
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplications of hydrogen as a collision and reaction cell gas for enhanced measurement capability applied to low level stable and radioactive isotope detection using ICP-MS/MS
B.
Russell
 *a,
S. L.
Goddard
b,
H.
Mohamud
a,
O.
Pearson
a,
Y.
Zhang
c,
H.
Thompkins
a and
R. J. C.
Brown
bd
*a,
S. L.
Goddard
b,
H.
Mohamud
a,
O.
Pearson
a,
Y.
Zhang
c,
H.
Thompkins
a and
R. J. C.
Brown
bd
aNuclear Metrology Group, National Physical Laboratory, Hampton Road, Teddington, UK. E-mail: ben.russell@npl.co.uk
bAir Quality and Aerosol Metrology Group, National Physical Laboratory, Hampton Road, Teddington, UK
cElectrochemistry Group, National Physical Laboratory, Hampton Road, Teddington, UK
dDepartment of Physics, University of Surrey, Guildford GU2 7XH, Surrey, UK
First published on 9th November 2021
Abstract
Tandem inductively coupled plasma mass spectrometry (ICP-MS/MS) with collision/reaction cell capability can achieve highly effective separation of spectral interferences. This improves confidence in measurement, reduces the detection limits achievable and expands the number of isotopes measurable in a range of industries, including semi-conductor, pharmaceutical, environmental, food and nuclear. Amongst the range of cell gases tested, hydrogen (H2) has proven to be an effective cell gas, either used alone or in combination with other gases. This study demonstrates the benefits of using H2 in combination with ICP-MS/MS for improved detection of stable and radioactive pollutants of interest to a number of technical disciplines. This is presented through several case studies considering stable (35Cl, 40Ca, 56Fe, Ni isotopes) and radioactive (36Cl, 41Ca, 63Ni and 93Mo) isotopes in stable element standards and sample matrices including air quality filters and aqueous nuclear decommissioning waste.
1. Introduction
ICP-MS is a routine analytical technique for a range of sectors, capable of multi-element detection, measurement times of only several minutes per sample and detection limits in the fg g−1 range. The flexibility of the instrument design means that measurement of gaseous, solid and, most frequently, aqueous samples is possible. In all cases, accurate measurement is dependent on the removal of several interferences. Firstly, isobaric overlap from an isotope of another element at a very similar mass to the analyte, which cannot be resolved by the detector. Secondly, polyatomic interferences formed by a combination of two or more isotopes in the high temperature plasma, with the product formed again having a very similar mass to the analyte. Finally, tailing is caused by an isotope at a significantly higher concentration one or two mass units either side of the analyte. Removal of these interferences is commonly achieved using chemical separation techniques such as solvent extraction, precipitation, ion exchange and/or extraction chromatography.As ICP-MS has developed, instrument-based interference removal capability has improved, reducing or even removing the need for relatively time consuming offline chemical separation prior to measurement. One such option is using a collision and/or reaction gas in a pressurised cell for selective removal of isobaric and polyatomic interferences. Collision–reaction cells are a powerful tool for removal of spectral interferences in quadrupole ICP-MS instruments. A reaction gas is used to remove the analyte from known interferences. The likelihood of the reaction proceeding depends on the enthalpy of reaction between the gas and the elements involved. If the enthalpy of reaction is negative (exothermic), the reaction will occur spontaneously when the element and gas meet in the cell, whilst a positive enthalpy of reaction (endothermic) means energy is required and the reaction is less likely to proceed.1 The bond energies and ionisation energies of the reactants and products can be used to calculate the enthalpy of reaction.
A collision gas selectively removes polyatomic interferences based on their larger size compared to the analyte of the same mass. The gas will collide more frequently with polyatomic ions due to their larger collisional cross section, resulting in higher energy losses relative to the analyte. A bias voltage at the cell exit excludes these low kinetic energy polyatomics from entering the quadrupoles whilst the majority of the analyte signal is retained thus dramatically improving the detection limits achievable for the analytes of interest. The cell gas flow rate and bias voltage must be optimised for interference removal and analyte sensitivity for each application.1
The commercial availability of tandem mass spectrometry (ICP-MS/MS) has been proven to expand measurement capabilities in fields including semiconductor, pharmaceutical, environmental monitoring, and nuclear decommissioning. The additional quadrupole mass filter improves tailing removal compared to single quadrupole designs, as well as filtering the ion beam prior to the collision/reaction cell, simplifying understanding of the cell chemistry.1 The collision–reaction cell can accommodate a range of cell gases (e.g. H2, O2, NH3, CH4 and CH3F) to support or even replace relatively time-consuming offline chemical separation. ICP-MS/MS has expanded the number of isotopes measurable as well as reducing measurement uncertainty and the detection limits achievable for elements that suffer from significant interferences.
Hydrogen has been applied effectively as a cell gas for interference reduction. Eiden et al. proposed H2 as a method for selective removal of Ar and other plasma matrix ions, noting significant Ar suppression due to charge transfer from Ar+ to form low m/z ions H2+ and H3+.2 The same authors noted the suitability of H2 in efficiently reducing the Ar signal to enable measurement of analytes including 40Ca and 40K (40Ar isobar), 56Fe (40Ar16O polyatomic) and 80Se (40Ar2 dimer).3 Several interference removal mechanisms have been proposed, including hydrogen atom transfer (eqn (1)), proton transfer (eqn (2)) and charge transfer (eqn (3)).4,5Table 1 gives some examples of elements that have benefitted from the use of H2 as a cell gas.
