
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantifying nanotherapeutic penetration using a hydrogel-based microsystem as a new 3D in vitro platform†
Saba
Goodarzi
 a,
Audrey
Prunet
a,
Fabien
Rossetti
a,
Guillaume
Bort
a,
Olivier
Tillement
a,
Erika
Porcel
b,
Sandrine
Lacombe
b,
Ting-Di
Wu
cd,
Jean-Luc
Guerquin-Kern
cd,
Hélène
Delanoë-Ayari
a,
François
Lux
ae and
Charlotte
Rivière
*ae
a,
Audrey
Prunet
a,
Fabien
Rossetti
a,
Guillaume
Bort
a,
Olivier
Tillement
a,
Erika
Porcel
b,
Sandrine
Lacombe
b,
Ting-Di
Wu
cd,
Jean-Luc
Guerquin-Kern
cd,
Hélène
Delanoë-Ayari
a,
François
Lux
ae and
Charlotte
Rivière
*ae
aUniversity of Lyon, Université Claude Bernard Lyon 1, CNRS, Institut Lumière Matière, F-69622, Villeurbanne, France. E-mail: charlotte.riviere@univ-lyon1.fr
bUniversité Paris-Saclay, CNRS, Institut des Sciences Moléculaires d'Orsay, 91405, Orsay, France
cInstitut Curie, Université PSL, Paris, France
dUniversité Paris-Saclay, CNRS, Inserm, Centre d'Imagerie Multimodale, 91401, Orsay, France
eInstitut Universitaire de France (IUF), France
First published on 7th June 2021
Abstract
The huge gap between 2D in vitro assays used for drug screening and the in vivo 3D physiological environment hampered reliable predictions for the route and accumulation of nanotherapeutics in vivo. For such nanotherapeutics, multi-cellular tumour spheroids (MCTS) are emerging as a good alternative in vitro model. However, the classical approaches to produce MCTS suffer from low yield, slow process, difficulties in MCTS manipulation and compatibility with high-magnification fluorescence optical microscopy. On the other hand, spheroid-on-chip set-ups developed so far require a practical knowledge of microfluidics difficult to transfer to a cell biology laboratory. We present here a simple yet highly flexible 3D model microsystem consisting of agarose-based microwells. Fully compatible with the multi-well plate format conventionally used in cell biology, our simple process enables the formation of hundreds of reproducible spheroids in a single pipetting. Immunostaining and fluorescence imaging including live high-resolution optical microscopy can be performed in situ, with no manipulation of spheroids. As a proof of principle of the relevance of such an in vitro platform for nanotherapeutic evaluation, this study investigates the kinetics and localisation of nanoparticles within colorectal cancer MCTS cells (HCT-116). The nanoparticles chosen are sub-5 nm ultrasmall nanoparticles made of polysiloxane and gadolinium chelates that can be visualized in MRI (AGuIX®, currently implicated in clinical trials as effective radiosensitizers for radiotherapy) and confocal microscopy after addition of Cy5.5. We show that the amount of AGuIX® nanoparticles within cells is largely different in 2D and 3D. Using our flexible agarose-based microsystems, we are able to resolve spatially and temporally the penetration and distribution of AGuIX® nanoparticles within MCTS. The nanoparticles are first found in both extracellular and intracellular space of MCTS. While the extracellular part is washed away after a few days, we evidenced intracellular localisation of AGuIX®, mainly within the lysosomal compartment, but also occasionally within mitochondria. Hence, our agarose-based microsystem appears as a promising 3D in vitro user-friendly platform for investigation of nanotherapeutic transport, ahead of in vivo studies.
Introduction
There is an ongoing effort to develop efficient therapeutics for cancer treatment including nanodrugs and nanoparticles; nevertheless, the clinical translation of these therapeutics has to overcome numerous challenges from the early stages of development to a successful translation.1,2 Currently, the standard pipeline for drug development consists of the following: (1) efficacy tests on 2D in vitro assays and (2) on rodent in vivo models, (3) regulatory toxicity tests on two animal species and (4) clinical trials.However, 2D in vitro assays do not replicate the 3D physiological environment encountered by the cells in vivo. This could be the underlying reason for the high rate of clinical failure in the development of new drugs. On the other hand, there is also a rising question as to the economical and ethical relevance of rodent animal models, in particular because such models are not fully representative of human specificity.3 Getting as close as possible to the in vivo situation in in vitro models is a key issue to truly understand and control cancer cell response, accompanied by reduction in animal usage. For the pharmaceutical industry, tackling this issue will enable better identification of relevant therapeutics by performing relevant screening on 3D models. For precision medicine, it will help physicians to adjust the therapeutic treatment to complement current clinical analysis.2,4 For fundamental research, it will allow deciphering cell response in a truly relevant context.
Many approaches have been developed during the past decade to set up various organ-on-a-chip or tumour-on-a-chip devices, integrating many different in vivo features in a miniaturized in vitro format.5,6 This is particularly important for emerging nanosized therapeutics.7 The presence of different physiological barriers, such as cell–cell compaction, tumour heterogeneity and dense extracellular matrix along with various cancer-associated cells, will decrease the amount of nanotherapeutics effectively reaching the targeted tumour cells.8,9 The lack of such a physiological context hampered reliable predictions for the route and accumulation of these nanoparticles in vivo10 and is a major limitation for the efficient development of novel therapeutic approaches.11 To move beyond the classical 2D plastic dishes, different 3D in vitro models have been developed to try to better replicate in vivo the complexity of the tumour microenvironment.12 Among them, multi-cellular tumour spheroids (MCTS) recapitulate many tumour features, including 3D cellular architecture, cellular heterogeneity, signalling pathways and physiochemical gradient, similar to real in vivo tumour micrometastasis (for spheroids >500 μm in diameter).13–17
MCTS could be prepared with various techniques13 such as using non-adherent surfaces,18 spinner flasks19 or hanging drop methods.20 Emerging attempts to integrate spheroids in microfluidic set-ups open up new possibilities to deal with the low yield and slow process of the classical approach.21,22 However, such spheroid-on-chip approaches require a practical knowledge of microfluidics that is difficult to transfer to a cell biology laboratory.
In addition, the polymeric materials commonly used for such devices (polydimethylsiloxane –PDMS) suffer from major limitations, precluding their usage for efficient drug screening under physiological conditions:23 large absorption of therapeutics24,25 (resulting in the underestimation of cell response to drugs), non-permeability to small water-soluble molecules (leading to fast medium conditioning if continuous flow is not provided otherwise), rigidity several orders of magnitude larger than the physiological condition (MPa vs. kPa range in vivo26).
To go beyond PDMS and its limitations, hydrogel-based microwell devices have been considered.27,28 Hydrogels are networks of cross-linked polymers with tuneable physical properties, a high capacity for water retention and interconnected pores enabling free diffusion of O2, nutrient and metabolic wastes, which make them favourable alternatives in micro-system applications. Various techniques using natural or synthetic hydrogels for MCTS formation have been developed.28–33 However, none of these set-ups meets all the criteria required for long-term time-lapse analysis (i.e. compatibility with high-resolution video-microscopy, efficient medium and oxygen renewal, in situ immunostaining/drug application, no reduction of the available drug dose, easy cell retrieval for further standard molecular analyses) within a physiological stiffness range.
We present here a simple yet highly flexible 3D model microsystem consisting of agarose-based microwells. This hydrogel with tuneable rigidity and great integrity presents several advantages, making it a suitable biomaterial in cell studies.34,35 The tuneable mechanical properties of the agarose can reproduce the in vivo microenvironment stiffness. Its porous nature enables the free diffusion of salt and small chemical species (hydrodynamic diameter <30 nm in 2% agarose,36 which is the case for most proteins). Our simple process enables the formation of hundreds of reproducible spheroids in a single pipetting, and its compatibility with multi-well plate formats conventionally used in cell biology can accelerate the screening of drugs in comparison with conventional 3D models. Of note, these microwells can also be manufactured on coverslips, opening the possibility for live high-resolution optical microscopy. In addition, hydrogel-based microwells provide a user-friendly platform for in situ immunostaining and can be used for in-depth analysis of cell phenotypic modifications after drug treatment.
As a proof of principle of the relevance of such an in vitro platform for the evaluation of nanoparticle screening, the aim of this study was to analyse the kinetics and localisation of these nanoparticles within colorectal cancer cell MCTS (HCT-116). The nanoparticles chosen for this proof-of-concept study are sub-5 nm ultrasmall nanoparticles made of polysiloxane and gadolinium (Gd) chelates that can be visualized in MRI and confocal microscopy (after functionalization by Cy5.5, a near-infrared fluorophore). These nanoparticles, called AGuIX®, are effective radiosensitizers for radiotherapy37 and are now implicated in three clinical trials associating radiotherapy with AGuIX® for treatment of multiple brain metastases by whole brain radiation therapy (NanoRad 2, phase II, multicentric), stereotactic radiosurgery (NanoStereo, phase II, multicentric) and cervical cancer (phase Ib, Gustave Roussy). The nanoparticle–cell interactions and internalization pathways of these nanoparticles have been assessed in vitro in 2D,38 but never in 3D, multicellular tumour spheroids.
We show in this study that the 3D cell arrangement highly impacts the amount of AGuIX® nanoparticles within cells. Using our flexible agarose-based microsystem, we were able to resolve spatially and temporally the penetration and distribution of AGuIX® nanoparticles within tumour spheroids. The nanoparticles were first found in both the extracellular and the intracellular space of spheroids, mostly within the lysosomal compartment, but also occasionally within mitochondria. Whereas the extracellular part was washed away after a few days, the colocalisation with lysosomes remained almost constant. Our agarose-based microsystem hence appears as a promising 3D in vitro platform for investigation of nanotherapeutic transport, ahead of in vivo studies.
