
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A microfluidic impedance-based extended infectivity assay: combining retroviral amplification and cytopathic effect monitoring on a single lab-on-a-chip platform†
Michaela
Purtscher‡
a,
Mario
Rothbauer‡
 b,
Sebastian Rudi Adam
Kratz
bc,
Andrew
Bailey
d,
Peter
Lieberzeit
e and
Peter
Ertl
*b
b,
Sebastian Rudi Adam
Kratz
bc,
Andrew
Bailey
d,
Peter
Lieberzeit
e and
Peter
Ertl
*b
aUniversity of Applied Sciences FH Technikum Wien, Höchstädtplatz 6, 1200 Vienna, Austria
bFaculty of Technical Chemistry, Vienna University of Technology (TUW), Getreidemarkt 9, 1060 Vienna, Austria. E-mail: peter.ertl@tuwien.ac.at
cInstitute of Pharmaceutical Technology and Buchmann Institute for Life Sciences, Goethe University, mAx-von-Laue-Straße 15, 60438 Frankfurt am Main, Germany
dViruSure GmbH, Donau-City-Straße 1, 1220 Vienna, Austria
eDepartment of Physical Chemistry, University of Vienna, Währingerstrasse 42, 1090 Vienna, Austria
First published on 1st February 2021
Abstract
Detection, quantification and monitoring of virus – host cell interactions are of great importance when evaluating the safety of pharmaceutical products. With the wide usage of viral based vector systems in combination with mammalian cell lines for the production of biopharmaceuticals, the presence of replication competent viral particles needs to be avoided and potential hazards carefully assessed. Consequently, regulatory agencies recommend viral clearance studies using plaque assays or TCID50 assays to evaluate the efficiency of the production process in removing viruses. While plaque assays provide reliable information on the presence of viral contaminations, they are still tedious to perform and can take up to two weeks to finish. To overcome some of these limitations, we have automated, miniaturized and integrated the dual cell culture bioassay into a common lab-on-a-chip platform containing embedded electrical sensor arrays to enrich and detect infectious viruses. Results of our microfluidic single step assay show that a significant reduction in assay time down to 3 to 4 days can be achieved using simultaneous cell-based viral amplification, release and detection of cytopathic effects in a target cell line. We further demonstrate the enhancing effect of continuous fluid flow on infection of PG-4 reporter cells by newly formed and highly active virions by M. dunni cells, thus pointing to the importance of physical relevant viral–cell interactions.
1. Introduction
In light of current and reoccurring viral outbreaks such as Ebola, Influenza, Zika and most recently SARS-Cov-2, improved detection systems capable of detecting ultralow levels of virus concentrations are of growing interest to the public and the medical community. Additionally, new methods that allow monitoring of virus–cell interactions are equally of importance in the development of virotherapy options, where either native or engineered viruses are used as alternatives in cancer therapies, immunotherapies and delivery vehicles in gene therapy applications.1,2 Furthermore, reliable and accurate identification of viral contaminations in pharmaceutical products constitutes an essential risk assessment strategy to evaluate the safety of drugs, to date. This safety issue has arisen with the widespread usage of mammalian cell lines in the production of biopharmaceuticals, which potentially can express and release endogenous derived retroviral-like particles. Viral clearance studies are therefore recommended by the Food and Drug Administration (FDA) to evaluate pharmaceutical purification processes.3 In virus clearance studies the biosafety of pharmaceutical products is determined by (a) selecting the most appropriate virus for the study and (b) assessing process steps that are effective in removing viruses. Among others, the murine leukemia virus as one of the best studied retrovirus is predominantly used in virus clearance studies.4,5 This virus can be readily propagated without adverse effects in the skin fibroblast cell line M. dunni (mus terricolour) resulting in the release of large quantities of infectious virus particles, which can be subsequently detected using focus forming and plaque assays.6,7 This means that in theory the presence of a single replication competent retrovirus can be detected in pharmaceutical products using a cell-based virus amplification strategy. In contrast to PCR-based strategies that are used as golden standard for quantification of viral content, focus forming assays are based on immunofluorescent labelling of antibodies detecting cytopathic effects of a viral titer on cells. In addition, plaque assays rely on infecting a secondary cell culture (reporter cell line) that in turn forms visual circular cell-free areas and are the golden standard to analyze virus infectiousness or replication.8,9 Here, the cat brain Moloney sarcoma virus-transformed PG-4 cell line is often used in viral clearance studies for detection of replication competent retroviruses in products and reagents for human use. Although proven effective and recommended by the FDA, the standard assay protocol is labor intensive involves multiple manual steps, is expensive due to the large volumes of cell culture media, reagents and consumables needed and can take up to two weeks to complete using trained biomedical analysts. As an example, a standard assay protocol involves initial virus amplification using M. dunni cell cultures, repeated collection of the supernatant, followed by preparation of several dilutions of the virus containing reagent and its addition to a semi-confluent grown PG-4 cell layers usually followed by the coverage with a solid or semisolid overlay to hinder the spread of the virus ad random. Once infected PG-4 