| Ar+ + H2 → ArH+ + H | (1) |
| ArH+ + H2 → H3+ + Ar | (2) |
| Ar+ + H2 → H2+ + Ar | (3) |
| Sample matrix | Analyte(s) | Interference(s) | Measured ion | Sample preparation | Limit of detection (pg g−1) | Reference |
|---|---|---|---|---|---|---|
| Ultrapure water | 40Ca | 40Ar | 40Ca | Acidified water | 0.041 (BEC) | 16 |
| Silicon wafer | 31P | 30SiH | 31PH4 | Silicon wafer dissolution in HF/HNO3 | 227 (BEC) | 9 |
| Titanium nanoparticles | 48Ti | 48Ca | 48Ti16O 48Ca16O1H | Dilution and sonication | ∼30 nm particle size | 11 |
| N-Methyl-2-pyrrolidone | 28Si 35Cl | 14N2, 12C16O16O18OH | 28Si 35ClH2 | Distillation and acidification | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 (BEC) 34200 (BEC) 800 (BEC) 34200 (BEC) |
10 |
| Gas standards e.g. silane, germane (contaminants in petrochemical and semiconductor) | 28Si | 14N2, 12C16O | 28Si | Balanced in Ar or H2, diluted in He GC-ICP-MS/MS | 0.009–0.2 | 17 |
| CRMs (leaves, pine needles, rice flour) | 111Cd | 95Mo16O | 111Cd | Microwave digestion and dilution | — | 11 |
| Ni alloy | Se isotopes | 40Ar38Ar, 62Ni16O (example for 78Se) | 78Se | None | 12.9 | 12 |
| Standard solutions, stainless steel, aqueous waste | 93Zr | 93Nb, 93Mo | 93Zr(NH3)6 | Dilution | 1.1–8.6 | 12 and 13 |
If used as a collision gas, there is a lower analyte energy loss using H2 compared to heavier He, improving sensitivity and potentially lowering the detection limits achievable.6 Hydrogen is known to work effectively for removal of argide based interferences, such as 38Ar1H on 39K, 40Ar12C on 52Cr and 40Ar40Ar on 80Se.5,7 In one example, 40Ar plasma gas was suppressed via a charge transfer reaction with H2 for measurement of 40Ca impurities in ultrapure water.8 This was supported by operating under cold plasma conditions (RF reduced to approximately 600 W compared to approximately 1300 W) to reduce the Ar signal due to its high ionisation energy, with the background equivalent concentration (BEC) calculated (0.041 pg g−1) as being two orders of magnitude lower than single quadrupole ICP-MS. Hydrogen has also been used as a reaction gas to measure 31P in the presence of high 30Si concentrations in Si wafers. The 31P signal was shifted to 31P1H4 whilst polyatomic 30Si1H remained on mass.9,10
Hydrogen has also been effectively used in combination with other reaction cell gases. In the case of improved interference removal for measurement of Ti nanoparticles, H2 was used in combination with O2 to measure Ti as 48Ti16O. The addition of H2 effectively shifted polyatomic 48Ca16O to 48Ca16O1H, whilst Ti remains as 48Ti16O. This is beneficial to a number of applications including paints, cosmetics and pharmaceuticals.11 The same combination of gases was also used for measurement of trace Se in Ni alloys. High concentrations of Se can lead to weakness in alloy composition, which is particularly significant in industrial applications where high temperature resistance is required e.g. turbine blades in aircraft. Argon-based interferences on Se isotopes (e.g.40Ar36Ar on 76Se and 40Ar38Ar on 78Se) can be removed using H2, whilst Ni matrix interferences (e.g.60Ni16O on 76Se and 62Ni16O on 78Se) can be removed using O2 to convert Se to SeO.12 The use of tandem ICP-MS/MS also removes water adducts that would otherwise enter the cell and lead to potential interferences such as 60Ni16O(H2O)+ on 78Se16O. As an example, 78Se instrument detection limits of 12.9 ng L−1 and 18.8 ng L−1 were achieved in H2 and O2 modes, respectively.
Iglesias et al. investigated multiple cell gases for removal of Ar-based interferences affecting measurement of Fe and Se.13 In both cases, a mixture of H2 and He returned the lowest detection limits, with values of 2 ng L−1 and 6 ng L−1 for 80Se and 56Fe, respectively. A mixture of gases was also used for separation of the long-lived radionuclide 93Zr from stable isobar 93Nb.14,15 Specifically, H2 was combined with NH3 to shift 93Zr to 93Zr(NH3)6, with no increase in background from 93Nb(NH3)6 at mass fractions up to 5 × 104 pg g−1, which was higher than the levels present in the stainless steel and aqueous waste samples measured. The presence of H2 increased the efficiency of cell product formation compared to using NH3 alone. Detection limits of 1.1 pg g−1 and 8.6 pg g−1 were calculated for steel and aqueous waste samples, respectively, which was several orders of magnitude below the regulatory limits.
This study demonstrates the combination of ICP-MS/MS with H2 for expanding the range of applicable measurement capabilities. Methods have been developed by the Air Quality and Aerosol Metrology, and Nuclear Metrology Groups at the National Physical Laboratory (NPL) for measurement of stable and radioactive isotopes. Several case studies are presented including for the measurement of new elements and improved sensitivity and detection limits when using H2 alone or in combination with other cell gases.
The focus for stable isotopes is the measurement of air quality filter samples, with the aim to develop methods with improved interference removal to (i) improve analyte sensitivity and detection limits, and (ii) enable isotope ratio measurement to aid source apportionment, focusing on Fe and Ni isotopes. NPL is the UK's National Metrology Institute and the current operator of the UK Metals Monitoring Network (hereafter referred to as ‘the Metals Network’) on behalf of the Environment Agency and the UK governmental Department for Environment, Food and Rural Affairs (Defra).18 The Metals Network consists of monitoring sites all around the UK that sample airborne particulate matter with an aerodynamic diameter less than 10 μm (PM10) onto filters. These filters are prepared for analysis by microwave acid digestion and analysed by ICP-MS for a suite of twelve metals.19 Results are reported to the EU to assess UK compliance with air quality legislation.20,21 For the current study, improvements to the determination of 56Fe were attempted. The development of a method to accurately measure all stable Ni isotopes was also investigated.
The focus for radioactive isotopes was to investigate radionuclides that must be accurately characterised in nuclear wastes and environmental samples to ensure the correct waste sentencing route, contributing to safe and cost-effective decommissioning and ensuring safety to the workforce and the public. ICP-MS/MS has proven a valuable part of the radioanalytical toolbox for rapid measurement of radionuclides including 90Sr, 93Zr, 129I, 135Cs/137Cs and 236U/238U.14,15,22–25 This study focuses on radionuclides that suffer from multiple spectral interferences that may benefit from the interference removal capabilities offered by ICP-MS/MS: 36Cl, 41Ca, 63Ni and 93Mo.
2. Experimental
2.1. Reagents and materials
Stable element standards (100–1000 μg g−1, Fluka Analytical) were used for initial instrument tuning. For Cl, calibration standards were prepared from gravimetric dilution of hydrochloric acid (Fisher Scientific, Trace Analysis Grade). Single radionuclide standards were provided by the NPL Nuclear Metrology Group and diluted to the required activity concentrations in a dedicated radioactive source preparation facility. All standards were prepared in nitric acid (Fisher Scientific, Trace Analysis Grade) diluted to 2% (v/v) in deionised water obtained using an ELGA purelab flex water purification system (ELGA, Veolia Water, Marlow, UK, 18 MΩ cm, 5 ppb Total Organic Carbon).Iron-56 was measured in a set of five calibration standards (with an approximate range of 1–10 ng g−1) and a matrix-matched acid blank (1% HNO3). The following isotopes were measured in a calibration standard containing 5 ng g−1 Fe, Ni, Cu, Zn and a matrix-matched acid blank (1% HNO3): 57Fe,58Ni, 60Ni, 63Cu, 64Ni and 66Zn.
Nickel-63 was measured in aqueous waste samples provided by Sellafield Ltd. Stable 40Ca and radioactive 41Ca was measured in several different concrete samples: inactive blank concrete prepared at NPL, bio-shield concrete obtained from SCK-CEN (Belgium), and 41Ca-spiked concrete from a European Metrology Research Programme project ‘Metrology for Radioactive Waste Management’ (MetroRWM).26 Additionally, 41Ca/40Ca standard solutions (ERM-AE701) supplied by the Institute of Reference Materials and Measurements (IRMM) (Geel, Belgium) were measured, with 41Ca/40Ca ratios ranging from 10−6 to 10−13.