Materials and methods
Hydrogel based microsystem
Agarose-based microsystems were prepared using moulding procedures. First, a silicon wafer mould was made using a classical photolithography technique (Fig. 1A). The mould consists of an array of 130 cylindrical wells of 200 μm in diameter and 250 μm in height, created using the SU8-2100 photosensitive resin.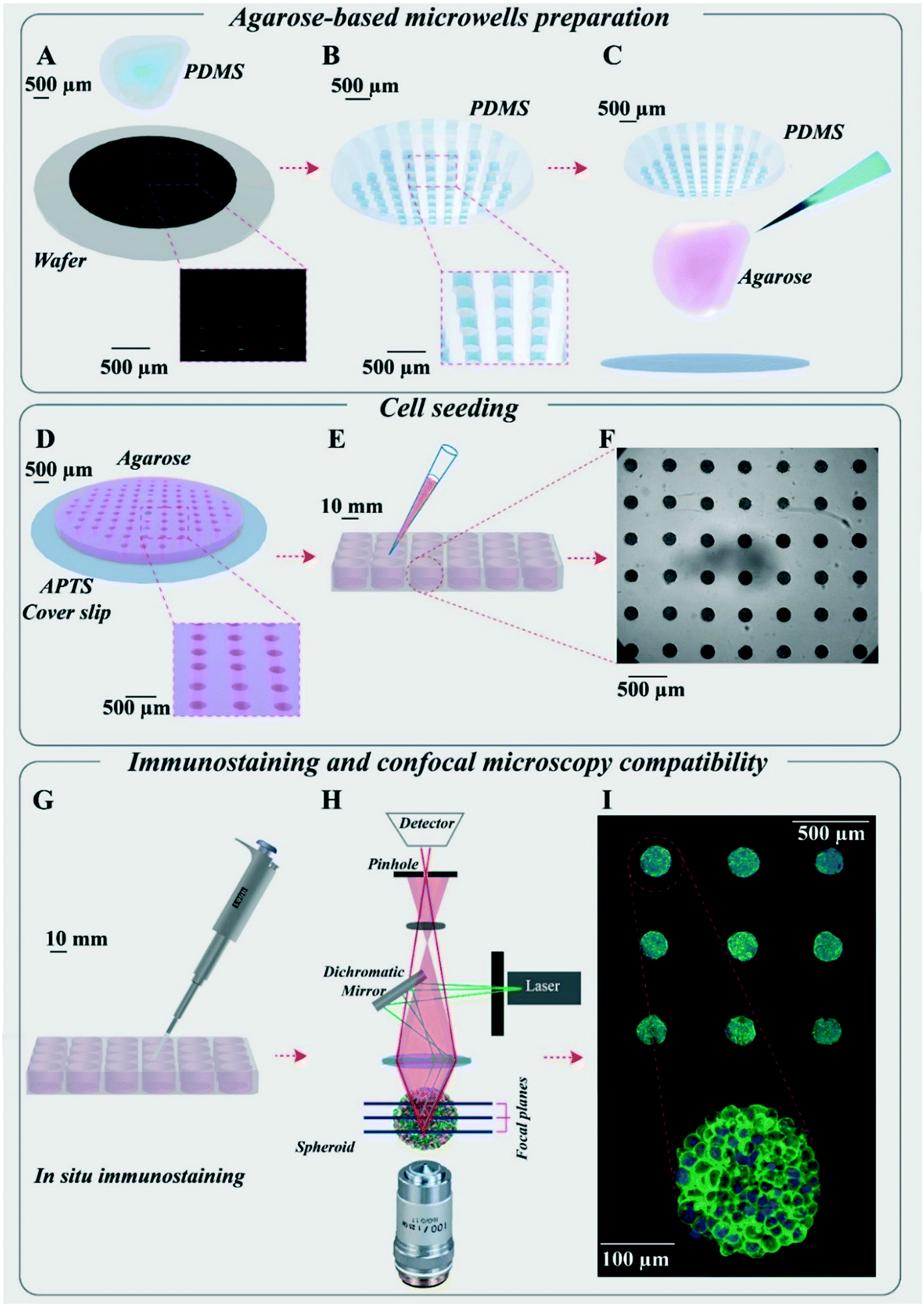 | ||
| Fig. 1 Presentation of the hydrogel-based microsystems for spheroid growth and follow-up. (A) Silicon wafer mould made by photolithography. (B) PDMS replica mould made from the silicon wafer mould. (C) Moulding of agarose using PDMS replica moulds on a coverslip functionalized by APTS to make the agarose microsystem adhesive on the coverslip. (D) Cylindrical agarose microwells with diameter and height of 200 μm for each microwell. (E) Cell seeding using the agarose microsystem in a 24-well plate for the preparation of spheroids (leading to the formation of 130 spheroids per well). (F) Optical microscopy images of several homogenous HCT-116 cell spheroids made in the agarose microsystem (5× magnification) at day 6. (G) In situ immunostaining of spheroids in microwells in a 24-well plate. (H) Diagram of confocal fluorescence microscopy. (I) Maximal image projection (MIP) of confocal fluorescence images of spheroids in the agarose microsystem labelled for actin (green) and nuclei (blue) (10× magnification) and an enlarged MIP of one of the spheroids (20× magnification). | ||
A polydimethylsiloxane (PDMS) replica mould was then casted on this master mould (Fig. 1B) and used for agarose moulding. The agarose moulding procedure differed depending on the aim of the experiments: (1) for imaging of fixed samples, the microwells were free-standing in each well of a multi-well plate, enabling easy retrieval and transfer (see detailed description below); (2) for time-lapse imaging, agarose moulding is performed on 3-aminopropyltriethoxisilane (APTS)-functionalised coverslips, enabling to directly bond the microwells to the coverslips and avoiding any drift during acquisition (Fig. 1D, patented process39).
Agarose solution (2%, w/v) was prepared by dissolving ultra-pure agarose powder (Invitrogen™) in water. An autoclave was used for the dissolution to avoid formation of bubbles (121 °C, 15 min).
Colorectal cancer cell line HCT-116 and culture conditions
HCT-116 colorectal carcinoma (CCL-247) cell line was purchased from the American Type Culture Collection (ATCC, Virginia, USA). All cells were cultured in Dulbecco's modified Eagle's medium (DMEM-Glutamax, Gibco™) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Sigma, St. Louis, Missouri, USA), 100 units/100 μg penicillin/streptomycin (Gibco™).Routinely, the HCT-116 cells were grown in T-25 cell culture flasks and were placed in an incubator at 37 °C with a 5% CO2 atmosphere. The culture medium was changed regularly, and cell passage was carried out at 70% confluency every 3 days. The cell passage was performed using recombinant cell-dissociation enzyme (TrypLE, Gibco™) to detach cells followed by neutralizing with culture medium. The cell suspension was centrifuged at 1000 rpm (equal to 106g) for 5 min, the supernatant was discarded, and the cell pellet was resuspended in 1 mL of complete culture medium. The number of cells was counted using a Neubauer chamber, and the final cell volume was adjusted to reach the desired cell concentration.
Multicellular tumour spheroids
MCTS of HCT-116 cells were formed in 24-well plates containing agarose in each well. After trypsinization and centrifugation, 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells in 1 mL complete medium was added in each well (each containing 1 microsystem). To encourage and accelerate cell aggregation, the 24-well plate was placed under orbital agitation (160 rpm) for 15 min in an incubator at 37 °C and 5% CO2. After 4 h, the plate was rinsed with fresh medium to remove cells that did not reach the microwells. After 2 days, spheroids were ready for incubation with nanoparticles.
000 cells in 1 mL complete medium was added in each well (each containing 1 microsystem). To encourage and accelerate cell aggregation, the 24-well plate was placed under orbital agitation (160 rpm) for 15 min in an incubator at 37 °C and 5% CO2. After 4 h, the plate was rinsed with fresh medium to remove cells that did not reach the microwells. After 2 days, spheroids were ready for incubation with nanoparticles.
Monolayer cell culture
After trypsinization and centrifugation of HCT-116 cells in culture, a cell suspension with 120000 cells in 1 ml was prepared. The cell suspension was added to tissue-treated coverslip plates (either 300 μL in an 8-well Ibidi® or 2 mL in a 12-well plate). Cells were incubated with nanoparticles 48 h after cell seeding.
Preparation of Cy5.5-conjugated gadolinium-based nanoparticles (AGuIX®-Cy5.5)
The Gd-based nanoparticles (AGuIX®) synthesized by NH TherAguix (Lyon, France) are composed of a polysiloxane matrix surrounded by covalently bound DOTAGA-Gd ((1,4,7,10-tetraazacyclodode-cane-1-glutaric acid-4,7,10-triacetic acid)-Gd). The synthesis process is already described in the literature.40 Briefly, AGuIX® nanoparticles are composed of a polysiloxane network surrounded by Gd chelates. The chemical composition of AGuIX® nanoparticles is (GdSi6.5N6C25O21H42·10H2O)n with a molar mass of around 10 kDa. The hydrodynamic diameter of the AGuIX® nanoparticles is close to 5 nm, and the AGuIX® nanoparticles are characterized by a zeta potential of 9.0 ± 5.5 mV at pH 7.2. These AGuIX® nanoparticles were further conjugated to cyanine-5.5 (Cy5.5) fluorophore to make them detectable by confocal fluorescence microscopy. They are referred to as AGuIX®-Cy5.5 nanoparticles in the rest of the article.Incubation of cells with AGuIX®-Cy5.5 nanoparticles
To incubate MCTS and monolayer cells with AGuIX®-Cy5.5 nanoparticles, an intermediate solution of AGuIX®-Cy5.5 nanoparticles with 100 mM concentration of Gd was prepared in distilled-water. From this intermediate solution, just before the incubation with cells, AGuIX®-Cy5.5 solutions were prepared in fresh DMEM with Gd concentrations of 0.8, 1.5 and 2 mM, respectively. The MCTS in all microsystems of a 24-well plate were incubated with 1 mL of AGuIX®-Cy5.5 nanoparticle solution. For cell monolayers, an Ibidi® 8-well plate or a 12-well plate was used, and cells were incubated with 200 μL or 2 mL AGuIX®-Cy5.5 solution, respectively.Inductively coupled plasma-mass spectrometry (ICP-MS)
The concentration of Gd was analysed using a validated inductively coupled plasma-mass spectrometry (ICP-MS) analysis. To prepare samples for this analysis, spheroids and monolayer cultured cells were incubated with AGuIX®-Cy5.5 nanoparticles with 0.8, 1.5 and 2 mM concentration in Gd for 24 h. After incubation, spheroids were rinsed three times with PBS for 15 min each and dissociated using trypsin + EDTA (Gibco). The number of cells in each microwell was evaluated using a Neubauer chamber. The cell suspensions in trypsin + EDTA of each sample were then centrifuged (900g for 5 min), the supernatants were discarded, and the cells pellets were dissolved in 150 μl 69% HNO3 (ROTH) at 80 °C for 3 h. The volume of samples was adjusted to 10 mL by adding ultra-pure water and the Gd concentration in each sample was measured using an ICP-mass spectrometer (PerkinElmer, NexION® 2000). A similar procedure was used for the monolayer cell culture (the cells were rinsed with PBS (3 × 5 min) and detached using trypsin (Gibco)).The mean value of the cell volume was calculated by measuring the cell diameter after detachment or dissociation using bright-field microscopy followed by image processing using ImageJ software.41 Accordingly, Gd concentrations obtained by ICP-MS measurements were divided by the calculated average cell volume.
Localisation of nanoparticles: fixation, permeabilization and immunostaining
First, cell nuclei and actin filaments in the cytoskeleton were labelled. After incubation with AGuIX®-Cy5.5 nanoparticles, the spheroids were rinsed with PBS (3 × 5 min), then fixed in paraformaldehyde (4%) for 20 min and permeabilized using 0.1% Triton X-100 (Acros) for 10 min. After blocking with 3% bovine serum albumin (BSA, Sigma-Aldrich) for 20 min, the samples were incubated with phalloidin-546 solution (Invitrogen™, A22283, 1:50 in PBS) containing NucGreen™-Dead 488 (Invitrogen™, R37109, 1 drop per 5 ml in PBS) at 4 °C overnight. The procedure ended with rinsing spheroids with PBS (3 × 5 min).
In a second series of experiments, to find out the precise intracellular localisation of AGuIX®-Cy5.5 nanoparticles, three antibodies were used to label the main cell compartments: EEA1 for early endosomes (Cell Signaling Technology, #3288), AIF for mitochondria (Cell Signaling Technology, #5318) and LAMP-1 for lysosomes (Cell Signaling Technology, #9091). After fixation in paraformaldehyde (4%) for 20 min and rinsing with PBS (3 × 5 min) according to the protocol proposed by the manufacturer, cells were blocked in a buffer (PBS/5% BSA/0.3% Triton™ X-100) for 60 min and rinsed with PBS (3 × 5 min). These samples, either spheroids in microwells or cell monolayers in Ibidi plates, were incubated with EEA1 (1:100), AIF (1:400) and LAMP1 (1:200) in a buffer (PBS/1% BSA/0.1% Triton™ X-100) overnight.
The incubation buffers were aspirated and cells were rinsed with PBS (3 × 5 min). For the secondary antibody, goat-anti rabbit IgG-Alexa 555 (Invitrogen™, A21428, 1:500, in PBS/1% BSA/0.1% Triton™ X-100) was used. All samples were then incubated with NucGreen™ Dead 488 (Invitrogen™, 1 drop per 5 ml in PBS) overnight for spheroids and 4 h for cell monolayers. In the last step, they were rinsed with PBS (3 × 5 min).
Spheroid clarification
Optical imaging of three-dimensional biological samples can be performed using confocal fluorescence microscopy which images these 3D samples via optical sectioning. However, this technique faces several limitations, including light scattering, attenuation of photons due to light absorption and local refractive index differences, limiting the light depth of penetration.42 Many different clarification techniques have been developed to overcome such issues.43,44 In the current study, the clearing efficiency of two methods was analysed using NucGreen™ signals in HCT-116 spheroids: RapiClear 1.52 (Sunjin Lab) and glycerol.45 Based on the quantification of fluorescence intensities (Fig. S1†), clarification with glycerol/PBS (80%/20%) was chosen to clear spheroids in this study.The solution for clarifying spheroids was prepared by mixing glycerol (99.5%, VWR Chemicals) with PBS in the ratio 80%/20%. A fresh solution was prepared for every experiment. To clarify spheroids, just after fixation, they were incubated in glycerol solution for 24 h. A detailed description of the mounting procedure used for imaging of live and fixed spheroids is described in Fig. S2.† For most experiments, the microsystems were incubated with a fresh glycerol solution and mounted between 2 coverslips separated by a 1 mm sticky spacer (2 × 0.5 mm thick Ispacer, SunJin Lab).
