
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Determination of the naturally occurring vanadium-complex amavadin in Amanita muscaria with HPLC-ICPMS†
Simone
Braeuer‡
 *,
Martin
Walenta
,
Lorenz
Steiner
and
Walter
Goessler
*,
Martin
Walenta
,
Lorenz
Steiner
and
Walter
Goessler
Institute of Chemistry, University of Graz, Universitaetsplatz 1, 8010 Graz, Austria. E-mail: simone.braeuer@uni-graz.at; Tel: +43 316 380 5318
First published on 19th March 2021
Abstract
Amanita muscaria, also known as the fly agaric mushroom, can accumulate vanadium (V), with up to several hundred mg V kg−1 dry mass. It is long known that V is present in A. muscaria as a complex called amavadin, but methods for the investigation of the distribution and biosynthesis of amavadin in mushrooms are missing. Here, we describe the development of the first sensitive method for the determination of amavadin and other V-containing compounds in environmental samples by employing high performance liquid chromatography (HPLC) and inductively coupled plasma mass spectrometry (ICPMS). A strong anion-exchange column serves as the stationary phase, and the mobile phase consists of an aqueous ammonium citrate buffer and ethylenediaminetetraacetate (EDTA). The concentration and pH of the mobile phase as well as the column temperature were evaluated to optimize the separation. With the final method, amavadin is eluted in less than 17 minutes, and its limit of detection is 0.05 μg V L−1. Moreover, the compound's two isomers are separated from each other and can be quantified independently. The method was applied to extracts of fruit-body samples of A. muscaria. The extraction efficiency was 74 ± 12%, and amavadin accounted for 75–96% of the extracted V. In addition, significant concentrations of other V species could be detected, which have never been described before. Our results demonstrate that V speciation in mushrooms is more complex than assumed until now and that more in-depth investigations on this matter are needed. The developed method enables the investigation of organic and inorganic V species in the environment, even at low concentrations.
 Simone Braeuer | Simone Braeuer obtained her PhD in 2018 from the University of Graz, Austria, in the research group of Prof. Walter Goessler. She stayed there for one more year and then started a Postdoctoral fellowship at Ghent University, Belgium, in the Atomic & Mass Spectrometry research group of Prof. Frank Vanhaecke. Her work in Graz focused on the development and application of ICPMS and HPLC-ICPMS methods for the determination of trace element species in the environmental context. For her PhD, her main focus was on arsenic in mushrooms. Since mushrooms also contain other elements, sometimes in surprising concentrations and species, she soon became interested in some of those as well. The fascination with ICPMS and mushrooms grew so big that she is still investigating mushrooms in her current Postdoctoral project, but now also via multi-collector ICPMS and laser ablation ICPMS. She thinks that multidisciplinary and international collaborations are both fascinating and essential for environmental research. She also believes that high-quality, state-of-the-art analytical chemistry is of key importance – for environmental as well as for many other (interdisciplinary) studies. |
1 Introduction
1.1 Vanadium
Vanadium (V) is widely distributed in nature, being the 22nd most abundant element in the earth's crust.1 The essentiality of V has been proven for some bacteria, terrestrial fungi, lichens and marine macro-algae,2 but for other living organisms, the role of V is still unclear.3 Because of the similarity of vanadate and phosphate, it has been suggested that V could play a role in the regulation of the phosphate metabolism. A variety of V compounds are under investigation as potential anti-diabetic drugs4 and for several other medical applications.5 On the other hand, V can also be toxic to living organisms.6,7 It is usually thought that vanadate, the inorganic pentavalent form of V, is more toxic than V in the oxidation state +4 and that organic V species are less toxic than inorganic ones.8 However, the adverse health effects of V exposure are usually only observed in laboratory animals and people working in the V-processing industry,3 with a few exceptions.9Ascidians (sea squirts) and some fan worms can accumulate incredible amounts of V. The highest concentrations, up to 350 mM (more than 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mg V kg−1), have been reported in Ascidia gemmata,10 where specialized cells store the element in its otherwise rather unstable trivalent form.11 Until now, the actual function of V in ascidians remains elusive.10,12
000 mg V kg−1), have been reported in Ascidia gemmata,10 where specialized cells store the element in its otherwise rather unstable trivalent form.11 Until now, the actual function of V in ascidians remains elusive.10,12
1.2 Amanita muscaria
Another living organism that accumulates V is the macrofungus Amanita muscaria (L.) Lam, also prominently known as fly agaric, along with a few closely related species. While most other mushrooms typically contain less than 0.5 mg V kg−1 dry mass (dm),13–15A. muscaria can easily take up more than 100 mg V kg−1 dm.13,16–18The highest concentrations, namely up to 1000 mg V kg−1 dm, have been found in the “bulb”, which is the lowest part of the stipe.17,19,20 This part of the fruit body is prone to contamination by soil, and no measures to minimize such contamination were described in the respective studies. However, the V concentrations in European top soils are reportedly between 1.28 and 537 mg kg−1, with a median of only 60 mg kg−1.9 This makes it unlikely that 1000 mg V kg−1 in a mushroom sample are solely to be attributed to remaining soil particles.
Concerning the other parts of the fruit body, the V concentrations in the stipe, cap skin and cap flesh were at least two times lower than in the corresponding bulb sample, while they were again a little higher in the gills (but still lower than in the bulb; the concentrations were only shown in a rough graph, so that no exact numbers can be cited).19 In the spores, only 1–5 mg V kg−1 dm were detected.19 Many other macrofungal species have been investigated for their V concentrations, including several members of the genus Amanita, but only A. regalis and A. velatipes accumulated as much V as A. muscaria.17,21
1.3 Amavadin
In 1972, Bayer and Kneifel found that V is present in A. muscaria as a negatively charged compound (pH 1–10), which they named “Amavadin”.22 They reported a tentative sum formula for the compound, and electron paramagnetic resonance (EPR) spectroscopy indicated that the oxidation state of V was +4. One year later, they published a first structure of the complex.23 Further structural investigations by EPR, infrared (IR), and other techniques followed, which have been reviewed a few times over the decades.24–26 EPR spectra suggested that the different parts of the fruit body contained the same compound.20,27 The first synthesis of amavadin was reported in 1986,28 and in the following years, a new structure of the complex was proposed29 and confirmed by crystallography, X-ray absorption spectroscopy and nuclear magnetic resonance (NMR) spectroscopy.34,35Amavadin, as shown in Fig. 1, contains a rather unusual non-oxo V(IV) ion (the vanadyl ion VO2+ is more common) and can exist in two isomers, namely Δ-amavadin and Λ-amavadin. The two tetradentate ligands can coordinate to the V center in two distinct orientations, as shown in Fig. 1. It was reported that synthetic and also natural amavadin were present in solution in an almost equal mixture of the Δ- and Λ-isomers, although it was noted that isomerization could theoretically occur after extracting or dissolving the compound.30,31 The first enantioselective synthesis of amavadin was carried out by Hubregtse et al., who found that the ratio of the diastereoisomers Λ:Δ was first 2.27, but later decreases to 0.8 at equilibrium.32,33
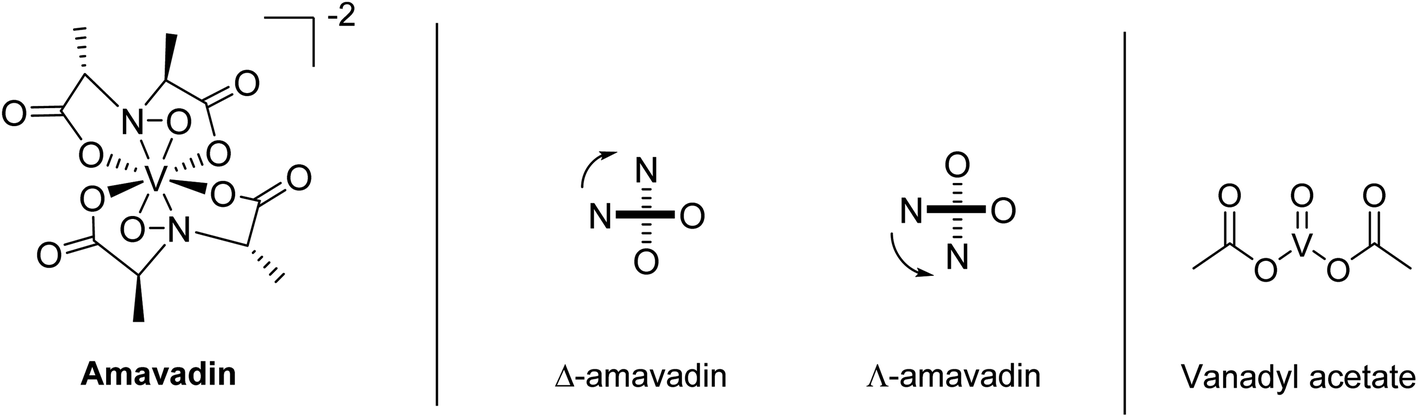 | ||
| Fig. 1 Structure of amavadin, schematic representation of its Δ- and Λ-helical forms, and structure of vanadyl acetate. | ||
Because of the reversible oxidation of V(IV) to V(V) in amavadin, several studies investigated its catalytic properties. It was found that a broad range of reactions can be catalyzed by the complex. This led to the synthesis and investigation of amavadin analogues with a different metal ion in the center or slightly modified ligands.35
The biological role of amavadin is unknown. A few proposals have been made, but none have been proven so far. It has been speculated that amavadin was part of an ancient, primitive enzyme, which has been outdated through evolution.26 Other suggestions are that it serves as a protective agent against external microbial pathogens or aids the self-regeneration of damaged fruit-body tissue by cross-linking thiol groups in proteins.32,34 The ability to mediate water oxidation gave the idea that amavadin is involved in the oxygen production in the fruit body.35 da Silva et al. speculated that amavadin could react with the side chains of amino acid residues of proteins, but also noted that the high coordination number of the complex makes a permanent coordination to proteins unfavorable.25 This was confirmed by a recent study, where Ugone et al. showed that only non-covalent interactions between amavadin and proteins are possible.36 Finally, the high stability of the complex over a broad pH range led to the suggestion that amavadin could be used as a catalyst in an acidic medium, and its chiral properties could be handy in asymmetric synthesis.25
Of the many old and recent studies on amavadin and derived complexes, most explored the compounds' physicochemical properties and their catalytic behavior. However, hardly anything is published on amavadin's occurrence, biosynthesis, and distribution in A. muscaria and other macrofungi. One exception is a study by Gillard et al., where EPR data suggested that all parts of the fruit body of A. muscaria contained the same V compound.20 The only other notable exception is the work of Koch et al. from 1987, where they extracted several different mushrooms of the genus Amanita and different fruit-body parts of A. muscaria with water at 60 °C and then analyzed with ion-pair chromatography and UV-detection.17 They, too, found that amavadin was present in all the investigated fruit-body parts of A. muscaria, with concentrations between 15 and 56 mg V kg−1 dm. The highest concentrations were detected in the bulb samples. The concentrations accounted for only 10 to 20% of the total V in the samples. As possible reasons for these low percentages, the authors speculated that the extraction was maybe not quantitative, that amavadin was degraded in the extract, or that amavadin was not the only V species in A. muscaria, although they deemed the last option not very likely.17 Furthermore, Koch et al. found that amavadin is not only present in A. muscaria, but also in A. regalis and A. velatipes.17 In the case of A. rubescens, A. parcivolvata and A. wellsii, no clear identification of amavadin was possible, due to the low concentrations.