cells undergo distinct morphological changes (e.g. rounding up and cell detachment) resulting in the formation of cell free areas during the process of viral release, which overtime leads to the formation of visible plaques in the cell layer. To enhance the contrast between cell layer and plaques, a potential cancerogenic and environmentally toxic staining procedure of the cell layer is usually performed at the end of the cultivation period (e.g. 9 days) and the number of plaque forming units (PFU ml−1) are microscopically analyzed and calculated in relation to serial dilutions.10,11To overcome some of the limitations associated with detecting virus–host cell interactions a variety of advanced cell-based technologies have been reported to improve automation, miniaturization and integration of biosensing strategies to eliminate tedious staining and endpoint detection. For instance, impedance measurements have been employed to determine viral titers by tracing cytopathic effects induced changes in cell cultures using the commercial available xCELLigence™ real-time cell analysis system (Agilent, USA) and ECIS™ systems (Applied BioPhysics Inc., Germany).12–14 Despite these efforts to improve outcomes, reduce time-to-result and costs, the complexity of the multistep cell-based assay prevails. As complementary technology to analyze virus–cell interactions microfluidic cell culture systems have been used to detect cell-to-cell, cell-to-matrix and cell-to-surface as well as material–biology interactions especially regarding biocompatibility and toxicology.15–17 Microfluidic cell culture systems are ideally suited to monitor cytopathic and cytolytic effects of viruses, since laminar flow enables improved particle–cell interactions with high spatiotemporal resolution creating a more controllable and precise microenvironment.18,19 In the current work we introduce an automated microfluidic-based extended infectivity assay where virus amplification and continuous release of newly formed virions by a primary producer cell line as well as the secondary cytopathic or cytolytic effect on an indicator cell line are monitored on a single lab-on-a-chip platform with non-invasive and dynamic monitoring using embedded impedance microsensors. Rapid detection of replication competent viral particles in samples is readily accomplished with our platform by initially increasing the viral load using the virus replication supporting M. dunni cell line, followed by infection of the indicator cell line PG-4 by the newly formed and active virions resulting in a loss of cell-surface integrity (onset of plaque formation). As visualized in Fig. 1 our platform combines the basic virus – host mechanisms in a dual compartment sensing system with an automated microfluidic assay principle to significantly cut down the assay timeline and automate tedious assay steps. An important aspect of our microfluidics approach is the application of constant fluid flow of 1.5 μl min−1 during virus uptake, amplification and release resulting in newly formed and active virions that readily infect the downstream PG-4 target cell line,20 where cell-surface detachment processes are readily investigated by cell impedance sensing.21,22 Our “Sample In Result Out” microfluidic infectivity assay reduces assay steps by (i) performing dual cell culture handling in parallel, (ii) eliminating supernatant collection and biased sample transfer by manual pipetting, (iii) maintaining higher numbers of replication competent virions and (iv) omitting staining procedures and end-point detection by read-out automation. Our automated microfluidic infectivity assay therefore provides results within a fewer time window and has the potential to reduce costs of QC measures based on standard microtiter plate-based plaque assays when integrated into existing biopharmaceutical work flows.
 | ||
| Fig. 1 A.) Schematic overview of the biological principles underlying the proposed extended infectivity assay. Left mouse silhouette indicates the M. dunni amplification cell line, that allows for non-cytopathic virus propagation. Middle shows the lab-on-a-chip platform for simultaneous dual cell cultures with embedded impedance sensors. Right cat silhouette indicates the PG-4 indicator cell line which upon superinfection with the model virus shows cytopathic effects and cells detach from sensor surface. B.) Representative outline of the process steps involved in a standard plaque assay protocol. C.) Rapid one step “sample in result out” microfluidic extended infectivity assay based on M. dunni cell-based x-MuLV virus amplification and release followed by infection of target PG-4 cells and detection of cytopathic effects using embedded impedance sensors. | ||
2. Materials and methods
Chip design and fabrication
The bottom of our lab-on-the-chip device consisted of a (30 × 30) mm2 borosilicate glass substrate (Schott Borofloat® D263Teco) containing interdigitated electrode structures (μIDES), 200 fingers with electrode finger width as well as a gap distance of 5 μm (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) and an IDES area of 2 mm2 (see ESI† Fig. S1). The fluidic layer consists of polydimethylsiloxane (Sylgard 184) consisting of 100 μm high and wide channels that connect both cell culture chambers. The culture chambers were 20 mm long and 5 mm wide featuring a cell culture area of 95 mm2 for each chamber. The microfluidic PDMS is layered between the bottom glass substrate containing the microelectrode arrays and a top object glass (VWR international) substrate containing 1 mm in diameter access holes to connect to external tubing. Final assembly of the two glass and PDMS layers were performed by thoroughly cleaning each single substrate followed by plasma activation and bonding.
:1 ratio) and an IDES area of 2 mm2 (see ESI† Fig. S1). The fluidic layer consists of polydimethylsiloxane (Sylgard 184) consisting of 100 μm high and wide channels that connect both cell culture chambers. The culture chambers were 20 mm long and 5 mm wide featuring a cell culture area of 95 mm2 for each chamber. The microfluidic PDMS is layered between the bottom glass substrate containing the microelectrode arrays and a top object glass (VWR international) substrate containing 1 mm in diameter access holes to connect to external tubing. Final assembly of the two glass and PDMS layers were performed by thoroughly cleaning each single substrate followed by plasma activation and bonding.
Bioimpedance spectroscopy
The dual-cell chip was placed onto an aluminium holder and heated to 37 °C, while contact pads located at the sensor substrate were connected via spring-laded pins and insulated wires to a VMP3 multi-channel potentiostat (BioLogic). Impedance spectra ranging from 400 Hz to 400 kHz were continuously recorded every 5 min (V peak to peak 140 mV) over a period of 3 to 5 days. For data analysis EC Lab software, Graph Prism 7.0a and OriginPro 8.5 and FlowJo10.3; Dean–Jett–Fox analysis was used. Impedance data were normalized according to ((IZI total column – IZI medium)/(IZI peak – IZI medium)).Cell culture handling
The cat brain Moloney sarcoma virus – transformed PG-4 cell line (European Collection of Cell Cultures, 94102703) and the normal mouse tail fibroblast M. dunni (Clone III8C) cell line (European Collection of Cell Culture, 94101211) were cultured at 37 °C in 5% CO2 humidified atmosphere (incubator; HeraCell). Cell expansion took place in 25 cm2 cell culture flaks (PAA Laboratories), where after reaching 70 to 80% confluence both cell lines were split at a ratio of 1:6 using 0.25% trypsin–EDTA (trypsin–ethylenediaminetetraacetic acid, fisher scientific) at 37 °C for 3 min for enzymatic cell detachment. The culture medium, McCoys (PAA Laboratories), was supplemented with 1% stabile L-glutamine (PAA Laboratories) and 5% FCS (PAA Laboratories) for the M. dunni cell line as well as 10% FCS for die PG-4 cell line. For on-chip experiments cells were transferred into a 6-well plate 24 h prior to their seeding into the microfluidic device at a density of approximately 70% to ensure comparability of the cellular state between experiments. The final cell culture medium was prepared with 10% FCS and complemented with 15 mM HEPES puffer (PAA Laboratories) for on-chip experiments.
On-chip cultivation – extended infectivity protocol
The retrovirus x-MuLV (ATCC®Vr-1447™) strain pNFS Th-1 was obtained from ViruSure GmbH (AT) and stored at −80 °C prior to usage. Prior to cell seeding, the microfluidic device was rinsed for a minimum of 1 h with 70% ethanol followed by a second rinsing step with PBS supplemented with 1% gentamycin for several hours to ensure sterility. This was followed by flushing the biochip with cell culture medium using a 10 mL gas-tight syringe at 15 μl min−1 using a pressure driven syringe pump (Nemesis) for several hours. Next, the flow was set to 1.5 μl min−1 and impedance baseline signals were recorded as described above. M. dunni and PG-4 cells were individually seeded into each cell chamber to cover 40% (6.6 × 104M. dunni cells) and 20% (1.32 × 105 PG-4 cells per 195 μl volume) of the 95 mm2 cell culture area, respectively. After 2 h of attachment time under static conditions, the culture medium flow rate was set to 1.5 μl min−1 and infection of the M. dunni cells with varying virus titres was performed after 13.5 h using 1 mL plastic syringe.Flow cytometry – Cell cycle analysis
M. dunni and PG-4 cells were seeded 24 h prior to the experiment into 6-well plates at an approximate density of 70%. For the experiment itself, both cell types where seeded into 12-well plates at a density of 20% complying to 1.05 × 105 cell per well. Both cell types were cultured under non-starvation, starvation and starvation-release conditions and samples were taken after 24, 48 and 72 h after seeding. After aspiration and rinsing with PBS, cells were detached using 300 μl of a trypsin–EDTA solution and 3 min of incubation at 37 °C. The cells were than resuspended in 1 mL culture medium and centrifuged for 5 min at an acceleration force of 170 g. The supernatant was discarded, and the cell pellet was resuspended in a 2% paraformaldehyde solution (PFA) for 10 min at room temperature. Samples were once more centrifuged for 5 min at an acceleration force of 170 g, the PFA solution was removed, the cells were resuspended in 1 mL blocking buffer (PBS + 0.2% BSA) and stored at 4 °C. Prior to the FACS analysis, cells were stained using the DNA binding dye DAPI. Cells were centrifuged for 5 min at 170 g and cells were resuspended in staining solution comprised of PBS with 0.2% BSA, 0.2% TritionX-100, 100 μg mL−1 RNAse and 2 ng mL−1 DAPI dilution and kept for 20 min in the dark before analysing via FACS.qPCR – virus sample preparation
Starting after the cell seeding on-chip, at an initial density of 40% confluence, the supernatant of the cell culture was periodically collected and stored at −80 °C. The cell culture was inoculated with a virus titer of 7.7 × 103 PFU ml−1 12 h following cell seeding and supernatants were collected after 6, 12, 24, 30, 36, 42, 48 and 60 h after virus inoculation. The frozen samples were shipped to ViruSure Inc. for qPCR analysis. RNA isolation and reverse transcription was performed according to standard protocols. The qPCR protocol was performed using the Applied Biosystems 7500 real-time PCR System with the following program. Reverse transcription for 15 min at 48 °C, enzyme activation for 10 min at 95 °C followed by 40 cycles of a denaturation phase of 15 s at 95 °C and annealing/extension phase of 1 min at 60 °C.Computational fluid dynamics – fluid modeling
The CFD simulation was performed by CFD Autodesk 2019. The CAD model of the chip was created in Fusion 360 (Autodesk). The fluid was modeled as water at room temperature. Furthermore, there was no heat exchange and gravity simulated. The fluid inlets were modeled by a defined 1.5 μl min−1 of volume flow. The outlets were modeled as openings with 0 pascal pressure (please see table). No further initial conditions were added, and the net was generated automatically by the software.3. Results and discussion
3.1. Characterization of microfluidic device
Initial microdevice characterization was performed using computational fluid dynamics (CFD) simulation and fluorescent imaging of labelled nanoparticles (diameter of 100 nm) within the two-chamber lab-on-a-chip system to analyse viral transport behaviour. Results shown in Fig. 2A revealed identical measurement conditions for both cell culture chambers, while nanoparticle movement from the upstream-located virial amplification cell culture chamber to the viral detection cell chamber required 20 min in the presence of 1.5 μl min−1 fluid flow. Therefore, it takes approx. 10 min for the nanoparticles to pass over sensor region 1 and 2 within a single chamber (see Fig. 2B), thereby ensuring sufficient time for virus to cell interactions. | ||
| Fig. 2 A.) Distribution and shear stress analysis by CFD simulation of the two-chamber lab-on-a-chip system. Flow velocity in both culture chambers show similar profiles with a higher shear stress levels in the area of ports and connectors while a fluid flow of 1.5 μl min−1 was applied. B.) Tracing of fluorescence labelled nanoparticle with a diameter of 100 nm over the area of (upstream) region 1 to region 2 (downstream) within a single cell culture chamber. Nanoparticle remain approximately 20 min within one culture chamber before passing over to the second chamber. | ||
Next, biosensor sensitivity, cell adhesion and growth curve dynamics of M. dunni and PG4 cell lines was assessed using the on-chip electrochemical impedance measurements. Since sensitivity of an cell-based impedance assays is influenced by sensor geometry, frequency analyses for both cell types were conducted.23,24Fig. 3A shows impedance spectra of confluent cell monolayers of M. dunni and PG-4 using the 5 × 5 μm gap-to-finger geometry. To verify that no significant signal maxima shifts are present due to cell line differences, frequency analyses were performed using infected M. dunni and PG-4 cells. Results shown in ESI† Fig. S2 point at similar frequency dependent sensor sensitivities of M. dunni (n = 11) and PG-4 (n = 10) cultures, which were infected by increasing titres of the x-MuLV prior measurements.
 | ||
| Fig. 3 Sensor sensitivity, cell adhesion and growth curve dynamics of the integrated impedance sensors: A) analysis of frequency dependent sensor sensitivity of PG-4 (n = 9) and M. dunni (n = 12) control cultures. B and C), Growth curve dynamics of M. dunni (n = 4) and PG-4 (n = 11) control culture over the time course of 100 h. | ||
Interestingly the lower impedance signal change-fold observed with infected PG-4 cells (indicator cell line) already indicates the occurrence of cytopathic effects of the retrovirus resulting in cell detachment and cell lyses. In a next set of experiments, cell attachment, spreading and proliferation of M. dunni cells (amplification cell line) were investigated to characterize on-chip growth behaviour of healthy cells. It is important to note that M. dunni cells need to be seeded at a low surface coverage of max. 40% to allow cells to proliferate and thereby undergo all cell cycle phases (G1, S, G2 and M). This is crucial to ensure effective integration and amplification of the retroviral genome into the proliferating M. dunni cell line. Impedance-time traces shown in Fig. 3B reveal a rapid signal increase within the first 4 h (e.g. cell attachment) after cell seeding followed by a slow signal decrease (e.g. cell spreading) of (1.55 ± 0.77) Ohm h−1 (n = 12) for 25 h resulting in stable impedance signals between 40 to 45 h, which indicates the establishment of fully covered sensor surfaces and stable cell layers up to 100 h of cultivation duration. ESI† Fig. S3A depicts the representative change in sensor coverage during impedance sensing of M. dunni cells over the whole culture period. Similarly, the establishment and stability of PG-4 indicator cell line over a period 100 h cultivation inside the microfluidic biochip was confirmed using the embedded impedance sensors. Fig. 3C shows a steady signal increase of in average (1.04 ± 0.62) Ohm h−1 (n = 9) over the first 65 h after which a plateau is reached that indicates sensor coverage by PG-cells (ESI† Fig. S3B). The fact that the chip-based PG-4 cell culture reaches confluency approximately a day later than M. dunni cells is ideally suited to perform dual cell cultivations in parallel to the infectivity assay, thus eliminating multiple cell-loading steps at different time points. It is of importance that the cell-based virus-amplification carried out by M. dunni cells takes place prior to reaching a confluent PG-4 monolayer to ensure effective integration of the retroviral genome and manifestation of its cytopathic effects.
3.2. Cell-based virus amplification and infection efficiency
A key aspect of any viral infectivity assay is the successful integration of retroviral DNA into the host genome, which takes place during cell mitosis in particular during the nuclear membrane breakdown. The production of x-MuLV viral progeny usually begins within 18 h to 24 h after infection (see also ESI† Fig. S4), which makes a time-oriented infection protocol an important issue in optimizing the on-chip co-cultivation protocol. Since mammalian cell mitosis last only for about 2 h, cell cycle synchronization of the entire cell culture may improve infection rates by increasing the number of cells present in the G2/M phase at the time of infection. To assess the impact of a 48 h pre-starvation period as a simple drug-free means to induce cell cycle arrest, on-chip cell culture phase synchronisation was analyzed using time-resolved FACS analyses. Fig. 4 shows cell cycle distributions over time in the absence and presence of starvation as well as “release of starvation” using both M. dunni and PG-4 cells. In the absence of cell starvation, M. dunni cell cycle distribution over a 72 h period grown in full media revealed that a majority of cells (>60%) within the cell population resides in the G1 phase, while a significant increase in G2/M phase occurred only after 13–17 h and 26 h, respectively (see Fig. 4A). This phenomenon can most likely be attributed to cells re-entering a proliferative state after a delay in cell cycle progression due to initial attachment and spreading events within the first 10 h. As a result, an increase of cells in G2/M phase up to 33% of the total cell population 13 h after cell seeding is repeatedly obtained. Interestingly, G2/M phase population remained stable over the next 17 h of cultivation, indicating that under regular growth conditions, virus addition is optimally performed at about 13 h of M. dunni on-chip cultivation. A direct comparison of cell cycle distribution using complete growth medium, serum depleted medium and release out of a 48 h starvation period is shown in Fig. 4B. Interestingly neither starvation using serum free medium nor release out of starvation after a 48 h pre-starvation period increased the amount of cells residing in the G2/M phase, which is reflected also by an average cell doubling time of 14.7 to 16.7 h of M. dunni cells. Since cell cycle synchronization did not yield any advantage over our regular cell culture protocol, virus addition was set at 13.5 h post cell ceding to ensure efficient integration of host cell genome in all subsequent on-chip experiments. These findings are further supported by time-resolved qPCR analyses, where the release of newly formed virions by M. dunni cells 12 h after infection with a x-MuLV titer of 7.7 × 103 PFU ml−1 was quantified. Results shown in ESI† Fig. S3 confirm constant virus propagation over time exhibiting a linear increase over 66 h in culture. Additional FACS results shown in Fig. 4C highlight that PG-4 cells are ideally suited to serve as indicator cell line for x-MuLV infection studies, since over 50% of the cell culture displays a G2/M phase at any given timepoint. In order words, PG-4 cells can be readily infected by freshly produced virus particles over the entire analysis period of our on-chip infectivity assay. | ||
| Fig. 4 Drug-free and starvation-induced cell cycle synchronisation analysis using FACS: A) M. dunni cells cultured in complete growth medium. B) Comparison of cell cycle populations of M. dunni cultures 13 h after cell seeding of cultures in complete growth medium as well as cultures starved by serum depletion and cultures 12 h after release from a 48 h starvation period. C) PG-4 cell cultures in complete growth medium. | ||
3.3. Application of the microfluidic sensor-integrated system as extended infectivity assay
Prior to the application of the extended infectivity assay, impedance signals in the presence of infected M. dunni cell cultures were investigated to determine possible viral induced side effects such as loss of sensor signals as a result of cell detachment processes. Fig. 5A shows impedance time-traces of healthy and M. dunni cells inoculated with a virus titer of 7.7 × 103 PFU ml−1, translating to 1.5 × 103 PFU per culture chamber. Although signal differences were observed for the first 4 h of cultivation, stable impedance-time traces are obtained for healthy and infected M. dunni cells for the remaining 96 h of on-chip cultivation. Interestingly, obtained impedance signals are in average by 2-fold higher in the presence of infected cells that constantly release newly formed virions as seen in Fig. 5B. However, regardless of infection status impedance signals reached a plateau already after 30 h in on-chip cultures (see Fig. 5A). Moreover, a successful infection of M. dunni cells, the amplification cell line, can also be recognized by an increase of impedance values. | ||
| Fig. 5 A) Comparison of M. dunni impedance time traces of single versus dual cell cultures. While the grey (n = 3) trace show M. dunni control cultures the black (n = 5) one show M. dunni coupled to PG-4 cultures which were additionally infected at a titer of 2.2 × 105 PFU ml−1. B) Absolute impedance changes (Ohm) over the culture period of 90 h. | ||
When finally coupling M. dunni cell cultures to the PG-4 indicator cell line similar growth dynamics (data not shown) are observed, indicating that reproducible co-culturing in parallel is feasible in the current microfluidic dual-cell chip system. To rule out any influence of the M. dunni culture upstream of the PG-4 reporter cell line, impedance curves of PG-4 in co-culture with M. dunni cells from the extended infectivity assay was compared with PG-4 monocultures (n = 11) with no discernable difference observable (see ESI† Fig. S7). In a next set of experiments, the ability of the microfluidic extended infectivity assay to rapidly detect cytopathic effects induced by virions in PG-4 indicator cells that were produced by the upstream located M. dunni cells is evaluated. Following individual loading of each cell culture compartments (e.g. 40% M. dunni and 20% PG-4) intended to prevent cell cross contaminations, the cell cultivation chambers were manually connected and a flow rate of 1.5 μl min−1 was adjusted, and impedance measurements were conducted over the entire assay period of 100 h. As shown in Fig. 6A the microfluidic extended infectivity assay containing embedded electrical microsensors already detected the onset of virus-induced cytopathic effects such as cell rounding and detachment of PG-4 cells after only 60 h (<3 days) in cultivation resulting in impedance decrease. Here, M. dunni cells were infected with x-MuLV at a concentration 2.2 × 105 PFU per culture chamber, which was more than sufficient to induce cell-based virus amplification and release. Final performance evaluation involved the effectiveness of the extended infectivity protocol using our dual cell chip set up over a single cell culture system. Results in Fig. 6b show obtained signal fold changes between 60 to 100 h of assay time. The 2.45 ± 0.69-fold signal decrease using the extended infectivity assay strongly points at increased PG-4 cell rounding, detachment and death rates following infection with newly formed virions by M. dunni cells. Interestingly, in the absence of M. dunni cell where PG-4 cells were directly infected with viral stock solutions at seeding (t = 0) or 13.5 h after seeding a significant difference were observed, thus highlighting the amplification effect of using a dual cell culture set up.
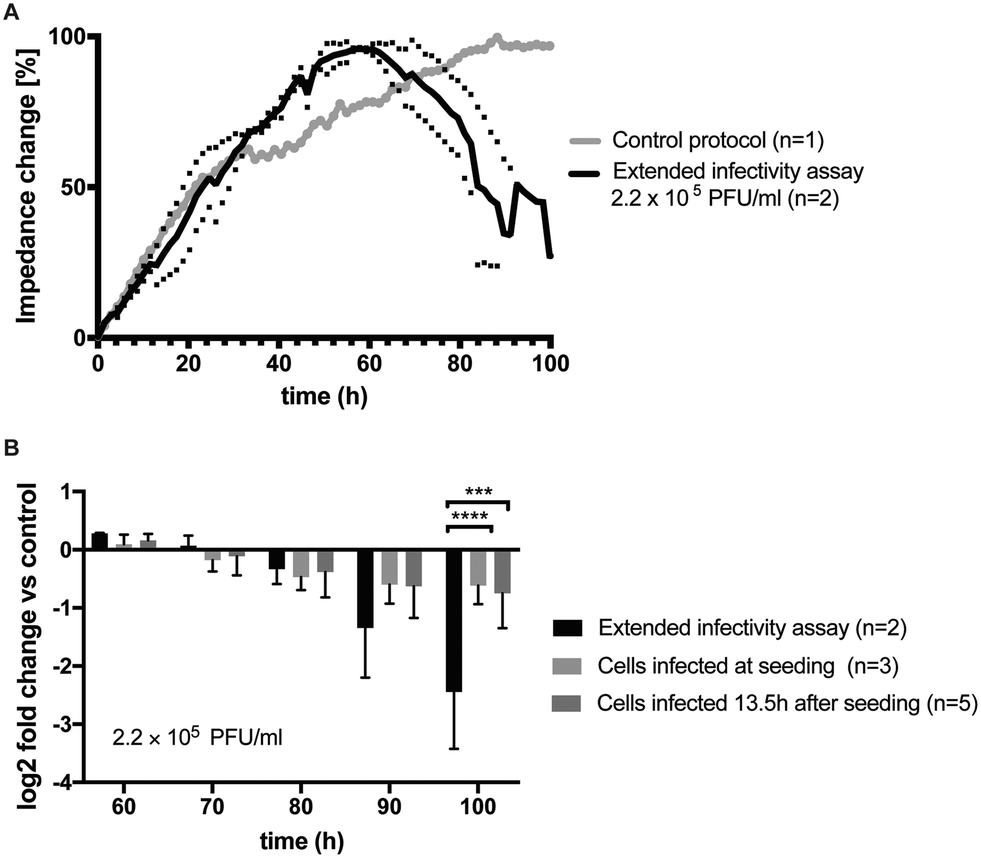 | ||
| Fig. 6 A) Impedance time trace of PG-4 cell cultures infected with 2.2 × 105 PFU ml−1 using the extended infectivity assay protocol (black, n = 2) in comparison to untreated control (grey, n = 1). B) Comparison of fold changes in impedance signal to control using different infection protocols. A −2.45 ± 0.69 fold change in impedance signals was observed after 100 h of assay time in the presence of the extended infectivity assay protocol. Also, significant differences to directly infected PG-4 cultures without in-line amplification through connected M. dunni cultures was evident (2-way ANOVA, Turkey's multiple comparison test, p = 0.0001 (***), p < 0.0001 (****)). | ||
Impedance-time traces (grey line) in control experiments showed typical growth curves and stable cell monolayer integrity of healthy PG-4 cells exhibiting a steady signal increase over time. Importantly, ESI† Fig. S5A shows best performance were obtained using the single-step co-culture system, further highlighting the importance of a timed assay protocol. In turn, ESI† Fig. S5B shows impedance-time traces of directly infected PG-4 cells using a 2.2 × 105 PFU ml−1 viral titer added after cell seeding and 13.5 h of culture, respectively. Results of this timed infection study revealed that cytopathic effects in PG-4 cell can be readily detected using impedance measurements, however obtained average signal decreases of 30% between 60 h to 70 h is less dominant than seen using a cell-based virus amplification strategy. This demonstrates that in the presence of similar viral titers, the directly produced and newly formed viruses in our co-culture system are highly potent, active and infectious. In a final control experiment, the microfluidic extended infectivity assay was exposed to heat inactivated x-MuLV titers to confirm that only replication-competent virus particles are detected in the dual-cell chip system. Impedance-time trace seen in ESI† Fig. S6 also shows a steady impedance signal increase reaching a plateau after 70 h, thus pointing at normal growth characteristics for the first three days followed by a rapid loss of impedance as a result of cytopathic events. The brief signal recovery at 80 h can be linked to fluid flow disruption during changing of the media supply syringe at the external pumping station. In summary, our results clearly demonstrate the potential benefits of downscaling, miniaturizing and integrating cell culture systems, since our microfluidic extended infectivity assay coupled with impedance sensors is able to provide results in less than 4 days instead of over one to two weeks required when using the standard plaque assay protocol.
4. Conclusion
The main principle of our microfluidic extended infectivity assay is based on the on-chip combination of cell-based viral amplification using M. dunni cells and detection of cytopathic effects of newly formed and highly active virions in target PG-4 cells using embedded impedance sensors. Only the application of an advanced microfluidic cell-based assays will eliminate multiple sequential cell culture handling steps and staining procedures needed for microscopic readouts. While simultaneous co-cultivation of both cell lines in a common lab-on-a-chip systems eliminates numerous and tedious cell culture handling steps, the inherent sensitivity of the bioimpedance sensors allows the time-resolved assessment of cytopathic effects such cell rounding and detachment events as of infected PG-4 cells. We have shown that time-to-result in viral clearance studies can be significantly reduced from over a week down to 4 days, using a simple cell culture and infection protocol. Another benefit of using a dynamic cell assay protocol is that new formed virions are actively transported to the indicator cell line, thus ensuring constant and effective virus – cell interactions.Despite the advantages of using an improved microfluidic dual cell culture system over standard plaque assays, some limitations still remain and are associated with its industrial integration. Key for a successful implementation for viral clearance studies is concerned with assay parallelization and increased throughput. In particular additional automation to generate virus titrations, perform sample and cell loading procedures as well as dose–response analysis routines still need to be integrated to ensure reliable adaptation into pharmaceutical safety evaluations and quality control measures. However, automation and parallelization can also be accomplished by increasing the numbers of cultivation chambers and integrating additional microfluidics components such concentration gradients and mixers. Also, replacement of the active flow control using external syringe pumps with passive flow strategies (e.g. refilling of reservoirs) may allow the application of robotic pipetting stations leading to improved sample throughput, thus presenting a real alternative to plague assays.
In summary, the developed microfluidic extended infectivity assay has shown to provide reliable results even when using low to medium viral titers. However, it is not entirely clear whether ultralow concentrations of replication-competent particles present in a final pharmaceutical product can be detected within a limited assay time of 4 days. A comparative analysis within an industrial QM setting is still needed to fully evaluate the potential of our microfluidic extended infectivity assay for viral safety evaluation in pharmaceutical production. Since a direct performance evaluation and benchmarking against standard plaque assays also requires a significant adaptation in biochip layout to deal with the increased sample volume, it is, however, beyond the current study and subject to future development.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dipl.Ing. Natascha Hodosi for her technical support with the performance of qPCR experiments. The authors gratefully acknowledge the financial support from the Austrian Research Promotion Agency (FFG: # 815477/13900) and ViruSure GmbH (Vienna, Austria).Notes and references
- E. Ylösmäki and V. Cerullo, Curr. Opin. Biotechnol., 2020, 65, 25–36 CrossRef.
- N. Slade, Period. Biol., 2001, 103, 139–143 CAS.
- FDA (Food and Drug Administration), Fda, 2020, 16.
- A. Rein, Adv. Virol., 2011, 2011, 403419 Search PubMed.
- J. W. Hartley, N. K. Wolford, L. J. Old and W. P. Rowe, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 789–792 CrossRef CAS.
- P. T. Peebles, Virology, 1975, 67, 288–291 CrossRef CAS.
- D. K. Haapala, W. G. Robey, S. D. Oroszlan and W. P. Tsai, J. Virol., 1985, 53, 827–833 CrossRef CAS.
- R. H. Bassin, S. Ruscetti, I. Ali, D. K. Haapala and A. Rein, Virology, 1982, 123, 139–151 CrossRef CAS.
- Z. Li, M. Blair and L. Thorner, J. Virol. Methods, 1999, 81, 47–53 CrossRef CAS.
- A. Baer and K. Kehn-Hall, J. Visualized Exp., 2014, 1–10 Search PubMed.
- M. J. Chiang, M. Pagkaliwangan, S. Lute, G. Bolton, K. Brorson and M. Schofield, Biotechnol. Bioeng., 2019, 116, 2292–2302 CrossRef CAS.
- C. Charretier, A. Saulnier, L. Benair, C. Armanet, I. Bassard, S. Daulon, B. Bernigaud, E. Rodrigues de Sousa, C. Gonthier, E. Zorn, E. Vetter, C. Saintpierre, P. Riou and D. Gaillac, J. Virol. Methods, 2018, 252, 57–64 CrossRef CAS.
- S. Lebourgeois, A. Fraisse, C. Hennechart-Collette, L. Guillier, S. Perelle and S. Martin-Latil, Front. Cell. Infect. Microbiol., 2018, 8, 335 CrossRef CAS.
- M. R. Pennington and G. R. Van de Walle, mSphere, 2017, 2, 1–12 CrossRef.
- J. Rosser, B. Bachmann, C. Jordan, I. Ribitsch, E. Haltmayer, S. Gueltekin, S. Junttila, B. Galik, A. Gyenesei, B. Haddadi, M. Harasek, M. Egerbacher, P. Ertl and F. Jenner, Mater. Today Bio, 2019, 4, 100023 CrossRef CAS.
- M. Rothbauer, V. Charwat, B. Bachmann, D. Sticker, R. Novak, H. Wanzenböck, R. A. Mathies and P. Ertl, Lab Chip, 2019, 19, 1916–1921 RSC.
- M. Rothbauer, I. Praisler, D. Docter, R. H. Stauber and P. Ertl, Biosensors, 2015, 5, 736–749 CrossRef CAS.
- G. Birnbaumer, S. Küpcü, C. Jungreuthmayer, L. Richter, K. Vorauer-Uhl, A. Wagner, C. Valenta, U. Sleytr and P. Ertl, Lab Chip, 2011, 11, 2753–2762 RSC.
- H. Zirath, M. Rothbauer, S. Spitz, B. Bachmann, C. Jordan, B. Müller, J. Ehgartner, E. Priglinger, S. Mühleder, H. Redl, W. Holnthoner, M. Harasek, T. Mayr and P. Ertl, Front. Physiol., 2018, 9, 1–12 Search PubMed.
- J. M. Coffin, S. H. Hughes and H. E. Varmus, Retroviruses, 1997 Search PubMed.
- I. Giaever and C. R. Keese, Nature, 1993, 366, 591–592 CrossRef CAS.
- W. Gu and Y. Zhao, Expert Rev. Med. Devices, 2010, 7, 767–779 CrossRef.
- N. S. Mazlan, M. M. Ramli, M. M. A. B. Abdullah, D. S. C. Halin, S. S. M. Isa, L. F. A. Talip, N. S. Danial and S. A. Z. Murad, AIP Conf. Proc., 2017, 1885, 020276 CrossRef.
- M. L. Gelsinger, L. L. Tupper and D. S. Matteson, Int. J. Biostat., 2020, 16, 1–12 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0lc01056a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |