
Rapid sample preparation for detection of antibiotic resistance on a microfluidic disc platform†
Alexandra
Perebikovsky‡
 *a,
Yujia
Liu‡
b,
Alexander
Hwu
c,
Horacio
Kido
d,
Ehsan
Shamloo
c,
Dian
Song
d,
Gabriel
Monti
e,
Oren
Shoval
d,
Daniel
Gussin
f and
Marc
Madou
*acd
*a,
Yujia
Liu‡
b,
Alexander
Hwu
c,
Horacio
Kido
d,
Ehsan
Shamloo
c,
Dian
Song
d,
Gabriel
Monti
e,
Oren
Shoval
d,
Daniel
Gussin
f and
Marc
Madou
*acd
aDepartment of Physics and Astronomy, University of California, 4129 Frederick Reines Hall, Irvine, CA 92697, USA. E-mail: aperebik@uci.edu; mmadou@uci.edu; Tel: 949 394 9238 Tel: 949 981 5672
bDepartment of Materials Science and Engineering, University of California, 544 Engineering Tower Irvine, Irvine, CA 92697, USA
cDepartment of Chemical and Biomolecular Engineering, University of California, 916 Engineering Tower, Irvine, California 92697, USA
dDepartment of Mechanical Engineering, University of California, 4200 Engineering Gateway, Room W3311, Irvine, CA 92697, USA
eDepartment of Urology, University of California, 200 Medical Plaza Driveway 140, Los Angeles, CA 90095, USA
fToolbox Medical Innovations, 1965 Kellogg Ave, Carlsbad, CA 92008, USA
First published on 8th December 2020
Abstract
Rapid, point-of-care (PoC) diagnostics for antibiotic susceptibility testing (AST) are critical in combating the antimicrobial resistance epidemic. While new, alternative technologies are capable of rapidly identifying antibiotic resistance, traditional AST methods, where a patient sample is incubated with different antibiotics, remain the most reliable and practical in determining antibiotic effectiveness. Here, we demonstrate a novel sample incubation technique on a microfluidic centrifugal disc (CD) as a proof of concept automated sample processing platform for AST. By using ribosomal RNA (rRNA) as a marker for cell growth, we demonstrated that incubation on the microfluidic CD was enhanced (>1.6 fold) for 11 out of 14 clinically relevant isolates of Escherichia coli compared to traditional shaker incubators. Finally, we utilize the system to identify antibiotic resistance of 11 E. coli isolates incubated with 5 different antibiotics in under 2 hours.
Antibiotics save lives every day by treating severe infections and allowing medical practitioners to perform life-saving surgical and medical procedures. However, antibiotic abuse has contributed to selection pressure and the emergence of new drug resistant and untreatable strains. On top of this, only two new classes of antibiotics have been developed since 1962,1 resulting in a global healthcare crisis. According to the WHO, antibiotic-resistant infections currently claim at least 50
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 lives each year across Europe and the US, with several hundred thousand more dying in other areas of the world.2 In the US alone there are currently 23000 deaths, 2.0 million illnesses, and $20 billion worth of direct extra costs. If antibiotic resistance continues to increase, microbial infections could kill 10 million people every year by 2050.3
000 lives each year across Europe and the US, with several hundred thousand more dying in other areas of the world.2 In the US alone there are currently 23000 deaths, 2.0 million illnesses, and $20 billion worth of direct extra costs. If antibiotic resistance continues to increase, microbial infections could kill 10 million people every year by 2050.3
The immediate solution is proper antibiotic stewardship which could control antibiotic resistance and prolong the effectiveness of our current antibiotics for generations to come.4 A critical component to antibiotic stewardship is rapid and effective antibiotic susceptibility testing (AST). Currently, it is estimated that up to 50% of antibiotics prescribed are not needed or are not optimally effective as prescribed. Typically, clinicians use broad spectrum antibiotics andbest guess methods for immediate antibiotic prescription due to the long turnaround times of conventional AST.5
Traditional phenotypic AST methods, such as broth dilution, agar disc diffusion, or ellipsometer test, are the gold standard in providing relevant diagnostic information on susceptibility since they look at the real-time growth and metabolic response of a bacterial population to an antibiotic. However, these tests are limited by the time it takes for visible colony growth to appear, with incubation times ranging from 18 to 24 hours. Furthermore, they require high starting volumes, involve sample transportation to a central lab, and require several intermediate handling steps performed by trained laboratory technicians, increasing costs and delaying accessibility to diagnostic information for days. State of-the-art genotypic ASTs utilize amplification techniques to create a genome sequence map and identify resistant genes. While they eliminate the need for lengthy bacteria culture and have been implemented commercially, they have not been widely adopted for clinical practice. The main limitations include their inability to identify multi-drug resistance, and predominance of both false positive results, caused by non-pathogenic microorganisms that contaminate the test, and false negative results, caused by new or unknown resistance mechanisms.6,7 As a result, phenotypic ASTs remain the most appropriate and reliable diagnostic tests for antibiotic prescription.
While microfluidic, lab-on-a-chip systems have significantly improved accessible diagnostics, successful integration of phenotypic AST in a point-of-care system has remained challenging due to the complex and lengthy nature of the sample preparation. Since the rate limiting step for traditional AST is the incubation of bacteria with antibiotics, many research groups attempt to circumvent this by using techniques such as microfluidic confinement, digital microfluidics, electrokinetics, and agarose microchannels to look at the response of individual cells to an antibiotic within the first few hours of incubation.8–11 For example, Pitruzzello et al. evaluated the morphology and hydrodynamic interactions between single E. coli cells and microfluidic traps to determine the bactericidal response to antibiotics within 1–3 hours after the onset of incubation. However their system is only compatible with motile bacteria species and requires traditional off-chip incubation. Furthermore, most single-cell trapping techniques are optimized based on a small number of pathogens and are unable to perform AST on polymicrobial samples.10 Li et al. addressed these issues by developing a microfluidic chip that can identify individually sorted bacteria species within a complex sample and rapidly incubate bacteria with antibiotics on-chip. However, the system is costly and bulky, requiring an expensive microscope capable of simultaneous incubation, high resolution imaging, and complex image analysis. Additionally, separating the different bacterial species prior to incubation may provide unreliable AST results that do not account for the codependent interactions among different species in a polymicrobial sample.11
To address these issues, we developed a portable, sample-to-answer CD microfluidic cartridge and prototype (SpinBox) that automates all sample processing steps to determine susceptibility results in under 2 hours while maintaining the standard phenotypic-based AST. Unlike other reported microfluidic AST prototypes, the SpinBox provides clinicians with a completely automated, point-of-care diagnostic tool capable of running the following assay steps: sample dilution and metering, bacteria incubation, cell lysis, sample mixing and neutralization, washing steps, and detection, see Fig. 1.
 | ||
| Fig. 1 The SpinBox design and components. | ||
By utilizing ribosomal RNA (rRNA) as a unique assay target, the SpinBox is able to process complex, polymicrobial samples much faster than traditional AST methods. There are several advantages of using rRNA over genotypic targets or single cell analysis techniques:7
1. rRNA has been shown to be an excellent indicator of bacterial growth and cellular metabolism, and responds rapidly to the presence of antibiotics.12,13
2. In ideal incubation conditions, the number of rRNA copies per cell increases significantly (between 104–106 copies), removing the need for further target amplification and shortening assay time.13–15
3. rRNA has both universal and species-specific sequences that are highly accessible to hybridization by probe pairs, enabling simple bacteria identification and analysis in polymicrobial samples.16,17
In order to accommodate the different bacteria found in complex clinical samples, we developed a universal cell lysis method capable of extracting rRNA from both Gram-positive and Gram-negative microbes. We show lysis on six unique Gram-positive and negative species with lysis efficiency comparable to commercial lysis systems, such as the Omnilyse.18,19 We also developed a never reported, small-volume bacteria incubation technique in a microfluidic CD that rapidly enhances bacterial growth. By controlling incubation chamber materials, geometry, temperature, and spin protocol, we demonstrate the ability to enhance bacterial incubation (>150% increase in rRNA copies) compared with normal laboratory plate shakers for multiple species of bacteria, including 13 different isolates of E. coli. Finally, we demonstrate the use of our sample preparation system in determining antibiotic resistance in 11 isolates of E. coli incubated with 5 different antibiotics in under 2 hours.
1 Materials and methods
1.1 Platform instrument fabrication
The portable platform consists of two key components: 1) the SpinBox, or instrument (Fig. 1), and 2) disposable microfluidic cartridges (Fig. 2). The instrumentation automates the sample processing in the microfluidic cartridge and was assembled primarily from modified off-the-shelf components and 3D-printable parts. The code controlling the various aspects of the instrumentation was written in Python 3, loaded into a Raspberry Pi 3B+ running NOOBS, and interfaced using a 7′′ Raspberry Pi Touchscreen. An in-house PCB was prototyped to distribute power to various components. More on the Python repository and CAD assembly can be found in the supplementary information. | ||
| Fig. 2 The microfluidic disc design. a) The disc consists of three layers: an inner feature disc made from COC with disc chambers and channels milled on both sides, and two laminating, gas-permeable adhesives that seal in the features. An optional microarray can be inserted for hybridization of rRNA with capture probes, b) a photo of the assembled disc, with all components inside, is shown, c) a cross section schematic of the world-to-chip interface is shown, d) the fully assembled cartridge, including the disc and world-to-chip interface. | ||
The enclosure was assembled from laser-cut PMMA (McMaster Carr) and secured to t-slot extrusions (McMaster Carr). The touchscreen and Raspberry Pi holder were 3D-printed in polycarbonate material on a Stratasys Fortus 450MC material extrusion printer. Forexperimental testing, a custom spinchuck (ESI† section 2) was machined in aluminum and uses two spring loaded balls to hold the microfluidic cartridge in place and enhance angular alignment. For the portable unit, a world-to-chip microfluidic spinchuck was prototyped and 3D printed using a Raise3D N2 printer, see Fig. 2c. The interface included a large central inlet port for sample loading and a conical chamber that allowed for facile loading of the sample into the disc by spinning. It was affixed to loading holes on the underside of the disc using double-sided adhesives (FLEXMOUNT DFM 200 clear V-95150 POLY H-9).
 | ||
| Fig. 3 The microfluidic disc design is shown with assay steps on right corresponding to fluidic chambers labeled on the disc. The middle column highlights critical features of important fluidic geometries on the disc. The red arrows in the right column indicates the flow sequence of liquid. | ||
1.2 Microfluidic disc fabrication
Three different types of microfluidic discs were fabricated in this work: prototype cyclic clefin copolymer (COC, TOPAS) discs manufactured using a CNC machine at microfluidic Chipshop GmbH (Jena, Germany), prototype poly(methyl methacrylate) (PMMA) discs fabricated using a laser cutter (Trotec Speedy 360), and cyclic olefin copolymer (COC, TOPAS) injection molded discs obtained from microfluidic Chipshop GmbH (Jena, Germany). NaOH was stored in a groove machined in the center of the lysis chamber and sealed using wax. A metal disc that facilitates cell lysis was placed on top of the wax seal to prevent premature release of NaOH. Equal quantities of lysis beads were dispensed into the lysis chambers using a slurry of silica–zirconia beads mixed with polyvinyl alcohol. The disc was then placed into a vacuum heater at 40 °C for 30 minutes. Dried down phosphbate buffer (1×) and detector probes were dispensed into the neutralization chamber prior to assembly. Finally, the COC inner feature layer was placed on an alignment jig and sandwiched between two transparent, gas-permeable, single-sided adhesives (MP Biomedicals 097640205) that were cut to shape using a polyvinyl cutter (Silhouette Cameo 2). A double-sided adhesive (3M 300 LSE) was affixed tothe cutout on the bottom of the detection chamber of the disc, creating a fluidic seal between an optional, modular microarray and the detection chamber of the disc.1.3 Bacteria, antibiotics, and media
E. coli clinical urine isolates were obtained from the University of California, Los Angeles (UCLA) clinical microbiology laboratory with approval from the UCLA and Veterans Affairs Institutional Review Boards and appropriate Health Insurance Portability and Accountability Act exemptions. Bacteria was inoculated into Mueller-Hinton (MH) broth with 12% glycerol (Becton, Dickinson, Sparks, MD) and stored at −80 °C.Experiments were conducted from the frozen isolates. Bacteria was defrosted and cultured overnight by inoculating 5 μL of freezer stock bacteria into 5 mL of cation-adjusted Mueller Hinton (MH2) broth (Sigma, St. Louis, MO) and incubating in a shaker incubator at 37 C. The following day, working samples were prepared by diluting the overnight culture with MH2 broth to ∼6 × 105 cfu per mL. The prepared cultures were pipetted into the incubation chamber on the disc or 96-well plate and immediately sealed. Bacteria was also plated on LB agar (MOBIO Laboratories Inc., Carlsbad, CA) to verify the colony forming units (cfu).
For rapid incubation experiments, either 75 μL or 200 μL of prepared cultures were pipetted into each chamber of the disc or 96-well plate and placed in the appropriate spinstand incubator or shaker incubator. Bacteria was incubated at 37 C and oscillated at 1 Hz through an angle of 180 on the spinstand or 400 rpm on the tabletop shaker. Sample was collected for plating or lysed for Luminex analysis at different time stamps, 0 min, 60 min and 90 min to be specific. Serial dilutions (10−6) were performed prior to triplicate plating and were incubated overnight prior to counting the following day. For Luminex readings, 70 μL of sample was removed from the chambers and mixed with 35 μL of 1 M NaOH for 5 minutes. The sample was neutralized by adding 105 μL of phosphate buffer solution. 150 μL of neutralized lysate was collected and characterized using a Luminex MagPix assay instrument with custom capture probes designed to hybridize with oligonucleotides on Luminex MagPlex-TAG microspheres.
In certain growth experiments, cultures were spiked with one of the following antibiotics diluted in (MH2) broth before incubation in the disc: 4 μg ml−1 ciprofloxacin (Sigma, St. Louis, MO), 32 μg ml−1 ceftriaxone, 512 μg ml−1 ampicillin, 16 μg ml−1 nitrofurantoin, or 64 μg ml−1 cefazolin.13
For cell lysis experiments, bacteria was spun down from the incubation chamber into the cell lysis chamber following sample entry.
2 Results and discussion
2.1 Microfluidic CD design and operation
The centrifugal force, shown in eqn (1), acts radially outward, providing unidirectional liquid pumping. The many chambers and channels milled into the disc create barriers to fluid flow. A narrow channel coming out of a chamber creates a pressure barrier that must be overcome before fluid can pass. By rotating the disc above a critical speed, the centrifugal force can overcome the pressure force and the fluid is free to burst to the next chamber.
 | (1) |
The Euler force, perpendicular to the centrifugal force, is proportional to the change in angular velocity per unit time and can be used to generate vortical flow and provide uniform mixing within a microfluidic chamber. The Euler force, per unit volume, is expressed in eqn (2)
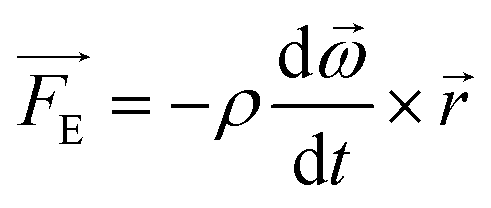 | (2) |
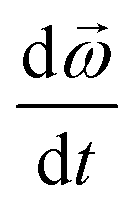 is the change in the angular velocity per unit time, and
is the change in the angular velocity per unit time, and ![[r with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0072_20d1.gif) is the mean distance of the liquid from the center of the disc.
is the mean distance of the liquid from the center of the disc.
