
Temporal analysis of ion arrival for particle quantification
Andrew M.
Duffin
 *,
Edward D.
Hoegg
,
Ryan I.
Sumner
,
Trevor
Cell
,
Gregory C.
Eiden
and
Lynn S.
Wood
*,
Edward D.
Hoegg
,
Ryan I.
Sumner
,
Trevor
Cell
,
Gregory C.
Eiden
and
Lynn S.
Wood
Pacific Northwest National Laboratory, USA. E-mail: andrew_duffin@pnnl.gov
First published on 18th November 2020
Abstract
Inductively coupled plasma mass spectrometry (ICP-MS) is a powerful technique for accurately and precisely measuring particles. Built on instrumentation developed for steady state signals, traditional detection and quantification of particles with ICP-MS instruments is based on signal intensity rising above background within an integration window. This research presents results with time stamp digitization of all single ion detection events with no predetermined integration windows. With this method particles are distinguished from background by the short burst of ions (10's to 100's of μs) associated with a particle passing through the plasma. That is, particle signal is identified by the timing between successive ion arrivals at the detector. The new method allows for fast, efficient, and sensitive detection of micro and nanoparticles and provides a powerful means to discriminate against non-particle backgrounds. We tested the method with standard gold nanoparticles, uranium particles from an aerosol created during laser ablation, and iron nanoparticles. The results show efficient detection of nanoparticles separated from interfering background. The timing based method also enabled the association of a single mass 236 count with 236U uranium in a particle rather than background.
Introduction
The increased production and use of nanomaterials in a wide array of products and industrial processes has a concomitant necessity for analytical instrumentation and methods that can rapidly and reliably identify and quantify nanoparticles.1–6 Similarly, sensitive particle analysis methods enable detection and quantification of single cells or large biomolecules.7–9 Scientific studies on the fate and transport of nanomaterials in the environment is also a field that benefits from methods for nanoparticle identification and quantification.10–12 Moreover, with sensitive and accurate quantification, the isotopic composition of nanoparticles could be used as a nuclear materials signature for safeguards applications.13 While there is considerable need for sensitive, rapid, and accurate analytical methods to detect and quantify nanoparticles, due to their diminutive size, both naturally occurring and man-made nanoparticles prove challenging to analyze.Over the last several decades single particle inductively coupled plasma mass spectrometry (SP-ICP-MS) has emerged as a sensitive and trusted method for characterization of particulate material.2,4,14–18 This method relies on the atomization and ionization of particles in the ICP followed by extraction of ions into a mass spectrometer where the ions are separated by mass-to-charge ratio and detected as they impinge on the detector. The traditional secondary electron multiplier (SEM) amplifies each ion signal producing an electronic pulse that can be recorded as a single count. Conventional ICP-MS signal processing counts ions within an integration (counting) window and the signals are typically reported as counts per second. With this method of data collection and processing, identification of a particle relies on the count rate for the integration window(s) containing the particle signal to rise significantly above the background count rate. Effort has been placed into optimizing the signal integration time for particle detection.14,19 Even with optimized integration time, the signature of a particle event is traditionally intensity (number of counts) based. In the current work, we show that precisely capturing the arrival times of all ions on a given SEM provides a temporal signature of a particle which is a sensitive means for particle identification and quantification. That is, the signature of a particle is based on the temporal structure of ion arrival times at the detector rather than intensity per se. In this way, smaller particles can be identified and quantified than would otherwise be possible and it is possible to pick particle signals out of even intense background interference signals.
The result of a particle transiting and being atomized and ionized in the ICP is an analyte ion cloud that, is transferred to and impinged on the detector in the tens to hundreds of microsecond time frame.15,20 The duration of this burst of ions depend on both the particle size and precisely where it transits the ICP (different radial locations and particle trajectories). The closely spaced arrival of ions from the particle constitutes the signature of the particle and differs from the Poisson ion arrival distribution of background ions or ions resulting from continuous sample introduction such as solution nebulization. Identification of particle signal can be accomplished by analyzing the time difference between successive ions. The time difference between ions can be compared to the background arrival rate to determine if it is statistically different than the background distribution. Alternatively, and more simply, a particle can be identified by successive ion arrival times that are faster than a threshold timing value. That is, the particle provides ions that repeatedly arrive faster than a threshold time value characteristic of particles. This method also requires a meeting a threshold number of ions that are each within the threshold time since the last ion arrival. This creates a background independent means to identify particle signal. That is to say the temporal signature of the particle persists and is unchanged regardless of the background count rate (within the limits of the detector) and the background count rate does not need to be re-established between successive particle events. The simpler method can be more applicable when high particle rates (as associated with laser ablation or highly concentrated solutions) do not allow the statistical establishment of background arrival frequency between particles. Very high background count rates may still require background subtraction.
Application of this rapid transient particle method to a multicollector ICP-MS instrument allows for highly sensitive and simultaneous detection of multiple elements or multiple isotopes in a particle-by-particle fashion. With multicollection, the temporal signature of the particle from an abundant isotope or element can be used to attribute ions counts to a minor isotope/element that would otherwise be impossible to measure above the background. In this manner single ion counts of minor isotopes/elements can be statistically significant.
The research presented here represents the initial development of the rapid transient method. The results were generated by precisely recording the arrival time of each ion after SEM detection. The method was tested against standard gold nanoparticle solutions as well as uranium containing particles aerosolized by laser ablation. It was also tested against Fe nanoparticles detected with and without polyatomic interferences. The results show highly sensitive detection of nanoparticles.
Experimental
These experiments utilized a modified Thermo NeptunePlus™ MC-ICP-MS. This particular instrument is equipped with a detector array optimized for uranium isotopic characterization.21 This actinide detector array is in addition to the standard 8 movable Faraday cups (labeled H1–H4 on the high mass side and L1–L4 on the low mass side) and the switchable central channel SEM and Faraday cup (FC). The uranium detector array consists of four SEMs: a stationary compact discrete dynode (CDD) in position for 233U, a retarding potential quadrupole (RPQ) fronted multiplier for 234U, and a full size multiplier for 235U. (236U is detected by deflecting this beam back into the central channel RPQ/SEM.) There is another CDD mounted on the low mass side of L3 allowing either the L4 FC or the L3 mounted CDD to be positioned to collect the 238U beam. There is an additional L5 Faraday cup positioned such that the 235U beam can be electronically switched between the electron multiplier and the FC. With this array all the common uranium isotopes can be simultaneously detected on electron multipliers and it is this configuration that was used for the uranium portions of the research. For the iron isotope analysis the L3 mounted CDD (54Fe) was positioned in relation to the central channel (56Fe) to allow two isotopes to be simultaneously detected on electron multipliers. Experiments with gold nanoparticles were collected on the central channel SEM.In the native configuration the electron multipliers on the NeptunePlus™ output a current pulse that is sent to an amplifier and discriminator card. The output of the discriminator is passed to the onboard computer for counting within the designated integration windows. We modified the detection of the instrument to pass the output of the discriminators to a Cronologic TimeTagger4-2G time-to-digital converter (TDC) capable of generating timestamps to 0.5 ns accuracy across 4 channels. The TDC was mounted into a PCIe ×1 slot in a Windows 10 computer and controlled via C program written with Microsoft Visual Studio. This program controlled the TDC operation, read the timestamp data stream from the TDC via DMA, reported instantaneous count rates, and wrote data to disk. Since the NeptunePlus™ detector housing floats at −10 kV during instrument operation, the TDC and its computer were placed entirely within the instrument protective housing. The computer was accessed remotely via optical coupling to the facility network. Alternatively, the output of the discriminators was split to feed both the time-to-digital converter and the NeptunePlus™ counting cards. This variation allowed for testing, but the protection circuitry in the NeptunePlus™ would shut off the detectors at high count rates associated with larger particles, so this configuration was used infrequently. The protection circuitry was bypassed when only the TDC was used for data collection. The TDC was placed in a mode where it would output a time stamp for each pulse emanating from the discriminator. The experimentally measured ion count data is thus a series of time-stamps rather than counts measured over an integration window. In this mode, particle detection is based on temporal characteristics of a particle event rather than relying on an intensity/count rate rise above background. Early versions of these experiments utilized a Struck SIS3316 digitizer for digitization, but this system could not continuously digitize ion arrival at high rates and over long time periods. However, the data shown in Fig. 3 was collected with this digitizer.
The time-stamp files were processed with scripts written in IgorPro. As mentioned above, particles can be identified by the statistical comparison of background arrival rates with a particle event that produces a burst of ions that deviates significantly from the established background distribution. However, more simply and practically, particle events can also be identified with a time and counting threshold such that a counting threshold number of ions all arriving within a certain time threshold of each other can be flagged as a particle event. This second method is simpler and remains applicable even when high particle rates (such as with a laser ablation aerosol) prevent the establishment of a background ion arrival statistics between successive particles. For the majority of the particle data presented in this work, this second method was employed for particle identification and quantification.
More specifically, the particle identification algorithm used in this research ran with three adjustable parameters; a boxcar number (optional), a time threshold, and a count threshold. Successive time-stamp data points from the TDC were subtracted to create an array of time differences between ions. The algorithm ran a forward-looking boxcar average (typically 3 points) over the time differences. When this average was less than the time threshold (typically 20 μs) the leading time-stamp would be noted and the box moved to successive time stamp. This process would proceed until the time threshold criteria was no longer met. If there were more than the count threshold number of successive points (∼5 for small particles but set higher for larger particles) then the algorithm would flag a particle. The time-stamps associated with the start and stop of the particle signal were then used as bookends to count ions for an individual particle. Fig. 1 shows some of the steps in the data processing used to identify particles. As mentioned above, other identification algorithms could be used, notably the boxcar average step is optional. The important feature of the rapid transient method is the utilization of individual arrival times for particle detection, information that is lost with digitization using integration windows of any time duration. With this method it is impossible for a particle to straddle integration windows as the particle time windows are defined by the temporal particle data.
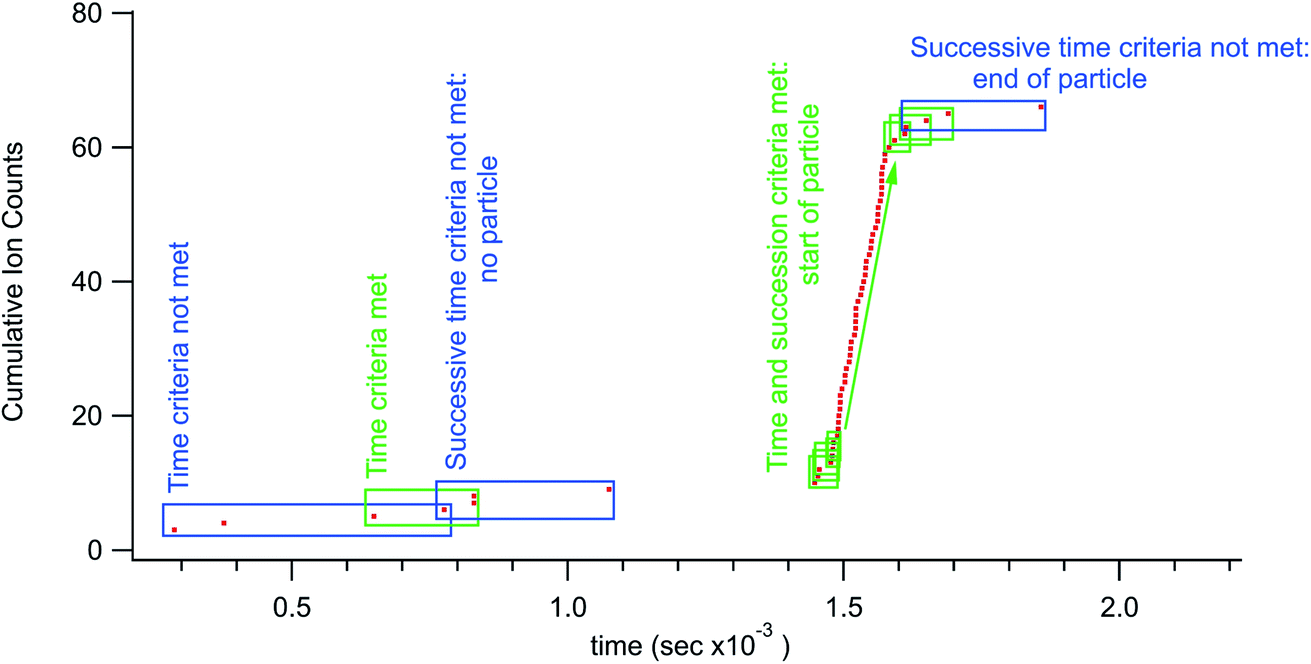 | ||
| Fig. 1 Rapid transient data processing example. The boxes around data points show representative boxcar positions. In this case averaging the time difference between 3 successive ions. To identify a particle signal there must be a minimum number (counting threshold) of successive boxes that all meet the time threshold. | ||
Nanoparticles were introduced to the NeptunePlus™ both via solution nebulization and with laser ablation. The ablation experiments used an Applied Spectra J200 UV femtosecond laser ablation system. Ablated samples included NIST 612 glass and powdered U015 uranium isotope ratio standard immobilized in a collodion matrix. All other samples were introduced with solution nebulization. The gold nanoparticles were purchased from Cytodiagnostics and diluted with a citrate buffer solution to reach a particle density of ∼103 per milliliter. This particle concentration was chosen to produce well separated particle events for experimentation and no efforts were made in this study to back calculate concentration. The Fe particles were synthesized by precipitating ferrous and ferric iron in the presence of a 1.5 M sodium hydroxide solution.22,23 The Fe particles were purified by magnetic decantation with a neodymium magnet. The Fe particles were further reacted with potassium hexacyanoferrate in a dilute HCl matrix to grow a Prussian blue shell around the particle giving the particles a core/shell structure. The particles were again purified by magnetic decantation with a neodymium magnet. Based on dynamic light scattering measurements (not shown) these particles are estimated to be ∼100 nm before the shell growth.
Results and discussion
The aim of the research presented here is to show sensitive detection of nanoparticles based on the arrival time of individual ions. Ion counts from particles can be identified by the fact that they occur as a burst corresponding to the duration of the ionization and ion transport process in the plasma. Traditional mass spectrometry relies on counting ions over a designated dwell time or integration window. With the TDC detection system there is no dwell time or integration window, rather ions are continuously detected for the entire duration of the measurement. It is the ability to deftly differentiate particle signal from background signal that sets the current method apart from traditional single particle ICP-MS. The scope of this study includes various case studies designed to display the capability of the rapid transient method to detect nanoparticles or minor isotopes. It should be noted that the cases presented here would be extremely challenging or impossible to detect with traditional single particle ICP-MS. However, the scope of this initial research does not extend to a detailed comparison with standard single particle ICP-MS methods. Also, particle quantification herein refers to measurement of ions per particles. It is straightforward to extend the detection of individual particles to determine particle concentration, but this is not pertinent to the present scope. In general, the same calibration methods used for traditional single particle ICP-MS will apply to this method as well.Au nanoparticles
Standardized gold nanoparticles proved an excellent sample set to test the rapid transient method. Fig. 2 shows example rapid transient signals from four nanoparticle sizes, 5, 10, 15 and 20 nm. For comparison the cumulative ion counts (left axis) have been reset to zero counts at the start of the particle signal. Also, for comparison the time scale (bottom axis) has also been adjusted for each particle example such that the rapid transient start time is slightly offset for each particle size. The salient features of the plot are the timing of the rapid transient particle signal, background ion distribution, and cumulative counts of the individual particle signals all of which are preserved and more conveniently visualized with the normalized (linearly shifted) axes. The inset in Fig. 2 shows a zoomed in region around the 5 nm Au particle transient signal. It is also easier to see the ion by ion digitization. As expected, the rapid transient particle signals extend over a few hundred microseconds and are easily distinguished from the background signals. The background ion arrival rate is ∼3000 counts per second in these gold nanoparticle experiments.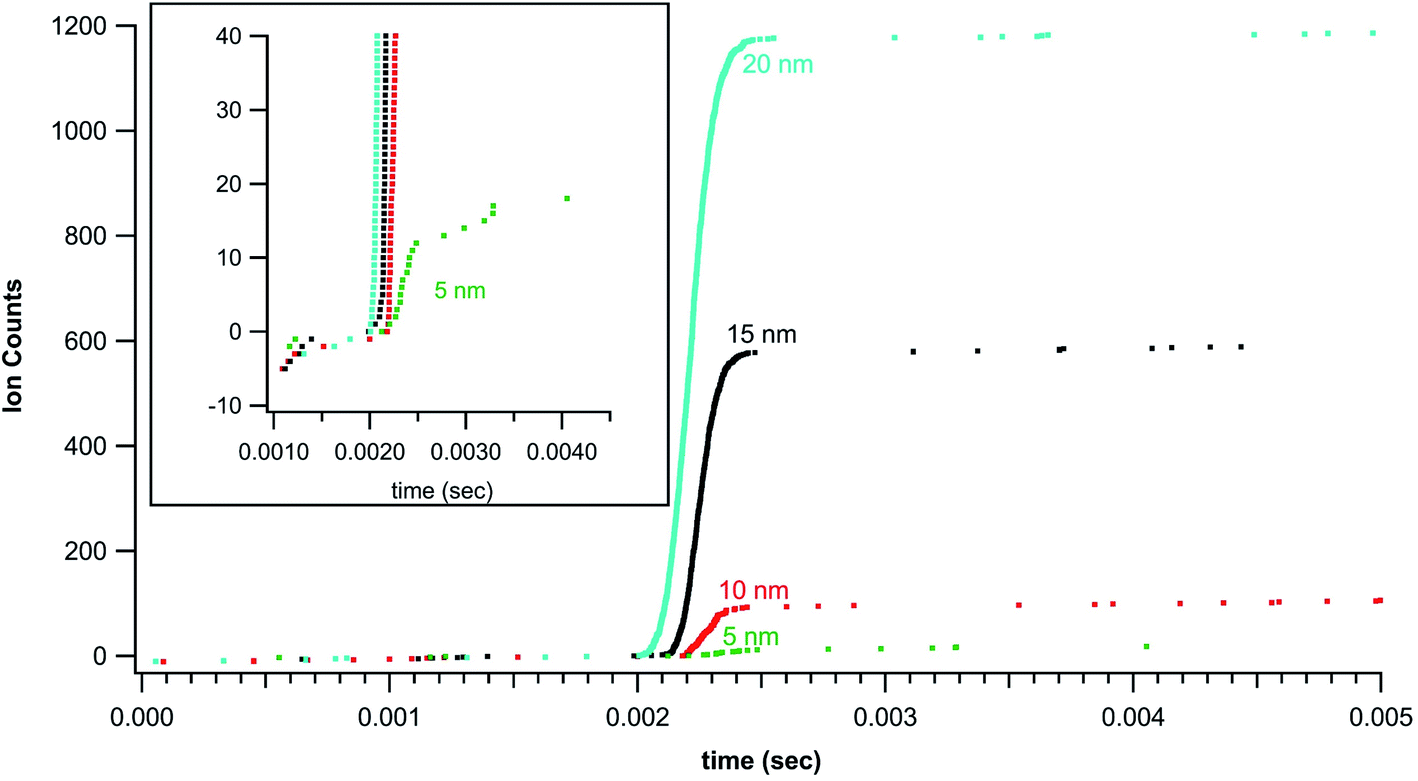 | ||
| Fig. 2 Cumulative counts of Au+ ions from ICP-MS digestion of nanoparticles (shifted to 0 counts at start of particle) as a function of time (shifted to particle start at ∼2 ms). The plots show the rapid burst of ion counts associated with particle detection. | ||
Fig. 2 introduces cumulative ion counts against time as a convenient means to visualize mass spectrometry particle data. The vertical climb relative to the slower background rise in the cumulative count is a visual indicator of particle signal. The individual ion arrival times as well as number of ion counts comprising the particle signal are more easily assessed with the cumulative count plots. The more traditional means to plot mass spectrometry data is to plot rate (counts measured within an integration window divided by the integration window time) against time. The timestamp data is less amenable to plotting rate as the only rate measurement is the inverse of the time difference between adjacent ion arrivals. This leads to noise related to closely spaced ions that are not associated with a particle. Nevertheless, Fig. 3 plots traditional rate data for a section of an experiment (40 nm Au particles) collected simultaneously on both the NeptunePlus™ data acquisition system and the digitizer. For comparison, the digitizer data was also binned into various length integration windows and plotted. In general, the 4 particles in this particular section of data are easily visualized with the digitizer data even when plotted as a rate.
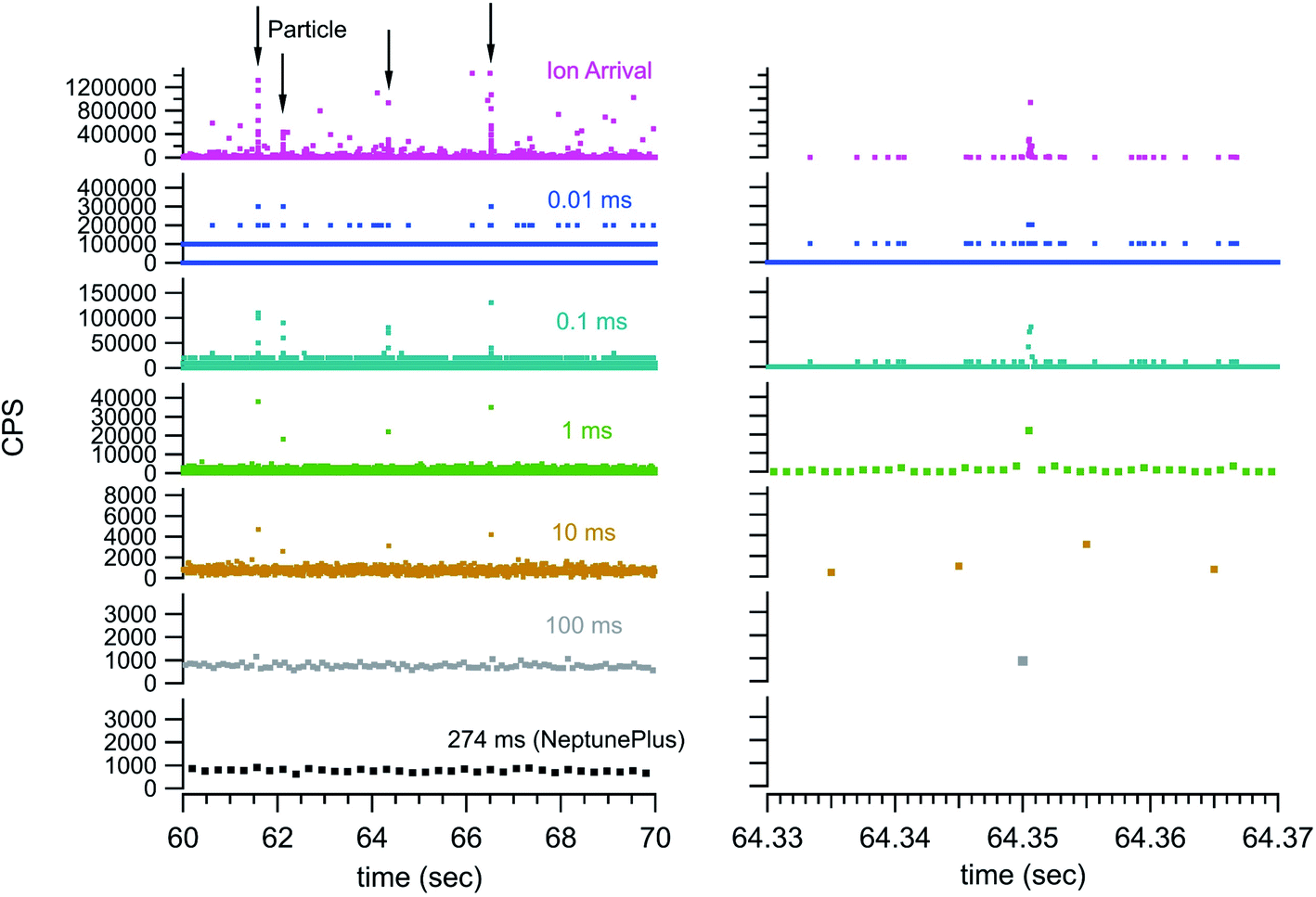 | ||
| Fig. 3 Experimental rate data (40 nm Au particle solution) simultaneously collected with the NeptunePlus digitization and TDC digitization. The TDC data was binned to show detection characteristics with a series of integration windows. The panel on the right shows a close in view of the 3rd particle indicated by the arrows on the left panel. The numbers above each trace indicate the integration window in milliseconds. | ||
Fig. 3 also shows some of the advantages and disadvantages of various integration window durations. The longer duration integration windows include too much background to pinpoint the particle signal. The shorter integration windows provide sufficient timing and exclusion of background to pinpoint a particle but begin to split the particle signal across multiple integration windows. This requires the detection system to have no blind time (time after an integration window where no ions are digitized) between integration windows for accurate quantification. In addition, the shorter integration windows can dramatically increase the data storage and handling requirements. For example, the 10 second window in Fig. 3 contains 7546 ion arrival time stamps from the TDC. However, the integration window data requires 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 data points at 100 μs integration windows and 1000000 data points at 10 μs integration windows. This is a heavy data handling tax while simultaneously eliminating exact timing information. The TDC data storage requirements are exactly matched to the signal intensity and retain the full detail of the measurement.
000 data points at 100 μs integration windows and 1000000 data points at 10 μs integration windows. This is a heavy data handling tax while simultaneously eliminating exact timing information. The TDC data storage requirements are exactly matched to the signal intensity and retain the full detail of the measurement.
Fig. 4 returns to the gold nanoparticle series and plots the average counts measured per particle as a function of the calculated number of atoms for each nominal size. There is a linear relationship for the smallest particles that falls off with particle size until reaching a stable value for the largest particles. The drop-off in linearity and the plateau value (∼104 counts) seen in Fig. 4 for the larger particles is an indication that the detection system is failing to quantify the rapid transient signals for the larger particles. While the rapid transient method is excellent for digitization and identification of particle signals it cannot alleviate challenges with pile-up at the detectors associated with measurements of larger particles. The linear fit (for the 5, 10, 15, and 20 nm particles) shows a 0.4% relationship between the measured and expected. This number is not a sample utilization efficiency but is a measure of detection efficiency for the particles and agrees with the typical efficiency of the MC instrument. Quadrupole instruments show a linear response up through 60 nm (ref. 17 and 24) and the lower linear dynamic range measured on the MC likely reflects this increased efficiency. In other words, the more efficient MC instrument more readily exhibits pile-up problems that cannot be effectively detected and/or digitized.
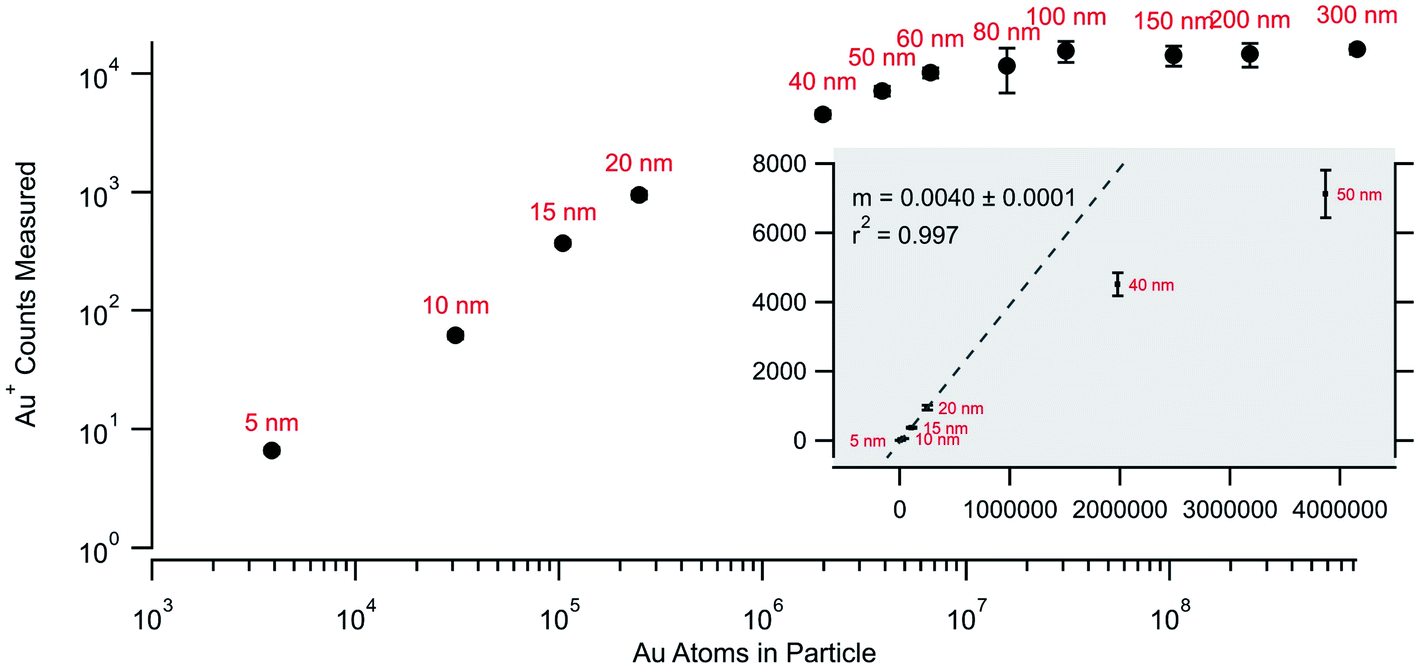 | ||
| Fig. 4 Measured average counts measured for each particle size as against the calculated number of atoms for each nominal size. | ||
Laser ablation aerosol particles
While standardized gold nanoparticles provided a means to test detection and quantification, the monoisotopic nature of gold cannot highlight the full utility of the temporally correlated signals. With the NeptunePlus™ MC-ICP-MS and the TDC digitizer it is possible to collect rapid transient data on the four most common isotopes of uranium on a particle-by-particle basis. Uranium or uranium containing aerosol particles were created via femtosecond laser ablation of uranium containing materials. The uppermost panel in Fig. 5 shows the results of ablation rastering 10 lines in NIST 612 SRM glass with a ∼35 second delay between successive lines. The lower panels show successively zoomed plots to highlight that the entire data set is composed of individual aerosol particles. It can be noted that the laser ablation aerosol presented particles to the plasma in rapid succession such that there was no time to establish a background rate between particles. With a background rate of approximately 0.5 counts per second with the laser off, 10's of seconds are required to establish the background. The ablation conditions were also sufficiently high such that the 238U could have/should have been measured on a Faraday cup detector. Even with the high particle density from the laser ablation aerosol, only occasionally were particles seen to temporally overlap in the data. In other words, there are typically distinct time/signal gaps between the successive particles.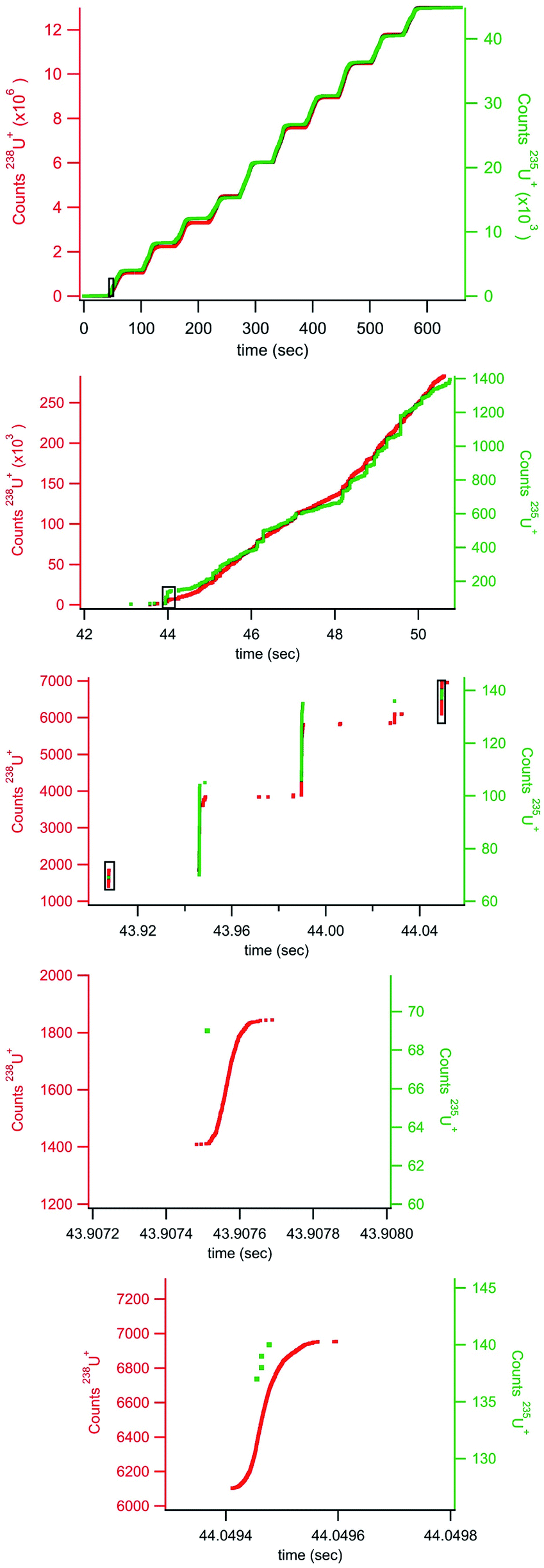 | ||
| Fig. 5 Rapid transient data collected during ablation of NIST 612 glass. The black boxes show approximate locations of enhanced areas in lower plots. | ||
With the rapid transient method, the laser ablation data can now be analyzed on a particle-by-particle basis. Fig. 6 plots the counts 235U against the counts 238U for each particle (with at least 20 counts 238U) identified in the data shown in Fig. 5. The 20 count cutoff was arbitrarily chosen to provide (with the known 235U/238U ratio of 0.002391) a roughly 5% chance of observing an associated 235U count. It should be noted that the total number of particles identified increases dramatically with a lower count cutoff; 80218 particles with a 20-count cutoff and 114252 particles with a 10-count cutoff. Fig. 6 also shows the expected 235U/238U ratio (red line) for NIST 612. The bulk of the particle data scatters around the expected line, but similar to the larger gold particles, there appears to be particles with pile-up problems on the 238U detector. As a result, insufficient 238U was measured for some particles, artificially increasing the 235U/238U ratio. A weighted average of all the particles returns a 235U/238U ratio of 0.0036 ± 0.0003. This number is higher due to the weighting, but in general agreement with traditional analysis of the laser ablation signals (integration of background subtracted signal from each rastered line) which returns a 235U/238U ratio of 0.0033 ± 0.0002 (1 × 107 total 238U counts). Both numbers are well above the expected ratio. However, the data in Fig. 6 indicate that particles with fewer than ∼700 238U counts do not exhibit pileup problems. This data, shown in the figure inset, are highly discretized, but the weighted average returns a 235U/238U ratio of 0.0025 ± 0.0008 (6.5 × 106 total 238U counts), consistent with the expected value. None of these ratios were mass bias or detector gain corrected but are presented as raw ratios and used to highlight the advantage of particle-by-particle processing.
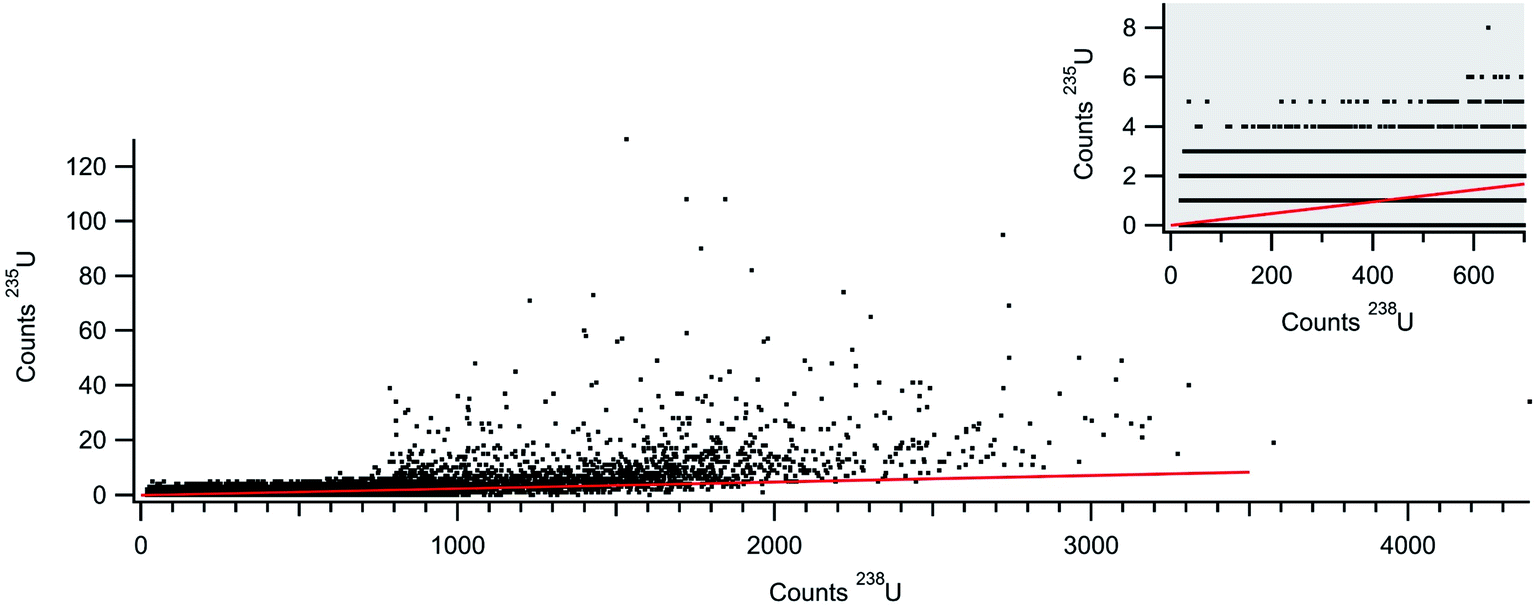 | ||
| Fig. 6 Counts 235U+ against counts 238U+ for NIST 612 laser ablation aerosol. Each data point plots counts from an individual particle. The inset shows lower count section of the particle data. | ||
U aerosol particles
In addition to the laser ablation of NIST 612 glass, Fig. 7 shows the rapid transient signal from a uranium particle collected after laser ablation analysis of SRM U015 powder embedded in a polymer film. This specific particle shown in Fig. 7 (1 count 236U, 14 counts 235U, and 779 counts 238U) gives a 235U/238U ratio of 0.018, close to the expected ratio of 0.01557. However, the salient feature of Fig. 7 is the single count of 236U within the timing envelop of the major isotope signal. This correlated signal gives added significance to the 236U count and illustrates the means by which the rapid transient method can pick minor component signal apart from background. In this case, the temporal correlation supports the detection of 236U, but with only a single count measured, a more precise isotope ratio requires measurement of a statistically significant particle population. The entire data set (not shown) gives a 236U/235U ratio of 0.011 ± 0.005 (with a total of 465 counts 236U and 40615 counts 235U) compared to the expected ratio of 0.0107. Again, this is a raw ratio, not corrected for mass bias or detector gain differences.
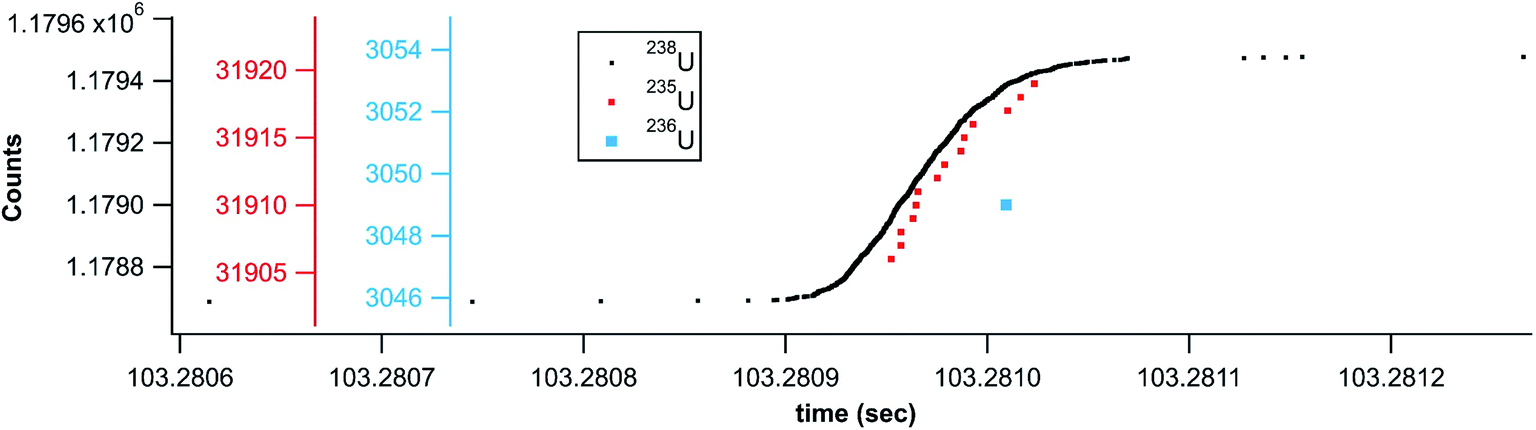 | ||
| Fig. 7 U015 rapid transient from ablation of U015 isotope standard. | ||
As with the gold particles and the NIST glass, the 238U signal from the U015 could not be accurately quantified for the larger particles. Fig. 8 shows an example of a U015 particle where the 238U detector fails to return counts during the most intense portion of the particle transient. The continuous 235U signal indicates that this is a single particle signal with a break in detection of 238U rather than two closely spaced particles. This pile-up related challenge is responsible for the falloff in dynamic range for the gold particles and difficulty measuring accurate isotope ratios for the larger NIST glass and U015 particles. Future efforts will attempt to interpolate the missing signal based on the measured temporal profile. Given the highly dynamic ion detection rate, this interpolation method based on the temporal signature of a particle may provide a more accurate means of pile-up (deadtime) correction compared to traditional rate based deadtime correction methods (for particles).
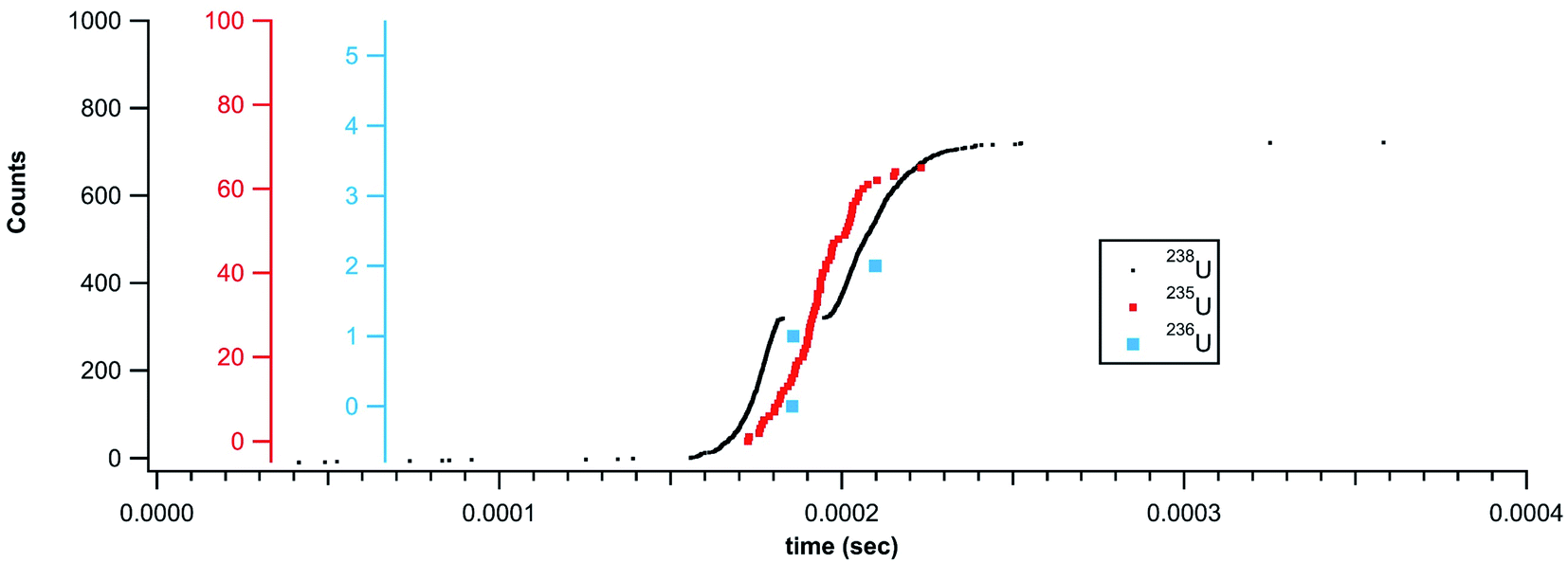 | ||
| Fig. 8 Rapid transient signal from a U015 particle showing a break in 238U detection due to pileup. | ||
Fe nanoparticles
As mentioned for the minor isotopes of uranium, the rapid transient method holds tremendous analytical potential for separating particle signal apart from background. For example, iron isotope ratio measurements on ICP-MS instruments typically require high mass resolution to separate polyatomic interferences from the iron signal. Fig. 9 shows a high-resolution mass scan of an iron solution showing simultaneous detection of 54Fe+ and 56Fe+. Typically, Fe ratios are measured with the magnet set to collect data on the Fe-only shoulder of the partially resolved peak. However, we collected high mass resolution data from iron nanoparticles with the magnet set to the Fe shoulder (excluding the polyatomic interferences) and set at a position to include polyatomic interferences. In both cases, analysis of only the nanoparticle signals returns an isotope ratio consistent with the known value (54Fe/56Fe = 0.0637). The rapid transient particle method effectively isolated the particle signals comprised of less than 2500 total counts from an intense (∼125 K CPS for the case of 56Fe) atmospheric polyatomic signal. Moreover, the isotope ratios measured from the particle signal were calculated without background subtraction. For an element like Fe, this method has potential beyond nanoparticles to make isotope ratio measurements on a cell-by-cell basis allowing for ratio determinations without time consuming separations and purifications or the need for high resolution. It would also allow for the detection (but not isotope ratio analysis) and quantification of Fe particles on lower resolutions single collector instruments.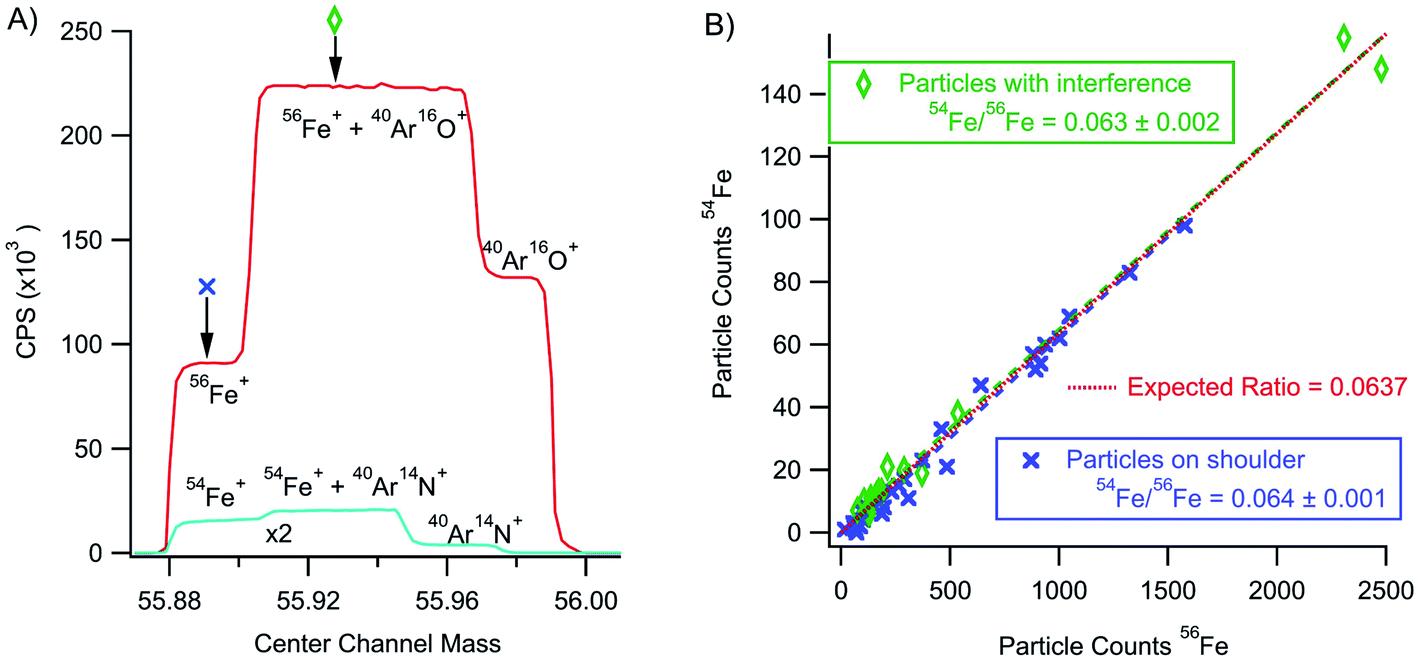 | ||
| Fig. 9 Panel (A) shows a high resolution scan over 56Fe+ and 54Fe+ masses along with partially resolved polyatomic interferences. Panel (B) shows the 54Fe+ particle signal against the 56Fe+ signal for particles collected without (on the resolved Fe shoulder) and with polyatomic interference. | ||
Conclusions
This research represents the initial development of the rapid transient method for detecting and accurately measuring nanoparticles with a high degree of sensitivity. The results show the ability to use characteristics of ion arrival times to discriminate particle signal from background and as a means for particle detection. The results presented here not only show that nanoparticles can be measured in the presence of a high background, but also the ability to measure minor isotopes within a particle by temporal correlation with a major component. Pileup remains a significant challenge for the rapid transient method and limits quantification to nanoparticles. Additional research into expanding the dynamic range of the detection systems will make the rapid transient method applicable to a wider range of particle sizes. Ultimately the rapid transient particle method has far reaching implications for the detection of both anthropogenic and naturally occurring particles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Nuclear Security Administration supported this work under an Interagency Agreement with the U.S. Department of Energy (DOE) under Contract DE-AC05-75RLO1830.References
- S. Thomas, N. Kalarikkal, A. M. Stephan and B. Raneesh, Advanced Nanomaterials: Synthesis, Properties, and Applications, Apple Academic Press, 2014 Search PubMed.
- W. W. Lee and W. T. Chan, J. Anal. At. Spectrom., 2015, 30, 1245–1254, 10.1039/C4JA00408F.
- L. Hendriks, B. Ramkorun-Schmidt, A. Gundlach-Graham, J. Koch, R. N. Grass, N. Jakubowski and D. Günther, J. Anal. At. Spectrom., 2019, 34, 716–728, 10.1039/C8JA00397A.
- D. Mozhayeva and C. Engelhard, J. Anal. At. Spectrom., 2020, 35, 1740–1783, 10.1039/C9JA00206E.
- K. Flores, R. S. Turley, C. Valdes, Y. Ye, J. Cantu, J. A. Hernandez-Viezcas, J. G. Parsons and J. L. Gardea-Torresdey, Appl. Spectrosc. Rev., 2019, 1–26, DOI:10.1080/05704928.2019.1694937.
- E. Bolea-Fernandez, A. Rua-Ibarz, M. Velimirovic, K. Tirez and F. Vanhaecke, J. Anal. At. Spectrom., 2020, 35, 455–460, 10.1039/C9JA00379G.
- Z. Sun, J. Fan, H. Li and H. Jiang, Appl. Sci., 2018, 8, 132 CrossRef.
- E. P. Gray, J. G. Coleman, A. J. Bednar, A. J. Kennedy, J. F. Ranville and C. P. Higgins, Environ. Sci. Technol., 2013, 47, 14315–14323, DOI:10.1021/es403558c.
- B. R. Li, H. Tang, R. Q. Yu and J. H. Jiang, Anal. Chem., 2020, 92, 2379–2382, DOI:10.1021/acs.analchem.9b05741.
- M. Bundschuh, J. Filser, S. Lüderwald, M. S. McKee, G. Metreveli, G. E. Schaumann, R. Schulz and S. Wagner, Environ. Sci. Eur., 2018, 30, 6, DOI:10.1186/s12302-018-0132-6.
- D. M. Schwertfeger, J. R. Velicogna, A. H. Jesmer, R. P. Scroggins and J. I. Princz, Anal. Chem., 2016, 88, 9908–9914, DOI:10.1021/acs.analchem.6b02716.
- R. Vogt, D. Mozhayeva, B. Steinhoff, A. Schardt, B. T. F. Spelz, A. Philippe, S. Kurtz, G. E. Schaumann, C. Engelhard, H. Schönherr, D. K. Lamatsch and J. Wanzenböck, Sci. Total Environ., 2019, 696, 134034, DOI:10.1016/j.scitotenv.2019.134034.
- G. Craig, Improving the utility of LA-ICP-MS for isotope ratio analyses of single particles with application to uranium oxide, thesis, Loughborough University, 2016, https://hdl.handle.net/2134/21518.
- D. Mozhayeva and C. Engelhard, J. Anal. At. Spectrom., 2019, 34, 1571–1580, 10.1039/C9JA00042A.
- J. W. Olesik and P. J. Gray, J. Anal. At. Spectrom., 2012, 27, 1143–1155, 10.1039/C2JA30073G.
- P. Shaw and A. Donard, J. Anal. At. Spectrom., 2016, 31, 1234–1242, 10.1039/C6JA00047A.
- L. A. Rush, M. C. Endres, M. Liezers, J. D. Ward, G. C. Eiden and A. M. Duffin, Talanta, 2018, 189, 268–273, DOI:10.1016/j.talanta.2018.06.071.
- M. D. Montaño, J. W. Olesik, A. G. Barber, K. Challis and J. F. Ranville, Anal. Bioanal. Chem., 2016, 408, 5053–5074, DOI:10.1007/s00216-016-9676-8.
- A. Hineman and C. Stephan, J. Anal. At. Spectrom., 2014, 29, 1252, 10.1039/C4JA00097H.
- I. Strenge and C. Engelhard, J. Anal. At. Spectrom., 2016, 31, 135–144, 10.1039/C5JA00177C.
- G. C. Eiden, A. M. Duffin, M. Liezers, J. D. Ward, J. W. Robinson, G. L. Hart, S. H. Pratt, K. W. Springer, A. J. Carman and D. C. Duckworth, International Atomic Energy Agency International Conference on Advances in Nuclear Forensics Proceedings, Vienna, Austria, 2014 Search PubMed.
- C. Thammawong, P. Opaprakasit, P. Tangboriboonrat and P. Sreearunothai, J. Nanoparticle Res., 2013, 15, 1689, DOI:10.1007/s11051-013-1689-z.
- F. Bao, J.-L. Yao and R.-A. Gu, Langmuir, 2009, 25, 10782–10787, DOI:10.1021/la901337r.
- J. Y. Liu, K. E. Murphy, R. I. MacCuspie and M. R. Winchester, Anal. Chem., 2014, 86, 3405–3414, DOI:10.1021/ac403775a.
| This journal is © The Royal Society of Chemistry 2021 |