
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrofluidic platform for serial mixing experiments with in operando nuclear magnetic resonance spectroscopy†
Marek
Plata
 abc,
William
Hale‡
a,
Manvendra
Sharma
a,
Jörn M.
Werner
bc and
Marcel
Utz
*ab
abc,
William
Hale‡
a,
Manvendra
Sharma
a,
Jörn M.
Werner
bc and
Marcel
Utz
*ab
aSchool of Chemistry, University of Southampton, SO17 1BJ, UK. E-mail: marcel.utz@gmx.net
bInstitute for Life Sciences, University of Southampton, SO17 1BJ, UK
cSchool of Biological Sciences, University of Southampton, SO17 1BJ, UK
First published on 4th March 2021
Abstract
We present a microfluidic platform that allows in operando nuclear magnetic resonance (NMR) observation of serial mixing experiments. Gradually adding one reagent to another is a fundamental experimental modality, widely used to quantify equilibrium constants, for titrations, and in chemical kinetics studies. NMR provides a non-invasive means to quantify concentrations and to follow structural changes at the molecular level as a function of exchanged volume. Using active pneumatic valving on the microfluidic device directly inside an NMR spectrometer equipped with a transmission-line NMR microprobe, the system allows injection of aliquots and in situ mixing in a sample volume of less than 10 μL.
1 Introduction
Serial mixing experiments, where two solutions are mixed in systematically varying proportion, are a widely used approach in chemistry and the life sciences. They are fundamental for the determination of binding constants,1 to acquire kinetic data,1,2 and for toxicity studies,3 to name but a few examples. Microfluidic implementation of such experiments offers automation of what is otherwise a very repetitive task, with advantages in cost and reliability. Also, miniaturisation is an advantage if one of the reactants involved is costly or only available in small quantities.Lab-on-a-Chip (LoC) devices allow efficient experimentation with sub-μL sample volumes, and quite complex experimental protocols can be implemented using pneumatically actuated valves,4 digital microfluidics,5 and ultrasound separation.6 Commonly, data readout in LoC systems is based on fluorescence spectroscopy,7,8 mass spectrometry,9 UV-vis spectroscopy,10 plasmonics11 or electrochemistry.12
In contrast, NMR has largely remained outside of the focus of microfluidics, despite its wide range of applications. The versatile, specific and non-invasive character of NMR allows to examine, in parallel, different characteristics of the same sample. For example, detailed reaction monitoring13 may be coupled with chemical analysis14 and molecular structure determination.15 Conventional liquid-state NMR uses relatively large samples, of the order of 600 μL, and reaches concentration limits of detection around 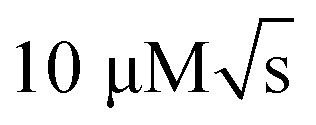 . Miniaturised receiver coils offer significantly improved mass limits of detection.16–18 A number of low-volume detectors19–21 and capillary flow NMR systems14,22–24 have been described on this basis. In some cases, systems with simple planar microfluidic devices that can be inserted and removed from the micro-NMR probe have been proposed.25–28 However, it is challenging to integrate non-trivial microfluidic functionality such as valving and pumping into such micro-NMR (μNMR) platforms.29 Complexity on the chip tends to lead to a loss in spectral resolution due to differences in magnetic susceptibility of the components.30 Moreover, ancillary hardware such as solenoid valves are not compatible with high magnetic fields, and the space inside the NMR bore is very constrained. A common approach has been to carry out most of the flow manipulation away from the spectrometer13,23,24,31,32 and deliver the final sample to the NMR detector by a several meters long capillary.
. Miniaturised receiver coils offer significantly improved mass limits of detection.16–18 A number of low-volume detectors19–21 and capillary flow NMR systems14,22–24 have been described on this basis. In some cases, systems with simple planar microfluidic devices that can be inserted and removed from the micro-NMR probe have been proposed.25–28 However, it is challenging to integrate non-trivial microfluidic functionality such as valving and pumping into such micro-NMR (μNMR) platforms.29 Complexity on the chip tends to lead to a loss in spectral resolution due to differences in magnetic susceptibility of the components.30 Moreover, ancillary hardware such as solenoid valves are not compatible with high magnetic fields, and the space inside the NMR bore is very constrained. A common approach has been to carry out most of the flow manipulation away from the spectrometer13,23,24,31,32 and deliver the final sample to the NMR detector by a several meters long capillary.
An exception is the work by Swyer, et al.33,34 who have successfully integrated electrically actuated digital microfluidics (DMF) with NMR. Based on electrowetting phenomena, DMF allows flexible manipulation of liquid droplets in contact with a dielectric surface. In the present work, we show that NMR can also be successfully combined with conventional, pressure-driven microfluidics and pneumatically actuated valves. While DMF is very versatile, pressure-driven microfluidics has advantages in certain circumstances. It is insensitive to the type of fluid being used, whereas DMF requires ionic solutions, or at least a solvent with high dielectric constant. Pressure-driven fluidic devices are simple, and can be produced cost-effectively through soft lithography or rapid prototyping techniques such as laser cutting and 3D printing. DMF devices require lithographic processes to define the electrodes, along with careful control of surface properties.
As shown in the following, conventional pneumatically actuated on-chip valving4,35 and pumping can be combined with high-resolution μNMR spectroscopy in order to implement serial mixing experiments. Controlled serial mixing of two liquids in order to systematically quantitate the response of chemical and biological systems to concentration changes has been implemented in microfluidic systems before; for example to characterise protein–substrate binding events36 and in toxicity assays based on microfluidic cell cultures.37,38 Mixing of the liquids is a common bottleneck for such systems. One effective solution is offered by circular micromixers,39,40 often with pneumatically actuated microvalves and pumps,4,35 which rely on deformation of elastomer membranes, usually polydimethylsiloxane (PDMS). Elastomer structures are problematic in context of NMR as they cause broad background signals that are difficult to suppress. It is therefore necessary to keep the elastomer away from the NMR detection area.41
The chip design presented here integrates a system of pneumatic microvalves into the liquid flow path. An external micro-syringe pump injects a small volume increment of a reagent to the working volume on the chip. After each injection, the working area is sealed off and the solution it contains is mixed by peristaltic action of the integrated valves. The chip is compatible with an optimised transmission-line NMR detector that has been characterised previously.28 The performance of this LoC system is demonstrated in the remainder of this paper by following the gradual mixing of two inert compounds monitored by proton NMR spectroscopy.
2 Design and operation
The system presented here exploits the design versatility of a transmission-line probe assembly (TLP),28 designed to accommodate generic microfluidic devices inside a standard NMR spectrometer. The TLP detects NMR signals from a 2.5 μL detection chamber on the microfluidic device with optimal sensitivity and high spectral resolution. The probe and microfluidic device are of a corresponding geometry, such that the detection chamber coincides with the area of maximum sensitivity of the TLP.27 Apart from the fixed size and position of the detection chamber the NMR system does not constrain the fluidic design, such that a wide range of functionalities can be implemented. In the present case, the chip accommodates a number of microvalves, which are actuated pneumatically using solenoid valves located outside of the NMR magnet. A micro-syringe pump, also kept externally, is used to inject precisely measured aliquots into the chip.The planar microfluidic chip shown in Fig. 1A contains six microvalves which are implemented by sandwiching the wider part of the chip with elastomer membranes between two 3D-printed holder structures, which also serve as interfaces for liquid and pneumatic tubing. A detailed description of the chip assembly and ancillary hardware is given in the ESI.† Manual intervention is required only for device assembly and filling with the sample liquid. After that, liquid flow on the chip is controlled completely through the micro-syringe pump and chip microvalves.
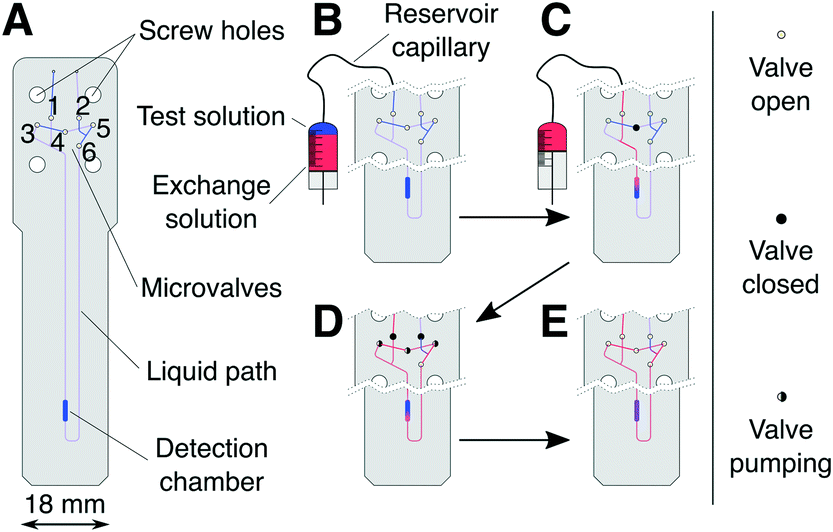 | ||
| Fig. 1 Schematic representation of the microfluidic chip designed for serial mixing experiments (A) and graphic representation of the liquid flow path during injection (B → C), mixing (D), and data acquisition (E). | ||
The microfluidic circuit consists of a detection chamber (2.5 μL), connected to the chip inlet and outlet through a pair of pneumatically actuated cutoff valves42 (1 and 2 in Fig. 1A). A bridge pathway, equipped with valves 3–6, allows peristaltic circulation (mixing) of the liquid contained on the chip. During injection and filling, the cutoff valves are kept open, whereas the bridge pathway is closed using valve 4. During mixing, the chip is isolated from the supply by closing the cutoff valves, and valves 3–5 are actuated periodically with a phase shift to create a peristaltic motion of the liquid.4 The total volume subject to circular peristaltic mixing Vs, consisting of the detector chamber, valves 3–6, and connecting channels, amounts to 10 μL.
At the start of a serial mixing experiment one solution (see Fig. 1B) is contained within the microfluidic chip and the other in the connecting reservoir capillary. The majority of the capillary volume and connected micro-syringe pump is filled with an inert working fluid, limiting the overall sample use to a few tens of microliters. Initially, only the test solution (blue) is present inside Vs (see Fig. 1B). Through the micro-syringe action, a volume increment Vi of the exchange solution (red) is injected into Vs (Fig. 1C), displacing an equivalent volume of the test solution out of the chip. Vs is then isolated using the cutoff valves, followed by a period of peristaltic mixing. The NMR spectrum of the solution in the detection chamber is then acquired, before the next increment Vi is injected.
The concentration of a compound A in the detection volume after the k-th injection/mixing cycle is expected to be
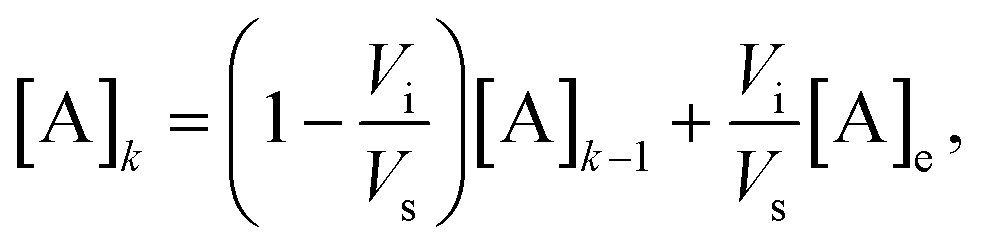 | (1) |
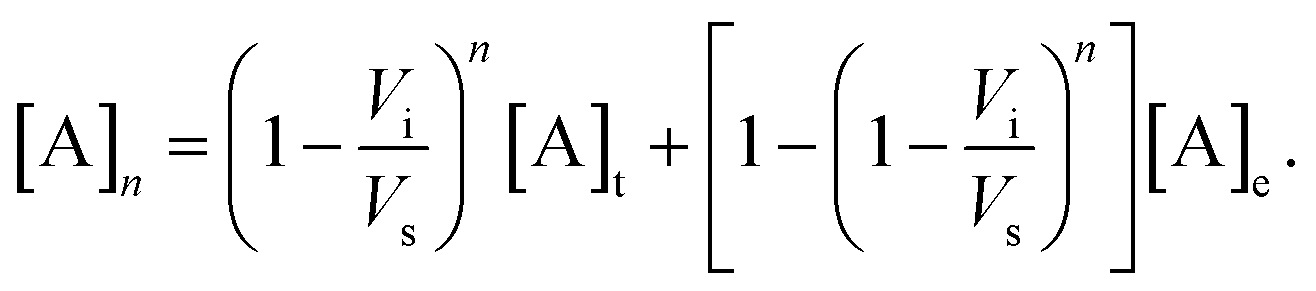 | (2) |
3 Experimental
3.1 Microfluidic device design, fabrication and assembly
All microfluidic chips were designed and manufactured on site. Each chip consists of a 500 μm thick polymethyl methacrylate (PMMA) layer that is sandwiched between two outer 200 μm thick layers. The design features of the PMMA layers were specified in an AutoCAD (Autodesk, US) file and cut from commercially available PMMA sheets (CLAREX, Japan) using a CO2 laser cutter (HPC Laser Ltd., UK). Adjusting the power and speed of the laser controls the depth and width of the cut. Microfluidic channels in the chip were produced to an approximate width and depth of 150 μm, while the valve elements were fully cut through. After cutting, the layers were washed with isopropanol, dry treated with O2 plasma (Diener, CH), and bonded with plasticiser (2.5% v/v dibutyl phalate in isopropyl alcohol) under heat and pressure, according to a previously established protocol.41 The device assembly was completed using three elastomer membranes, two 200 μm thick PDMS, and a single 100 μm thick nitrile rubber (NBR), and two holder blocks that cover the upper part of the microfluidic chip (Fig. S2†). Like the PMMA layers, the elastomer membranes were cut from the respective sheets (Shielding Solutions, UK; Ansell Healthcare, Belgium) with a CO2 laser. The chip holder blocks were designed in SolidWorks (Dassault Systèmes, France) and produced from Accura® Xtreme™ 200 Plastic by Stereolitography 3D-printing (Protolabs, UK). The PDMS membranes were placed directly on the chip as a gasket. The NBR membrane was placed between the chip holder and the PDMS membrane, preventing the pressurised air permeating through the PDMS layer. The microvalves use a plunger architecture42 (Fig. S3†). The holders house liquid and gas channels, and the microports that allow to connect the device to external pressure and liquid supply systems. The assembly is held together by four M3 screws.3.2 Sample loading
A 80 μm inner diameter polyethylene ether ketone (PEEK) capillary connects the microfluidic device to a SPS01 micro-syringe stepper-motor pump (LabSmith, US). Initially, the syringe and capillary volume was entirely filled with a perfluorinated oil (Fluoinert™ FC-43, Sigma-Aldrich, UK). The sample solution was loaded into the capillary from a length-modified pressure resistant NMR tube (Wilmad-LabGlass, US) serving as the primary reservoir. The loading end of the capillary was immersed in the exchange solution and 3 bar of pressure were supplied to the NMR tube. Using the micro-syringe at the other end of the capillary, 20 μL of the exchange solution were then drawn into the capillary. The same procedure was repeated for loading of the test solution. Subsequently, the loading end of the reservoir capillary was connected to the assembled microfluidic device and 3 bar back pressure was applied to the device outlet. The microfluidic chip was then filled with the test solution by the micro-syringe action, such that the boundary between the test and exchange solutions coincided with the chip inlet.3.3 Microvalve control
The microvalves on the chip are actuated via ∼4 m leads operating at 5 bar air pressure. Using a common pneumatic manifold, a single pressure input is separated into six leads that extend to the individual microvalves. The manifold holds six solenoid valves (Festo, Germany), controlled by an Arduino Mega 2560 (Arduino, US) microcontroller.3.4 Software
Firmware controlling the solenoid valves was written in Arduino IDE (Arduino, US) and allows for individual and sequential actuation of the valves with varying patterns and frequency. The micro-syringe is operated using manufacturer supplied uProcess™ software (LabSmith, US).3.5 Chemicals
Sample solutions were prepared by dissolving 43.9 mg sodium 3-trimethylsilyl-1-propanesulfonate (DSS) and 23.5 mg fumaric acid, respectively, in a stock solution of 4 mL 50 mM sodium acetate in H2O. In both solutions the pH was adjusted to 13 ± 0.5 using 50 mM sodium acetate/1.5 M NaOH.3.6 Data acquisition & processing
All NMR measurements were performed at 11.7 T in a 500 MHz Bruker Active Shield II WB magnet equipped with a Bruker AVANCE III console. 8 transients were recorded for each spectrum with a repetition delay of 8 s and an acquisition time of 2.0 s. Water signals were suppressed by 3 s of presaturation with a nutation frequency of 100 Hz. Spectra were processed with 0.5 Hz of Lorentzian line broadening and the baseline was automatically corrected using a first order polynomial. All processing was done using the NMR.jl (https://github.com/marcel-utz/NMR.jl) package for the Julia programming language.434 Results and discussion
4.1 Microvalve function
During the serial mixing experiments the microvalves serve two distinct functions: (i) closing of specific valves determines the flow path of the sample liquid, and (ii) sequential valve triggering pattern creates the peristaltic flow needed for sample mixing. Valve closing is achieved when the applied pressure to the valves exceeds the pressure in the liquid, pushing the elastomer membranes against the valve floor. The required pressure was determined experimentally for each of the valves. A water filled capillary was connected to the device inlet and the flow was generated with application of 4 bar air pressure at the capillary end. Flow was observed at the device outlet. For all tested chips, 5 bar air pressure applied to each of the valves was effective to stop the flow of the liquid through the device. Function of the microvalves was unaffected by the long (∼4 m) pneumatic tubing.Due to the asymmetrical circuit design it is possible to distinguish two opposite directions of the flow inside the Vs. Here, we refer to those as clockwise and anticlockwise. While the flow is created by sequential actuation of valves 3, 4, 5 (see Fig. 1), valve 6 remains open during this routine and contributes to the fluidic capacitance of the circuit. In the clockwise direction valve 6 is positioned downstream from valves 3, 4, 5, and upstream in the anticlockwise direction. This asymmetry is reflected in the liquid flow observed inside Vs. In a series of experiments carried out outside of the NMR spectrometer, injection of bromophenol blue (BpB) dye to Vs was carried out in order to visualise the flow, and to evaluate the time needed to reach sample homogeneity. These results are shown in Fig. 2. At t = 0, 1.5 μL of the BpB exchange solution was injected into the chip. The injection flow is downward, as indicated by an arrow in the figure. The boundary between the exchange and test solution is clearly visible in the detection chamber. Peristaltic flow in the clockwise direction is then started at a frequency of 0.75 Hz, leading to an upward movement of the liquid in the image (arrow). After 40 s, the blue boundary between the fluids reaches the detection volume from below. The liquid continues to flow around the mixing volume Vs, gradually getting mixed in the process. After 60 s, the colour gradient is still visible, while it has disappeared after 120 s of mixing.
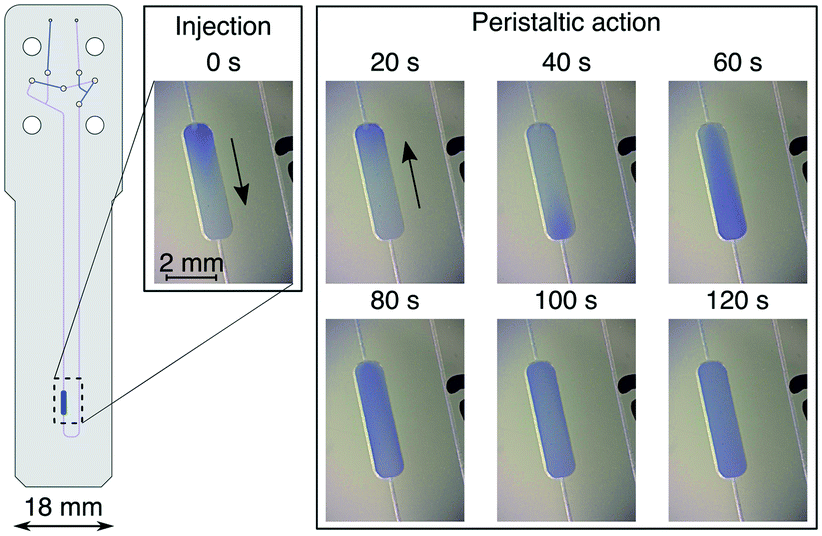 | ||
| Fig. 2 Visualisation of the flow inside the sample chamber of the microfluidic chip. Images were taken at specified time points after initial injection of the BpB dye. Arrows indicate the direction of the flow at each step. | ||
From the known volume of the detection chamber of 2.5 μL and the circulation time, the flow rates reached by peristaltic pumping were estimated. We obtained average values of 5 ± 2.5 μL min−1 clockwise, and 1.5 ± 0.7 μL min−1 anticlockwise. The variability is likely due to the fabrication tolerances of the microfluidic chips. In the clockwise direction, a mixing time of 120 s was sufficient to reach a condition where no more changes to the sample colour could be observed (see Fig. 2). For all NMR detected experiments the mixing time was conservatively extended to 240 s to ensure sample homogeneity.
4.2 NMR detected serial mixing
Serial mixing of fumaric acid and DSS solutions inside the microfluidic device was monitored by the intensity changes of the corresponding NMR signals. The two compounds, together with sodium acetate used as internal concentration standard, were chosen because they do not react and have well separated 1H chemical shifts. The main resonances appear at 0 ppm, 1.92 ppm, and 6.52 ppm for the nine equivalent methyl protons on DSS, three methyl protons on sodium acetate, and two equivalent methine protons on fumaric acid, respectively. Secondary signals due to methylene protons on DSS at 0.62 ppm, 1.77 ppm, and 2.92 ppm can be observed above ∼5 mM.After filling the reservoir capillary with 20 μL each of the fumarate (exchange) and DSS (test) solutions, the experiment was started by injecting the device with 18 ± 1 μL at a flow rate of 15 μL min−1. Since this is in excess of the mixing volume of Vs = 10 μL, it leads to complete filling of the chip with the test solution. The first NMR spectrum shown in Fig. 3 (V = 0) shows only the acetate and DSS signals, and no fumaric acid peak. For each subsequent spectrum, an additional Vi = 2 ± 0.25 μL were injected, followed by 240 s of peristaltic mixing before acquisition. 3 bar of back pressure applied at the device outlet was employed to ensure the Vi is equal to the volume displaced by the movement of the micro-syringe plunger at the opposite end of the reservoir capillary. The gradual reduction in intensity for DSS, and increase for fumaric acid can clearly be observed in all subsequent spectra shown in Fig. 3.
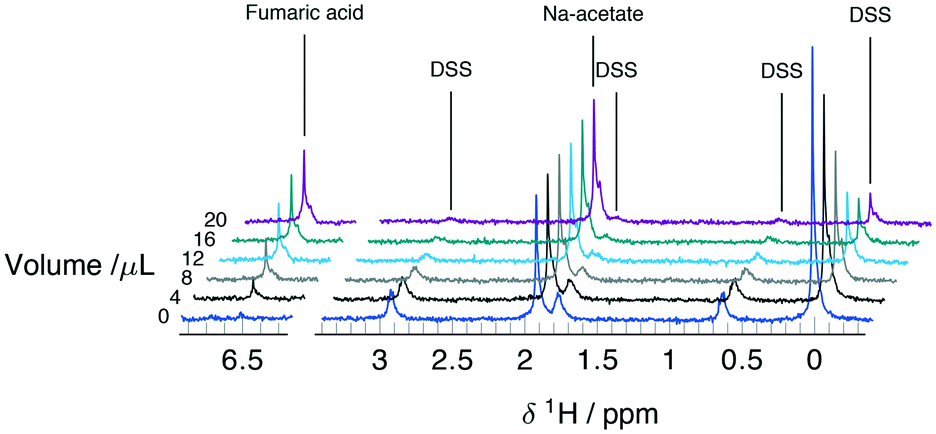 | ||
| Fig. 3 Series of 1H detected spectra obtained during the serial mixing experiment between 35 mM DSS and 40 mM fumaric acid solutions in 50 mM sodium acetate at pH 13. Spectra are referenced to 0 ppm using the main DSS resonance and the traces are stacked according to the combined injection volume. | ||
Concentrations of DSS and fumaric acid were computed from the spectra based on the known concentration of acetate [Ac], according to
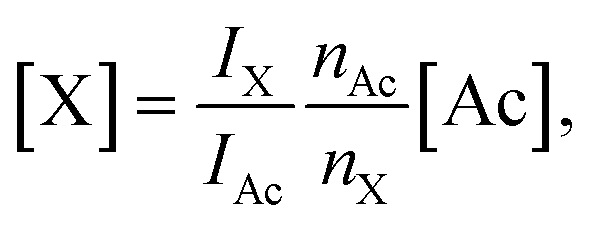 | (3) |
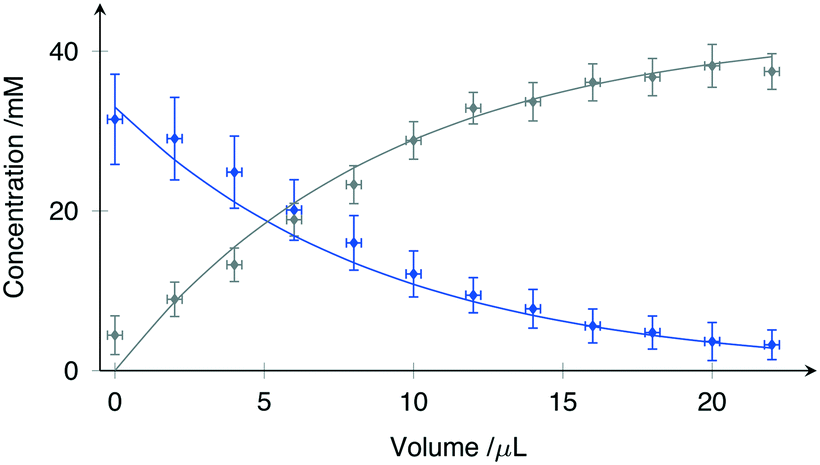 | ||
| Fig. 4 Concentration changes observed during serial mixing of DSS (blue) and fumaric acid solutions (gray). All values are normalised with respect to the signal intensity of the internal standard (sodium acetate). Scattered points represent the experimental data while the line-plots demonstrate the best fit model as specified by (2). | ||
The solid lines in Fig. 4 represent a least-squares fit of (2) to the data, where Vs has been treated as the fitting parameter. While the microfluidic chips were designed for a nominal value of Vs = 10 μL, fabrication tolerances invariably lead to small differences.
Using the Levenberg–Marquardt method for nonlinear least-square fitting, Vs was evaluated from the data shown in Fig. 4 as 9.7 ± 0.6 μL at 95% confidence intervals. The root mean square error (RMSE) of the fit was 1.23 mM, well within the expected concentration error range.
5 Conclusions
We have presented a versatile microfluidic system for serial mixing experiments with in operando detection by NMR spectroscopy. The device is capable of repeated sample manipulation, including injection and mixing, in a constant volume of approximately 10 μL while inside the NMR spectrometer. The entire experimental procedure going from 100% test solution to 100% exchange solution, with multiple exchange steps, can be completed with under 50 μL of sample. This value could be reduced further by utilising soft lithography for chip fabrication. At the same time the complexity of the fluidic circuit and the precision of the metering and valving could be increased by more sophisticated manufacturing, without inflating the overall working volume. However, the ultimate scaling limit is dictated by the NMR limit of detection, which requires increasing concentrations of analytes as the detection volume gets smaller. The current design is optimised for 2.5 μL detection volume, which leads to concentration limits of detection around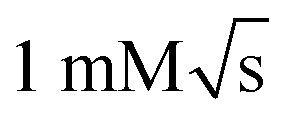 .28 Significantly smaller volumes could be used in systems that support higher analyte concentrations.
.28 Significantly smaller volumes could be used in systems that support higher analyte concentrations.
Ongoing development is aimed to make this a generic reactor for chemical analysis, and with improved microfabrication methods practically unlimited number of inlets and microvalves could be implemented on chip to monitor complex chemical reactions. As such, we expect this platform to be well suited for thermodynamic characterisation of in vitro chemical equilibria, with application in e.g. phase separation, macromolecular assembly, or for kinetic analysis by systematically varying catalyst concentration. In protein work specifically, serial mixing experiments with NMR detection are uniquely placed to study protein folding or protein–ligand interactions. Other uses can be envisaged for cellular systems, such as microfluidic cultures of cells, organoids and tissues, for dose–response analysis in toxicology or metabolic pathway analysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the “TISuMR” project (grant no. 737043) funded through the Future and Emerging Technologies (FETOPEN) call of the EU Horizon 2020 research framework. MP gratefully acknowledges support from the Institute for Life Sciences, University of Southampton, Southampton, UK.Notes and references
- E. C. Hulme and M. A. Trevethick, Br. J. Pharmacol., 2010, 161, 1219–1237 CrossRef CAS PubMed.
- A. R. Mukhametgalieva, I. V. Zueva, A. R. Aglyamova, S. V. Lushchekina and P. Masson, Biochim. Biophys. Acta, Proteins Proteomics, 2020, 1868, 140270 CrossRef CAS PubMed.
- Z. Gerdes, M. Hermann, M. Ogonowski and E. Gorokhova, Sci. Rep., 2019, 9, 10695 CrossRef PubMed.
- M. A. Unger, H.-P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–116 CrossRef CAS PubMed.
- E. Samiei, M. Tabrizian and M. Hoorfar, Lab Chip, 2016, 16, 2376–2396 RSC.
- J. Nilsson, M. Evander, B. Hammarström and T. Laurell, Anal. Chim. Acta, 2009, 649, 141–157 CrossRef CAS PubMed.
- S. Huang, C. Li, B. Lin and J. Qin, Lab Chip, 2010, 10, 2925–2931 RSC.
- D. Migliozzi, B. Pelz, D. G. Dupouy, A.-L. Leblond, A. Soltermann and M. A. M. Gijs, Microsyst. Nanoeng., 2019, 5, 1–12 CrossRef PubMed.
- M. C. Mitchell, V. Spikmans and A. J. de Mello, Analyst, 2001, 126, 24–27 RSC.
- A.-L. Liu, Z.-Q. Li, Z.-Q. Wu and X.-H. Xia, Talanta, 2018, 182, 544–548 CrossRef CAS PubMed.
- A. Yang, T. B. Hoang, M. Dridi, C. Deeb, M. H. Mikkelsen, G. C. Schatz and T. W. Odom, Nat. Commun., 2015, 6, 6939 CrossRef CAS PubMed.
- A. Fernández-la-Villa, D. F. Pozo-Ayuso and M. Castaño-Álvarez, Curr. Opin. Electrochem., 2019, 15, 175–185 CrossRef.
- M. Maiwald, H. H. Fischer, Y.-K. Kim, K. Albert and H. Hasse, J. Magn. Reson., 2004, 166, 135–146 CrossRef CAS PubMed.
- G. R. Eldridge, H. C. Vervoort, C. M. Lee, P. A. Cremin, C. T. Williams, S. M. Hart, M. G. Goering, M. O'Neil-Johnso and L. Zeng, Anal. Chem., 2002, 74, 3963–3971 CrossRef CAS PubMed.
- P. R. L. Markwick, T. Malliavin and M. Nilges, PLoS Comput. Biol., 2008, 4, e1000168 CrossRef PubMed.
- D. L. Olson, T. L. Peck, A. G. Webb, R. L. Magin and J. V. Sweedler, Science, 1995, 270, 1967–1970 CrossRef CAS.
- A. M. Wolters, D. A. Jayawickrama and J. V. Sweedler, Curr. Opin. Chem. Biol., 2002, 6, 711–716 CrossRef CAS PubMed.
- V. Badilita, R. C. Meier, N. Spengler, U. Wallrabe, M. Utz and J. G. Korvink, Soft Matter, 2012, 8, 10583–10597 RSC.
- Z. Miao, M. Jin, X. Liu, W. Guo, X. Jin, H. Liu and Y. Wang, Anal. Bioanal. Chem., 2015, 407, 3405–3416 CrossRef CAS PubMed.
- S. Rezzi, F. A. Vera, F.-P. J. Martin, S. Wang, D. Lawler and S. Kochhar, J. Chromatogr., B, 2008, 871, 271–278 CrossRef CAS PubMed.
- P. Rossi, G. V. T. Swapna, Y. J. Huang, J. M. Aramini, C. Anklin, K. Conover, K. Hamilton, R. Xiao, T. B. Acton, A. Ertekin, J. K. Everett and G. T. Montelione, J. Biomol. NMR, 2010, 46, 11–22 CrossRef CAS PubMed.
- M. Kakuta, D. A. Jayawickrama, A. M. Wolters, A. Manz and J. V. Sweedler, Anal. Chem., 2003, 75, 956–960 CrossRef CAS PubMed.
- M. Khajeh, M. A. Bernstein and G. A. Morris, Magn. Reson. Chem., 2010, 48, 516–522 CrossRef CAS PubMed.
- P. Hentschel, K. Holtin, L. Steinhauser and K. Albert, Chirality, 2012, 24, 1074–1076 CrossRef CAS PubMed.
- H. Ryan, S.-H. Song, A. Zaß, J. Korvink and M. Utz, Anal. Chem., 2012, 84, 3696–3702 CrossRef CAS PubMed.
- N. Spengler, A. Moazenzadeh, R. C. Meier, V. Badilita, J. G. Korvink and U. Wallrabe, J. Micromech. Microeng., 2014, 24, 034004 CrossRef CAS.
- G. Finch, A. Yilmaz and M. Utz, J. Magn. Reson., 2016, 262, 73–80 CrossRef CAS PubMed.
- M. Sharma and M. Utz, J. Magn. Reson., 2019, 303, 75–81 CrossRef CAS PubMed.
- S. S. Zalesskiy, E. Danieli, B. Blümich and V. P. Ananikov, Chem. Rev., 2014, 114, 5641–5694 CrossRef CAS PubMed.
- H. Ryan, A. Smith and M. Utz, Lab Chip, 2014, 14, 1678–1685 RSC.
- Y. Lin, S. Schiavo, J. Orjala, P. Vouros and R. Kautz, Anal. Chem., 2008, 80, 8045–8054 CrossRef CAS PubMed.
- F. Dalitz, M. Cudaj, M. Maiwald and G. Guthausen, Prog. Nucl. Magn. Reson. Spectrosc., 2012, 60, 52–70 CrossRef CAS PubMed.
- I. Swyer, R. Soong, M. D. M. Dryden, M. Fey, W. E. Maas, A. Simpson and A. R. Wheeler, Lab Chip, 2016, 16, 4424–4435 RSC.
- I. Swyer, S. von der Ecken, B. Wu, A. Jenne, R. Soong, F. Vincent, D. Schmidig, T. Frei, F. Busse, H. J. Stronks, A. J. Simpson and A. R. Wheeler, Lab Chip, 2019, 19, 641–653 RSC.
- W. H. Grover, A. M. Skelley, C. N. Liu, E. T. Lagally and R. A. Mathies, Sens. Actuators, B, 2003, 89, 315–323 CrossRef CAS.
- T. W. Herling, D. J. O'Connell, M. C. Bauer, J. Persson, U. Weininger, T. P. Knowles and S. Linse, Biophys. J., 2016, 110, 1957–1966 CrossRef CAS PubMed.
- S. Messner, I. Agarkova, W. Moritz and J. M. Kelm, Arch. Toxicol., 2013, 87, 209–213 CrossRef CAS PubMed.
- F. Yu, S. Zhuo, Y. Qu, D. Choudhury, Z. Wang, C. Iliescu and H. Yu, Biomicrofluidics, 2017, 11, 034108 CrossRef PubMed.
- M. Du, Z. Ma, X. Ye and Z. Zhou, Sci. China: Technol. Sci., 2013, 56, 1047–1054 CrossRef CAS.
- H.-Y. Tseng, C.-H. Wang, W.-Y. Lin and G.-B. Lee, Biomed. Microdevices, 2007, 9, 545–554 CrossRef PubMed.
- A. Yilmaz and M. Utz, Lab Chip, 2016, 16, 2079–2085 RSC.
- J. Y. Baek, J. Y. Park, J. I. Ju, T. S. Lee and S. H. Lee, J. Micromech. Microeng., 2005, 15, 1015–1020 CrossRef.
- J. Bezanson, A. Edelman, S. Karpinski and V. B. Shah, SIAM Rev., 2017, 59, 65–98 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0lc01100b |
| ‡ Current address (from Jan 2020): Dept. of Chemistry, University of Florida, FL 32611, USA. |
| This journal is © The Royal Society of Chemistry 2021 |