This force dominates when the disc is subject to changes in velocity, such as cycles of unidirectional acceleration-and-deceleration or changes in the direction of rotation. Turbulent mixing, and bacterial incubation, is thus dependent on chamber geometry (height and inner and outer chamber radii, r1 and r2 in Fig. 3), acceleration/deceleration rate, and angular span of the mixing chamber.20 Oscillation is accomplished by cycling between two angles of the disc at a specific frequency. This motion takes advantage of the Euler force to create chaotic advection within the sample, effectively mixing it.
Table 1 demonstrates the fluidic sequence for the microfluidic disc. To simplify sample loading, we prototyped a world-to-chip (WTC) interface to dispense liquid into the first chamber of each section of the disc simultaneously. The sample is loaded into the main compartment, designed to store liquid buffer, and the centrifugal disc is secured using the self-sealing “top-piece”. Baffles were incorporated at the bottom of the WTC to improve mixing during oscillation. By spinning at a critical RPM (>5000), the centrifugal force drives the mixed sample up the inner walls of the WTC and through the outlet holes connected to each incubation chamber. Metering is accomplished by taking advantage of chamber geometry and having overflow channels, shown in Fig. 3. The volume of the sample entry chamber dictates the amount of sample used in the disc and the rest runs off to the waste chamber via an overflow channel on the bottom of the disc.
| Step | Protocol | Details |
|---|---|---|
| Incubation | Oscillation | 2 Hz, 180° |
| Valve 1 | Laser + spin | 2000 RPM |
| Cell lysis | Spin | 200 RPM |
| Valve 2 | Laser + spin | 2000 RPM |
| Neutralization | Oscillation | 2 Hz, 180° |
| Valve 3 | Laser + spin | 2000 RPM |
| Hybridization | Oscillation | 2 Hz, 180° |
| Valve 4 | Laser + spin | 4000 RPM |
| Wash 1 | Spin | 1000 RPM |
| Wash 2 | Spin | 1000 RPM |
| Detection | Position + camera | 0 RPM |
An oscillation protocol is used to incubate bacteria once in the incubation chamber (Fig. 3). The incubation chamber is subject to cycles of rapid acceleration/deceleration to generate vigorous mixing. The robust mixing provides even distribution of macromolecules (e.g. antibiotics and nutrients) and ensures sufficient sample aeration, required for bacterial growth. Oscillation is also used in the neutralization chamber to resuspend dried down phosphate buffer.
The lysis chamber utilizes a combination of mechanical and chemical lysis to effectively release genomic data from a number of bacterial pathogens. As the microfluidic disc is rotated at a low angular velocity (<500 RPM), the metal disc interacts with the array of permanent magnets, moving back and forth in the lysis chamber and mixing the sample with the silica–zirconia beads and the chemical lysis reagent (1 M NaOH). The mechanical forces generated between the metal disc and beads provide shear forces that break open cell walls.
The neutralization chamber contains dried down PBS and fluorescently labeled detector probes, which are resuspended during oscillation of the disc. The PBS neutralizes the NaOH from the lysis chamber, preventing degradation of rRNA sequences. At this point, the sample can be either removed from the disc, for detection on a separate system (e.g. Luminex Mag-Pix or other fluorescent microscope), or can continue on for hybridization and detection directly in the disc if a microarray is inserted into the detection chamber.
The hybridization and detection chamber contains a sticky adhesive on one side, allowing a glass or polymer slide functionalized with detector probes (microarray) to snap into the back of the disc. A 9 × 5 mm window exposes the detector probes to the rest of the chamber. Following neutralization, neutralized lysate slowly and completely fills up the chamber. Diffusion, combined with oscillatory mixing, allows rRNA to hybridize and bind with the complementary detector probes. Following hybridization, two wash buffers are released, flowing through the chamber to wash away any unbound rRNA into a waste chamber, eliminating false positive results. After these washing steps, the detection chamber is imaged using the camera and fluorescent lenses present in the SpinBox.
A series of active valves are used as gates between different chambers connected by narrow fluidic channels, see Fig. 4. Since the inner feature disc is fabricated from a black COC polymer, these valves are made by milling half of a channel on one side of the feature disc, and the subsequent half on the other side of the disc, leaving a 0.25 mm thick piece of COC overlapping the two half-channels, see Fig. 4a. To open a valve, a laser is focused on the valve position (Fig. 4b), melting away the plastic covering (Fig. 4c and allowing free flow of liquid between the adjacent half-channels (Fig. 4d). The energy required for opening each valve is nearly 1 Joule, estimated using eqn (3).
| Qtot = QT + QF + QV = mcpΔT + mLF + mLV | (3) |
 | ||
| Fig. 4 The sequence for opening one of the six laser valves on the disc is shown: a) liquid in channel 1 (top side of disc and located at smaller radius than channel 2) is blocked from passing to channel 2 (bottom side of disc and located at larger radius than channel 1) by the black COC polymer, b) a laser is focused on the valve location, c) the COC polymer melts, creating an opening between channel 1 and 2, and d) liquid is free to flow from channel 1 to channel 2. | ||
2.2 Universal cell lysis
The bacteria cell wall has evolved to become one of the most resilient barriers against harsh environmental conditions filled with toxic contaminants. In fact, the origin of antibiotic resistance stems from this evolutionary response. For example, Gram-negative bacteria are naturally more resilient to antibiotics than Gram-positive bacteria due to their dual-layered cell envelope. The additional outer-layer, absent in Gram-positives, consists of an asymmetric lipid membrane formed by exterior-facing lipopolysaccharides and interior-facing phospholipids.Due to the thicker cell wall of Gram-positive bacteria, a combination of mechanical and alkaline chemical lysis is needed to release rRNA from cells. The mechanical component of our lysis system helps puncture and tear open the tough outer cell membrane of Gram-positive bacteria by causing shear and friction forces between the cell wall and 100 μm sized silica/zirconia beads (Fig. 5b). Once the cell wall is obliterated, NaOH denatures the ribosomal complex, allowing the 16 s rRNA fragment to be recovered (Fig. 5c). Finally, the sample is neutralized with PBS to prevent rRNA hydrolysis by prolonged NaOH exposure (Fig. 5d).
 | ||
| Fig. 5 Universal lysis schematic. a) the thick cell wall of Gram-positive bacteria means both chemical and mechanical lysis are required, b) mechanical lysis with metal discs and silica/zirconia beads punctures cell walls, releasing genomic data, c) alkaline lysis (1 M NaOH) denatures ribosomal complex, allowing target rRNA fragmets to be recovered, d) PBS neutralizes NaOH, preventing further RNA hydrolysis. | ||
To optimize the lysis protocol for bacteria, we varied a number of parameters, including lysis time, spin speed of the disc (which determined the oscillation of the metal disc in the lysis chamber), silica/zirconia bead slurry amount (which determined the amount of grinding material for mechanical lysis), and lysis puck geometry. The results are shown for Gram positive isolate Staphylococcus aureus (SA15-21-08) in (Fig. 6) and expressed as a percentage of the Luminex signal obtained using the Omnilyse commercial lysis system. As a control, chemical lysis (1 M NaOH) is shown as well. The results of the optimization experiments were used to determine an optimal universal lysis system, with characteristics summarized in Table 2.
| Characteristic | Optimal value |
|---|---|
| Lysis time | 6 min |
| Spin speed | 100 RPM |
| Bead amount | 120 μL |
| Puck geometry | Double |
 | ||
| Fig. 6 Optmizing lysis efficiency. Both chemical lysis and chemical + mechanical lysis of E. coli were tested against the Omnilyse cartridge for a series of parameters. All measurements are shown as a percentage of the omnilyse cartridge signal, which has been normalized to 100% (dotted green line). a) Total lysis time was varied from 4–10 minutes. b) Total spin RPM was varied from 50–400 RPM. c) Total silica/zirconia bead slurry was varied from 90–120 μL pipetted into the lysis chamber. d) Several puck geometries were tested, including a single or double thin steel disc, a magnetic stir bar, or a spherical steel disc. | ||
Finally, to demonstrate our universal lysis system, we lysed several Gram-positive bacteria, including 2 strains of Staphylococcus aureus (SA), a strain of coagulas negative staphylococcus (CNS), a strain of Streptococcus agalactiae (Sagal), and a strain of Enterococcus (EFs), see Fig. 7. While lysis efficiency on the disc varied, all strains showed lysis efficiency at least 75% of the Omnilyse system.
 | ||
| Fig. 7 6 Gram positive isolates were compared with chemical lysis alone and chemical + mechanical lysis, including 2 Staph. aureus (SA), 1 coagulas negative staphylococcus (CNS), 1 Strep. agalactiae (Sagal), and 1 Enterococcus (EFs). All measurements are shown as a percentage of the omnilyse cartridge signal, which has been normalized to 100% (dotted green line). | ||
2.3 Rapid bacteria incubation
For liquid bacterial cultures, rapid and healthy growth depends on three main parameters: 1.) sample aeration: a bacteria sample needs to have plenty of access to fresh oxygen for growth, 2.) nutrient availability: a sample needs to be thoroughly mixed in order to provide nutrients equally to all parts of the culture, and 3.) prevention of biofilms and clumping: Shaking and agitation can prevent bacteria culture from settling to the bottom of a chamber and forming biofilms or clumps that hinder reproduction.The low Reynold numbers in microfluidic systems exhibit laminar flow regimes, which are dominated by viscous, rather than inertial forces. The absence of turbulence makes healthy and rapid bacterial growth particularly challenging and forces microfluidic devices to rely on either passive molecular diffusion or external energy sources for mixing.21,22 Furthermore, the small enclosed volumes characteristic of microfluidic systems restricts access to fresh oxygen, making sample aeration difficult without bulky or complex external pumps that bubble oxygen through a sample.23
To optimize incubation, while keeping within the available real estate of the portable disc, the incubation chamber characteristics in Table 3 were chosen. The angular span was maximized while still keeping 6 multiplexed sections on the disc, and r1 and r2 were restricted by the need for fitting all assay steps onto the chosen disc radius of 60 mm. The incubation chamber was also milled completely through the inner feature disc, allowing for a large surface area of the chamber tobe covered by a gas-permeable membrane.
