formation in channels ✗ Swelling of the substrate due to the absorption of hydrophobic solvents ✗ Altering local concentration of biofluids and declining the sensitivity of biosensors due to the non-specific adsorption |
|
| Oxygen/air plasma2,15,35UVO17,62 Piranha etching18,85,86 H2O2/HCl27 Corona discharge17,69,87 |
Hydroxyl | • Negatively charging surfaces for electrostatic adsorption• Forming hydrogen bonds for immobilization • Rendering the surface hydrophilic • Providing OH groups for further chemical modification |
NA | ✓ Addressing the problems associated with hydrophobic surfaces✓ Improving the adsorption of ECMs35 ✓ Simplicity ✓ Time efficiency ✗ High chance of dissociation of the adsorbed biomolecules ✗ Hydrophobic recovery specially after plasma treatment ✗ Need for advanced |
|
| Allylamine plasma110,119Ammonia plasma28,125 Nitrogen plasma APTES16,19 APTMS54,68 Amine functional parylenes84,155 PAH39 PEI29 Am-PAM26 |
Amine | • Rendering surfaces hydrophilic• Positively charging surfaces for electrostatic adsorption • Providing functional groups for covalent immobilization • Enhancing biocompatibility and cell adhesion |
• DCC• DIC170 • EDC • EDC + NHS16 • EDC + sulfo-NHS • Glutaraldehyde • Thiophosgene27 |
✓ Fast process for plasma treatment methods✓ Producing stable functional groups ✓ Am-PAM can render surfaces non-fouling ✗ Instability of the functional groups after plasma treatment ✗ Chemical processes are time consuming ✗ Possibility of protein aggregation by carbodiimide chemistry ✗ Toxicity of glutaraldehyde |
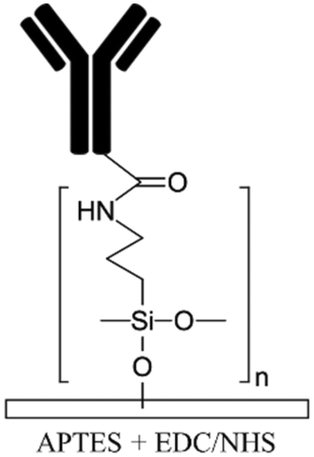
|
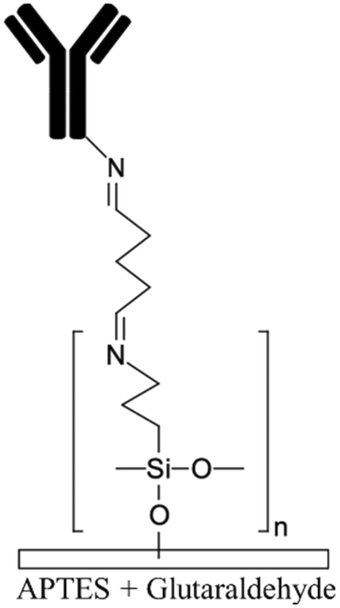
|
|||||
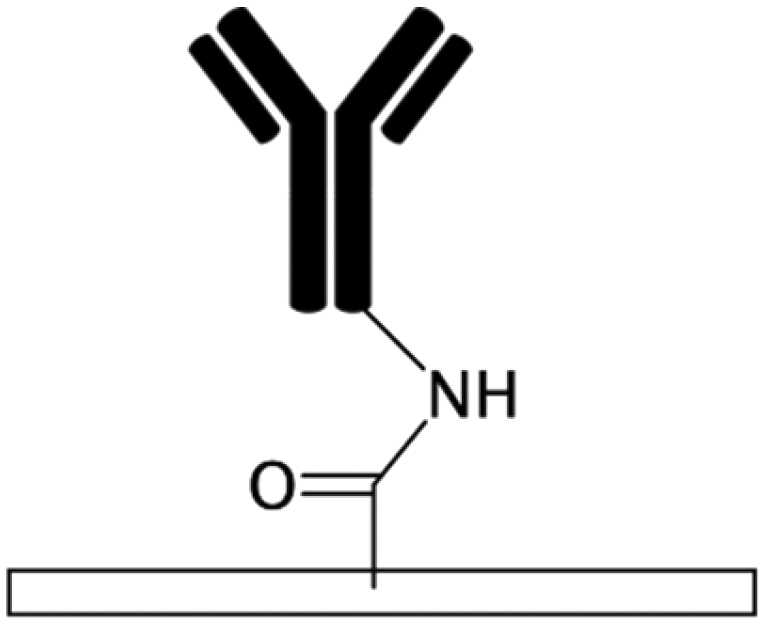
| PAA72,122CO2 plasma treatment58 Alginate109,150 Acrylic acid158 |
Carboxylic | • Rendering surfaces hydrophilic• Negatively charging surfaces in PAA • Providing functional groups for covalent immobilization • Alginate enhances cell adhesion |
• DCC• DIC170 • EDC • EDC + NHS16 • EDC + sulfo-NHS |
✓ Producing stable functional groups✓ Capability of surface patterning by use of mask in UV grafting in PAA and acrylic acid ✓ The crosslinkers do not need to be mixed by the biomolecules (preventing aggregation) ✗ Complexity of the process |
|
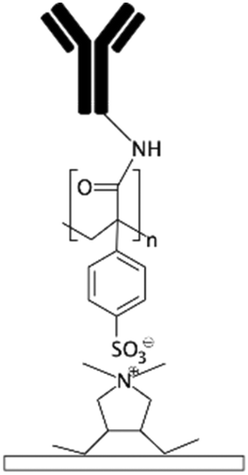
| PDADMAC/PAA multilayers133 | Carboxylic if PAA is the final layer | • Rendering surfaces hydrophilicNegatively/positively charging surfaces depending on the final layer | • DCC• DIC170 • EDC • EDC + NHS16 • EDC + sulfo-NHS |
✓ The multilayers could be used for cell culture without further modification✗ Complexity of the process |
|
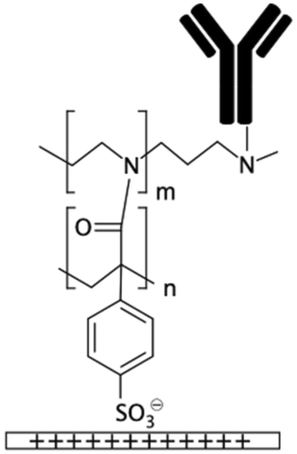
| PEI/PAA multilayers29PAH/PAA multilayers39 | Carboxylic or amine depending on the final layer | • Rendering surfaces hydrophilic and anti-fouling• Negatively/positively charging surfaces depending on the final layer • Providing functional groups for covalent immobilization |
• DCC• DIC170 • EDC • EDC + NHS16 • EDC + sulfo-NHS • Glutaraldehyde • Thiophosgene27 |
✓ They layers could be bonded together either electrostatically or covalently via carbodiimide chemistry✓ Covalent binding of biomolecules ✗ Complexity of the process |
|
| 4-Formyl para cyclophane66Keto functional parylene155 Other parylenes21,155 |
Aldehyde | Providing functional groups for covalent immobilization | Binds to hydrazine/hydrazide groups without crosslinker | ✓ Producing stable and uniform coating✗ Need for advanced CVD equipment ✗ The uniformity depends on the channel geometry |
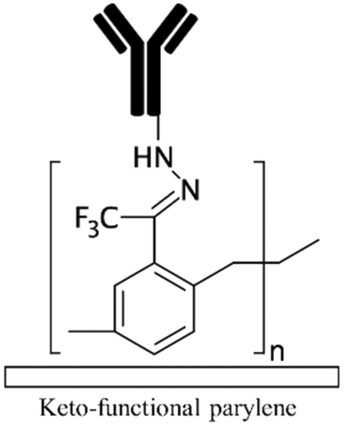
|
| Ketone | |||||
| NA | Preventing adsorption of biomolecules | NA | |||
| CPTES54 | Nitrile | • Rendering the surface hydrophilic• Enhancing cell adhesion | NA | ✓ Low hydrophobic recovery✗ Time consuming |

|
| GLYMO171 | Epoxy | Providing functional groups for covalent immobilization | Binds to amine groups without crosslinker | ✓ Producing stable and uniform coating✗ Complexity and time consuming |
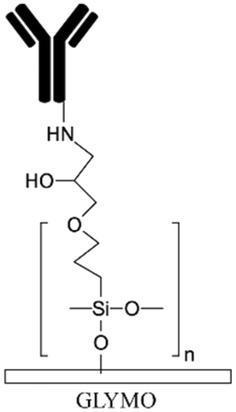
|
| PEG-silane20,27PEG-DA71,88,127,148,150 | PEG | • Rendering the surface hydrophilic• Achieving antifouling | NA | ✓ Robust blocking agent✓ Possibility of surface patterning via use of mask for PEG-DA ✗ It is challenging to immobilize biomolecules on the coating in order to create a biointerface |
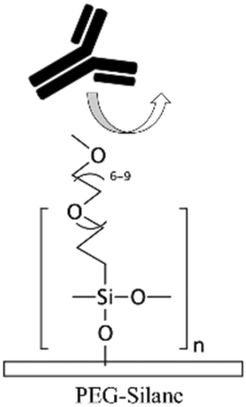
|
| Biotin–PEG:mPEG71,127 | Biotin | • PEG-based modification renders the surface hydrophilic and antifouling• Binding streptavidin-conjugated biomolecules | NA | ✓ PEG-based modification provides robust blocking agent✓ Capability for robust immobilization of biomolecules ✗ Streptavidin conjugation is required for biomolecule immobilization |
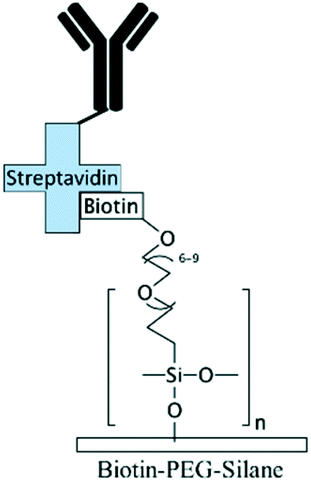
|
| NHS-PEG-acrylate29,130 | NHS | • Achieving antifouling and hydrophilicity• Crosslinking to biomolecules | NA | ✓ Robust blocking agent✓ Capability for covalent immobilization of biomolecules ✗ Usually, should be accompanied with other chemicals such as PEG-DA to obtain good antifouling properties |
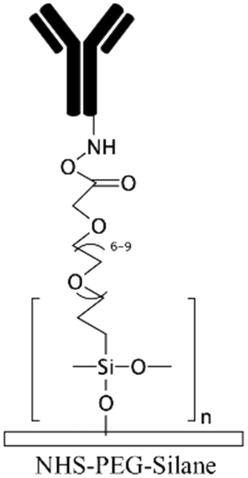
|
| Acrylate-PEG–protein G29 | Protein G | • Achieving antifouling and hydrophilicity• Crosslinking to biomolecules | NA | ✓ Robust blocking agent✓ Capability for immobilization of antibodies with preferred orientation ✗ Complexity of the process |
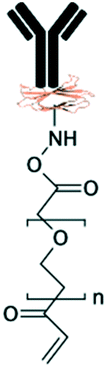
|
| PNIPAAm71,127,131,132 | NA | • Achieving “on–off” switchable hydrophilicity and anti-fouling properties in response to temperature• Collecting and releasing beads, cells, and biomolecules • Switchable microvalves |
NA | ✓ Possibility of surface patterning via embedding micro-heaters or use of mask in UV grafting✗ Needs to be accompanied by a heating system ✗ The beads should be coated with PNIPAAm |
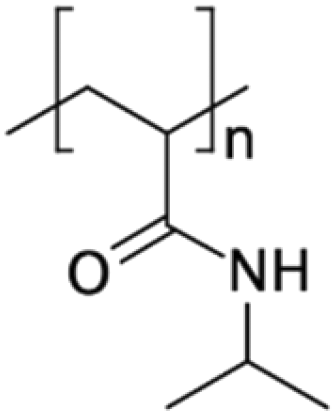
|
| P[NIPAAm-co-AAc]71 | NA | Affinity separations and diagnostic assays | NA | ✓ Tunable wettability in response to pH and temperature✓ Possibility of surface patterning via embedding micro-heaters or use of mask in UV grafting ✗ Needs to be accompanied by a heating system or specific buffer for changing the pH |
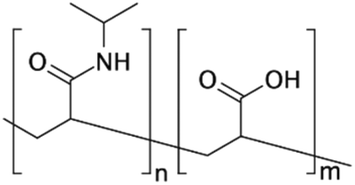
|
| PEMA125POMA125 PVP160 |
NA | Rendering the surface hydrophilic and anti-fouling | NA | ✓ Stable layer by covalent binding the layers to the substrate✗ Pre-amine treatment is required which adds complexity |
|
| HPG110 | Hydroxyl (NHS and carboxylic after mod.) | • Negatively charging surfaces• Achieving antifouling and hydrophilicity • Covalent immobilization of biomolecules |
Succinic anhydride followed by hydrolyzation in water to create both NHS esters and carboxylic groups | ✓ Capability to bind to both amin and carboxylic groups of biomolecules (carbodiimide chemistry is required for immobilization through carboxylic groups)✗ Complexity of the process |
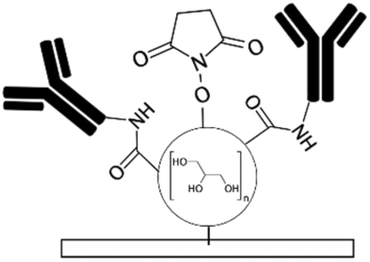
|
| PVA79,159 | NA | • Rendering the surface hydrophilic• Generation of oil-in-water droplets | NA | ✓ Quick and low-cost✓ Possibility of patterning of hydrophilic regions ✗ Lack of functional groups for biomolecules immobilization |
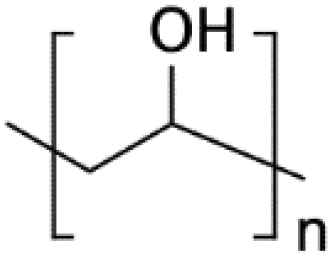
|
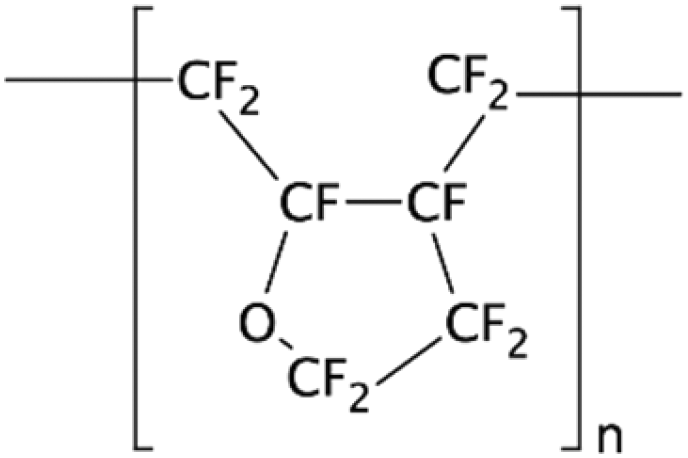
| CYTOP134 | NA | • Creating a chemical inert layer• Organic synthesis applications • Coating for thermal fusion bonding |
NA | ✓ Resistant to non-polar organic solutions✓ No deformation or damage seen after a long run ✗ Pre-amine treatment with APTES is required which adds complexity |
|
| MPTMS122 | Sulfhydryl (thiol) | • Covalent immobilization of biomolecules• Immobilization of gold particles • Increase the cell adhesion |
GMBS | ✓ Durable immobilization technique✗ Time consuming process |
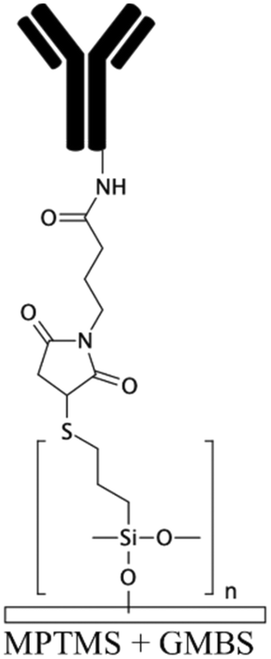
|
| NCO-sP(EO-stat-PO)28,147 | Isocyanate | • Rendering the surface hydrophilic• Covalent immobilization of biomolecules | NA | ✓ No need for a crosslinker for covalent immobilization✓ Highly stable coating ✓ Could be incorporated with RGD peptides for cell attachment ✗ Complexity of the process ✗ Isocyanate groups are functional for a short time |
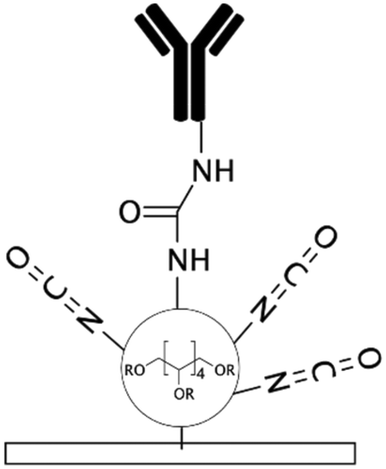
|
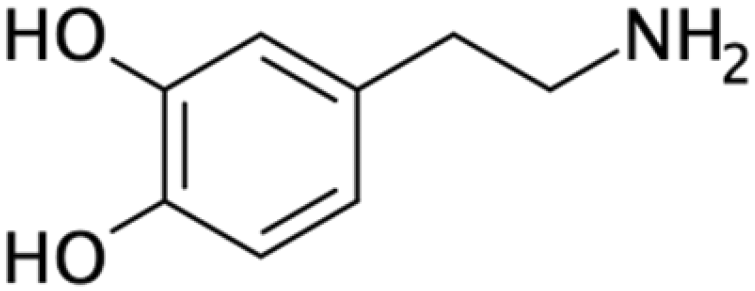
| Dopamine26,104,172 | NA | • Rendering the surface hydrophilic• Promoting cell adhesion and proliferation | NA | ✓ Can be spontaneously polymerized and self-organized✓ Can serve as a bridge for further surface modification ✓ Strong adhesion with many substrates ✗ Time consuming process |
|
| Au, SiO2, titania, zirconia, and vanadia particles22,151,152,164Glass coating153,154 | NA | • Rendering the surface hydrophilic and antifouling• Creating a protective and chemically inert layer | NA | ✓ Uniformity using sol–gel method✓ Capable of further functionalization through silane chemistry ✓ Ability for in situ coating ✗ Complexity of the process |
|
Conclusion and future trends
Novel low-cost methods for fabrication of PDMS-microfluidics and introducing functional interfaces in the channels have brought about a tremendous breakthrough in the implementation of these devices for biomedical applications and bioanalytical systems via the miniaturization and simplification of current therapeutic and diagnostic devices.Soft lithography of PDMS using silicon master moulds followed by oxygen plasma bonding is the most promising approach reported in the literature for the fabrication of PDMS microfluidics. The master moulds are conventionally fabricated via photolithography techniques. However, the high cost of the facilities and time-consuming fabrication needed for this process hinders the opportunities for scaling up and commercialization of PDMS microfluidics. As discussed in this review paper, using portable LED light instead of high-voltage mercury lamps as well as nail polish MA instead of SU-8 could reduce the cost of fabrication. Moreover, mask-free procedures such as direct laser writing, electron-beam, and focused-ion beam, two photon polymerization, and digital micro mirrors could eliminate the cost of mask fabrication. Other strategies such as lamination of ADEX dry films, 3D printing techniques with further fluorosilanization step, and laser ablation or micromachining of PMMA substrates could eliminate the need for clean-room environment. We also explained the possibility of directly forming microchannels in PDMS slabs using low-energy electron beam or CO2 laser ablation which omits the master mould fabrication steps. As alternatives to plasma treatment, more cost-effective methods such as uncured or partially cured PDMS, adhesive films, and flame-treatment could be utilized for PDMS bonding and enclosing the device. Moreover, irradiation of an electron beam or 60Co γ-rays allows for simultaneous bonding of a stack of PDMS layers already aligned on top of each other, which is useful for batch production of 3D microfluidics. These fabrication substitutes for photolithography can significantly reduce the time and cost required for mass production of PDMS microfluidics and help with commercialization of these devices. Nevertheless, due to some limitations associated with the alternative strategies, such as lack of good resolution and precision for the produced channels and intrinsic low-speed of the mask-free approaches, implementation of these strategies for scaling up is still quite challenging and there is room for further improvements in this respect. Interestingly, in a protocol proposed by Bhattacharjee et al. the entire PDMS microchannels were 3D printed using a desktop stereolithography printer.157 They used PDMS-methacrylate macromers as the resin for 3D printing and succeeded in obtaining transparent PDMS channels suitable for cell culture after extraction of unreacted compounds using different solvents. Gonzalez et al. also 3D printed a PDMS-like microchannels via digital light processing (DLP) printing using a formulation based on acrylate PDMS.158 Despite all these limitations, PDMS still remains as the most attractive choice for prototyping of microfluidic devices for research and development.
Regarding the functionalization of PDMS-based microchannels, graft polymerization is proven to be the best method to achieve long-term hydrophilicity and antifouling properties on PDMS microfluidics. In Gonzalez et al.'s work, since there was still unreacted acrylic double-bonds after the 3D printing step, they could induce carboxylic groups onto the surface using acrylic acid via a “grafting to” polymerization method under UV light.158 They also showed the possibility of patterning hydrophobic/hydrophilic areas inside microfluidic channels using the grafting solution in combination with silicon oil.
Amine treatment of these microfluidic systems using either APTES silanization or grafting amine-terminated polymers is one the most effective strategies for the covalent immobilization of biomolecules. Primary amine groups are able to covalently bind to several functional entities such as epoxy, aldehyde, and carboxylic groups (via carbodiimide chemistry).
One suggestion for the generation of functional groups in microfluidic channels is to use CO2 plasma treatment as a simple and fast surface modification approach.58 The CO2 plasma treated glass substrate can irreversibly bind to a PDMS platform to form the microchannel. Moreover, the carboxylic groups can remain functional inside the channel for further activation via EDC–NHS chemistry in order to covalently immobilized biomolecules. This eliminates the need for surface functionalization through wet chemistry methods (such as APTES/APTMS treatment) prior to biomolecule immobilization. It is also possible to covalently pattern PDMS channels with biomolecules using microcontact printing. This strategy can potentially be tuned to create robust bio-interfaces in polymeric microfluidic channels in a simple manner.
Lin et al. intriguingly suggested casting PDMS on a dried shark skin to achieve superhydrophobic properties.159 In their design, after casting and producing a negative PDMS mould, the mould was platinum coated and used for another run of PDMS casting to finally replicate the protrusions of the shark skin on the PDMS. The final superhydrophobic PDMS membrane was able to effectively resist bacteria attachment. They also produced a superhydrophilic PDMS layer by coating a PDMS membrane with PVA and 2-methacryloyloxyethyl phosphorylcholine (MPC). The combination of two super- hydrophobic/hydrophilic layers was used for wound healing applications to simultaneously suppress the bacteria attachment and absorb the wound exudates, while MPC could enhance the wound repairs. Although they did not use the system for microfluidics, but this technique could be integrated with microfluidic applications where superhydrophobicity for bacteria repellency is required. In another study, poly(N-vinylpyrrolidone) (PVP) was coated on an amine treated PDMS substrate to drop the contact angle of PDMS from 116° to only 14°.160 PVP was initially functionalized with catechol to be able to be self-adhered onto PDMS through the reaction with the amine groups of PDMS. Implementation of this strategy into microfluidics could be advantageous as it can reduce the amount of hydrophobic adsorption of proteins.
In order to increase the specificity of the created bio-interfaces, lubricant-infused surface technology can be a promising candidate to effectively repel all sort of non-specific adsorption, thereby enhancing the sensitivity and efficacy of microfluidic devices.113,161,162 Our group has shown that the combination of lubricant-infused surfaces with micropatterning strategies in microfluidics with glass substrates can result in the creation of localized bio-interfaces with high sensitivity in capturing secondary antibodies or cells.163 Integration of this protocol with PDMS microfluidics can bring about flexible biosensors with significantly high sensitivity due to the presence of lubricant.
An increased biosensing sensitivity on PDMS microfluidic platforms can also be achieved via coating the channels with gold nanoparticles (AuNPs). AuNPs could simply be functionalized with capture antibodies or other biomolecules through thiol reaction using either crosslinkers such as MPTMS or thiol-conjugated biomolecules. Therefore, they provide a substantially large surface area for detection of target biomolecules. Zhu et al. demonstrated the immobilization of AuNPs on amine treated PDMS, which were subsequently conjugated with thiol-terminal Staphylococcus aureus (S. aureus) aptamers in order to detect S. aureus via surface-enhanced Raman scattering (SERS).164 Applying this surface functionalization technique to PDMS microfluidics can potentially enhance the limit of detection.
Recently, Banerjee et al. presented a self-healable PDMS hydrogel with antifouling capability through use of zwitterionic PDMS polymersomes and amine functionalized PDMS polymersomes.165 They entrapped curcumin drug in the hydrogel for therapeutic applications for eye diseases. The self-healing and antifouling properties of their PDMS-based hydrogel are highly beneficial for long-lasting PDMS microchannels if this technology could be incorporated in microfluidics. Moreover, Lamers et al. succeeded in achieving different mechanical properties out of PDMS (ranging from a viscous PDMS to a brittle PDMS) via functionalizing the PDMS backbone with hydz, benzene-1,3,5-carboxamide (BTA), and ureidopyrimidinone (UPy).166 They also showed the recyclability of the modified PDMSs via solvation, compression moulding, or thermal recycling. This capability, if embedded in microfluidics, could raise a new generation of environmentally friendly recyclable PDMS microchannels.
Furthermore, patterning of the biomolecules in PDMS microfluidic channels allows for multiplex detection of different biomarkers for high throughput applications. Incorporating simple methods such as microcontact printing via use of PDMS stamps with microfluidic fabrication58,163 could significantly simplify the method compared to other patterning techniques such as non-contact printing with piezoelectric inkjet printing technology,167 photolithographic patterning using a proper photomask,168 and multifaceted tree-shape designs for the generation of 2 dimensional (2D) concentration gradients.169 Benefiting from hydrodynamic resistances induced by channel geometries, an innovative, simple microfluidic design has also been demonstrated to generate 1D and 2D concentration gradients with a small footprint size,117 which could potentially be used in PDMS microfluidic applications.
In conclusion, PDMS microfluidics have represented a revolutionary breakthrough in the search for more efficacious biomedical devices and point-of-care diagnostics. The large body of literature that has explored means by which its physical and chemical properties can be modified for specific microfluidic applications has further cemented its position as a premier material of choice. That being said, there are still a number of technological barriers such as reproducibility and durability hindering its full implementation on a commercial scale. These barriers need to be overcome to address the fast-evolving role of microfluidic systems in the biomedical industry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant, Ontario Early Researcher Award Grant, and McMaster start-up funds to T. F. Didar.References
- J. Zhou, A. V. Ellis and N. H. Voelcker, Electrophoresis, 2010, 31, 2–16 CrossRef CAS PubMed.
- J. C. McDonald and G. M. Whitesides, Acc. Chem. Res., 2002, 35, 491–499 CrossRef CAS PubMed.
- K. J. Regehr, M. Domenech, J. T. Koepsel, K. C. Carver, S. J. Ellison-Zelski, W. L. Murphy, L. A. Schuler, E. T. Alarid and D. J. Beebe, Lab Chip, 2009, 9, 2132–2139 RSC.
- K. Liu, J. Xiang, Z. Ai, S. Zhang, Y. Fang, T. Chen, Q. Zhou, S. Li, S. Wang and N. Zhang, Microsyst. Technol., 2017, 23, 1937–1942 CrossRef CAS.
- N. Keller, T. M. Nargang, M. Runck, F. Kotz, A. Striegel, K. Sachsenheimer, D. Klemm, K. Länge, M. Worgull, C. Richter, D. Helmer and B. E. Rapp, Lab Chip, 2016, 16, 1561–1564 RSC.
- S. Torino, B. Corrado, M. Iodice and G. Coppola, Inventions, 2018, 3, 65 CrossRef.
- J. C. McDonald, D. C. Duffy, J. R. Anderson, D. T. Chiu, H. Wu, O. J. A. Schueller and G. M. Whitesides, Electrophoresis, 2000, 21, 27–40 CrossRef CAS PubMed.
- S. Su, G. Jing, M. Zhang, B. Liu, X. Zhu, B. Wang, M. Fu, L. Zhu, J. Cheng and Y. Guo, Sens. Actuators, B, 2019, 282, 60–68 CrossRef CAS.
- J. H. Lee, S. K. Kim, H. H. Park and T. S. Kim, J. Biomed. Mater. Res., Part A, 2015, 25, 035032 Search PubMed.
- C. Séguin, J. M. McLachlan, P. R. Norton and F. Lagugné-Labarthet, Appl. Surf. Sci., 2010, 256, 2524–2531 CrossRef.
- P. Fürjes, E. G. Holczer, E. Tóth, K. Iván, Z. Fekete, D. Bernier, F. Dortu and D. Giannone, Microsyst. Technol., 2014, 21, 581–590 CrossRef.
- J. N. Lee, C. Park and G. M. Whitesides, Anal. Chem., 2003, 75, 6544–6554 CrossRef CAS PubMed.
- A. Shakeri, S. Rahmani, S. M. Imani, M. Osborne, H. Yousefi and T. F. Didar, in Bioelectronics and Medical Devices: From Materials to Devices - Fabrication, Applications and Reliability, ed. K. Pal, H.-B. Kraatz, A. Khasnobish, S. Bag, I. Banerjee and U. Kuruganti, Woodhead Publishing, Cambridge, Massachusetts, 2019, pp. 635–699 Search PubMed.
- Z. L. Zhang, C. Crozatier, M. Le Berre and Y. Chen, Microelectron. Eng., 2005, 78–79, 556–562 CrossRef CAS.
- C. Priest, P. J. Gruner, E. J. Szili, S. A. Al-Bataineh, J. W. Bradley, J. Ralston, D. A. Steele and R. D. Short, Lab Chip, 2011, 11, 541–544 RSC.
- H. Li, M. Zhao, W. Liu, W. Chu and Y. Guo, Talanta, 2016, 147, 430–436 CrossRef CAS PubMed.
- H. J. Kim, D. Huh, G. Hamilton and D. E. Ingber, Lab Chip, 2012, 12, 2165–2174 RSC.
- K. S. Koh, J. Chin, J. Chia and C. L. Chiang, Micromachines, 2012, 3, 427–441 CrossRef.
- A. Siddique, T. Meckel, R. W. Stark and S. Narayan, Colloids Surf., B, 2017, 150, 456–464 CrossRef CAS PubMed.
- K. M. Kovach, J. R. Capadona, A. Sen Gupta and J. A. Potkay, J. Biomed. Mater. Res., Part A, 2014, 102, 4195–4205 Search PubMed.
- H. Sasaki, H. Onoe, T. Osaki, R. Kawano and S. Takeuchi, Sens. Actuators, B, 2010, 150, 478–482 CrossRef CAS.
- C. W. Li, Y. Zhu, J. Zhan, J. Ma, L. Gu, Y. Fang and C. Yi, Microchim. Acta, 2017, 184, 2227–2239 CrossRef CAS.
- T. Shay, M. D. Dickey and O. D. Velev, Lab Chip, 2017, 17, 710–716 RSC.
- P. S. Nunes, P. D. Ohlsson, O. Ordeig and J. P. Kutter, Microfluid. Nanofluid., 2010, 9, 145–161 CrossRef CAS.
- G. Perozziello, G. Simone, N. Malara, R. La Rocca, R. Tallerico, R. Catalano, F. Pardeo, P. Candeloro, G. Cuda, E. Carbone and E. Di Fabrizio, Electrophoresis, 2013, 34, 1845–1851 CrossRef CAS PubMed.
- T. T. Vu, M. Fouet, A. M. Gue and J. Sudor, Sens. Actuators, B, 2014, 196, 64–70 CrossRef CAS.
- G. Sui, J. Wang, C. C. Lee, W. Lu, S. P. Lee, J. V. Leyton, A. M. Wu and H. R. Tseng, Anal. Chem., 2006, 78, 5543–5551 CrossRef CAS PubMed.
- A. M. Greiner, P. Hoffmann, K. Bruellhoff, S. Jungbauer, J. P. Spatz, M. Moeller, R. Kemkemer and J. Groll, Macromol. Biosci., 2014, 14, 1547–1555 CrossRef CAS PubMed.
- W.-C. Sung, H.-H. Chen, H. Makamba and S.-H. Chen, Anal. Chem., 2009, 81, 7967–7973 CrossRef CAS PubMed.
- J. Xu and K. K. Gleason, Chem. Mater., 2010, 22, 1732–1738 CrossRef CAS.
- J. Christoffersson and C.-F. Mandenius, in Methods in Molecular Biology, ed. C.-F. Mandenius and J. A. Ross, Springer New York, New York, NY, 2019, pp. 227–233 Search PubMed.
- G. Kaur, M. Tomar and V. Gupta, Sens. Actuators, B, 2018, 261, 460–466 CrossRef CAS.
- Y. Li, P. Wu, Z. Luo, Y. Ren, M. Liao, L. Feng, Y. Li and L. He, J. Micromech. Microeng., 2015, 25, 055020 CrossRef.
- H. T. Nguyen, H. Thach, E. Roy, K. Huynh and C. M. T. Perrault, Micromachines, 2018, 9, 1–10 Search PubMed.
- A. Bubendorfer, X. Liu and A. V. Ellis, Smart Mater. Struct., 2007, 16, 367–371 CrossRef CAS.
- T. G. Oyama, K. Oyama and M. Taguchi, Lab Chip, 2020, 20, 2354–2363 RSC.
- M. Zhang, J. Wu, L. Wang, K. Xiao and W. Wen, Lab Chip, 2010, 10, 1199–1203 RSC.
- Z. Yan, X. Huang and C. Yang, Microfluid. Nanofluid., 2017, 21, 1–9 CrossRef CAS.
- C. H. Choi, H. Lee and D. A. Weitz, ACS Appl. Mater. Interfaces, 2018, 10, 3170–3174 CrossRef CAS PubMed.
- J. Lee, W. Choi, K. Lee and J. Yoon, J. Micromech. Microeng., 2008, 18, 125015 CrossRef.
- M. Fenech, V. Girod, V. Claveria, S. Meance, M. Abkarian and B. Charlot, Lab Chip, 2019, 19, 2096–2106 RSC.
- M. A. Smith, S. Berry, L. Parameswaran, C. Holtsberg, N. Siegel, R. Lockwood, M. P. Chrisp, D. Freeman and M. Rothschild, J. Micro/Nanolithogr., MEMS, MOEMS, 2019, 18, 043507 CAS.
- D. Lai, J. M. Labuz, J. Kim, G. D. Luker, A. Shikanov and S. Takayama, RSC Adv., 2013, 3, 19467–19473 RSC.
- S. Brittman, S. Z. Oener, K. Guo, H. Āboliņš, A. F. Koenderink and E. C. Garnett, J. Mater. Chem. C, 2017, 5, 8301–8307 RSC.
- L. Filipponi, P. Livingston, O. Kašpar, V. Tokárová and D. V. Nicolau, Biomed. Microdevices, 2016, 18, 9 CrossRef PubMed.
- R. Saive, C. R. Bukowsky and H. A. Atwater, 2017, arXiv preprint arXiv:1702.04012v1.
- H. Li, D. Kang and M. G. Blamire, Nanotechnology, 2003, 14, 220–223 CrossRef CAS.
- M. Ziauddin, A. I. Mourad and S. A. Khashan, in Advances in Science and Engineering Technology International Conferences (ASET), IEEE, 2018, pp. 1–3 Search PubMed.
- Y. Lin, C. Gao, D. Gritsenko, R. Zhou and J. Xu, Microfluid. Nanofluid., 2018, 22, 97 CrossRef.
- Y. Lin and J. Xu, Adv. Opt. Mater., 2018, 6, 1701359 CrossRef.
- I. Weiss and D. M. Marom, in 2015 International Conference on Optical MEMS and Nanophotonics (OMN), 2015, pp. 1–2 Search PubMed.
- A. Rammohan, P. K. Dwivedi, R. Martinez-duarte, H. Katepalli, M. J. Madou and A. Sharma, Sens. Actuators, B, 2011, 153, 125–134 CrossRef CAS.
- P. Mukherjee, F. Nebuloni, H. Gao, J. Zhou and I. Papautsky, Micromachines, 2019, 10, 192 CrossRef PubMed.
- C.-C. Wu, C.-Y. Yuan and S.-J. Ding, Surf. Coat. Technol., 2011, 205, 3182–3189 CrossRef CAS.
- J. W. Song, W. Gu, N. Futai, K. A. Warner, J. E. Nor and S. Takayama, Anal. Chem., 2005, 77, 3993–3999 CrossRef CAS PubMed.
- T. F. Didar, A. Dolatabadi and R. Wüthrich, Mater. Lett., 2009, 63, 51–53 CrossRef CAS.
- M. Villegas, Z. Cetinic, A. Shakeri and T. F. Didar, Anal. Chim. Acta, 2018, 1000, 248–255 CrossRef CAS PubMed.
- A. Shakeri, S. M. Imani, E. Chen, H. Yousefi, R. Shabbir and T. F. Didar, Lab Chip, 2019, 19, 3104–3115 RSC.
- K. Kang, S. Oh, H. Yi, S. Han and Y. Hwang, Biomicrofluidics, 2018, 12, 14105 CrossRef PubMed.
- D. N. H. Kim, K. T. Kim, C. Kim, M. A. Teitell and T. A. Zangle, Microfluid. Nanofluid., 2017, 22, 2 CrossRef PubMed.
- G. A. Giridharan, M.-D. D. Nguyen, R. Estrada, V. Parichehreh, T. Hamid, M. A. Ismahil, S. D. Prabhu and P. Sethu, Anal. Chem., 2010, 82, 7581–7587 CrossRef CAS PubMed.
- Y. Berdichevsky, J. Khandurina, A. Guttman and Y. H. Lo, Sens. Actuators, B, 2004, 97, 402–408 CrossRef CAS.
- N. C. A. Van Engeland, A. M. A. O. Pollet, J. M. J. Den Toonder, C. V. C. Bouten, O. M. J. A. Stassen and C. M. Sahlgren, Lab Chip, 2018, 18, 1607–1620 RSC.
- S. Jeong, S. Kim, J. Buonocore, J. Park, C. J. Welsh, J. Li and A. Han, IEEE Trans. Biomed. Eng., 2018, 65, 431–439 Search PubMed.
- M. Li, S. Li, J. Wu, W. Wen, W. Li and G. Alici, Microfluid. Nanofluid., 2012, 12, 751–760 CrossRef CAS.
- T. G. Oyama, B. Jeremiah, D. Barba, Y. Hosaka and M. Taguchi, Appl. Phys. Lett., 2018, 112, 213704 CrossRef.
- P. Yang and W. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 3759–3770 CrossRef CAS PubMed.
- A. Jain, A. D. van der Meer, A. L. Papa, R. Barrile, A. Lai, B. L. Schlechter, M. A. Otieno, C. S. Louden, G. A. Hamilton, A. D. Michelson, A. L. Frelinger and D. E. Ingber, Biomed. Microdevices, 2016, 18, 73 CrossRef PubMed.
- M. Adiraj Iyer and D. T. Eddington, Lab Chip, 2019, 19, 574–579 RSC.
- C. H. Wang, C. J. Chang, J. J. Wu and G. Bin Lee, Nanomedicine, 2014, 10, 809–818 CrossRef CAS PubMed.
- M. Ebara, J. M. Hoffman, P. S. Stayton and A. S. Hoffman, Radiat. Phys. Chem., 2007, 76, 1409–1413 CrossRef CAS.
- S. W. Han, E. Jang and W. G. Koh, Sens. Actuators, B, 2015, 209, 242–251 CrossRef CAS.
- C. Bergemann, A. Quade, F. Kunz, S. Ofe, E. D. Klinkenberg, M. Laue, K. Schröder, V. Weissmann, H. Hansmann, K. D. Weltmann and B. Nebe, Plasma Processes Polym., 2012, 9, 261–272 CrossRef CAS.
- B. Mészáros, G. Járvás, L. Hajba, M. Szigeti, A. Dallos and A. Guttman, Sens. Actuators, B, 2018, 258, 1184–1190 CrossRef.
- C. F. Chen and K. Wharton, RSC Adv., 2017, 7, 1286–1289 RSC.
- C. fu Chen, J. Adhes. Sci. Technol., 2018, 32, 1239–1252 CrossRef.
- Y. A. Alzahid, P. Mostaghimi, A. Gerami, A. Singh, K. Privat, T. Amirian and R. T. Armstrong, Sci. Rep., 2018, 8, 15518 CrossRef PubMed.
- M. A. Eddings, M. A. Johnson and B. K. Gale, J. Micromech. Microeng., 2008, 18, 067001 CrossRef.
- T. Trantidou, Y. Elani, E. Parsons and O. Ces, Microsyst. Nanoeng., 2017, 3, 16091 CrossRef CAS PubMed.
- D. T. Eddington, J. P. Puccinelli and D. J. Beebe, Sens. Actuators, B, 2006, 114, 170–172 CrossRef CAS.
- J. Bacharouche, H. Haidara, P. Kunemann, M. F. Vallat and V. Roucoules, Sens. Actuators, A, 2013, 197, 25–29 CrossRef CAS.
- C. Yang and Y. J. Yuan, Appl. Surf. Sci., 2016, 364, 815–821 CrossRef CAS.
- F. Darain, P. Yager, K. L. Gan and S. C. Tjin, Biosens. Bioelectron., 2009, 24, 1744–1750 CrossRef CAS PubMed.
- H. Y. Chen, A. A. McClelland, Z. Chen and J. Lahann, Anal. Chem., 2008, 80, 4119–4124 CrossRef CAS PubMed.
- H. Xu, A. Gomez-Casado, Z. Liu, D. N. Reinhoudt, R. G. H. Lammertink and J. Huskens, Langmuir, 2009, 25, 13972–13977 CrossRef CAS PubMed.
- D. Maji, S. K. Lahiri and S. Das, Surf. Interface Anal., 2012, 44, 62–69 CrossRef CAS.
- K. Haubert, T. Drier and D. Beebe, Lab Chip, 2006, 6, 1548–1549 RSC.
- C. L. Lewis, Y. Lin, C. Yang, A. K. Manocchi, K. P. Yuet, P. S. Doyle and H. Yi, Langmuir, 2010, 26, 13436–13441 CrossRef CAS PubMed.
- L. Engel, C. Liu, N. M. Hemed, Y. Khan, A. C. Arias, Y. Shacham-Diamand, S. Krylov and L. Lin, J. Micromech. Microeng., 2018, 28, 105005 CrossRef.
- L. G. Villa-Diaz, Y. S. Torisawa, T. Uchida, J. Ding, N. C. Nogueira-De-Souza, K. S. O'Shea, S. Takayama and G. D. Smith, Lab Chip, 2009, 9, 1749–1755 RSC.
- M. Chu, T. T. Nguyen, E. K. Lee, J. L. Morival and M. Khine, Lab Chip, 2017, 17, 267–273 RSC.
- A. Lai, N. Altemose, J. A. White and A. M. Streets, J. Micromech. Microeng., 2019, 107001 CrossRef CAS.
- C. F. Carlborg, T. Haraldsson, M. Cornaglia, G. Stemme and W. van der Wijngaart, J. Microelectromech. Syst., 2010, 19, 1050–1057 CAS.
- S. Satyanarayana, R. N. Karnik and A. Majumdar, J. Microelectromech. Syst., 2005, 14, 392–399 Search PubMed.
- H. Y. Tan, W. K. Loke and N. T. Nguyen, Sens. Actuators, B, 2010, 151, 133–139 CrossRef CAS.
- S. H. Tamrin, A. S. Nezhad and A. Sen, Sci. Rep., 2021, 11, 4821 CrossRef PubMed.
- S. Kim, J. Micromech. Microeng., 2019, 29, ab27d3 Search PubMed.
- J. Kim, R. Surapaneni and B. K. Gale, Lab Chip, 2009, 9, 1290–1293 RSC.
- C. S. Thompson and A. R. Abate, Lab Chip, 2013, 13, 632–635 RSC.
- R. Ghaemi, M. Dabaghi, R. Attalla, A. Shahid, H. H. Hsu and P. R. Selvaganapathy, J. Micromech. Microeng., 2018, 28, aabd29 CrossRef.
- S. Farris, S. Pozzoli, P. Biagioni, L. Duó, S. Mancinelli and L. Piergiovanni, Polymer, 2010, 51, 3591–3605 CrossRef CAS.
- C. S. Effenhauser, G. J. M. Bruin, A. Paulus and M. Ehrat, Anal. Chem., 1997, 69, 3451–3457 CrossRef CAS PubMed.
- J. H. L. Beal, A. Bubendorfer, T. Kemmitt, I. Hoek and W. Mike Arnold, Biomicrofluidics, 2012, 6, 036503 CrossRef PubMed.
- S. H. Ku, J. S. Lee and C. B. Park, Langmuir, 2010, 26, 15104–15108 CrossRef CAS PubMed.
- K. J. Land, M. B. Mbanjwa, K. Govindasamy and J. G. Korvink, Biomicrofluidics, 2011, 5, 036502 CrossRef PubMed.
- M. Rafat, D. R. Raad, A. C. Rowat and D. T. Auguste, Lab Chip, 2009, 9, 3016–3019 RSC.
- in Characterization of Biomaterials, ed. M. Jaffe, W. Hammond, P. Tolias and T. Arinzeh, Woodhead Publishing, 2013, pp. 182–223 Search PubMed.
- P. Tengvall, in Comprehensive Biomaterials, ed. P. Ducheyne, K. E. Healy, D. W. Hutmacher, D. W. Grainger and C. J. Kirkpatrick, Elsevier, Oxford, 2011, pp. 63–73 Search PubMed.
- C. Cha, E. Antoniadou, M. Lee, J. H. Jeong, W. W. Ahmed, T. A. Saif, S. A. Boppart and H. Kong, Angew. Chem., Int. Ed., 2013, 52, 6949–6952 CrossRef CAS PubMed.
- P. Y. Yeh, N. A. A. Rossi, J. N. Kizhakkedathu and M. Chiao, Microfluid. Nanofluid., 2010, 9, 199–209 CrossRef CAS.
- S. Ilyas, N. Joseph, A. Szymczyk, A. Volodin, K. Nijmeijer, W. M. De Vos and I. F. J. Vankelecom, J. Membr. Sci., 2016, 514, 322–331 CrossRef CAS.
- T. F. Didar, A. M. Foudeh and M. Tabrizian, Anal. Chem., 2012, 84, 1012–1018 CrossRef CAS PubMed.
- M. Badv, S. M. Imani, J. I. Weitz and T. F. Didar, ACS Nano, 2018, 12, 10890–10902 CrossRef PubMed.
- T. F. Didar and M. Tabrizian, Lab Chip, 2012, 12, 4363–4371 RSC.
- T. F. Didar, K. Bowey, G. Almazan and M. Tabrizian, Adv. Healthcare Mater., 2014, 3, 253–260 CrossRef CAS PubMed.
- A. Shakeri, D. Yip, M. Badv, S. M. Imani, M. Sanjari and T. F. Didar, Materials, 2018, 11, 1003 CrossRef PubMed.
- A. Shakeri, N. Sun, M. Badv and T. F. Didar, Biomicrofluidics, 2017, 11, 044111 CrossRef PubMed.
- A. Shakeri, N. A. Jarad, A. Leung, L. Soleymani and T. F. Didar, Adv. Mater. Interfaces, 2019, 1900940, 1900940 CrossRef.
- M. Crespin, N. Moreau, B. Masereel, O. Feron, B. Gallez, T. Vander Borght, C. Michiels and S. Lucas, J. Mater. Sci.: Mater. Med., 2011, 22, 671–682 CrossRef CAS PubMed.
- B. Asadishad, S. Ghoshal and N. Tufenkji, Environ. Sci. Technol., 2011, 45, 8345–8351 CrossRef CAS PubMed.
- A. Sarkar and T. Daniels-Race, Nanomaterials, 2013, 3, 272–288 CrossRef CAS PubMed.
- C. Launiere, M. Gaskill, G. Czaplewski, J. H. Myung, S. Hong and D. T. Eddington, Anal. Chem., 2012, 84, 4022–4028 CrossRef CAS PubMed.
- D. Xiao, T. Van Le and M. J. Wirth, Anal. Chem., 2004, 76, 2055–2061 CrossRef CAS PubMed.
- T. Tugulu and H. A. Klok, Macromol. Symp., 2009, 279, 103–109 CrossRef.
- A. L. Cordeiro, S. Zschoche, A. Janke, M. Nitschke and C. Werner, Langmuir, 2009, 25, 1509–1517 CrossRef CAS PubMed.
- P. Kim, H. E. Jeong, A. Khademhosseini and K. Y. Suh, Lab Chip, 2006, 6, 1432–1437 RSC.
- M. Ebara, J. M. Hoffman, A. S. Hoffman and P. S. Stayton, Lab Chip, 2006, 6, 843–848 RSC.
- H. S. Park, W. Lee and Y. S. Nam, BioChip J., 2016, 10, 48–55 CrossRef CAS.
- H. Lee, T. Kyung, J. Yoo, T. Kim, C. Chung, J. Y. Ryu, H. Lee, K. Park, S. Lee, W. D. Jones, D. Lim, C. Hyeon, W. Do Heo and T. Yoon, Nat. Commun., 2013, 4, 1505 CrossRef PubMed.
- D. Patel, Y. Gao, K. Son, C. Siltanen, R. M. Neve, K. Ferrara and A. Revzin, Lab Chip, 2015, 15, 4614–4624 RSC.
- D. L. Huber, R. P. Manginell, M. A. Samara, B. Il Kim and B. C. Bunker, Science, 2003, 301, 352–354 CrossRef CAS PubMed.
- X. Cheng, Y. Wang, Y. Hanein, K. F. Böhringer and B. D. Ratner, J. Biomed. Mater. Res., Part A, 2004, 70, 159–168 CrossRef PubMed.
- S. Demming, G. Peterat, A. Llobera, H. Schmolke, A. Bruns, M. Kohlstedt, A. Al-Halhouli, C. P. Klages, R. Krull and S. Büttgenbach, Biomicrofluidics, 2012, 6, 034106 CrossRef PubMed.
- T. Yang, J. Choo, S. Stavrakis and A. de Mello, Chem. – Eur. J., 2018, 24, 12078–12083 CrossRef CAS PubMed.
- G. Y. Huang, L. H. Zhou, Q. C. Zhang, Y. M. Chen, W. Sun, F. Xu and T. J. Lu, Biofabrication, 2011, 3, 12001 CrossRef PubMed.
- S. Zhao, Y. Chen, B. P. Partlow, A. S. Golding, P. Tseng, J. Coburn, M. B. Applegate, J. E. Moreau, F. G. Omenetto and D. L. Kaplan, Biomaterials, 2016, 93, 60–70 CrossRef CAS PubMed.
- K. Kinoshita, M. Iwase, M. Yamada, Y. Yajima and M. Seki, Biotechnol. J., 2016, 11, 1415–1423 CrossRef CAS PubMed.
- J. A. Burdick and G. D. Prestwich, Adv. Mater., 2011, 23, H41–H56 CrossRef CAS PubMed.
- M. R. DeWitt and M. N. Rylander, in Targeted Drug Delivery Methods and Protocols, ed. R. W. Sirianni and B. Behkam, Springer, US, New York, NY, 2018, pp. 159–178 Search PubMed.
- S. Cosson and M. P. Lutolf, Sci. Rep., 2014, 4, 4462 CrossRef CAS PubMed.
- P. Domachuk, K. Tsioris, F. G. Omenetto and D. L. Kaplan, Adv. Mater., 2010, 22, 249–260 CrossRef CAS PubMed.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- Z. Zhu and C. J. Yang, Acc. Chem. Res., 2017, 50, 22–31 CrossRef CAS PubMed.
- Y. Shin, S. Han, J. S. Jeon, K. Yamamoto, I. K. Zervantonakis, R. Sudo, R. D. Kamm and S. Chung, Nat. Protoc., 2012, 7, 1247–1259 CrossRef CAS PubMed.
- Y. Wang, L. Wang, Y. Guo, Y. Zhu and J. Qin, RSC Adv., 2018, 8, 1677–1685 RSC.
- N. Annabi, Š. Selimović, J. P. Acevedo Cox, J. Ribas, M. Afshar Bakooshli, D. Heintze, A. S. Weiss, D. Cropek and A. Khademhosseini, Lab Chip, 2013, 13, 3569–3577 RSC.
- J. Fiedler, J. Groll, E. Engelhardt, P. Gasteier, C. Dahmen, H. Kessler, M. Moeller and R. E. Brenner, Int. J. Mol. Med., 2011, 27, 139–145 CAS.
- Y. Piao, D. J. Han, M. R. Azad, M. Park and T. S. Seo, Biosens. Bioelectron., 2015, 65, 220–225 CrossRef CAS PubMed.
- A. Sheikhi, J. de Rutte, R. Haghniaz, O. Akouissi, A. Sohrabi, D. Di Carlo and A. Khademhosseini, Biomaterials, 2019, 192, 560–568 CrossRef CAS PubMed.
- S. Guo, G. Kang, D. T. Phan, M. N. Hsu, Y. C. Por and C. H. Chen, Sci. Rep., 2018, 8, 2245 CrossRef CAS PubMed.
- G. T. Roman, T. Hlaus, K. J. Bass, T. G. Seelhammer and C. T. Culbertson, Anal. Chem., 2005, 77, 1414–1422 CrossRef CAS PubMed.
- G. T. Roman and C. T. Culbertson, Langmuir, 2006, 22, 4445–4451 CrossRef CAS PubMed.
- J. B. Orhan, V. K. Parashar, J. Flueckiger and M. A. M. Gijs, Langmuir, 2008, 24, 9154–9161 CrossRef CAS PubMed.
- A. R. Abate, D. Lee, T. Do, C. Holtze and D. A. Weitz, Lab Chip, 2008, 8, 516–518 RSC.
- H. Y. Chen, Y. Elkasabi and J. Lahann, J. Am. Chem. Soc., 2006, 128, 374–380 CrossRef CAS PubMed.
- Y. J. Chuah, Y. T. Koh, K. Lim, N. V. Menon, Y. Wu and Y. Kang, Sci. Rep., 2016, 5, 18162 CrossRef PubMed.
- N. Bhattacharjee, C. Parra-cabrera, Y. T. Kim, A. P. Kuo and A. Folch, Adv. Mater., 2018, 30, 1800001 CrossRef PubMed.
- G. Gonzalez, A. Chiappone, K. Dietliker, C. F. Pirri and I. Roppolo, Adv. Mater. Technol., 2020, 5, 2000374 CrossRef CAS.
- Y. Lin, Y. Ting, B. Chen, Y. Cheng and T. Liu, Surf. Coat. Technol., 2020, 391, 125663 CrossRef CAS.
- T. Le and C. Lee, Appl. Biochem. Biotechnol., 2020, 191, 29–44 CrossRef CAS PubMed.
- M. Villegas, Y. Zhang, N. Abu Jarad, L. Soleymani and T. F. Didar, ACS Nano, 2019, 13, 8517–8536 CrossRef CAS PubMed.
- M. Osborne, A. Aryasomayajula, A. Shakeri, P. R. Selvaganapathy and T. F. Didar, ACS Sens., 2019, 4, 687–693 CrossRef CAS PubMed.
- S. M. Imani, M. Badv, A. Shakeri, H. Yousefi, D. Yip, C. Fine and T. F. Didar, Lab Chip, 2019, 19, 3228–3237 RSC.
- A. Zhu, S. Ali, Y. Xu, Q. Ouyang and Q. Chen, Biosens. Bioelectron., 2021, 172, 112806 CrossRef CAS PubMed Contents.
- S. L. Banerjee, S. Samanta, S. Sarkar and N. K. Singha, J. Mater. Chem. B, 2020, 8, 226–243 RSC.
- B. A. G. Lamers, M. L. Ślęczkowski, F. Wouters, T. A. P. Engels, E. W. Meijer and A. R. A. Palmans, Polym. Chem., 2020, 11, 2847–2854 RSC.
- B. Feyssa, C. Liedert, L. Kivimaki, L. S. Johansson, H. Jantunen and L. Hakalahti, PLoS One, 2013, 8, e68918 CrossRef CAS PubMed.
- T. Vong, S. Schoffelen, S. F. M. Van Dongen, T. A. Van Beek, H. Zuilhof and J. C. M. van Hest, Chem. Sci., 2011, 2, 1278–1285 RSC.
- G. A. Cooksey, C. G. Sip and A. Folch, Lab Chip, 2009, 9, 417–426 RSC.
- G. Simone, P. Neuzil, G. Perozziello, M. Francardi, N. Malara, E. Di Fabrizio and A. Manz, Lab Chip, 2012, 12, 1500–1507 RSC.
- S. Mccormick, Z. Tong, A. Ivask, M. Morozesk, N. H. Voelcker, E. Lombi and C. Priest, Biofabrication, 2018, 10, 014101 CrossRef PubMed.
- B. Hong, P. Xue, Y. Wu, J. Bao, Y. J. Chuah and Y. Kang, Biomed. Microdevices, 2016, 18, 21 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2021 |