
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCryopreservable arrays of paper-based 3D tumor models for high throughput drug screening†
Bisan
Samara
a,
Muhammedin
Deliorman‡
a,
Pavithra
Sukumar‡
a and
Mohammad A.
Qasaimeh
 *ab
*ab
aDivision of Engineering, New York University Abu Dhabi (NYUAD), Abu Dhabi, 129188 United Arab Emirates. E-mail: mohammad.qasaimeh@nyu.edu
bDepartment of Mechanical and Aerospace Engineering, New York University, Brooklyn, NY, 11201 USA
First published on 22nd February 2021
Abstract
Three-dimensional (3D) tumor models have gained increased attention in life-science applications as they better represent physiological conditions of in vivo tumor microenvironments, and thus, possess big potential for guiding drug screening studies. Although various techniques proved effective in growing cancer cells in 3D, their procedures are typically complex, time consuming, and expensive. Here, we present a versatile, robust, and cost-effective method that utilizes a paper platform to create cryopreservable high throughput arrays of 3D tumor models. In the approach, we use custom 3D printed masks along with simple chemistry modifications to engineer highly localized hydrophilic ‘virtual microwells’, or microspots, on paper for 3D cell aggregation, surrounded by hydrophobic barriers that prevent inter-microspot mixing. The method supports the formation and cryopreservation of 3D tumor arrays for extended periods of storage time. Using MCF-7 and MDA-MB-231 breast cancer cell lines, we show that the cryopreservable arrays of paper-based 3D models are effective in studying tumor response to cisplatin drug treatment, while replicating key characteristics of the in vivo tumors that are absent in conventional 2D cultures. This technology offers a low cost, easy, and fast experimental procedure, and allows for 3D tumor arrays to be cryopreserved and thawed for on-demand use. This could potentially provide unparalleled advantages to the fields of tissue engineering and personalized medicine.
Introduction
Three-dimensional (3D) tumor models are essential in life sciences applications, especially in pharmaceutical research, mainly because they better represent physiological conditions of tumor microenvironments. 3D tumor models surpass conventional two-dimensional (2D) cell culture systems in terms of spatial arrangement of tissues and cell–cell/cell–extracellular matrix (ECM) communication. It has been proven that growing cells in 3D influences their morphology, polarity, signaling pathways, nutrient gradients, secretome profile, and gene expression.1–3 Yet, to date, the majority of drug testing is conducted using overly simplified 2D cancer cell models before moving to animal models. Although animal models may give insight into key biological responses, the anatomy and physiology of animals are profoundly different from those of humans, and there are ethical concerns regarding their use in scientific research. Moreover, the lack of mimicry between in vitro 2D cell culture conditions and in vivo 3D tumor microenvironments contributes significantly to high failure rates of drugs during clinical trials,4 causing delays in development and crippling pharmaceutical companies with big financial losses.A key characteristic of 3D tumor models is cell aggregation (i.e. bringing cells in close proximity to facilitate communication through paracrine signaling or direct cell–cell contact). Among existing approaches for 3D aggregate formation, common ones include hanging drop, microwells, low adherence plates, and magnetic manipulation.5 In these approaches, cell aggregation is achieved either by settling cells due to gravity or by ‘forcing’ them to remain in a suspended state due to buoyant/magnetic forces. However, they generally suffer from lengthy preparation protocols, complicated setups, and the need for dedicated cell manipulation tools. Furthermore, the throughput is limited due to technological constraints, thus hindering scalability and commercialization.
Recently, engineered paper platforms have emerged as attractive simple tools for 3D cell culture formation.6 Indeed, paper has an inherent cellulose-based microenvironment made of interconnecting fibers that provides a porous, biocompatible niche for 3D cell growth. Moreover, paper platforms can be easily manufactured to have varying physical (e.g. pore and fiber sizes), mechanical (e.g. paper strength and reinforcement), and chemical (e.g. adhesion and wetting) properties,7 making them versatile scaffolds for diverse cell applications. Yet, the majority of existing paper-based 3D culture platforms either require equipment that is not readily available in laboratory settings, or more notably, lack throughput. For instance, chromatography paper was utilized to study the cellular response to oxygen and nutrient concentration gradients.8 This was performed by first manually pipetting cell cultures onto the paper and then stacking the paper layers to create tissue-like constructs. Despite being a valuable platform for concentration gradient studies within a 3D microenvironment, its success and throughput rates are bound to the availability of automated liquid handlers and subject to error due to user variability. In a more recent study, a 3D culture system made of rolls of coffee filter paper was developed to analyze the cell behavior under hypoxic conditions.9 The system was validated with different cell types and was well-designed for various biological analyses. However, the protocol required complex manufacturing of custom parts to support the rolls, and the overall throughput was limited. Several other studies also utilized paper platforms for secretome analysis, drug screening, personalized medicine, and tissue engineering applications.6 Nevertheless, practicality and throughput limitation in these approaches remain prominent, highlighting the pressing need for other strategies related to culture and formation of paper-based cell models.
Another obstacle hindering the use of 3D cell culture systems in clinical/industrial settings is the lack of off-the-shelf models. While conventional cryopreservation of cell suspensions is relatively well-established, cryopreservation of 3D cell tissues is complex due to differences in heat and mass transfer across the tissue.10 This makes it difficult to prevent ice formation leading to tissue disruption during the cooling/thawing processes, a fundamental reason behind the failure of many tissue cryopreservation procedures.11 Interestingly, increasing the cryoprotectant concentration or reducing the freezing solution has been demonstrated to be successful strategies for preserving 3D hepatocyte cultures with enhanced viability and functions compared to suspended cells.12 However, the former approach is generally not desirable since it can be toxic for cells,13 and the latter approach requires the use of engineered porous substrates.14 Remarkably, paper has a high surface area to volume ratio, making it possible to minimize the volume of freezing solution. Moreover, its 3D mesh of fibers provides a mechanical support for the tissue, thus protecting it from damage caused by ice crystals formed during freezing.
Previous studies used paper as a 2D substrate to support the cryopreservation of embryos.15 Also, in our recent work, we showed that paper platforms can also be effectively utilized for the cryopreservation of various suspended mammalian cell types with recovery rates comparable to conventional slow freezing methods.16,17 In this work, we present a robust method to create high throughput arrays of cell aggregates (3D tumor models) on a single paper platform for cryopreservation and drug screening applications. The technology allows for long term storage of the 3D tumor arrays, and subsequent thawing for drug testing. It first utilizes the inexpensive, bench-top 3D printing technology to create masks with through-holes to selectively pattern silanized paper, via simple chemistry alterations, with an array of highly localized hydrophilic ‘virtual microwells’. Cell aggregation is then facilitated within the microwells, hereafter referred to as microspots, which are surrounded by hydrophobic regions, or ‘virtual barriers’, that prevent cell inter-microspot mixing. We show that the generated paper-based 3D tumor arrays can be successfully cryopreserved and thawed for on-demand use, which could also be potentially utilized in the future as off-the-shelf components, stacked together for building advanced and sophisticated tumor models.