Confocal fluorescence microscopy
Image acquisition of spheroids and cell monolayers was carried out with a confocal microscope (Leica SP5) using either a 20× dry objective (NA = 0.7), a 25× water immersion objective (NA = 0.95), or a 40× oil immersion objective (NA = 1.25). Image acquisition in the Z direction was performed using a 1 μm z-step. Automatic image acquisitions for a large number of spheroids were performed (about 4 h for 30 spheroids using 30% power for AGuIX®-Cy5.5 nanoparticles (λexcitation = 633 nm)).Image processing
Images obtained by confocal fluorescence microscopy were analysed using a dedicated MATLAB routine. While spheroids were imaged using optical sectioning in the Z direction, it was useful to quantify the average signal intensity along the radius of each spheroid.To do this, the entire surface of each spheroid at each imaging depth was first segmented using the intensity signals coming from every nucleus (labelled with NucGreen™-488). From this segmentation, the segmented spheroid slices were first fitted into a perfect circle for each imaging depth, followed by fitting each spheroid z-stack into a perfect sphere. By changing the coordinates of analysis from Cartesian (x, y, z) to spherical (R, theta, phi) coordinates, the mean intensity of AGuIX®-Cy5.5 nanoparticles was averaged along theta and phi angles. The obtained averaged intensity was normalised with the maximum grey value of images obtained and plotted as a function of the distance from the periphery.
For cell monolayers, the maximum Z-projection of each field of view imaged by confocal microscopy (obtained by ImageJ) was used and analysed with a MATLAB script to quantify the mean intensity in these images. For each sample, the average of the mean intensity computed in the different fields of view was calculated.
Colocalisation quantification
To quantify the colocalisation of AGuIX®-Cy5.5 nanoparticles with cell organelles from confocal fluorescence images, a dedicated routine was developed to calculate the Pearson correlation coefficient, indicating the degree of colocalisation between fluorophores. Briefly, for each image of the acquired stack, a mask of the spheroid was automatically defined using nucleus staining. The correlation between the far-red and red channels (corresponding to AGuIX®-Cy5.5 and organelle-immunostaining, respectively) was then computed using the corr2 MATLAB function. Using this routine, the Pearson correlation coefficient was calculated in the spheroid area for each image along the Z-direction (same as acquisition).NanoSIMS cellular imaging
To prepare samples for NanoSIMS cellular imaging, HCT-116 cell spheroids were incubated with 2 mM AGuIX® nanoparticles for 72 h and then fixed with 2% glutaraldehyde in cacodylate buffer (0.1 M, pH 7.8) for 60 min followed by rinsing with PBS (3 × 5 min). Samples were then postfixed with 1% osmium tetroxide followed by uranyl acetate staining and gradually dehydrated in ethanol (30% to 100%) and embedded in Epon.A 0.2 μm relatively thick section was deposited onto a clean Si chip and dried in air before being introduced into a NanoSIMS-50 Ion microprobe (CAMECA, Gennevilliers, France) operating in scanning mode.46,47 For the present study, a tightly focused Cs+ primary ion beam at an impact energy of 16 keV was used to monitor up to five secondary ion species in parallel from the same sputtered volume: 12C−, 12C14N−, 28Si−, 31P−, as well as 35Cl−. The primary beam steps over the surface of the sample to create images for these selected ion species. The primary beam intensity was 3 pA with a typical probe size of ≈200 nm. The raster size was 60 μm with an image definition of 512 × 512 pixels. The acquisition was carried out in multiframe mode with a dwell time of 0.5 ms per pixel and 220 frames were recorded. The image processing was performed using ImageJ software.41 Successive image frames were properly aligned using the TOMOJ plugin48 with 12C14N− images as reference to correct the slight image field shift during the 8 h signal accumulation before a summed image was obtained for each ion species.
Results and discussion
A hydrogel-based microsystem was developed to generate uniform-sized multicellular tumour spheroids (Fig. 1). The design of these microwells was meant to meet the following goals: (1) to make homogenous and uniform cell spheroids, (2) to increase the throughput in drug screening and (3) to be compatible with in situ treatment, immunostaining and image acquisition as well as ex situ characterization techniques. First, a silicon wafer mould was designed and made using a classical photolithography technique (Fig. 1A). From this silicon wafer mould, counter moulds in polydimethylsiloxane (PDMS) were prepared (Fig. 1B), which could be used several times to replicate microwells with agarose hydrogel. To prepare agarose microwells, 2% ultra-pure agarose solution was poured on the PDMS moulds (Fig. 1C) and after gelation, they were placed on APTS-functionalized coverslips (Fig. 1D) or directly transferred to any classical multi-well plate (Fig. 1E).This method enabled us to generate hundreds of homogenous spheroids per microsystem in each well of a multi-well plate (Fig. 1E and F). Thanks to the hydrogel nature of the microwells, many experimental steps including rinsing, changing the medium, spheroid fixation and immunostaining could be implemented in the same multi-well plate with no manipulation of spheroids, which resulted in the treatment and labelling of several spheroids simultaneously (Fig. 1G). The advantage of the agarose microwells was the efficient transfer of medium and solutions through it. The exchange rate has been quantified by following up the removal of the FITC dye and AGuIX®-Cy5.5 nanoparticles from the agarose microwells via time-lapse image acquisition using confocal microscopy (Fig. S3†). All curves were exponentially decreasing with a characteristic time of 25 min for FITC (23–27 min depending on the depth) and of 1 to 2 hours for AGuIX®-Cy5.5 nanoparticles depending on the depth of the focal plane.
A plateau is reached after two hours for FITC (at 25 ± 5%) and after 10 hours for AGuIX®-Cy5.5 nanoparticles (at 5 ± 3%).
Of note, the compatibility of the hydrogel-based microsystem with coverslips enabled in situ quantification of nanoparticle penetration and their 3D distribution within spheroids with high-resolution optical microscopy such as confocal fluorescence microscopy (Fig. 1H). All spheroids were within the same focal plane, giving access to easy parallelization of 3D spheroid imaging (Fig. 1I). This is an important aspect compared to already proposed hydrogel microwells, where spheroids need to be transferred to a dedicated microscopy plate for high-resolution 3D imaging.27,49–51 Such a transfer first increases the complexity in terms of handling and imaging, and second may induce fusion between spheroids or deformation of spheroids, which in turn may introduce biases in the analysis. Our original and simple process (Biocompatible hydrogel microwell plate, under patent39) bridges an important gap for in-depth optical spheroid analysis.
Moreover, these microwells are compatible with time-lapse optical microscopy, facilitating follow-up of spheroid growth for several days (Fig. S4 and Movie S1†). The system enables us to produce very homogenous spheroids (Fig. S5†), which gives access to the heterogeneity of cell response, with no bias induced by size heterogeneity. In our study, nanoparticle penetration was mainly evaluated using fluorescence intensity obtained from 3D confocal image acquisition. Taking advantage of the large statistics provided by our microsystems, we assessed the minimum number of spheroids required to get reliable results (Fig. S6†). A minimum of N = 30 spheroids is recommended to obtain reliable results at an imaging depth corresponding to the first quarter of the spheroids (0–50 μm from the periphery). This number rises to N = 70 spheroids for an accurate analysis close to the equatorial plane.
Cellular uptake of AGuIX®-Cy5.5 nanoparticles in 2D and 3D
As a proof of concept of the relevance of this new hydrogel-based microsystem, the penetration and distribution of AGuIX®-Cy5.5 nanoparticles (DH = 5 nm) within spheroids was investigated using the colorectal cancer cell line HCT-116. In a previous study, it has been proven that the localisation of Gd-based nanoparticles tagged with Cy5.5 is the same as that of label-free nanoparticles in U87 cells.52 After 48 h of growth within 200 μm agarose microwells, spheroids were incubated with three different concentrations of AGuIX®-Cy5.5, 0.8, 1.5 and 2 mM in Gd, selected according to previous studies performed in 2D cell culture.53 In Fig. 2A, fluorescence images show the distribution of AGuIX®-Cy5.5 nanoparticles in spheroids after 24 h of incubation with the three different concentrations at different depths. These images showed qualitatively that the number of nanoparticle clusters in spheroids directly increases with the increase in initial concentration of AGuIX®-Cy5.5 nanoparticles. For 0.8 mM, very few nanoparticle clusters could be observed, while the number of clusters increased in 1.5 and 2 mM concentrations. For 2 mM concentration, nanoparticles were detected within the deeper layers of spheroids. Taking the spherical geometry of the sample into account to quantify the fluorescence in each image of spheroids, the mean intensity was calculated by averaging the intensity along theta and phi angles in the direction of the radius. The in situ fluorescence analysis of Fig. 2B enabled us to decipher the relative differences in nanoparticle penetration in the range of concentrations analysed. Consistent with the fluorescence images in Fig. 2A, the mean intensity increased as the incubation concentration increased. From the outermost layer to the centre of the spheroids, the mean intensity decreased differently depending on the concentration (from 34 ± 8% to 28 ± 14% for 0.8 mM, from 68 ± 8% to 46 ± 15% for 1.5 mM and from 66 ± 5% to 60 ± 24% for 2 mM). For the largest concentration (2 mM), deep penetration was possible, while the penetration decreased exponentially with the depth for 1.5 mM, with a characteristic length of 44 ± 2 μm. Such a difference could be attributed to the higher number of nanoparticles reaching the centre of the spheroids for an incubation with 2 mM Gd. The relative independence of fluorescence intensity with depth for the lowest concentration (0.8 mM) could be attributed to a level close to noise, with no real penetration of nanoparticles in the periphery or in the centre of the spheroids.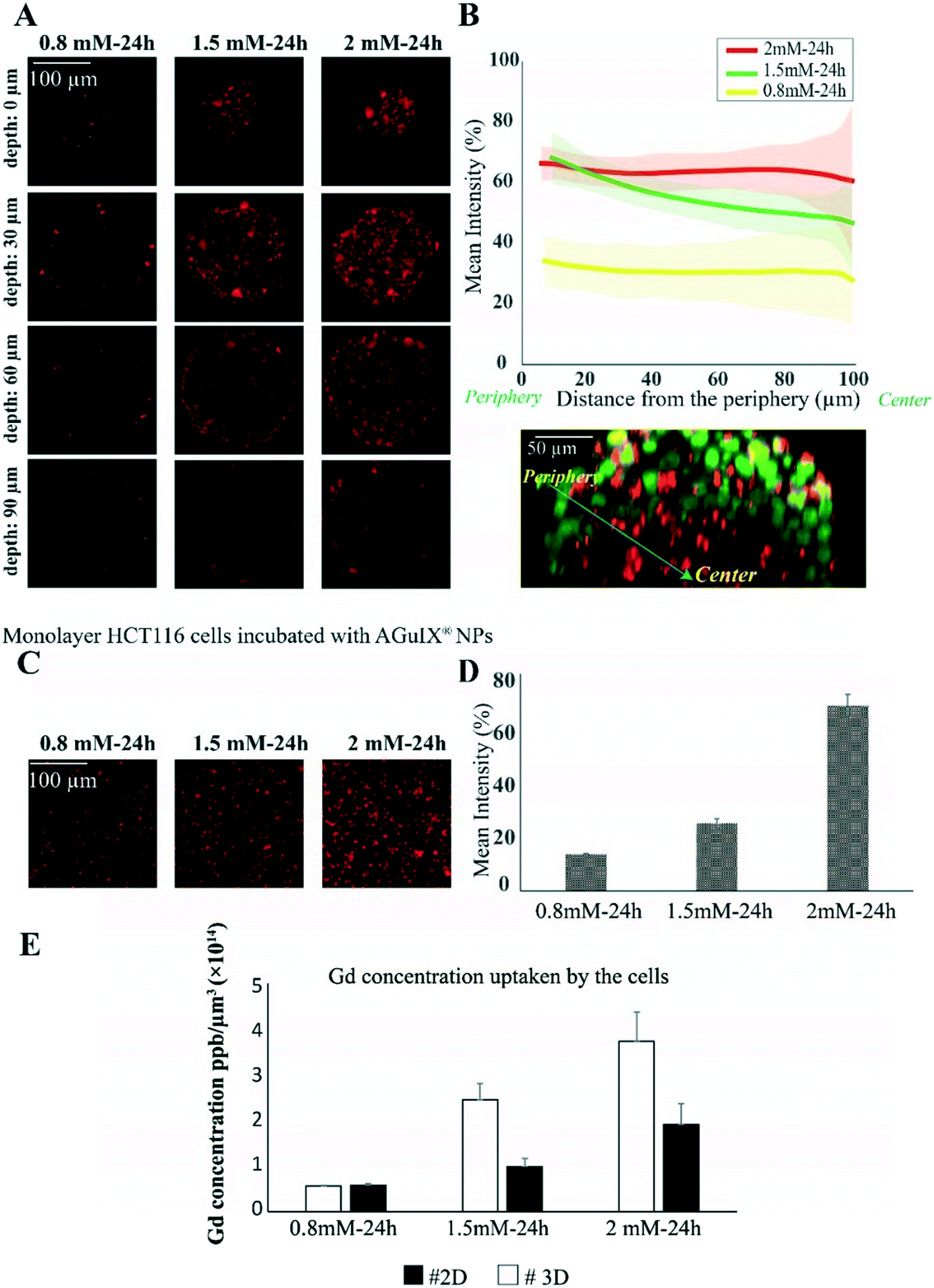 | ||
| Fig. 2 Quantification of penetration and cellular uptake of AGuIX®-Cy5.5 nanoparticles in HCT-116 tumour spheroids and monolayer cell culture. (A) Representative confocal fluorescence images of HCT-116 spheroids incubated with 0.8, 1.5 and 2 mM concentration of AGuIX®-Cy5.5 for 24 h for four different depths (0, 30, 60 and 90 μm). (B) Mean intensity along with standard deviation (light colors) of AGuIX®-Cy5.5 as a function of the distance from the spheroid periphery (see the orthogonal view in the inset, green = nuclei, red = AGuIX®-Cy5.5) for 0.8 mM (yellow, N = 73), 1.5 mM (green, N = 68) and 2 mM (red, N = 121); three independent experiments. (C) Representative confocal fluorescence images of monolayer HCT-116 cells exposed to AGuIX®-Cy5.5 nanoparticles with 0.8, 1.5 and 2 mM concentration. (D) Quantification of the mean intensity of AGuIX®-Cy5.5 nanoparticles in maximal projection of confocal fluorescence images of monolayer cells after 24 h of incubation with different AGuIX®-Cy5.5 concentrations: 0.8 mM (yellow, N = 40), 1.5 mM (green, N = 40) and 2 mM (red, N = 40); three independent experiments. Error bars represent the standard deviations. (E) Mean and standard deviation of the concentration of Gd (ppb μm−3) uptaken by the cells after incubation with 0.8, 1.5 and 2 mM concentration of AGuIX® for 24 h in HCT-116 cell spheroids and monolayer cell culture measured with ICP-MS (N = 6, two independent experiments). | ||
To be sure that the presence of agarose in our microsystem does not affect the distribution and cellular uptake of AGuIX®-Cy5.5 nanoparticles within spheroids, a control experiment was made using an ultralow adhesion 96-well plate and 2 mM AGuIX®-Cy5.5 nanoparticle concentration (Fig. S7†). Similar results concerning the penetration of the nanoparticles were obtained: the same normalised intensity range and similar evolution as a function of distance from the periphery.
Deep penetration of small nanoparticles (<12 nm) within deep interstitial space has already been reported in vivo.54 The in vitro platform described in the current study enables assessing more quantitatively such penetration. Hence, it will be a valuable tool to relate such penetration with therapeutic efficacy in future studies.
To make a direct comparison with cellular uptake in 2D cell culture, monolayers of HCT-116 cells were incubated with the same concentrations of AGuIX®-Cy5.5 nanoparticles (Fig. 2C). As expected, the number of AGuIX®-Cy5.5 clusters increased as the initial concentration increased and the quantification of fluorescence images of cell monolayers (based on the mean intensity of AGuIX®-Cy5.5) confirmed that the uptake of nanoparticles increased with the concentration of AGuIX®-Cy5.5 in the incubation medium (Fig. 2D, from 14.0 ± 0.3% for 0.8 mM, 25.8 ± 1.8% for 1.5 mM to 70.8 ± 4.4% for 2 mM). This mean intensity evolution was hence different from the one obtained in 3D in the periphery. However, as a true quantitative comparison is not possible using fluorescence analysis, elemental analysis by ICP-MS was performed concurrently to obtain a quantitative analysis of Gd content within cells for both 2D and 3D models (Fig. 2E). While the average nanoparticle uptake per cell in 2D and 3D was similar for 0.8 mM ((0.580 ± 0.006) × 10−14 ppb μm−3 in 2D vs. (0.59 ± 0.05) × 10−14 in 3D), the uptake was two-fold higher in 2D compared to 3D for both 1.5 mM ((2.5 ± 0.5) × 10−14 ppb μm−3 in 2D vs. (1.0 ± 0.2) × 10−14 in 3D) and 2 mM ((3.8 ± 0.9) × 10−14 ppb μm−3 in 2D vs. (1.9 ± 0.6) × 10−14 ppb μm−3 in 3D).
One of the reasons for the reduction in effectiveness of therapeutics in vivo compared to monolayer cell cultures is the lack of efficient penetration and distribution of therapeutics throughout the tumour tissue.55 This is what we also observed here, with a large reduction of nanoparticle uptake in 3D compared to 2D cell culture.
Another approach was used to compare the cellular uptake of AGuIX®-Cy5.5 nanoparticles in 2D and 3D: 2D cells were treated with 2 mM AGuIX®-Cy5.5 nanoparticles for 24 h, then spheroids were made from these AGuIX®-Cy5.5 labelled cells using the usual protocol (Fig. S8†). Interestingly, the distribution of nanoparticles differs when spheroids are made with already labelled cells compared to direct incubation with already formed spheroids, further highlighting the difference in nanoparticle availability between 2D and 3D models.
Kinetics of AGuIX®-Cy5.5 nanoparticle transport into spheroids
One of the crucial parameters in nanoscale design is the pharmacokinetics of nanoparticles and understanding this aspect of cell–nanoparticle interactions has a great importance.56,57 The kinetics of penetration of AGuIX®-Cy5.5 nanoparticles within HCT-116 cell spheroids grown for 48 h were assessed by analysing confocal images obtained for different incubation times (1, 24 and 72 h) for the highest concentration investigated (2 mM) (Fig. 3A). After 1 h of incubation, the AGuIX®-Cy5.5 nanoparticles were mostly residing in the peripheral layer of the spheroids, especially in the extracellular space. After 24 h, clusters of nanoparticles were found throughout the spheroids. At 72 h, the number of clusters was increasing for all depths up to the equatorial plane (100 μm).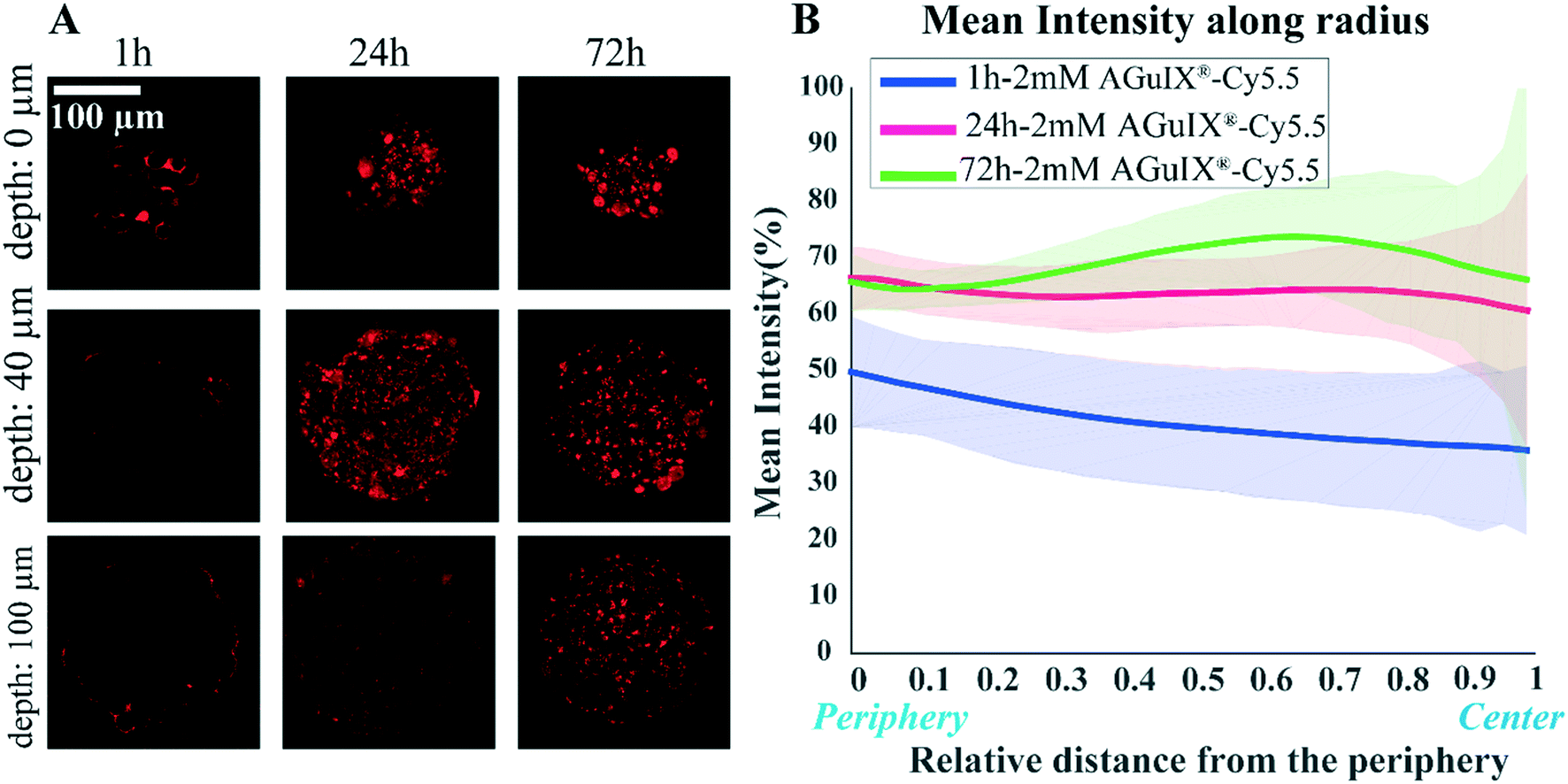 | ||
| Fig. 3 Kinetics of penetration of AGuIX®-Cy5.5 nanoparticles in HCT-116 cell spheroids. (A) Representative confocal fluorescence images of HCT-116 grown for 48 h and exposed to 2 mM concentration of AGuIX®-Cy5.5 nanoparticles for 1, 24 and 72 h. (B) Mean intensity along with standard deviation (light colours) as a function of relative distance from the periphery for 1 h (blue, N = 50), 24 h (magenta, N = 121) and 72 h (green, N = 63; three independent experiments. | ||
The average intensity exhibited a different evolution with the distance from the periphery, depending on the incubation time (Fig. 3B). At the periphery, the average intensity was lower for 1 h incubation (52.2 ± 1.3%) than for 24 h and 72 h that exhibited similar values (68 ± 1% and 65.8 ± 0.7%, respectively). When we moved to the centre of the spheroids, the mean intensity was slightly lowered for 1 h and 24 h incubation (from 52.2 ± 1.3% down to 38.7 ± 2.7% for 1 h, from 68 ± 1% down to 59.5 ± 1.9% for 24 h). Accordingly, in 1 h incubation the mean intensity was less than that of 24 h and 72 h samples in all regions of the spheroids. Surprisingly, for 72 h, the average intensity exhibited a non-monotonous evolution with the distance from the periphery, with an intensity larger for the middle layers than at the periphery (66 ± 5% vs. 73.8 ± 8.5% at a depth of 60 μm). This may be the result of the increased number of clusters found for intermediate layers after 72 h of incubation time.
To follow the distribution and the transport of nanoparticles within spheroids, an experiment was designed to assess changes in AGuIX®-Cy5.5 nanoparticle distribution before and after rinsing steps (Fig. 4). In this experiment, 2 days after cell seeding (Fig. 4, step I), the formed HCT-116 spheroids were incubated with 2 mM AGuIX®-Cy5.5 solution for 72 h (Fig. 4, step II). Spheroids were then imaged in incubation medium (Fig. 4, step III) and after three washing steps of 15 min each (Fig. 4, step IV). Spheroids were kept in the incubator for an additional 24 h and then imaged before (Fig. 4, step V) and after (Fig. 4, step VI) another washing procedure. Confocal fluorescence microscopy of living spheroids showed that AGuIX®-Cy5.5 nanoparticle fluorescence signals of the surrounding background (fluorescence signal outside spheroids) was decreasing gradually with the different washing steps for all depths (Fig. 4A–D). This is confirmed by the quantification of the mean intensity along the spheroid radius (Fig. 4E): while the mean intensity before washing (red curve) at the periphery was around 92 ± 10%, it was decreasing to 59 ± 10% at 55 μm distance from the periphery. After the first washing step (yellow curve), the mean intensity at the periphery reduced to 66 ± 6% and reached a similar intensity level to the one before washing at 55 μm distance from the periphery (58 ± 4%). Additional washing steps further reduced the mean intensity at the periphery (64 ± 14% and 54 ± 14% before and after the second washing step), while the mean intensity obtained for deeper layers exhibited similar levels. The second washing (blue curve) led to a steady value of mean intensity (∼54%) close to the mean intensity obtained at 55 μm distance from the periphery of spheroids for all washing steps. This mean intensity should correspond to the signal coming from nanoparticles that are internalized by the cells, as all nanoparticles residing in the extracellular space have been washed away.
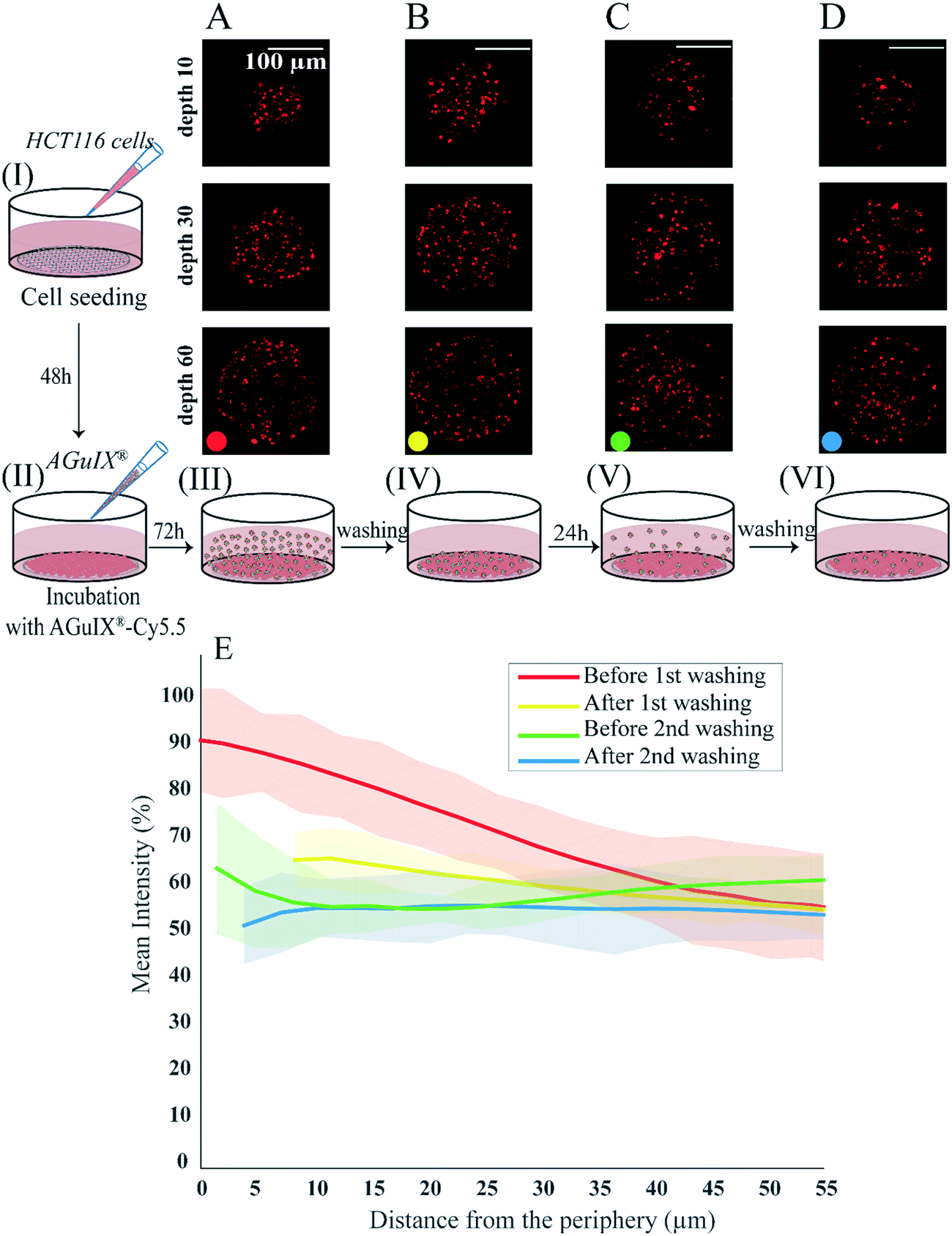 | ||
| Fig. 4 Dynamic analysis of AGuIX®-Cy5.5 nanoparticle transport and localisation within spheroids. HCT-116 cell spheroids were prepared using the agarose microsystem (step I). After 48 h of growth, they were exposed to 2 mM AGuIX®-Cy5.5 solution for 72 h (step II). (A) Spheroids were imaged in the incubation medium (step III). (B) Spheroids were then rinsed with fresh medium three times for 15 min each and were imaged again (step IV). (C) Spheroids were allowed to grow for an additional 24 h (in an incubator at 37 °C and 5% CO2) before imaging (step V). (D) Spheroids were rinsed again with fresh medium (3 × 15 min) before imaging (step VI). (E) Quantification of AGuIX®-Cy5.5 nanoparticle mean intensity along the distance from the periphery (N = 25). Bold lines represent the mean intensities, averaged for all spheroids. Light colours represent the standard deviations. | ||
Localisation of AGuIX®-Cy5.5 nanoparticles in spheroids using their chemical signature
Due to the limit of resolution using standard confocal optical microscopy (200 nm in the best imaging conditions), only clusters of nanoparticles can be detected. In addition, we cannot rule out that the distribution of the fluorophores does not truly represent the distribution of the nanoparticles themselves. To confirm the presence of nanoparticles, nanoscale secondary ion mass spectrometry (nanoSIMS) was performed on spheroid sections (Fig. 5). This analytical technique allows the acquisition of elemental composition maps with a spatial resolution down to 50 nm. The images of 12C− (see Fig. S7†) and 35Cl− (data not shown) indicate the absence of defect in the sample section. Any damage, even tiny holes, would appear with high contrast in the signal, and such a signal was not observed. This validated that the signal measured originated from the sample and not from the subjacent pure silicon substrate. The image of 12C14N− showed the histological aspect of the cell (data not shown), while the one of 31P− (Fig. 5A) highlights the cell nucleus. Since AGuIX®-Cy5.5 nanoparticles are mainly made of Si, the images of 28Si− allowed the observation of the chemical signature of the nanoparticles (Fig. 5B). Thereby, the nanoparticles were found unequivocally inside the spheroid, exclusively in the cytoplasm of the cells. Of note, again our microsystems enabled an easy sample preparation, as all spheroids were within the same sectioning plane.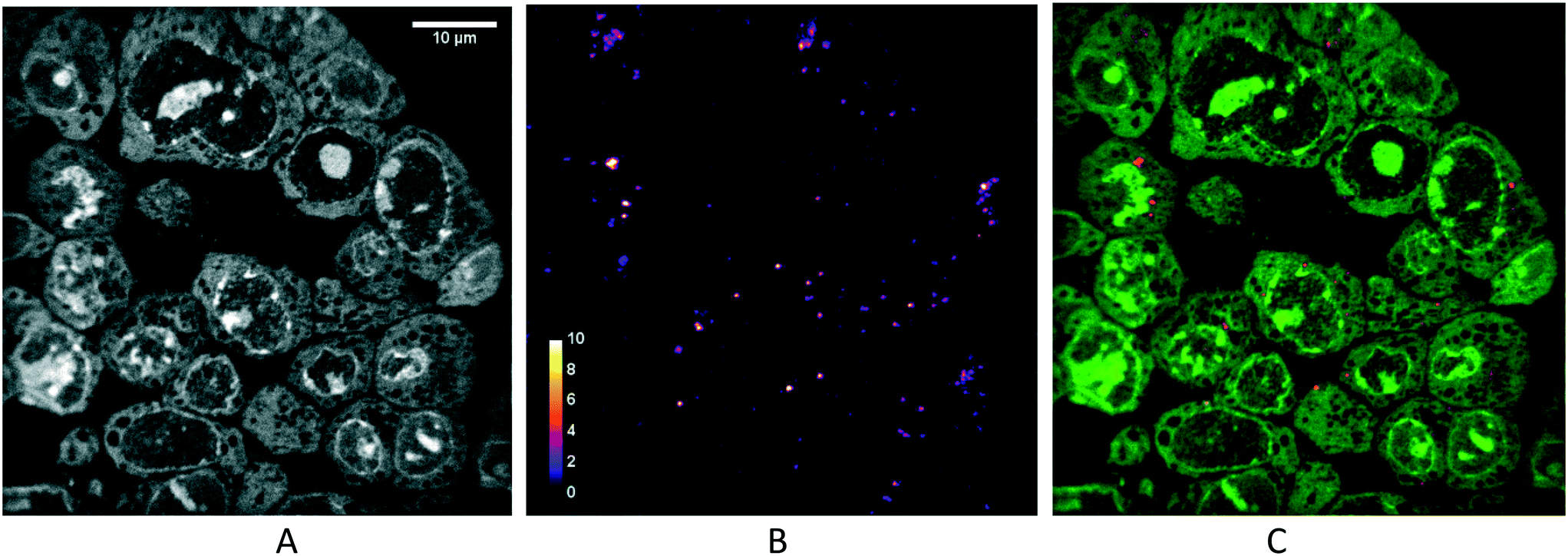 | ||
| Fig. 5 Localisation of the AGuIX®-Cy5.5 nanoparticles in spheroids using NanoSIMS. NanoSIMS images of HCT-116 spheroids loaded with AGuIX®-Cy5.5 nanoparticles. (A) Corresponds to the signal of 31P− showing the cell structure. (B) Highlights the signal of 28Si− representing the intracellular location of AGuIX®-Cy5.5 nanoparticles. (C) Merged image of 28Si− and 31P−. Scale bar: 10 μm. | ||
Localisation of AGuIX®-Cy5.5 within cells in 2D and 3D using immunostaining
Thanks to the full compatibility of the microsystems with in situ immunostaining, it was possible to assess the localisation of nanoparticles in 2D cells (Fig. 6) and multicellular tumour spheroids (Fig. 7) using confocal fluorescence microscopy.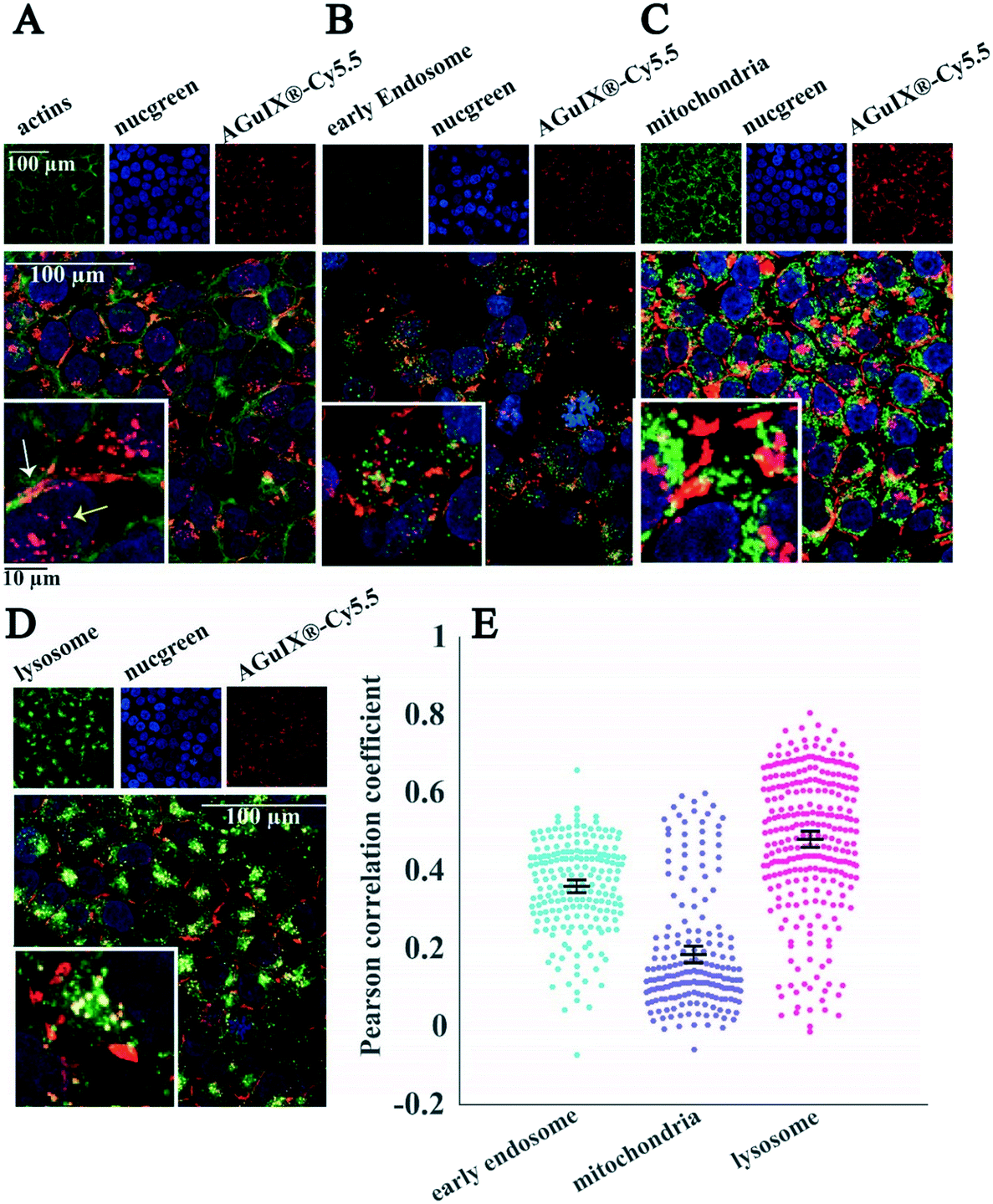 | ||
| Fig. 6 Localisation of the AGuIX®-Cy5.5 nanoparticles in 2D monolayers. Fluorescence images of HCT-116 cells incubated with AGuIX®-Cy5.5 nanoparticles (2 mM, 24 h) and immunostained with antibodies to find the colocalization of nanoparticles inside cells. In all images, the red channel and blue channel represent AGuIX®-Cy5.5 nanoparticles and cell nuclei, respectively. (A) The green channel depicts phalloidin, a marker of actin in cells, which demonstrates nanoparticles localising both inside cells (yellow arrow) and in the space between cells (white arrow). (B) The green channel shows early endosome in the cells, with no colocalisation with AGuIX®-Cy5.5 nanoparticles. (C) The green channel shows mitochondria and reveal very low colocalisation with AGuIX®-Cy5.5 nanoparticles (yellow colour). (D) The green channel shows the lysosomes, and colocalisation is demonstrated by the yellow colour. White scale bar, 100 μm; black scale bar, 10 μm. (E) Distribution of Pearson correlation coefficients in the different fields of view to quantify the colocalisation of AGuIX®-Cy5.5 nanoparticles with the three different cell organelles investigated. Error bars represent the standard errors of the mean (SEM) of Pearson correlation coefficient values obtained for all fields of view and all available depths for three independent experiments. It was plotted as scatter plots using the MATLAB UnivarScatter function (©Manuel Lera Ramírez, 2015, available in MATLAB exchange files). | ||
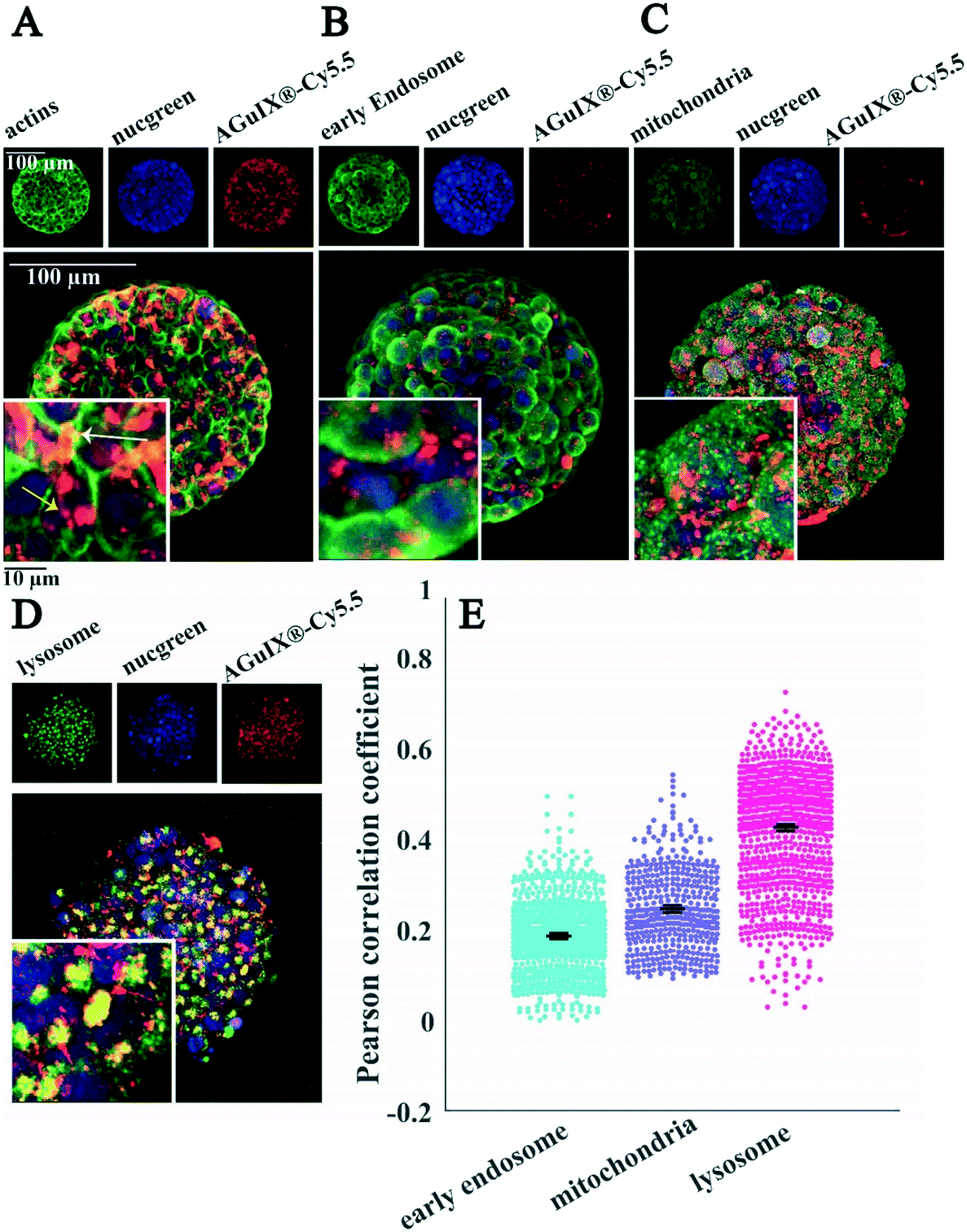 | ||
| Fig. 7 Localisation of AGuIX®-Cy5.5 nanoparticles within spheroids. Fluorescence images of HCT-116 cell spheroids incubated with AGuIX®-Cy5.5 nanoparticles (2 mM, 24 h) and immunostained with antibodies to find the colocalisation of nanoparticles inside cells. In all images, red and blue channels represent AGuIX®-Cy5.5 nanoparticles and cell nuclei, respectively. (A) The green channel depicts phalloidin, a marker of actin in cells, which demonstrates that nanoparticles localise both inside cells (yellow arrow) and in the extracellular space of spheroids (white arrow). (B) EEA1 antibody in the green channel shows early endosome; with very low colocalisation with nanoparticles (yellow colour). (C) AIF antibody labelled mitochondria are shown in green, with very low colocalisation with AGuIX®-Cy5.5 nanoparticles (yellow colour). (D) LAMP1 antibody in green channel stains lysosomes. Yellow colour represents the colocalisation of nanoparticles in red and lysosomes in green. White scale bar, 100 μm; black scale bar, 10 μm. (E) Quantification of the colocalisation of AGuIX®-Cy5.5 nanoparticles with the three different cell organelles investigated using Pearson correlation coefficient. Distribution obtained for all imaged spheroids and all imaging depths for the three different cell organelles. Error bars represent standard errors of the mean (SEM) of Pearson correlation coefficient values obtained for all fields of view and all available depths for three independent experiments. It was plotted as scatter plots using the MATLAB UnivarScatter function (©Manuel Lera Ramírez, 2015, available in MATLAB exchange files). | ||
Labelling of cell organelles confirmed that nanoparticles were present in both extracellular and intracellular space in 2D cells (Fig. 6A) and in 3D spheroids (Fig. 7A). Very low colocalisation of AGuIX®-Cy5.5 with early endosomes (Fig. 6B and E in 2D and Fig. 7B and E in 3D) or mitochondria (Fig. 6C and E in 2D and Fig. 7C and E in 3D) was evidenced by immunostaining, while a large colocalisation with lysosomes was observed in both 2D (Fig. 6D and E) and 3D environments (Fig. 7D and E). The Pearson correlation coefficient in both 2D cells and 3D spheroids (Fig. 6E in 2D and Fig. 7E in 3D) showed a higher value for lysosomes (0.48 ± 0.18 and 0.42 ± 0.12 for 2D and 3D, respectively, compared to 0.36 ± 0.12 and 0.18 ± 0.09 for early endosomes in 2D and 3D and 0.19 ± 0.15 and 0.24 ± 0.09 for mitochondria in 2D and 3D), showing the main intracellular localisation of AGuIX®-Cy5.5 nanoparticles. It is noteworthy that this colocalisation was not total and some nanoparticles were still residing in between cells. These outcomes are in accordance with previous studies showing localisation of nanoparticles in the endocytic pathway and in lysosomes.38,52
The internalization mechanisms of AGuIX® have been thoroughly investigated in 2D.38 It has been shown that the entry of such sub-5 nm nanoparticles is different depending on nanoparticle concentration: passive diffusion and eventually macropinocytosis, in case of formation of nanoparticle clusters at the surface of the cell. It is known that the internalization pathway for a specific nanoparticle can differ between cell lines.58 For the HCT-116 cell line used in this study, localisation of AGuIX®-Cy5.5 nanoparticles in lysosomes and in smaller amounts in early endosomes confirms that they were likely internalized by an endocytic mechanism.59 Despite dominant colocalisation for both 2D and 3D with lysosomes, in 2D images the Pearson correlation coefficient average value for early endosomes is higher than that for mitochondria (Fig. 6B and E), which contrasts with these values in 3D (Fig. 7B and E). One explanation for this difference is that for spheroids, cells have varying access to the nanoparticles depending on their spatial position within spheroids, which could lead to different internalization processes. In 3D spheroids, AGuIX®-Cy5.5 nanoparticles were confronted by barriers to reach the cells in deeper layers; therefore they reach deeper layers in a lower amount (Fig. 2B) and with a delay (Fig. 3B), which can change their intracellular fate. This is another argument in favour of the 3D system for nanoparticle transport analysis. In 2D, all cells are submitted to the same homogeneous concentration of nanoparticles, while in 3D, there is a large difference in nanoparticle availability between cells that are at the periphery and cells in the centre of the spheroids. In addition, in spheroids, similar to natural tumours, there is a gradient of pH, oxygen and metabolites,60 which might affect internalization and intracellular trafficking of nanoparticles in deeper layers.61
As highlighted by the overall mean intensity decrease with the washing procedure for the peripheral layers of the spheroids (Fig. 4), the extracellular nanoparticles were efficiently washed away after a long washing procedure (Fig. S10,† no extracellular nanoparticles were detected with immunostaining). Similar to results obtained after 72 h of incubation, colocalisation with lysosomes was still the major localisation of nanoparticles after this extensive washing procedure (Fig. 8A–C for lysosomes compared to Fig. 8D–F for mitochondria). The comparison of the Pearson correlation coefficient of AGuIX®-Cy5.5 with both lysosomes and mitochondria before and after washing suggests minor intracellular trafficking and/or exocytosis of nanoparticles over time (Fig. 8B for lysosomes and 8E for mitochondria). The Pearson correlation coefficient of AGuIX®-Cy5.5 with lysosomes remained within a similar range before and after washing (mean values of 0.41 ± 0.03 vs. 0.40 ± 0.11, respectively), while a decrease in the Pearson correlation coefficient of AGuIX®-Cy5.5 with mitochondria is observed after washing, particularly in outer layers (0.26 ± 0.02 vs. 0.18 ± 0.04 at 10 μm depth before and after washing, respectively), reaching very low values for inner layers (0.20 ± 0.04 vs. 0.18 ± 0.02 at 60 μm depth before and after washing, respectively). Such a decrease could be attributed to the removal of a few AGuIX®-Cy5.5 clusters residing in mitochondria or possible intracellular trafficking during the washing procedure. Hence, we could say that the washing procedure had lesser effect on AGuIX®-Cy5.5 nanoparticles residing in lysosomes.
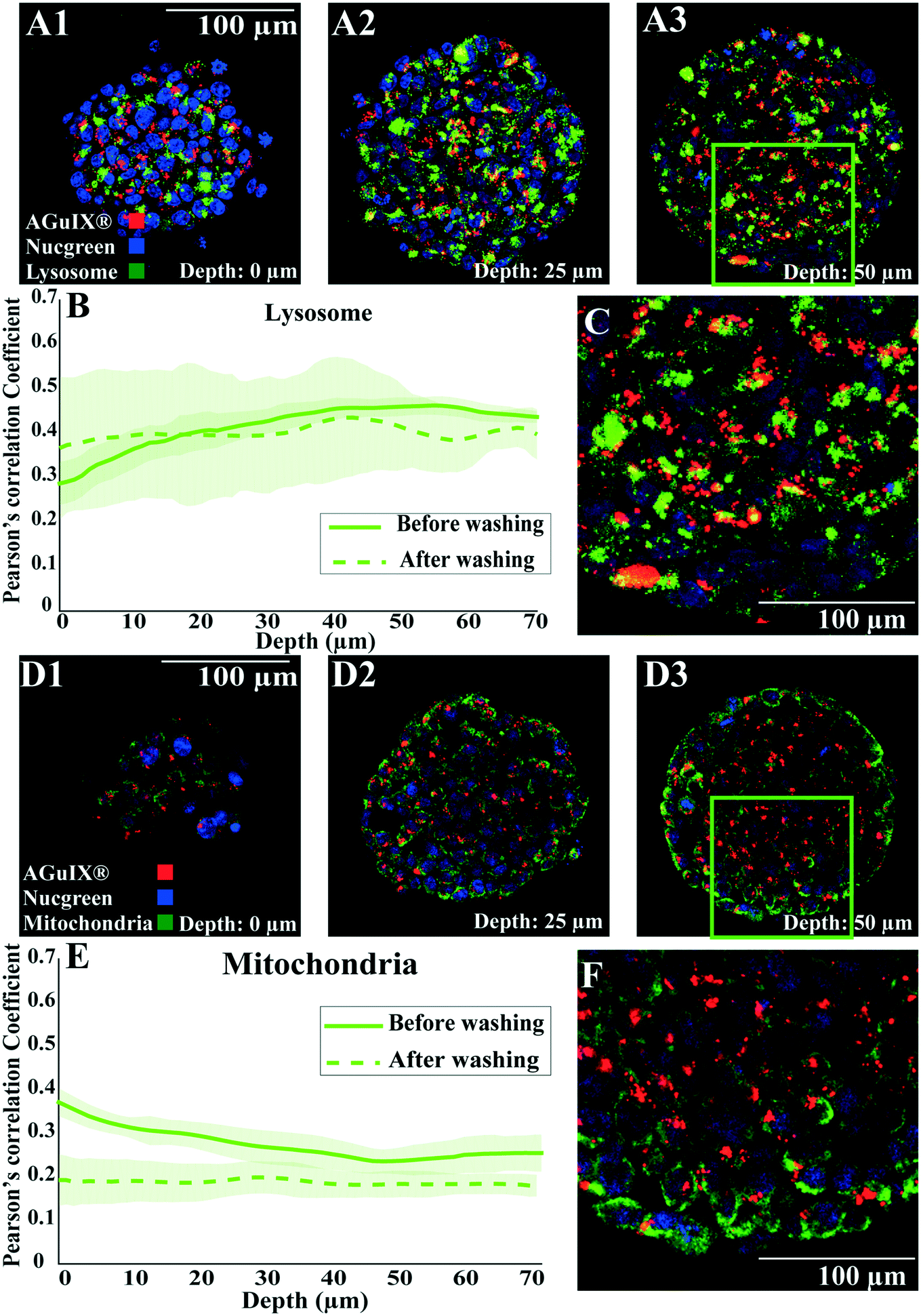 | ||
| Fig. 8 Localisation of Aguix®-Cy5.5-nanoparticles after an extensive washing procedure. Confocal fluorescence images of HCT-116 spheroids incubated with AGuIX®-Cy5.5 nanoparticles for 72 h with 2 mM AGuIX® solution and washed according to the procedure mentioned in Fig. 4, then fixed and immunostained with antibodies to find the colocalisation of nanoparticles in spheroids. For all images, red and blue channels are stained AGuIX®-Cy5.5 and nuclei, respectively. (A1–A3) Representative images of lysosome immunostaining obtained at various depths (A1, 0 μm; A2, 25 μm; A3, 50 μm. Green channel = lysosome [LAMP1 antibody], yellow colour = possible colocalisation of AGuIX®-Cy5.5 nanoparticles with lysosomes). (B) Pearson correlation coefficient for AGuIX®-Cy5.5 nanoparticles with lysosomes along with standard error of the mean (light colour) as a function of depth (n = 27 spheroids, 3 independent experiments before washing, n = 5 spheroids after washing). (C) Zoomed-in portion of merged image at a depth of 50 μm (square in A3). (D1–D3) Representative images of mitochondria immunostaining obtained at various depths (D1, 0 μm; D2, 25 μm; D3, 50 μm. Green channel = mitochondria [AIF antibody], yellow colour = possible colocalisation of AGuIX®-Cy5 nanoparticles with mitochondria). (E) Pearson correlation coefficient for AGuIX®-Cy5.5 nanoparticles and mitochondria along with standard error of the mean (light colour) as a function of depth (n = 22 spheroids, 3 independent experiments before washing, n = 5 spheroids after washing). (F) Zoomed-in portion of merged image at a depth of 50 μm (square in D3). | ||
Conclusion and outlook
We show in this study a simple agarose-based microsystem to quantitatively track nanoparticle penetration and subcellular localisation within a 3D cell culture model. The reproducibility of the spheroid size obtained with such a procedure dispenses the use of sophisticated automatic procedures to choose and pick the appropriate spheroids. Of note, our microsystems can be manufactured on conventional multi-well plates. It is hence fully compatible with available multi-well automated strategies.62 In the present study, the proof of concept was validated using spheroids made with the classical colorectal cell line HCT-116. Nevertheless, our approach is fully compatible with primary cells from patients that could be grown as organoids63 in our microsystems, combining full optical microscopy compatibility, size and shape reproducibility, and large statistics. Combined with optical and digital clearing,64 our approach opens up the possibility to resolve tumour heterogeneity, at the single cell-level, in a physiological context.In the present study, the standard agarose used for the preparation of the microsystems provides a cell-repellent surface, with a stiffness in the 150 kPa range.65,66 In future studies, the mechanical properties of the agarose gels will be adjusted using different concentrations and types of agarose. Elastic hydrogels as soft as 1 kPa can be obtained using low concentrations of ultra-low agarose,65 matching the physiological range of stiffness. It now calls for dedicated studies to assess how nanoparticle penetration and therapeutic efficacy is affected by the size of the 3D cell assembly, the presence of an extracellular matrix of different stiffness and composition, and the presence of associated tumour cells.67
Author contributions
Saba Goodarzi: methodology, investigation, validation, formal analysis, visualization, writing – original draft; Audrey Prunet: methodology, investigation, visualization, writing – review and editing; Fabien Rossetti: methodology, investigation; Guillaume Bort: writing – review and editing; Olivier Tillement: conceptualization; Erika Porcel: methodology, investigation, validation, visualization, writing – review and editing; Sandrine Lacombe: methodology, writing – review and editing; Ting-Di Wu: methodology: writing – review and editing; Jean-Luc Guerquin-Kern: methodology, investigation, writing – review and editing; Hélène Delanoë-Ayari: methodology, formal analysis; François Lux: conceptualization, supervision, writing – review and editing; Charlotte Rivière: conceptualization, supervision, writing – original draft, writing – review and editing.Conflicts of interest
CR, AP and HDA have to disclose the patent FR3079524A1. This patent protects the process to produce agarose-based microsystems on coverslips described in this publication. FL and OT have to disclose the patent WO2011/135101. This patent protects the AGuIX® NPs described in this publication. FL and OT possess shares of this company.Acknowledgements
This work was supported by the “Institut Universitaire de France” (IUF). We thank L. Fuoco for her help in the early development of this project, R. Fulcrand for his support in photolithography, M. G. Blanchin for her help in spheroid preparation protocols for electron microscopy as well as the CTμ platform (Centre Technologique des Microstructures) for resin embedding of spheroids.References
- S. Hua, M. B. C. de Matos, J. M. Metselaar and G. Storm, Front. Pharmacol., 2018, 9, 1–14 CrossRef PubMed.
- L. J. Bray, D. W. Hutmacher and N. Bock, Front. Bioeng. Biotechnol., 2019, 7, 1–36 CrossRef PubMed.
- S. E. Gould, M. R. Junttila and F. J. De Sauvage, Nat. Med., 2015, 21, 431–439 CrossRef CAS PubMed.
- G. Lazzari, P. Couvreur and S. Mura, Polym. Chem., 2017, 8, 4947–4969 RSC.
- A. Sontheimer-Phelps, B. A. Hassell and D. E. Ingber, Nat. Rev. Cancer, 2019, 19, 65–81 CrossRef CAS PubMed.
- S. Peel, A. M. Corrigan, B. Ehrhardt, K. J. Jang, P. Caetano-Pinto, M. Boeckeler, J. E. Rubins, K. Kodella, D. B. Petropolis, J. Ronxhi, G. Kulkarni, A. J. Foster, D. Williams, G. A. Hamilton and L. Ewart, Lab Chip, 2019, 19, 410–421 RSC.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- D. Rosenblum, N. Joshi, W. Tao, J. M. Karp and D. Peer, Nat. Commun., 2018, 9, 1410 CrossRef PubMed.
- M. Zanoni, F. Piccinini, C. Arienti, A. Zamagni, S. Santi, R. Polico, A. Bevilacqua and A. Tesei, Sci. Rep., 2016, 6, 1–11 CrossRef PubMed.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- E. J. Guggenheim, S. Milani, P. J. F. Röttgermann, M. Dusinska, C. Saout, A. Salvati, J. O. Rädler and I. Lynch, NanoImpact, 2018, 10, 121–142 CrossRef.
- W. Asghar, R. El Assal, H. Shafiee, S. Pitteri, R. Paulmurugan and U. Demirci, Mater. Today, 2015, 18, 539–553 CrossRef CAS PubMed.
- S. Nath and G. R. Devi, Pharmacol. Ther., 2016, 163, 94–108 CrossRef CAS PubMed.
- M. Millard, I. Yakavets, V. Zorin, A. Kulmukhamedova, S. Marchal and L. Bezdetnaya, Int. J. Nanomed., 2017, 12, 7993–8007 CrossRef CAS PubMed.
- F. Hirschhaeuser, H. Menne, C. Dittfeld, J. West, W. Mueller-klieser and L. A. Kunz-schughart, J. Biotechnol., 2010, 148, 3–15 CrossRef CAS PubMed.
- H. L. Ma, Q. Jiang, S. Han, Y. Wu, J. C. Tomshine, D. Wang, Y. Gan, G. Zou and X. J. Liang, Mol. Imaging, 2012, 11, 487–498 CrossRef CAS PubMed.
- S. Huo, H. Ma, K. Huang, J. Liu, T. Wei, S. Jin, J. Zhang, S. He and X. J. Liang, Cancer Res., 2013, 73, 319–330 CrossRef CAS PubMed.
- A. Virgone-Carlotta, M. Lemasson, H. C. Mertani, J. J. Diaz, S. Monnier, T. Dehoux, H. Delanoë-Ayari, C. Rivière and J. P. Rieu, PLoS One, 2017, 12(11), e0188100 CrossRef PubMed.
- B. Rodday, F. Hirschhaeuser, S. Walenta and W. Mueller-Klieser, J. Biomol. Screening, 2011, 16, 1119–1124 CrossRef PubMed.
- J. M. Kelm, N. E. Timmins, C. J. Brown, M. Fussenegger and L. K. Nielsen, Biotechnol. Bioeng., 2003, 83, 173–180 CrossRef CAS PubMed.
- Y.-C. Chen, P. N. Ingram, S. Fouladdel, S. P. McDermott, E. Azizi, M. S. Wicha and E. Yoon, Sci. Rep., 2016, 6, 27301 CrossRef CAS PubMed.
- M. Akay, J. Hite, N. G. Avci, Y. Fan, Y. Akay, G. Lu and J. J. Zhu, Sci. Rep., 2018, 8, 1–9 CAS.
- R. Mukhopadhyay, Anal. Chem., 2007, 79, 3249–3253 Search PubMed.
- B. J. van Meer, H. de Vries, K. S. A. Firth, J. van Weerd, L. G. J. Tertoolen, H. B. J. Karperien, P. Jonkheijm, C. Denning, A. P. IJzerman and C. L. Mummery, Biochem. Biophys. Res. Commun., 2017, 482, 323–328 CrossRef CAS PubMed.
- M. W. Toepke and D. J. Beebe, Lab Chip, 2006, 6, 1484–1486 RSC.
- D. T. Butcher, T. Alliston and V. M. Weaver, Nat. Rev. Cancer, 2009, 9, 108–122 CrossRef CAS PubMed.
- J. M. Lee, D. Y. Park, L. Yang, E. J. Kim, C. D. Ahrberg, K. B. Lee and B. G. Chung, Sci. Rep., 2018, 8, 1–10 Search PubMed.
- Y. Li and E. Kumacheva, Sci. Adv., 2018, 4, 1–11 Search PubMed.
- X. Gong, C. Lin, J. Cheng, J. Su, H. Zhao, T. Liu, X. Wen and P. Zhao, PLoS One, 2015, 10, e0130348 CrossRef PubMed.
- J. Dahlmann, G. Kensah, H. Kempf, D. Skvorc, A. Gawol, D. A. Elliott, G. Dräger, R. Zweigerdt, U. Martin and I. Gruh, Biomaterials, 2013, 34, 2463–2471 CrossRef CAS PubMed.
- D. L. Priwitaningrum, J. B. G. Blondé, A. Sridhar, J. van Baarlen, W. E. Hennink, G. Storm, S. Le Gac and J. Prakash, J. Controlled Release, 2016, 244, 257–268 CrossRef CAS PubMed.
- G. Fang, H. Lu, A. Law, D. Gallego-Ortega, D. Jin and G. Lin, Lab Chip, 2019, 19, 4093–4103 RSC.
- X. Hu, X. Hu, S. Zhao, S. Zhao, Z. Luo, Y. Zuo, Y. Zuo, F. Wang, F. Wang, J. Zhu, J. Zhu, L. Chen, L. Chen, D. Yang, Y. Zheng, Y. Zheng, Y. Cheng, F. Zhou, Y. Yang and Y. Yang, Lab Chip, 2020, 20, 2228–2236 RSC.
- V. Normand, D. L. Lootens, E. Amici, K. P. Plucknett and P. Aymard, Biomacromolecules, 2000, 1, 730–738 CrossRef CAS PubMed.
- T. H. Jovic, G. Kungwengwe, A. C. Mills and I. S. Whitaker, Front. Mech. Eng., 2019, 5, 19 CrossRef.
- A. Pluen, P. A. Netti, R. K. Jain and D. A. Berk, Biophys. J., 1999, 77, 542–552 CrossRef CAS PubMed.
- F. Lux, V. L. Tran, E. Thomas, S. Dufort, F. Rossetti, M. Martini, C. Truillet, T. Doussineau, G. Bort, F. Denat, F. Boschetti, G. Angelovski, A. Detappe, Y. Crémillieux, N. Mignet, B. T. Doan, B. Larrat, S. Meriaux, E. Barbier, S. Roux, P. Fries, A. Müller, M. C. Abadjian, C. Anderson, E. Canet-Soulas, P. Bouziotis, M. Barberi-Heyob, C. Frochot, C. Verry, J. Balosso, M. Evans, J. Sidi-Boumedine, M. Janier, K. Butterworth, S. Mcmahon, K. Prise, M. T. Aloy, D. Ardail, C. Rodriguez-Lafrasse, E. Porcel, S. Lacombe, R. Berbeco, A. Allouch, J. L. Perfettini, C. Chargari, E. Deutsch, G. Le Duc and O. Tillement, Br. J. Radiol., 2019, 92, 109320180365 Search PubMed.
- W. Rima, L. Sancey, M. T. Aloy, E. Armandy, G. B. Alcantara, T. Epicier, A. Malchère, L. Joly-Pottuz, P. Mowat, F. Lux, O. Tillement, B. Burdin, A. Rivoire, C. Boulé, I. Anselme-Bertrand, J. Pourchez, M. Cottier, S. Roux, C. Rodriguez-Lafrasse and P. Perriat, Biomaterials, 2013, 34, 181–195 CrossRef CAS PubMed.
- C. Riviere, A. Prunet, L. Fuoco and H. Delanoë-Ayari, Patent, FR3079524A1, 2018, https://patents.google.com/patent/FR3079524B1/en Search PubMed.
- G. Le Duc, S. Roux, A. Paruta-Tuarez, S. Dufort, E. Brauer, A. Marais, C. Truillet, L. Sancey, P. Perriat, F. Lux and O. Tillement, Cancer Nanotechnol., 2014, 5, 1–14 CrossRef PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- L. Le Roux, A. Volgin, D. Maxwell, K. Ishihara, J. Gelovani and D. Schellingerhout, Mol. Imaging, 2008, 7, 214–221 CrossRef PubMed.
- J. F. Dekkers, M. Alieva, L. M. Wellens, H. C. R. Ariese, P. R. Jamieson, A. M. Vonk, G. D. Amatngalim, H. Hu, K. C. Oost, H. J. G. Snippert, J. M. Beekman, E. J. Wehrens, J. E. Visvader, H. Clevers and A. C. Rios, Nat. Protoc., 2019, 14, 1756–1771 CrossRef CAS PubMed.
- T. Silva Santisteban, O. Rabajania, I. Kalinina, S. Robinson and M. Meier, Lab Chip, 2018, 18, 153–161 RSC.
- E. Nürnberg, M. Vitacolonna, J. Klicks, E. von Molitor, T. Cesetti, F. Keller, R. Bruch, T. Ertongur-Fauth, K. Riedel, P. Scholz, T. Lau, R. Schneider, J. Meier, M. Hafner and R. Rudolf, Front. Mol. Biosci., 2020, 7, 1–19 CrossRef PubMed.
- J. L. Guerquin-Kern, T. D. Wu, C. Quintana and A. Croisy, Biochim. Biophys. Acta, 2005, 1724, 228–238 CrossRef CAS PubMed.
- G. Slodzian, B. Daigne, F. Girard, F. Boust and F. Hillion, Biol. Cell, 1992, 74, 43–50 CrossRef CAS.
- C. MessaoudiI, T. Boudier, C. O. S. Sorzano and S. Marco, BMC Bioinf., 2007, 8, 1–9 CrossRef PubMed.
- M. Singh, D. A. Close, S. Mukundan, P. A. Johnston and S. Sant, Assay Drug Dev. Technol., 2015, 13, 570–583 CrossRef CAS PubMed.
- L. B. Sims, L. T. Curtis, H. B. Frieboes and J. M. Steinbach-Rankins, J. Nanobiotechnol., 2016, 14, 1–12 CrossRef PubMed.
- A. R. Kang, H. I. Seo, B. G. Chung and S. H. Lee, Nanomedicine, 2015, 11, 1153–1161 CrossRef CAS PubMed.
- L. Štefančíková, E. Porcel, P. Eustache, S. Li, D. Salado, S. Marco, J. L. Guerquin-Kern, M. Réfrégiers, O. Tillement, F. Lux and S. Lacombe, Cancer Nanotechnol., 2014, 5, 1–15 CrossRef PubMed.
- L. Sancey, F. Lux, S. Kotb, S. Roux, S. Dufort, A. Bianchi, Y. Crémillieux, P. Fries, J.-L. Coll, C. Rodriguez-Lafrasse, M. Janier, M. Dutreix, M. Barberi-Heyob, F. Boschetti, F. Denat, C. Louis, E. Porcel, S. Lacombe, G. Le Duc, E. Deutsch, J.-L. Perfettini, A. Detappe, C. Verry, R. Berbeco, K. T. Butterworth, S. J. McMahon, K. M. Prise, P. Perriat and O. Tillement, Br. J. Radiol., 2014, 87, 20140134 CrossRef CAS PubMed.
- T. Stylianopoulos, L. L. Munn and R. K. Jain, Trends Cancer, 2018, 4, 292–319 CrossRef CAS PubMed.
- K. Carver, X. Ming and R. L. Juliano, Mol. Ther. – Nucleic Acids, 2014, 3, e153 CrossRef CAS PubMed.
- H. Kang, S. Mintri, A. V. Menon, H. Y. Lee, H. S. Choi and J. Kim, Nanoscale, 2015, 7, 18848–18862 RSC.
- K. Raza, P. Kumar, N. Kumar and R. Malik, Pharmacokinetics and biodistribution of the nanoparticles, Elsevier Ltd, 2017 Search PubMed.
- V. Ivošev, G. J. Sánchez, L. Stefancikova, D. A. Haidar, C. R. González Vargas, X. Yang, R. Bazzi, E. Porcel, S. Roux and S. Lacombe, Nanotechnology, 2020, 31, 13 CrossRef PubMed.
- N. D. Donahue, H. Acar and S. Wilhelm, Adv. Drug Delivery Rev., 2019, 143, 68–96 CrossRef CAS.
- C. A. Lyssiotis and A. C. Kimmelman, Trends Cell Biol., 2017, 27, 863–875 CrossRef CAS PubMed.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218–4244 RSC.
- J. Kondo, T. Ekawa, H. Endo, K. Yamazaki, N. Tanaka, Y. Kukita, H. Okuyama, J. Okami, F. Imamura, M. Ohue, K. Kato, T. Nomura, A. Kohara, S. Mori, S. Dan and M. Inoue, Cancer Sci., 2019, 110, 345–355 CrossRef CAS PubMed.
- S. E. Park, A. Georgescu and D. Huh, Science, 2019, 364, 960–965 CrossRef CAS PubMed.
- A. Ahmed, S. Goodarzi, C. Frindel, G. Recher, C. Riviere and D. Rousseau, bioRxiv, 2021, DOI:10.1101/2021.01.31.428996.
- A. Prunet, S. Lefort, H. Delanoë-Ayari, B. Laperrousaz, G. Simon, C. Barentin, S. Saci, F. Argoul, B. Guyot, J.-P. Rieu, S. Gobert, V. Maguer-Satta and C. Rivière, Lab Chip, 2020, 20, 4016–4030 RSC.
- I. F. Rizzuti, P. Mascheroni, S. Arcucci, Z. Ben-Mériem, A. Prunet, C. Barentin, C. Rivière, H. Delanoë-Ayari, H. Hatzikirou, J. Guillermet-Guibert and M. Delarue, Phys. Rev. Lett., 2020, 125, 128103 CrossRef CAS PubMed.
- A. Albanese, A. K. Lam, E. A. Sykes, J. V. Rocheleau and W. C. W. Chan, Nat. Commun., 2013, 4, 1–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1lc00192b |
| This journal is © The Royal Society of Chemistry 2021 |