
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSensitive determination of mercury by magnetic dispersive solid-phase extraction combined with flow-injection-cold vapour-graphite furnace atomic absorption spectrometry†
J. C.
García-Mesa
,
P.
Montoro-Leal
,
S.
Maireles-Rivas
,
M. M.
López Guerrero
 * and
E.
Vereda Alonso
*
* and
E.
Vereda Alonso
*
Department of Analytical Chemistry, Faculty of Sciences, University of Malaga, Campus of Teatinos, 29071 Málaga, Spain. E-mail: eivereda@uma.es; mmlopez@uma.es
First published on 31st March 2021
Abstract
Mercury is a non-essential trace element that is toxic to humans due to the bioaccumulation effect. In this work, a ferrofluid based on Fe3O4@graphene oxide nanospheres together with an ionic liquid was used to develop a magnetic dispersive solid-phase extraction (MDSPE) method for the extraction of the complex formed between the chelating agent methyl thiosalicylate (MTS) and mercury. This MDSPE methodology was combined with an automatic analysis by flow injection-cold vapour-graphite furnace atomic absorption spectrometry (FI-CV-GFAAS). The developed semiautomatic method was applied to the determination of ultra-trace amounts of Hg(II) in biological and environmental samples. Several analytical parameters for MDSPE and FI-CV-GFAAS, such as pH, MTS concentration, eluent composition, extraction time, etc., were optimized by uni and multivariate methodologies. Under the optimized conditions, the %RSD, detection limit and determination limit were 2.9%, 0.25 ng L−1 and 4.9 ng L−1, respectively. The achieved preconcentration factor with the MDSPE methodology was 250. The accuracy of the proposed method was verified using a Standard Reference Material (Mussel Tissue SRM 2976) and by determining the analyte content in spiked seawater and tap water samples collected from Málaga and Cádiz (Spain). The determined values were in good agreement with the certified values and the recoveries for the spiked samples were close to 100% in all cases. The results showed that the proposed method is simple, rapid, environmentally friendly, highly sensitive and accurate for determination of mercury in biological and environmental samples.
1. Introduction
Mercury is one of the most toxic elements, and its presence in the environment arises from anthropogenic and natural sources such as geothermal activity, volcanic events, weathering of rocks, coal and oil combustion, waste incineration, agricultural activities, metal refining and manufacturing.1–3 Upon exposure to mercury above the permissible limits, the human nervous system, immune system, brain, kidneys, heart, stomach and lungs can be seriously damaged.4 The World Health Organization (WHO) has set a limit of 1.6 μg per kg body weight per week for total Hg due to the high bioaccumulation factor.5 For this reason, determination of mercury is an important subject for analytical scientists and environmentalists. For example, the Spanish government has established a maximum Hg concentration of 0.07 μg L−1 for continental superficial waters (RD 817/2015).6 Moreover, some environmental samples present complex and highly saline matrices (seawater), with other transitional metals and noble metals,7 which may seriously affect the results. Due to the trace or ultra-trace level concentrations of mercury in environmental samples, highly sensitive analytical techniques are required, which can be combined with preconcentration/separation treatments in order to improve the sensitivity of the detection technique and remove matrix interferences. Mercury determination is performed by using different methods, such as inductively coupled plasma mass spectrometry (ICP-MS), cold vapour atomic fluorescence spectrometry (CV-AFS) and cold vapour atomic absorption spectrometry (CV-AAS).8 Thus, the most common pre-treatment used for mercury determination is cold vapour generation (CV)9 due to its simplicity, speed and possible automation of the detection system.10 In addition to CV, a huge number of green analytical procedures have been reported in the bibliography for the preconcentration treatments, for example, solid-phase extraction (SPE)11–13 and liquid–liquid extraction (LLE).14,15The SPE methods present excellent properties such as rapid phase separation, high selectivity, low cost, less use of organic solvents, simple extraction, high recovery, high preconcentration factor and automation of more detection techniques.16–19 Magnetic SPE (MSPE) is a new type of SPE developed by Šafaříková and Šafarík for enriching pollutants with magnetic materials.20 In MSPE, a magnetic adsorbent is added to the solution containing the target analytes. After adsorption of the analytes, the adsorbent is separated from the solution using an external magnetic field. Thus, filtration and centrifugation processes are avoided.21–23
The exploration of new magnetic nanomaterials by combining magnetic inorganic materials and non-magnetic adsorbents is an active research area in MSPE. The best adsorbents are nanomaterials due to their main advantages such as high surface-to-volume ratio, easy derivation procedures and unique properties. Among non-magnetic adsorbents, graphene oxide (GO) is characterized by being cheap and easily scalable to a high-volume production. In addition, GO is well-suited for chemical modification and subsequent processing.24 This sorbent possesses a large surface area and a high density of oxygen-containing polar functional groups on the surface (epoxy, carboxylic acid, carbonyl and hydroxyl groups), as well as a rich delocalized π–π electron systems that make it interact strongly with organic compounds with benzene rings.25 GO can be modified with magnetic nanoparticles (MNPs) for use in MSPE, which can reduce the equilibrium time due to the fast mass transfer. Different magnetic nanomaterials based on graphene have been used successfully as adsorbents for preconcentration and determination of mercury, such as Fe3O4/GO26 and graphene/ZnFe2O4.27 On the other hand, nowadays, the preconcentration steps tend towards miniaturization, so solid-phase microextraction (SPME),28 dispersive liquid–liquid microextraction (DLLME),29 single-drop microextraction,30 and so on are being widely used as extraction techniques.7
Magnetic dispersive solid-phase extraction (MDSPE) was applied for the first time in 2013 by Farahani et al.31 In this technique, an adequate volume of a ferrofluid is quickly injected into an aqueous sample using a syringe. The large contact surface between the two phases accelerates the mass transfer processes and improves the extraction kinetics; in addition, the phase separation is facilitated with the aid of an external magnetic field. Ferrofluids are stable colloidal suspensions of magnetic nanomaterials in an ionic liquid, showing both magnetic and fluid properties. Ionic liquids (ILs) are solvents with unique physicochemical properties including negligible vapour pressure and ability to be miscible in water and organic solvents, and have attracted a lot of attention for use as extractants in microextraction techniques. ILs can bind to the carbon network structures of GO via π–π electronic interactions causing strong connections by physical crosslinking.32 Metal ions tend to stay in the aqueous phase, but their hydrophobicity will be increased upon complexation with a suitable ligand. The complex formed can be quickly extracted in the ferrofluid.