2.2. Instrumentation
Experimental work was carried out using an Agilent 8800 and Agilent 8900 ICP-MS/MS. Compared to the Agilent 8800, the newer generation 8900 benefits from an upgraded collision/reaction cell and the capability to apply an axial acceleration voltage to the octopole rods in the reaction cell. This increases the transmission rate and sensitivity for product ions. This is especially useful when analyte ions lose energy due to collisions/reactions with cell gases, as the product ions can be re-accelerated and sensitivity regained, potentially improving sensitivity and detection limits.The instruments are both equipped with two quadrupole mass filters (Q1 and Q2) separated by a collision–reaction cell and fitted with a quartz double-pass spray chamber, MicroMist nebuliser (Glass Expansion) and the X-lens setup. The 8800 utilised nickel sample and skimmer cones; the 8900 was fitted with platinum tipped cones (Crawford Scientific). Four cell gas lines were fitted to each instrument, connected between the supply and the instrument by approximately 1 m of 2.1 mm internal diameter stainless steel tubing: dedicated hydrogen and helium lines, a corrosive gas line (10% NH3 balanced in 90% He) and a non-corrosive line (O2). All cell gases and Ar plasma gas were provided by BOC with a purity of N6.0. Hydrogen was supplied to both instruments from a Linde NM Plus generator. To reduce the risk of high water and O2 content from the use of a generator, an Agilent Gas Clean Filter was fitted between the supply and the instrument.
The instruments were conditioned with the cell gases of interest overnight prior to use to ensure the gas lines were fully purged. The instrument was tuned daily in Single Quadrupole mode (only Q2 operating) using a 1 ng mL−1 standard solution consisting of Be, Y, Ce and Tl in 2% (v/v) HNO3. The sensitivity and repeatability were assessed at low (9Be), medium (89Y) and high (205Tl) mass, whilst the CeO and doubly charged Ce formation was measured as the 156/140 and 70/140 ratio. Each monitored element had to reach a threshold sensitivity with an uncertainty <5%, and oxide and doubly charged formation had to be less than 2%. The 8900 instrument was conditioned and tuned in the same way, with the exception that the 1 ng mL−1 standard solution contained 7Li for low mass tuning.
2.3. Methodology
For all applications, the ramp cell gas function was used to give an indication of the optimal gas flow rate with regards to interference removal and analyte sensitivity. When using NH3 as a reaction gas, the product ion scan function was used to assess potential cell products. In this setting, Q1 is set to a single mass and Q2 scans the entire mass range. This setup simplifies understanding of the cell chemistry and facilitates the identification of the various NH-based products that can form. For both the ramp cell gas and the product ion scan, the instrument background was first assessed using blank samples, with any cases of elevated background discussed in Section 3.For all studies, the instrument background, analyte sensitivity, (BEC) and detection limit (LOD) were considered. The BEC is the instrument blank response expressed as a concentration, calculated by dividing the counts per second (CPS) in the blank by the sensitivity determined from a calibration curve. The BEC is monitored as part of the ramp cell gas function described above, highlighting the change in sensitivity and the blank as a function of cell gas flow rate. The LOD is calculated as the equivalent concentration of three times the standard deviation of the instrument blank.
For all applications, initial testing was carried out using single element standards, followed by mixed standards of the analyte and potential interferences. After daily tuning, the impact of operating in Single Quad and MS/MS mode (both Q1 and Q2 operating) on analyte sensitivity and interference removal was investigated, as was the impact of different collision and reaction cell gases. The instrument was operated in custom tune mode, enabling users to modify conditions, focusing in this study on the cell gas flow rate, as well as the cell energy discrimination and octopole bias voltages.
For radionuclide applications, the optimised method was validated where possible using real samples. Aqueous nuclear waste samples were measured as received, focusing on detection of 63Ni. Calcium-41 was measured in concrete samples following borate fusion dissolution and multi-stage radiochemical separation.27 In the absence of an active tracer 95Mo was investigated as a stable analogue of 93Mo.
3. Results and discussion
3.1. Impact of H2 on analyte sensitivity and instrument background
An increase in H2 flow rate reduced the instrument background, shown for several masses in Fig. 1. In agreement with previous studies (eqn (1)–(3) and Table 1), H2 was effective in reducing the 40Ar background at m/z = 40, with the signal dropping by a factor of 2 × 105 from 7.3 × 107 CPS at 1 mL min−1 to 400 CPS at 10 mL min−1. Significant reductions in background were also observed at m/z = 29 (28Si1H and 28Si) by a factor of 4 × 105 to <5 CPS at 10 mL min−1 over the same flow rate range. At m/z = 37 (37Cl), the signal was reduced from 7.4 × 105 CPS to <5 CPS. | ||
| Fig. 1 Impact of H2 flow rate on instrument background (data shown for Agilent 8800). | ||
A multi-element standard solution was run at H2 flow rates from 1–10 mL min−1 at 1 mL increments to assess the impact of flow rate on sensitivity. Fig. 2 and 3 shows the impact of collisional focusing, where an increase in H2 flow rate can focus the analyte ions closer to the centre of the beam, improving sensitivity. As the flow rate increases beyond the optimal level, scattering of the ion beam reduces the sensitivity. As the analyte mass increases, the optimum flow rate for collisional focusing also increases. This is because heavier ions are already closer to the centre of the beam than lighter ions and can tolerate higher cell gas flow rates. For example, the highest sensitivity for 24Mg is recorded at a H2 flow rate of 2 mL min−1, increasing to 6 mL min−1 for 175Lu and 8 mL min−1 for 238U. The results highlight the importance of custom tuning the cell gas flow rate for each analyte.
 | ||
| Fig. 2 Impact of H2 flow rate on analyte sensitivity. Grey bars show the H2 flow rate corresponding to peak sensitivity at different mass-to-charge ratios (m/z). | ||
 | ||
| Fig. 3 Correlation between hydrogen flow rate at peak sensitivity and mass-to-charge (m/z) ratio. | ||
The remainder of this section focuses on several case studies (Table 2) where H2 has been effectively used alone or in combination with another reaction gas to reduce the impact of spectral interferences.
| Analyte | Instrument (Agilent 8800/8900) | Q1/Q2 value | H2 flow rate (mL min−1) | Other cell gases (mL min−1) | Sensitivity (CPS per ng g−1) | Instrument detection limit (pg g−1) |
|---|---|---|---|---|---|---|
| a Sensitivity and limit of detection calculated from stable analogues. | ||||||
| 36Cl | 8800 | 36/38 | 3.5 | — | 100 | 81.2 |
| 41Ca | 8800 | 41/41 | 5.0 | NH3 (7.5), He (1.0) | 1300 | 99.4 |
| 56Fe | 8800 | 56 (Single Quad) | 5.5 | — | 20300 |
400 |
| 8900 | 2.0 | He (3.0) | 24800 |
192 | ||
| Stable Ni | 8900 | 58/109 | 6.5 | NH3 (4.0), He (1.0) | 77600 |
4.6 |
| 60/111 | 33200 |
4.8 | ||||
| 63Ni | 8800 | 63/114 | 3.0 | NH3 (1.0), He (1.0) | 6100 | 0.3 |
| 93Moa | 8800 | 93/127 | 3.5 | NH3 (4.3), He (1.0) | 1100 | 45.6 |
3.2. Case studies
The sequential reaction with H2 was applied to the measurement of 35Cl as the formation of 35Cl1H2 to assist with a project assessing the corrosion susceptibility of 304L SS materials with a novel coating intended to be applied on fuel racks that are used to store Advanced Gas-Cooled Reactor (AGR) fuel under an NaOH (pH 11.4) pond solution. The storage environment is commonly controlled in alkaline conditions with caustic dosing to maintain pH at 11.4. However, transient changes in local chemical/electrochemical environment is not uncommon to observe, such as increase in chlorides, decrease in pH and increase in oxidation potentials due to radiolysis. Therefore, it is important to measurement traceable chloride concentrations in the μg g−1 to ng g−1 concentration range in the test environment to correlate with the corrosion resistance of coated 304L SS in aqueous solution.
Samples were measured as received in dilute NaOH solution. The instrument was first conditioned with deionised water for one hour, followed by a NaOH blank solution for one hour. The optimal H2 gas flow rate was determined to be 3.5 mL min−1. Initial tests show the blank level to vary between runs, with the lowest values achieved after cleaning the sample introduction system and interface cones. A sensitivity of around 100 CPS was achieved for a 1 ng g−1 standard, which is approximately three orders of magnitude lower than the majority of other elements under the same conditions, due to the high first ionisation energy of Cl. Despite the low sensitivity, it was possible to quantify Cl in all samples, with the results showing good agreement with those measured in parallel using ion chromatography.
The results for stable Cl highlighted the potential for measurement of radioactive 36Cl, not previously measured by ICP-MS. It is a long-lived radionuclide (half-life 3.02(4) × 105 years) and is formed by neutron activation of stable 35Cl, which is present as an impurity in concrete and other reactor components.30,31 Chlorine-36 can be measured by liquid scintillation counting (LSC), with detection limits on the order of 10–15 mBq g−1 achievable, equivalent to 8.2–12.3 pg g−1.32 The long half-life makes 36Cl theoretically suitable for ICP-MS measurement, with a higher sample throughput compared to decay counting techniques.
The same sequential H2 shift was applied, with Q1 and Q2 set to m/z = 36 and 38, respectively. Using this approach, isobaric interferences from 36S (36.0% abundance) and 36Ar from the plasma gas (0.3% abundance) do not form 36S1H2 or 36Ar1H2 and are therefore minimised, as is polyatomic 35Cl1H, which is not expected to form 35Cl1H3. There was no increase in background up to stable Cl concentrations of 80 μg g−1. The optimal H2 flow rate for 36Cl was determined to be 4.5–5.0 mL min−1, with an instrument background of <20 CPS, a sensitivity of approximately 2300 CPS for a 9.9 Bq g−1 (8.1 × 103 pg g−1) 36Cl standard and a calculated detection limit of 81.2 pg g−1 (equivalent to 98.8 mBq g−1). Future work will focus on measurement of decommissioning samples containing 36Cl.
Following on from the successful detection of 40Ca by ICP-MS/MS in several studies, it was recently proven that the interference removal capabilities made 41Ca detection feasible at 41Ca/40Ca ratios of 10−6 and 10−7, with an instrument detection limit of 0.1 ng g−1 (0.3 Bq g−1).27 The addition of H2 to the cell improved the suppression of 40Ar1H and 40Ar interferences compared to using NH3 alone, or indeed compared to any other single or mixed cell gas combination. Using an Agilent 8800 at the optimised flow rates of 7.5 mL min−1 and 5.0 mL min−1 for NH3 and H2, respectively, the background at m/z = 41 was reduced to <10 CPS, compared to ∼2.5 × 109 CPS at 0.5 mL min−1 NH3. This gas flow rate had the highest signal-to-noise ratio of any of the gas flow rates tested (1.7 × 105), with a sensitivity of 1300 CPS for a 1 ng g−1 solution based on stable 40Ca standards.
One limitation of this study was the low sensitivity preventing measurement below 41Ca/40Ca ratios of 10−7, which will be required for decommissioning samples and can be achieved by LSC, AMS and RIMS. Stable Ca standards were run on an Agilent 8900 over the same range of cell gas conditions to see if ICP-MS/MS capability for 41Ca could be improved. At NH3 and H2 flow rates of 7.5 mL min−1 and 5.0 mL min−1, respectively, the sensitivity was 9700 CPS for a 1 ng g−1 solution, which was 7.5 times higher than that achieved for the 8800. As was observed with Fe (Section 3.2.3), the instrument background was also higher when using the newer generation 8900 instrument. However, the optimal signal to noise ratio achieved using the 8900 was 9.0 × 105, compared to 1.7 × 105 for the 8800. Whilst this improvement will not bring ICP-MS/MS in line with AMS, RIMS and LSC capability, it does demonstrate the improvements made with the latest generation instruments, and that detection of challenging radionuclides such as 41Ca is now feasible.
Of the twelve metals analysed by NPL for the Metals Network, the detection limits for Fe are the highest, even with the use of He as a collision gas to reduce the 40Ar16O signal at m/z 56. In this study, H2 was investigated as an alternative and results compared from the 8800 and 8900 instruments. The joint aims were to (i) find a method to improve analyte sensitivity and detection limits for air quality samples and (ii) assess the sensitivity improvements offered by the 8900.
Iron-56 was measured on-mass in Single Quad mode. Helium and H2 were compared for their effectiveness as collision gases to remove polyatomic 40Ar16O. Operating in MS/MS mode did not offer any improved interference removal, as the 40Ar16O interference is formed in the plasma before Q1. These measurements were performed on both the 8800 and 8900 instruments.
The ramp cell gas determined the optimal He and H2 flow rates for the 8800 instrument to be 5 mL min−1 and 5.5 mL min−1, respectively (Table 3). For the 8900 instrument, the optimised flow rates were slightly higher at 5.5 mL min−1 for He and 6.5 mL min−1 for H2.
| 8800 He | 8800 H2 | 8900 He | 8900 H2 | 8800 H2 + He | 8900 H2 + He | |
|---|---|---|---|---|---|---|
| Cell gas flow rate (mL min−1) | 5.0 (He) | 5.5 (H2) | 5.5 (He) | 6.5 (H2) | 1.5 (H2) | 2.0 (H2) |
| 3.0 (He) | 3.0 (He) | |||||
| Sensitivity (CPS ng g−1) | 10500 |
20300 |
9300 | 31400 |
22260 |
24800 |
| Average acid blank (CPS) | 9600 | 7400 | 9300 | 16300 |
9360 | 3920 |
| Background equivalent (BEC: ng g−1) | 0.93 | 0.36 | 1.02 | 0.53 | 0.52 | 0.16 |
| Detection limit (LOD) (ng g−1) | 1.00 | 0.40 | 1.20 | 0.56 | 0.91 | 0.19 |
Table 3 shows that the use of H2 as the collision gas results in a more sensitive method for Fe determination compared to He. The sensitivity was higher by a factor of two with the 8800 (10500 CPS per ng g−1 for He, compared to 20300 CPS with H2), and a factor of three with the 8900 (9300 CPS per ng g−1 with He, compared to 31400 CPS with H2). The detection limits and background equivalents were also lower with H2. In terms of comparing the two ICP-MS instruments, the 8800 and 8900 provided comparable results for Fe when He was used as the collision gas. When H2 was used the sensitivity was 1.5 times higher with the 8900; however, a lower background equivalent (0.36 ng g−1) and detection limit (0.40 ng g−1) was achieved with the 8800. It seems that the improved sensitivity of the 8900 extends below the detection limit to the background signal.
Following on from a study by Iglesias et al.,13 a combination of H2 and He was also investigated. For the 8800 instrument, the detection limit was not improved compared to using H2 alone. However, for the 8900, a detection limit of 0.19 ng g−1 was calculated at the optimised H2 and He flow rates of 2.0 and 3.0 mL min−1, respectively (Table 3). This is an improvement over the 0.56 ng g−1 calculated using H2 only.
Using the 8900, the combination of H2 and He resulted in a lower Fe sensitivity compared to H2 alone (24800 CPS per ng g−1 compared to 31400 CPS). However, the difference in count rate for the different acid blanks used was significant using H2 (16300 CPS) compared to H2 and He (3920 CPS).
This study highlighted the importance of closely controlling the cleanliness of reagents used. An acid blank count rate of 14600 CPS at m/z = 56 was recorded on the 8800 when an online internal standard solution containing indium was connected via a mixing block. This was reduced to 7400 CPS without the internal standard solution. It was concluded that preparation of reagents must be carefully controlled when measuring Fe, as in this case the internal standard solution prepared was contaminated with significant levels of Fe, so could not be used. Further consideration should also be given to the long-term variation in background at m/z = 56, as changes in the Ar plasma gas flow rate, cell gas flow rate or other background ion contributions could all contribute to variations in the signal.
The ICP-MS method for Metals Network samples measures 60Ni (26.2% abundance) to avoid the isobaric interference from 58Fe on the most abundant Ni isotope at m/z = 58 (68.1% abundance). However, the capability to make accurate measurements of all Ni isotopes to enable isotope ratio analysis could provide useful data for source apportionment in this context. The challenge is to overcome isobaric interferences from Fe and Zn at m/z = 58 and 64, respectively (Table 4). There could also be tailing interferences from 63Cu and 65Cu on the neighbouring nickel isotopes 62Ni (3.6% abundance) and 64Ni (0.9% abundance).39
| Ni isotope (% abundance) | Isobaric interference | Polyatomic interference | Tailing interference |
|---|---|---|---|
| 58Ni (68.1) | 58Fe | 57Fe1H | 59Co |
| 60Ni (26.2) | — | 59Co1H | 59Co |
| 61Ni (1.1) | — | 60Ni1H | 60Ni |
| 62Ni (3.6) | — | 61Ni1H | 63Cu |
| 64Ni (0.9) | 64Zn | 63Cu1H | 63Cu, 65Cu |
Product ion scans were performed on the 8900 instrument to investigate reaction-cell separation of 57Fe,58Ni, 60Ni, 63Cu, 64Ni and 66Zn. The first method used NH3 as the cell gas to facilitate a mass shift, whilst the second method utilised a combination of H2 and NH3 cell gases. The reaction of NH3 with several analytes including Ni has been noted previously,45 leading to trace level determination not being advised. However, online interference removal opens the possibility of isotopic ratio measurements without the need for relatively time-consuming offline separation.
Using only NH3 cell gas at a flow rate of 3 mL min−1, the product ion scan results showed formation of several cell products that could potentially be used to separate Ni from interferences (m/z + 17, +34 and +51). Of these, m/z + 51 was the most promising, equivalent to Ni(NH3)3. The sensitivity for the signal at m/z + 51 for Ni was equivalent to 17% of the total signal, compared to 1% for Fe and <1% for Cu and Zn. A mass shift of m/z + 17 (NH3) was not considered suitable; the signal for Ni was equivalent to just 2% of the total signal, Zn was 6%, Cu 2% and Fe was 1%. A mass shift of m/z + 34 (NH3)2 provided different but still undesirable results, with the Ni signal equivalent to 11% of the total, and 28%, 19%, and 3% for Cu, Fe and Zn, respectively. For all cell products, the trends for these four elements agreed with data from the instrument manufacturer, which also did not detect any other cell products other than m/z + 17, +34 and +51.1
The addition of H2 along with NH3 enhanced the formation of Ni(NH3)3, whilst having a limited impact on the same mass shifts for Fe, Cu and Zn (Fig. 4). The optimised NH3 and H2 flow rates were determined to be 4.0 mL min−1 and 6.5 mL min−1, respectively. Of the total signals, for both 60Ni and 58Ni/58Fe (on-mass and mass shift channels +17, +34, +51), 51% was observed on the m/z + 51 channel, compared to 37% remaining on-mass. The 57Fe(NH3)3 response at m/z + 51 remains significantly lower than on-mass, just 3.2% of the total compared to 56% on-mass and 40% at m/z + 34. Combined with the fact that the isotopic abundance of 58Fe is only 0.28%, it can be assumed that the response at m/z = 109 is attributable to Ni(NH3)3. The responses for 63Cu(NH3)3 and 64Zn(NH3)3 ions also remain below on-mass levels. The response for 64Zn/Ni at m/z + 51 is also much less than on-mass (3.3% of total signal at m/z + 51, compared to 80% on-mass), despite the presence of the Ni isotope. However, the natural abundance of 64Ni is only 0.9%, so would be lost amid any shifted signal from 64Zn (48.6% natural abundance).
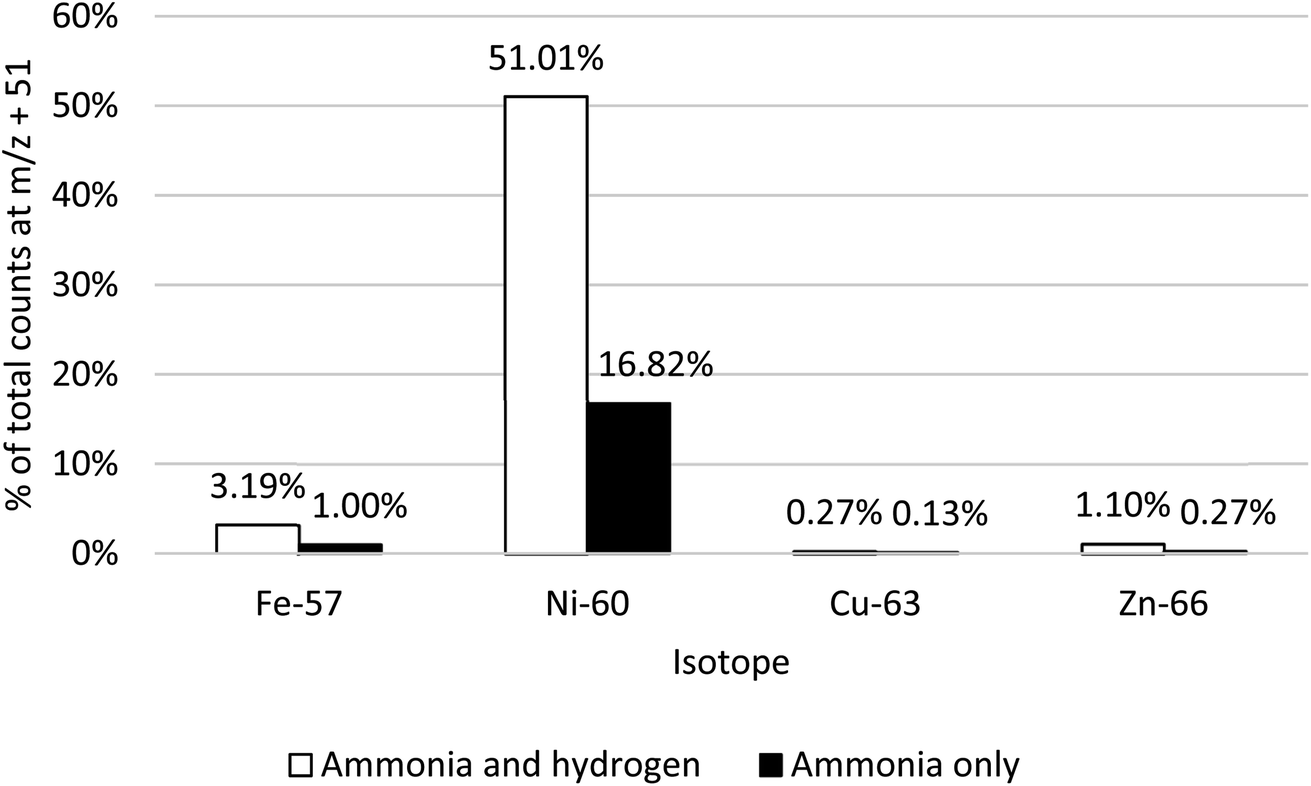 | ||
| Fig. 4 Responses of 57Fe, 60Ni, 63Cu and 66Zn using NH3 only and H2 and NH3 cell gases to facilitate a mass shift of m/z + 51 (NH3)3. | ||
A further Ramp Cell Gas optimisation using H2 with NH3 was performed on the 8800. As with the 8900, it was observed that for all elements, an increase in H2 flow rate increases the formation of (NH3)3 cell products compared to NH3 alone, and the formation of Ni(NH3)3 is favoured over the products of Fe, Cu and Zn.
At optimised NH3 and H2 flow rates of 2 mL min−1 and 2.5 mL min−1 respectively, the Ni(NH3)3 signal was equivalent to 77.7% of the signal measured on-mass (based on 58Ni). This compares to 7.5% for 57Fe, 0.9% for 63Cu and 2.9% for 64Zn, suggesting cell-based separation is possible. As the H2 flow rate increased above 2.5 mL min−1, the Ni(NH3)3 signal decreased, whilst the signals for Fe(NH3)3, Cu(NH3)3 and Zn(NH3)3 increased until flow rates of 5.0, 3.5 and 6.5 mL min−1, respectively.
The optimised NH3 and H2 flow rates differed significantly between the 8800 (2 mL min−1 and 2.5 mL min−1 respectively) and 8900 (4.0 mL min−1 and 6.5 mL min−1 respectively). This is likely due to the increased sensitivity of the 8900 allowing for greater tolerance of higher flow rates without adversely affecting sensitivity. Additionally, the optimisation of the 8900 gas flow rates focussed on the 60Ni signal, so fine tuning to minimise the signals for the other elements could also explain the optimised flow rate discrepancy.
The effective combination of NH3 and H2 is auspicious for the development of an interference-free method for the determination of the majority of Ni isotopes. It is hoped that this method will prove suitable for isotope ratio analysis of air quality filter samples, as well as for other applications such as improved understanding of the early formation of the solar system and as a geochemical tracer in magmatic processes.46,47 It must be considered that more complex sample matrices may result in additional and higher concentrations of interferences that would likely require modification of the instrument setup presented in this study.
The same cell gas combination was successfully used for detection of radioactive 63Ni (half-life 98.70(24) years), formed by neutron activation of stable 62Ni and a radionuclide of significant interest in nuclear waste characterisation and decommissioning. The formation of 63Ni(NH3)3 provided cell-based separation from isobaric 63Cu and polyatomic 62Ni1H, whilst MS/MS mode effectively removed 62Ni tailing. A product ion scan using NH3 gas at a flow rate of 3 mL min−1 was initially tested using stable 58Ni and 63Cu. The 58Ni(NH3)3/63Cu(NH3)3 formation rate was 35.4 based on the difference in CPS for each isotope, which increased at lower gas flow rates at the expense of 58Ni(NH3)3 sensitivity, which was >99% lower at 0.5 mL min−1 compared to 3 mL min−1. Compared to NH3 alone, the addition of H2 gas increased Ni(NH3)3 formation up to a flow rate of 1.5 mL min−1, with a comparatively low increase in Cu(NH3)3. The 58Ni(NH3)3/63Cu(NH3)3 formation rate increased to around 100 at 1 mL min−1 NH3 and 3 mL min−1 H2, with a sensitivity of 61000 CPS for a 10 ng g−1 Ni solution. This compares to a similar separation factor of ∼90 at 1 mL min−1 NH3 without H2, but with a low Ni sensitivity of 145 CPS for a 10 ng g−1 solution.
The optimised cell conditions were tested on 63Ni standards, with an instrument detection limit of 0.25 pg g−1 (0.52 Bq g−1), with the background at m/z = 63 of <10 CPS, compared to ∼500 CPS in no-gas mode. When tested on aqueous waste samples without chemical separation, the method detection limit was calculated as 12.1 pg g−1 (25.6 Bq g−1) due to the elevated 63Cu concentrations. This could be improved by offline chemical separation such as ion exchange or extraction chromatography.48–50 However, this demonstrates that direct measurement of 63Ni in real samples is feasible.
The relatively short half-life of 63Ni means the detection limits achievable by ICP-MS/MS are higher than those of liquid scintillation counting. However, the measurement time of ICP-MS/MS of several minutes per sample offers an improved sample throughput, which is further improved by cell based separation reducing the reliance on relatively time consuming offline separation. This also reduces the analyst working time and the amount of secondary waste generated through the use of separation reagents and materials.
The Agilent 8900 used in this study was not able to run radioactive materials. However, the higher sensitivity compared to the 8900 achieved using stable Ni standards could potentially improve the detection limits achievable. Future work will look at the potential of ICP-MS/MS for measurement of longer-lived 59Ni (half-life 76(5) × 103 years), which would have an additional advantage of 59Ni/63Ni ratio measurements for source attribution. Accurate measurement requires effective removal of isobaric 59Co (100% abundance), which was not achieved using the NH3 + H2 method presented in this study, as well as polyatomic 58Ni1H and 58Ni tailing interferences.
The starting point for this work was the successful use of NH3 + H2 for measurement of 93Zr as 93Zr(NH3)6 in previous studies.14,15 In these studies, interference removal of 93Mo was the focus, rather than measurement of this radionuclide. Stable 90Zr, 93Nb and 95Mo were run through product ion scans with Q1 set to 90, 93 and 95, respectively, using only NH3 at a flow rate of 3 mL min−1. The results identified a mass shift of 34 (equivalent to (NH3)2) as potentially offering separation of Mo from Zr and Nb. The majority of the Mo signal (85.22%) stayed on mass, compared to approximately 2% for Zr and Nb, which produced a range of product ions (Fig. 5). At a mass shift of m/z + 34, 12% of the total Mo signal was detected, compared to <0.1% for Zr and Nb. There were no other significant cell products (>1% of the total signal) detected for Mo.
 | ||
| Fig. 5 Results of product ion scan for Mo, Zr and Nb. | ||
The NH3 ramp cell gas result suggested that the peak Mo(NH3)2 sensitivity was achieved at a flow rate of 4.0–4.5 mL min−1, with no significant change in instrument background from Zr(NH3)2 or Nb(NH3)2 over all NH3 flow rates. At a fixed NH3 flow rate of 4.3 mL min−1, the cell gas ramp was repeated for varied H2 flow rates. This had no impact on the Nb(NH3)2 or Zr(NH3)2 signal, however, the signal for Mo(NH3)2 increased by approximately 10% compared to using NH3 alone. The optimal H2 flow rate was determined to be 3.5 mL min−1. Under these optimised conditions, the Mo(NH3)2 signal was 1100 CPS for a 1 ng g−1 solution, compared to 900 CPS for Mo measured on mass.
Based on the measurement of stable Mo standards, an instrument detection limit of 45.6 pg g−1 (1.6 Bq g−1) was calculated based on measurement of Mo(NH3)2. The concentrations of Zr and Nb were increased and showed <5 CPS from Zr(NH3)2 or Mo(NH3)2 until the concentration exceeded 1 ng g−1. At a concentration of 50 ng g−1, the signal from Zr(NH3)2 and Mo(NH3)2 was 160 CPS and 10 CPS, respectively, compared to 4.3 × 107 CPS from Mo(NH3)2 at the same concentration. This shows that ICP-MS/MS can reduce the reliance on relatively time-consuming offline chemical separation prior to measurement. At this stage, the method has only been tested using stable isotopes of Mo, Zr and Nb, due to the absence of a 93Mo tracer, the production of which is the next stage of this work. As with 63Ni, the relatively short half-life of 93Mo for ICP-MS measurement means testing of active 93Mo using an Agilent 8900 could offer benefits for low-level measurements. Online separation of Mo from interfering elements also has the potential for stable Mo isotope ratio measurements for applications including past redox conditions in water,51 and metabolism pathways relating to biochemistry and environmental chemistry.52
4. Conclusions
ICP-MS/MS combined with H2 as a collision or reaction gas (either alone or mixed with other gases) has been proven to offer enhanced interference removal for a number of analytes. This study expands the applications of H2 for a number of stable and radioactive pollutants relevant to two groups at the National Physical Laboratory, UK. Stable Fe and Ni isotopes are measured as part of a programme of work investigating metal concentrations in air filter samples to meet regulatory requirements. Hydrogen improved polyatomic 40Ar16O removal for accurate 56Fe measurement, whilst H2 combined with NH3 selectively shifted Ni to Ni(NH3)3, offering removal from isobaric and polyatomic interferences from Fe, Cu and Zn, opening the possibility of Ni isotope ratio measurements for source attribution. The same method was effective for detecting radioactive 63Ni as Ni(NH3)3, whilst the reaction with isobaric 63Cu did not proceed. A combination of H2 and NH3 was effective in minimising the background at m/z = 41, enabling detection of radioactive 41Ca in concrete samples relevant to nuclear decommissioning. Hydrogen was effectively used as the sole gas for measurement of 35Cl as 35Cl1H2, with the results showing the first known ICP-MS/MS measurement of the long-lived radionuclide 36Cl. The potential of ICP-MS/MS for measurement of 93Mo was also investigated using a stable 95Mo analogue, with selective formation of 95Mo (NH3)3 achieving good interference removal from stable and radioactive isobars 93Nb and 93Zr, respectively. The outcomes of this study show enhanced measurement capability for multiple analytes, and in some cases the possibility of isotope ratio measurement for source attribution, which will have benefits for applications beyond this study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The funding of the National Measurement System by the UK's Department for Business, Energy & Industrial Strategy is gratefully acknowledged. The measurement of 41Ca was supported by the European Metrology Research Program (EMRP) joint research project ‘in situ metrology for decommissioning nuclear facilities’ (MetroDECOM II). The European Metrology Research Programme (EMRP) is jointly funded by the EMRP participating countries within EURAMET and the European Union. The Nuclear Metrology Group would like to thank the University of Birmingham for collaborating on the 63Ni work through student placements.References
- Reaction data for 70 elements using O2, NH3 and H2 Gases with the Agilent 8800 Triple Quadrupole ICP-MS, https://www.agilent.com/cs/library/technicaloverviews/public/5991-4585EN_TechNote8800_ICP-QQQ_reactiondata.pdf, accessed 16/10/21 Search PubMed.
- G. C. Eiden, C. J. Barinaga and D. W. Koppenaal, J. Anal. At. Spectrom., 1996, 11, 317–322 RSC
.
- D. W. Koppenaal, G. C. Eiden and C. J. Barinaga, J. Anal. At. Spectrom., 2004, 19, 561–570 RSC
- S. D. Tanner, J. Anal. At. Spectrom., 1995, 10, 905–921 RSC
- I. Feldmann, N. Jakubowski and D. Stuewer, Fresenius. J. Anal. Chem., 1999, 365, 422–428 CrossRef CAS
- T. Vincent, Thermo Sci. Appl, Note 43442, http://tools.thermofisher.com/content/sfs/brochures/AN-43442-ICP-MS-Selenium-Hydrogen-CRC-AN43442-EN.pdf, accessed 10/08/21 Search PubMed.
-
F. Vanhaecke, Agilent Technologies Application Handbook, 2015, vol. 2, pp. 1–92 Search PubMed
- A. Lee, V. Yang, J. Hsu, E. Wu, R. Shih and K. Mizobuchi, Agilent Application Note, https://www.agilent.com/cs/library/applications/5991-1693EN_AppNote_ICP-MS_8800_semicon_Ca_ultrapure_water.pdf, accessed 10/08/21 Search PubMed.
- J. Takahashi, Agilent Application Note, https://www.agilent.com/cs/library/applications/appcompendium_icp-qqq-5991-2802en-us-agilent.pdf, accessed 10/08/21 Search PubMed.
-
N. Sugiyama, Applications Using the Agilent 8800 and 8900, 4th edn, 2020, pp. 27–30 Search PubMed
-
M. Yamanaka, Agil. 8800 ICP-QQQ Appl. Handbook. Prim, 2013, pp. 218–220 Search PubMed
- S. Manecki, M. lofthouse, P. Boening and S. McSheehy Ducos, Thermo Scientific Application Note, https://assets.thermofisher.com/TFS-Assets/CMD/Application-Notes/AN-43285-ICP-MS-Arsenic-Selenium-Sediment-Rock-AN43285-EN.pdf, accessed 10/08/21 Search PubMed.
- M. Iglesias, N. Gilon, E. Poussel and J. M. Mermet, J. Anal. At. Spectrom., 2002, 17, 1240–1247 RSC
- P. Petrov, B. Russell, N. Douglas and H. Goenaga-infante, Anal. Bioanal. Chem., 2018, 410, 1029–1037 CrossRef CAS PubMed
- H. Thompkins, B. Russell and S. Goddard, Agilent Application Note, https://www.agilent.com/cs/library/applications/application_zr-93_icp-qqq_8800_8900_5994-1532en_us-agilent.pdf, accessed 10/08/21 Search PubMed.
- A. Lee, V. Yang, J. Hsu, E. Wu, R. Shih and K. Mizobuchi, Agilent Application Note, https://www.agilent.com/cs/library/applications/5991-1693EN_AppNote_ICP-MS_8800_semicon_Ca_ultrapure_water.pdf, accessed 10/08/21 Search PubMed.
-
W. Geiger, Applications Using the Agilent 8800 and 8900, 4th edn, 2020, pp. 37–40 Search PubMed
-
DEFRA, UK Statutory Instrument No. 0000: The Environmental Permitting England and Wales (Amendment) Regulations 2018, National Radiological Protection Board, 2018 Search PubMed
-
S. Goddard, R. J. Brown, D. Butterfield, E. McGhee, C. Robins, A. Brown, S. Beccaceci, A. Lilley, C. Bradshaw and S. Brennan, Annual Report for 2014 on the UK Heavy Metals Monitoring Network, Queen’s Printer and Controller of HMSO, 2015 Search PubMed
-
European Commission Council, Directive 2004/107/EC of the European Parliament and of the Council of 15 December 2004 Relating to Arsenic, Cadmium, Mercury, Nickel and Polycyclic Aromatic Hydrocarbons in Ambient Air, 2004 Search PubMed
-
European Parliament, Council of the European Union, Directive 2008/50/EC of the European Parliament and of the Council of 21 May 2008 on Ambient Air Quality and Cleaner Air for Europe, 2008 Search PubMed
- M. A. Amr, A. F. I. Helal, A. T. Al-Kinani and P. Balakrishnan, J. Environ. Radioact., 2016, 153, 73–87 CrossRef CAS PubMed
- T. Ohno, Y. Muramatsu, Y. Shikamori, C. Toyama, N. Okabe and H. Matsuzaki, J. Anal. At. Spectrom., 2013, 28, 1283–1287 RSC
- J. Zheng, K. Tagami, W. Bu, S. Uchida, Y. Watanabe, Y. Kubota, S. Fuma and S. Ihara, Environ. Sci. Technol., 2014, 48(10), 5433–5438 CrossRef CAS PubMed
- M. Tanimizu, N. Sugiyama, E. Ponzevera and G. Bayon, J. Anal. At. Spectrom., 2013, 28, 1372–1376 RSC
- Metrology for Radioactive Waste Management, No Title, https://www.euramet.org/research-innovation/search-research-projects/details/project/metrology-for-radioactive-wastemanagement/?L=0%26tx_eurametctcp_project%5Baction%5D=show%26tx_eurametctcp_project%5Bcontroller%5D=Project%26cHash=31533f8774a0bc6b299635ee4f2519f6, accessed 16/10/2021 Search PubMed.
- B. Russell, H. Mohamud, M. G. Miranda, P. Ivanov, H. Thompkins, J. Scott, P. Keen and S. Goddard, J. Anal. At. Spectrom., 2021, 36, 845–855 RSC
- K. Nakano, Agilent Application Note, https://www.agilent.com/cs/library/applications/5991-6852EN.pdf, accessed 10/08/21 Search PubMed.
- J. Nelson, Agilent Application Note, https://www.agilent.com/cs/library/applications/application_cl-crude-icp-ms-8900-5994-1094en_us_agilent.pdf, accessed 10/08/21 Search PubMed.
- L. Ashton, P. Warwick and D. Giddings, Analyst, 1999, 124, 627–632 RSC
- M. Baxter, L. Castle, H. M. Crews, M. Rose, C. Garner, G. Lappin and D. Leong, Food Addit. Contam., Part A, 2009, 26, 139–144 CrossRef CAS PubMed
- X. Hou, L. F. Østergaard and S. P. Nielsen, Anal. Chem., 2007, 79(8), 3126–3134 CrossRef CAS PubMed
- X. Hou, Radiochim. Acta, 2005, 93, 611–617 CrossRef CAS
- P. E. Warwick, I. W. Croudace and D. J. Hillegonds, Anal. Chem., 2009, 81, 1901–1906 CrossRef CAS PubMed
- D. Hampe, B. Gleisberg, S. Akhmadaliev, G. Rugel and S. Merchel, J. Radioanal. Nucl. Chem., 2013, 296, 617–624 CrossRef CAS
- P. Müller, B. A. Bushaw, K. Blaum, S. Diel, C. Geppert, A. Nähler, N. Trautmann, W. Nörtershäuser and K. Wendt, Fresenius’ J. Anal. Chem., 2001, 370, 508–512 CrossRef PubMed
- S. Merchel, L. Benedetti, D. L. Bourlès, R. Braucher, A. Dewald, T. Faestermann, R. C. Finkel, G. Korschinek, J. Masarik, M. Poutivtsev, P. Rochette, G. Rugel and K. O. Zell, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268, 1179–1184 CrossRef CAS
- X. Hou and P. Roos, Anal. Chim. Acta, 2008, 608, 105–139 CrossRef CAS PubMed
- J. R. De Laeter, K. G. Heumann, R. C. Barber, I. L. Barnes, J. Cesario, T. L. Chang, T. B. Coplen, J. W. Gramlich, H. R. Krouse, I. A. Lebedev, T. J. Murphy, K.J.R. Rosman, M. P. Seyfried, M. Shima, K. Wade, P. De Bievre, R.L. Martin and H. S. Peiser, International Union of Pure and Applied Chemistry, Pure Appl. Chem., 1991, 63(7), 991–1002 Search PubMed
- E. McCurdy and G. Woods, J. Anal. At. Spectrom., 2004, 19, 607–615 RSC
- M. S. Wheal, E. Decourcy-Ireland, J. R. Bogard, S. H. Thilsted and J. C. R. Stangoulis, Food Chem., 2016, 201, 222–229 CrossRef CAS PubMed
- L. Poirier, J. Nelson, D. Leong, L. Berhane, P. Hajdu and F. Lopez-Linares, Energy Fuels, 2016, 30(5), 3783–3790 CrossRef CAS
- W. Castro, T. Trejos, B. Naes and J. R. Almirall, Anal. Bioanal. Chem., 2008, 392, 663–672 CrossRef CAS PubMed
- V. Dufailly, L. Noël and T. Guérin, Anal. Chim. Acta, 2006, 565(2), 214–221 CrossRef CAS
- S. D. Tanner and V. L. Baranov, J. Am. Soc. Mass Spectrom., 1999, 10, 1083–1094 CrossRef CAS
- S. M. Chernonozhkin, S. Goderis, L. Lobo, P. Claeys and F. Vanhaecke, J. Anal. At. Spectrom., 2015, 30, 1518–1530 RSC
- S. Liu, Y. Li, Y. Ju, J. Liu, J. Liu and Y. Shi, Geochim. Cosmochim. Acta, 2018, 222, 1–16 CrossRef CAS
- E. Holm, P. Roos and B. Skwarzec, Int. J. Radiat. Appl. Instrum., Part A, 1992, 43(1–2), 371–376 CrossRef CAS
- P. E. Warwick and I. W. Croudace, Anal. Chim. Acta, 2006, 567(2), 277–285 CrossRef CAS
- O. Rosskopfová, M. Galamboš and P. Rajec, J. Radioanal. Nucl. Chem., 2011, 289(1), 251–25654 CrossRef
- K. Horan, R. G. Hilton, A. J. McCoy-West, D. Selby, E. T. Tipper, S. Hawley and K. W. Burton, Geochemical Perspect. Lett., 2020, 13, 1–6 Search PubMed
- D. Malinovsky and N. A. Kashulin, Anal. Methods, 2018, 10, 131–137 RSC
| This journal is © The Royal Society of Chemistry 2021 |