Results
Paper patterning
The entire process for paper patterning and consequent cell loading involves simple steps and takes only 15 to 20 min, as opposed to days in the majority of existing techniques for cell aggregation.3 The overall flow of the approach to create high throughput arrays of 3D tumor models on paper platforms is shown in Fig. 1.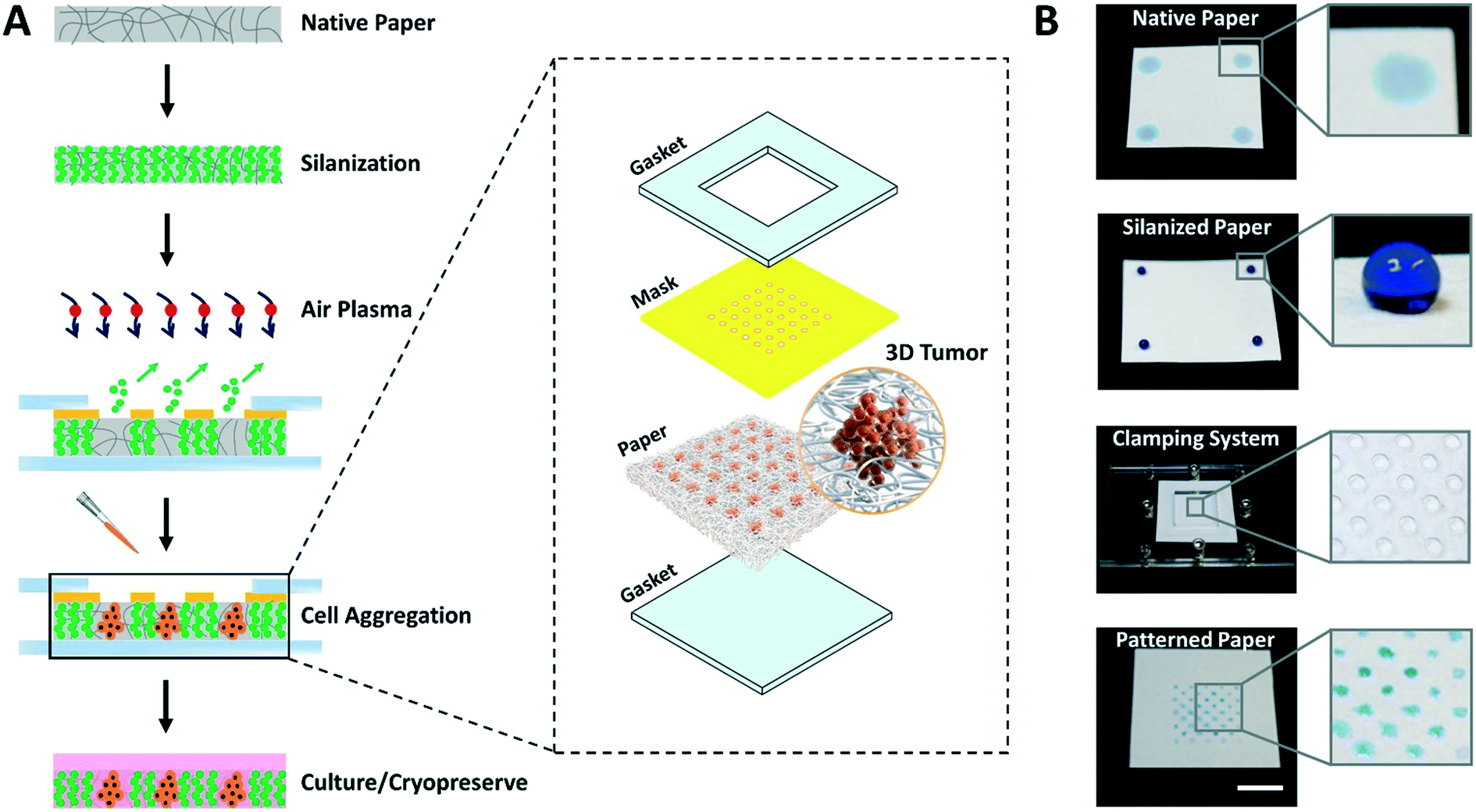 | ||
| Fig. 1 Concept of paper patterning for microspot array formation and consequent cell loading. (A) The paper patterning process starts with vapor silanization of autoclaved filter paper to make it entirely hydrophobic. This is followed by selective exposure (using a custom 3D printed mask clamped to paper through acrylic-based clamps) of silane molecules to air plasma to create an array of highly localized hydrophilic microspots. Next, the cell suspension is loaded and the paper platform containing cells is submerged in growth medium to facilitate cell culture. The paper platform is then used to culture/cryopreserve 3D tumor models. Inset: Illustration of individual parts of the system used in paper patterning. (B) Top to bottom photographs show each step involved in the paper patterning process: the native filter paper, the hydrophobic paper after silanization, the clamping system, and the final array of microspots, respectively. The blue solution is food color dye used for illustration purposes. Scale bar is 1 cm. | ||
First, the autoclaved filter paper is made entirely hydrophobic by vapor deposition of trichloro(1H,1H,2H,2H-perfluorooctyl)silane molecules. Second, a custom 3D printed mask with through-holes is placed on top of the silanized paper and sandwiched within acrylic-based gaskets using clamps. Third, the clamped setup is exposed to air plasma to selectively ablate silane molecules within the unmasked paper regions by highly energetic gaseous ions. In this step, the hydrophobic paper is engineered with hydrophilic microspots comprising patterns based on the size and shape of through-holes in the mask. Fourth, the cell suspension is loaded, the mask is removed, and the paper containing cells is submerged in culture medium facilitating the growth of cell aggregates within the microspots. Finally, depending on the need, cell aggregates are either cryopreserved or continued to grow in culture.
The dimensions of the through-holes in the 3D printed mask define the size of the microspots as well as the rate of cell loading into the paper. Despite the huge potential of commercial bench-top 3D printers, limitations related to resolution and precision have been reported.18 Hence, it was important to understand the capabilities of the 3D printer used in this work (i.e. Asiga MAX X27 UV). We were able to successfully print arrays of through-holes of different shapes (Fig. 2A), and replicate these holes on paper as patterns of various geometric shapes, including channels and labels (Fig. 2B). This showed the applicability of the patterning method for a wide range of paper-based microfluidic applications. Among the shapes, however, triangle, pentagon and square appeared to have less well-defined edges, likely due to limitations imposed by the printer's resolution especially on printing high definition concave corners. Therefore, in this work circular through-holes were adopted for optimization purposes.
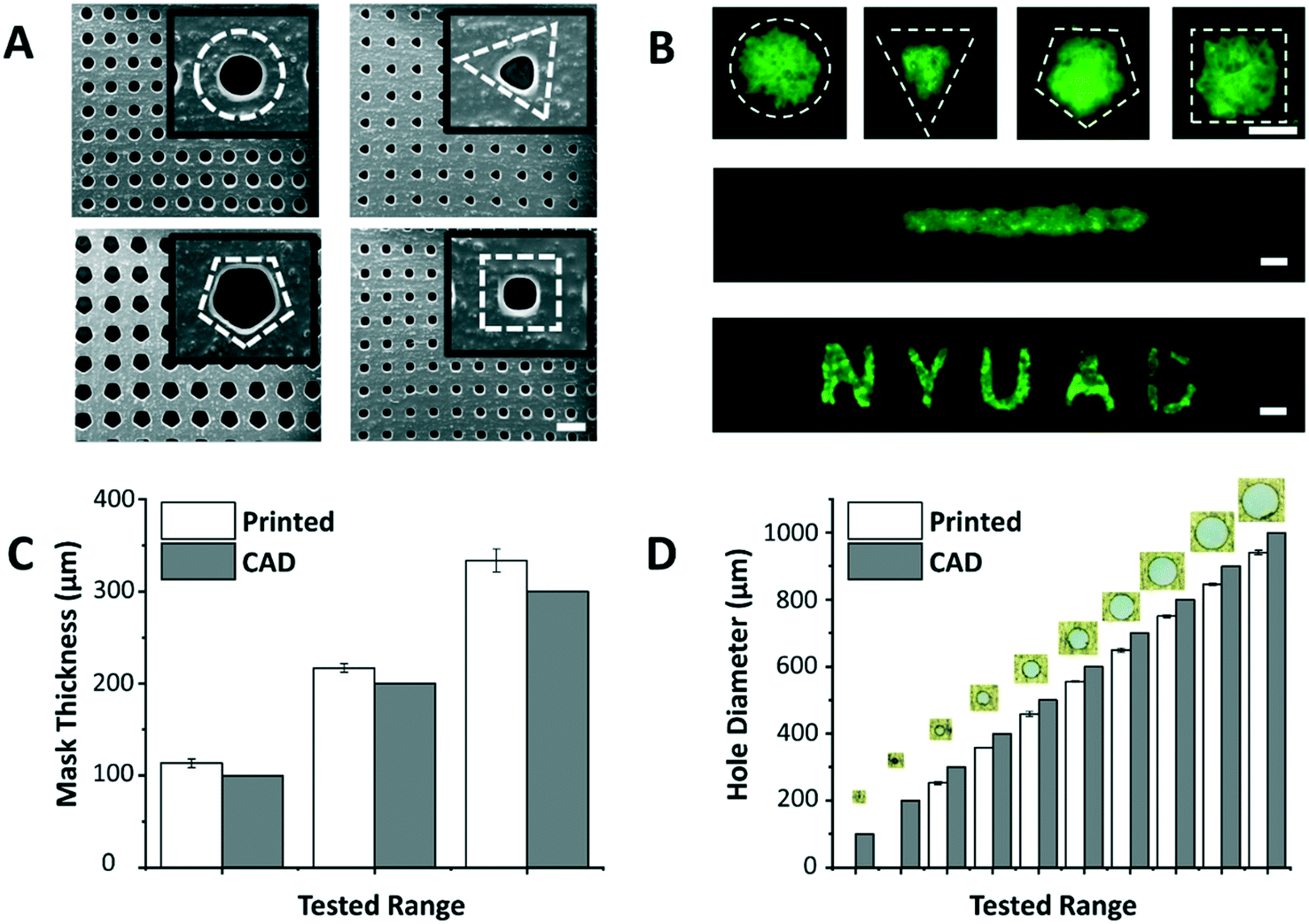 | ||
| Fig. 2 Custom 3D printed masks. (A) SEM images show 3D printed masks with arrays of holes having different shapes (from left to right: circle, triangle, pentagon, and square). Insets: The magnified images of the shapes (dashed white lines for illustration). (B) Microscopy images show fluorescein dyes (green) entrapped and localized within hydrophilic microspots of circle, triangle, pentagon, and square shapes (top, dashed white lines), a line pattern (middle), and an N Y U A D text (bottom) generated on the paper platform. Dimensions of the (C) mask thickness and (D) circular through-holes matched fairly (margin error <15.8%) with the values provided in the CAD model. Values and error bars represent mean ± SD (n = 3). Scale bars are 500 μm. | ||
The thickness of the mask affects penetration of the loaded cell suspension to the paper underneath, and the thicker the mask is, the higher the risk of air bubble entrapment within the holes, which stop cells from penetrating to the paper platform. Thus, the ability of the 3D printer to generate masks of varying thicknesses was tested (Fig. 2C). Results revealed that the thinnest mask we could achieve had a thickness of 113.3 ± 4.7 μm, which was very susceptible to tearing. A mask of 216.7 ± 4.7 μm in thickness, on the other hand, provided a decent compromise between sturdiness of handling the mask and facilitating cell loading into the paper, and hence, was chosen as the base in the follow-up paper patterning experiments.
Another important aspect is the dimensions of the through-holes in the mask, which directly influence the efficiency of the cell loading. To test this, we challenged the capability of the 3D printer to produce circular through-holes with CAD diameters ranging from 100 to 1000 μm (Fig. 2D). The dimensions of the printed circular through-holes were in good agreement with the CAD model (15.8% or less margin error), with the least attainable hole diameter being 252.6 ± 4.7 μm. Diameters less than 200 μm failed to print open through-holes at the given mask thickness (∼200 μm), potentially due to curing of residual resin in the hole vicinity during the printing process. For future work, through-hole resolution can be enhanced by adding photoabsorbers to the resin formulation. This can limit the depth of UV light penetration and prevent undesired curing in the vertical axis. Alternatively, 3D printers with higher end specifications can be used.
The optimal mask and hole dimensions allowed us to print highly dense arrays of 221 circular through-holes, thereby increasing the throughput of the platform. Theoretically, by using the minimum possible hole diameter (∼250 μm) and the maximum possible overall mask dimensions (∼25 × 50 mm2), which are limited to the size of the printer's head used in this study, ∼7000 through-holes can be printed per mask. However, our optimization experiments with fluorescein revealed that 25 × 25 mm2 square masks give improved patterning results since they provide a better seal when clamped with the paper underneath. Moreover, flat margins (without holes) helped the acrylic clamps in having a better grip, and thus, were also incorporated in the mask design. It is noteworthy, however, to mention that the dimensions of the mask and through-holes can easily be modulated for a versatile range of applications.
With the overall mask dimensions, we were able to print two masks at a time in about 5 min, covering the entire printer's head surface. Additionally, once printed, each mask was suitable for patterning of at least 15 paper chips. The masks can also be oriented vertically relative to the printer's head, which would allow printing more masks per print. However, this would require adding structural supports to prevent the masks from collapsing, and will significantly increase material consumption and printing time.
Platform characterization
After optimizing the mask dimensions and the patterning process, the paper platform was next characterized for additional functional parameters (Fig. 3). To start with, the choice of paper type is important since cells will be cultured within its 3D microenvironment, where the ultimate intention will be to contain cells within the pores of the paper over extended periods of culture while allowing for cell aggregation and 3D tissue formation. In our previous work, Whatman grade 114 filter paper with 25 μm pore sizes was mainly used for cell cryopreservation studies, with the end goal being the efficient release of cells from the paper after thawing.16,17 When using the Whatman grade 114 filter paper for cell aggregation, we found that many cells are released or move across the paper thickness without proper entrapment within the patterned microspots. This observation was attributed to the larger pore sizes of the paper. To confirm this, Whatman grade 1 filter paper platforms with 11 μm pore sizes were tested by passing beads with diameters comparable to those of MCF-7 breast cancer cells, and bead entrapment results were compared with those using Whatman grade 114 filter paper. Indeed, the 11 μm pore paper resulted in a significantly lower concentration of passed beads than the 25 μm pore paper (Fig. 3A), indicating a substantial amount of entrapped beads within the paper volume. Therefore, Whatman grade 1 filter paper was used in the next experiments.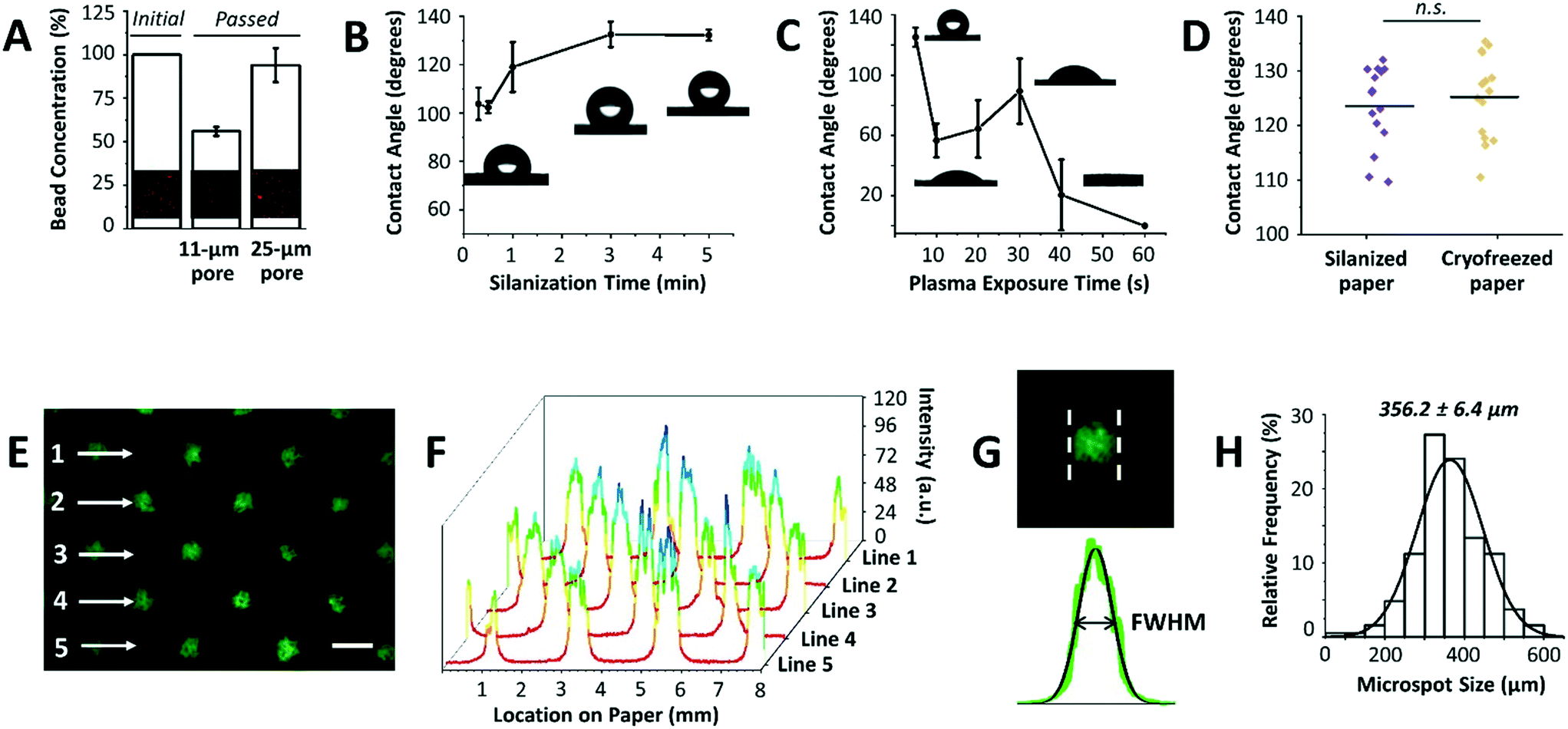 | ||
| Fig. 3 Platform characterization. (A) Comparison of bead entrapment within Whatman grade 1 (pore size = 11 μm) and grade 114 (pore size = 25 μm) filter paper with respect to the initial concentration (inset: respective fluorescence images). The enhanced entrapment of beads within Whatman grade 1 paper is due to its pore size being comparable to that of beads. Values and error bars represent mean ± SD (n = 3). (B) Effect of silanization time on paper hydrophobicity and (C) effect of air plasma exposure time on silane desorption as measured from water contact angles (insets: respective optical images). Values and error bars represent mean ± SD (n = 3). (D) Contact angle distribution of silanized paper (control) and silanized paper incubated in culture medium for 3 days followed by cryopreservation for 5 days. n.s.: not significant at P < 0.05 using two-sided Student's t-test. (E) The fluorescence image shows paper patterned with fluorescein salt solution using circular microspots. Scale bar is 500 μm. (F) 3D plot of fluorescence intensities across lines 1–5 in (E). (G) Analysis of the full width at half maximum (FWHM) of the peaks reveals (H) a mean peak of 356.2 ± 6.4 μm when the mask with a hole diameter of 252.6 ± 4.7 μm is used. The solid black line is the data fit to the Gaussian probability density function (n = 18 from 6 independent experiments). | ||
Another important parameter in the paper patterning process is silanization. When silane molecules are vaporized with pressure and heat, they couple to the cellulose fibers across the paper volume. This turns the paper platform to be entirely hydrophobic, with water contact angles ranging from 103.8° ± 6.7° to 132.2° ± 2.3° for 0.3 to 5 min incubation time with silane, respectively (Fig. 3B). The optimal silanization time, which forms hydrophobic barriers that are strong enough to prevent inter-microspot mixing and provides good desorption efficiency, was experimentally determined to be 3 min, resulting in hydrophobic paper with a contact angle of 132.6° ± 5.2°. Then, air plasma was used to remove silane molecules in the unmasked regions, creating highly localized hydrophilic microspots. The desorption of silane was observed by a noticeable drop in the contact angle following ∼10 s of plasma exposure (Fig. 3C). 20 s and 30 s of plasma exposure, on the other hand, temporarily caused silane molecules to self-condense (by forming Si–O–Si bonds),19 as revealed by a slight increase in the contact angle from 64.4° ± 19.0° to 89.4° ± 21.6°, respectively. Whereas, 40 s and 60 s of plasma exposure turned the paper volume into an entirely hydrophilic one, where the added water droplets immediately wetted the paper (Fig. 3C). This was attributed to oxidation of silane molecules to silanols (Si–O–H).19 Silanized paper was additionally incubated in culture medium for 3 days (n = 3), and then cryofrozen for 5 days. Contact angle measurements revealed that the change in hydrophobicity was not significant compared to that in directly silanized paper (control), with contact angle values of 125.2° ± 7.6° to 123.5° ± 7.4°, respectively (Fig. 3D).
Furthermore, it has been reported that plasma exposure increases the surface roughness of cellulose fibers and causes them to shrink.20 Therefore, and to avoid microstructural alteration to the paper fibers, plasma exposure was performed at low radio frequency discharge voltage and pressure and the exposure time was kept to the minimum (10 s), thus neither compromising the hydrophobic barriers between the microspots nor influencing the fiber structure (ESI† Fig. S1).
A number of effective techniques have been applied to create hydrophobic barriers on paper platforms for cell culture studies within patterned hydrophilic regions, including wax printing,21 wax dipping,22 and polydimethylsiloxane (PDMS) printing.23 Moreover, sacrificial 3D printing was recently employed to embed microchannels in paper-based devices.24,25 Although these techniques reasonably cut down the turn-around times and offer good control over the patterning process, they require specialized printers and materials. Paper patterning with hydrophobic silane followed by UV-based selective desorption has also been previously reported.26 However, utilizing the more powerful air plasma in this work reduces the exposure time from hours to a few seconds while effectively ablating the silane molecules within the paper.
The final parameter in the paper patterning process is the uniform loading of cell suspension. For consistency and homogeneity check, a fluorescein solution was loaded onto the patterned paper and the fluorescence intensities across rows of microspots were measured (Fig. 3E). The results showed peak values of similar intensities (Fig. 3F), and the full width at half maximum of the peaks (FWHM) followed a Gaussian distribution with a mean of 356.2 ± 6.4 μm (Fig. 3G and H) when the mask with a hole diameter of 252.6 ± 4.7 μm was used. The histogram was generated by analyzing the FWHM across lines of microspots from 6 independent paper platforms, with 3 different fields of view per platform. The relatively low standard deviation (±6.4 μm) indicated high consistency among the microspot sizes within one paper chip, as well as among different paper chips. Although the resulting average microspot diameter in these experiments was larger than that of the mask, which is due to fluorescein solution wicking on paper, such patterning variation in size is insignificant when loading cells. This is expected as lateral diffusion of liquid is considerably larger than that of micron-sized cells. When patterning proteins for example, this size variation is expected due to wicking. Therefore, the sizes of the microspots can be readily modulated by changing the dimensions of the 3D printed through-holes in the mask (ESI† Fig. S2).
Culture and cryopreservation of 3D cell aggregates
Following platform characterization, our next aim was to investigate the effect of silane molecules that may have remained within the microspots after plasma exposure on the formation of cell aggregates and culture. For this, MCF-7 breast cancer cells were cultured for a duration of three days within native paper (control) and paper microspots, followed by measuring their metabolic activity by the WST-1 assay. However, the WST-1 assay failed to detect the changes in the metabolic activity of cells grown within microspots of ∼350 μm in diameter because the cell count was below the assay's detection limit. Therefore, for all the WST-1 measurements, the diameter of microspots was increased to ∼1000 μm; thus, increasing the cell count to a level detectable by the assay. Nevertheless, results revealed that residual silane molecules within the microspots do not compromise overall cellular functions (Fig. 4A). In all cell aggregation experiments, fibronectin was added to enhance cell adhesion onto paper fibers. Our preliminary experiments on growing tumor models without fibronectin, on the other hand, had no significant influence on their proliferation.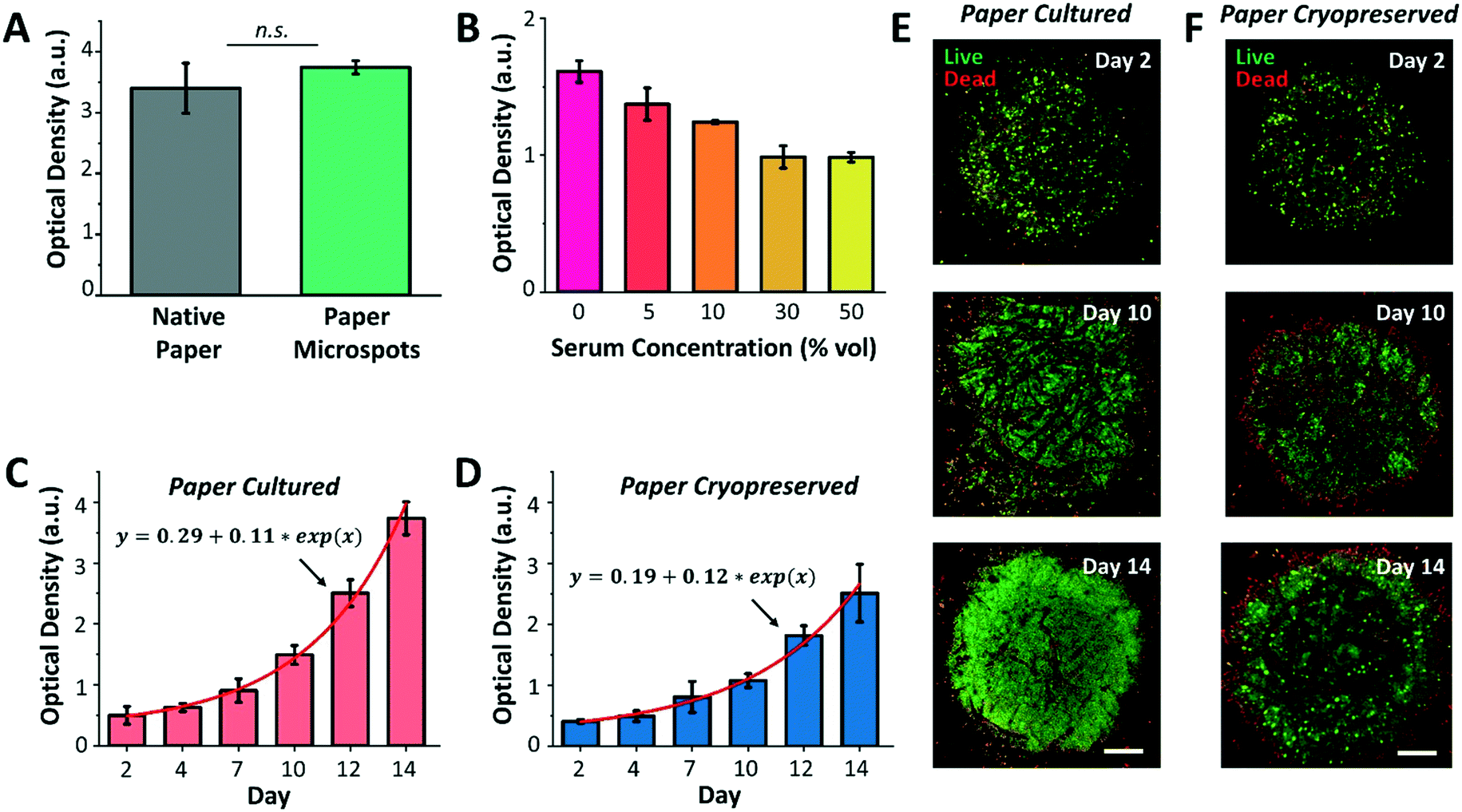 | ||
| Fig. 4 Culture and cryopreservation of paper patterned 3D cell aggregates. (A) Comparison of the growth of MCF-7 3D culture within native paper (control) and paper high throughput microspots, using the WST-1 assay, proves the biocompatibility of the paper patterning method. n.s.: not significant at P < 0.01 using two-sided Student's t-test. (B) Optimization of serum concentration in growth medium reveals that serum-free medium is the most effective in supporting the growth of MCF-7 cell aggregates within paper microspots, evaluated using the WST-1 test. (C) Exponential growth (solid red line is the data fit) of MCF-7 3D models is observed (WST-1 assay) within microspots in serum-free medium when directly culturing the paper platform and when (D) the platform is cultured following cryopreservation and thawing steps. Confocal microscopy images of MCF-7 3D models following (E) their direct culture and (F) culture after cryopreservation and thawing reveal more than 90% viable aggregates within the microspots (live/dead assay). Values and error bars represent mean ± SD (n = 5). Scale bars are 250 μm. | ||
We then proceeded with optimizing the culture medium composition and assessing the growth of 3D cell culture within paper microspots over time. Despite the fact that animal-derived serum contains a cocktail of growth factors and hormones essential for the growth of cells, its use has been controversial due to multiple reasons: i) it possesses a non-deniable batch-to-batch variation, ii) it has not been adequately characterized, iii) it is a potential source of microbial contamination, and iv) it faces ethical concerns related to its extraction and use. Thus, several studies reported the use of serum-free media for the growth of spheroids, including MCF-7, and showed enhanced 3D cell culture compared to serum-containing media.27 Our results were in agreement with these findings, where the growth of MCF-7 3D culture in serum-free media, measured by the WST-1 assay, was the highest compared to their growth rates in 5, 10, 30, and 50% serum media (Fig. 4B).
Notably, serum-free media also supported exponential growth of MCF-7 3D culture for a duration of two weeks, with more than 7-fold increase when the paper platform is directly cultured (Fig. 4C) and about 6-fold increase after their thawing and culture when the paper platform is cryopreserved (Fig. 4D). This result is consistent with our previous finding, where slow freezing (at a rate of −1 °C min−1) and cryopreserving (for 3 days) of paper-based MCF-7 cultures maintained the integrity and viability of cells for extended culture (8 days) after thawing.16 The confocal images of MCF-7 3D models (directly cultured, Fig. 4E, or cultured after their cryopreservation and thawing, Fig. 4F) further revealed more than 90% viable aggregates within the microspots, with dead aggregates mostly seen at the perimeter. Here, the hydrophobic edges of microspots may have prevented cells in close proximity within the aggregates from forming focal adhesions, thus inducing anoikis, a form of apoptosis for anchorage dependent cells that is induced when the interaction between the cell and its ECM is weak or lost. As for cells already attached to edge fibers, silane molecules may have formed a rigid barrier that constrained the movement of their plasma membrane. This may have limited the extent of actin polymerization in the cells, thus leading to the activation of apoptotic signals.28 Additional imaging in bright field mode did not provide a useful approach in viewing cells (ESI† Fig. S3), which was mainly due to the paper thickness that prevented the light from effectively passing through. Nonetheless, the peripheral dead cells had no noticeable influence, both qualitatively and quantitatively as measured by the WST-1 assay, on the overall growth of cultures. Moreover, the fluorescence images in Fig. 4 and ESI† Fig. S3 further revealed that extended incubation of paper in culture medium (up to 14 days) does not significantly change the size of the microspots both for paper cultured and paper cryopreserved conditions, thus, confirming durable hydrophobicity within their surroundings.
Drug testing
Next, we investigated the sensitivity of two different human breast cancer cell lines, MCF-7 and MDA-MB-231, to chemotherapeutic drug cisplatin. Both MCF-7 and MDA-MB-231 cell lines were 3D cultured, cryopreserved, thawed, and drug tested under identical conditions. To this end, both cell lines were cultured, using serum-free medium, in 96-well plates as 2D monolayers of cells (control) and within paper microspots as 3D tumor models. Varying concentrations of cisplatin (10, 20, 30 and 50 μg mL−1) were then introduced and their effect on relative cell survival was measured by the WST-1 assay after incubation for 24 and 48 hours. The effectiveness of cisplatin on the growth of aggregates thawed and cultured after their paper cryopreservation was also investigated (Fig. 5). Table 1 summarizes the differences between the two cell lines, which includes the tissue they were initially derived from, the cell subtype and morphology, and the expression of three major receptors used to classify the breast cancer, namely estrogen receptor (ER), progesterone receptor (PR), and human epithelial receptor 2 (HER2).29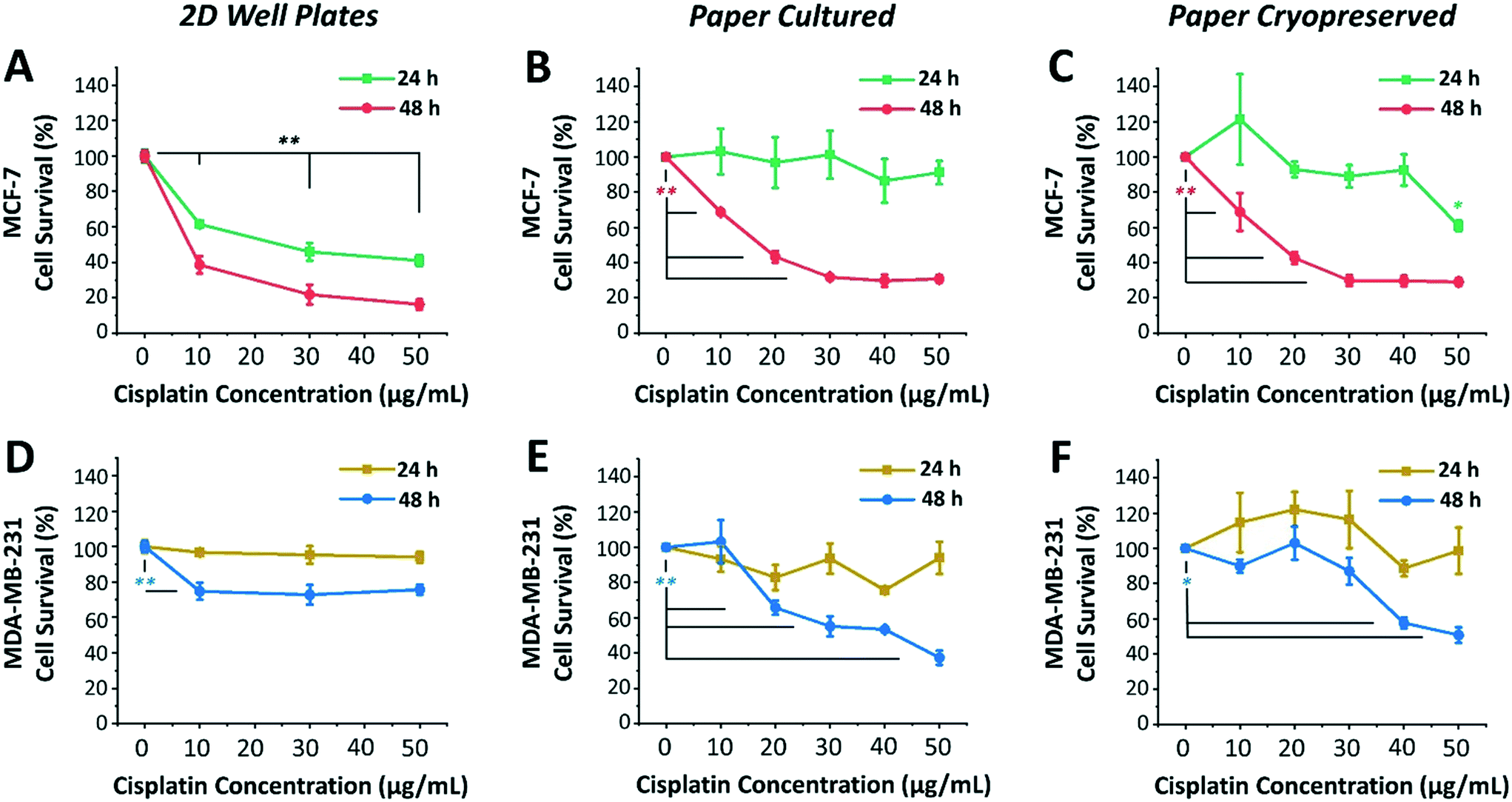 | ||
| Fig. 5 Response of MCF-7 and MDA-MB-231 breast cancer tumor models to cisplatin-induced cytotoxicity. Effect of cisplatin-induced cytotoxicity on survival of (A–C) MCF-7 and (D–F) MDA-MB-231 cells grown in 96-well plates as a control 2D cell monolayers (first column) and within paper microspots as 3D models, directly cultured (second column) and cultured after their cryopreservation and thawing (third column). Values and error bars represent mean ± SD (n = 5). The significance was evaluated using two-sided Student's t-test, where respective time points were compared to no drug conditions (i.e. 100% cell survival). Values of *P < 0.05 were considered significant and those of **P < 0.01 highly significant. | ||
| MCF-7 | MDA-MB-231 | |
|---|---|---|
| Tissue | Ductal carcinoma | Adenocarcinoma |
| Subtype | Laminal A | Basal |
| Morphology | Epithelial | Mesenchymal-like |
| Receptors | ER+, PR+, HER2− | ER−, PR−, HER2− |
Among the two cell lines, cisplatin exerted a greater time- and dose-dependent cytotoxic effect on 2D cells of MCF-7 compared to the 3D culture formed within paper microspots (Fig. 5A–C).
For example, treating 2D cells with 10 μg mL−1 concentration for 24 and 48 hours resulted in ∼61% and 38% cell survival, respectively, while 50 μg mL−1 concentration further reduced these numbers to ∼40% and 16% (Fig. 5A). As for the paper-based 3D culture, the cytotoxic effect of tested cisplatin concentrations was to a lesser extent when incubated for 24 hours than for 48 hours (Fig. 5B), indicating multicellular time-dependent drug resistance.30 Moreover, for 48 hours of incubation, 20 μg mL−1 cisplatin concentration resulted in ∼43% cell survival, which then plateaued at ∼30% for 30 μg mL−1 or higher concentrations. This response to cisplatin-induced cytotoxicity is considered saturable passive, whereby the levels of various factors (e.g. drug uptake, obligate targets, and proapoptotic factors) required for drug efficacy are reduced in cells at higher doses.31 Moreover, the relative effect of cisplatin was also remarkably consistent for 3D aggregates thawed and cultured after their cryopreservation (Fig. 5C).
MDA-MB-231 cells, on the other hand, exhibited clear resistance to cisplatin in both 2D cultures and 3D models (Fig. 5D–F). This was not surprising, since this cell line is regarded as multidrug resistant, where specifically the P-glycoprotein present in their plasma membrane acts as a drug efflux pump.32 For 2D cultures, the relative cisplatin-induced cytotoxic effect was only time-dependent, reducing the survival of cells by ∼5% and 30% when incubated for 24 hours and 48 hours, respectively (Fig. 5D). For paper cultured and paper cryopreserved 3D models, on the other hand, the cytotoxicity of cisplatin was time- and dose-dependent. For 48 hours of incubation, the difference was more evident at cisplatin concentrations of 30 μg mL−1 or higher for paper cultured (Fig. 5E) and of 40 μg mL−1 and higher for paper cryopreserved (Fig. 5F) 3D models, resulting in ∼55% to 37% and ∼57% to 50% cell survival, respectively.
Compared to 2D cultures, paper-based tumor models had larger variations in their response to cisplatin due to the heterogeneous nature of 3D cultures as opposed to cells growing on flat surfaces of the well plates. In some cases, survival rates after cisplatin addition exceeded 100%, indicating insensitivity to the drug at the respective concentration, with the expected healthy growth of cells being unaffected. Moreover, the response curves of cryopreserved paper models were in good agreement but not identical to those of directly cultured paper models. This could be attributed to the potential loss of some cells during the freeze/thaw processes, or the need for a longer recovery time after thawing to exhibit more similar responses. Nevertheless, overall, the complexity and heterogeneity introduced by the cryopreservable paper-based 3D tumor models recapitulated key aspects of the in vivo microenvironment, making them very good candidates for performing drug screening assays.
Discussion
Breast cancer is the most frequently diagnosed cancer in more than 80% of the countries around the world. In the United States alone, ∼12% of American women develop invasive breast cancer over the course of their lives,33 with a mortality rate above 70% in its advanced stages.34 Despite tremendous efforts in the investigation of effective drugs to treat cancer, several challenges persist. Most of all, the drug development process suffers from slow progress, low success rates, and high costs.4 Moreover, existing cancer cell models to evaluate drug candidates in preclinical stages often lack relevance to human physiology, resulting in false propagation of some candidates to more laborious and expensive clinical trials. A key characteristic observed in several in vivo tumors is drug resistance, which is typically in orders of magnitude higher than that of in vitro 2D cell monolayers.35 Thus, for successful drug response studies on cancer cells, development of alternative in vitro platforms that efficiently generate 3D tumor models is a continuous necessity. To cover this need, in this work we describe a robust, time efficient, and cost-effective method to create cryopreservable high throughput arrays of 3D tumor models on paper platforms.In relation to generating 3D tumor models, the developed paper platform offers several major advantages. First, the patterning procedure on paper is simple and scalable, and takes no longer than 20 min. The printed mask is reusable (at least 15 times) for patterning, and the generated arrays of 3D tumor models cost less than US$5 per paper chip. Second, the platform supports extended cell culture. In other reported approaches (e.g. hanging drop and low adhesion plates), changing the growth medium poses a challenge, as there is a good chance of disturbing the clusters with the pipette pressure.36 Meanwhile in our platform, this process is more robust where medium exchange is achieved only by aspirating the medium from the flasks/tubes containing paper, and then replacing it with a fresh one. During the exchange, the tumor models are structurally protected within the paper fibers and hydrated with readily absorbed medium in the paper, thus avoiding disruption of aggregates with pipette pressure and preventing cultures from drying. Third, the paper itself acts as an inherent vascularized 3D network when delivering fresh medium to all parts of the aggregates, even when the size of the microspots exceeds the diffusion limit of oxygen and nutrients (which is typically 200 μm).37 In our platform, this exhibited excellent biocompatibility apparent from healthy cell proliferation over extended durations of culture. Fourth, the cell aggregates within the platform can be efficiently cryopreserved for longer storage durations (months) and thawed when needed. As evidenced from our previous results, where long-term (up to 6 months) paper-cryopreserved cells exhibited a viability of >90% with minimum change in their morphology and metabolic activity,16 this advantage is particularly important, as it saves a significant amount of preparation time and effort, and ultimately cost, while allowing for off-the-shelf 3D tumor models to be readily available for on-demand use. Moreover, it offers an advantage from a commercial perspective, where cryopreserved 3D tumor models can be efficiently supplied to research laboratories around the world in dry ice or liquid nitrogen as commonly followed by cell biobanks. A future extension of this method could also be applied to cancer cells derived directly from patient biopsies, allowing for easy and stream-lined drug screening studies applicable to precision medicine.
In addition to offering high throughput arrays of 3D tumor models, the developed paper platform also provides in vitro microenvironments for exploring the cytotoxic effects of chemotherapeutic drugs. By studying the effect of cisplatin-induced cytotoxicity on 3D models of MCF-7 and MDA-MB-231 breast cancer cell lines, we show that our patterned paper chip can potentially accelerate drug testing, and increase accuracy and cost efficiency. In view of the drug-resistant characteristics of 3D tumors, for example, the platform enables growing cells in a biomimetic 3D niche that is close to the in vivo environment. This way, generated 3D tumors manifest key drug-resistant characteristics of native tumors that are otherwise absent in currently widely used 2D culture systems.35 In view of preserving 3D tumors, on the other hand, the platform enables successful cryopreservation of cells with viability comparable to conventional methods.16 In our experiments, the applicability of these two important attributes was evident with comparison against time- and dose-dependent responses of 2D cells and 3D models of MCF-7 and MDA-MB-231 after cisplatin treatment.
In this work, the presented paper patterning technology was largely directed at growing cryopreservable 3D tumor models for high throughput drug testing. For example, the reported 221 microspots per paper platform allow for significant statistical sampling of the drug efficacy results. Additionally, the paper patterning could be transformed into various other trending therapeutic applications, such as measuring tumor response to drug concentration gradients38 and investigating cancer cell signaling pathways.39 Moreover, higher throughput generation of 3D tumor models could be easily accomplished by employing 3D printers of higher resolution in patterning the microspots, and the technology can be combined with high-precision multiplexed drug dispensing using advanced robotics. By rolling, folding, or stacking layers of paper arrays, the platform can also be easily modified for in vitro investigation of chemotactic migration and penetration of 3D tumor models.40 For this, different cell types (e.g. HeLa, PC3, tumor-associated fibroblasts, and primary cells) can be grown as co-aggregates to test their multidrug response at each spot or line of spots. With slight modifications, paper patterning of 3D tumor models can also be easily integrated to origami-based immunoassay applications.41
In conclusion, this work presents a paper patterning method for high throughput culture, cryopreservation, and drug testing of 3D tumor models. Compared to existing approaches, we show that our technology offers a lower cost, easier and faster experimental procedure. We also show that the patterned 3D tumor arrays can be cryopreserved for prolonged periods and thawed for on-demand use, providing a great advantage for tissue engineering and personalized medicine applications.
Materials and methods
3D printed mask and the clamping system
Designs of the mask were created using commercial SolidWorks CAD software (Dassault Systèmes, France). Masks were printed using PlasCLEAR resin and a MAX X27 UV stereolithography 3D printer with 27 μm pixel resolution (Asiga, Australia). A custom made, laser-cut, acrylic-based clamping system was used to sandwich the paper and the mask. The overall mask dimensions were 25 × 25 × 0.2 mm2 (l × w × t) containing a margin of 5 mm on the sides, where the acrylic clamps were placed. The inner 15 × 15 mm2 area of the mask was filled with through-holes of different shapes.Paper patterning
Whatman grade 1 (11-μm pore size) and grade 114 (25-μm pore size) cellulose filter paper chips (Sigma-Aldrich) were first autoclaved and then made hydrophobic by vapor deposition of trichloro(1H,1H,2H,2H-perfluorooctyl)silane molecules (Sigma-Aldrich). In the process, 200 μL of silane solution was added to a glass Petri dish inside a small vacuum desiccator. With the help of metal blocks heated at 360 °C, the desiccator was allowed to heat up for 10 min. Then, the paper chips, cut into 3 × 3 cm2, were placed opposite to the Petri dish to maximize silanization efficiency. Next, vacuum was applied and the paper chips were left inside the desiccator for 3 min. The silanization time was optimized such that it gives enough hydrophobicity to the paper, as measured from contact angles, while allowing the hydrophobicity to be reversed within the localized microspots. After silanization, the 3D printed mask with through-holes was added on top of the hydrophobic paper and clamped between the acrylic gaskets. The clamped system was then exposed to air plasma using a plasma cleaner (Harrick Plasma, USA) for 10 s at a pressure of 700 mTorr using low radiofrequency discharge voltage, where silane molecules desorbed in the unmasked regions by the energetic plasma molecules, creating hydrophilic microspots. Subsequently, the paper was loaded with 1 mg mL−1 fluorescein salt in PBS for characterization of the microspots.Contact angle measurement
The hydrophobicity of the silanized paper was evaluated by contact angle measurements based on the sessile drop method using an OCA 15EC (DataPhysics Instruments, Germany). A 3 μL drop of distilled water was dispensed on different parts of the silanized or plasma exposed paper at a rate of 1 μL s−1, and the images of each droplet were used to measure the corresponding contact angles.Scanning electron microscopy
A Quanta 450 FEG field emission scanning electron microscope (Thermo Fisher) was used to image the 3D printed masks and the cell clusters on the paper. Prior to imaging, the samples were coated with a thin conductive gold layer using a sputter coater 108auto (Cressington, UK). Images were taken under high vacuum conditions (chamber pressure = 15 × 10−4 Torr) at 20 keV beam accelerating voltage and 4.0 beam spot size.Cell culture and cryopreservation
MCF-7 and MDA-MB-231 breast cancer cell lines (Cell Line Services, Germany) were cultured in growth medium (Dulbecco's modified essential medium, Gibco) supplemented with 10% fetal bovine serum (Sigma-Aldrich) and 1% penicillin–streptomycin (Sigma-Aldrich). The cells were then incubated in a humidified environment at 37 °C and 5% CO2. Cells suspended in growth medium with 10 μg mL−1 concentration of fibronectin human plasma (Sigma-Aldrich) were loaded into the paper. Other ECM proteins can also be incorporated at this step to accommodate different applications. Subsequently, the paper chips loaded with cells were submerged in serum-free media and kept in culture for two weeks, or cultured for 1–3 days before cryopreservation. For cryopreservation, the procedure explained in our previous work was followed.16,17 Briefly, the paper chip containing cell aggregates was rolled and placed inside a standard cryotube, submerged in full growth medium supplemented with 10% dimethyl sulfoxide (Sigma-Aldrich), and then frozen conventionally at a rate of −1 °C min−1 overnight using a Mr. Frosty container (Thermo Fisher) before being transferred to a −196 °C liquid nitrogen tank.Cell viability and proliferation assays
The viability of cell aggregates in culture, or after cryopreservation and thaw, was assessed with a live/dead cell viability imaging kit (Invitrogen). Briefly, the samples were submerged in a solution containing 0.2% ethidium homodimer and 0.05% calcein in PBS and incubated for 20–30 min at room temperature in the dark. Then, fluorescence microscopy images were used to qualitatively assess cell viability. Because of the proximity of cells to each other within the aggregates, it was difficult to identify cell–cell boundaries to count the number of individual live and dead cells in the microspots. Therefore, for quantification of live/dead cell aggregates, the green and red fluorescence images were analyzed separately in ImageJ after 3D reconstructing them using maximum intensity Z-projection comprising at least 6 planes. Following their conversion to grayscale images, covered areas for live and dead aggregates were calculated based on their intensity distribution at each pixel.For quantitative assessment of cell proliferation and metabolic activity, the WST-1 assay (Sigma-Aldrich) was used. Tetrazolium salt WST-1 cleaves into formazan through dehydrogenase produced by the mitochondria of metabolically active cells. Briefly, the WST-1 reagent was added to the wells containing paper samples in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 (vol/vol) and incubated for 2–4 hours, followed by the use of a Synergy H1 plate reader (BioTek Instruments, USA) to measure the optical density (OD) for formazan at 440 nm. Simultaneously, the OD at 640 nm was subtracted from each measurement as the background.
:10 (vol/vol) and incubated for 2–4 hours, followed by the use of a Synergy H1 plate reader (BioTek Instruments, USA) to measure the optical density (OD) for formazan at 440 nm. Simultaneously, the OD at 640 nm was subtracted from each measurement as the background.
Fluorescence microscopy
Confocal images were acquired with an inverted confocal two-photon FV1000MPE FluoView microscope (Olympus, Japan). Lasers at excitation wavelengths of 488 nm (green) or 612 nm (red) were used. All images were taken with a 10× air objective. Imaris software (Bitplane, Switzerland) was used for the 3D reconstruction of images. Images with a wider field of view were taken with an SMZ18 stereo microscope (Nikon, Japan) modulating the magnification between 0.75 and 13.5×. Fluorescence intensity measurements from microscopy images were conducted using Fiji software (NIH, USA).Drug testing
Antineoplastic cisplatin (Sigma-Aldrich) was prepared at 3 mg mL−1 concentration by diluting the drug stock in sterile deionized water containing 0.9% sodium chloride to enhance its stability. The solution was then additionally sterilized with a 0.22 μm filter and stored at 4 °C in the dark. For drug testing, cryopreserved paper chips were thawed and put in culture for 4–14 days prior to the experiments. At the time of the testing, the drug stock was further diluted in cell culture media to working solutions in the μg mL−1 range. The paper platforms with the 3D cell cultures were then submerged in cisplatin solutions at different concentrations and incubated for 24 or 48 hours. The cytotoxic effect of cisplatin on the metabolic activity of cell aggregates was measured by the WST-1 assay as described earlier.Data analysis
All values are reported as the mean ± standard deviation from at least three replicates per experiment. Statistical significance was evaluated using two-sided Student's t-test. Values of P < 0.05 were considered significant and those of P < 0.01 were considered highly significant. All plots, data fitting, and statistical analysis were generated using data analysis and graphing software (OriginLab, USA).Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This study was financially supported by NYU Abu Dhabi Internal Fund as well as the NYU Abu Dhabi Grants for Publication Program. The authors acknowledge the technical support from the Core Technology Platforms at NYU Abu Dhabi, especially Mr. Vijay Dhanvi and Dr. Rachid Rezgui. The authors also thank Ms. Jumaanah ElHashemi for the scientific illustration (inset in Fig. 1A). The icons in the Graphical Abstract image were generated using science illustration software (BioRender, Canada).References
- K. Duval, H. Grover, L.-H. Han, Y. Mou, A. F. Pegoraro, J. Fredberg and Z. Chen, Physiology, 2017, 32, 266–277 CrossRef CAS.
- B. M. Baker and C. S. Chen, J. Cell Sci., 2012, 125, 3015–3024 CrossRef CAS.
- E. C. Costa, A. F. Moreira, D. de Melo-Diogo, V. M. Gaspar, M. P. Carvalho and I. J. Correia, Biotechnol. Adv., 2016, 34, 1427–1441 CrossRef.
- M. J. Waring, J. Arrowsmith, A. R. Leach, P. D. Leeson, S. Mandrell, R. M. Owen, G. Pairaudeau, W. D. Pennie, S. D. Pickett, J. Wang, O. Wallace and A. Weir, Nat. Rev. Drug Discovery, 2015, 14, 475–486 CrossRef CAS.
- D. Liu, S. Chen and M. Win Naing, Biotechnol. Bioeng., 2021, 118, 542–554 CrossRef CAS.
- K. Ng, B. Gao, K. W. Yong, Y. Li, M. Shi, X. Zhao, Z. Li, X. Zhang, B. Pingguan-Murphy, H. Yang and F. Xu, Mater. Today, 2017, 20, 32–44 CrossRef CAS.
- K. Yamada, H. Shibata, K. Suzuki and D. Citterio, Lab Chip, 2017, 17, 1206–1249 RSC.
- R. Derda, A. Laromaine, A. Mammoto, S. K. Y. Tang, T. Mammoto, D. E. Ingber and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18457–18462 CrossRef CAS.
- D. Rodenhizer, T. Dean, B. Xu, D. Cojocari and A. P. McGuigan, Nat. Protoc., 2018, 13, 1917–1957 CrossRef CAS.
- J. O. Karlsson and M. Toner, Biomaterials, 1996, 17, 243–256 CrossRef CAS.
- D. E. Pegg, CryoLetters, 2001, 22, 105–114 CAS.
- J.-H. Lee, D.-H. Jung, D.-H. Lee, J.-K. Park and S.-K. Lee, Transplant. Proc., 2012, 44, 1015–1017 CrossRef CAS.
- Y. C. Song, B. S. Khirabadi, F. Lightfoot, K. G. M. Brockbank and M. J. Taylor, Nat. Biotechnol., 2000, 18, 296–299 CrossRef CAS.
- H. Miyoshi, T. Ehashi, N. Ohshima and A. Jagawa, Artif. Organs, 2010, 34, 609–614 CrossRef.
- K.-H. Lee, J.-C. Sun, C. Chuang, S.-F. Guo, C.-F. Tu and J.-C. Ju, Cryobiology, 2013, 66, 311–317 CrossRef CAS.
- R. Alnemari, P. Sukumar, M. Deliorman and M. A. Qasaimeh, Adv. Biosyst., 2020, 4, 1900203 CrossRef.
- M. Deliorman, P. Sukumar, R. Alnemari and M. A. Qasaimeh, Bio-Protoc., 2020, 10, e3764 Search PubMed.
- A. K. Au, W. Lee and A. Folch, Lab Chip, 2014, 14, 1294–1301 RSC.
- S. Liu, H. Zhou, H. Wang, Y. Zhao, H. Shao, Z. Xu, Z. Feng, D. Liu and T. Lin, Adv. Mater. Interfaces, 2017, 4, 1700027 CrossRef.
- S. Sapieha, A. M. Wrobel and M. R. Wertheimer, Plasma Chem. Plasma Process., 1988, 8, 331–346 CrossRef CAS.
- E. Carrilho, A. W. Martinez and G. M. Whitesides, Anal. Chem., 2009, 81, 7091–7095 CrossRef CAS.
- T. Songjaroen, W. Dungchai, O. Chailapakul and W. Laiwattanapaisal, Talanta, 2011, 85, 2587–2593 CrossRef CAS.
- H. Juvonen, A. Määttänen, P. Laurén, P. Ihalainen, A. Urtti, M. Yliperttula and J. Peltonen, Acta Biomater., 2013, 9, 6704–6710 CrossRef CAS.
- F. Cheng, X. Cao, H. Li, T. Liu, X. Xie, D. Huang, S. Maharjan, H. P. Bei, A. Gomez, J. Li, H. Zhan, H. Shen, S. Liu, J. He and Y. S. Zhang, Nano Lett., 2019, 19, 3603–3611 CrossRef CAS.
- H. Li, F. Cheng, W. Li, X. Cao, Z. Wang, M. Wang, J. A. Robledo-Lara, J. Liao, C. Chávez-Madero, S. Hassan, J. Xie, G. Trujillo-de Santiago, M. M. Álvarez, J. He and Y. S. Zhang, Biofabrication, 2020, 12, 045027 CrossRef CAS.
- Q. He, C. Ma, X. Hu and H. Chen, Anal. Chem., 2013, 85, 1327–1331 CrossRef CAS.
- Y. Zhao, E. Xiao, W. Lv, X. Dong, L. He, Y. Wang and Y. Zhang, Stem Cells Int., 2020, 2020, 1031985 Search PubMed.
- S. S. Martin and P. Leder, Mol. Cell. Biol., 2001, 21, 6529–6536 CrossRef CAS.
- X. Dai, H. Cheng, Z. Bai and J. Li, J. Cancer, 2017, 8, 3131–3141 CrossRef.
- A. Frankel, R. Buckman and R. S. Kerbel, Cancer Res., 1997, 57, 2388–2393 CAS.
- D. J. Stewart, Crit. Rev. Oncol. Hematol., 2007, 63, 12–31 CrossRef.
- J. Chen, L. Lu, Y. Feng, H. Wang, L. Dai, Y. Li and P. Zhang, Cancer Lett., 2011, 300, 48–56 CrossRef CAS.
- U.S. Breast Cancer Statistics, https://www.breastcancer.org/symptoms/understand_bc/statistics, (accessed August 15, 2020).
- Breast Cancer - Statistics, https://www.cancer.net/cancer-types/breast-cancer/statistics, (accessed August 15, 2020).
- N. T. Elliott and F. Yuan, J. Pharm. Sci., 2011, 100, 59–74 CrossRef CAS.
- V. Velasco, S. A. Shariati and R. Esfandyarpour, Microsyst. Nanoeng., 2020, 6, 1–13 CrossRef.
- R. K. Jain, P. Au, J. Tam, D. G. Duda and D. Fukumura, Nat. Biotechnol., 2005, 23, 821–823 CrossRef CAS.
- B. Hong, P. Xue, Y. Wu, J. Bao, Y. J. Chuah and Y. Kang, Biomed. Microdevices, 2016, 18, 21 CrossRef.
- C.-H. Huang, K. F. Lei and N.-M. Tsang, Lab Chip, 2016, 16, 2911–2920 RSC.
- T. J. Puls, X. Tan, M. Husain, C. F. Whittington, M. L. Fishel and S. L. Voytik-Harbin, Sci. Rep., 2018, 8, 13039 CrossRef CAS.
- C.-A. Chen, W.-S. Yeh, T.-T. Tsai, Y.-D. Li and C.-F. Chen, Lab Chip, 2019, 19, 598–607 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0lc01300e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |