
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The material-enabled oxygen control in thiol-ene microfluidic channels and its feasibility for subcellular drug metabolism assays under hypoxia in vitro†
Iiro
Kiiski
 a,
Päivi
Järvinen
a,
Elisa
Ollikainen
a,
Ville
Jokinen
b and
Tiina
Sikanen
*a
a,
Päivi
Järvinen
a,
Elisa
Ollikainen
a,
Ville
Jokinen
b and
Tiina
Sikanen
*a
aFaculty of Pharmacy, Drug Research Program, Division of Pharmaceutical Chemistry and Technology, University of Helsinki, P.O. Box 56 (Viikinkaari 5E), Helsinki, FI-00014, Finland. E-mail: tiina.sikanen@helsinki.fi
bDepartment of Materials Science and Engineering, School of Chemical Engineering, Aalto University, Espoo, FI-02150, Finland
First published on 22nd March 2021
Abstract
Tissue oxygen levels are known to be critical to regulation of many cellular processes, including the hepatic metabolism of therapeutic drugs, but its impact is often ignored in in vitro assays. In this study, the material-induced oxygen scavenging property of off-stoichiometric thiol-enes (OSTE) was exploited to create physiologically relevant oxygen concentrations in microfluidic immobilized enzyme reactors (IMERs) incorporating human liver microsomes. This could facilitate rapid screening of, for instance, toxic drug metabolites possibly produced in hypoxic conditions typical for many liver injuries. The mechanism of OSTE-induced oxygen scavenging was examined in depth to enable precise adjustment of the on-chip oxygen concentration with the help of microfluidic flow. The oxygen scavenging rate of OSTE was shown to depend on the type and the amount of the thiol monomer used in the bulk composition, and the surface-to-volume ratio of the chip design, but not on the physical or mechanical properties of the bulk. Our data suggest that oxygen scavenging takes place at the polymer-liquid interface, likely via oxidative reactions of the excess thiol monomers released from the bulk with molecular oxygen. Based on the kinetic constants governing the oxygen scavenging rate in OSTE microchannels, a microfluidic device comprising monolithically integrated oxygen depletion and IMER units was designed and its performance validated with the help of oxygen-dependent metabolism of an antiretroviral drug, zidovudine, which yields a cytotoxic metabolite under hypoxic conditions.
Introduction
Oxygen partial pressure is an essential parameter in many biological processes in the human body, including signaling pathways affecting, e.g., stem cell differentiation1 and cancer pathophysiology.2 The physiological oxygen levels vary from 10–13% in the arteries and lungs (most oxygenated tissues) to 1–4% in the liver and colon.3 Oxygen also plays a crucial role with respect to the safety and efficacy of therapeutic drugs, as their hepatic clearance (metabolism) may be oxygen-dependent. For example, the optimum oxygen concentration of cytochrome P450 (CYP) enzymes, which are the main elimination pathway for majority of therapeutic drugs,4 is known to be enzyme-dependent and different between drug substrates.5,6 Under normoxic conditions in the liver (4% O2),2,3 oxygen is not the limiting factor with respect to drug clearance, because the half-maximal CYP activity (KO2) is typically reached at oxygen concentrations in the range of 10 μM (1%).5,6 However, upon aging and in the context of liver diseases, the hepatic oxygen uptake may be impaired and lead to deficient metabolic clearance (therapeutic level exceeded)7 or formation of cytotoxic metabolites only produced in hypoxic conditions.8–10 The current state-of-the-art in vitro techniques are well established for high-throughput screening of metabolites using human liver microsomes (vesicle-like artifacts of the endoplasmic reticulum) under atmospheric oxygen pressure, but the impact of oxygen on drug metabolism is much less explored. The oxygen concentration in cell cultures can be controlled using hypoxia incubators,11 but these do not enable rapid changes in the oxygen levels and are thus not well feasible for identification of hypoxic metabolites in short-term (<1 h) incubations on microsomal enzymes.12 Even more demanding is the rapid screening of the impact of oxygen concentration on the metabolic profile of a drug compound in a cost-effective manner. As a result, the largely ignored hypoxic metabolites of new drug candidates may give rise to unexpected hepatotoxicity in clinical trials or even post-marketing. Bridging this technology gap would substantially improve the preclinical in vitro–in vivo extrapolation of drug safety and efficacy.13In recent years, increasing number of microfluidic oxygen modulation approaches have been developed to improve the control of oxygen microenvironment, primarily in cell-based in vitro assays.14,15 The microfluidic platforms often rely on multi-compartment designs comprising a cell culture assay unit and an “oxygen control” unit, typically separated by a gas-permeable membrane to enable precise adjustment of the gas composition via controlled oxygen diffusion through the membrane.16–18 The gas content of the oxygen control compartment is usually adjusted by an external gas supply17,19 or supplementary reducing agents, such as sodium sulfite.18,20,21 While these approaches enable precise control of the oxygen microenvironment, they often require massive auxiliary instruments (external gas regulators and off-chip mixers) or the use of reagents that are not only highly reactive with molecular oxygen but also prone to attack biological macromolecules.
Recently, microfluidic devices made of off-stoichiometric thiol-enes (OSTE) were shown to possess an inherent capacity for depleting oxygen from aqueous media,22 which could be used as an alternative to supplementary oxygen scavengers, thus enabling substantially simpler microfluidic chip designs. OSTE refers to a group of polymers prepared via UV-initiated radical reaction between monomers containing thiol and allyl (‘ene’) functional groups.23 Using thiol and allyl monomers in an off-stoichiometric ratio enables straightforward tuning of both the surface and bulk properties of the resulting polymer.24 This facilitates, for instance, straightforward bonding25 and further surface biofunctionalization via click chemistry.24 In the previous work, also the inherent oxygen scavenging capability was associated with the use of thiol excess in the bulk polymer.
In the present study, the mechanistic basis of OSTE-induced oxygen scavenging was examined in detail with the aim of establishing in-depth understanding of the rate-limiting steps and deriving the kinetic constants that govern oxygen depletion in OSTE-based microfluidic channels prepared by UV-replica molding in the absence of photoinitiators or other additives. With the help of the kinetic constants, new design principles were drawn for OSTE microdevices so that normoxic and hypoxic conditions could be reproduced on demand in a single microfluidic biological experiment. As a proof-of-concept, an OSTE-based microfluidic device comprising two monolithically integrated compartments, an oxygen depletion unit and an immobilized enzyme reactor (IMER) unit, was designed and its performance validated with the help of the oxygen-dependent metabolism of an antiretroviral drug, zidovudine.
Experimental
Materials and reagents
The monomers used for fabricating the microfluidic chips included pentaerythritol tetrakis(3-mercaptopropionate) (PETMP) (≥95.0%), trimethylolpropane tris(3-mercaptopropionate) (TMPTMP) (>95.0%), and triallyl-1,3,5-triazine-2,4,6(1H,3H,5H)-trione (TATATO) (≥98.0%), and were purchased from Sigma-Aldrich (St. Louis, MO; TMPTMP, TATATO) or Bruno Bock (Marschacht, Germany; ThioCure® PETMP). The microchip masters were fabricated from SU-8 100 (micro resist technology, Berlin, Germany) and the poly(dimethyl siloxane) (PDMS) molds from Sylgard 184 (Down Corning Corporation, Midland, MI).Magnesium chloride, biotin-PEG4-alkyne, ethylene glycol, 5,5-dithio-bis-(2-nitrobenzoic acid) (DTNB), β-nicotinamide adenine dinucleotide 2′-phosphate (NADPH) reduced tetrasodium salt hydrate, uridine 5′-diphosphoglucuronic acid trisodium salt (UDPGA), Trizma® base and phosphate buffered saline (PBS, pH 7.4) were purchased from Sigma Aldrich. Zidovudine (AZT), zidovudine glucuronide (AZT-G) and 3′-amino-3′-deoxythymidine (AMT) were from Toronto Research Chemicals (Ontario, Canada). Irgacure® TPO-L photoinitiator (ethyl phenyl[2,4,6 trimethylbenzoyl]phosphinate, 84![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 434-11-7) was obtained from BASF (Ludwigshafen, Germany). Streptavidin (Alexa Fluor® 488 conjugate) was from Life Technologies (Eugene, OR) and the lipids used for preparation of fusogenic liposomes were from Avanti Polar Lipids (Alabaster, AL), including 1,2-dioleoyl-3-trimethylammonium-propane (chloride salt) (DOTAP), 1,2-dioleoyl-sn-glycero-3-phophoethanolamine (DOPE), 1,2-dioleoyl-sn-glycero-3-phophoethanolamine-N2 (Cap-biotinyl) (sodium salt) (biotin-cap-DOPE) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt) (lissamine rhodamine B-DOPE). The human liver microsomes (HLM, Corning® Gentest, 20-Donor Pool) were from Corning (Wiesbaden, Germany). All chemicals were of analytical grade unless otherwise stated. Water was purified with a Milli-Q water purification system (Millipore, Molsheim, France).
434-11-7) was obtained from BASF (Ludwigshafen, Germany). Streptavidin (Alexa Fluor® 488 conjugate) was from Life Technologies (Eugene, OR) and the lipids used for preparation of fusogenic liposomes were from Avanti Polar Lipids (Alabaster, AL), including 1,2-dioleoyl-3-trimethylammonium-propane (chloride salt) (DOTAP), 1,2-dioleoyl-sn-glycero-3-phophoethanolamine (DOPE), 1,2-dioleoyl-sn-glycero-3-phophoethanolamine-N2 (Cap-biotinyl) (sodium salt) (biotin-cap-DOPE) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt) (lissamine rhodamine B-DOPE). The human liver microsomes (HLM, Corning® Gentest, 20-Donor Pool) were from Corning (Wiesbaden, Germany). All chemicals were of analytical grade unless otherwise stated. Water was purified with a Milli-Q water purification system (Millipore, Molsheim, France).
Fabrication of off-stoichiometric thiol-ene microfluidic chips
The microchip designs used in the oxygen depletion assays included “unpacked” rectangular microchannels (30 × 2 × 0.2 mm) with surface-to-volume ratio of A/V ≈ 6 mm−1 (73 mm2/12 mm3), and microchannels (30 × 4 × 0.2 mm) comprising an array of ca. 14400 micropillars (Ø 50 μm, in a hexagonal lattice with center-to-center interpillar distance of 100 μm) with A/V ≈ 30 mm−1 (691 mm2/23 mm3). The microfabrication protocol (Fig. 1) consisted of the following four steps: (i) microfabrication of SU-8 masters by photolithography, (ii) soft lithography of the PDMS molds with the help of the SU-8 masters, (iii) UV-replica molding of the microchannel and cover layers in OSTE with the help of the PDMS molds, and (iv) bonding of the two OSTE layers together by lamination. The SU-8 master (step i) was fabricated similar to previous work.26 The PDMS molds (step ii) were prepared by mixing the base elastomer and the curing agent in a weight ratio of 10:1, casting the prepolymer onto the SU-8 master and curing in the oven either at 80 °C for 3 h or at 65 °C overnight before detaching the PDMS mold from the SU-8 master. The preparation of the OSTE layers (step iii) consisted of mixing the thiol (TMPTMP or PETMP) and allyl (TATATO) monomers and casting the mixture on the PDMS mold, before curing under UV for 5 min (Dymax 5000-EC series UV flood exposure lamp, nominal power 225 mW cm−2, Dymax Corporation, Torrington, CT). After UV-curing, the two layers were detached from the PDMS mold, heated above their glass transition temperature in the oven (ca. 60–70 °C), laminated together, and exposed to UV through the cover layer for 2 min to finalize the bonding (step iv). The structural fidelity of the microfluidic chips was investigated using a FEI Quanta™ 250 FEG scanning electron microscope (Hillsboro, OR). For this purpose, the samples were attached onto the sample stage with a carbon-coated double-sided tape and sputtered with platinum for 25 seconds (30 mA) yielding a 5 nm-thick coating.
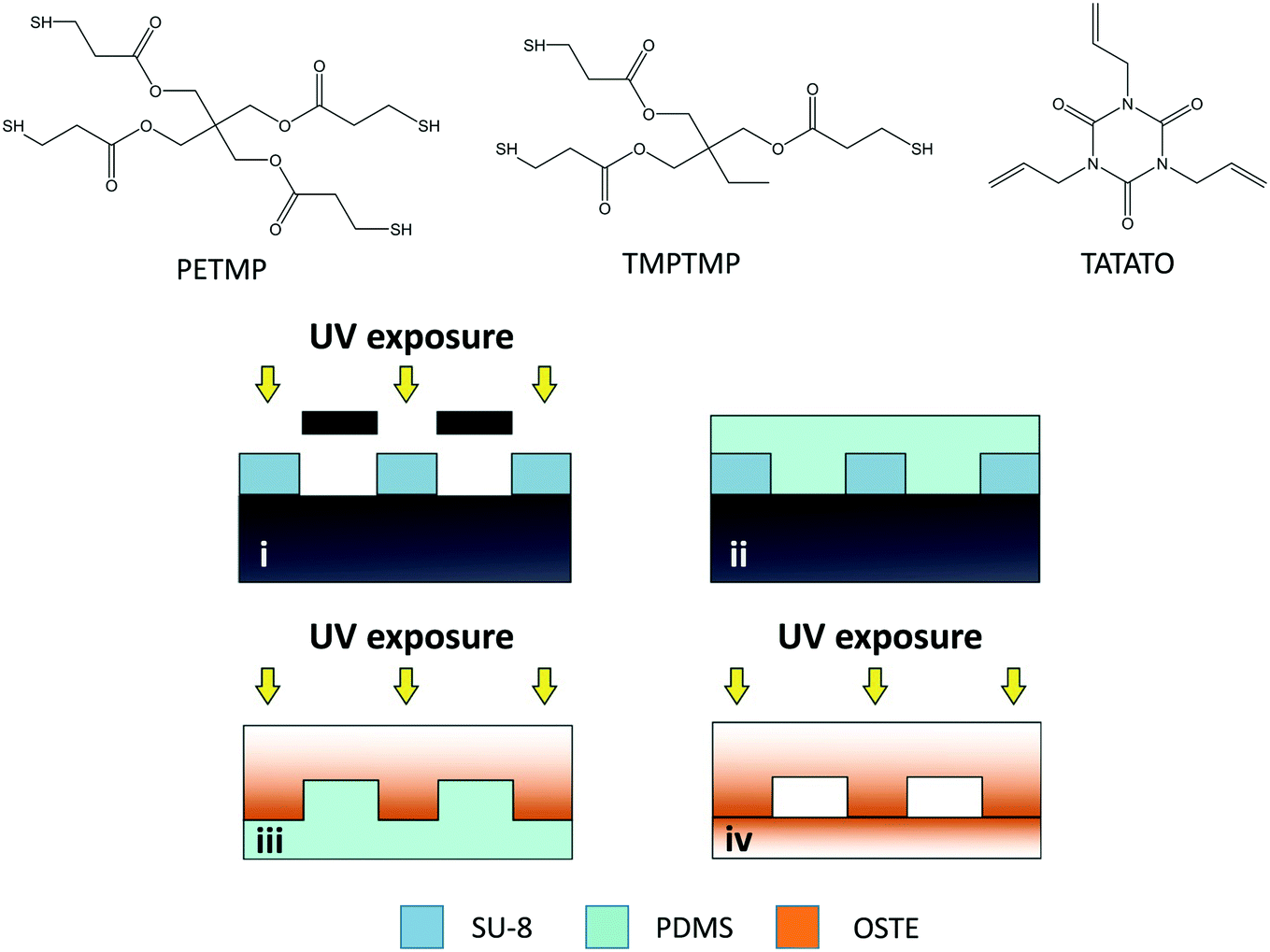 | ||
| Fig. 1 Molecular structures of the thiol (TMPTMP or PETMP) and allyl (TATATO) monomers used for fabricating the microfluidic chips and schematic illustration of the microfabrication workflow: (i) patterning of the negative photoresist SU-8 through a photomask, (ii) replica molding of PDMS negative mold using the SU-8 master, (iii) UV-induced replica molding of OSTE using the PDMS mold, and (iv) UV-induced bonding of OSTE layers. The color gradient in OSTE parts visualizes the monomer migration within the bulk and the resultant gradient in the chemical composition of the bulk polymer: intense orange indicates low crosslinking degree (high concentration of uncrosslinked monomers) and faint orange indicates high crosslinking degree (low concentration of uncrosslinked monomers). TMPTMP = trimethylolpropane tris(3-mercaptopropionate), PETMP = pentaerythritol tetrakis(3-mercaptopropionate), TATATO = triallyl-1,3,5-triazine-2,4,6(1H,3H,5H)-trione. | ||
Differential scanning calorimetry
The glass transition temperatures (Tg) of native and heat treated (110 °C overnight) OSTE polymers were determined with DSC 823e differential scanning calorimeter (Mettler-Toledo, Switzerland) using nitrogen as the purge gas. For this purpose, OSTE polymer of the chosen composition was cured against the PDMS mold, cut manually to suitably small pieces, and one representative sample per composition was analyzed. The Tg determination consisted of three steps: 1) cooling the sample to −35 °C, 2) isothermal sequence at −35 °C for 3 min, and 3) heating from −35 °C to 100 °C at a rate of 20 °C min−1. The glass transition temperatures were determined using STARe software.Oxygen permeability
For oxygen permeability measurements, thin OSTE membranes were fabricated by pipetting a small amount of the uncured monomer mixture of the chosen composition between two microscope glass slides (25 × 75 mm2) pre-coated with PDMS (base elastomer-curing agent 10:1, w/w) by spincoating (4000 rpm, 60 s). The glass slides were fixed together by two layers of Scotch double-sided tape, resulting in an interplate gap of ca. 100–200 μm, which defined the thickness of the OSTE membrane. The OSTE membranes were cured through the glass plate under UV for 5 min (Dymax) and then carefully detached from the PDMS coated glass slides. The oxygen flux (cc m−2 per day) through the membrane at 37 °C (100% humidity) was determined twice for each sample (two replicates of each composition) using an 8101e OxySense® oxygen transmission rate analyzer equipped with a coulometric detector (Systech Instruments Ltd, Thame, UK). The average oxygen permeability (cc cm m−2 per day atm−1) of the four technical replicates per composition was then calculated accounting for the measured thickness of the membrane.
Oxygen level measurements
Oxygen levels inside the microchannels were monitored with the help of commercial nanoparticles incorporating a fluorescent indicator dye (OXNANO, Pyro Science, Aachen, Germany), diluted to 1 mg mL−1 particle concentration in de-ionized MQ water (feed solution). The fluorescence lifetime of the particles was measured through the optically transparent OSTE layer using a Piccolo2 oxygen meter (Pyro Science, Aachen, Germany), as illustrated in Fig. 2A and B. Before use, a two-point calibration (0% and 100% air saturation) of the oxygen meter was performed according to the manufacturer's instructions using glucose oxidase (0.1 mg mL−1) and glucose (10 mg mL−1) to obtain oxygen-free water. Following this protocol, the nominal detection limit of the sensor was 0.1% and the resolution 0.05% (air saturation). With a view to biological applications, all measurements were performed at physiological temperature (37 °C) using an aluminum heating block element affixed under the OSTE chip. The temperature on top of the chip was monitored with a thermocouple and kept at the target temperature with the help of proportional–integral–derivative (PID) controller.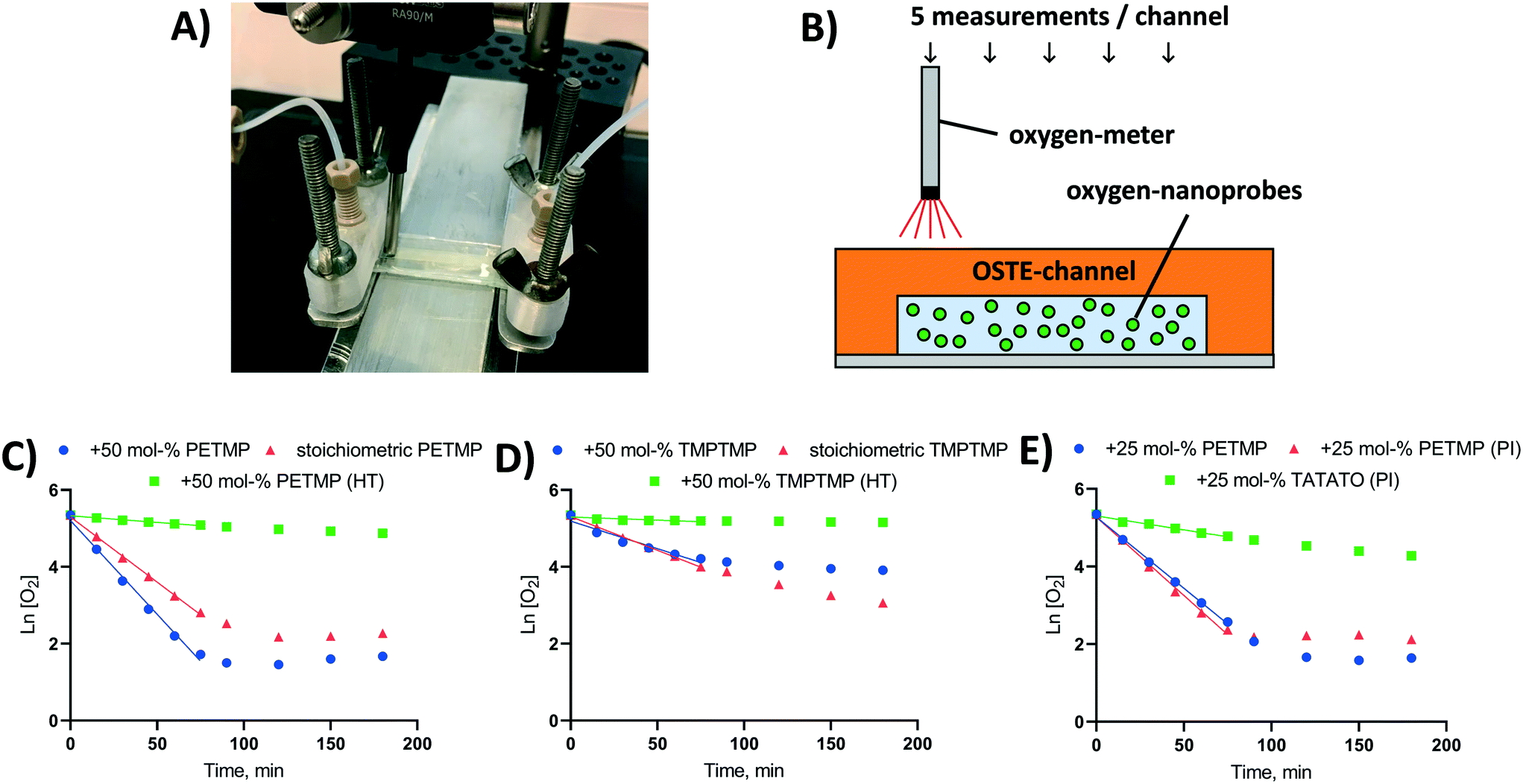 | ||
| Fig. 2 Oxygen depletion in OSTE microchannels in static conditions. (A) A photograph and (B) schematic of the measurement setup used for determining the on-chip oxygen concentration in the OSTE channels in static conditions with the help of OXNANO particles. (C–E) The oxygen scavenging kinetics in crosslinked, rectangular thiol-ene microchannels (30 × 2 × 0.2 mm) fabricated using stoichiometric and thiol-rich (+50 mol%) bulk compositions made of (C) tetrafunctional PETMP or (D) trifunctional TMPTMP as the thiol component and TATATO as the allyl component. HT refers to heat treatment (110 °C, overnight) after UV-curing. (E) The oxygen scavenging kinetics in crosslinked rectangular thiol-ene microchannels fabricated using either thiol-rich (+25 mol%) or allyl-rich (+25 mol%) bulk compositions made of PETMP and TATATO. PI refers to added photoinitiator (Irgacure® TPO-L, 0.1% m/v) in the bulk composition. Data points are averaged from n = 5 measurements per time point from n = 4–8 individual microchannels per sample group. The standard error between replicates was within 0.2–7.4% RSD, but due to the logarithmic scale the error bars are not visible. | ||
The effects of monomer type (TMPTMP or PETMP) and bulk polymer composition as well as the impacts of different post-fabrication treatments on oxygen depletion rates were first determined in static conditions using rectangular thiol-ene channels (2 × 30 × 0.2 mm) fabricated as described above and bonded onto a glass substrate. The microchannels (n = 4–8 each composition/treatment) were filled with the OXNANO nanoparticle solution, the channel openings were carefully sealed with Parafilm (Bemis Company, Oshkosh, WI), and the oxygen concentration was measured through the cover layer at five different locations along the channel length at indicated time points. To ensure a saturated environment, the channel was kept under a Petri dish cover with moistened paper.
To study oxygen scavenging under flow-through conditions, the microchannels were connected to a syringe pump using commercial nanoports (IDEX Corp., Lake Forest, IL) and 3D-printed nanoport holders prepared in-house. The instant oxygen concentration on chip was measured by increasing the flow rate step-wise from 2.5 to 20 μL min−1 as soon as the system reached steady state. The total duration of the flow-rate experiment was ca. 2.5 h.
In addition to crosslinked OSTE chips, the oxygen scavenging ability of free TMPTMP and PETMP monomers was separately examined by dissolving either monomer at different concentrations with the OXNANO nanoparticles (0.5 mg mL−1) in de-ionized water. In this case, the test solution (1 mL) was placed in a 1.5 mL Eppendorf tube and the oxygen level over time was measured through the wall of the tube using the Piccolo2 oxygen meter.
Quantitation of thiol monomers in OSTE microchannels
The amount of thiol monomers released from the bulk OSTE polymer into the microchannel per time was quantitated by titration using Ellman's reagent (5,5′-dithiobis[2-nitrobenzoic acid], DNTB).27 These experiments were performed with micropillar arrays (n = 6, A/V ≈ 30 mm−1) fabricated from PETMP containing composition with 25% molar excess of thiol functional groups over allyl functional groups. First, the free surface thiols were passivated by incubating a 1 mM DNTB solution (aq) in the channel for 30 min in static conditions. Next, a fresh DNTB solution was fed into the microchannel and incubated in the channel for 10, 20, or 30 min in static conditions, after which the reaction product, 2-nitro-5-thiobenzoate (TNB), produced in an equimolar ratio compared with free thiol functional groups, was quantitated by UV absorbance (λ = 412 nm) using Varioskan LUX Microplate Reader (ThermoScientific, Vantaa, Finland). The concentration of the thiol functional groups was determined using a molar extinction coefficient of ε = 14150 M−1 cm−1 for TNB28 and the in-built pathlength correction function of Varioskan LUX.
Fabrication and functionalization of the micropillar arrays for on-chip drug metabolism assays
For conducting drug metabolism assays under flow-through conditions on-chip, a microdevice with monolithically integrated oxygen depletion and immobilized enzyme microreactor (IMER) units was designed and fabricated from PETMP and TATATO containing composition with 25% molar excess of thiol over allyl functional groups. The oxygen depletion unit comprised of six serially connected micropillar channels of the same type as those used in the oxygen depletion assays. The IMER unit was a similar micropillar array and was serially connected, as the seventh element, with the oxygen depletion unit through a Y-junction. The micropillars of the IMER unit were functionalized sequentially with biotin, streptavidin, and pre-biotinylated HLM similar to the previous work.26 The first biotin layer was achieved by crosslinking biotin-PEG4-alkyne (0.1 mM in ethylene glycol, with 1% Irgacure® TPO-L) with the surface thiols under UV for 1 min (λ = 365 nm, LED, nominal intensity 14 mW cm−2). After rinsing sequentially with methanol and water (≥5 mL each), the micropillar array of the IMER unit was filled with 0.5 μg mL−1 streptavidin (in PBS), incubated at room temperature for 30 min and rinsed with ≥5 mL PBS. Last, the micropillar array of the IMER unit was filled with pre-biotinylated human liver microsomes (b-HLM), prepared as described by in the previous work.30 Next, the inlets and outlets were sealed with Parafilm and the b-HLM was allowed to immobilize onto the streptavidin-coated micropillars at 4 °C overnight. Before use, the micropillar array of the IMER unit was rinsed with the run buffer (0.1 mM Tris buffer, 5 mM MgCl2, pH 7.5), after which the entire integrated chip was filled with fresh run buffer.Microfluidic drug metabolism assays under controlled oxygen environment
The drug metabolism assays were performed with the integrated chip design comprising of the oxygen depletion and the IMER units using an antiretroviral drug, zidovudine (azidothymidine, AZT), as the model substrate. In addition to zidovudine (3 mM), the feed solution contained 1 mM NADPH (endogenous co-substrate of cytochrome P450 reactions) and 1 mM UDPGA (endogenous co-substrate of UDP-glucuronosyltransferase reactions) in 0.1 mM Tris buffer (5 mM MgCl2, pH 7.5). This solution was fed to the oxygen depletion unit (feed Q1, from the first syringe pump). The feed of the IMER unit, in turn, comprised of the anoxic flow from the oxygen depletion unit (Q2) and air-saturated flow from the other end of the Y-junction (Q3, second syringe pump with the same feed solution). The two syringe pumps were programmable and interconnected so that the total flow rate of the feed to the IMER unit (Q2 + Q3) could be kept constant. In this study, a flow rate of 5 μL min−1 was used in the IMER unit. The metabolites were quantitated by collecting 50 μL fractions of the elute of the IMER unit (Q4) with the help of an automated refrigerated fraction collector (CMA 470, CMA Microdialysis AB, Kista, Sweden). The concentrations of the zidovudine metabolites were quantitated using an ACQUITY UPLC™ liquid chromatograph (LC, Waters, Milford, MA) and Xevo TQ-S triple quadrupole mass spectrometer (MS, Waters, Manchester, UK). The data were analyzed using MassLynx V4.1 software. The LC and MS parameters and the method validation results for AMT and AZT-G quantitation are provided in the ESI† (Tables S1–S3 and Fig. S1 and S2).Statistics
All results are presented as mean ± standard deviation from repeated experiments, the number of which is indicated in the context of each dataset. Statistical analysis and curve fitting was performed using GraphPad Prism (GraphPad Software, San Diego, CA) version 8.4.3.Results and discussion
The inherent oxygen-scavenging capability of OSTEs has been previously associated with the oxidation of thioether-linkages in the polymer backbone of an OSTE+ formulation.22 The commercial OSTE+ formulation (Mercene Labs) includes a heat-curable epoxy component in addition to the thiol and allyl components. Similar thiol and allyl monomers are also common in many custom OSTE formulations used in microfluidic devices, including both cell-based and subcellular assays.26,29–31 In the previous work,22 the oxygen scavenging rate was shown to significantly decrease upon postprocessing of the bulk polymer by heat, which increases the glass transition temperature (Tg), and thus decreases the oxygen permeability, of the bulk polymer.22 The oxygen depletion was thereby hypothesized to result from the diffusion of the molecular oxygen to the bulk and its subsequent reaction with the thioether groups in the polymer backbone. However, the mechanism proposed for the OSTE+ formulation22 cannot readily explain the oxygen scavenging observed in microchannels made of custom OSTE formulations, which only contain the thiol and allyl monomers, but not the thermo-responsive epoxy component of the OSTE+. According to our preliminary findings, the oxygen depletion rate in custom (only thiol- and allyl-containing) OSTE microhannels is similarly impacted by heat treatment, even if the bulk properties (Tg, oxygen permeability) do not change upon heating. Thus, in the present study, we examined the OSTE-induced oxygen scavenging in greater depth with the aim of understanding its mechanistic basis, which is critical to both evaluating its feasibility to biological microfluidics and drawing design principles for microfluidic devices that exploit this unique property of OSTE polymers.Overall, photostructuring of OSTE-based microfluidic devices can be done by, for instance, UV lithography,32 UV-replica molding,29 3D printing,33 or roll-to-plate manufacturing.34 Some of these techniques tend to result in microscale compositional gradients upon monomer diffusion in the bulk polymer yielding zones of densely crosslinked polymer at highly illuminated areas and less quantitatively crosslinked, highly off-stoichiometric polymer at low-illuminated areas.32 In the UV-replica molding technique used in this study, the monomer diffusion seemingly occurs in vertical direction from the top surface (closer to the UV source) toward the bottom surface (against the PDMS mold), as illustrated in Fig. 1 (step iv). This conclusion was supported by the fact that the off-stoichiometric compositions typically showed two distinct glass transition temperatures (Tg), whereas stoichiometric compositions gave only a single Tg. It has also been experimentally shown in the previous literature that the uncrosslinked, excess monomers may be released from the bulk polymer, when incubated or flushed with aqueous solvents.35,36 On the basis of the high reactivity of thiols, we hypothesized that the inherent oxygen scavenging capability of thiol-rich OSTE microdevices could be the consequence of reactions between the released thiols and molecular oxygen in the microchannel.
Mechanism of oxygen depletion by crosslinked thiol-enes
First, the impact of bulk polymer composition on the oxygen depletion rate in UV-replica molded OSTE channels (2 × 30 × 0.2 mm, w × h × L) was addressed by varying both the thiol-monomer type and the thiol/allyl monomer ratio in the bulk. In our experiments, we used tri-functional TATATO as the allyl component and either a tri-functional TMPTMP or a tetra-functional PETMP as the thiol component. The monomer ratio varied from stoichiometric composition to a 50% molar excess of thiol functional groups over allyl groups (Fig. 2C and D). Under static (no flow) conditions, the oxygen depletion in OSTE microchannels made from either TMPTMP- or PETMP-containing polymers followed first order kinetics (Ln [O2] = −k × t + [O2]0). The rate constant of oxygen depletion (k) was strongly dependent on the thiol/allyl monomer ratio and increased along with increasing excess of the thiol component in the bulk (ESI,† Table S4). The rate constant (k) was also shown to be greater for PETMP-containing polymers (Fig. 2C) than TMPTMP-containing polymers (Fig. 2D).Comparison of different bulk compositions gave further support to our initial hypothesis that the mechanism of oxygen depletion is likely associated with the oxidation of uncrosslinked thiol monomers and the oxygen depletion rate correlates with the amount of free (uncrosslinked) monomers in the bulk. To confirm the hypothesis, the amount of the uncrosslinked PETMP monomers was further manipulated by using opposite excess (+25 mol%) of thiol and allyl functional groups in the bulk (Fig. 2E). As expected, the use of allyl-rich formulation decreased the rate constant substantially, as the amount of uncrosslinked thiols in the bulk reduced to minimum. On the other hand, adding photoinitiator (Irgacure® TPO-L, 0.1% m/v) to the thiol-rich (+25 mol%) formulation did not significantly change the oxygen depletion rate in thiol-rich channels (Fig. 2E), even if the Tg was substantially increased with the addition of the photonitiator (13 → 42 °C). These observations confirmed that the oxygen scavenging rate in custom OSTE channels was mostly governed by the chemical composition of the bulk, particularly the amount of uncrosslinked thiol monomers, and was rather independent on the mechanical bulk properties, such as Tg. In the absence of photoinitiator, the monomer conversion (crosslinking) is typically incomplete even for stoichiometric compositions,37,38 which could explain the relatively high oxygen depletion rate in microchannels fabricated with stoichiometric monomer ratios. With the replication method used in this study, the inner surface of the microchannel is furthest away from the UV light source during the curing step, as illustrated in Fig. 1. It is also known that the monomer conversion decreases as a function of increasing layer depth.39 This likely results in a relatively high amount of free monomers next to the microchannel surface, which facilitates their release to the microchannel and subsequent reaction with molecular oxygen in the aqueous sample solution.
Similar to the previous work,22 heat treatment of the microchannels (110 °C, overnight) after initial crosslinking substantially reduced the oxygen depletion rate (Fig. 2C and D). However, in the present study, this was associated with the vaporization of uncrosslinked thiol monomers from the bulk, instead of changes in the oxygen permeability of the bulk as proposed in the previous study.22 For example, the oxygen permeabilities of polymer compositions featuring 50 mol% excess of thiols were 3.0 ± 0.007 cc cm m−2 per day atm−1 (TMPTMP–TATATO) and 1.0 ± 0.62 cc cm m−2 per day atm−1 (PETMP–TATATO). If oxygen diffusion from inside the microchannel to the bulk polymer was the most critical rate limiting step, the TMPTMP-containing polymer should have showed a greater rate constant (k) for oxygen depletion compared with the PETMP-containing polymer, which is opposite to our observations (Fig. 2C and D). Furthermore, the impact of the heat treatment on Tg, which is known to correlate with the oxygen permeability of the bulk,40 was practically insignificant in case of both TMPTMP-containing (2.2 → −3.1 °C) and PETMP-containing (25.9 → 26.6 °C) polymers (both +50% molar excess of thiols).
These results further confirmed the hypothesis that oxygen scavenging in OSTE microdevices is associated with the reactions between uncrosslinked thiols, released from the bulk, and molecular oxygen in the microchannel. Overall, the oxygen permeabilities of the OSTE compositions used in this study were approximately three orders of magnitude lower than those reported for gas-permeable PDMS,41 which further confirms that the oxygen diffusion within the crosslinked OSTE network is negligible. Theoretically, however, the greater oxygen permeability of TMPTMP-containing polymer may improve the diffusion of ambient oxygen to the microchannel compared with PETMP-containing polymers, which could result in minor differences in the lowest achievable oxygen partial pressures between compositions in static conditions (Fig. 2Cvs. D).
The rate-limiting step of OSTE-induced oxygen depletion
The apparent rate constants (k) of oxygen scavenging in the OSTE microchannels for each polymer composition and post-curing treatment tested are summarized in the ESI† (Table S4). The data suggests that the rate of oxygen depletion is dependent on the monomer-specific reaction kinetics or the monomer diffusion kinetics from the bulk to the microchannel, or both. To further examine the rate-limiting step of oxygen depletion in OSTE microchannels, we determined the rate constants for the reactions between molecular oxygen and free TMPTMP or PETMP monomers dissolved in an aqueous media in plastic microtubes (in constant 1 mL sample volume). This experiment was performed at high micromolar concentrations (100 and 1000 μM) and revealed that the oxygen depletion rate does not markedly depend on the thiol monomer concentration (Fig. 3A and B), until the thiol component is completely consumed (e.g., at approximately 120 min in case of 100 μM PETMP, Fig. 3A).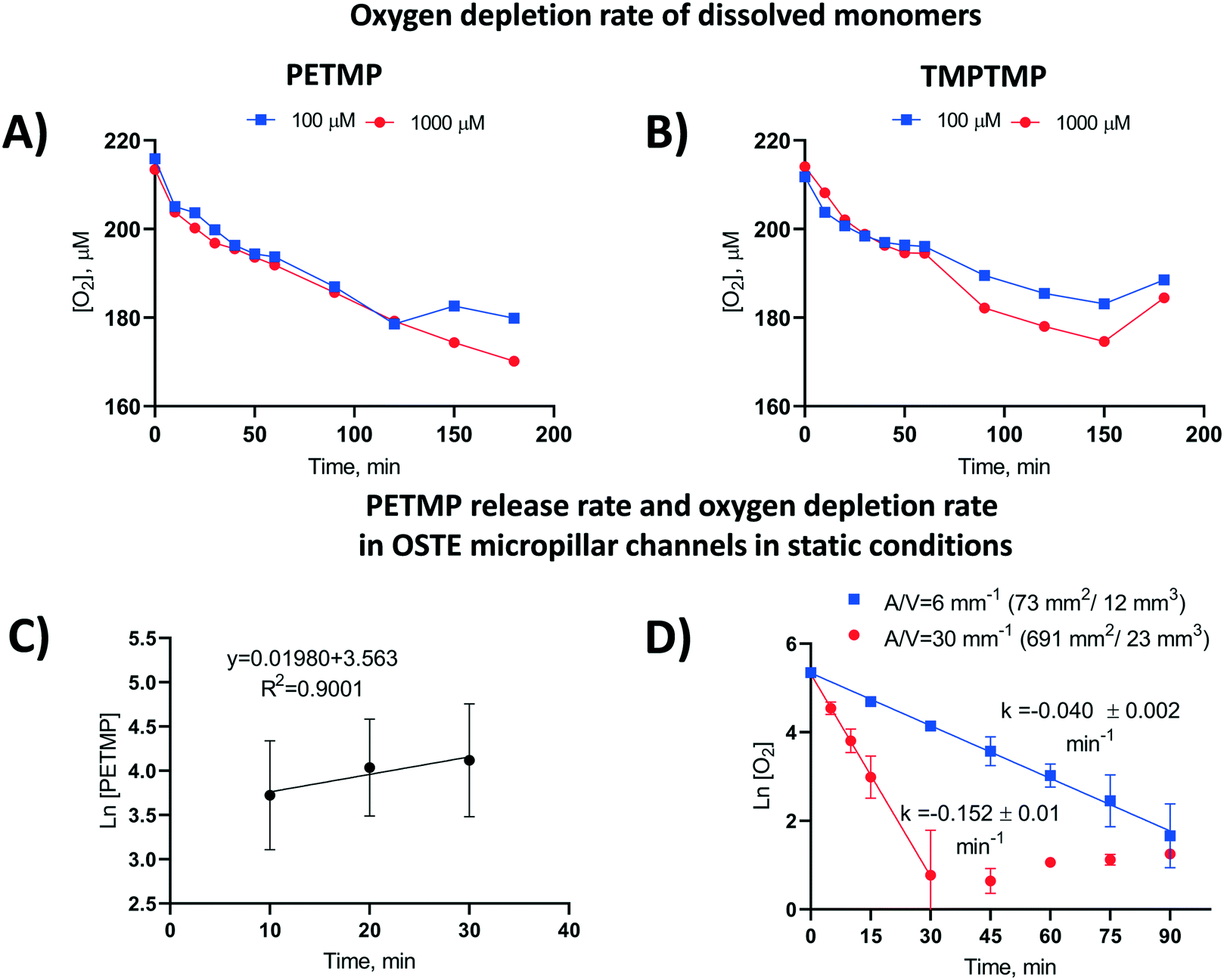 | ||
| Fig. 3 Oxygen depletion rate in water solution in a closed container (V = 1 mL) by dissolved (A) PETMP and (B) TMPTMP monomers as a function of time. (C) PETMP release rate in OSTE micropillar channels (A/V = 691 mm2/23 mm3 = 30 mm−1) made of PETMP and TATATO monomers with 25% molar excess of thiol functional groups. The free thiol functional groups were quantitated in static conditions by using DTNB (5,5′-dithiobis[2-nitrobenzoic acid], “Ellman's reagent”), of which the molar concentration of the released tetrafunctional PETMP monomer was calculated. Error bars denote standard deviation from n = 6 individual micropillar channels. (D) The effect of surface-to-volume ratio (A/V) on oxygen depletion kinetics in OSTE channels in static conditions. Each data point represents the average of n = 3 individual channels. Both types of microchannels were fabricated using PETMP and TATATO monomers with 25% molar excess of thiol functional groups. | ||
Next, the cumulative amount of thiols released from the bulk polymer to the OSTE microchannel over time was determined in a similar static (no flow) setting as the oxygen depletion kinetics. These experiments were performed with microchannels (n = 6) made of PETMP-containing polymer with 25% molar excess of thiol functional groups. The amount of thiols was titrated with the Ellman's reagent31 after 10, 20, and 30 min incubation, which confirmed that the mass transfer of PETMP released from the bulk follows first-order kinetics (Fig. 3C), similar to oxygen depletion in OSTE microchannels in static conditions. On the basis of these results, it was hypothesized that the thiol diffusion across the polymer-liquid interface is the rate limiting step for oxygen depletion in OSTE microchannels, and that the diffusion rate is most dependent on the amount of uncrosslinked thiols in the bulk. Consequently, the oxygen depletion rate should also depend on the chip surface-to-volume ratio. This hypothesis was further confirmed by comparison of the rate constants derived from ‘unpacked’ microhannels with A/V = 6 mm−1 and micropillar channels with A/V = 30 mm−1, both made of PETMP and TATATO using 25% molar excess of thiol functional groups. Comparison of the oxygen depletion kinetics between these two designs, in static conditions, confirmed that the rate coefficient k increased with increasing surface-to-volume ratio (Fig. 3D). The data also suggests that the oxygen depletion rate correlates well with the surface-to-volume ratio, with somewhat similar average rate constants (kA/V) between the micropillar channels (5.1 ± 0.3 × 10−3 mm min−1) and the unpacked channels (6.7 ± 0.3 × 10−3 mm min−1), when proportioned to A/V.
Impact of flow rate on the oxygen depletion rate
The mechanistic studies of oxygen depletion in static conditions laid the basis for rational microsystem design by enabling precise control of the oxygen concentration on chip with the help of microfluidic flow. Under flow conditions, the system will not saturate in a similar manner as in static conditions, but the flow rate also affects the reaction time between molecular oxygen and the thiol monomers, and thus defines the lowest achievable oxygen concentration. The impact of flow rate on the oxygen concentration was determined using two different designs of micropillar arrays (A/V = 30 mm−1), including a 30 mm-long channel (Fig. 4A) and its sextuplicate (L = 6 × 30 mm), in which six identical micropillar arrays were monolithically integrated on a single platform and serially connected via 10 mm-long transfer channels (Fig. 4B). Both designs were made of PETMP-containing polymer with 25% molar excess of thiol functional groups. This experiment confirmed that, besides bulk polymer composition, the oxygen concentration on chip can be manipulated by controlling the residence time with the help of the flow rate and/or the microchannel length (Fig. 4A and B). On the basis of our results, the hypoxic conditions could be maintained for at least 2.5 h under flow-through conditions and the oxygen scavenging properties were not markedly altered by storing of the microdevices at room temperature prior to use for at least five days.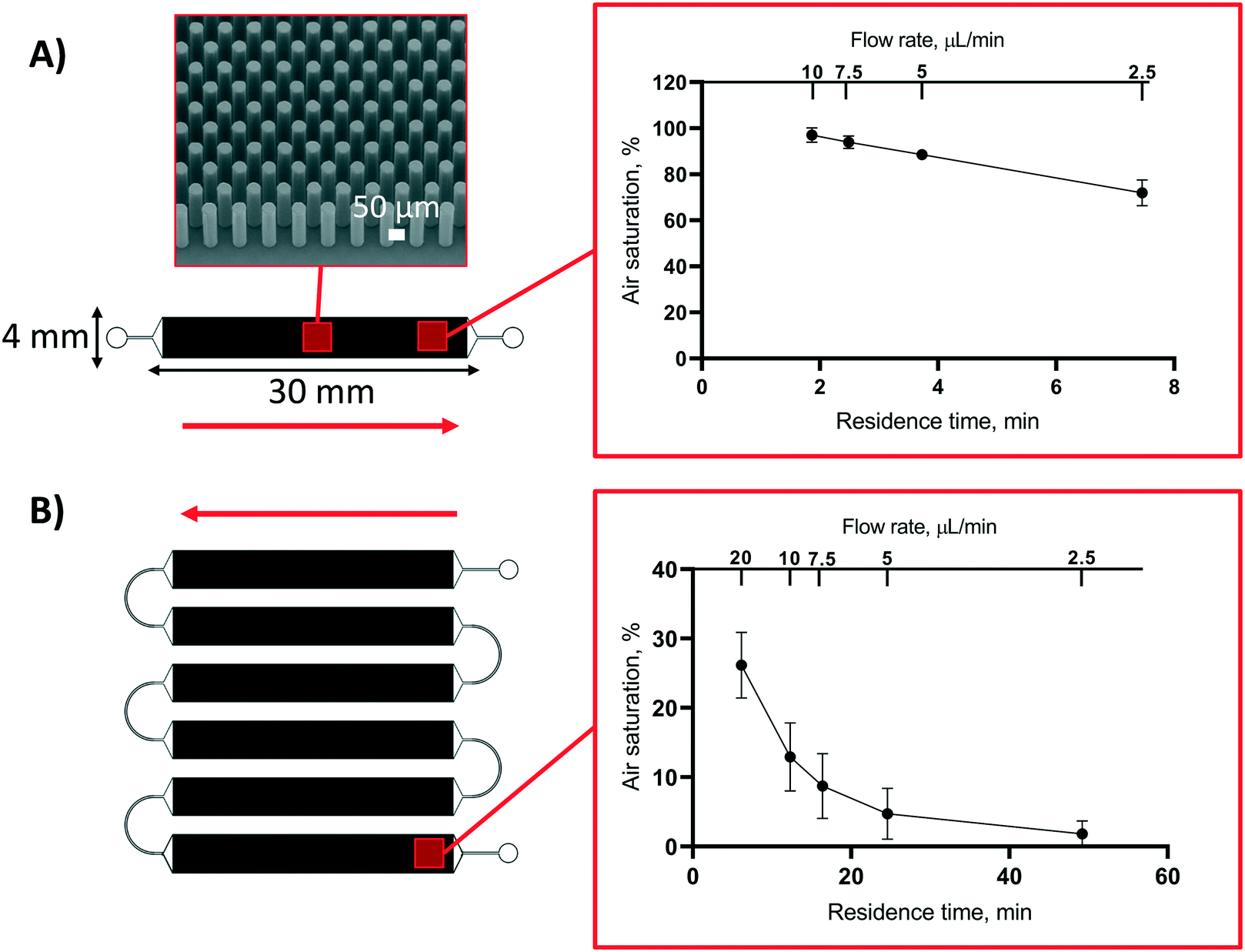 | ||
| Fig. 4 The impact of flow rate and residence time on the air saturation level (%) at the end of (A) a single micropillar array (L = 30 mm) and (B) six serially interconnected micropillar channel units (L = 6 × 30 mm). Red arrows denote the direction of the feed solution and the scanning electron micrograph shows the detail of the micropillar array. Both types of microchannels were fabricated using PETMP and TATATO monomers with 25% molar excess of thiol functional groups. Error bars denote standard deviation from n = 3 micropillar channels of each type. | ||
Recreating physiological oxygen levels on a microfluidic device for in vitro drug metabolism assays
Based on the material and design characterizations, an integrated, OSTE-based microdevice was developed for controlling the on-chip oxygen concentration on demand within a single microfluidic experiment. This chip design featured six serially connected micropillar channels (oxygen depletion unit) monolithically integrated, through a Y-junction, with a seventh micropillar channel, which served as the biological assay unit (Fig. 5A). With a view to drug metabolism assays under hypoxic conditions, the biological assay unit was functionalized sequentially with biotin, streptavidin, and pre-biotinylated human liver microsomes similar to previous work,26 to create an immobilized enzyme reactor (IMER) that incorporates the key microsomal enzymes that contribute to hepatic drug metabolism. In the IMER unit, the flow rate also defines the enzymatic reaction rate. According to our previous work, flow rate of 5 μL min−1 is optimal for drug metabolism assays in a flow-through setting with similar A/V.42 In this study, the total flow rate in the IMER unit was thus kept constant at 5 μL min−1 and its oxygen concentration was adjusted by changing the flow rate ratio of the anoxic solution (Q2, 0% air saturation, from the oxygen depletion unit) and an oxygen-rich solution (Q3, 100% air saturation, from the other inlet of the Y-junction) to meet the desired level (Fig. 5B). According to Fig. 4B, nearly anoxic conditions (2.2 ± 0.9% air saturation) were achieved in the oxygen depletion unit at a flow rate of 5 μL min−1, which represents the maximum feed of Q2 (and zero-feed from Q3). Lowering the flow rate in the oxygen depletion unit increases the residence time and results in even lower air saturation level so that at flow rates <5 μL min−1 the feed from Q2 was always anoxic. Thus, the oxygen concentration of the combined flow could be increased in a controlled manner simply by increasing the relative share of the air-saturated Q3 feed solution. Further oxygen scavenging within the IMER unit was considered negligible, because the residence time of the combined flow (5 μL min−1) in the 30 mm-long IMER unit was so short that the air saturation level decreased less than 10% (Fig. 4A). Furthermore, the surface biofunctionalization in the IMER unit also decreased the oxygen depletion rate compared with native surfaces (ESI,† Table S4). The mixing efficiency of Q2 and Q3 was further confirmed by measuring the air saturation % in the IMER unit at different ratios of Q2 and Q3, and comparing it with the theoretical air saturation% calculated according to eqn (1):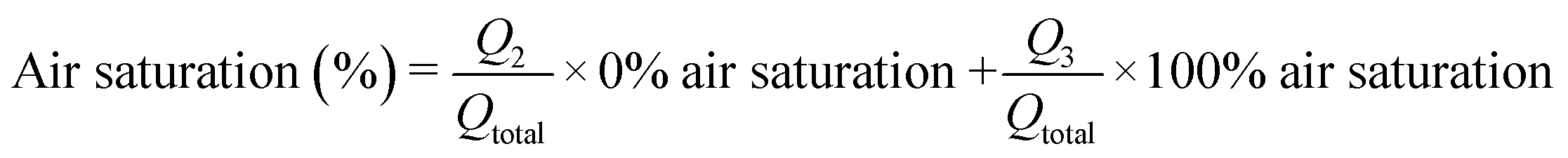 | (1) |
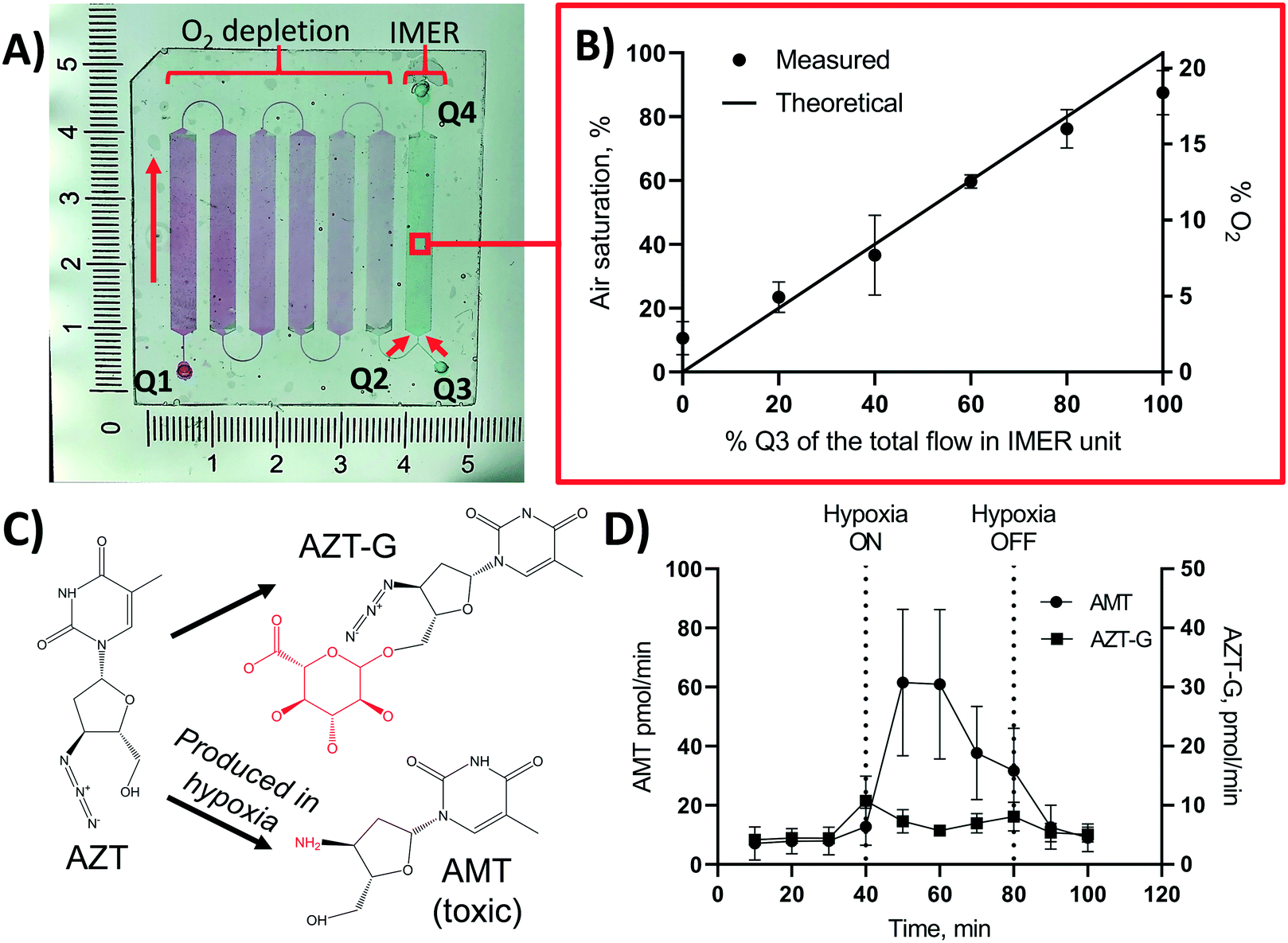 | ||
| Fig. 5 Monolithic integration of an on-chip oxygen depletion unit with a microfluidic immobilized enzyme reactor (IMER). (A) Photograph of the chip design comprising an oxygen depletion unit (filled with red dye), with an inlet Q1 and an outlet Q2, monolithically integrated with an immobilized enzyme microreactor (IMER) unit (filled with green dye) with a Y-junction providing air-saturated feed solution from inlet Q3 and an outlet Q4 for collecting metabolite samples. Oxygen level inside the microreactor was controlled by tuning the flow rate ratio of flows Q2 (oxygen-depleted) and Q3 (air-saturated). Red arrows denote the direction of the flow. The artificial color gradient of the red dye aims to visualize oxygen scavenging in the oxygen depletion unit and the green dye the constant oxygen concentration in the IMER unit. (B) Measured vs. theoretical oxygen concentration inside the IMER unit as a function of the percentual share of the air-saturated feed (Q3) of the total feed (Q2 + Q3) to the IMER unit at constant total flow rate of 5 μL min−1 (in the IMER unit). Error bars denote standard deviation from n = 3 chips. (C) Schematic of the oxygen-sensitive metabolism of zidovudine (AZT). AMT = 3′-amino-3′-deoxythymidine, AZT-G = zidovudine glucuronide. (D) Concentration of the oxygen-dependent metabolite (AMT) and the oxygen-independent metabolite (AZT-G) of zidovudine (AZT) at ambient (0–40 min and 80–100 min) and hypoxic (40–80 min) conditions. Hypoxic conditions were created by changing the feed from air-saturated flow (from inlet Q3) to oxygen-depleted flow (from Q2) at t = 40 min. At 80 min, the oxygen supply was re-initiated by changing the feed back to air-saturated flow from Q3. Flow rate during the experiment was constantly 5 μL min−1. Error bars denote standard deviation of n = 4 chips. | ||
As a proof of concept, the feasibility of the designed microfluidic chip (Fig. 5A) for recreation of biologically relevant oxygen concentrations in flow-through drug metabolism assays was demonstrated with the help of an antiviral drug, zidovudine (AZT). While the majority of AZT is eliminated through glucuronidation into zidovudine-glucuronide (AZT-G) in vivo, part of AZT is also metabolized into a myelotoxic metabolite 3′-amino-3′-deoxythymidine (AMT) by the reduction of the drug's azide group into an amine (Fig. 5C).43 AMT production is known to elevate in hypoxic conditions and it has been hypothesized to be responsible for, e.g., the bone marrow suppression encountered in patients receiving AZT therapy.8,44 To demonstrate the impact of oxygen on AZT metabolism, the on-chip oxygen level was controlled by switching the feed solution at the entrance of the IMER unit from 100% of Q3 (air-saturated flow, 0–40 min) to 100% of Q2 (oxygen-depleted flow, 40–80 min) and back to 100% of Q3 (air-saturated flow, 80–120 min). The total flow rate (Q2 + Q3) was kept constant at 5 μL min−1. When conducting the flow-through metabolism assays at ambient oxygen concentration (feed from Q3 only), AZT was effectively glucuronidated to AZT-G with an average rate of 6.2 ± 2.0 pmol min−1. As expected, elimination of oxygen from the IMER feed (feed from Q2 only) increased the production rate of the cytotoxic AMT by approximately 10-fold from initial 7.2 ± 4.2 pmol min−1 to 61 ± 25 pmol min−1 (Fig. 5D). When the IMER feed was switched back to the air-saturated flow (Q3 only), the AMT production rate dropped back to its initial low level (Fig. 5D). In addition to AMT, the formation of the AZT glucuronide was monitored over the entire experiment as an internal control. As expected, the impact of oxygen concentration on the AZT glucuronidation was shown to be somewhat negligible (Fig. 5D).
The proof-of-concept demonstration with AZT highlights the importance of oxygen control in biological assays. The chip design developed in this study could pave the way for higher throughput screening of the metabolic profiles of drug candidates at varying oxygen conditions in vitro. For example, the production rate of the toxic metabolite of AZT is significantly underestimated if only determined in ambient oxygen conditions using conventional in vitro assays. The possibility to adjust the oxygen concentration independent of the total flow rate, as demonstrated in this study, is particularly useful with a view to enzyme kinetic determinations under flow-through conditions, which necessitate precise control over residence (reaction) times.42 In this study, a total flow rate of 5 μL min−1 was used in the biological assay unit, but the same assay configuration is generally applicable to a wide range of flow rates, by adjusting the design (residence time, A/V) of the oxygen depletion unit so that oxygen is fully scavenged before the biological assay unit. If needed, the capacity of the oxygen depletion unit may be further adjusted by manipulating the bulk composition, which defines the rate constant of oxygen depletion.
Conclusion
This work addressed the mechanistic basis of material-induced oxygen depletion in aqueous solutions on microfluidic devices made of OSTE polymers. The inherent oxygen scavenging of the OSTE compositions used in this study was strongly associated with the reactions of the uncrosslinked thiol monomers, released from the bulk polymer, with molecular oxygen at the polymer-liquid interface in the microchannel. The oxygen depletion in OSTE microchannels was shown to follow first order kinetics, with the rate constant being dependent on the type and relative amount of the thiol monomer used in the bulk composition. The rate-limiting step of oxygen depletion was shown to be the kinetics of the thiol monomer diffusion from the bulk to the microchannel. Thus, under flow-through conditions, the oxygen depletion rate constant was proportional to the surface-to-volume ratio and the residence time determined by the microfluidic flow rate. This eventually enabled the rational design of OSTE-based microfluidic devices for controlling the on-chip oxygen concentration on demand.This study specifically addressed the craving need for new in vitro tools that could facilitate rapid screening of drug metabolism (molecular biotransformations) in normoxic and hypoxic conditions using microsomal enzymes (subcellular assays). For this purpose, we developed an integrated microfluidic chip comprising distinct oxygen depletion and immobilized enzyme microreactor units, and demonstrated the impact of oxygen on the formation of the cytotoxic metabolite of zidovudine in a single microfluidic experiment within 2 h. Similarly fast temporal control over oxygen level is difficult to achieve in traditional laboratory settings and thus the impact of oxygen on drug metabolism is often ignored in the current state-of-the art (static) microsomal in vitro assays. Theoretically, similar chip configuration may be exploited to microfluidic cell-based assays as well, although care should be taken to ensure that the thiol monomers released from the bulk do not interfere with cell viability and proliferation.
Author contributions
Iiro Kiiski: conceptualization, methodology, investigation, validation, formal analysis, writing – original draft; Päivi Järvinen: conceptualization, investigation, supervision, writing – review & editing; Elisa Ollikainen: methodology, writing – review & editing; Ville Jokinen: conceptualization, supervision, writing – review & editing; Tiina Sikanen: conceptualization, supervision, writing – review & editing, project administration, funding acquisition.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the Academy of Finland (grants 309608, 314303, 308911) and the Doctoral Programme in Drug Research, Doctoral School in Health Sciences, University of Helsinki (UH). The Electron Microscopy Unit of the Institute of Biotechnology, UH is acknowledged for providing access to the scanning electron microscope. We thank the DDCB core facility supported by the University of Helsinki and Biocenter Finland for providing access to the plate reader. Dr Hanna Koivula, Faculty of Agriculture and Forestry, UH, is acknowledged for help with the oxygen permeability measurements and Dr Sari Tähkä, Faculty of Pharmacy, UH, for help with Tg determinations.References
- A. Mohyeldin, T. Garzón-Muvdi and A. Quiñones-Hinojosa, Cell Stem Cell, 2010, 7, 150–161 CrossRef CAS.
- G. L. Semenza, N. Engl. J. Med., 2011, 365, 537–547 CrossRef CAS.
- A. Carreau, B. E. Hafny-Rahbi, A. Matejuk, C. Grillon and C. Kieda, J. Cell. Mol. Med., 2011, 15, 1239–1253 CrossRef CAS PubMed.
- U. M. Zanger and M. Schwab, Pharmacol. Ther., 2013, 138, 103–141 CrossRef CAS PubMed.
- D. P. Jones, Biochem. Pharmacol., 1981, 30, 1019–1023 CrossRef CAS PubMed.
- D. P. Jones, T. Y. Aw and X. Shan, Drug Metab. Rev., 1989, 20, 247–260 CrossRef CAS PubMed.
- D. G. Le Couteur and A. J. McLean, Clin. Pharmacokinet., 1998, 34, 359–373 CrossRef CAS PubMed.
- V. A. Eagling, J. L. Howe, M. J. Barry and D. J. Back, Biochem. Pharmacol., 1994, 48, 267–276 CrossRef CAS PubMed.
- L. R. Pohl, R. D. Schulick, R. J. Highet and J. W. George, Mol. Pharmacol., 1984, 25, 318–321 CAS.
- B. Wen, K. J. Coe, P. Rademacher, W. L. Fitch, M. Monshouwer and S. D. Nelson, Chem. Res. Toxicol., 2008, 21, 2393–2406 Search PubMed.
- J.-P. Gagner, M. Lechpammer and D. Zagzag, in Glioblastoma: Methods and Protocols, ed. D. G. Placantonakis, Springer, New York, NY, 2018, pp. 111–123 Search PubMed.
- O. Pelkonen, M. Turpeinen, J. Uusitalo, A. Rautio and H. Raunio, Basic Clin. Pharmacol. Toxicol., 2005, 96, 167–175 Search PubMed.
- L. Donovan, S. M. Welford, J. Haaga, J. LaManna and K. P. Strohl, Sleep Breath. Schlaf Atm., 2010, 14, 291–298 Search PubMed.
- M. D. Brennan, M. L. Rexius-Hall, L. J. Elgass and D. T. Eddington, Lab Chip, 2014, 14, 4305–4318 RSC.
- P. E. Oomen, M. D. Skolimowski and E. Verpoorte, Lab Chip, 2016, 16, 3394–3414 RSC.
- S. Barmaki, V. Jokinen, D. Obermaier, D. Blokhina, M. Korhonen, R. H. A. Ras, J. Vuola, S. Franssila and E. Kankuri, Acta Biomater., 2018, 73, 167–179 CrossRef CAS PubMed.
- S. C. Oppegard, A. J. Blake, J. C. Williams and D. T. Eddington, Lab Chip, 2010, 10, 2366–2373 RSC.
- M. Skolimowski, M. W. Nielsen, J. Emnéus, S. Molin, R. Taboryski, C. Sternberg, M. Dufva and O. Geschke, Lab Chip, 2010, 10, 2162–2169 RSC.
- H. E. Abaci, R. Devendra, R. Soman, G. Drazer and S. Gerecht, Biotechnol. Appl. Biochem., 2012, 59, 97–105 CrossRef CAS PubMed.
- C.-C. Peng, W.-H. Liao, Y.-H. Chen, C.-Y. Wu and Y.-C. Tung, Lab Chip, 2013, 13, 3239–3245 RSC.
- L. Li, Y. Li, Z. Shao, G. Luo, M. Ding and Q. Liang, Anal. Chem., 2018, 90, 11899–11907 CrossRef CAS PubMed.
- D. Sticker, M. Rothbauer, J. Ehgartner, C. Steininger, O. Liske, R. Liska, W. Neuhaus, T. Mayr, T. Haraldsson, J. P. Kutter and P. Ertl, ACS Appl. Mater. Interfaces, 2019, 11, 9730–9739 CrossRef CAS PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- C. F. Carlborg, T. Haraldsson, K. Öberg, M. Malkoch and W. van der Wijngaart, Lab Chip, 2011, 11, 3136–3147 RSC.
- T. M. Sikanen, J. P. Lafleur, M.-E. Moilanen, G. Zhuang, T. G. Jensen and J. P. Kutter, J. Micromech. Microeng., 2013, 23, 037002 CrossRef CAS.
- I. M. A. Kiiski, T. Pihlaja, L. Urvas, J. Witos, S. K. Wiedmer, V. P. Jokinen and T. M. Sikanen, Adv. Biosyst., 2019, 3, 1800245 CrossRef PubMed.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS PubMed.
- P. Eyer, F. Worek, D. Kiderlen, G. Sinko, A. Stuglin, V. Simeon-Rudolf and E. Reiner, Anal. Biochem., 2003, 312, 224–227 CrossRef CAS PubMed.
- S. Tähkä, J. Sarfraz, L. Urvas, R. Provenzani, S. K. Wiedmer, J. Peltonen, V. Jokinen and T. Sikanen, Anal. Bioanal. Chem., 2019, 411, 2339–2349 CrossRef PubMed.
- J. P. Lafleur, R. Kwapiszewski, T. G. Jensen and J. P. Kutter, Analyst, 2013, 138, 845–849 RSC.
- D. Sticker, M. Rothbauer, S. Lechner, M.-T. Hehenberger and P. Ertl, Lab Chip, 2015, 15, 4542–4554 RSC.
- M. Hillmering, G. Pardon, A. Vastesson, O. Supekar, C. F. Carlborg, B. D. Brandner, W. van der Wijngaart and T. Haraldsson, Microsyst. Nanoeng., 2016, 2, 1–7 Search PubMed.
- K. K. Childress, M. D. Alim, J. J. Hernandez, J. W. Stansbury and C. N. Bowman, Polym. Chem., 2019, 11, 39–46 RSC.
- S. Senkbeil, J. Aho, L. Yde, L. R. Lindvold, J. F. Stensborg, J. Rantanen, J. P. Lafleur and J. P. Kutter, J. Micromech. Microeng., 2016, 26, 075014 CrossRef.
- S. M. Tähkä, A. Bonabi, V. P. Jokinen and T. M. Sikanen, J. Chromatogr. A, 2017, 1496, 150–156 CrossRef PubMed.
- F. Ejserholm, J. Stegmayr, P. Bauer, F. Johansson, L. Wallman, M. Bengtsson and S. Oredsson, Biomater. Res., 2015, 19, 19 CrossRef PubMed.
- N. B. Cramer and C. N. Bowman, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3311–3319 CrossRef CAS.
- N. B. Cramer, J. P. Scott and C. N. Bowman, Macromolecules, 2002, 35, 5361–5365 CrossRef CAS.
- A. Vitale, M. G. Hennessy, O. K. Matar and J. T. Cabral, Adv. Mater., 2015, 27, 6118–6124 CrossRef CAS PubMed.
- L. Kwisnek, S. Nazarenko and C. E. Hoyle, Macromolecules, 2009, 42, 7031–7041 CrossRef CAS.
- T. C. Merkel, V. I. Bondar, K. Nagai, B. D. Freeman and I. Pinnau, J. Polym. Sci., Part B: Polym. Phys., 2000, 38, 415–434 CrossRef CAS.
- I. Kiiski, E. Ollikainen, S. Artes, P. Järvinen, V. Jokinen and T. Sikanen, Eur. J. Pharm. Sci., 2021, 158, 105677 CrossRef CAS PubMed.
- G. J. Veal and D. J. Back, Gen. Pharmacol., 1995, 26, 1469–1475 CrossRef CAS PubMed.
- S. Fayz and T. Inaba, Antimicrob. Agents Chemother., 1998, 42, 1654–1658 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0lc01292k |
| This journal is © The Royal Society of Chemistry 2021 |