1.4 Vanadium speciation analysis
Speciation analysis of V is mainly focused on the determination of V(IV) and V(V) in water and soil extracts,37,38 V porphyrins in crude oil,39 and molecular size fractions in blood and organ samples, usually after the administration of different V compounds.40Inductively coupled plasma mass spectrometry (ICPMS) is nowadays typically used as a sensitive, V-selective detector, but other techniques have been used in the past as well, like electrothermal atomic absorption spectroscopy, inductively coupled plasma optical emission spectroscopy or even UV.38
For blood and organ samples, size exclusion chromatography (SEC), often in combination with anion-exchange chromatography (AEC) has been used to distinguish between low-molecular mass fractions, high molecular mass fractions and transferrin-associated V.41,42 Nischwitz et al. used electrospray ionization tandem mass spectrometry in addition to SEC and tentatively identified a divanadate-phosphate derivate in liver cells that were exposed to an antidiabetic V species.43
The V speciation in water and soil extracts usually only consists of inorganic V(V) and V(IV) ions.37 The speciation analysis of these samples is nevertheless challenging, because of the lability of the V species. If the samples are not stabilized properly, species interconversion can easily take place, especially the oxidation of V(IV) to V(V).61 For this reason, most methods add a chelating agent to the samples or the mobile phase, for example ethylenediaminetetraacetate (EDTA).44,45 The separation of the formed vanadyl and vanadate complexes is typically achieved via high performance liquid chromatography (HPLC), with ion-pair or ion-exchange chromatography.46–51 Capillary electrophoresis has been employed as well, but only in very few studies.52 Alternatively, some publications use selective extraction one of the two V species or solid phase extraction with selective elution, and apply the developed methods to water, soil and plant samples.53–55
There are also examples of V speciation analysis with X-ray absorption spectroscopy, more precisely with X-ray absorption near edge structure (XANES) spectroscopy, for example of V compounds in mycelium56 or ascidians.57 These studies were able to deliver information on the element's oxidation state and in some cases on its spatial distribution in the sample.
There are only a few publications that do not only distinguish between V(IV) and V(V) or between rough molecular mass fractions, or investigate other sample types than the mentioned ones. Jensen-Fontaine et al. used HPLC-ICPMS with AEC to determine V species in the benthic invertebrate Hyalella azteca, exposed to V.58 Apart from V(IV) and V(V) ions, they also found an unknown V compound. They speculated that it could be V associated or bound to a protein or a smaller cellular molecule of H. azteca, but this still remains to be investigated in more detail. Kütter et al. used SEC and AEC-UV-ICPMS as well as matrix-assisted laser desorption/ionization (MALDI) time of flight mass spectrometry for V speciation analysis in plankton.59 They found a V-containing compound with a mass of 8–16 kDa, but concluded that it was not possible to further identify this compound. Finally, in 1987, Koch et al. used ion-pair chromatography with UV detection to determine amavadin in different parts of Amanita muscaria, as already described earlier.17 This is the only described chromatographic method for the investigation of amavadin until now, but it is neither sensitive nor selective, nor was it validated. The authors themselves write that the unambiguous identification of amavadin is not possible in samples with low V concentrations, due to a high background and “not very characteristic UV spectra”.17 While this work was useful for a rough estimate of the amavadin concentration in the samples of Amanita muscaria back in 1987, it does not meet the criteria for a modern, state-of-the-art analytical method in 2021.
Thus, we developed a sensitive method for the detection of amavadin and related V species with HPLC-ICPMS. The method was validated and applied to three fruit-body samples of A. muscaria, divided into their different parts.
2 Experimental
2.1 Chemicals and instruments
Ultrapure water (18.2 MΩ cm) was used throughout the study. All chemicals were purchased from common chemical suppliers and used as received without further purification, if not described otherwise. A detailed description of all chemicals, including the purity and supplier, can be found in the ESI (Section 1 “Chemicals and instruments”†). The employed instruments are also listed in more detail in the ESI (Section 1†) and are only described in an abbreviated version here.Air and moisture sensitive reactions were carried out following the standard Schlenk technique with Ar as an inert atmosphere. NMR spectra were recorded on a Bruker Advance III 300 MHz NMR spectrometer in deuterated solvents. IR spectra were recorded on a Bruker Alpha FTIR spectrometer.
Electrospray mass spectrometry (ESMS) was performed with a 6120 Quadrupole LC/MS (Agilent Technologies). A 1260 Infinity HPLC (Agilent Technologies) and a Nucleodur C18 Pyramid column (Macherey-Nagel) were used to separate the compounds for further analysis with ESMS. Total V concentrations were determined with a 7700x ICPMS (Agilent Technologies).
For speciation analysis, a 1260 Infinity HPLC (Agilent Technologies) was coupled to a 7700x ICPMS or a tandem ICPMS (ICPMS/MS, 8900, Agilent Technologies). A silica-based strong anion-exchange column (Zorbax SAX, 4.6 × 150 mm, 5 μm, Agilent Technologies) was used for the separation of the V species.
The water content of the mushroom samples was determined with a SMART Trac II Moisture & Fat Analyzer (CEM). An Ultraclave IV (MLS GmbH) was used for microwave assisted digestions. Extracts were centrifuged on a Rotina 420 R (Hettich Lab Technology). Nylon syringe filters (0.22 μm, Simplepure) were used for filtering the extracts prior to analysis.
2.2 Synthesis
1H NMR (300 MHz, CDCl3) δ = 5.79 (s, 1H), 5.29 (s, 2H), 3.71 (q, J = 6.9, 2H), 3.70 (s, 6H), 1.33 (d, J = 6.9, 6H). 13C NMR (75 MHz, CDCl3) δ = 173.2, 60.9, 53.4, 52.2, 12.6.
1H NMR (300 MHz, D2O) δ = 4.02 (q, J = 7.0, 2H), 1.41 (d, J = 7.0, 6H). 13C NMR (75 MHz, D2O) δ = 174.5, 62.7, 11.6. ESMS: m/z calc for [M + 1]+: 178, found: 178. IR: 1747 (νCO) cm−1.
ESMS: m/z calculated for [M + 1]+: 186; m/z found: 158, corresponding to [VO(OOCH)2 + H]+ – owing to the formic acid in the mobile phase. IR: 1508 (νCO), 894 (νVO) cm−1.
ESMS: m/z calc for [M + 3H]+: 402, found: 402. IR: 1561 (νCO) cm−1.
2.3 Confirmation of the structure and concentration
| ICPMS measurements | |
| Instrument | Agilent 7700x |
| Collision gas mode | He (5 mL min−1) |
| Monitored m/z | 51 (V), 74 (Ge, internal standard) |
| Integration time | 0.1 s per m/z |
| Intensity of 1 μg V L−1 | 40 kcps |
| Oxide ratio (m/z 156/140, 140Ce16O+/140Ce+) | 0.5% |
| Doubly charged ratio (m/z 70/140; 140Ce2+/140Ce+) | 1.3% |
| Signal stability (RSD) | 2–3% |
|
|
| Optimized HPLC-ICPMS method | |
| Instruments | Agilent HPLC 1260 Infinity and ICPMS 7700x or ICPMS/MS 8900 |
| Collision gas mode | He (5 mL min−1) |
| Monitored m/z | 51 (V) |
| Integration time | 0.3 s |
| Column | Zorbax SAX, 4.6 × 150 mm, 5 μm |
| Mobile phase | 10 mM citric acid, 10 mM Na2EDTA, pH adjusted to 5.0 with aqueous ammonia |
| Flow rate | 1.0 mL min−1 |
| Column temperature | 13 °C |
| Injection volume | 25 μL |
| Injection speed | 100 μL min−1 |
|
|
| Evaluated parameters during HPLC-ICPMS method optimization (starting values in bold) | |
| Concentrations | 2, 5, 10, 20, 30, 40, 50 mM citric acid |
| pH values | 4.0, 4.5, 5.0, 5.3, 5.5, 6.0, 6.5, 7.0, 7.5 |
| Temperatures | 5, 10, 13, 15, 20, 25, 30. 35, 40, 45 °C |
Ge (m/z 74, 200 μg L−1 in 10% nitric acid) was used as the internal standard. It was added online via a T-piece in front of the nebulizer to the calibration standards and the sample. Its tubing's diameter was 1/4 of the sample tubing.
2.4 HPLC-ICPMS method development
Because amavadin carries two negative charges, an anion-exchange column (Zorbax SAX) and an aqueous mobile phase with ammonium citrate (citric acid, pH adjustment with concentrated aqueous ammonia) were employed. Na2EDTA (10 mM) was added to the mobile phase for complexing all present smaller V species and thus prohibiting unwanted precipitation or interconversion. Different column temperatures (5–45 °C), concentrations of citric acid (2–50 mM) and pH values (4.0–7.5) were tested to optimize the separation method. The evaluated parameters and also the final method are summarized in Table 1. A standard containing 10 μg V L−1 of amavadin and vanadyl acetate was used for the experiments, dissolved and diluted in the initial mobile phase (Table 1, “starting values”).The peaks were integrated at the baseline, with a vertical division at the valley between the two amavadin isomers.
The resolution R of the amavadin's two isomers was calculated according to the following formula, where tR1 and tR2 are the retention times at the peak maxima, and w0.5 h1 and w0.5 h2 are the peak widths at 50% peak height.
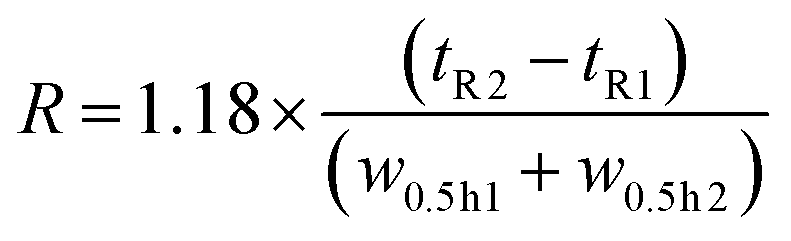
The optimized method was validated by performing a calibration from 0.1 to 100 μg V L−1 (calibration points: 0, 0.1, 0.5, 1, 5, 10, 50, and 100 μg V L−1) of amavadin (sum of both isomers) and vanadyl acetate. The two standards were mixed together and diluted with the mobile phase (Table 1). The lowest standard was injected 5 times to determine the limit of detection (LOD) and limit of quantification (LOQ). The LOD was calculated as three times the standard deviation, and the LOQ ten times the standard deviation of the lowest standard. A standard solution with 10 μg V L−1 of amavadin and vanadyl acetate was injected six times over the course of a 24 hour long measurement to evaluate the method's stability. The SRM 1640a (n = 1) was diluted 1 + 1 with the mobile phase and analyzed with the developed method to determine the method's accuracy.
2.5 Mushroom samples
 | ||
| Fig. 2 Different parts of the fruit body of Amanita muscaria. *The cap flesh is beneath the skin and not visible. | ||
For V speciation analysis, around 2 g of cap, stipe and gills and 1 g of bulb and skin were extracted with 20 mL (10 mL in the case of the bulb and skin) of mobile phase (Table 1). The parts of sample A were extracted in triplicates, the parts of samples B and C only one time each, and three blank samples were prepared. After the addition of the mobile phase, the samples were thoroughly mixed, put in an ultrasonic bath for 20 minutes, centrifuged for 10 minutes, and then filtered through 0.22 μm Nylon filters. The extracts were subjected to HPLC-ICPMS analysis without further dilution. For the determination of the extraction efficiency, the extracts were diluted 1 + 99 with 10% (v/v) nitric acid.
2.5.3 Determination of total V
The total V concentrations in the digests and in the diluted extracts were measured with ICPMS, as already described earlier for the amavadin standard (Section 2.3.2). In addition to the digested SRM 1573a, the SRM 1640a (n = 1) was diluted 1 + 9 (10% (v/v) nitric acid) and analyzed as well.The identity and accuracy of amavadin and vanadyl acetate in the mushroom extracts were confirmed by spiking one sample (gills A) online via a HPLC autosampler with standard solutions. For the confirmation of amavadin, 2 μL of extract were spiked with 20 μL of 100 μg V L−1 of amavadin (see ESI, Fig. S11†). In the case of vanadyl acetate, 20 μL of extract were spiked with 1 μL of a 5 μg V L−1 solution of vanadyl acetate. The ratios were selected to achieve a doubling of the peaks in question.
3 Results and discussion
3.1 Synthesis
Amavadin was synthesized employing a slightly modified version of the procedure reported by Hubregtse et al.32 The ligand was prepared from lactic acid methyl ester, starting with the conversion of the hydroxyl moiety into the corresponding triflate with triflic anhydride in the presence of lutidine, and subsequent nucleophilic substitution with hydroxylamine to give 1 (see Scheme 1). The hydrolysis of the methyl esters in aqueous KOH and precipitation with zinc acetate gave the desired ligand as the corresponding zinc salt. Ion exchange chromatography yielded a free ligand (2), that reacted with vanadyl acetate to form amavadin. A sample of 243 mg of dark blue crystals was obtained this way. Later analyses with ICPMS, HPLC-ICPMS and co-chromatography with vanadyl acetate revealed that 26% (w/w) of the obtained solid still was vanadyl acetate, and 74% (w/w) was amavadin (see also Section 3.2). This means that amavadin was synthesized with a yield of 180 mg, or 73%.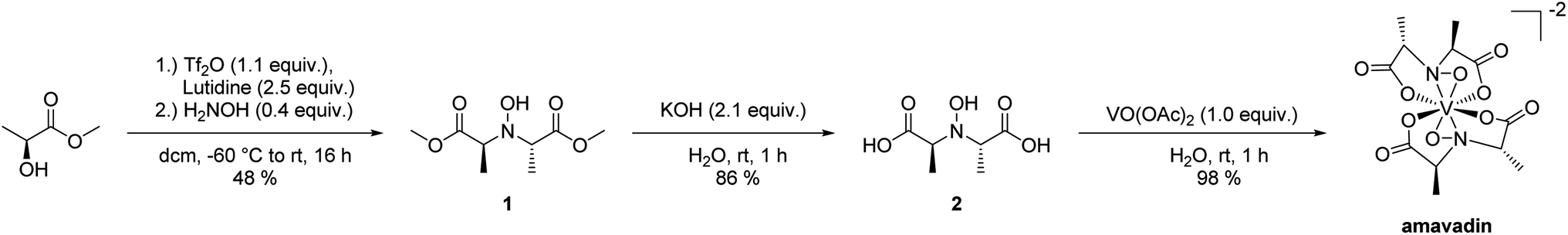 | ||
| Scheme 1 Synthesis of amavadin starting from (L)-lactic acid methyl ester. | ||
Due to the d1 electron configuration of the V(IV) center, amavadin and vanadyl acetate both are paramagnetic, and thus NMR could not be performed on the product. The oxidation of V(IV) to V(V) in DMSO was attempted, as reported in the literature.31 A sample of amavadin in DMSO-d6 was exposed to the atmosphere for several weeks, which caused the solution to turn from blue to red; however still no NMR spectrum was obtained. It is unclear whether amavadin could not be oxidized completely or the presence of paramagnetic vanadyl acetate made the sample immeasurable by standard NMR techniques.
The successful synthesis of amavadin was proven by ESMS and IR-spectroscopy. The IR spectrum (ESI, Fig. S7†) shows distinct peaks at wavenumbers reported in the literature.32 The mass spectrum of amavadin is shown in ESI, Fig. S8 and S9.† The molecule peak for [M + 3H]+ can be seen at m/z 402, besides lower peaks at m/z 401 and 403, and also sodium adducts at m/z 423, 424 and 425 are visible.
3.2 Method development
Because of the negative charges of amavadin, even at very low pH values,22 anion-exchange chromatography was selected as the best option for its chromatographic separation. Citric acid at pH 5.3 (adjusted with ammonia) in ultrapure water was selected as the starting point for the mobile phase. At this pH, citric acid has a dianionic character (pKa values: 3.13, 4.76 and 6.4061), like amavadin, which should increase the elution strength of the buffer.
Amavadin was at first dissolved in ultrapure water. During method development, it was found that there was another V-containing compound in the amavadin standard. The first logical candidate was vanadyl acetate, the precursor that was used in the synthesis of amavadin. Thus, vanadyl acetate was dissolved in the initial mobile phase (Table 1, “starting values”) and analyzed along with amavadin. The first results from ESMS and HPLC-ICPMS showed that the impurity was indeed vanadyl acetate, which was subsequently included in the calibration standards. Quantification via ICPMS and HPLC-ICPMS (in the latter case with vanadyl acetate as the quantification standard and SRM 1640a as the reference material) showed that 43% of the total V of the “amavadin standard” was present in the form of vanadyl acetate (meaning that 26% of the solid material is vanadyl acetate).
Due to the poor solubility of vanadyl acetate in pure water, the amavadin standard was henceforth also dissolved in the mobile phase, to ensure that the vanadyl acetate moiety in the amavadin standard was quantitatively dissolved. In order to have similar concentrations of the two V species, additional vanadyl acetate was added to the amavadin standard. Over the course of method development, the relative peak areas of both species remained constant, which indicates that no interconversion of the two species was taking place.
Na2EDTA was included in the mobile phase to ensure a quantitative elution of vanadyl acetate. This can also be beneficial for other small V species, which were not a target of the developed method, but can nevertheless occur in environmental samples. Vanadyl acetate (that is to say, very likely vanadyl complexed by EDTA) was eluting significantly earlier than amavadin as a clean, symmetric peak at all settings of the method development and will not be further discussed in this context.
Finally, 20 mM citric acid at pH 5.3 (adjusted with ammonia) with 10 mM Na2EDTA was selected as the starting point (see also Table 1). The column oven was set at 25 °C, and the flow rate was 1 mL min−1. With these first settings, a double peak appeared in the chromatogram at 7.1 and 7.5 minutes, corresponding to the two isomers of amavadin. To achieve a good resolution of these two peaks, the buffer concentration and pH value, as well as the column temperature, were optimized. The evaluated conditions are summarized in Table 1, and the resulting chromatograms are shown in Fig. 3.
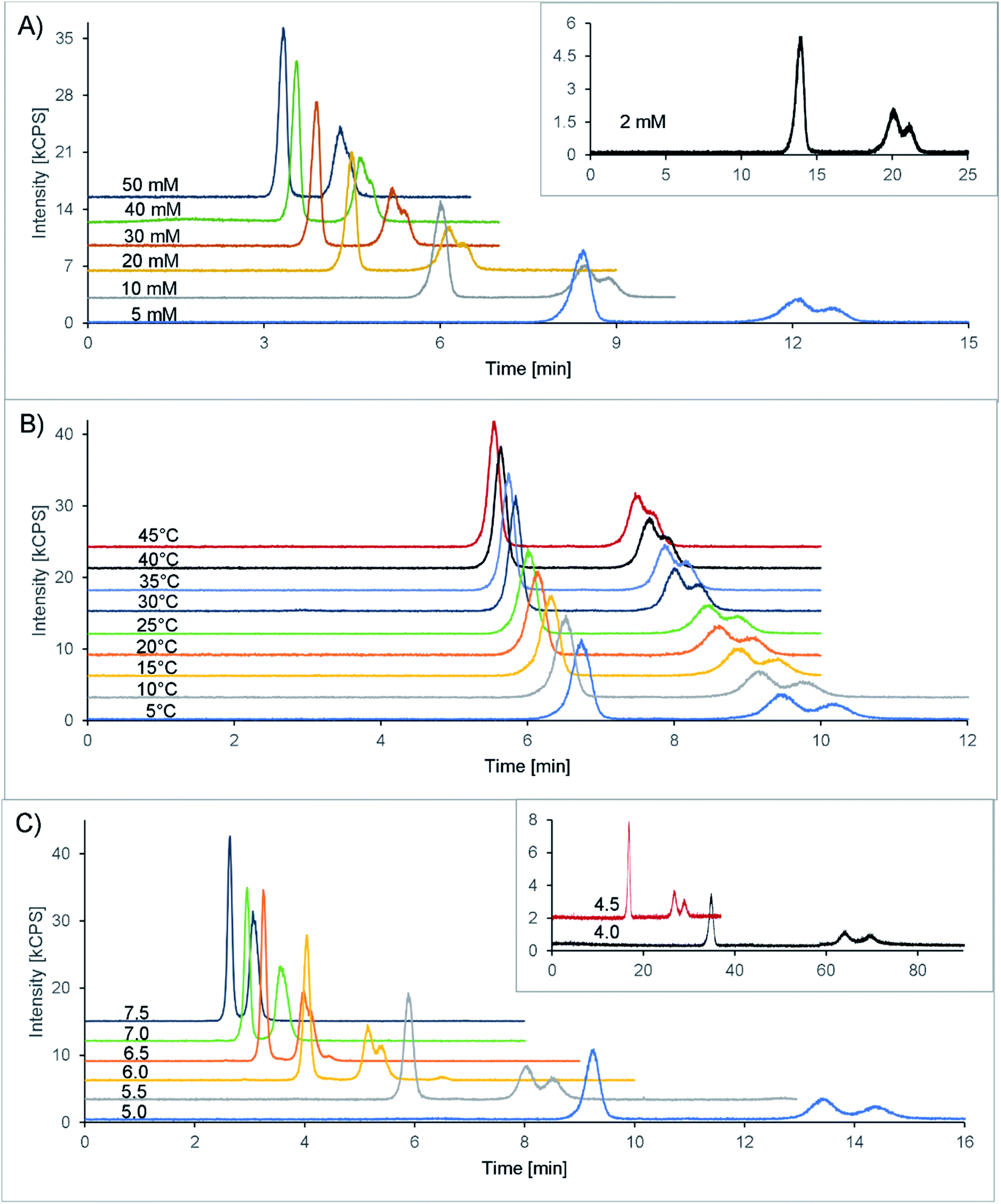 | ||
| Fig. 3 HPLC-ICPMS chromatograms (V at m/z 51) of amavadin (5 μg V L−1, later-eluting double peak, corresponding to the two isomers) and vanadyl acetate (5 μg V L−1, first peak) during method optimization. (A) Varying concentration of citric acid, at 25 °C and pH 5.3. The lowest concentration, 2 mM, is shown in the inset due to the much longer retention times. (B) Varying temperature, at 10 mM citric acid and pH 5.3. (C) Varying pH, at 10 mM citric acid and 10 °C. The chromatograms at pH 4.0 and 4.5 are shown in the inset due to the much longer retention times. Offsets of the y-axis in A, B and C: +3 kcps from the previous line. Exception: +1.5 kcps for pH 4.5 in the inset of C. | ||
The concentration of citric acid was varied between 2 and 50 mM. As can be seen from the resolution values depicted in Fig. 4A and the chromatograms in Fig. 3A, lowering the concentration of the buffer improved the separation of the two isomers. However, this also increased the retention time significantly, up to 20 and 21 minutes at 2 mM citric acid. Based on these findings, we found that a concentration of 10 mM provides a good balance between the retention time (second isomer: 8.9 min at the peak maximum, resulting in a total chromatogram time of 10 min) and resolution (R = 0.56) of the amavadin peaks.
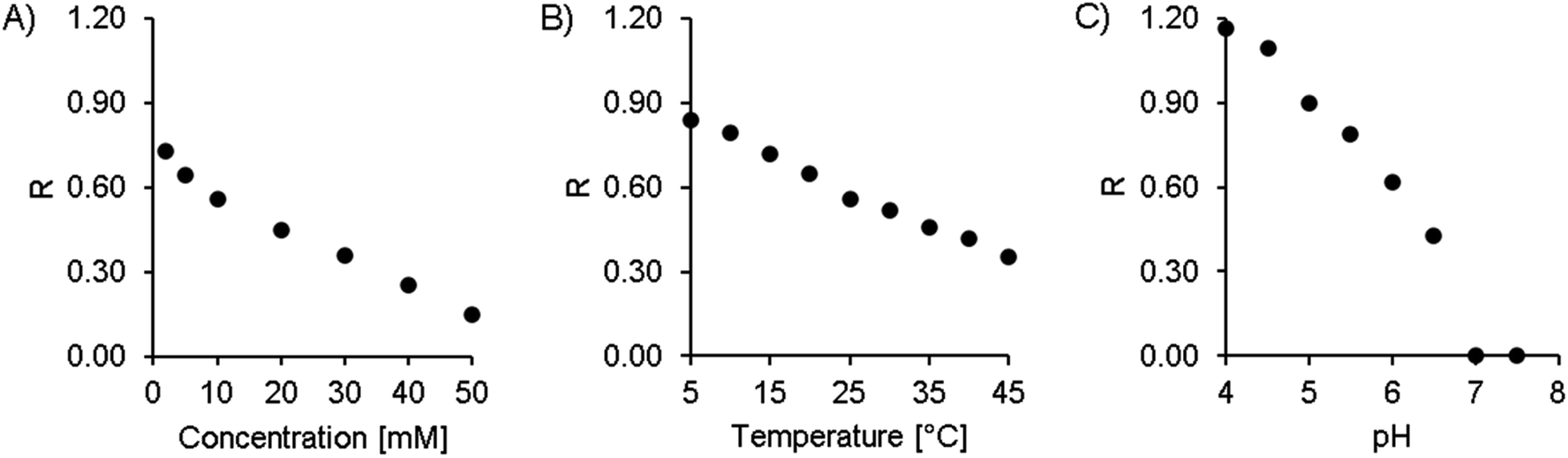 | ||
| Fig. 4 Resolution of the two amavadin isomers at different chromatographic settings. (A) Varying concentration of citric acid, at 25 °C and pH 5.3. (B) Varying temperature, at 10 mM citric acid and pH 5.3. (C) Varying pH, at 10 mM citric acid and 10 °C. | ||
As the next step, the column temperature was varied between 5 and 45 °C. With colder settings, the two peaks were better separated (Fig. 3 and 4B) but also slightly broadened. There was not much difference between 5 and 10 °C, and so 10 °C was selected as the best option (R = 0.80). However, it was later observed that under the given laboratory conditions longer experiments at 10 °C led to condensation in the column oven, which eventually resulted in a leak error in the HPLC. To overcome this problem, the temperature was slightly increased to 13 °C. This enabled a stable measurement for more than 24 hours, with still good resolution.
Finally, the influence of the pH value on the separation was evaluated. As can be seen in Fig. 3C and 4C, the two isomers were not separated any more at pH values above 6.5. Lower pH values led to a higher resolution, but while the retention times were 13.4 and 14.4 minutes at pH 5.0 (R = 0.90), they increased dramatically to over 26 minutes at pH 4.5 and more than 60 minutes at pH 4.0. This is not surprising, because citric acid will only have one negative charge at those pH values, while amavadin will still have a charge of −2.
In summary, as can also be seen in Table 1, the final method included a Zorbax SAX (4.6 × 150 mm 5 μm) silica-based anion-exchange column and a mobile phase of 10 mM citric acid, pH 5.0, and 10 mM Na2EDTA. The pH was adjusted with ammonia. The flow rate was 1.0 mL min−1 and the column temperature 13 °C. The injection volume was set to 25 μL, and the injection speed to 100 μL min−1.
Exemplary chromatograms of 10 and 0.1 μg V L−1 are drawn in Fig. 5. Note that the retention times are a bit longer than in the chromatograms obtained during method development (Fig. 3). An already used column was employed during method development. To make sure that no previous use had led to a modification of the stationary phase and therefore influence the separation of amavadin, a virgin column was used for method validation and application to mushroom samples. As mentioned, this led to an increase in the retention times, but the resolution of the two isomers was not affected.
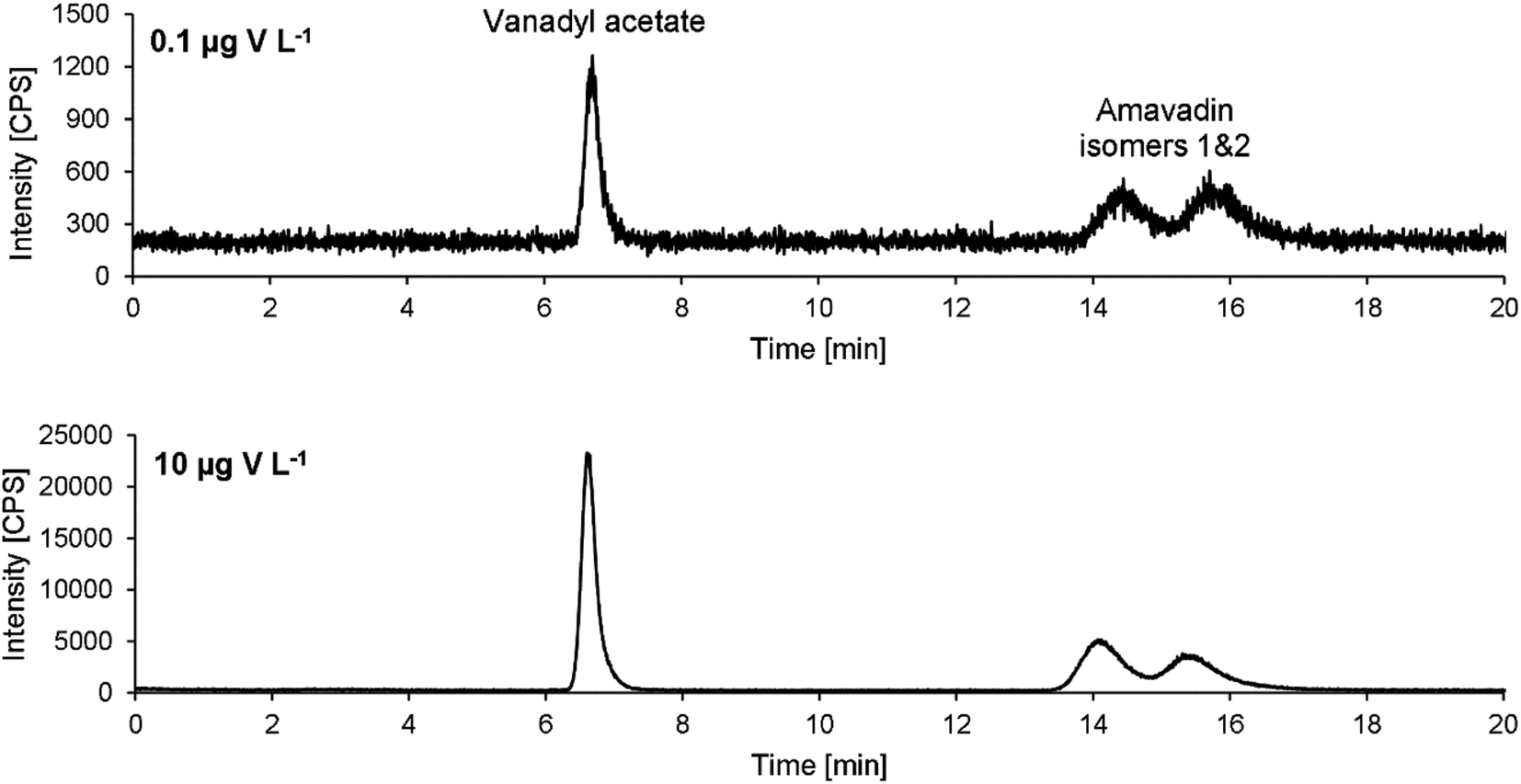 | ||
| Fig. 5 HPLC-ICPMS chromatograms of V (m/z 51) of the lowest (0.1 μg V L−1) and a medium concentrated (10 μg V L−1) standard of amavadin and vanadyl acetate, obtained with the optimized method parameters. | ||
3.3 Method validation
Although the two isomers of amavadin were almost baseline-separated, they were considered as one compound for method validation and calibration. The values obtained for the individual isomers were used to calculate the ratio of the two isomers.The method's figures of merit are listed in Table 2. They include the LOD, LOQ, correlation coefficient R, stability of a 10 μg V L−1 standard during a 24 hour measurement and the average repeatability of three replicates of the five mushroom parts of sample A. The low LODs of 0.04 and 0.05 μg V L−1 for vanadyl acetate and amavadin, respectively, allow the detection of traces of amavadin or similar species. With a dilution factor of 10 (due to the extraction process), the LODs for mushroom samples are still below 1 μg V kg−1 (namely 0.4 and 0.5 μg V kg−1).
| LOD [μg V L−1] | LOQ [μg V L−1] | R | Stability [%] | Repeatability [%] | Recovery [%] | |
|---|---|---|---|---|---|---|
| Vanadyl acetate | 0.04 | 0.13 | 0.9999 | 2.8 | 4.6 ± 2.1 | 90 |
| Amavadin | 0.05 | 0.17 | 0.9999 | 2.6 | 3.7 ± 1.7 | 105 |
The method is very stable; the detected amavadin concentrations only vary by around 2.6% (2.8% for vanadyl acetate) in the re-measured standard. Also, for the mushroom samples, the repeatability is lower than 5%. The amavadin concentrations in the three replicates of sample A varied between 1.7 and 5.6% for the five different fruit-body parts, while the variations of the vanadyl acetate concentrations were 4.6 ± 2.1%. The highest relative standard deviations (RSDs) were found in the skin sample (5.6% and 7.8% for amavadin and vanadyl acetate, respectively), which can be explained by the difficult homogenization of this part of the mushroom. The accuracy of the measured SRM, NIST 1640a, was excellent, namely 98.4% of the total V. The V speciation in this SRM is not defined, but V had the same retention time as vanadyl acetate (probably eluted as vanadyl EDTA). The method's accuracy was further confirmed by the recoveries after online spiking of an extract (gills A) with amavadin and vanadyl acetate, which were 90 and 105%, respectively. Another indirect proof of the method's accuracy is the quantitative column recovery of the mushroom extracts (see below, Section 3.4.3).
3.4 Mushroom samples
Concerning the samples of Amanita muscaria, the highest V concentrations, namely 38–97 mg kg−1, were found in the bulb samples. In the other samples, the V concentrations were between 6.4 and 33 mg kg−1. Within each mushroom, the concentrations in the stipe and gills were very similar. Slightly lower concentrations were found in the skin, and the caps had the lowest V concentrations among all fruit-body parts. All total V concentrations, along with the extractable V concentrations and V species, are listed in Table 3.
| Sample | Total V | Extractable V | Extraction efficiency | Amavadin | Vanadyl acetatea | Unknown 1 | Other species |
|---|---|---|---|---|---|---|---|
| a Species detected at the same retention time as the vanadyl acetate standard (vanadyl EDTA). b Traces: between the LOD and LOQ (0.0004 and 0.0013 mg V kg−1). | |||||||
| Skin A | 8.5 ± 0.6 | 4.2 ± 0.3 | 49 ± 4 | 4.0 ± 0.2 | 0.0040 ± 0.0003 | 0.27 ± 0.01 | 0.32 ± 0.03 |
| Cap A | 6.4 ± 0.3 | 5.1 ± 0.3 | 80 ± 5 | 4.8 ± 0.2 | Tracesb | 0.32 ± 0.03 | 0.19 ± 0.02 |
| Gills A | 13.5 ± 0.3 | 9.3 ± 0.1 | 69 ± 1 | 8.1 ± 0.1 | 0.0035 ± 0.0001 | 0.50 ± 0.03 | 0.20 ± 0.01 |
| Stipe A | 16.4 ± 2.2 | 11.0 ± 0.8 | 67 ± 5 | 10.3 ± 0.6 | 0.037 ± 0.001 | 0.48 ± 0.04 | 0.28 ± 0.09 |
| Bulb A | 38.1 ± 0.4 | 31.0 ± 1.0 | 81 ± 3 | 26.2 ± 0.8 | 0.60 ± 0.01 | 1.21 ± 0.03 | 0.59 ± 0.13 |
| Skin B | 18.0 ± 1.3 | 11 | 63 | 11 | 0.0088 | 0.63 | 0.26 |
| Cap B | 13.1 ± 0.4 | 11 | 85 | 11 | 0.0030 | 0.63 | 0.17 |
| Gills B | 25.6 ± 0.8 | 20 | 79 | 16 | 0.0048 | 0.92 | 0.27 |
| Stipe B | 25.3 ± 0.4 | 18 | 70 | 17 | 0.075 | 0.71 | 0.31 |
| Bulb B | 58.1 ± 1.6 | 51 | 88 | 38 | 1.8 | 1.6 | 1.1 |
| Skin C | 19.5 ± 0.9 | 12 | 60 | 11 | < 0.0004 | 0.92 | 0.35 |
| Cap C | 18.1 ± 0.5 | 13 | 73 | 12 | 0.0033 | 0.66 | 0.34 |
| Gills C | 33.0 ± 1.2 | 23 | 71 | 22 | 0.0086 | 1.1 | 0.58 |
| Stipe C | 27.5 ± 0.7 | 23 | 84 | 21 | 0.068 | 0.82 | 0.51 |
| Bulb C | 97.4 ± 0.7 | 90 | 93 | 83 | 1.4 | 2.9 | 1.2 |
Overall, 74 ± 12% of the total V was extractable with the mobile phase. The repeatability of the samples of fruit-body A was between 1 and 7% (three replicate extractions per sample). The bulb samples had the highest extraction efficiencies of 81–93%, while the lowest extraction efficiencies were found for the skin samples, with 50–63%. It will be interesting to see in future studies whether an extended extraction time or higher temperature can lead to an increased extraction efficiency in Amanita muscaria samples.
Besides amavadin, other V-containing compounds could be detected in the extracts as well. A typical chromatogram of a bulb sample is drawn in Fig. 6, and the concentrations are given in Table 3.
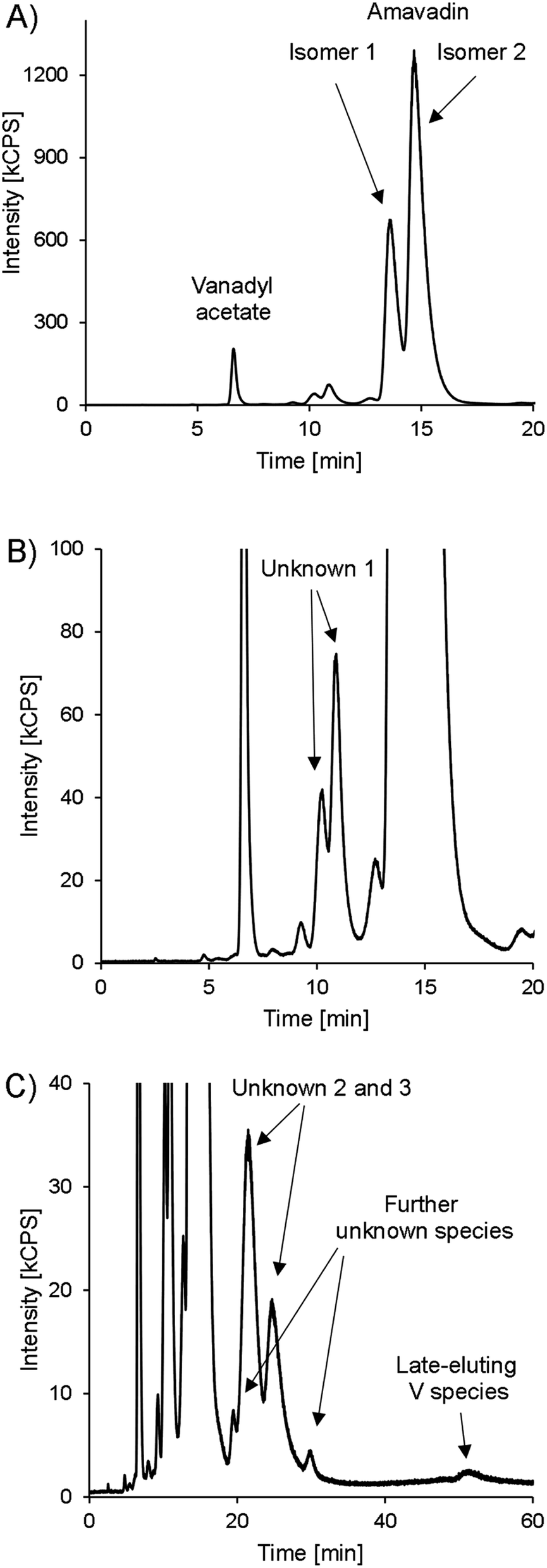 | ||
| Fig. 6 HPLC-ICPMS chromatogram of V (m/z 51) in bulb A, presented in different variants. (A) 0–20 minutes. (B) 0–20 minutes, zoomed in to see lower abundant species. (C) 0–60 minutes, zoomed in. | ||
The peak representing vanadyl acetate was present in most samples, but usually at low concentrations, accounting for only 0.02–0.43% of the extractable V except in the bulb samples, where it accounted for up to 1.8 mg V kg−1 (3.4% of the extractable V). As already mentioned earlier, the vanadyl moiety of vanadyl acetate was very likely complexed by EDTA. In real samples, the same will apply to any present vanadyl ions, and they will show the same retention behavior as vanadyl acetate. This was confirmed by preliminary experiments in our lab. For future investigations on environmental V speciation, it will be important to evaluate the behavior of small inorganic V species with our method systematically, but it was beyond the scope of the present study.
At around 10 minutes, another double peak can be found (Fig. 6B). It accounted for 3.1–4.4% of the extractable V (“unknown 1”). The highest relative concentrations were found in the skin samples, with 5.6–7.8% of the extractable V, while it accounted for only 3.4 ± 0.4% of the extracted V in the bulb and 4.0 ± 0.4% in the stipe samples. In gills and cap samples, between 4.6 and 6.2% of the extractable V was present as unknown 1. The peak's shape was very similar to that of amavadin, albeit at a much lower concentration. The chemical structure of this compound(s) is unknown, but it can be speculated that it is related to amavadin, for example a precursor or homologue, and is also present in two isomeric forms. The ratio of the second to the first peak in bulb and stipe samples is a little lower than in the case of amavadin, namely 1.6–2.1. In the cap, gills and skin samples, however, the ratio is between 2.2 and 2.6.
During the measurement of the first samples, it was observed that the extracts also contain V species that are retained much longer on the column than amavadin. Therefore, replicate 1 of each part of fruit-body A was injected again and monitored for 60 minutes instead of 20. The whole chromatogram of the bulb sample is shown in Fig. 6C, and the other chromatograms can be found in ESI, Fig. S11.† In all parts of Amanita muscaria, we detected one to two peaks between around 20 and 30 minutes (“unknown 2 and 3”). They only accounted for around 0.15% of the extracted V in the skin, cap and gills, but were more abundant in the stipe (1.8% of the extracted V) and bulb (5.7%), where also two more small peaks appeared at around 20 and 30 minutes, respectively. Finally, at 52 minutes, a small peak could be found in all five chromatograms. The compound's V concentration was very low, only 0.01–0.13% of the extracted V, but it would still be interesting to explore the identity of this extremely strong retained species in the future.
4 Conclusion and outlook
We synthesized amavadin and developed a sensitive anion-exchange HPLC-ICPMS method for the separation and detection of amavadin and its two diastereoisomers with a limit of quantification of 0.17 μg V L−1.We applied the method on the extracts of fruit-body samples of Amanita muscaria. 75–96% of the extracted V was found in the form of amavadin. We also found other V species in the extracts, of which one was eluting with a similar double-peak profile to the two diastereoisomers of amavadin. Additionally, long retained V containing signals were detected in all the investigated parts of the mushroom (bulb, stipe, cap, skin, and gills).
Our results add valuable and unique information to our knowledge on amavadin in mushrooms. The new method allows vanadium speciation analysis at trace concentrations in mushrooms and other environmental samples.
Until now, it was assumed that only Amanita muscaria and its few closely related species contain amavadin, because of their extraordinary V concentrations. With the developed method, it can be explored whether traces of amavadin are present in other environmental samples as well. This will help in elucidating the biogeochemical pathway of V and its role in the environment.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors want to thank Evelyn Waldhauser, Antonia Toedling and Linda Maierhofer for their support in the lab. The authors acknowledge the financial support by the University of Graz.References
- Agency for Toxic Substances and Disease Registry (ATSDR), Toxicological Profile for Vanadium, U.S. Department of Health and human Services, Public Health Service, Atlanta, GA, 2012 Search PubMed.
- D. Rehder, Vanadium in health issues, ChemTexts, 2018, 4, 20 CrossRef.
- D. Rehder, The future of/for vanadium, Dalton Trans., 2013, 42, 11749–11761 RSC.
- S. Treviño and A. Diaz, Vanadium and insulin: Partners in metabolic regulation, J. Inorg. Biochem., 2020, 208, 111094 CrossRef PubMed.
- D. Rehder, The potentiality of vanadium in medicinal applications, Inorg. Chim. Acta, 2020, 504, 119445 CrossRef CAS.
- T. I. Fortoul, V. Rodriguez-Lara, A. González-Villalva, M. Rojas-Lemus, G. Cano-Gutiérrez, M. Ustarroz-Cano, L. Colín-Barenque, P. Bizarro-Nevares, I. García-Pealez, L. F. Montaño, R. S. Jimenez-Martinez, N. Lopez-Valdez, M. L. Ruiz-Guerrero, N. A. Meléndez-García, F. A. García-Ibarra, V. Martínez-Baez, D. Z. Alfaro, A. Muñiz-Rivera-Cambas, L. S. López-Zepeda, E. M. Quezada-Maldonado and S. Cervantes-Yépez, Inhalation of vanadium pentoxide and its toxic effects in a mouse model, Inorg. Chim. Acta, 2014, 420, 8–15 CrossRef CAS.
- F. L. Assem and L. S. Levy, A review of current toxicological concerns on vanadium pentoxide and other vanadium compounds: Gaps in knowledge and directions for future research, J. Toxicol. Environ. Health, Part B, 2009, 12, 289–306 CAS.
- K. Gruzewska, A. Michno, T. Pawelczyk and H. Bielarczyk, Essentiality and toxicity of vanadium supplements in health and pathology, J. Physiol. Pharmacol., 2014, 65, 603–611 CAS.
- J. P. Gustafsson, Vanadium geochemistry in the biogeosphere –speciation, solid-solution interactions, and ecotoxicity, Appl. Geochem., 2019, 102, 1–25 CrossRef CAS.
- T. Ueki, N. Yamaguchi, Romaidi, Y. Isago and H. Tanahashi, Vanadium accumulation in ascidians: A system overview, Coord. Chem. Rev., 2015, 301–302, 300–308 CrossRef CAS.
- J. Costa Pessoa, E. Garribba, M. F. A. Santos and T. Santos-Silva, Vanadium and proteins: Uptake, transport, structure, activity and function, Coord. Chem. Rev., 2015, 301–302, 49–86 CrossRef CAS.
- D. Rehder, The role of vanadium in biology, Metallomics, 2015, 7, 730–742 CrossRef CAS PubMed.
- Z. Řanda and J. Kučera, Trace elements in higher fungi (mushrooms) determined by activation analysis, J. Radioanal. Nucl. Chem., 2004, 259, 99–107 CrossRef.
- P. Kalač, Trace element contents in European species of wild growing edible mushrooms: A review for the period 2000–2009, Food Chem., 2010, 122, 2–15 CrossRef.
- J. Vetter, Vanadium content of some common edible, wild mushroom species, Acta Aliment., 1999, 28, 39–48 CAS.
- J. Falandysz, T. Kunito, R. Kubota, K. Lipka, A. Mazur, J. J. Falandysz and S. Tanabe, Selected elements in fly agaric Amanita muscaria, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2007, 42, 1615–1623 CrossRef CAS PubMed.
- E. Koch, H. Kneifel and E. Bayer, Das Vorkommen von Amavadin in Pilzen der Gattung Amanita [in German], Z. Naturforsch., 1987, 873–878 CrossRef CAS.
- J. Vetter, Mineral composition of basidiomes of Amanita species, Mycol. Res., 2005, 109, 746–750 CrossRef CAS PubMed.
- H.-U. Meisch, J. A. Schmitt and W. Reinle, Schwermetalle in Höheren Pilzen, III * Vanadium und Molybdän/Heavy Metals in Higher Fungi, II I. Vanadium and Molybdenum, Z. Naturforsch., C: J. Biosci., 1978, 33, 1–6 CrossRef.
- R. D. Gillard and R. J. Lancashire, Electron spin resonance of vanadium in Amanita muscaria, Phytochemistry, 1984, 23, 179–180 CrossRef CAS.
- H.-U. Meisch, W. Reinle and J. A. Schmitt, High vanadium content in mushrooms is not restricted to the fly agaric (Amanita muscaria), Sci. Nat., 1979, 66, 620–621 CrossRef CAS.
- E. Bayer and H. Kneifel, Isolation of Amavadin, a Vanadium Compound Occurring in Amanita Muscaria, Z. Naturforsch., B: Anorg. Chem., Org. Chem., Biochem., Biophys., Biol., 1972, 27, 207 CrossRef CAS.
- H. Kneifel and E. Bayer, Determination of the Structure of the Vanadium Compound, Amavadin, from Fly Agaric, Angew. Chem., Int. Ed., 1973, 12, 508 CrossRef.
- C. D. Garner, E. M. Armstrong, R. E. Berry, R. L. Beddoes, D. Collison, J. J. A. Cooney, S. N. Ertok and M. Helliwell, Investigations of Amavadin, J. Inorg. Biochem., 2000, 80, 17–20 CrossRef CAS PubMed.
- J. A. L. da Silva, F. da Silva, J. R. João and A. J. L. Pombeiro, Amavadin, a vanadium natural complex: Its role and applications, Coord. Chem. Rev., 2013, 257, 2388–2400 CrossRef CAS.
- F. J. J. R. da Silva, Vanadium in biology—the case of the Amanita toadstools, Chem. Speciation Bioavailability, 1989, 1, 139–150 CrossRef.
- P. Krauß, E. Bayer and H. Kneifei, Elektronenspinresonanz-Untersuchungen an Amavadin, einem vanadiumhaltigen Naturstoff/Electron Spin Resonance Investigations of Amavadin, a Natural Product Containing Vanadium, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1984, 39, 829–832 CrossRef.
- H. Kneifel and E. Bayer, Stereochemistry and total synthesis of amavadin, the naturally occurring vanadium compound of Amanita muscaria, J. Am. Chem. Soc., 1986, 108, 3075–3077 CrossRef CAS.
- E. Bayer, E. Koch and G. Anderegg, Amavadin, an Example for Selective Binding of Vanadium in Nature: Studies of Its Complexation Chemistry and a New Structural Proposal, Angew. Chem., Int. Ed. Engl., 1987, 26, 545–546 CrossRef.
- E. M. Armstrong, R. L. Beddoes, L. J. Calviou, J. M. Charnock, D. Collison, N. Ertok, J. H. Naismith and C. D. Garner, The Chemical Nature of Amavadin, J. Am. Chem. Soc., 1993, 115, 807–808 CrossRef CAS.
- E. M. Armstrong, D. Collison, N. Ertok and C. D. Garner, NMR studies on natural and synthetic Amavadin, Talanta, 2000, 53, 75–87 CrossRef CAS PubMed.
- T. Hubregtse, E. Neeleman, T. Maschmeyer, R. A. Sheldon, U. Hanefeld and I. W. C. E. Arends, The first enantioselective synthesis of the amavadin ligand and its complexation to vanadium, J. Inorg. Biochem., 2005, 99, 1264–1267 CrossRef CAS PubMed.
- T. Hubregtse, H. Kooijman, A. L. Spek, T. Maschmeyer, R. A. Sheldon, I. W. C. E. Arends and U. Hanefeld, Study on the isomerism in meso-amavadin and an amavadin analogue, J. Inorg. Biochem., 2007, 101, 900–908 CrossRef CAS PubMed.
- C. M. M. Matoso, A. J. L. Pombeiro, J. J. R. F. da Silva, M. F. C. G. da Silva, J. A. L. da Silva, J. L. Baptista-Ferreira and F. Pinho-Almeida, in Vanadium Compounds, ed. A. S. Tracey and D. C. Crans, American Chemical Society, Washington, DC, 1998, vol. 711, pp. 241–247 Search PubMed.
- M. Domarus, M. L. Kuznetsov, J. Marçalo, A. J. L. Pombeiro and J. A. L. da Silva, Amavadin and Homologues as Mediators of Water Oxidation, Angew. Chem., Int. Ed., 2016, 55, 1489–1492 CrossRef CAS PubMed.
- V. Ugone, D. Sanna, G. Sciortino, D. C. Crans and E. Garribba, ESI-MS Study of the Interaction of Potential Oxidovanadium(IV) Drugs and Amavadin with Model Proteins, Inorg. Chem., 2020, 59, 9739–9755 CrossRef CAS PubMed.
- J.-H. Huang, F. Huang, L. Evans and S. Glasauer, Vanadium: Global (bio)geochemistry, Chem. Geol., 2015, 417, 68–89 CrossRef CAS.
- K. Pyrzyńska and T. Wierbicki, Determination of vanadium species in environmental samples, Talanta, 2004, 64, 823–829 CrossRef PubMed.
- G. P. Dechaine and M. R. Gray, Chemistry and Association of Vanadium Compounds in Heavy Oil and Bitumen, and Implications for Their Selective Removal, Energy Fuels, 2010, 24, 2795–2808 CrossRef CAS.
- T. Jakusch, J. Costa Pessoa and T. Kiss, The speciation of vanadium in human serum, Coord. Chem. Rev., 2011, 255, 2218–2226 CrossRef CAS.
- T. Iglesias-González, C. Sánchez-González, M. Montes-Bayón, J. Llopis-González and A. Sanz-Medel, Absorption, transport and insulin-mimetic properties of bis(maltolato)oxovanadium (IV) in streptozotocin-induced hyperglycemic rats by integrated mass spectrometric techniques, Anal. Bioanal. Chem., 2012, 402, 277–285 CrossRef PubMed.
- K. De Cremer, M. van Hulle, C. Chéry, R. Cornelis, K. Strijckmans, R. Dams, N. Lameire and R. Vanholder, Fractionation of vanadium complexes in serum, packed cells and tissues of Wistar rats by means of gel filtration and anion-exchange chromatography, J. Biol. Inorg Chem., 2002, 7, 884–890 CrossRef CAS PubMed.
- V. Nischwitz, J. T. Davies, D. Marshall, M. González, J. L. Gómez Ariza and H. Goenaga-Infante, Speciation studies of vanadium in human liver (HepG2) cells after in vitro exposure to bis(maltolato)oxovanadium(IV) using HPLC online with elemental and molecular mass spectrometry, Metallomics, 2013, 5, 1685 CrossRef CAS PubMed.
- N. Kilibarda, S. E. Afton, J. M. Harrington, F. Yan and K. E. Levine, Rapid speciation and determination of vanadium compounds using ion-pair reversed-phase ultra-high-performance liquid chromatography inductively coupled plasma-sector field mass spectrometry, J. Chromatogr. A, 2013, 1304, 121–126 CrossRef CAS PubMed.
- X. S. Li and X. C. Le, Speciation of vanadium in oils and coke and bacterial culture by high performance liquid chromatography inductively coupled plasma mass spectrometry, Anal. Chim. Acta, 2007, 602, 17–22 CrossRef CAS PubMed.
- T. V. Komarova, O. N. Obrezkov and O. A. Shpigun, Ion chromatographic behaviour of anionic EDTA complexes of vanadiurn(IV) and vanadium(V), Anal. Chim. Acta, 1991, 254, 61–63 CrossRef CAS.
- L. Minelli, E. Veschetti, S. Giammanco, G. Mancini and M. Ottaviani, Vanadium in Italian waters: monitoring and speciation of V(IV) and V(V), Microchem. J., 2000, 67, 83–90 CrossRef CAS.
- F. Aureli, S. Ciardullo, M. Pagano, A. Raggi and F. Cubadda, Speciation of vanadium(IV) and (V) in mineral water by anion exchange liquid chromatography-inductively coupled plasma mass spectrometry after EDTA complexation, J. Anal. At. Spectrom., 2008, 23, 1009–1016 RSC.
- M. A. Larsson, M. D'Amato, F. Cubadda, A. Raggi, I. Öborn, D. B. Kleja and J. P. Gustafsson, Long-term fate and transformations of vanadium in a pine forest soil with added converter lime, Geoderma, 2015, 259–260, 271–278 CrossRef CAS.
- M. Colina, P. H. E. Gardiner, Z. Rivas and F. Troncone, Determination of vanadium species in sediment, mussel and fish muscle tissue samples by liquid chromatography-inductively coupled plasma-mass spectrometry, Anal. Chim. Acta, 2005, 538, 107–115 CrossRef CAS.
- Z. L. Chen and G. Owens, Trends in speciation analysis of vanadium in environmental samples and biological fluids—A review, Anal. Chim. Acta, 2008, 607, 1–14 CrossRef CAS PubMed.
- Z. Chen and R. Naidu, On-column complexation and simultaneous separation of vanadium(IV) and vanadium(V) by capillary electrophoresis with direct UV detection, Anal. Bioanal. Chem., 2002, 374, 520–525 CrossRef CAS PubMed.
- K. Pyrzyńska and T. Wierzbicki, Solid-phase extraction for preconcentration and separation of vanadium species in natural waters, Microchim. Acta, 2004, 147, 59–64 CrossRef.
- J.-Y. Yang and Y. Tang, Accumulation and Biotransformation of Vanadium in Opuntia microdasys, Bull. Environ. Contam. Toxicol., 2015, 94, 448–452 CrossRef CAS PubMed.
- D. Wang and S. A. Sañudo-Wilhelmy, Development of an analytical protocol for the determination of V(IV) and V(V) in seawater: Application to coastal environments, Mar. Chem., 2008, 112, 72–80 CrossRef CAS.
- M. Žižić, T. Dučić, D. Grolimund, D. Bajuk-Bogdanović, M. Nikolic, M. Stanić, S. Križak and J. Zakrzewska, X-ray absorption near-edge structure micro-spectroscopy study of vanadium speciation in Phycomyces blakesleeanus mycelium, Anal. Bioanal. Chem., 2015, 407, 7487–7496 CrossRef PubMed.
- P. Frank, K. Kustin, W. E. Robinson, L. Linebaugh and K. O. Hodgson, Nature and Ligation of Vanadium within Whole Blood Cells and Henze Solution from the Tunicate Ascidia ceratodes, As Investigated by Using X-ray Absorption Spectroscopy, Inorg. Chem., 1995, 34, 5942–5949 CrossRef CAS.
- M. Jensen-Fontaine, W. P. Norwood, M. Brown, D. G. Dixon and X. C. Le, Uptake and Speciation of Vanadium in the Benthic Invertebrate Hyalella azteca, Environ. Sci. Technol., 2014, 48, 731–738 CrossRef CAS PubMed.
- V. T. Kütter, M. Montes-Bayón, S. M. Sella, A. Sanz-Medel and E. V. Silva-Filho, Vanadium-binding protein in marine plankton from tropical south atlantic ocean, J. Braz. Chem. Soc., 2014, 25, 1116–1123 Search PubMed.
- D. C. Crans, J. J. Smee, E. Gaidamauskas and L. Yang, The Chemistry and Biochemistry of Vanadium and the Biological Activities Exerted by Vanadium Compounds, Chem. Rev., 2004, 104, 849–902 CrossRef CAS PubMed.
- A. M. N. Silva, X. Kong and R. C. Hider, Determination of the pKa value of the hydroxyl group in the α-hydroxycarboxylates citrate, malate and lactate by 13C NMR: implications for metal coordination in biological systems, BioMetals, 2009, 22, 771–778 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ja00518e |
| ‡ Current address: Atomic & Mass Spectrometry Research Unit, Department of Chemistry, Ghent University, Krijgslaan 281-S12, 9000 Ghent, Belgium. |
| This journal is © The Royal Society of Chemistry 2021 |