
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn-chip perivascular niche supporting stemness of patient-derived glioma cells in a serum-free, flowable culture†
Magda
Gerigk
ab,
Harry
Bulstrode
c,
HaoTian Harvey
Shi
 da,
Felix
Tönisen
ea,
Camilla
Cerutti
f,
Gillian
Morrison
g,
David
Rowitch
h and
Yan Yan Shery
Huang
*ab
da,
Felix
Tönisen
ea,
Camilla
Cerutti
f,
Gillian
Morrison
g,
David
Rowitch
h and
Yan Yan Shery
Huang
*ab
aDepartment of Engineering, University of Cambridge, UK. E-mail: yysh2@cam.ac.uk
bThe Nanoscience Centre, University of Cambridge, UK
cDepartment of Clinical Neuroscience, University of Cambridge, UK
dDepartment of Mechanical & Industrial Engineering, University of Toronto, Canada
eDepartment of Cell Biology, Radboud Institute for Molecular Life Sciences, Radboudumc, Netherlands
fRandall Centre of Cell & Molecular Biophysics, King's College London, UK
gCentre for Regenerative Medicine, University of Edinburgh, UK
hDepartment of Paediatrics, University of Cambridge, UK
First published on 5th May 2021
Abstract
Glioblastoma multiforme (GBM) is the most common and the most aggressive type of primary brain malignancy. Glioblastoma stem-like cells (GSCs) can migrate in vascular niches within or away from the tumour mass, increasing tumour resistance to treatments and contributing to relapses. To study individual GSC migration and their interactions with the perivasculature of the tumour microenvironment, there is a need to develop a human organotypic in vitro model. Herein, we demonstrated a perivascular niche-on-a-chip, in a serum-free condition with gravity-driven flow, that supported the stemness of patient-derived GSCs and foetal neural stem cells grown in a three-dimensional environment (3D). Endothelial cells from three organ origins, (i) human brain microvascular endothelial cells (hCMEC/D3), (ii) human umbilical vein endothelial cells (HUVECs) and, (iii) human lung microvascular endothelial cells (HMVEC-L) formed rounded microvessels within the extracellular-matrix integrated microfluidic chip. By optimising cell extraction protocols, systematic studies were performed to evaluate the effects of serum-free media, 3D cell cultures, and the application of gravity-driven flow on the characteristics of endothelial cells and their co-culture with GSCs. Our results showed the maintenance of adherent and tight junction markers of hCMEC/D3 in the serum-free culture and that gravity-driven flow was essential to support adequate viability of both the microvessel and the GSCs in co-culture (>80% viability at day 3). Endpoint biological assays showed upregulation of neovascularization-related genes (e.g., angiopoietins, vascular endothelial growth factor receptors) in endothelial cells co-cultured with GSCs in contrast to the neural stem cell reference that showed insignificant changes. The on-chip platform further permitted live-cell imaging of GSC – microvessel interaction, enabling quantitative analysis of GSC polarization and migration. Overall, our comparative genotypic (i.e. qPCR) and phenotypic (i.e. vessel permeability and GSC migration) studies showed that organotypic (brain cancer cells–brain endothelial microvessel) interactions differed from those within non-tissue specific vascular niches of human origin. The development and optimization of this on-chip perivascular niche, in a serum-free flowable culture, could provide the next level of complexity of an in vitro system to study the influence of glioma stem cells on brain endothelium.
Introduction
Glioblastoma multiforme (GBM) is the most common and aggressive form of primary brain tumour associated with poor survival. Following the diagnosis of a GBM, the current standard of care – surgical resection, chemotherapy, and radiation – together yield a median patient survival of fewer than 15 months.1 Although standard treatments can modestly extend survival, they fail to address molecular inter- and intra-tumour heterogeneity of GBM cells, as well as the dynamic regulation of the tumour microenvironment (TME) that actively supports tumour progression and evolves treatment resistance. Moreover, tumours exhibit inherent chemo-resistance, which has been attributed to a subpopulation of cancer cells termed GBM and glioma stem-like cells (GSCs).2 Additionally, glioblastomas can often grow and progress without angiogenesis (formation of new blood vessels) and thus escape anti-angiogenic therapies.3 Neovascularization has long been implicated as a key feature of glioblastoma progression and could be achieved through different mechanisms, including vascular co-option, angiogenesis, vasculogenesis, vascular mimicry, and glioblastoma–endothelial cell transdifferentiation.4–6 Therefore, the development of a 3D in vitro model to study the vasculature in the presence of brain cancer cells could potentially aid the discovery of new strategies to target GBM neovascularization.Substantial progress has been made recently to develop in vitro 3D GBM culture models through organ-on-a-chip platforms.7,8 For example, several microfluidic models have been developed to study brain cancer cells interfacing with one or more elements of the TME (Table S1†). Ayuso et al. have created a microfluidic platform that allowed real-time monitoring of oxygen and glucose levels within the device to study their effects on the proliferation of a GBM cell line.9 In their subsequent work, Ayuso et al. focused on mimicking the pseudopalisade formation in the GBM microenvironment via nutrient starvation.10 Ma et al. recreated a coherent GBM microenvironment by means of embedding multicellular spheroids within a collagen matrix accompanied by a perfused culture.11 While early studies from Ayuso et al.9,10 and Ma et al.11 employed long-term established GBM cell lines such as U87 and U251, recent work by Truong et al.12 and Xiao et al.13 demonstrated the potential of using patient-derived glioma cell lines co-cultured with endothelial cells (HUVECs) in microfluidic settings, to overcome the limitations of the established GBM cell lines such as genetic drifts14 or unknown origin.15
To further enhance the biological relevance of a perivascular niche model for GBM, it is important to consider, firstly, the use of serum-free media in the on-chip system, and secondly, how different organotypic vessels of human origin could impact on the GBM in vitro model. With regards to the first point, although glioma had been traditionally cultured in fetal bovine serum (FBS) enriched media, studies of cancer stem cells in GBM pointed to the concerning effects of serum on GSC differentiation and gene expression changes,16 highlighting the need for optimising a serum-free microvessel co-culture for maintaining GSC stemness which enabling direct observations on GSC behaviours. Considering the second point, since the tumour microenvironment in each type of cancer is unique to the organ, the regulation of vascular niches is likely to be organ-specific. However, in previous studies involving GSCs with human endothelial cells,12,13 only HUVECs were used to form the microvessels, which are not derived from the brain, and therefore may not represent organotypic GBM-microvasculature crosstalk.
Here, we perform comparative studies on the interactions between patient-derived glioma stem-like (GSC) cell lines and flowable microvessels generated from human microvascular endothelial cells (hCMEC/D3), human umbilical vein endothelial cells (HUVECs) and human lung microvascular endothelial cells (HMVEC-L). Although more complex 3D brain vasculature mimicking blood–brain barrier characteristics or 3D organoid models have been demonstrated,17,18 the focus of our work is to investigate fundamentally: (1) Can we establish a serum-free culture supporting adequate cell viability and characteristics in a microvessel–GSC co-culture system? And (2) To what extent assay format and tissue-specific vessel–glioma interaction matter for creating a perivascular niche for studying GBM?19–21 Validation to the above questions could hold practical importance towards designing biologically-relevant on-chip devices for glioma drug testing applications, balancing biological assay fidelity versus process simplicity, standardization and costs. With the above goal, a single channel microvessel-on-chip platform was used to support GSC–microvessel co-culture in three-dimensional (3D) extracellular matrices (ECM). We demonstrated the in vitro model design in a serum-free flowable culture, performed biological characterisation of the culture system under different experimental conditions, and compared the GSC cellular behaviour for the three vascular niches.
Experimental
Microfluidic device fabrication
The microfluidic device used to construct the microvessel-on-a-chip experiments was adapted from our previous design22 and is schematically shown in Fig. 1. Briefly, the microfluidic device was designed using AutoCAD software. The design contains two side channels that have a width of 180 μm and a height of 100 μm. It also includes three channels in the middle, where two channels have a width of 400 μm and a height of 100 μm, and one channel that has a width of 120 μm and a height of 100 μm. A glass slide was used as a bottom part of the device to provide better image quality.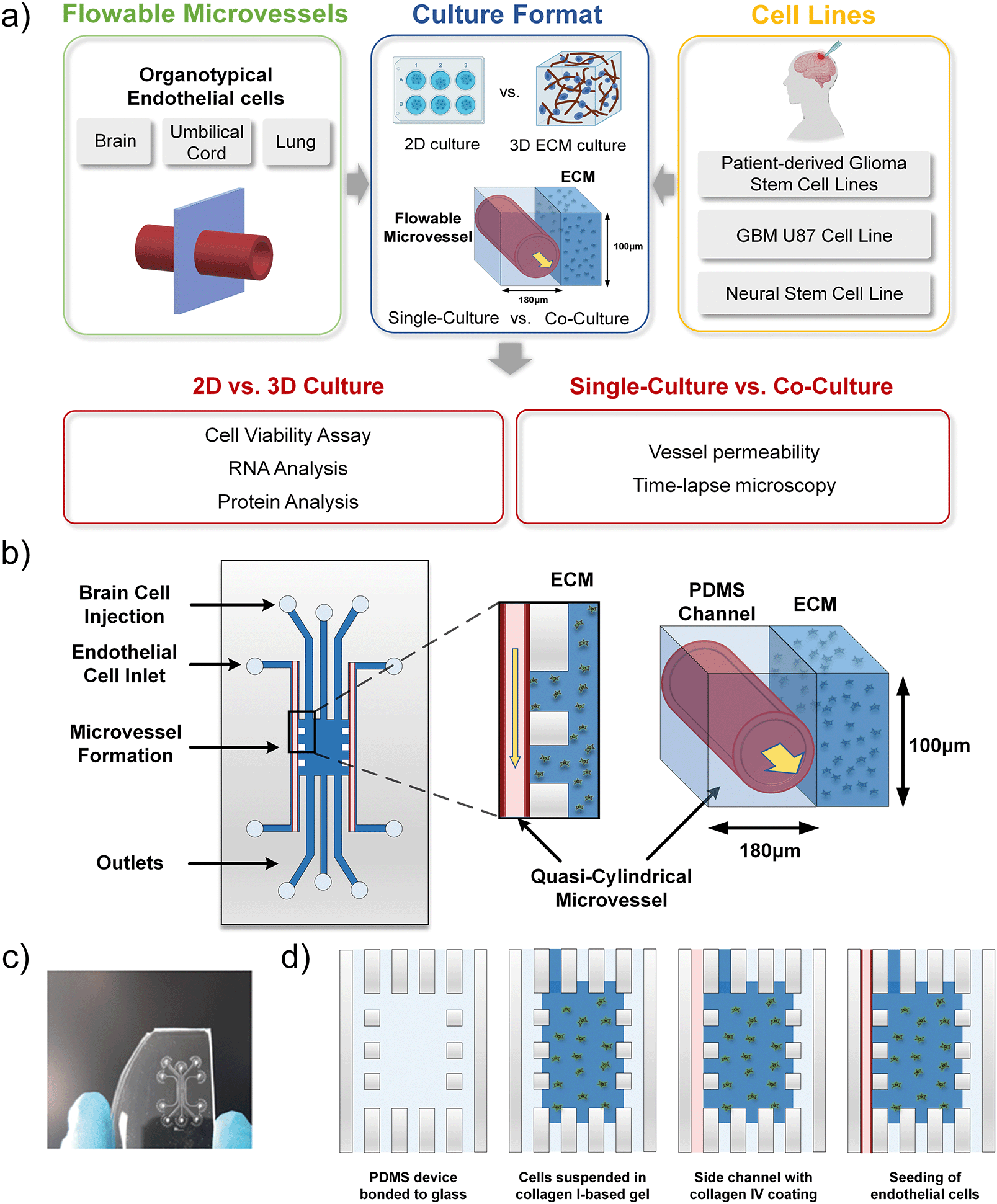 | ||
| Fig. 1 a) Schematics demonstrating physical attributes of on-chip perivascular niche with patient-derived glioma cells. b) Microfluidic device design and co-culture configuration. The design scheme of the microfluidic device, where the ECM-integrated PDMS channel shelters the quasi-cylindrical microvessel formed with various endothelial cells with flow; c) photograph of the actual microfluidics channel fabricated for microvessel-on-a-chip implementation; d) cell suspension injection leads to a central 3D ECM cell culture and two vacant side channels, then seeding of endothelial cells within a side channel forms a microvessel. | ||
The microfluidic master was fabricated using the soft lithography process. SU-8 negative photoresist (MicroChem) was coated onto a 6′′ silicon wafer via spinning coating. Mask was then patterned using standard ultraviolet exposure. The microfluidic channels were fabricated by moulding pre-crosslinked PDMS on the master. PDMS resin (Sylgard, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 w/w ratio between elastomer and curing agent) was poured on the master and desiccated to remove the bubbles formed during the mixing process. The PDMS was then placed in an oven for 3 h at 65 °C until fully cured. Afterwards, the PDMS was peeled off, and inlet and outlet holes with diameters of 0.75 mm were punched. Foreign particles were removed from the PDMS surfaces using adhesive tape, washing with ethanol, and then blow-drying. The 3 mm thick PDMS was bound to a round glass coverslip with a diameter of 22 mm by air-plasma treatment (Femto Science; 15 s, 25 sccm, 10 power), forming the microfluidic device.
:1 w/w ratio between elastomer and curing agent) was poured on the master and desiccated to remove the bubbles formed during the mixing process. The PDMS was then placed in an oven for 3 h at 65 °C until fully cured. Afterwards, the PDMS was peeled off, and inlet and outlet holes with diameters of 0.75 mm were punched. Foreign particles were removed from the PDMS surfaces using adhesive tape, washing with ethanol, and then blow-drying. The 3 mm thick PDMS was bound to a round glass coverslip with a diameter of 22 mm by air-plasma treatment (Femto Science; 15 s, 25 sccm, 10 power), forming the microfluidic device.
Coating of microfluidic channels
Plasma treatment conferred hydrophilic properties to the channel walls, making the subsequent poly-D-lysine (PDL) coating process feasible. The microfluidic channels were coated with PDL (1 mg ml−1 in deionized water (DIW)) for 5 h at 37 °C. After coating, residual PDL was washed multiple times using DIW to remove any excess molecules that could cause cellular damage. The device was then placed at 50 °C for 18 h to recover the surface hydrophobicity. The hydrophobic recovery was necessary to successfully insert collagen I gel in the central region of the device, forming vertical barriers facing the cell culture channels.Incorporation of an extracellular matrix
Collagen gel was incorporated in the PDMS-based device to establish a 3D extracellular matrix for cancer cell culture. While levels of collagens in the normal adult brain are relatively low, in glioma, collagen levels are elevated and play an important role in driving the tumour progression.23 The ECM of brain tumours consists of the basement membrane components, collagen IV, laminin and fibronectin lining the blood vessels, as well as collagen I, tenascin-C, vitronectin and hyaluronan surrounding the tumour.23–25 Thus, collagen I-based hydrogel, mixed with laminin, and collagen IV coating for the outermost side channels were chosen for the microfluidic platform constructed.Hydrogel injection and side channel coating
After restoring hydrophobicity in a PDL-coated device, the microfluidic chip was glued with a silicon rubber compound (RS Components, 692-542) to a new, bottomless polystyrene dish, and collagen I gel was prepared and carefully injected from the access port of one of the channels directly facing the side channels. In terms of the gel preparation, DIW, collagen I rat protein (Gibco, A1048301), 10x phosphate-buffered saline (PBS; Gibco) with phenol red and 0.5 N NaOH (Sigma-Aldrich) solutions were kept on ice. By using a wide orifice pipette tip, 27.2 μl of collagen was added to a 0.5 ml tube kept on ice. 5 μl of 10x PBS with phenol red and 3 μl of 0.5 N NaOH were added in a new 0.5 ml tube and mixed. 5.4 μl of DIW was then added and mixed until the gel appeared to be uniform in colour. Following the above procedure, the collagen gel concentration was 2 mg ml−1 and the pH around 7.5, resulting in a pink colour solution. Next, the gel solution was carefully injected into the access port of one of the channels directly facing the microvessel channels and interfaces were created between the cell culture channels and the central region of the device. Gel cross-linking was performed at room temperature for 1 h. The gel was able to distribute in the central region and in the three channels connected to it, leaving the two side channels empty for endothelial cell seeding. Vertical interfaces could be created at the gaps between the pillars separating the side channels from the central region. 10 min after gel insertion, drops of cell medium were positioned on top of the access ports of all the channels, except for the outermost side channels. After gel cross-linking, collagen IV (Sigma, C5533, 1 mg ml−1 in DIW) was inserted inside the side channels to perform a second coating of the channel surfaces with a substrate suitable for cell seeding. Cell medium was added to the polystyrene dish, which surrounds the device to avoid solution evaporation inside the channels, and the device was left inside an incubator, at 37 °C, for 1.5 h. After collagen IV coating, the side channels were washed several times with the cell medium to remove any excess of uncoated collagen. Next, the device was completely submerged in the cell medium and stored inside the cell incubator for endothelial cell seeding.Cell cultures
| Glioma cell line name | Classification |
|---|---|
| GCGR-E13 | Classical |
| GCGR-E17 | Classical |
| GCGR-E28 | Classical |
| GCGR-E35 | Mesenchymal |
| G166 | Classical |
Vessel flow
Vessel perfusion was achieved through gravitational-driven flow by the following procedure. A 3D-printed stage (Fig. S1a)†) was designed to fit a ∅100 mm polystyrene dish as a medium reservoir and two ∅35 mm with microfluidic devices, which were connected by tubing. Flow rates in the outermost channels were found experimentally and showed to be reproducible and reliable within the same devices and between devices. The results showed that a height difference of 4 mm could be used to achieve a flow rate of around 1.5 μl min−1 (Fig. S1b)†). Shear stresses were calculated using computational fluid dynamics over a range of flow rates based on the results generated empirically. The results demonstrated that the flow velocities and shear stresses over the collagen interface were within the desired range. Shear stresses between 1.4 and 0.4 dyne per cm2 were calculated within the side channel of the device with velocities around 1–2 mm s−1 as discussed previously.22Cell viability tests
For testing cell viability, LIVE/DEAD™ Viability/Cytotoxicity kit (Life Technologies, L3224) was used. Briefly, 20 μl of 2 mM ethidium homodimer-1 (EthD-1; enters cells with damaged membranes) stock solution was mixed in 10 ml of sterile PBS. Next, 5 μl of 4 mM calcein AM (CAM; live cells with ubiquitous intracellular esterase activity have the ability to enzymatically convert the virtually non-fluorescent CAM) stock solution was added to the 10 ml EthD-1 solution and mixed. That resulted in a final concentration of 4 μM EthD-1 and 2 μM CAM. 200 μl of the prepared solution was then added on top of the microfluidic device and perfused through the lateral microchannels. After 25 min of incubation at room temperature, cells were visualized by confocal microscopy – viable cells (CAM-positive) staining green and dead cells (EthD-1-positive) staining red. Cell viability profiles were then evaluated by analysing the fluorescence intensity of the viable/dead cells across the microchambers and calculating the ratio of live to dead cells. 21 devices (3 devices per day) for each cell line in triplicate (n = 3, different days) were analysed. All confocal images were taken at different focal planes (2.5 μm step size) with subsequent image analysis performed using FIJI software.RNA analysis
000 rpm. 150 μl of the colourless top phase (containing DNA) was transferred to a new tube. Next, RNA was precipitated by adding 500 μl of isopropanol. The sample was vortexed and incubated at room temperature for 10 min, then centrifuged at 12000 rpm for 10 min at 4 °C to obtain a pellet, which was then washed with 1 ml of 75% ethanol. After centrifugation at 12000 rpm for 3 min, the sample was air-dried for 5 min at room temperature. Finally, the pellet was dissolved in a maximum of 30 μl of RNase-free water.
Protein analysis
For experimental sets where live/dead cells were being studies, the images were analysed by quantifying labelled cells and by measuring the intensity of fluorescence. The analysis was carried out via FIJI. To quantify the number of marker-positive cells, the plugin cell counter was used after gating out weakly stained cells with the threshold. To quantify fluorescence intensity single cells were outlined and measured with the measure tool, which displayed the values for area, mean, internal density and raw internal density. As a control, sections of the background were outlined as well. To calculate fluorescence, the following formula was utilised:
| Integrated Density (sample) − (Mean (ctrl) × Area (ctrl)) = Fluorescence Intensity |
Live-cell imaging and image analysis
An in-house image analysis programme to pre-process the time-lapse images and create a database to automatically classify cell interactions with the regional microenvironments was used. This novel program utilizes several open-source applications, such as FIJI and CellProfiler (FIJI: https://imagej.net/Fiji; Cell Profiler: https://cellprofiler.org), and merges them with focus stack (z-stack) image analysis to enable cell tracking and segmentation. The experiments used brightfield (or transmission) images and green fluorescence protein (GFP) images. The intensity of GFP expression of the z-stack images acquired during the time-lapse experiments was projected onto one x–y plane at each time point; thus, the projected cell area, that is, the 2D projection of the 3D cell shape, was obtained at each time point. Cell speed, velocity and polarization were measured and analysed using the image-assisted microfluidic platform. The parameters were calculated for 3D cancer cell culture in the presence of a microvessel or in a reference system – without endothelial lining. Cancer cell migration velocity (measured between two consecutive time points) was quantified. The direction of the cancer cell migration was quantified by velocity components in the x-direction (perpendicular to the endothelium–collagen matrix barrier) and y-direction (along the channel direction). The presence of asymmetric shape features in cancer cells with respect to the position of the cell nucleus was described by using the cell polarization parameter. The position of the cell centre of mass was calculated by the CellProfiler software, weighting the x and y coordinates of the pixels within the projected cell shape with their GFP intensity; a computational error of about 20% was made due to misrecognition of the projected cell shape profile during automatic segmentation.Vessel permeability measurement by dextran diffusion
Upon the formation of a microvessel on-chip, the diffusive permeability was measured with fluorescently labelled dextran (fluorescein isothiocyanate-dextran, FITC-dextran) in the culture medium. The permeability measurements and calculations were adapted from Funamoto et al.27 Briefly, an intact endothelial monolayer gave rise to an intensity drop between the channel and the gel region once the fluorescently labelled dextran was introduced and persisted over time as dextran slowly diffused across the monolayer into the gel. Hence, the microvessel permeability was assessed by observing the diffusion of 70 kDa FITC-dextran (Sigma, 46945; 7 mg ml−1 in PBS, diluted further 1:9 in cell culture medium) from the microvessel channel into the collagen gel. The diffusion experiments were performed in the environmentally controlled chamber (set at 37 °C, 5% CO2) of a fluorescent microscope (Leica, CTR6500). The dextran solution was perfused inside the microvessel channel, and images were acquired every 30 s over 30 min. Beyond 30 min, the geometry of the device (e.g., pillars and the limited size of the gel chamber) broke down the perfect sink condition assumed in the calculation. Obtained fluorescent images were analysed using open-source software (FIJI).
Statistical methodology
Data were analysed using GraphPad Prism software and its built-in tests, or RStudio. Data were tested for normality with Shapiro–Wilk test and subsequently analysed with suitable parametric or non-parametric tests. In order to assess significance, unpaired or paired t-test or ANOVA with Tukey's post hoc test was used, depending on the experiment in question. The following asterisk rating system for p-value was used: *p ≤ 0.05, **p ≤ 0.01.Results and discussion
Protocol optimization to study gene and protein expression in cells cultured in 3D microfluidics
On-chip vascular niche (Fig. 1) was fabricated to investigate the interactions between patient-derived glioma cell lines and different human organ-specific endothelium. With this device configuration, GSCs can be cultured in the 3D ECM gel in proximity to a quasi-circular, perfusable microvessel. Despite the advantages of microfluidic cell culture, which would better emulate in vivo conditions than conventional 2D cell cultures, the integration of these techniques as a research tool in mainstream cancer research has been slow.28 Lack of in-depth biological characterisation accompanied by limited biological readouts of the assay has been central issues. Genomic or proteomic analysis post microfluidic cell culture requires protocol optimisation to retrieve cells in 3D culture from the microfluidic device. Here, we optimized several protocols for biological analysis on cells post-3D culture in a microfluid device, as summarized in Fig. 1. This makes our platform flexible and adaptable to commonly used biological analysis, such as protein/gene expression analysis coupled with functional analysis such as vessel permeability and live-cell imaging of cancer cell migration.The study of the glioma-associated neovascularization mechanisms is important as microvascular proliferation was declared a pathological hallmark of malignant high-grade gliomas.29,30 We developed a quick and straightforward method to recover endothelial cells from our on-chip vascular niche coupled to microfluidic assay, to perform qPCR analysis. After cell isolation, we used a commercial RNA isolation kit, and the RNA concentration and purity were assessed by absorbance measurement (260/280 ratio). RNA from cells recovered from microfluidic devices and 2D control samples was checked to ensure pure RNA from cells was obtained. The results passed Shapiro–Wilk normality test and were then subjected to a t-test. Our analysis between the 2D and 3D endothelial cell RNA quality test shows no significant difference, indicating that our RNA extraction and purification protocol is comparable to 2D assay routinely used in research. Using our optimized method, it was possible to extract up to 75 ng μl−1 of RNA per device. If the concentration of RNA in one sample was lower than 50 ng μl−1, RNA from 2 or more samples from the same condition and the same run of a given experiment were pooled. The extracted cells from the 3D microfluidic device are pure endothelial cell populations, as confirmed by qPCR analysis. It has shown that they did not contain any GFAP, a particular marker for the GBM cells.
Brain cancer cells in 3D ECM
U87 cells, a commonly used GBM model cell line, was used in our study for initial protocol optimisation, and also as a reference. In previous in vivo studies, it has been reported that U87 cell type failed to accurately model human GBM compared to patient-derived tumour stem cells.15 U87 cells, cultured in 2D in vitro cell culture with FBS-enriched medium, exhibit reduction in neural stem cell marker levels, such as nestin, SOX2, and CD133.31,32 Here, we performed studies to culture U87 cells in 3D-collagen gel with laminin, in three comparative media conditions: FBS-supplemented DMEM (standard U87 culture media), FBS-supplemented EGM-2MV (standard endothelial cell media), and serum-free EGM-2MV. ESI† Fig. S2 and S3 show that switching FBS-supplemented DMEM medium with FBS-supplemented EGM-2MV medium had negligible impacts on the U87 viability, and the selected glial markers (e.g. EGFR, GFAP), in either 2D or 3D culture formats.33 However, 3D culture significantly increased the stemness markers of U87 cells (CD44, CD133, nestin, olig2 and sox2 measured at day 3), which is consistent previous reports on GBM-derived cell lines in 3D collagen culture.34–36 In a 3D collagen culture, maintaining the U87 cells in serum-free EGM-2MV further enhances the expression of sox2 and nestin, compared to the serum-supplemented media, which echoes with prior reports that serum-free cultures enhanced stemness related genes.31,32 For all the culture format and media conditions studied, cellular viability of over 85% was seen for day 3, confirming the suitability of these culture conditions for assay purposes.3D collagen-I gel culture preserves glioma stem cell-like phenotype in patient-derived cell lines. Considering the complexity of both inter- and intra-tumour cellular, genetic and molecular heterogeneity of GBMs,37,38 it is of interest to design an in vitro model with human cells that can be easily used for culturing different patient-derived cancer cell lines and to dissect whether glioblastoma sub-types influence the process of neovascularisation. We used five patient-derived cell lines, containing glioma stem-like cell (GSC) population (G166, GCGR-E13, GCGR-E17, GCGR-E28 and GCGR-E35). The FNS cell line GCGR-NS6FB_A was used as a healthy brain control for stemness markers, and the U87 cell line were used as technical control for glial markers. Since GSCs are standardly cultured in serum-free conditions in 2D, it was crucial to test whether our 3D ECM and serum-free condition would influence the cellular characteristics of these five GBM patient-derived cell lines.
All the tested cell populations were successfully cultured in a serum-free condition within a 3D collagen gel/laminin for 3 days with the expected cell morphology (Fig. 2a)). Relative gene expression (mRNA level) analysis was performed to study the differences between 2D and 3D culture in proliferation, stemness and differentiation, in a manner similar to the U87 data shown above. First, we did not detect the glial fibrillary acidic protein (GFAP, a marker of astrocytic differentiation) in the G166, GCGR-E35 and FNS cell lines in either 2D and 3D cultures,39,40 by qPCR. The fact that GFAP was not detected in FNS (as a negative control) but in U87 (as a positive control), confirms the fidelity of our cell extraction and analysis process. In all cell lines, proliferation (Ki67 marker41) in 3D ECM culture was slightly decreased compared to 2D. Similar to the U87 results demonstrated above, 3D collagen culture enhances the expression of stemness-related markers (CD44, CD133, nestin, olig2, sox2) for all patient-derived GSC lines, compared to 2D culture, while inducing insignificant changes of GFAP expression for the selected cell lines with GFAP expression (Fig. 2a)).
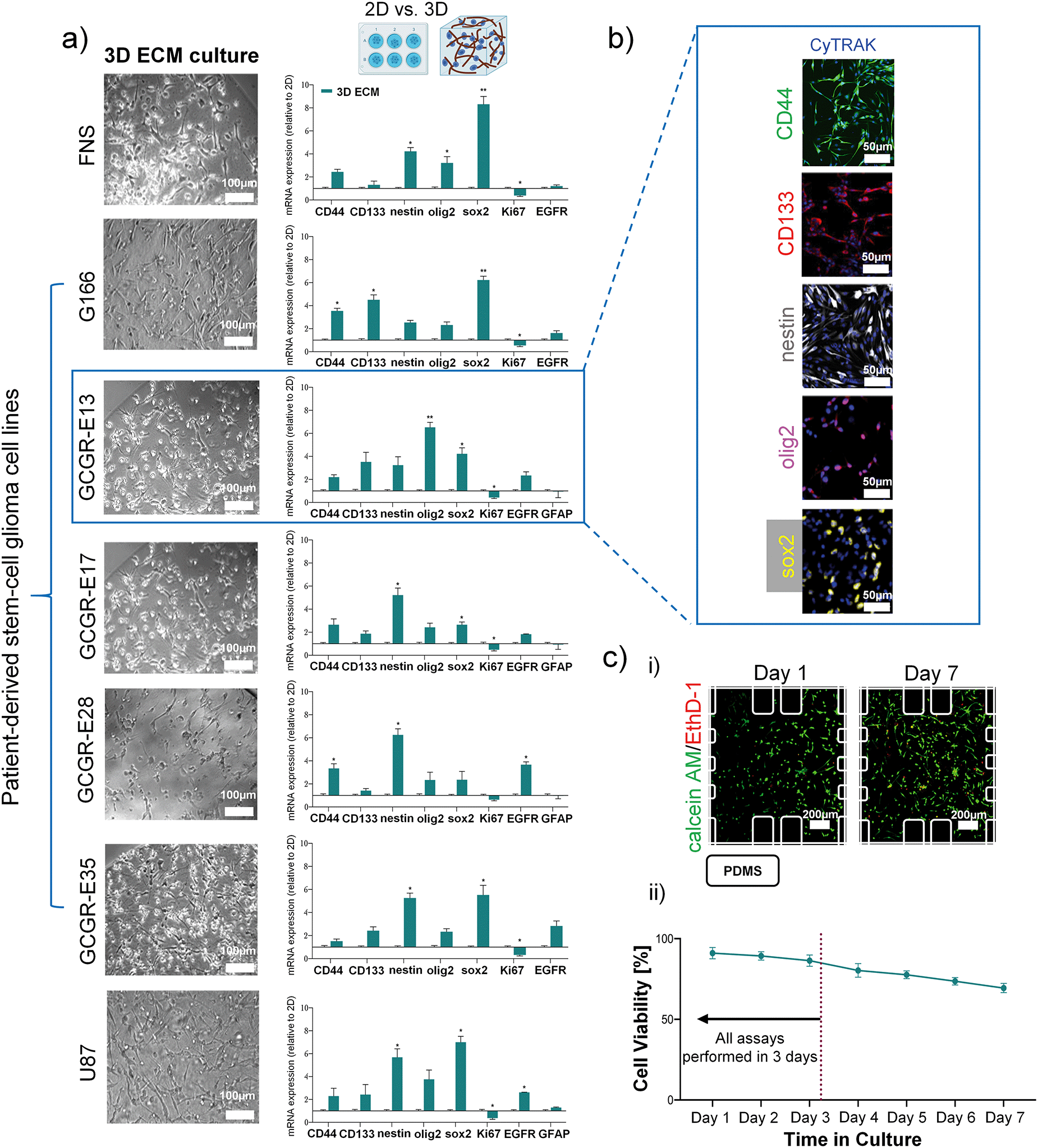 | ||
| Fig. 2 Patient-derived stem cell lines are characterised by stemness-related genes whilst cultured in 3D collagen I-based ECM for three days. a) Representative bright-field images of each GBM cell type cultured in 3D ECM acquired by light microscopy. mRNA expression of stemness-related gene in GBM cells normalised to 2D samples. Results for the 3D samples obtained from three independent experimental runs are presented by mean ± SEM. Two-tailed t-test was used for significance. Note that for FNS, G166 and GCGR-E35, GFAP was not detected by qPCR; b) example immunostaining images of GCGR-E13 cells grown in 3D for stemness-related markers: CD44, CD133, nestin, olig2, sox2. All were counter-stained for nucleus (CyTRAK); c) the viability of GBM cells in 3D culture. i) Example images of calcein AM/EthD-1 staining of GCGR-E13 cells at day-1 and day-7 culture, inside a microfluidic device; ii) graph presenting the cellular viability over seven days in cell culture. Data presented mean ± SD (percentage of live cells). | ||
The stemness-related markers that we selected have been indicated for their role in the maintenance of cell renewal and multi-lineage differentiation, contributing to phenotypic plasticity of GBM cells.42,43 Furthermore, epidermal growth factor receptor (EGFR) expression was upregulated in selected patient-derived cell lines (i.e. GCGR-E13, GCGR-E28). Overall, our mRNA level data are consistent with previously published data in GBM-derived cell lines derived from other sources, cultured in collagen gels.34–36
Then, we confirm the localization and protein expression of CD44, CD133, nestin, olig2 and sox2 in GCGR-E13 cells cultured in 3D ECM by immunostaining (Fig. 2b)). The multi-level analysis of factors contributing to GBM phenotype has shown comparable trends in the detection of mRNA transcripts and protein between 2D and 3D ECM cell culture. Based on the above results, we suggest the 3D collagen-I culture condition created for GSCs is suitable for maintaining their phenotype for 3 days. Finally, cell viability assays were performed in order to ensure the system is robust enough to support prolonged cell culture of various cell types. Each cell type was cultured within microfluidic devices for up to 7 days. Cells were cultured in a serum-free medium and in the presence of gravity-driven flow. Example images of calcein AM/EthD-1 staining of GCGR-E13 cells were shown in Fig. 2c) i). As shown in Fig. 2c) ii) and S4,† it was found that the cells have over 80% viability at day-3 in 3D culture, suitable for follow-on experiments.
Microvessel-on-a-chip formed by different organotypic endothelial cell types
Next, we investigated endothelial cells from the human brain (hCMEC/D3), umbilical cord (HUVECs) or the lung (HMVEC-L) in terms of microvessel formation in the microfluidic device. Fig. 3a) shows that the application of collagen IV coating is the prime factor controlling the expression of the endothelial junction and tightness-related genes, while the effect of the endothelial layer format (e.g. 2D vs. 3D microvessel) is secondary. Tight junction ZO-1 and actin (phalloidin) staining revealed that all three cell types were able to form a microvessel structure with a quasi-circular cross-section under 3D culture condition by immunofluorescence, shown in Fig. 3b). Using our 3D ECM device, HUVEC and hCMEC/D3 cells formed a more defined circular microvessel structure compared to HMVEC-L cells. qPCR revealed that hCMEC/D3 is characterized by significantly higher levels of ZO-1 expression than the other two cell types (Fig. 3b) ii)), where ZO-1 is required for tight junction formation44–46 in brain endothelial cells forming the blood brain barrier. Other tight-junctions-related molecules, claudin-5 and occludin47 were also shown to be higher in hCMEC/D3 compared to HUVEC or HMVEC-L, shown in Fig. S5a).† As the process of neovascularization is important in the context of GBM, we further compared the RNA levels of neovascularisation-related genes across the three cell types forming circular microvessels, and no significant differences in mRNA relative levels were detected, as shown in Fig. S5b).†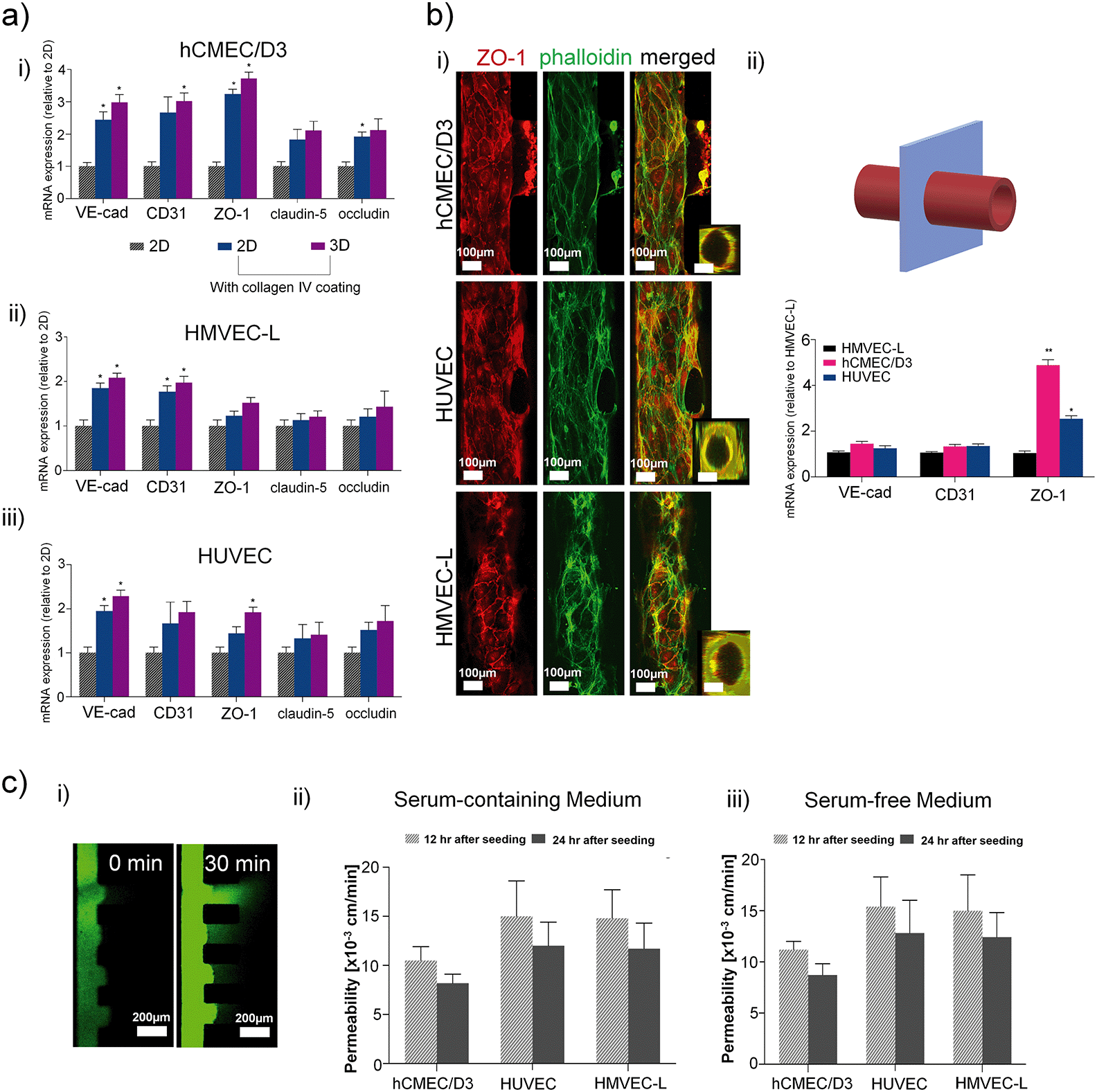 | ||
| Fig. 3 Endothelial cell 3D culture with ECM is important for tightness-related gene expression and barrier function, while serum reduction has negligible effects on permeability. a) A comparison of tightness-related gene expression in each endothelial cell type in 2D and 3D: i) hCMEC/D3; ii)HMVEC-L; and iii) HUVEC. mRNA expression normalised to samples from 2D without collagen IV coating; results obtained from three independent experimental runs are presented by mean ± SEM. One-way ANOVA with Tukey's post hoc test was used for significance; b) i) microvessel lumen formed by various types of microvessels in contact with ECM. Staining of junction protein ZO-1, cytoskeleton by phalloidin, and merged image. In all panels, on the right, a 3D reconstruction of the microvessel cross-section at a location along the vessel lumen (y-axis); ii) hCMEC/D3 express higher levels of endothelial tightness-related genes than HUVECs and HMVECs-L in the microfluidic setup. mRNA expression normalised to HMVEC-L (results obtained from three independent experimental runs are presented by mean ± SEM. One-way ANOVA with Tukey's post hoc test used for significance); c) diffusion measurements showed appropriate level of permeability for the hCMEC/D3 vessel; i) typical images from an permeability experiment showing 70 kDa FITC-dextran diffusion after 30 min; endothelial permeability measurements for cells ii) in serum-containing medium 12 and 24 h after seeding; and iii) in serum-free medium 12 and 24 h after seeding. The graphs show results from three independent experimental runs as mean ± SEM. t-Test shows insignificant difference for permeability in serum-containing versus serum-free cultures. | ||
The barrier function of the three microvessel types was measured by the permeability of fluorescent dextran. In particular, Fig. 3c) and Table S2† showed that in a serum-free condition, which is important for the subsequent co-culture for maintaining GSC stemness, induces insignificant changes in vessel permeability compared to the serum-supplemented condition. The permeability values showed that the brain endothelial cell line, hCMEC/D3, forms the tightest barrier compared to HUVECs or HMVECs, and the permeability on the microvessel decreases after 24 h of culture in the 3D device. These findings further confirmed that the hCMEC/D3 cells form a more favourable microvessel than HUVECs or HMVECs-L to study brain vasculature and GBM cells as they are specific to the brain, and that they are adequate to be used in the specific setup we described here. Nonetheless, all three cell types were able to create vessels of acceptable permeability, as compared to other microvessel-on-a-chip models where hCMEC/D3, HUVECs or HMVECs were used.48–51
Perivascular niche with GSCs on a chip
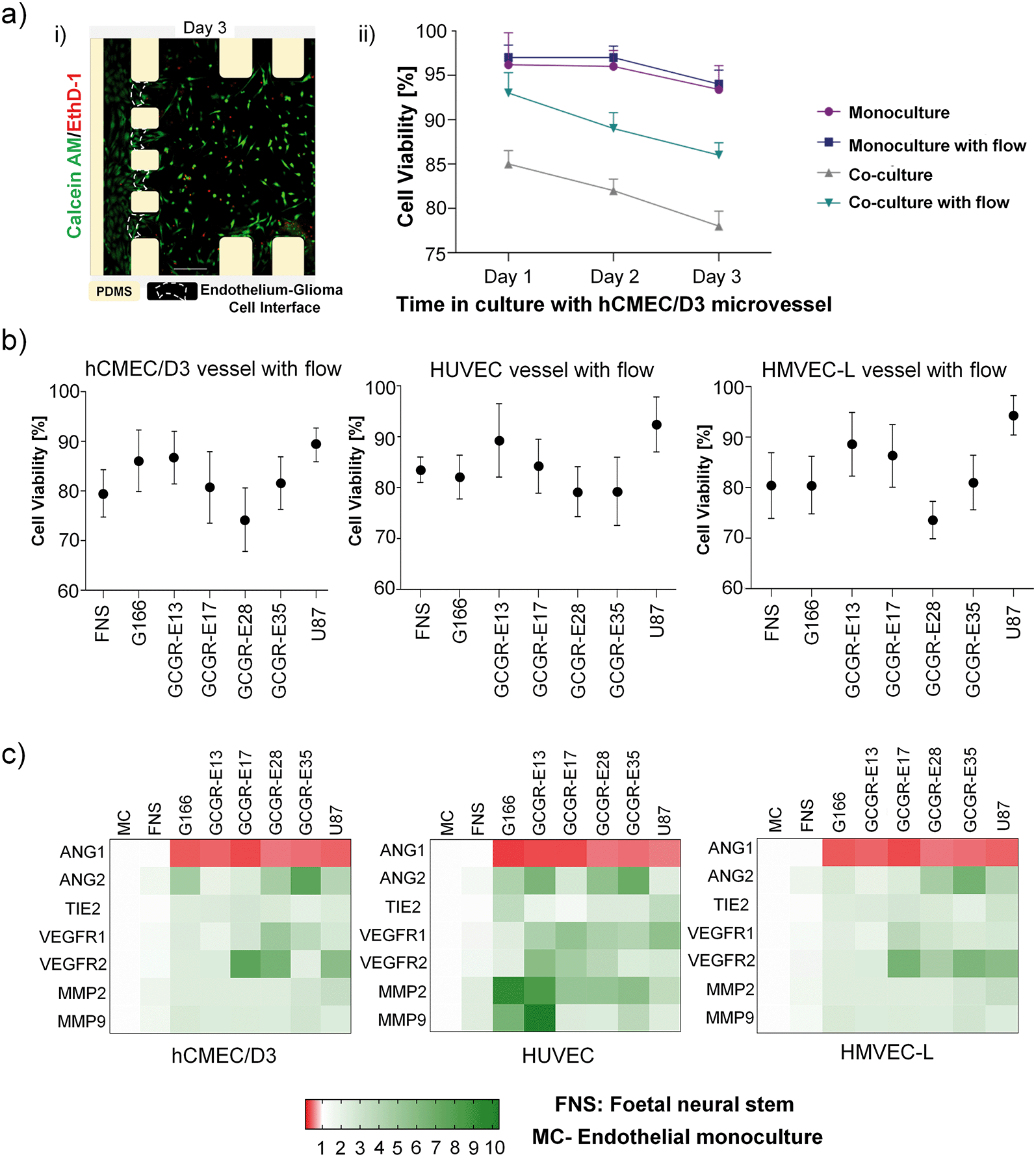 | ||
| Fig. 4 Viability of 3D GSCs culture with or without a microvessel, in the absence or presence of flow (∼1 dyne per cm2). a) i) Example image of calcein AM/EthD-1 staining of GCGR-E13 cells at day three in co-culture inside a microfluidic device with gravity-driven flow; ii) three-day GSC cell viability for the four different configurations. Data obtained from three independent experimental runs and presented as mean ± SD (percentage of live cells); b) graphs presenting viability of six different GBM cell lines, a FNS line, and the U87 cell line at day-3 culture with different microvessel types under flow. Data obtained from three independent experimental runs and presented as mean ± SD (percentage of live cells). c) GBM and FNS cells induce different response patterns in different endothelial cells under flow. Heatmaps were plotted in GraphPad Prism software after the analysis of mRNA expression in relation to GAPDH. Reference pattern was established from RNA isolated from endothelial cells cultured without the presence of brain cancer cells in microfluidic devices (MC = microvessel monoculture). Results obtained from three independent experimental runs. Green indicates upregulated expression, and red indicates downregulated expression. | ||
To determine how GSCs influence the microvessels, endothelial cells were recovered from microfluidic devices after 24 h of co-culture to establish an expression pattern of neovascularization-related genes at the mRNA level (heatmap in Fig. 4c) and unprocessed data in Fig. S6†). Neovascularization-related genes mRNA levels were comparable for FNS either in a mono-culture, or in co-culture with a microvessel (hCMEC/D3, HUVECs or HMVECs-L cells), that we defined as the reference conditions. In contrast, most of the neovascularization-related gene mRNA levels increased when endothelial cells are co-cultured with all glioma cells studied. Ang1 and Ang2 are modulators of endothelial permeability and barrier function via Tie2.52–54 In all cases, Ang1 was downregulated, suggesting disruption of vascular stability. VEGFR1 and VEGFR2 upregulation were detectable in selected samples, consistent with the previous report for endothelial cells of the tumour neovasculature and in normal brain vessels adjacent to tumours.55 On the other hand, the production of matrix-degrading proteases, particularly MMPs, by endothelial cells is a critical event during angiogenesis.56 It is interesting that MMP-2 and MMP-9 were noticeably upregulated in HUVEC, but not in the hCMEC/D3 and HMVECs-L co-cultures. Cross-comparing the heat map patterns for the three endothelial cells, we found that the response of endothelial cells to the co-cultures was GSC-specific and endothelial cell-specific. In particular, HUVECs seem to show more neovascularization susceptibility compared to hCMEC/D3 and HMVEC-L. These findings might reflect the nature of the endothelial cell types described by Uwamori et al., where the authors reported that HUVECs had greater potential than brain microvessel endothelial cells in terms of vascular formation.57
Cancer cell migration dynamics studied by live-cell imaging
We coupled the 3D microfluidic co-culture system with live-cell imaging to track cell behaviours in specific microvessel types (Fig. 5a)). GFP-tagged patient-derived GBM cells (G166) were used to observe their behaviour in an artificial vascular niche. In every 3D device, we monitored the behaviour of 5–15 cancer cells, in a field of view localized at the interface between the outermost side channel and collagen I-based hydrogel chamber. The cancer cell speed (micron/min) in the 3D device with a microvessel is higher compared to a microvessel-free device (Fig. 5b)). Between the three organ-specific endothelial cells, a hCMEC/D3 microvessel induced the fastest movement of cancer cells. We also measured to see if the entire cancer cell population (in the FOV) had a preferred direction of migration towards the endothelial cells (right to left). The velocity components perpendicular to the ECM–endothelium barrier (x-axis as shown in Fig. 5a)) and along the ECM chamber were analysed (Fig. 5c)). Cancer cells in the device with a microvessel were shown to move towards the channel interface direction more (i.e. with more negative x-velocity component) compared to the microvessel-free control (Fig. 5c)). But there was no difference detected in the y-direction (Fig. 5d)). Comparing the data among the three microvessel types, a slight enhancement in directed migration was observed for the cancer cells facing the hCMEC/D3 vessels. | ||
| Fig. 5 G166 cells cultured in the presence of a hCMEC/D3 microvessel with flow are characterised by higher speed, velocity, and preferential polarisation in the direction towards the microvessel. a) Sample images showing tracking of the cell movement in 3 h time intervals. b) Speed of the cancer cells for the reference and microvascular systems; c) cancer cell velocity component along the x-axis; d) cancer cell velocity component along the y-axis; e) polarisation magnitude of the cancer cells in the collagen matrix; f) cellular polarisation component along the x-axis; g) cellular polarisation component along the y-axis. x-Axis is situated perpendicular to the barrier, pointing away from the barrier, and y-axis is along the ECM gel chamber. Scale bar = 100 μm. Note that ‘Ref’ samples are measured in microvessel-free device and all data are obtained from three independent experimental runs. | ||
Cancer cell shape axial asymmetries were also investigated during cellular migration inside the collagen I matrix. Brain tumour cells in the presence of hCMEC/D3 microvessel were characterized by the highest median of polarization parameter, as a scalar quantity (Fig. 5e)). As illustrated in Fig. 5f) and g), investigation of cell polarization in x and y directions further indicated that cancer cells appear to favour shifting their mass centre towards the microvessel. According to the mRNA expression pattern shown in Fig. 4c), G166 induces insignificant angiogenic expression modification to hCMEC/D3 and HMVEC-L, in contrast to that of HUVECs (which shows elevated angiogenic expression). Thus, based on the G166 migration when co-cultured with three types of microvessel, we probed the relative effects of elevated angiogenic expression, versus ‘organ-specificity’ of the vessel, could potentially drive cell migration. Interestingly, based on the migration assay shown in Fig. 5c) and f), organ specificity of the microvessel seems to overwhelm the effects of the elevated angiogenic expression of the microvessel.
Microvessel permeability in co-culture
The permeability of the microvessel serves as an indicator for the endothelial barrier changes due to the presence of cancer cells. Here, we measured the changes in the permeability of vessels formed by the three organ-specific endothelial cell types cultured in 3D ECM microfluidic devices in the presence of GSCs (G166 or GCGR-E13) or ECM gel only.We used the G166 cancer cells as we assessed the cell movement using this cell line, and the GCGR-E13 cells because of the previous characterization for their stemness-related gene expression in 3D ECM culture. Permeability of all three types of endothelium increased in the co-culture setup in comparison to a mono-culture of microvessel, as shown in Fig. 6a) i) and b) i).
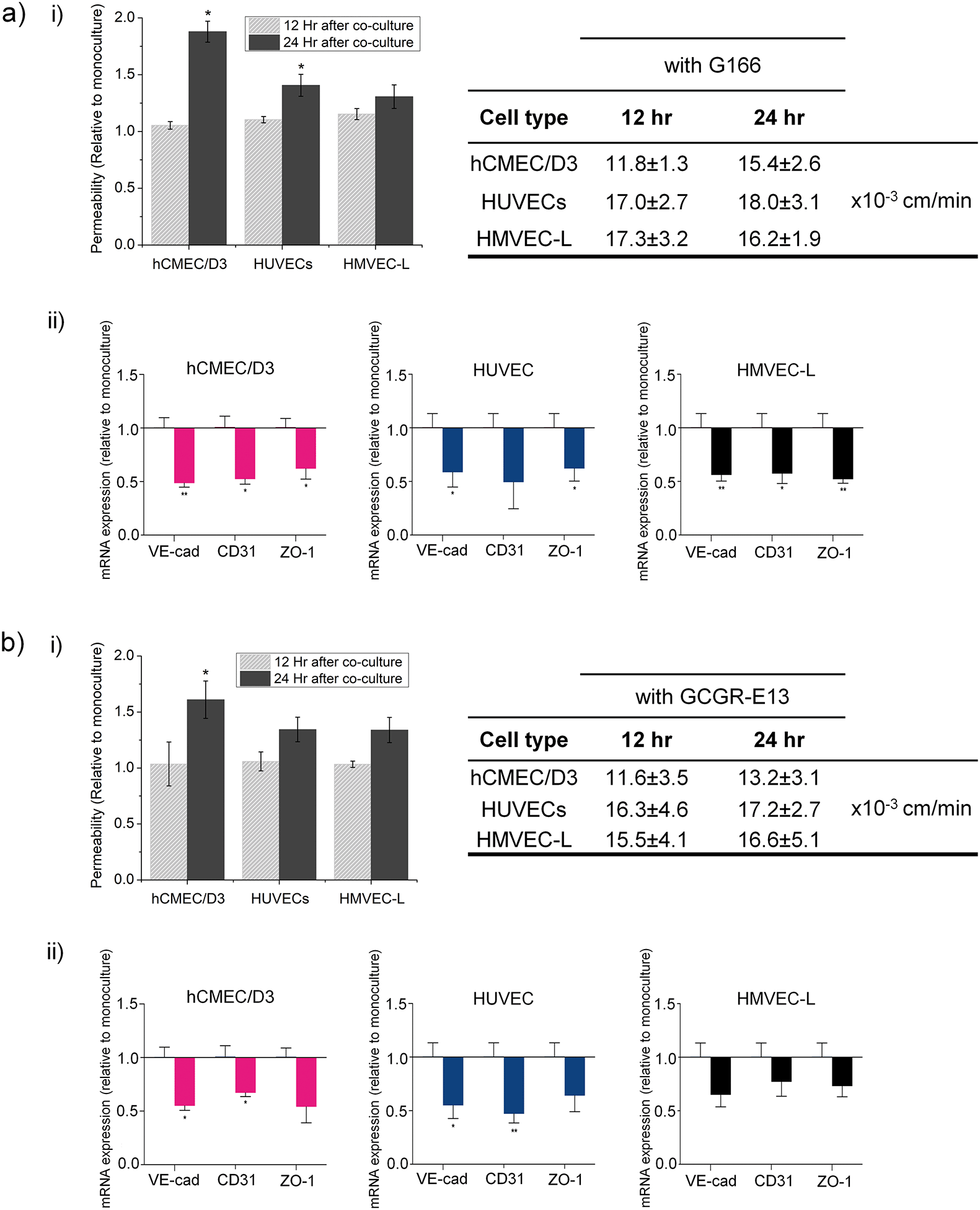 | ||
| Fig. 6 Increased endothelial permeability and the downregulation in mRNA expressions for junction proteins VE-cad, CD31, and ZO-1 in co-cultures with a) G166 and b) GCGR-E13 cancer cells. i) There is an increase in endothelial permeability in co-cultures with G166 and GCGR-E13 cancer cells. The graph shows results from three independent experimental runs as mean ± SEM. Two-tailed t-test was used for significance. The permeability increase correlates with downregulation of tightness-related genes in vessels formed by different vessel types; ii) mRNA expression normalised to samples of endothelial cells cultured without brain cancer cells; results obtained from three independent experimental runs are presented as mean ± SEM. Two-tailed t-test was used for significance. | ||
Since permeability in all microvessel types increased in the presence of cancer cells in the microfluidic devices, it was also of interest to learn how that corresponded to disruption in junction connections between endothelial cells by gene expression. Therefore, after 24 h of cultures, RNA was isolated from endothelial cells recovered from microfluidic chips cultured with or without GBM cells. For consistency, G166 and GCGR-E13 cells were used to conduct those experiments. Analysis of mRNA levels of VE-cad, CD31 and ZO-1 revealed downregulation of those genes in all three types of endothelial cells in the co-culture setup (Fig. 6a) ii) and b) ii)). This confirms the disruption in the tightness of the microvessels seen in the permeability studies.
Several mechanisms exist which could explain changes in permeability due to tumour cell interactions, such as tumour cells locally disrupting endothelial monolayer by contact58–60 or through secretion of chemical factors, which then compromises the endothelial barrier function. At the early stages, no cell-endothelium contact was observed during live-cell imaging in the microfluidic system presented here. Thus, we speculate that the changes in endothelial permeability could result from the secretion of certain cytokines.3,61–63 In future work, it would be necessary to analyse the secretome, as well as the cargo carried in extracellular vesicles released by given glioblastoma stem-like cells, to understand what triggers the changes in the permeability of endothelial cells in co-culture.
Conclusions and perspective
To summarize, the interactions between patient-derived GSC cell lines and microvessels generated from human endothelial cells of hCMEC/D3, HUVECs and HMVEC-L were studied utilizing a microvessel-on-chip platform. A serum-free condition, along with a gravity-driven flow system, was used to maintain endothelium–cancer cell co-culture, and protocols for cell retrieval and live-cell imaging were optimized, thus creating a microfluidic-based assay that could be used for studying GSCs and microvessel interaction. Within a 3D culture, hCMEC/D3 was observed to form a tighter, and therefore more favourable, artificial microvessel than HUVECs or HMVECs-L. It was also shown that endothelial cells grown in the serum-free medium created a tubular structure in the microfluidic devices, and the formed layer was characterized by tight junction expression and was acceptable for artificial microvessel permeability; thus, overcoming the drawback of using two different media with various growth factors in one experimental setup.Having optimized the microfluidic platform for GSC–microvessel co-culture, it was found that different cell types (patient-derived GSCs, U87, foetal neural stem cells) influence gene expression patterns in endothelial cells of three tissue origins, in that various signalling pathways, might be involved in initial neovascularization processes depending on brain cell type. Moreover, live-cell imaging and image analysis revealed that brain tumour cells are characterized by increased speed and velocity in the direction of the endothelial lining. Analysis of cell polarization further confirmed an asymmetric shift in cell bodies direction towards the vessel. Noticeably, GSCs cultured in the presence of hCMEC/D3 migrated with a higher speed towards the endothelial barrier than in the reference or other microvessel systems.
The presented 3D co-culture microsystem, optimized for both endpoint analysis and live-cell imaging, would be beneficial for future studies of vessel co-option. This proposed microfluidic model, with rapid co-culture formation (∼1 day), might be used for improved throughput of various patient-derived GBM cells and to monitor their behaviour in a model perivascular niche in a highly controllable environment. Further, quantifying parameters, like molecular markers of GBM cells, their speed, polarization, and influence on endothelium, might provide answers on the relation between, for instance, specific genetic and epigenetic alterations of GBM cells and the mechanisms of neovascularization they utilize.
The optimisation of the customizable microvessel-on-a-chip device provided additional complexity for in vitro systems and the opportunity to study the influence of glioma cells on normal brain endothelium. Our microfluidic protocol for forming in vitro models of patient-specific glioblastoma–vascular niches has demonstrated the possibilities to conduct comparative studies to dissect the influence of 3D culture, microvessel architecture and organotypic vessel types on glioma cells' stemness and migration. The possibility to create various customized tumour microenvironments (e.g., choice of ECM components and stiffness, microvessel size and flow rate) might allow clinicians to have the tool to study the tumour responses in differing microenvironment cues, which could lead to swift testing of new drugs and therapeutic approaches to expand GBM treatment options.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council (EPSRC, EP/S009000/1), European Research Council (ERC-StG 758865, ERC advanced grant REP-789054-1), National Institute for Health Research (NIHR), Cancer Research UK (CRUK), Academy of Medical Sciences, and Cambridge NIHR BRC. M. G. was a recipient of the CRUK Multidisciplinary Studentship. H. B. was a recipient of the CRUK Innovator Award. The GBM cell lines were obtained from the Glioma Cellular Genetics Resource (http://www.gcgr.org.uk) funded by Cancer Research UK. We thank Prof. Steve Pollard in facilitating the materials transfer. We would also like to thank Charles Cullen for his assistance in establishing the gravity-driven flow for the experiments and Prof. Anne Ridley for advice on the endothelial cell culture. Biorender was used for schematics.References
- M. Quaresma, M. P. Coleman and B. Rachet, Lancet, 2015, 385, 1206–1218 CrossRef.
- A. Roos, Z. Ding, J. C. Loftus and N. L. Tran, Front. Oncol., 2017, 7, 1 Search PubMed.
- R. K. Jain, Cancer Cell, 2014, 26, 605–622 CrossRef CAS PubMed.
- M. E. Hardee and D. Zagzag, Am. J. Pathol., 2012, 181, 1126–1141 CrossRef CAS PubMed.
- T. Donnem, A. R. Reynolds, E. A. Kuczynski, K. Gatter, P. B. Vermeulen, R. S. Kerbel, A. L. Harris and F. Pezzella, Nat. Rev. Cancer, 2018, 18, 323–336 CrossRef CAS PubMed.
- G. Seano and R. K. Jain, Angiogenesis, 2020, 23, 9–16 CrossRef PubMed.
- S. Nath and G. R. Devi, Pharmacol. Ther., 2016, 163, 94–108 CrossRef CAS.
- F. Weeber, S. N. Ooft, K. K. Dijkstra and E. E. Voest, Cell Chem. Biol., 2017, 24, 1092–1100 CrossRef CAS PubMed.
- J. M. Ayuso, M. Virumbrales-Muñoz, A. Lacueva, P. M. Lanuza, E. Checa-Chavarria, P. Botella, E. Fernández, M. Doblare, S. J. Allison, R. M. Phillips, J. Pardo, L. J. Fernandez and I. Ochoa, Sci. Rep., 2016, 6, 36086 CrossRef CAS.
- J. M. Ayuso, R. Monge, A. Martínez-González, M. Virumbrales-Muñoz, G. A. Llamazares, J. Berganzo, A. Hernández-Laín, J. Santolaria, M. Doblaré, C. Hubert, J. N. Rich, P. Sánchez-Gómez, V. M. Pérez-García, I. Ochoa and L. J. Fernández, Neuro-Oncology, 2017, 19, 503–513 CAS.
- J. Ma, N. Li, Y. Wang, L. Wang, W. Wei, L. Shen, Y. Sun, Y. Jiao, W. Chen and J. Liu, Biomed. Microdevices, 2018, 20, 80 CrossRef PubMed.
- D. Truong, R. Fiorelli, E. S. Barrientos, E. L. Melendez, N. Sanai, S. Mehta and M. Nikkhah, Biomaterials, 2019, 198, 63–77 CrossRef CAS PubMed.
- Y. Xiao, D. Kim, B. Dura, K. Zhang, R. Yan, H. Li, E. Han, J. Ip, P. Zou, J. Liu, A. T. Chen, A. O. Vortmeyer, J. Zhou and R. Fan, Adv. Sci., 2019, 6(8), 1801531 CrossRef PubMed.
- A. Torsvik, D. Stieber, P. Ø. Enger, A. Golebiewska, A. Molven, A. Svendsen, B. Westermark, S. P. Niclou, T. K. Olsen and M. Chekenya Enger, Cancer Med., 2014, 3, 812–824 CrossRef CAS.
- M. Allen, M. Bjerke, H. Edlund, S. Nelander and B. Westermark, Sci. Transl. Med., 2016, 8, 354re3 CrossRef PubMed.
- P. F. Ledur, G. R. Onzi, H. Zong and G. Lenz, Oncotarget, 2017, 8, 69185 CrossRef PubMed.
- M. Campisi, Y. Shin, T. Osaki, C. Hajal, V. Chiono and R. D. Kamm, Biomaterials, 2018, 180, 117–129 CrossRef CAS.
- S. I. Ahn, Y. J. Sei, H.-J. Park, J. Kim, Y. Ryu, J. J. Choi, H.-J. Sung, T. J. MacDonald, A. I. Levey and Y. Kim, Nat. Commun., 2020, 11, 1–12 Search PubMed.
- G. Adriani, D. Ma, A. Pavesi, R. D. Kamm and E. L. K. Goh, Lab Chip, 2017, 17, 448–459 RSC.
- M. S. Choe, J. S. Kim, H. C. Yeo, C. M. Bae, H. J. Han, K. Baek, W. Chang, K. S. Lim, S. P. Yun and I. Shin, FASEB J., 2020, 34(12), 16464–16475 CrossRef CAS PubMed.
- S. Bian, M. Repic, Z. Guo, A. Kavirayani, T. Burkard, J. A. Bagley, C. Krauditsch and J. A. Knoblich, Nat. Methods, 2018, 15, 631–639 CrossRef CAS.
- C. Bertulli, M. Gerigk, N. Piano, Y. Liu, D. Zhang, T. Müller, T. J. Knowles and Y. Y. S. Huang, Sci. Rep., 2018, 8, 12480 CrossRef PubMed.
- I. J. Huijbers, M. Iravani, S. Popov, D. Robertson, S. Al-Sarraj, C. Jones and C. M. Isacke, PLoS One, 2010, 5, e9808 CrossRef PubMed.
- S. K. Chintala, Z. L. Gokaslan, Y. Go, R. Sawaya, G. L. Nicolson and J. S. Rao, Clin. Exp. Metastasis, 1996, 14, 358–366 CrossRef CAS PubMed.
- A. C. Bellail, S. B. Hunter, D. J. Brat, C. Tan and E. G. Van Meir, Int. J. Biochem. Cell Biol., 2004, 36, 1046–1069 CrossRef CAS PubMed.
- S. M. Pollard, K. Yoshikawa, I. D. Clarke, D. Danovi, S. Stricker, R. Russell, J. Bayani, R. Head, M. Lee and M. Bernstein, Cell Stem Cell, 2009, 4, 568–580 CrossRef CAS PubMed.
- K. Funamoto, D. Yoshino, K. Matsubara, I. K. Zervantonakis, K. Funamoto, M. Nakayama, J. Masamune, Y. Kimura and R. D. Kamm, Integr. Biol., 2017, 9, 529–538 CrossRef CAS PubMed.
- Y. Liu, E. Gill and Y. Y. Shery Huang, Future Sci. OA, 2017, 3, FSO173 CrossRef CAS PubMed.
- S. Brem, R. Cotran and J. Folkman, J. Natl. Cancer Inst., 1972, 48, 347–356 CAS.
- D. N. Louis, H. Ohgaki, O. D. Wiestler, W. K. Cavenee, P. C. Burger, A. Jouvet, B. W. Scheithauer and P. Kleihues, Acta Neuropathol., 2007, 114, 97–109 CrossRef PubMed.
- R. Galli, E. Binda, U. Orfanelli, B. Cipelletti, A. Gritti, S. De Vitis, R. Fiocco, C. Foroni, F. Dimeco and A. Vescovi, Cancer Res., 2004, 64, 7011–7021 CrossRef CAS PubMed.
- X. Hong, K. Chedid and S. N. Kalkanis, Int. J. Oncol., 2012, 41, 1693–1700 CrossRef CAS PubMed.
- A. Kiviniemi, M. Gardberg, J. Frantzén, R. Parkkola, V. Vuorinen, M. Pesola and H. Minn, J. Neuro-Oncol., 2015, 124, 237–245 CrossRef CAS PubMed.
- D. Lv, S. C. Yu, Y. F. Ping, H. Wu, X. Zhao, H. Zhang, Y. Cui, B. Chen, X. Zhang, J. Dai, X. W. Bian and X. H. Yao, Oncotarget, 2016, 7, 56904–56914 CrossRef PubMed.
- W. Jia, X. Jiang, W. Liu, L. Wang, B. Zhu, H. Zhu, X. Liu, M. Zhong, D. Xie, W. Huang, W. Jia, S. Li, X. Liu, X. Zuo, D. Cheng, J. Dai and C. Ren, Int. J. Oncol., 2018, 52, 1787–1800 CAS.
- M. Virumbrales-Muñoz, J. M. Ayuso, A. Lacueva, T. Randelovic, M. K. Livingston, D. J. Beebe, S. Oliván, D. Pereboom, M. Doblare, L. Fernández and I. Ochoa, Sci. Rep., 2019, 9, 6199 CrossRef PubMed.
- A. Vartanian, S. K. Singh, S. Agnihotri, S. Jalali, K. Burrell, K. D. Aldape and G. Zadeh, Neuro-Oncology, 2014, 16, 1167–1175 CrossRef PubMed.
- D. V. Brown, S. S. Stylli, A. H. Kaye and T. Mantamadiotis, in Advances in Experimental Medicine and Biology, Springer, New York LLC, 2019, vol. 1139, pp. 1–21 Search PubMed.
- V. Bramanti, D. Tomassoni, M. Avitabile, F. Amenta and R. Avola, Front. Biosci., 2010, 2, 558–570 Search PubMed.
- S. Bien-Möller, E. Balz, S. Herzog, L. Plantera, S. Vogelgesang, K. Weitmann, C. Seifert, M. A. Fink, S. Marx, A. Bialke, C. Venugopal, S. K. Singh, W. Hoffmann, B. H. Rauch and H. W. S. Schroeder, Stem Cells Int., 2018, 2018, 9628289 Search PubMed.
- G. Cattoretti, M. H. G. Becker, G. Key, M. Duchrow, C. Schlüuter, J. Galle and J. Gerdes, J. Pathol., 1992, 168, 357–363 CrossRef CAS PubMed.
- J. D. Lathia, S. C. Mack, E. E. Mulkearns-Hubert, C. L. L. Valentim and J. N. Rich, Genes Dev., 2015, 29, 1203–1217 CrossRef CAS PubMed.
- D. Garnier, O. Renoult, M. C. Alves-Guerra, F. Paris and C. Pecqueur, Front. Oncol., 2019, 9, 118 CrossRef PubMed.
- A. Griveau, G. Seano, S. J. Shelton, R. Kupp, A. Jahangiri, K. Obernier, S. Krishnan, O. R. Lindberg, T. J. Yuen and A.-C. Tien, Cancer Cell, 2018, 33, 874–889 CrossRef CAS PubMed.
- K. Ebnet, C. U. Schulz, M. K. Meyer Zu Brickwedde, G. G. Pendl and D. Vestweber, J. Biol. Chem., 2000, 275, 27979–27988 CrossRef CAS PubMed.
- A. S. Fanning and J. M. Anderson, Ann. N. Y. Acad. Sci., 2009, 1165, 113–120 CrossRef CAS PubMed.
- C. Greene, N. Hanley and M. Campbell, Fluids Barriers CNS, 2019, 16, 3 CrossRef PubMed.
- T. D. Brown, M. Nowak, A. V. Bayles, B. Prabhakarpandian, P. Karande, J. Lahann, M. E. Helgeson and S. Mitragotri, Bioeng. Transl. Med., 2018, 4(2), e10126 Search PubMed.
- I. K. Zervantonakis, S. K. Hughes-Alford, J. L. Charest, J. S. Condeelis, F. B. Gertler and R. D. Kamm, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13515–13520 CrossRef PubMed.
- A. D. Wong and P. C. Searson, Cancer Res., 2014, 74, 4937–4945 CrossRef CAS PubMed.
- J. S. Jeon, I. K. Zervantonakis, S. Chung, R. D. Kamm and J. L. Charest, PLoS One, 2013, 8, e56910 CrossRef CAS PubMed.
- N. Jones, Z. Master, J. Jones, D. Bouchard, Y. Gunji, H. Sasaki, R. Daly, K. Alitalo and D. J. Dumont, J. Biol. Chem., 1999, 274, 30896–30905 CrossRef CAS.
- I. Kim, H. G. Kim, J. N. So, J. H. Kim, H. J. Kwak and G. Y. Koh, Circ. Res., 2000, 86, 24–29 CrossRef CAS PubMed.
- H. T. Yuan, E. V. Khankin, S. A. Karumanchi and S. M. Parikh, Mol. Cell. Biol., 2009, 29, 2011–2022 CrossRef CAS.
- J. H. Harmey and D. Stefanik, in VEGF and Cancer, 2004, pp. 72–82 Search PubMed.
- G. Taraboletti, S. D'Ascenzo, P. Borsotti, R. Giavazzi, A. Pavan and V. Dolo, Am. J. Pathol., 2002, 160, 673–680 CrossRef CAS.
- H. Uwamori, Y. Ono, T. Yamashita, K. Arai and R. Sudo, Microvasc. Res., 2019, 122, 60–70 CrossRef CAS.
- Y. H. Li and C. Zhu, Clin. Exp. Metastasis, 1999, 17, 423–429 CrossRef CAS.
- S. Kumar and V. M. Weaver, Cancer Metastasis Rev., 2009, 28, 113–127 CrossRef.
- C. T. Mierke, J. Biol. Chem., 2011, 286, 40025–40037 CrossRef CAS PubMed.
- S. Bao, Q. Wu, R. E. McLendon, Y. Hao, Q. Shi, A. B. Hjelmeland, M. W. Dewhirst, D. D. Bigner and J. N. Rich, Nature, 2006, 444, 756–760 CrossRef CAS PubMed.
- C. Folkins, Y. Shaked, S. Man, T. Tang, C. R. Lee, Z. Zhu, R. M. Hoffman and R. S. Kerbel, Cancer Res., 2009, 69, 7243–7251 CrossRef CAS PubMed.
- L. Treps, R. Perret, S. Edmond, D. Ricard and J. Gavard, J. Extracell. Vesicles, 2017, 6, 1359479 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1lc00271f |
| This journal is © The Royal Society of Chemistry 2021 |