To apply ferrofluids to the extraction/preconcentration of mercury and increase the efficiency towards mercury extraction during the preconcentration treatment, thiolate ligands can be used as mercury chelating agents. Thiosalicylic acid is an interesting heterodifunctional ligand. Combination of both hard (O) and soft (S) donor atoms and the ability of both carboxylate and thiolate groups to bridge two metal centres33,34 provide a multitude of bonding opportunities to metals in either their mono- or doubly deprotonated states.35,36 It is well known that thiosalicylic acid and its derivatives are organic ligands commonly used for medical purpose to treat mercury poisoning.37 The formed organic mercury complex can be easily extracted from the matrix during the MDSPE process. In this work, a magnetic sorbent material was fabricated by coupling magnetic iron nanoparticles (MNPs) and graphene oxide (GO), resulting in shell structured Fe3O4@graphene oxide nanospheres called magnetic graphene oxide (MGO). The material was suspended in the ionic liquid (IL) 1-n-butyl-3-methylimidazolium tetrafluoroborate [BMIM][BF4], resulting in a ferrofluid with excellent adsorbent properties. Thus, a MDSPE/CV-GFAAS method was optimized for the determination of ultra-trace amounts of Hg in environmental water and biological samples, using the ferrofluid described and the chelating agent methyl thiosalicylate (MTS). The preconcentration efficiency of the developed method, due to MDSPE and CV, resulted in excellent detection and determination limits compared with other similar methods reported in the bibliography.
2. Experimental
2.1. Equipment
A Perkin Elmer Zeeman AAnalyst 600 atomic absorption spectrometer (Norwalk, CT, USA) with a longitudinal Zeeman effect background correction system was used for the determination of Hg. A Perkin Elmer electrodeless discharge lamp (EDL) was used as the radiation source. The mercury absorbance was measured at 253.7 nm with a 0.7 nm spectral band pass. The employed atomizer was a transversely heated graphite tube with an integrated pyrolytic graphite platform. A Perkin Elmer FIAS-400 AS System, which consists of two peristaltic pumps with PVC tubing of various diameters, a five-port way rotary and a gas–liquid separator with a PTFE membrane of 0.5 mm pore diameter for hydride generation, was used as the flow injection (FI) accessory controlled by the instrument software. The FIAS-400 AS System was connected directly from the gas–liquid separator to the AAnalyst 600 using 26 cm long PTFE tubing (1.75 mm i.d.). The FI system and the GFAAS instrument were coupled and operated completely synchronously. Measurements were carried out in peak area mode (read time of 5 s). The graphite furnace temperature program is shown in the ESI, Table S1.† The graphite tubes were covered with Ir as a permanent modifier following the treatment described elsewhere.38A pH meter and a conductivity meter Hatch (Loveland, CA, USA) were employed for pH and ionic strength adjustments, respectively.
An ultrasonic bath VWR (West Chester, PA, USA) Unique, model USC 2800, 40 kHz, and a Vortex VWR (West Chester, PA, USA), model UV-2500, multi tube vortex mixer were also employed.
For the evaluation of the accuracy of the proposed method, a reference material was digested in a Milestone ultraWAVE microwave oven (Sorisole, Italy) equipped with 25 mL PTFE/TFM vessels.
2.2. Reagents and solutions
High purity reagents were used in all experiments. All glassware was cleaned with hot concentrated nitric acid and stored soaked in 10% (wt/wt) nitric acid, and was rinsed several times with water immediately before use. Doubly de-ionized water (18 MΩ cm) obtained from a Millipore Milli-Q water system (Bedford, MA, USA) was used throughout.Hg stock standard solution, 1000 mg L−1, from Merck (Darmstadt, Germany) was used. Standards of working strength were made immediately prior to use by appropriate dilution as required. In order to adjust the pH of standards and samples, a 1 M solution of hydrochloric acid was prepared from hydrochloric acid, 37% wt/wt, Merck (Darmstadt, Germany), art. number 113386. Finally, a 0.2% (wt/v) sodium tetrahydroborate(III), Acros Organics (Geel, Belgium), solution prepared in 0.1% (wt/v) NaOH, Sigma Aldrich Chemie (Steinheim, Germany), was used as a reductant for the generation of Hg cold vapour.
For the synthesis of MGO, ferric chloride hexahydrate (FeCl3·6H2O), ferrous chloride tetrahydrate (FeCl2·4H2O), ammonium hydroxide 30% (wt/wt), methanol, sodium chloride and H2SO4 98% were purchased from Merck (Darmstadt, Germany) and H2O2 35% from Scharlab (Barcelona, Spain). 3-Aminopropyltriethoxysilane was obtained from Fluka (Buchs, Switzerland). Brij 76C18EO10, tetraethoxysilane (TEOS), N,N′-dicyclohexylcarbodiimide (DCC), graphite, NaNO3 and KMnO4 were acquired from Aldrich Chemie (Steinheim, Germany). Ethanol was supplied by Carlo Erba (Milano, Italy).
For the Hg-complex (Hg–MTS), methyl thiosalicylate from Sigma Aldrich Chemie (Steinheim, Germany) was employed. The ionic liquid 1-n-butyl-3-methylimidazolium tetrafluoroborate ([BMIM][BF4]) was purchased from Merck (Darmstadt, Germany).
The Standard Reference Material (SRM 2976) analysed to determine the accuracy of the proposed procedure was from the National Institute for Standard & Technology (NIST): SRM 2976 Mussel Tissue. Seawater and tap water samples were collected in glass bottles (previously cleaned by soaking for 24 h in 10% (wt/wt) nitric acid and finally rinsed thoroughly with ultrapure water before use). Samples were immediately filtered by using a membrane of 0.45 mm pore size cellulose nitrate filters from Millipore (Bedford, MA, USA). After that, the samples were stored at 4 °C as recommended by Method 3010B from the Environmental Protection Agency (USA), for less than 3 days until analysis. Nitric acid 65% (wt/wt) was supplied by Merck (Darmstadt, Germany), art. number 100452.
2.3. Synthesis of MGO and preparation of the ferrofluid
The synthesis and detailed characterization of MGO (Fig. S1, ESI†) and the preparation of the ferrofluid are described elsewhere.39 A description of the synthesis and characterization of MGO is given in the ESI.†2.4. Sample preparation
The dissolution of the Standard Reference Material (SRM 2976) was carried out as follows: the sample was accurately weighted directly on a digest vessel (0.4–0.5 g of the previously dried sample according to the provider’s instructions). Then 4.0 mL of concentrated nitric acid, 4.0 mL of 30% hydrogen peroxide, and 1.0 mL of concentrated hydrochloric acid were added to the vessel. This mixture was subjected to microwave digestion with a power of 1400 W for 20 min, and a cooling time of 15 min. Finally, the pH was adjusted by decreasing the acidity with NaOH and by adding 1 M HCl solution and the solution was made up to 500 mL with Milli-Q water in a volumetric flask.For seawater and tap water samples, aliquots of 5 mL and 20 mL of sample, respectively, were placed in a 50 mL volumetric flask, then 5 mL of 1 M HCl (pH 1), 5 g of NaCl, and 350 μL of 1% MTS (v/v) were added, and de-ionized water was added up to the mark.
All samples were analysed immediately after preparation.
2.5. Extraction procedure
Accurate volumes of sample or standard solutions of Hg were poured into adequate volumetric flasks to have a final concentration in the eluent of 10 μg L−1 Hg(II), then MTS 1% v/v in ethanol and NaCl up to a final concentration of 0.007% (v/v) and 10% (wt/v), respectively, were poured. Finally, the pH was adjusted to 1.0 with 1 M HCl and the volumetric flasks were filled up to the mark with deionized water. The content of the flasks was then poured into polyethylene tubes. Then, the MDSPE was performed by injecting rapidly 220 μL of the ferrofluid into the sample solution with a 0.5 mL syringe (equipped with a suitable needle). The ferrofluid forms a homogeneous suspension within the sample, and the contact was kept for 1 min in an ultrasonic bath for the extraction of the Hg–MTS complex. In the following step, a magnet was placed at the bottom of the tube for the sorbent magnetic decantation and the solution turned clear in 1 min. The supernatant was decanted.Mercury was eluted from the sorbent by adding 2 mL of eluent (HNO3 0.5% and thiourea 0.5%) and stirring by vortexing for 1 min. Finally, the sorbent was separated using a magnet and the supernatant was poured into a polyethylene tube for mercury determination by FI-CV-GFAAS.
To determine the extraction process efficiency, a 100 ng L−1 Hg solution was prepared and extracted under optimal conditions. The supernatant was filtered and analysed by ICP-MS, showing a Hg concentration below the LOD. Then, the extraction efficiency was considered close to 100%.
The FI system configuration is shown in Fig. 1, and it was operated as follows: during the 11 s sample loading period, with the valve in the “fill” position, a 4.1 mL min−1 flow of sample (standard or blank) at pH 1.0 was pumped (via pump P1) through the 500 μL loop located in the valve. Then, the valve position was changed to inject position and P1 was stopped, while P2 pumped water at a rate of 1.8 mL min−1 through the loop dragging the sample to the chemical vapour generator. Thus, the mercury merges with 0.6 mL min−1 flow of the reductant in the mixing coil, where direct generation of mercury vapour takes place. The gas generated and the solvent were then passed into the gas–liquid separator which separates gases from liquid. The liquid was drained, and the generated vapour was swept into the graphite furnace through the 26 cm tubing, until the tip of the autosampler arm, by a stream of 250 mL min−1 argon. In this procedure, the FI system and the GFAAS instrument were coupled and operated completely synchronously.
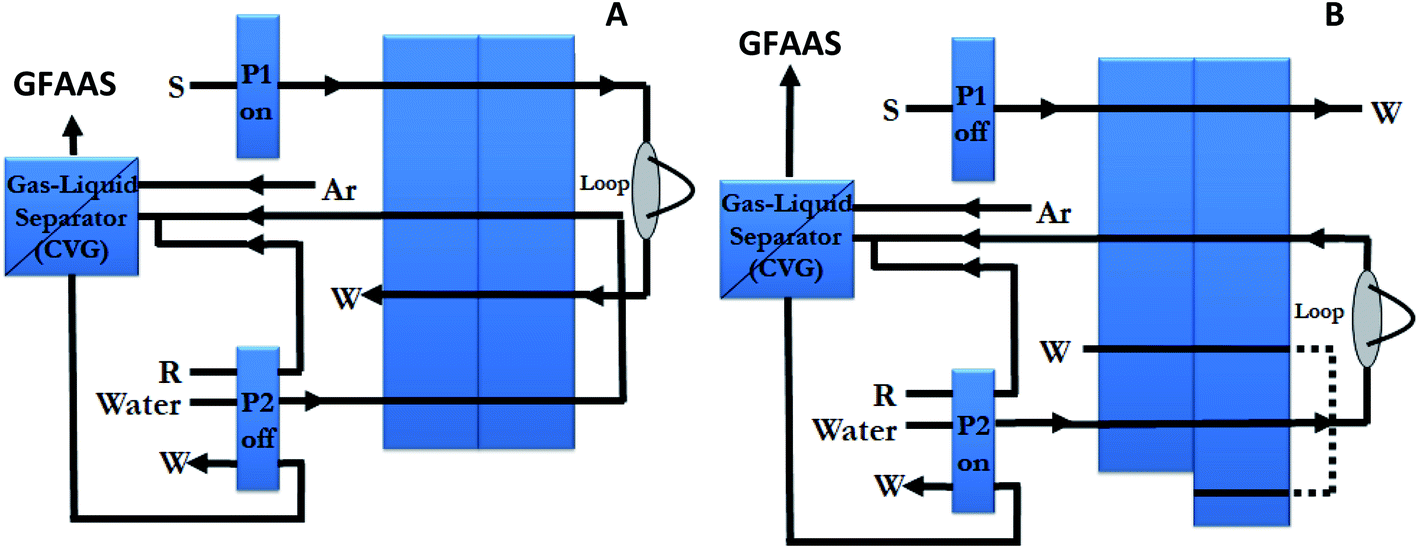 | ||
| Fig. 1 FI system schematic diagram for the loading step (A) and elution step (B). P1 and P2, peristaltic pumps; W, waste; S, sample; R, reductant. | ||
2.6. Optimization strategy
The determination of ultra trace amounts of mercury in environmental water samples is difficult not only due to the low analyte concentrations present but also due to the complexity of the sample matrices. Following the chemical vapour generation, only the analyte vapours are conducted to the graphite tube, with the risk of interference being very small. The best signal of the analyte was used as optimization criteria. The chemical and FI system (Fig. 1) variables of the used manifold, which affect the pre-concentration and mercury vapour generation, were optimized using a final concentration of 10.0 μg L−1 Hg(II) in the eluent solution. The scanning of each standard, blank and sample was repeated three times.Two different strategies were used: a one-at-a-time method (changing one parameter at a time while keeping the others constant) and a multivariate response surface experiment design.
Some parameters relevant to the optimization were elution of Hg in the manifold and reaction conditions for CV (reagent concentrations). For that reason, a response surface design was performed. The variables to be optimized were the concentrations of NaBH4, thiourea and HNO3. The lower and upper values given for each factor were 0.0% and 4.0% for NaBH4 concentration, 0.0% and 5.0% for thiourea concentration and 0.0% and 5.0% for HNO3 concentration. The response surface design used was a rotatable central composite design (CCD) which includes a 23 factorial design (8 experiments), a 2 × 3 star design (6 experiments) and 3 centre points (3 experiments). The resulting 17 experiments required for that design were randomly performed, and as response function, the Hg signal (peak area) was chosen. The results of the experiments were processed using the statistical software Statgraphics Centurion XVI. The significance of the effects was checked by analysis of the variance (ANOVA) and using p-value significance levels. This value represents the probability of the effect of a factor being due solely to random error. Thus, if the p-value is less than 5%, the effect of the corresponding factor is significant.
Once the concentrations of the reductant and eluent solutions were optimized, the rest of the experimental parameters for the MDSPE/CV-GFAAS were optimized by the one-at-a-time method in order to obtain the best peak area signal.
3. Results and discussion
3.1. Optimizing operating parameters
The graphite furnace temperature programs were optimized by univariate way in experiments conducted with 10 μg L−1 Hg(II), while running a blank in parallel. Ir was used as a permanent modifier. The optimized heating program for Hg can be seen in Table S1, ESI.†3.2. Effect of pH on the collection of the analyte
Since the solution pH affects the extent of complexation with MTS, which in turn determines the percentage of analyte retained by the MTS, the pre-concentration of 10 μg L−1 Hg(II) ions from solutions buffered at different pH was studied. The pH was varied between 1.0 and 11.0. pH 1 was obtained with 1 M HCl, and the pH was adjusted from 2.0 to 5.0 by using glycine–HCl or sodium acetate–acetic acid buffer, from 5.0 to 10.0 by using NaOH–boric acid buffer and pH > 10.0 was obtained with NaOH. As can be seen in Fig. 2, the maximum value of absorbance was observed at pH 1.0. This phenomenon can be explained considering the electronic interactions.39 At basic pH, oxygen groups from the MGO surface are deprotonated, while the complex Hg–TMS is still neutral. Therefore, under basic conditions, the sorbent presents a negatively charged surface and the adsorption of an aromatic neutral complex is not favoured. Thus, acid pH maintains a non-charged surface which leads to more effective adsorption. So, in order to accomplish the determination of mercury, a pH value of 1 was chosen. For subsequent experiments, a solution of 1 M HCl was used to obtain this pH.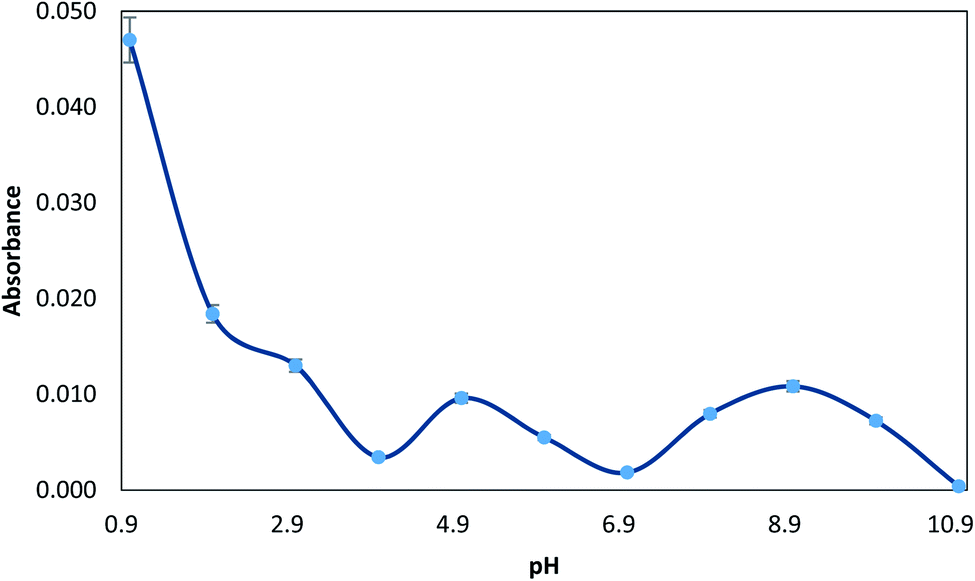 | ||
| Fig. 2 Effect of pH on the extraction of 10 μg L−1 Hg(II), prepared with 0.01% (v/v) MTS, 0.5% (wt/v) NaCl, and the respective buffer (pH 1–11). | ||
3.3. Optimization of the MDSPE/CV-GFAAS procedure
As described above (Section 2.6), the concentrations of the eluent and reductant solutions were optimized through a CCD, and the rest of the experimental parameters for the MDSPE/CV-GFAAS were optimized by the one-at-a-time method in order to obtain the best peak area signal.The following parameters of the MDSPE were optimized: (a) concentration of MTS; (b) influence of ionic strength; (c) extraction time; (d) elution conditions and CV generation. Respective data and figures are given in the ESI.† The following experimental conditions were found to give the best results: (a) a MTS concentration of 0.007% (Fig. S2, ESI†), (b) a NaCl concentration of 10.0% (Fig. S3, ESI†), (c) an extraction time between MGO and MTS–Hg complex of 1 min (Fig. S4, ESI†) and (d) elution conditions and CV generation: 0.5% thiourea, 0.5% HNO3 and 0.5% NaBH4 (Fig. S6, ESI†), and a reductant flow rate of 0.6 mL min−1 (Fig. S7, ESI†).
3.4. Study of sample volume
Different aqueous phase (sample) volumes were studied from 100 to 600 mL (0.007% MTS). All the experiments were carried out with 220 μL of ferrofluid and 2 mL of eluent (HNO3 0.5%, thiourea 0.5%). Considering that the volume of eluent was 2 mL, the ratio between sample and eluent volumes ranged from 50 to 300. Therefore, the preconcentration factor was from 50 to 300. In Fig. 3, it can be seen that the extraction efficiency was found constant up to an aqueous sample volume of 500 mL, and then a decrease was observed. This fact could be due to an incomplete elution of Hg from the ferrofluid at higher ratio of sample volume/eluent volume or an incomplete adsorption of Hg from a higher sample volume with the same adsorbent volume (220 μL). Using the maximum aqueous phase volume (500 mL) and an eluent volume of 2 mL, the preconcentration factor (PF) for the MDSPE method was 250. Moreover, considering the CV generation step, it can be said that the preconcentration factor of this method is over 250.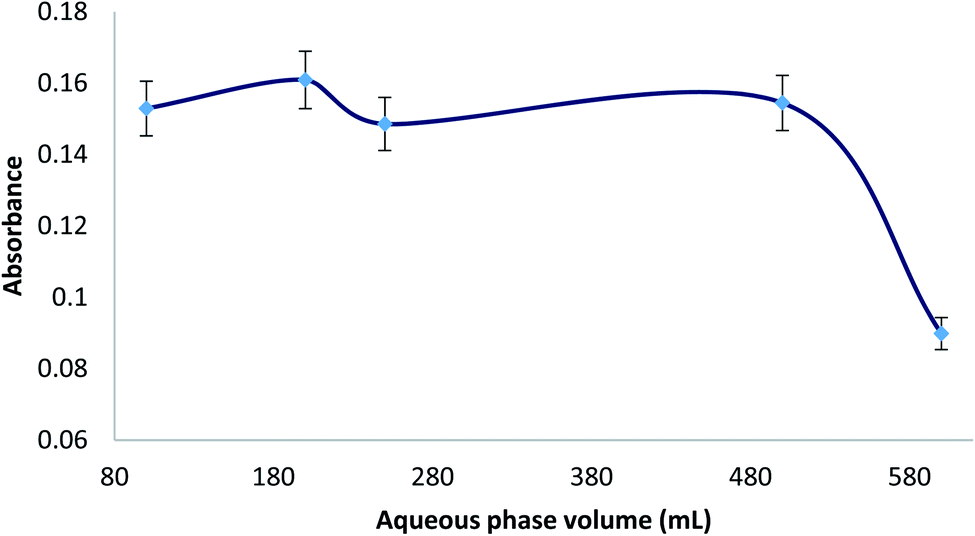 | ||
| Fig. 3 Study of the aqueous phase volume for extraction of 10 μg L−1 Hg(II), prepared with 0.007% (v/v) MTS, 10% (wt/v) NaCl, and 1 M HCl until pH = 1.0. | ||
3.5. Figure of merit
Under the optimal conditions described above, linear calibration graphs were obtained from 2 to 200 ng L−1 for Hg with reference to the original sample and 500 mL sampling volume. Thus, Hg was pre-concentrated 250 times. Correlation coefficients always better than 0.9903 were obtained. In order to determine the limits of detection and quantification (LOD and LOQ), a calibration graph in the range of 2–100 ng L−1 was built with six standards. The calibration equation was y = 0.00050x (μg L−1) + 0.0038, with confidence intervals of ±0.00009 for the slope and ±0.0005 for the intercept. The LOD and LOQ, calculated as the concentration of the analyte giving signals equivalent to three and ten times, respectively, the standard deviation of the blank plus the net blank intensity, were 0.25 ng L−1 (LOD) and 4.9 ng L−1 (LOQ) for Hg. The signal and standard deviation of the blanks (absorbance signals) used for these determinations were 0.00358 ± 0.00008.The precision of the whole method was evaluated in terms of inter-day precision, using the relative standard deviation (RSD), calculated as the average of relative standard deviations of 2, 50 and 100 ng L−1 standards measured on three days. The calculated inter-day precisions were 2.9, 2.2 and 1.4%, respectively. The preconcentration factor calculated as the ratio of sample volume to eluent volume and considering that the extraction process efficiency was close to 100% (Section 2.5) was 250.
3.6. Validation of the method
The accuracy of the method was evaluated by using a Standard Reference Material (SRM 2976). Moreover, recovery experiments were carried out in real samples: tap water, Tarifa seawater and Malaga seawater. As shown in Table 1, the recoveries (%) at different spiked concentrations were between 86.0 and 103.0%, providing satisfactory results in terms of precision, even in real samples presenting complex matrices. No significant differences were observed for p = 0.05 between the results and the certified value and the added concentrations according to the t-test for a confidence level of 95% (tcalculated = 1.19 and ttabulated = 2.57). All determinations were performed by external calibration with aqueous standards prepared by the same extraction procedure as for the samples. Although real samples have included trace elements such as transition metals, it can be said that there is no interference from these metals at μg mL−1 concentrations. Therefore, the proposed procedure was validated and demonstrated to be accurate in Hg determination in a wide variety of sample matrices.| Sample | Added (μg L−1) | Found ± standard error (μg L−1) | Recovery (%) |
|---|---|---|---|
| Tap water | — | — | — |
| Spike 1 | 0.02 | 0.020 ± 0.002 | 100.0 |
| Spike 2 | 0.06 | 0.056 ± 0.003 | 93.3 |
| Tarifa seawater | — | 0.018 ± 0.006 | — |
| Spike 1 | 0.04 | 0.053 ± 0.006 | 87.5 |
| Spike 2 | 0.12 | 0.140 ± 0.007 | 101.7 |
| Malaga seawater | — | 0.0075 ± 0.0002 | — |
| Spike 1 | 0.04 | 0.042 ± 0.003 | 86.3 |
| Spike 2 | 0.12 | 0.1297 ± 0.0004 | 101.8 |
| NIST 2976 Mussel Tissue | Certificate total value | ||
| 61.0 ± 3.6 μg kg−1 | 63 ± 5 (μg kg−1) | 103.3 | |
Hg(II) was found in seawater samples due to the high sensitivity of the method, and the concentrations (0.018 ± 0.006 and 0.0075 ± 0.0002 μg L−1 in Tarifa and Málaga seawater, respectively (Table 1)) were below the allowed limits by Spanish legislation (RD 817/2015)6 and within the normal concentration ranges.
For comparison purposes, the analytical performance data of similar methods reported in the literature are listed in Table 2. A direct comparison of the figures of merit for the developed method with results from other workers is difficult due to the different experimental conditions. All the methods presented in Table 1 consist of preconcentration and determination procedures combined with AAS for the determination of Hg(II). As can be seen, the analytical performances, such as LOD, RSD, and PF, of the method reported in this work are the best. The preconcentration method on the ferrofluid was easy and the Hg preconcentration required only one minute and another minute for elution, being a very efficient procedure, with relative recoveries between 86 and 103%. Besides the preconcentration on the ferrofluid, another preconcentration occurs on the graphite tube thanks to the Ir cover. To our knowledge, this is the first reported method that combines MDSPE and CV-GFAAS for Hg(II) determination. The use of CV generation and the preconcentration in the Ir permanent modifier explain the better results in the analytical performance of the method compared with recent literature methods.
| Method | Solid phase/reusability | Samples | Linear range (μg L−1) | Analytical performance | Relative recoveries (%) | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| LOD (μg L−1) | RSD (%) | PF | ||||||
| a DLLME: dispersive liquid–liquid microextraction. b G/ZnFe2O4: graphene/ZnFe2O4 nanocomposite adsorbent. c AFS: atomic fluorescence spectroscopy. d g-C3N4/Fe3O4: magnetic graphitic carbon nitride nanocomposites. e AMA: advanced mercury analyser. f μ-SPE: micro solid phase extraction. g UV-PVG/μ-CCP-OES: ultraviolet photochemical vapor generation/capacitively coupled plasma microtorch optical emission spectrometry. | ||||||||
| DLLMEa/GFAAS | — | Blood | 0.3–60 | 0.1 | 3.7 | 112 | 90–109 | 14 |
| DLLME/GFAAS | — | Biological | 0.5–50 | 0.1 | 6.2 | 68 | 90.5–108.0 | 15 |
| MDSPE/CV/AAS | MGO/— | River water, cow milk, omega 3 and lipstick | 1–200 | 0.57 | 6.5 | 21 | 86–105 | 8 |
| MDSPE/CV/AAS | MGO/— | Seafood | 1–85 | 0.025 | 4.0 | 17 | 85 | 26 |
| MDSPE/CV/AAS | G/ZnFe2O4b/50 cycles | Biological and well, tap and wastewater | 0.25–10 | 0.01 | 2.7 | 30 | 91–107 | 27 |
| MDSPE/CV/AFSc | g-C3N4/Fe3O4d/4 cycles | Natural water | 0.01–0.6 | 0.0014 | 4.6 | 40 | 85.0–116.7 | 28 |
| DSPE/AAS/AMAe | Graphene/— | Environmental water including seawater | 0.00038–1.038 | 0.00038 | 3.0 | — | — | 40 |
| MDSPE/CV/AFS | Au NP–Fe3O4/— | Environmental water | 0.005–0.2 | 0.0015 | 3.7 | 80 | 92.5–108.7 | 41 |
| DLLME/CV/AAS | — | Blood | 0.15–85 | 0.03 | <4 | 6.6 | >97 | 42 |
| μ-SPEf/CV/AFS | IL–3D graphene–Ni foam/250 | Environmental water, tap water, mineral water | 0.01–8 | 0.0036 | 4.1 | 180 | 101–105 | 43 |
| UV-PVG/μCCP OESg | — | Tap water, pool water, well water, bottled water, food | 0–1 | 0.0001 | 2.6–12.7 | 41 | 82–108 | 44 |
| MDSPE/CV/GFAAS | MGO/— | Biological and environmental water including seawater | 0.002–0.200 | 0.00025 | 2.9 | 250 | 86–103 | This work |
4. Conclusion
In this work, a ferrofluid based on Fe3O4@graphene oxide nanospheres together with an ionic liquid was used to develop a MDSPE method for the extraction of the complex formed between the chelating agent methyl thiosalicylate (a ligand used to treat mercury poisoning) and mercury. The procedure has resulted to be highly efficient in mercury extraction, being rapid (1 + 1 minute, pre concentration + elution); easy, the separation between the ferrofluid and the solution is achieved only with the use of a magnet and decantation; and green, since only small amounts of a non-toxic solvent are necessary. The combination of MDSPE with CV-GFAAS has resulted in high sensitivity towards mercury, and the analytical performances of the method reported in this work, such as LOD, RSD, and PF, compared very well to similar methods found in the bibliography. The method was validated by the analysis of a SRM of Mussel Tissue, and the analysis was performed with external calibration of Hg spiked environmental water samples including seawater. The relative recoveries were between 86 and 103%, showing the selectivity of the method because these samples have complex and highly saline matrices including trace elements such as transition metals.Author contributions
E. I. Vereda Alonso: conceptualization, methodology, formal analysis, writing – original draft, writing – review & editing, software, data curation, supervision, resources, funding acquisition. M. M. López Guerrero: conceptualization, methodology, formal analysis, writing – original draft, writing – review & editing, data curation, supervision, resources. S. Maireles-Rivas: investigation, methodology. P. Montoro Leal: investigation, methodology, writing – review & editing, software, formal analysis, data curation. J. C. García-Mesa: investigation, methodology, validation, software, formal analysis, writing – review & editing, data curation. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The author(s) declare that they have no competing interests.Acknowledgements
The authors thank the University of Malaga (Proyecto Puente UMA) and Junta de Andalucía, Project UMA18FEDERJA060 for supporting this study and the Spanish Ministerio de Ciencia y Tecnología for the fellowship FPU18/05371.References
- J. Wang, X. Feng, C. W. N. Anderson, Y. Xing and L. Shang, J. Hazard. Mater., 2012, 221–222, 1 CAS.
- Z. Tang, F. Fan, S. Deng and D. Wang, Ecotoxicol. Environ. Saf., 2020, 202(110950), 1 Search PubMed.
- S. A. Silyutin, S. A. Epshtein and T. O. Gushchina, Mining Informational and Analytical Bulletin, 2020, 5, 5 CrossRef.
- K. H. Kim, E. Kabir and S. A. Jahan, J. Hazard. Mater., 2016, 306, 376 CrossRef CAS PubMed.
- A. B. Shabestari, B. A. Adergani, M. Shekarchi and S. M. Mostafavi, Ekoloji, 2018, 27, 1935 Search PubMed.
- BOE núm. 217 de 11/09/2015. Real Decreto 817/2015 de 11 de septiembre, por el que se establecen los criterios de seguimiento y evaluación del estado de las aguas superficiales y las normas de calidad ambiental, 2015, pp. 80582–80677 Search PubMed.
- M. M. López Guerrero, E. Vereda Alonso, J. M. Cano Pavón, M. T. Siles Cordero and A. Garcia de Torres, J. Anal. At. Spectrom., 2016, 31, 975 RSC.
- S. Jamshidi, M. K. Rofouei, S. Seidi and Å. Emmer, Sep. Sci. Technol., 2019, 55, 1505 CrossRef.
- T. Zangmo and A. Siripinyanond, Anal. Chim. Acta, 2019, 1085, 29 CrossRef CAS PubMed.
- N. Ferrúa, S. Cerutti, J. A. Salonia, R. A. Olsina and L. D. Martinez, J. Hazard. Mater., 2007, 141, 693 CrossRef PubMed.
- M. M. López Guerrero, M. T. Siles Cordero, E. Vereda Alonso, A. García de Torres and J. M. Cano Pavón, Microchem. J., 2017, 132, 274 CrossRef.
- M. M. López Guerrero, M. T. Siles Cordero, E. Vereda Alonso, J. M. Cano Pavon and A. García de Torres, J. Anal. At. Spectrom., 2015, 30, 1169 RSC.
- J. C. Garcia-Mesa, P. Montoro Leal, M. M. Lopez Guerrero and E. I. Vereda Alonso, Microchem. J., 2019, 150, 104141 CrossRef CAS.
- R. Akramipour, M. R. Golpayegani, S. Gheini and N. Fattahi, Talanta, 2018, 186, 17 CrossRef CAS PubMed.
- M. Pirsaheb and N. Fattahi, Anal. Methods, 2015, 7, 6266 RSC.
- W. A. W. Ibrahim, L. I. A. Ali, A. Sulaiman, M. M. Sanagi and H. Y. Aboul-Enein, Crit. Rev. Anal. Chem., 2014, 44, 233 CrossRef PubMed.
- L. Zhang, X. Chang, Z. Hu, L. Zhang, J. Shi and R. Gao, Microchim. Acta, 2010, 68, 79 CrossRef.
- E. Yavuz, S. Tokalıoglu, H. Sahan and S. Patat, Food Chem., 2016, 194, 463 CrossRef CAS PubMed.
- Y. Zhai, S. Duan, Q. He, X. Yang and Q. Han, Microchim. Acta, 2010, 169, 353 CrossRef CAS.
- M. Šafaříková and I. Šafarík, J. Magn. Magn. Mater., 1999, 194, 108 CrossRef.
- Q. Han, Z. Wang, J. Xia, S. Chen, X. Zhang and M. Ding, Talanta, 2012, 101, 388 CrossRef CAS PubMed.
- H. A. Zadeh and Z. Talleb, Talanta, 2015, 134, 387 CrossRef PubMed.
- J. P. Wasylka, N. Szczepańska, M. de la Guardia and J. Namieśnik, TrAC, Trends Anal. Chem., 2016, 77, 23 CrossRef.
- X. Gao, J. Jang and S. Nagese, J. Phys. Chem. C, 2009, 14, 832 Search PubMed.
- D. R. Dreyer, A. D. Todd and C. W. Bielawski, Chem. Soc. Rev., 2014, 43, 5288 RSC.
- S. Seidi and M. Fotouhi, Anal. Methods, 2017, 9, 803 RSC.
- E. Yavuz, Ş. Tokalıoğlu and Ş. Patat, Microchem. J., 2018, 142, 85 CrossRef CAS.
- M. Shi, X. Yang and W. Zhang, Anal. Chim. Acta, 2019, 1074, 33 CrossRef CAS PubMed.
- A. N. Anthemidis and K. I. G. Ioannou, Talanta, 2009, 80, 413 CrossRef CAS PubMed.
- M. Mirzaei, M. Behzadi, N. Mahmoud Abadi and A. Beizaei, J. Hazard. Mater., 2011, 186, 1739 CrossRef CAS PubMed.
- M. D. Farahani, F. Shemirani and M. Gharehbaghi, Talanta, 2013, 109, 121 CrossRef PubMed.
- J. Lee and T. Aida, Chem. Commun., 2011, 47, 6757 RSC.
- M. Nayaka, A. K. Singhb, P. Prakashb, R. Kantc and S. Bhattacharyaa, Inorg. Chim. Acta, 2020, 501, 119263 CrossRef.
- T. Wehr-Candler and W. Henderson, Coord. Chem. Rev., 2016, 313, 111 CrossRef CAS.
- C. Ma, Q. Zhang, R. Zhang and D. Wang, Chem.–Eur. J., 2006, 12, 420 CrossRef PubMed.
- J. G. Melnick, K. Yurkerwich, D. Buccella, W. Sattler and G. Parkin, Inorg. Chem., 2008, 47, 6421 CrossRef CAS PubMed.
- S. Asadi, B. Zhang, Z. Weng, A. Angelidou, D. Kempuraj, K. D. Alysandratos and T. C. Theoharides, Int. J. Immunopathol. Pharmacol., 2010, 23, 1015 CrossRef CAS PubMed.
- E. Vereda Alonso, M. M. López Guerrero, P. Colorado Cueto, J. Barreno Benítez, J. M. Cano Pavón and A. García de Torres, Talanta, 2016, 153, 228 CrossRef CAS PubMed.
- X. Jia, D. Gong, J. Wang, F. Huang, T. Duan and X. Zhang, Talanta, 2016, 160, 437 CrossRef CAS PubMed.
- B. Duval, A. Gredilla, S. Fdez-Ortiz de Vallejuelo, E. Tessier, D. Amouroux and A. de Diego, Microchem. J., 2020, 154, 104549 CrossRef CAS.
- W. Zhang, C. Sun and X. Yang, Anal. Methods, 2014, 6(9), 2876 RSC.
- H. Shirkhanloo, A. Khaligh, H. Z. Mousavi, M. M. Eskandari and A. A. Miran-Beigi, Chem. Pap., 2015, 69(6), 779 CAS.
- A. C. Sotolongo, M. M. Messina, F. J. Ibanez and R. G. Wuilloud, Talanta, 2020, 210, 120614 CrossRef CAS PubMed.
- E. Covaci, M. Senila, C. Tanaselia, S. B. Angyus, M. Ponta, E. Darvasi, M. Frentiu and T. Frentiu, J. Anal. At. Spectrom., 2018, 33(5), 799 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ja00516a |
| This journal is © The Royal Society of Chemistry 2021 |