| Characteristic | Value |
|---|---|
| Inner radius (r1) | 10 mm |
| Outer radius (r2) | 15 mm |
| Acceleration/deceleration rate | 240 rad s−1 |
| Angular span (θI) | 45° |
We compared the growth using our chosen CD chamber design and oscillation protocol to cultures grown in a 96-well plate and CD placed into a standard incubator shaker (BRAND HERE) at 400 RPM. Observation of fluid within the different configurations showed more turbulence and advection in the disc in the SpinBox than in either the 96-well plate or disc in the SpinBox. Fig. 8 shows a significant increase in rRNA growth of the disc on the spinstand at both 60 and 90 minutes compared with the other configurations. At 90 minutes, an average 162% increase in rRNA was observed in the incubator spinstand compared with a 96-well plate on the plate shaker incubator. The incubator disc in the SpinBox showed an average 122% increase in rRNA after 90 minutes compared to the 96-well plate, demonstrating that both the type of mixing and aeration have an effect on bacterial growth.
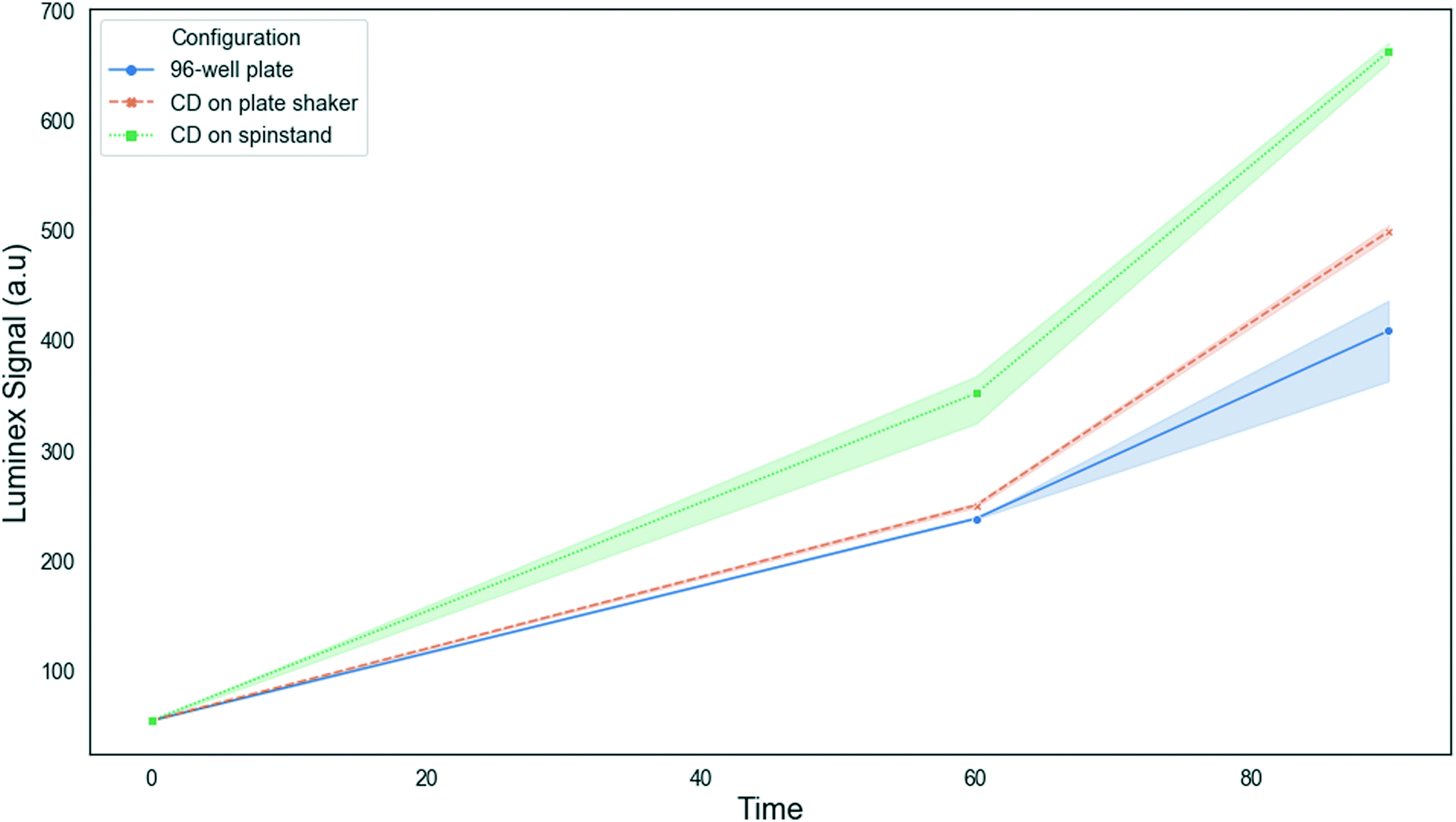 | ||
| Fig. 8 Comparing E. coli growth on the 96-well plate in the plate shaker incubator, the disc in the plate shaker incubator, and the disc on the incubator spinstand. Growth is shown as luminex signal intensity. Shading represents error bars. | ||
To ascertain that the effect was not isolated to a single E. coli strain, we compared growth on the disc and the 96-well plate in 14 different strains of E. coli, see Fig. 9. The graph compares cfu per mL on the disc (y-axis) vs. the 96-well plate (x-axis). The red dotted line has a slope of 1 (y = x) and represents bacteria growing equally on the plate and on the disc. All data points above the red dotted line represent strains that grew better on the disc and data points below the red dotted line represent strains that grew better on the 96-well plate. The majority of the isolates tested showed more than 2× better growth on the disc than the 96-well plate. There were 2 strains of E. coli that showed worse growth on the disc than on the 96-well plate. This might be either an outlier or might represent bacterial strains that favor alternative growth conditions. However, despite the growth characteristics of these E. coli strains, the measured cfu/mL, compared with the 0 minute time point, is sufficient to test antibiotic susceptibility.
 | ||
| Fig. 9 Growth for 14 different strains of E. coli in the disc on the incubator spinstand are compared with growth on the 96-well plate in the plate shaker incubator. The dotted red line (y = x) has a slope of 1, with data points above it showing greater growth on the disc and data points below it showing greater growth on the plate. Growth is shown in colony forming units per milliliter (cfu per mL). | ||
2.4 Antibiotic susceptibility testing
To demonstrate our system was efficient in determining antibiotic susceptibility, 11 different E. coli isolates were incubated with a total of five commonly prescribed, broad-spectrum antibiotics using the microfluidic disc and compared to the 96-well plate. We chose three antibiotics whose mechanism of action inhibits the bacterial cell wall (ampicillin, cefazolin, and ceftriaxone) and two antibiotics that target bacteria DNA functions, thereby preventing replication (nitrofurantoin and ciprofloxacin).Fig. 11 shows the ratio from the rRNA signals of bacteria incubated with and without antibiotics (Abx/No Abx) for both the disc (Fig. 11a) and the plate (Fig. 11b). If the ratio is large (near or above 1), the bacteria is resistant since its growth is similar to the no antibiotic control, see Fig. 10. If the ratio is small (closer to or below 0), the bacteria is susceptible since its growth is suppressed by the antibiotic and much less than the no antibiotic control, see Fig. 10. The red and blue lines in Fig. 11 show the average of all resistant or susceptible bacteria on either the disc or plate (error shown as a gradient). While both the plate and the disc showed significant differences in average ratios between the resistant and susceptible bacteria (p < 0.01), the resistant bacteria showed higher average ratios on the disc than the plate (p < 0.05) indicating an improved ability to distinguish antibioticsusceptibility on the disc.
 | ||
| Fig. 10 Conceptual drawing showing how the ratio is calculated for each signal. The ratio of the signal from the bacteria incubated with antibiotics to the positive control, where the same bacteria is incubated without antibiotics, is close to one for resistant bacteria and close to 0 for susceptible bacteria. | ||
 | ||
| Fig. 11 Ratios of signal intensity of each bacteria incubated with and without antibiotics for the disc (a) and the plate (b). The blue and red lines represent the average of all ratios for resistant (red) and susceptible (blue) bacteria. | ||
In Fig. 12, an average ratio for the response of multiple bacteria to each antibiotic was calculated. In all bacteria/antibiotic combinations, the average ratios from the disc incubation were comparable to those on the 96-well plate. However, on the disc, susceptible bacteria incubated with antibiotics showed very low uniform average ratios, with no statistically significant differences between antibiotic effectiveness. Interestingly, on the plate, susceptible bacteria exposed to the three antibiotics that target the cell wall (ampicillin, cefazolin, and ceftriaxone), showed a lower average ratio than those exposed to antibiotics that target bacterial DNA functions (nitrofurantoin and ciprofloxacin) (<0.05). This uncertainty could contribute to false positives when interpreting susceptibility data. Therefore, results from the disc incubation provide clinicians more confidence in their prescription of antibiotics.
 | ||
| Fig. 12 Average ratio of signal intensity of E. coli strains incubated with and without each antibiotic. The graph on the right shows ratios for resistant and susceptible bacteria grown on the plate and the left shows resistant and susceptible bacteria grown on the disc. | ||
3 Conclusion
We demonstrated a never reported method for enhancing bacteria incubation on a microfluidic platform capable of determining bacterial response in the presence of five antibiotics. Robust mixing of a liquid bacteria sample in the microfluidic chamber on the CD provides rapid growth by ensuring adequate sample aeration, nutrient dispersion, and prevention of biofilm formation. The well mixed sample is capable of determining bacterial response to antibiotics quicker compared to the off-chip incubation when using rRNA as the detection mechanism. The design of the incubation chamber on the CD was optimized to include further downstream sample processing, including a universal Gram positive and negative cell lysis system, capable of lysing 6 different types of Gram positive and negative bacteria, interchangeable microarray detection chamber, two washing chambers, and waste chamber. The CD was integrated with a custom designed SpinBox capable of automating all sample processing steps after the initial sample dilution in the world-to-chip interface. The system serves as a powerful step towards improving rapid AST PoC diagnostics by minimizing sample handling. In reality, the rapid incubation on a microfluidic CD can be adapted and combined with a variety of unexplored detection mechanisms to further improve AST.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the financial support provided by MicrobeDX, CARB-X, and SBIR (R43AI127015) for making this work possible. The authors also thank RapidTech at UC Irvine for use of their facilities for prototyping instrument housing and electronic circuits as well as Microfluidic Chipshop for helping with the design and fabrication of the injection molded microfluidic discs used in this report.Notes and references
- A. R. Coates, G. Halls and Y. Hu, Br. J. Pharmacol., 2011, 163, 184–194 CrossRef CAS.
- P. R. Shankar and R. Balasubramanium, Med. J. Aust., 2014, 7, 237 Search PubMed.
- R. O. A. Resistance, Antimicrobial resistance: tackling a crisis for the health and wealth of nations, Review on Antimicrobial Resistance, 2014 Search PubMed.
- C. L. Ventola, P T, 2015, 40, 277–283 Search PubMed.
- T. P. M. Editors, PLoS Med., 2016, 13, 1–3 Search PubMed.
- A. Sundsfjord, G. S. Simonsen, B. C. Haldorsen, H. Haaheim, S.-O. Hjelmevoll, P. Littauer and K. H. Dahl, APMIS, 2004, 112, 815–837 CrossRef CAS.
- M. Davenport, K. E. Mach, L. M. D. Shortliffe, N. Banaei, T.-H. Wang and J. C. Liao, Nat. Rev. Urol., 2017, 14, 296–310 CrossRef.
- S. Kim, F. Masum, J.-K. Kim, H. J. Chung and J. S. Jeon, Lab Chip, 2019, 19, 959–973 RSC.
- S. Brosel-Oliu, O. Mergel, N. Uria, N. Abramova, P. V. Rijn and A. Bratov, Lab Chip, 2019, 19, 1436–1447 RSC.
- G. Pitruzzello, S. Thorpe, S. Johnson, A. Evans, H. GadÃłlha and T. F. Krauss, Lab Chip, 2019, 19, 1417–1426 RSC.
- H. Li, P. Torab, K. E. Mach, C. Surrette, M. R. England, D. W. Craft, N. J. Thomas, J. C. Liao, C. Puleo and P. K. Wong, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10270–10279 CrossRef CAS.
- R. Mohan, K. E. Mach, M. Bercovici, Y. Pan, L. Dhulipala, P. K. Wong and J. C. Liao, PLoS One, 2011, 6, e26846 CrossRef CAS.
- C. Halford, R. Gonzalez, S. Campuzano, B. Hu, J. T. Babbitt, J. Liu, J. Wang, B. M. Churchill and D. A. Haake, Antimicrob. Agents Chemother., 2013, 57, 936–943 CrossRef CAS.
- K. Zwirglmaier, W. Ludwig and K.-H. Schleifer, Mol. Microbiol., 2004, 51, 89–96 CrossRef CAS.
- M. Ouyang, R. Mohan, Y. Lu, T. Liu, K. E. Mach, M. L. Y. Sin, M. McComb, J. Joshi, V. Gau, P. K. Wong and J. C. Liao, Analyst, 2013, 138, 3660–3666 RSC.
- H. Stender, M. Fiandaca, J. J. Hyldig-Nielsen and J. Coull, J. Microbiol. Methods, 2002, 48, 1–17 CrossRef CAS.
- K. E. Mach, C. B. Du, H. Phull, D. A. Haake, M.-C. Shih, E. J. Baron and J. C. Liao, J. Urol., 2009, 182, 2735–2741 CrossRef CAS.
- M. Brown, R. Doebler, B. Irvine, T. Ferguson and G. Blackburn, US Pat., 8,663,974, 2014 Search PubMed.
- E. K. Heiniger, J. R. Buser, L. Mireles, X. Zhang, P. D. Ladd, B. R. Lutz and P. Yager, J. Microbiol. Methods, 2016, 128, 80–87 CrossRef CAS.
- L. X. Kong, A. Perebikovsky, J. Moebius, L. Kulinsky and M. Madou, J. Lab. Autom., 2016, 21, 323–355 CrossRef.
- K. Ward and Z. H. Fan, J. Micromech. Microeng., 2015, 25, 094001 CrossRef.
- H. E. H. Meijer, M. K. Singh, T. G. Kang, J. M. J. D. Toonder and P. D. Anderson, Macromol. Symp., 2009, 279, 201–209 CrossRef CAS.
- M. D. Brennan, M. L. Rexius-Hall, L. Jane Elgass and D. T. Eddington, Lab Chip, 2014, 14, 4305–4318 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0lc00838a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |