
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combining acid-based deep eutectic solvents and microwave irradiation for improved chestnut shell waste valorization†
José
González-Rivera‡
 ab,
Angelica
Mero‡
a,
Elena
Husanu
a,
Andrea
Mezzetta
a,
Carlo
Ferrari
b,
Felicia
D'Andrea
a,
Emilia
Bramanti
c,
Christian S.
Pomelli
a and
Lorenzo
Guazzelli
*a
ab,
Angelica
Mero‡
a,
Elena
Husanu
a,
Andrea
Mezzetta
a,
Carlo
Ferrari
b,
Felicia
D'Andrea
a,
Emilia
Bramanti
c,
Christian S.
Pomelli
a and
Lorenzo
Guazzelli
*a
aDepartment of Pharmacy, University of Pisa, Via Bonanno, 6, 56126 Pisa, Italy. E-mail: lorenzo.guazzelli@unipi.it
bNational Institute of Optics, (INO-CNR)-UOS Pisa, Via G. Moruzzi 1, 56124 Pisa, Italy
cInstitute of chemistry of organometallic compounds (ICCOM-CNR) Pisa, Via G. Moruzzi 1, 56124 Pisa, Italy
First published on 26th November 2021
Abstract
A microwave (MW)/deep eutectic solvent (DES)-assisted (MWDA) extraction process for obtaining value-added compounds from chestnut shell waste (CSW) is presented. DESs were used as green solvents featuring both high biomass dissolution ability and good response to MW irradiation. A survey of different acid-based DESs led to the identification of Choline Chloride (ChCl)–oxalic acid as the system most sensitive to experimental design variations, especially when considering the total phenolic content (TPC) and the total mass amount recovered (yield wt%). Hence, the MW absorption properties of ChCl–oxalic acid dihydrate and ChCl–oxalic acid DESs, as single components and in mixture with water, were assessed by the experimental determination of their MW heating response. Moreover, the extraction efficiency for the best MW responding system, ChCl–oxalic acid dihydrate, was evaluated at different MW-irradiation times and extraction temperatures. The isolated polyphenols from CSW were further characterized by HPLC-DAD analysis. Gallic acid, ellagic acid, catechin hydrate and procyanidin B2 were identified and quantified, with their relative ratios varying as a function of the MWDA extraction conditions. Lignin, hemicellulose and cellulose compositions of the solid residues of CSW after the MWDA extraction were assessed using a cross validation model obtained by partial least squares regression (PLS) of their FTIR spectra. The chemometrics results, corroborated by SEM analyses, highlighted the ability of the oxalic acid-based DES to simultaneously remove lignin during polyphenol extraction. Overall, the MWDA extraction process presented here enables the fast, cheap and tunable processing of food waste yielding a high amount of valuable bioactive compounds.
Introduction
Microwave (MW) assisted chemical processes are nowadays a mature and well-established research field. Dielectric heating promoted by MW irradiation has been applied to various chemical processes such as reactions,1 extractions,2,3 materials synthesis4–6 and biomass processing.7 MW assisted heating significantly reduces processing time, energy consumption, costs, environmental impact and equipment size with respect to conventional heating methods.1,8The use of MWs is therefore considered as a powerful tool during the development of modern green chemical approaches for the sustainable processing and valorization of food waste.
However, the employment of MW irradiation in biomass conversion processes is not widespread on account of the intrinsic heterogeneity of the material and of the different microwave absorption properties that the partners involved in the chemical process (e.g. solvents, reagents, catalysts, and products) may have.8 Despite this, the selective heating of specific components of the reaction system via MW tuning may reduce the energy consumption and enhance the product selectivity.9
Economically viable and energy efficient approaches for biomass valorization using MW must satisfy two main points: (1) the biomass should preferably come from low-cost raw materials such as food waste or lignocellulosic matter and, (2) when a solvent is required, the latter must display strong MW absorption properties since it represents the highest energy-consuming mass percent.
Concerning the first aspect, chestnut shell waste (CSW) presents many positive features. A considerable amount of CSW (e.g. 5300 tons per year just in Italy) is generated during the peeling of nuts before food product preparation (flour, marron glacé, etc.).10 Chestnut shell is a rich feedstock containing high amounts of polyphenols, tannins, lignin, cellulose and sugars.11 In the past, the shells along with the generated waste were burned for energy production, but the high moisture content of the shells made this practice not efficient. This practice has been gradually abandoned leading to mounting costs for CSW disposal.
A green and potentially cost-effective alternative consists of CSW valorization for polyphenols and tannins extraction.10–33 Extracts from chestnut shells have been tested and displayed biological activities as antioxidants22 and anticancer16 and antimicrobial agents.12 From an economic point of view, the valorization of CSW for the recovery of polyphenols is clearly a business opportunity, considering that the global market of polyphenols was about USD 1.28 billion in 2018 with an estimated growing rate of 7.2% in the following 5 years.34
Efforts for the valorization of extracts from biomass have been focused on the reduction of extraction time and energy consumption as well as on the increase of the yields. The latter depends on the extraction conditions (temperature, time and solvent) used and on the feedstock properties. Chestnut shell extraction protocols include the use of common organic solvents and water solutions.10,12,13–20,23,27,28,30,32,33 Conventional heating using water as the solvent has been the most common extraction approach reported.10,18,19,22 The classical Soxhlet extraction using hexane has also been studied,27 while innovative extraction approaches concerning the use of MWs,25,32 ultrasound24,28 or subcritical fluid21,26 extraction and hydro-alcoholic mixtures have recently begun to be investigated.
As for the second essential feature for MW exploitation in biomass treatment mentioned above, deep eutectic solvents (DESs) represent potential matching partners. Indeed, DESs have been used during the last years for the valorization of different lignocellulosic and food waste biomasses, among which is CSW.35–44 DESs are composed of a hydrogen bond acceptor (HBA), often quaternary ammonium salts, and a hydrogen bond donor (HBD), such as carboxylic acids, polyols, amides and carbohydrates. The mixture of the HBA and the HBD at a specific molar ratio causes the decrement of the freezing temperature at the eutectic point below that expected for an ideal behavior.45 DESs are considered green solvents because of their low cost of preparation, low vapor pressure with respect to organic solvents46 and good recycling and reuse properties. Additionally, we have recently demonstrated the strong ability of DESs to interact with an electromagnetic field, which makes them strong MW absorbing solvents.47
The performance of DESs in biomass fractionation and separation can be tuned by modulating their composition, which allows for the high dissolution of biopolymers,35 polyphenol isolation48 and biomass delignification.49–51
Thus, the combined use of DESs and MWs appears to be a promising strategy for the separation/recovery of lignin49,50 and polyphenols52–58 in relatively short processing times.
In this work, we explored the combined use of acidic ChCl-based DESs and MWs for the valorization of CSW through the isolation of polyphenols and the subsequent recovery of the solid residue. First, acid-based DESs and DES–water mixtures were tested to identify the most versatile system in terms of the total amount of soluble polyphenols recovered. Then, the MW heating response of this best-performing system and its single components was assessed, along with the effect of the reaction time and extraction temperature. The isolated polyphenols were further characterized by HPLC-DAD analysis.
To determine the effect of the MWDA extraction process on the recovered solid residues, these latter were characterized by applying FTIR spectroscopy together with a chemometric method previously developed.3,59 The morphological characterization and thermal analysis of the solid residues have been carried out by SEM imaging and thermogravimetric analysis, respectively. The entire developed MWDA extraction process is illustrated in Fig. 1.
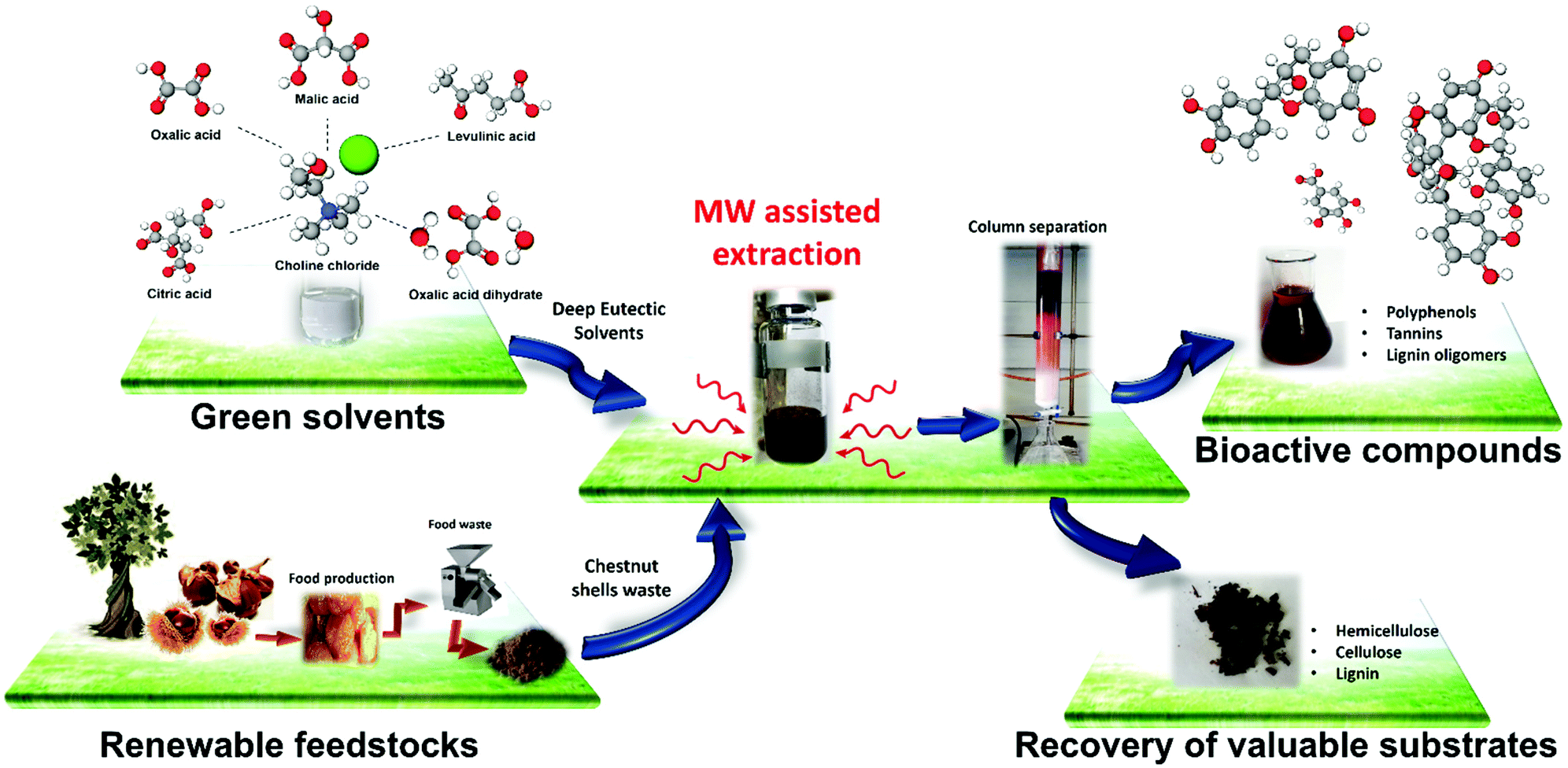 | ||
| Fig. 1 Overview of the MWDA extraction process developed herein for polyphenol and lignocellulose residue isolation and recovery from chestnut shell waste (CSW) using different acid-based deep eutectic solvents (DES). | ||
Experimental
Materials
Acid-based DESs were prepared using the following reagents: Choline chloride (ChCl, 98%) and DL-malic acid (98%) were purchased from Alfa Aesar and Thermo Fisher (Germany). Citric acid (99.5%), oxalic acid dihydrate (99%), levulinic acid (98%) and oxalic acid (98%) were purchased from Sigma Aldrich (Milan, Italy). Deionized water obtained with a Milli-Q 50 system (Millipore, Bedford, MA, USA) was used for the preparation of water–DES mixtures. Phloretin, ellagic acid, resveratrol, chlorogenic acid, cyanin chloride, coumarin, quercetin, tannic acid, resorcinol, pyrocatechol, pyrogallol, (−)-epicatechin, gallic acid, (−)-epigallocatechin gallate, trans-ferulic acid, caffeic acid, 1,3,5-trihydroxybenzene dehydrate, salicylic acid, acetylsalicylic acid, (+)-abscisic acid, vanillin, pinoresinol, (+)-catechin and 3-hydrotyrosol, tyrosol were HPLC analytical standards purchased from Merck (Milan, Italy). Tyrosol, hydroxytyrosol, oleuropein, syringic acid, luteolin and apigenin were purchased from EXTRASYNTHESE (Cedex, France). Deionized water obtained with a Milli-Q system (Purelab Pro + Purelab Classic, Millipore, USA) was used as a solvent for all the extractions. Ethanol (EtOH for HPLC, ≥99.8%, Fluka), methanol (MeOH for HPLC ≥99%, Merck), dimethyl sulfoxide (DMSO for GC ≥99.5%, Merck), sodium hydroxide (NaOH 0.1 M, Merck) and formic acid (≈98%, Fluka) were used as solvents for the preparation of standard solutions of polyphenols. The chestnut shell waste was provided by “Ortofrutticola del Mugello S.R.L.” (Italy), the material was ground into fine powder (0.5–1 mm) and dried at 60 °C in an oven for 16 h before use.DES preparation
DES preparation was carried out by mixing the corresponding HBD and HBA as reported elsewhere.47 Briefly, ChCl was dried under vacuum for 6 h at 80 °C and immediately weighed to prevent water absorption (due to its strong hygroscopic character). The corresponding carboxylic acids as HBD were weighed and transferred to the same glass vessel which was sealed. ChCl and the appropriate HBD were mixed until a homogeneous transparent liquid was formed. ChCl–malic acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), ChCl–oxalic acid (1:1) and ChCl–citric acid (1:1) were prepared at 80 °C while ChCl–levulinic acid (1:2) and ChCl–oxalic acid dihydrate (1:1) were prepared at room temperature. After DES formation, no further purification steps were needed and all DESs were kept at room temperature in sealed vessels until their use and characterization. The DES composition and purity were monitored by NMR spectroscopy and are reported elsewhere.36,47
:1), ChCl–oxalic acid (1:1) and ChCl–citric acid (1:1) were prepared at 80 °C while ChCl–levulinic acid (1:2) and ChCl–oxalic acid dihydrate (1:1) were prepared at room temperature. After DES formation, no further purification steps were needed and all DESs were kept at room temperature in sealed vessels until their use and characterization. The DES composition and purity were monitored by NMR spectroscopy and are reported elsewhere.36,47
Microwave/DES-assisted (MWDA) extraction of polyphenols
The extraction of polyphenols from CSW was carried out as follows: 5 g of DES were loaded into a 20 mL glass vial equipped with a magnetic stirrer and 0.5 g of dry CSW were then added and mixed. The vial was closed, and the mixture was stirred for 2 min at room temperature. The MW-assisted extraction was performed using a commercial Microwave synthesizer Initiator+ Biotage. The temperature of the extraction (65 °C, 75 °C or 85 °C) was set and controlled by the modulation of the microwave-applied power (source of MWs was a magnetron oscillator 0–400 W of continuous MW irradiation power at a frequency of 2.45 GHz). Once the temperature was reached, the MW irradiation time was kept at different extraction times (text = 5, 15, 30 or 60 min). After the established extraction time was reached, the MW power supplied was switched off and the vial was quenched to room temperature with pressurized air. The polyphenols isolation was carried out as follows: the undissolved solid residue contained in the suspension obtained after the reaction was filtered by vacuum filtration and washed with MeOH/H2O (70/30 v/v) until a clear solution came out, which was recovered and dried in an oven at 65 °C for 72 h. The yield % of solid residues was calculated: wt% of solid residues = (mass of recovered dried solid residues/initial mass of CSW) × 100. The filtrate, consisting of a mixture of extracted polyphenols, DESs and methanol–water washing solution, was collected, and concentrated under reduced pressure for methanol removal. The recovered methanol can be recycled and reused. After this, a separation column was prepared loading a polymeric resin (Amberlite XAD-7) to adsorb and separate the phenolic compounds from the recovered water–DES phase. Prior to use, the resin was washed and activated by stirring it with acidified water (HCl 0.01 M) for 30 min. The filtrate was then slowly added to the Amberlite XAD-7-column and DES was separated and recovered at the desorption exit of column. The Amberlite XAD-7-column with the adsorbed polyphenols was rinsed several times with water until neutral pH, dried with an airflow, and the extracted polyphenols were desorbed from the resin with MeOH until a clear solution came out (∼150 mL). After, the extracted polyphenols were recovered by methanol evaporation under reduced pressure. The isolated extracts were further dried at 65 °C overnight under vacuum to complete solvent removal, weighed and the yield of recovered polyphenols was calculated: wt% = (mass of dried phenolic extracts/initial mass of dried CSW) × 100. The extracts were then dissolved in 10 mL of MeOH, stored at 4 °C and protected from light for further analysis (total polyphenol content and HPLC quantification).Characterization
For the analysis, a proper amount of the dyes was dissolved in the solvent. Subsequently, the absorbance was measured with an UV-vis spectrophotometer at room temperature. The solvatochromic parameter was calculated using the following equations:
| π* = 0.314 × (27.52 − νNEt2) | (1) |
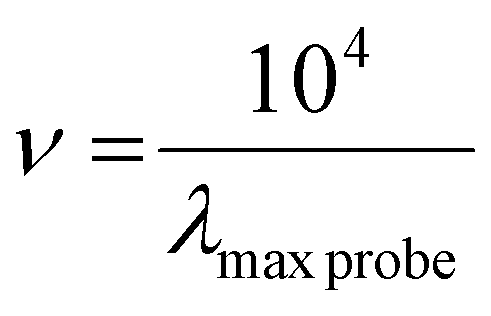 | (2) |
| A = (As − Ab) − (Ac − A0) | (3) |
Catechin hydrate was employed to prepare a calibration curve (y = 0.00197x, R2 = 0.99824), and the results were expressed as mg of catechin hydrate equivalents per g of dry biomass. For each sample, the vanillin–HCl assay was performed in triplicate.
The mobile phases for the determination of polyphenols in standard solutions and extraction samples were 0.2% formic acid in water (eluent A) and methanol (eluent B). A gradient elution was performed as follows: 0–5 min, 5% B; 5–55 min, linear gradient up to 95% B; 55–65 min, 95% B; 65–67 min, linear gradient up to 5% B (post-run time = 15 min). Elution was performed at a solvent flow rate of 1.0 mL min−1. Gallic acid, catechin hydrate, procyanidin B2, vanillic acid, (−)-epicatechin, syringic acid, p-coumaric acid, trans-ferulic acid, propyl gallate, ellagic acid hydrate, 3,5-di-tert-butyl-4-hydroxybenzaldehyde, ethoxyquin, quercetin, nordihydroguaiaretic acid, 3-tert-butyl-4-hydroxyanisole, 2,6-di-tert-butyl-4-hydroxymethyl phenol, octyl gallate, lauryl gallate, and tert-butylhydroquinone were used as analytical standards for HPLC quantification. The chromatographic peaks of analytes were confirmed by comparing UV spectra and their retention times with those of the reference compounds.
A ChromQuest™ 4.2 Chromatography Data System was used to control HPLC-DAD, for data acquisition and data analysis.
Results and discussion
MWDA polyphenol extraction using acid-based DESs
First, the performance of DESs based on ChCl as the HBA and five different carboxylic acids as the HBD (levulinic, oxalic, malic, and citric acid and oxalic acid dihydrate) was investigated. Key parameters in these studies were the yield mass percentage and total phenolic content (TPC) of isolated soluble extracts at fixed MW assisted extraction conditions (65 °C and 30 min of MW extraction time). All the DESs considered were tested using a solid to liquid weight ratio of 1:10. Fig. 2 shows the yield of polyphenols as mass percentage and TPC (mg GAE per g dry CSW) obtained using pure ChCl–levulinic acid (1:2 mol ratio), ChCl–oxalic acid (1:1 mol ratio), ChCl–malic acid (1:1 mol ratio) and ChCl–oxalic acid dihydrate (1:1 mol ratio) DESs. ChCl–citric acid (1:1 mol ratio) was used in combination with water as the co-solvent (25 wt%) due to the high viscosity and difficult handling of this DES in pure form. The effect of adding water to DESs was further investigated also for ChCl–oxalic acid, ChCl–malic acid and ChCl–levulinic acid. The addition of water to highly viscous DESs (DES–H2O system) has indeed been described as beneficial for the extraction process of bioactive compounds from biomass.36,61
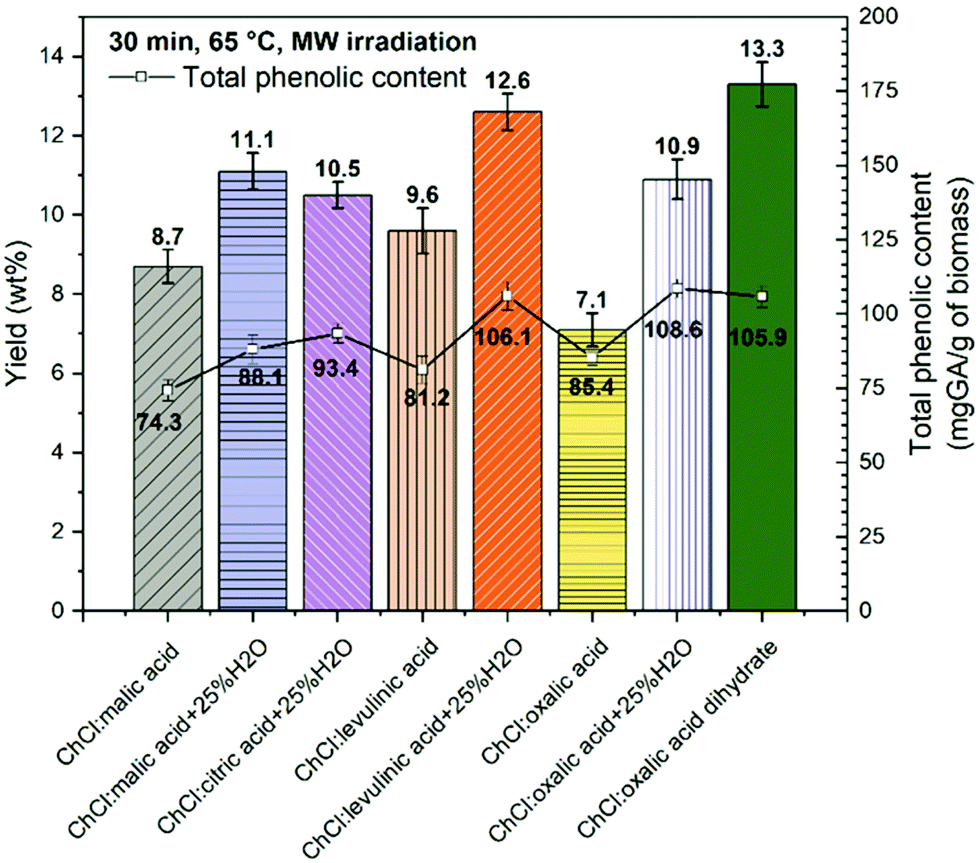 | ||
| Fig. 2 Yield wt% (vertical bars, left axis) and the total phenolic content (square dots, right axis) obtained at 65 °C and 30 min of MW extraction using acid-based DESs with or without the addition of water. | ||
The yield wt% of polyphenols extracted from CSW ranged between 13.3 ± 0.6 wt% obtained using ChCl–oxalic acid dihydrate and 7.1 ± 0.4 wt% obtained by using ChCl–oxalic acid.
As expected, the lowest yields were observed for the most viscous systems (ChCl:oxalic acid and ChCl:malic acid) followed by ChCl:levulinic acid, a less viscous medium already without the addition of water. High viscosity is one of the main drawbacks of most NADES which limits their use in pure form on account of the subsequent reduced mass transfer during the extraction process. Unsurprisingly, enhancements on extraction yield wt% were observed for all DES–H2O systems with respect to the corresponding pure DESs, in line with the results obtained by others.36,61 A higher yield was also obtained for ChCl:citric acid + 25% H2O. It is interesting to observe that the extraction yield wt% using ChCl–oxalic acid dihydrate was the highest obtained among all pure DESs and DES–H2O systems investigated.
This is notable given that its intrinsic content of water is about 13.5 wt%, which is about half of that associated with the corresponding DES–H2O systems studied. It can be speculated that oxalic acid's crystallization water promotes a different arrangement/interaction within the DES. The use of pure water as the solvent was also tested and yielded the lowest quantity of polyphenols (yield = 5.2 ± 0.4 wt%) under the same extraction conditions.
The TPC values ranged from 74.3 ± 3.5 to 108.6 ± 3.1 mg GAE per g of dry CSW for ChCl:malic acid and ChCl:oxalic acid, respectively (Fig. 2). The spectrophotometric assay appeared less sensitive to the kind of DES or DES–water system employed, nevertheless similar trends to those found for yield wt% were observed. Indeed the lowest values of the TPC were recorded for the three pure DES systems, while slight increments were noticeable with the addition of water. Unlike yields, for TPC values no significant differences were registered between ChCl:oxalic acid dihydrate and ChCl:oxalic acid + 25% H2O. A similar value was also obtained for ChCl:levulinic acid + 25% H2O.
Based on these results, DESs based on ChCl as the HBA and oxalic acid (anhydrous + 25% H2O and dihydrate) as the HBD were identified as the systems most sensitive to the MWDA extraction conditions and thus selected for further studies.
Insights into the MWDA extraction mechanism through the MW absorption response and heating behavior of ChCl–oxalic acid dihydrate
An explanation for the high performance of ChCl–oxalic acid dihydrate in the MWDA extraction of polyphenols may lie in its MW absorption properties. Therefore, a comparison of the experimental MW heating response of the following solvent systems was carried out:1. ChCl–oxalic acid dihydrate DES as the best-identified extraction solvent (based on yield wt% and the TPC of extracts obtained).
2. ChCl–oxalic acid as the identical DES system concerning the HBD (organic acid) and HBA (ionic character) compositions.
3. Pure DES components (HBD or HBA).
4. Single HBD or HBA in aqueous solutions.
5. Water as the reference MW absorbing solvent.
6. Concerning hydrogen bond arrangement promoted by the nature of water:
a. ChCl–oxalic acid DES + 13.5 wt% H2O, water added after DES formation.
b. ChCl–oxalic acid DES + 13.5 wt% H2O, water added before DES formation, at the initial stage together with ChCl and oxalic acid.
c. ChCl–oxalic acid dihydrate DES, water present during DES formation due to the presence of lattice water from oxalic acid dihydrate
Fig. 3 shows the experimental temperature profiles of all the solvent systems investigated obtained under uniform, unidirectional and constant MW applied power (30 W in Fig. 3a and 10 W in Fig. 3b at 2.54 GHz) for a fixed MW irradiation time interval (60 s). The MW heating profiles shown in Fig. 3 represent an indirect approach for the rapid estimation of the MW absorption properties. Although the quantification of the microwave interaction with solvents is usually determined through the complex permittivity (ε*), this indirect approach is straightforward and fast, and it allows for easily identifying good matching solvents for MW assisted processes.47
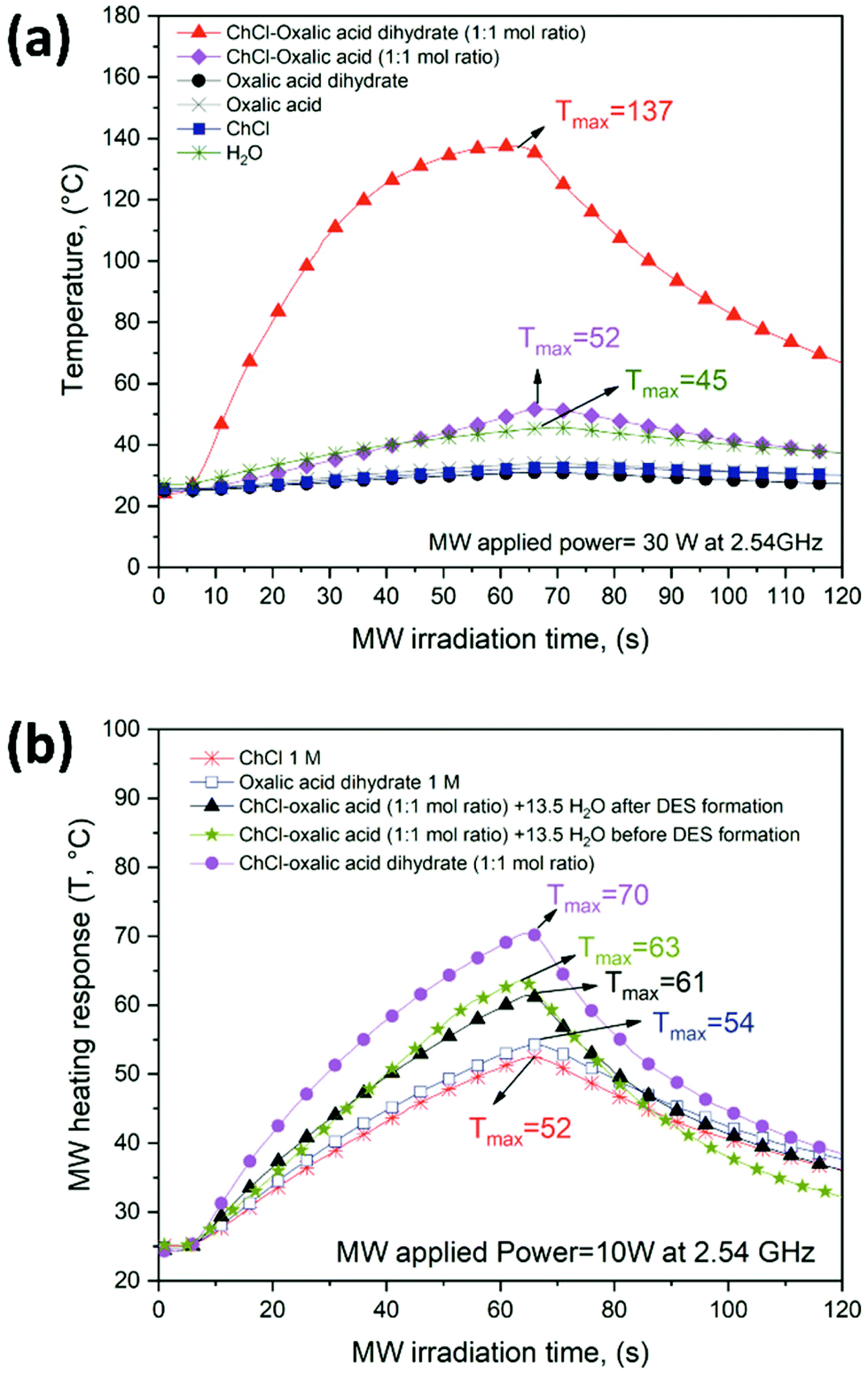 | ||
| Fig. 3 Temperature profiles of oxalic acid-based DESs and the different solvent systems (1–6) recorded under (a) 30 W and, (b) 10 W at a frequency of 2.54 GHz. | ||
Among all six investigated solvent systems, ChCl–oxalic acid dihydrate showed the highest response to MW irradiation due to the fast-heating rate (Fig. 3a, full triangle curve). The following three main heating steps were identified:
1. A fast continuous temperature increment (linear slope) during the first 30 s (from room temperature up to about 100 °C).
2. A decrease of the heating rate (change and reduction of the curve slope) during the last 30 s of MW irradiation (from about 100 °C to Tmax = 137 °C).
3. The natural cooling profile, once the MW applied power is stopped (from 60 to 120 s).
The first two heating steps (obtained under an electromagnetic field) are both related to the MW absorption properties. The strong slope change in the curve (above 100 °C) is probably due to DES decomposition, thermal breakdown or reconfiguration of the hydrogen bond network promoted by water migration/evaporation. A similar behavior was observed for polyol-based DESs under electromagnetic irradation.47 The MW heating response of ChCl–oxalic acid dihydrate was dependent on the MW applied power. Fig. S1 of the ESI† shows the temperature profiles of ChCl–oxalic acid dihydrate at three different MW powers (10, 20 and 30 W). The maximum temperatures reached with increments of the MW power applied increased, showing a good MW absorber solvent behavior.
ChCl–oxalic acid showed a significantly lower MW heating response achieving a Tmax (52 °C) almost three times lower than that of ChCl–oxalic acid dihydrate (Fig. 3a, full diamond curve). However, both ChCl–oxalic acid and ChCl–oxalic acid dihydrate showed higher affinity to MWs than water (Tmax = 45 °C), which is already considered a good MW absorbing solvent. Single DES components (oxalic acid, oxalic acid dihydrate and ChCl) were instead very poor MW absorbers, behaving basically as MW-transparent materials (see heating profiles in Fig. 3a).
The remarkably different MW heating response of ChCl–oxalic acid dihydrate and ChCl–oxalic acid can be associated with a different hydrogen bond network arrangement promoted by the presence of water. However, Fig. 3b shows that under the same MW heating conditions and water amount, ChCl–oxalic acid dihydrate still showed the best MW heating response (Tmax = 70 °C), higher than that of ChCl–oxalic acid containing 13.5 wt% of water. Importantly, water is present in the same amount in all systems but is added before (Tmax = 63 °C) or after DES formation (Tmax = 61 °C). We thus hypothesized that the crystallization water of oxalic acid dihydrate is likely a part of the hydrogen bond network arrangement within the DES system.62 Conversely, it can be speculated that water added at the initial stage or after the ChCl–oxalic acid DES formation gives rise to a so-called water-in DES system.63,64 Consequently, the first more homogeneous solvent system improves the MW heating response. To the best of our knowledge, this is the first time where the nature of the water molecules in a DES has a documented impact on its behavior. The properties of the pure HBA and HBD components in water were also explored. Aqueous solutions of ChCl (1 M) or oxalic acid dihydrate (1 M) were slightly less sensitive to MW heating achieving Tmax = 52 °C and Tmax = 54 °C, respectively (Fig. 3b). Since the ionic moiety in these DESs is represented by ChCl, ionic conduction is likely not the predominant factor in determining the MW absorption properties of oxalic acid-based DESs (the heating profile of ChCl–H2O is lower than that of ChCl–oxalic acid dihydrate). The MW dielectric heating of DESs can be greatly affected by the dipolar polarization which, along with the ionic properties, is the main factor accountable for the MW dielectric heating response.9 A correlation between π* (the dipolarity/polarizability solvatochromic parameter of a solvent) and the MW absorption properties of DESs was recently observed. It has been shown that the MW absorption intensified when π* increased due to the ability of the solvent to stabilize the charge or a dipole through its dielectric effect.65 Indeed, ChCl–oxalic acid dihydrate displays a higher dipolarity/polarizability (π* = 1.21)66 than ChCl–oxalic acid (π* = 1.16), which is in agreement with the experimental MW heating responses observed.
The thermal behavior of ChCl–oxalic acid dihydrate, ChCl–oxalic acid, ChCl–oxalic acid + 13.5 (wt%) H2O highlights again the differences among these systems due to the different nature of the water molecules involved. Fig. S2a, S2b, S2c and S2d of the ESI† show the TG and DTG curves of ChCl–oxalic acid dihydrate, ChCl–oxalic acid, ChCl–oxalic acid + 13.5 (wt%) H2O (added before DES formation), ChCl–oxalic acid + 13.5 (wt%) H2O (added after DES formation) and their pure components, respectively.
Table S1 of the ESI† summarizes the different thermal degradation steps (identified as the Tpeak from the DTG curves) and the corresponding mass loss % for the three DESs and their pure components analyzed.
The thermal decomposition of ChCl–oxalic acid dihydrate, ChCl–oxalic acid, ChCl–oxalic acid + 13.5 (wt%) H2O (added before DES formation) and ChCl–oxalic acid + 13.5 (wt%) H2O (added after DES formation) agrees with what is generally observed for DESs, where the thermal stability is between that of the pure components, with the HBDs which are less thermally stable than ChCl.47 However, the mass loss % of each step is different, which indicates perhaps the presence of a different hydrogen bond network within each DES system. Furthermore, although the mass loss % related to dehydration for ChCl–oxalic acid dihydrate, ChCl–oxalic acid + 13.5 (wt%) H2O (added before DES formation) and ChCl–oxalic acid + 13.5 (wt%) H2O (added after DES formation) accounts for the amount expected for these systems (13.5%), the different temperature observed for this step (110 °C, 74 °C and 67 °C, respectively) is likely caused by stronger interactions within the water lattice in the former DES. This corroborates the hypothesis that water molecules are present in a highly organized network in ChCl–oxalic acid dihydrate.
In the case of ChCl–oxalic acid, the first small mass loss can be ascribed to the water absorbed from the environment during DES preparation or handling (due to the hygroscopicity of ChCl).
The nature of the water molecules within these materials thus appears to be an important aspect for DESs containing oxalic acid as the HBD concerning the dipolarity/polarizability value, and consequently the MW heating response, as well as the thermal decomposition behavior.
Optimization of the conditions of MWDA extraction of polyphenols using ChCl–oxalic acid dihydrate
Fig. 4 shows the yield wt%, the total polyphenol content and the condensed tannin content of extracts obtained from CSW using ChCl–oxalic acid dihydrate. These experiments were run at a constant solid to liquid weight ratio (1:10), four different MW extraction times (text = 5, 15, 30 and 60 min) and three extraction temperatures (65, 75 and 85 °C). The best extraction performance led to a yield of 28.6 ± 1.1 wt% at text = 60 min and 85 °C (Fig. 4a).
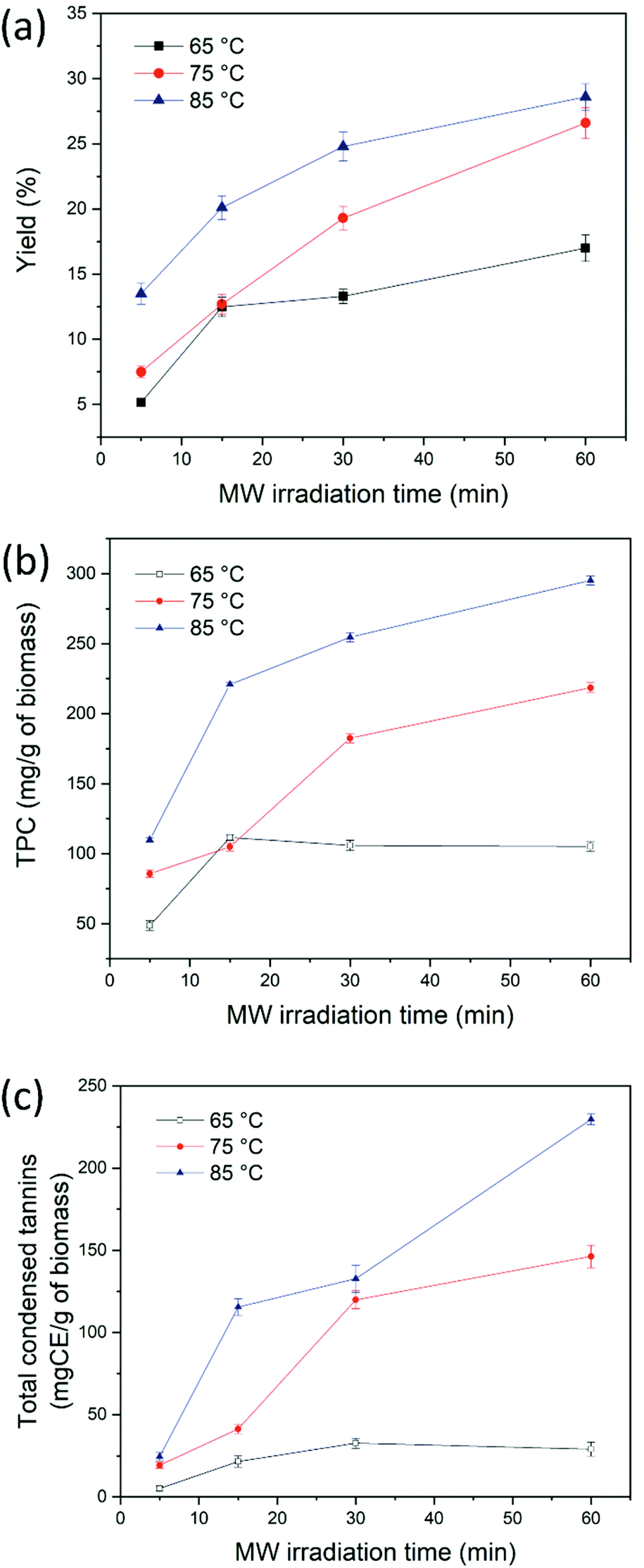 | ||
| Fig. 4 Effect of the temperature and MW irradiation time on yield wt% (a) total phenolic content (b) and condensed tannin content (c) of polyphenols extracted using ChCl–oxalic acid dihydrate. | ||
Fig. 4b shows the TPC obtained under the different extraction conditions explored. TPC values showed the same trend witnessed for the yield wt% of extracted polyphenols, where the most significant parameter was the temperature. At 65 °C the TPC was slightly influenced by the extraction time, while increments of temperature (from 65 °C to 85 °C) caused a considerable gain in the TPC (from 48.7 ± 3.5 to 109.9 ± 1.7 mg GAE per g dry CSW, from 111.6 ± 2.1 to 220.9 ± 1.3 mg GAE per g dry CSW, from 105.9 ± 3.6 to 254.6 ± 3.3 mg GAE per g dry CSW and from 105.1 ± 3.5 to 295.2 ± 3.2 mg GAE per g dry CSW for text = 5 min, 15 min, 30 min and 60 min, respectively). The highest TPC amount was obtained at 85 °C and 60 min (295.2 ± 3.2 mg GAE per g dry CSW). Subsequently, the presence of condensed tannins in the extracts was confirmed with the HCl–vanillin assay. Indeed, condensed and hydrolyzable tannins are known phenolic components contained in chestnut derived biomass. Their quantification revealed that condensed tannins were the main class of phenolic compounds present in the extracts, in agreement with our previous work employing the heating-stirring approach (HSA)36 and with the data reported by Squillaci et al.10 The colorimetric assay showed a similar trend to that of the TPC assay, with the highest values registered when increasing both temperature and extraction time. Moreover, the values obtained at high temperatures and long reaction times can partly be affected by the degradation of biomass lignin into low molecular weight phenolic compounds reactive to the assay. As mentioned above, the ability of acidic DESs to depolymerize and fractionate the lignin component of lignocellulosic biomass, as well of hemicellulose, is well known.67–71
Although a literature comparison of the quantity of extracted biomolecules is of limited value on account of the different biomass sources, extraction protocols, and pretreatment carried out on the waste (for instance brulage or boiling), the here reported MWDA process using DES ChCl–oxalic acid dihydrate represents an appealing extraction system. This holds true both in terms of yield and TPC as well as for its innovative use of an alternative energy source. Of particular interest is the comparison of our technology with the previously developed HSA36 with the same DES (ChCl:oxalic acid dihydrate) as the extraction solvent and the same batch of CSW: the MWDA approach gives a similar yield (33 ± 1.9% for HSA versus 28.6 ± 1.1% for MWDA process) albeit higher values of the TPC (197.4 ± 5.9 mg GAE per g of dry CSW for HSA versus 295.2 ± 3.2 mg GAE per g of dry CSW for MWDA process) and total condensed tannins (189.6 ± mg CE per g of dry CSW for HSA versus 229.6 ± 3.4 mg CE per g of dry CSW for MWDA process) with the additional advantage of very short extraction times (60 min versus 24 h for the HSA method).
Qualitative and quantitative characterization of polyphenols extracted by HPLC-DAD analysis
The polyphenols extracted from CSW were further characterized by HPLC-DAD chromatographic analysis.36 Four main polyphenol compounds were identified by the comparison of both UV absorbance spectrum and retention time (tR) with their corresponding analytical standards. Gallic acid (tR = 5.7 min), catechin (tR = 17.6 min), procyanidin B2 (tR = 19.5 min) and ellagic acid (tR = 30.4 min) were detected in the absorbance chromatograms at 280 nm as reported in Fig. S3 of the ESI† for the extracts obtained at different extraction times at 65 °C (a), 75 °C (b) and at 85 °C (c). The highest concentration of polyphenols was obtained, in general, with extractions performed at 75–85 °C for 30–60 min (Fig. 5). As observed for TPC values, longer extraction times and higher temperatures gave better yields of polyphenols. However, the most forceful conditions (85 °C, 60 min) were detrimental to the extraction process of some polyphenols. In particular, the quantification of gallic acid highlighted a strong dependence on both temperature and extraction time. Gallic acid concentrations in the 5–60 min text interval were 751.2 ± 42.5–2235.9 ± 67.1 μg per g of dry CSW at 65 °C, 1134.6 ± 64.1–5112.8 ± 153.4 μg per g of dry CSW at 75 °C and 1631.6 ± 58.9–3877.2 ± 116.3 μg per g of dry CSW at 85 °C.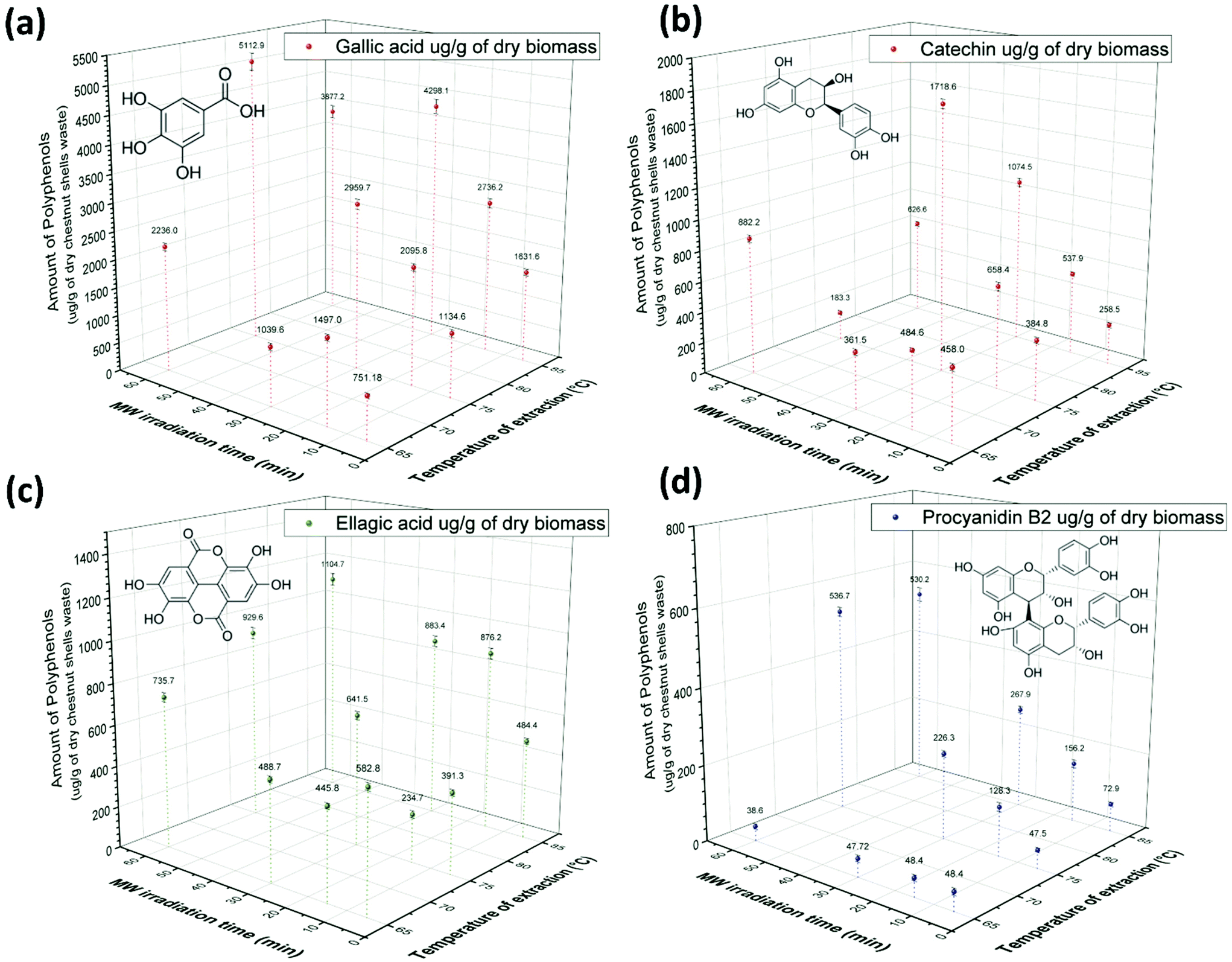 | ||
| Fig. 5 Polyphenol concentration values quantified by HPLC-DAD at different extraction times and temperatures using DES ChCl–oxalic acid dihydrate: (a) gallic acid, (b) catechin, (c) ellagic acid and (d) procyanidin B2. | ||
Catechin hydrate was the second identified polyphenol in the extracts and its concentration ranged from 458.1 ± 21.45 to 882.2 ± 22.1 μg per g of dry CSW at 65 °C, from 384.8 ± 19.6 to 183.3 ± 14.5 μg per g of dry CSW at 75 °C and from 258.5 ± 16.5 to 626.6 ± 15.7 μg per g of dry CSW at 85 °C (text from 5 to 60 min) (Fig. 5b). The highest concentration of catechin (1718.6 ± 28.9 μg per g of dry CSW) was obtained in the polyphenols extracted at 75 °C for 30 min. In this case, a remarkable lowering in catechin concentration was again observed for longer extraction times.
The concentration of ellagic acid ranged between 582.8 ± 17.5 and 735.8 ± 22.1 μg per g of dry CSW at 65 °C, 391.3 ± 14.7–929.6 ± 27.9 μg per g of dry CSW at 75 °C and 484.4 ± 14.5–1104.7 ± 30.1 μg per g of dry CSW at 85 °C (Fig. 5c). For this compound, longer extraction times and higher temperatures resulted in a better performance, with the highest yield recorded when the extraction was carried out at 85 °C for 60 min.
Procyanidin B2 was the fourth main polyphenol identified in dry CSW extracts, and its concentration ranged between 38.6 ± 7.4 and 48.4 ± 8.7, 47.5 ± 5.7 and 536.7 ± 11.8, and 72.9 ± 5.6 and 530.2 ± 18.6 μg per g of dry CSW at 65 °C, 75 °C and 85 °C, respectively. The strong effect of temperature and text changes was evident from the data on procyanidin B2 concentration in the extract: the amount obtained at 85 °C for 60 min was more than an order of magnitude higher than that obtained at 65 °C for 5 min.
It is worth noting the detrimental effect of long extraction times and high temperatures on the extract concentration of gallic acid and catechin on one side, and the beneficial effects of the same process variations on the concentration of procyanidin B2 and ellagic acid on the other. These opposite effects can be rationalized taking into account the different stability of the phenolic compounds here considered. Indeed, the different behavior of gallic and ellagic acid when subjected to acid/base/neutral hydrolysis, oxidation, dry heat and photolysis stress has already been reported by Damle et al.72 In this study, ellagic acid proved to be stable while gallic acid gave rise to degradation products under acid, alkali and oxidative conditions. Furthermore, the aqueous thermal degradation of gallic acid to pyrogallol via decarboxylation was already reported by Boles et al.73
Similarly, Yuan et al.74 studied the microwave-induced decomposition of the extracted target compounds and specifically the degradation of gallic acid and resveratrol via the formation of free radicals. This phenomenon was affected by various extraction conditions, namely solvent type and amount, MW irradiation power, extraction temperature and time.
The process parameters could be optimized to improve the extraction yield and at the same time reduce the potential degradation of phenolic compounds in the MW-mediated process. Finally, the thermal degradation of catechin was reported and it was proven that high temperatures (above 80 °C) shortened its half-life. Drawing from these literature results, it seems reasonable to conclude that the lower amount of gallic acid and catechin recovered in some of our test runs is related to their degradation, which is favored by the strong acidic extraction conditions (ChCl–oxalic acid dihydrate), long extraction times and high temperatures. Conversely, procyanidin B2 and ellagic acid are stabler compounds, able to withstand harsh conditions.
The main polyphenols identified here perfectly match the compounds previously reported in the literature when different solvents, source of chestnut and extraction heating approaches were used.10,15–17,21,24–26,30–32,36 Indeed, gallic and ellagic acid are typical phenols derived from hydrolyzable tannins (ester of gallic/ellagic acid and glucose), one of the most abundant class of polyphenols in chestnut biomass together with the more complex condensed tannins.
A large unresolved hump ascribable to the latter class of compounds was visible in all chromatograms. This is in agreement with our previous results, when ChCl–oxalic acid dihydrate was employed as the extraction solvent in HSA. The presence of complex condensed tannins was further confirmed by the HCl–vanillin assay (Fig. 4c).
The HPLC characterization and quantification of the main polyphenols extracted from CSW using ChCl–oxalic acid dihydrate in the MWDA process highlighted how the final chemical composition depends on the extraction conditions employed. It is therefore worth stressing that the same process set-up and the same feedstock could target different extract compositions by adjusting the time and temperature of the extraction process.
Morphological characterization and composition analysis of lignocellulosic by-products
The morphological characterization and composition analysis of solid residues after polyphenol extraction was carried out by SEM imaging, thermogravimetric analysis and FTIR spectroscopy. In order to assess the impact of the extraction conditions, the solid residues obtained after applying the best polyphenol extraction conditions in terms of yield wt% and TPC value (i.e. ChCl–oxalic acid dihydrate, 85 °C, 30 min of MW extraction time) and the solid residues obtained using the same MW extraction time (30 min) at 65 °C and 75 °C were compared. Moreover, the solid residues that were recovered after the treatment with ChCl–oxalic acid and ChCl–oxalic acid dihydrate (at 65 °C, 30 min) were also compared to assess any effect of the crystallization water contained in the DES.In Fig. 6, the SEM images of CSW before and after the treatment with the oxalic acid-based DES can be seen. Fig. 6a shows the SEM image of raw CSW that is characterized predominantly by fibrous structures with an apparently smooth surface. Also, smaller particles with irregular shapes were observed. Fig. 6b–f show SEM images of solid residues after the MWDA extraction of polyphenols. The SEM image in Fig. 6b depicts the solid residues after a 65 °C, 30 min MW extraction of polyphenols in water, thus under neutral conditions. The clear changes in morphology, likely due to the effects of MW irradiation, are small with respect to raw CSW, except for the presence of bigger aggregates of irregular particles characterized by a rough surface.
 | ||
| Fig. 6 SEM images of raw CSW (a). Solid residues after the MW-assisted extraction of polyphenols using as solvent H2O (b), ChCl–oxalic acid (c), ChCl–oxalic acid dihydrate (d–f). MW assisted extraction conditions: 65 °C, 30 min of MW extraction time for (a, b, c and d), 75 °C for (e), 85 °C for (f), respectively. Solid to liquid ratio = 1:10 (w/w) in all cases. | ||
In contrast, the effect of ChCl–oxalic acid or ChCl–oxalic acid dihydrate in conjunction with MW in the extraction process is evident: the large fibrous-like particles of the raw CSW are broken, yielding numerous solid residues of irregular shapes and higher aggregation patterns. This outcome was promoted by increased temperatures and extraction times (Fig. 6c, e, f, and Fig. S4 of the ESI†). The most extreme extraction conditions explored (60 min of MW irradiation time at 85 °C) gave drastic morphological changes in the size and surface of the solid residue (Fig. S4 of the ESI†).
The original fibrous-like particles were broken and highly aggregated particles with rough, irregular surfaces were formed. Indeed, broken and aggregated particles are also observed in other types of biomass when lignin and amorphous cellulose are removed totally or partially.75 Even more it is well known that acid-based DESs are good candidates for improving the crystallinity of cellulose by partial depolymerization of lignin and hydrolysis of amorphous cellulose.76
SEM imaging analysis confirms the effectiveness of the MWDA process using acid-based DESs.
The solid residues obtained after the extraction of polyphenols from CSW represent the by-products with the highest mass percentage. Fig. 7a shows the wt% yield of solid residues recovered after the extraction of polyphenols using the MWDA extraction with ChCl–oxalic acid dihydrate under the different conditions explored.
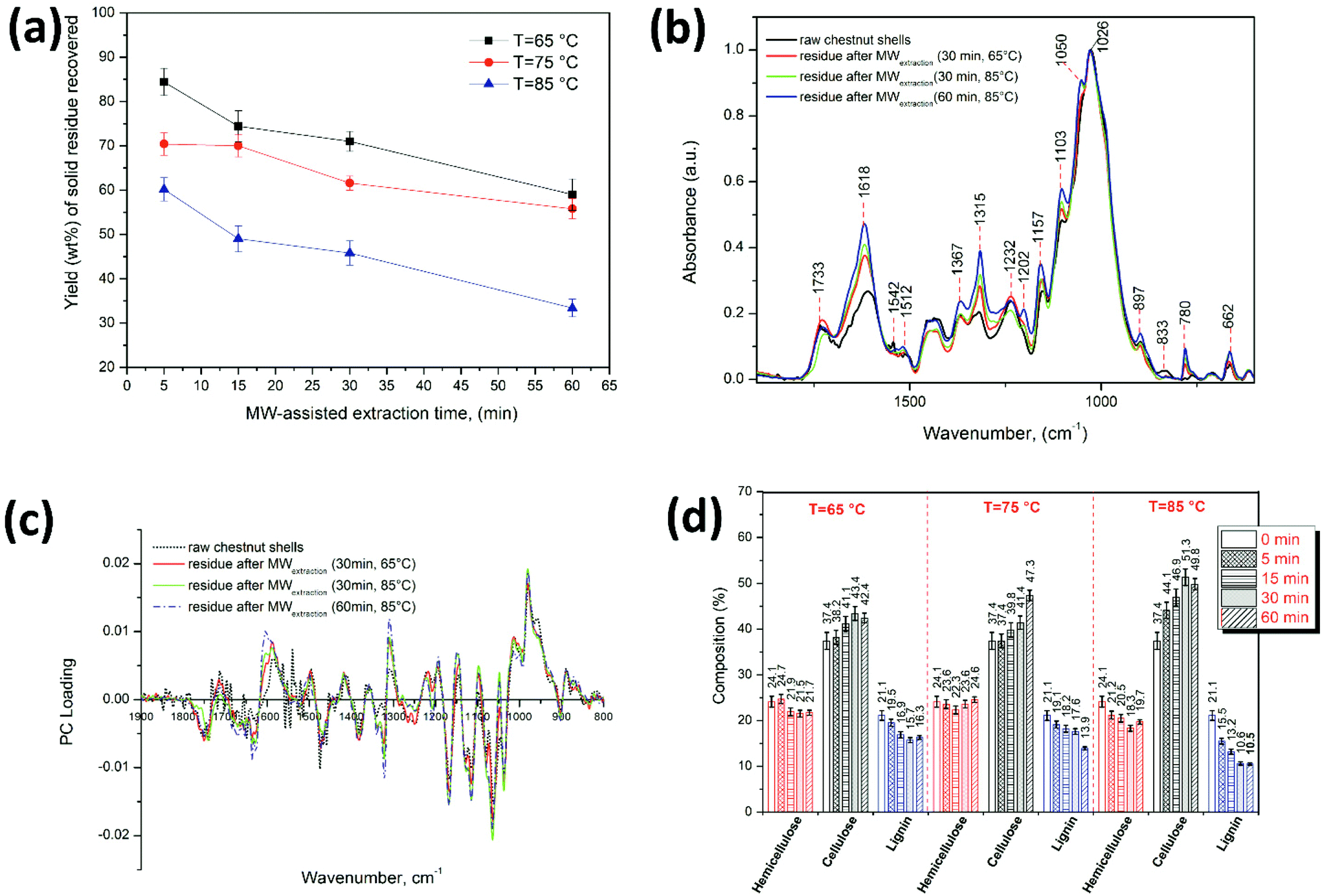 | ||
| Fig. 7 (a) wt% yield of solid residues recovered after polyphenol extraction, (b) ATR-FTIR spectra of raw CSW and solid residues after polyphenol extraction using ChCl–oxalic acid dihydrate under the different MWDA extraction conditions (30 min at T = 65 °C and at T = 85 °C, and 60 min at T = 85 °C). (c) Loading profile (FTIR first derivative) of the solid residue for the cross-validation-PLS prediction model and (d) the predicted chemical composition of cellulose, hemicellulose and lignin in dry solid residues obtained after the MW-assisted extraction of polyphenols using ChCl–oxalic acid dihydrate under the different extraction conditions. | ||
The yield wt% of solid residues decreased when more forcing conditions in terms of both temperature and time were employed, following an almost reversed trend if compared with the yields of the extracted polyphenols. At 85 °C and 60 min extraction time, the wt% of polyphenols was 28.6 ± 1.1%, the wt% of solid residues was 33.4 ± 2.1%, while the “missing” mass was likely due to unidentified, soluble by-products of the hydrolysis of biopolymers (starch, sugars, etc.). Thus, the dried recovered solid residues still represent a very considerable and valuable percentage of CSW that can be further exploited and valorized.
Subsequently, qualitative and quantitative evaluation of the solid residues was carried out via thermogravimetric analysis and FTIR spectroscopy. TGA curves and the related DTG show a variation of the relative hemicellulose/cellulose/lignin ratio in the solid residue recovered from the MWDA extractions, which qualitatively corroborates the lignin depolymerization hypothesis when this process is carried out under the harsher conditions (please refer to Fig. S5 of the ESI† for TGA curves). However, the presence of several parallel CSW polymer degradation phenomena does not allow for their easy quantification. Hence, in depth FTIR analyses and FTIR data elaboration were carried out. Fig. 7b shows the FTIR spectra in the fingerprint region (1900–800 cm−1) of the raw CSW compared with the spectra of three representative solid residues obtained after the MWDA extraction of polyphenols: (i) 30 min at 65 °C, namely soft conditions, (ii) 30 min at 85 °C (the optimal conditions) and, (iii) 60 min at 85 °C (harsh extraction conditions). The peak assignments are based on previous reports for the FTIR characterization of lignocellulosic materials, hard and softwoods.77–81 The FTIR spectra for solid residues obtained at 75 °C (30 min) using ChCl–oxalic acid dihydrate and at 65 °C (30 min) using ChCl–oxalic acid are shown in Fig. S6a and S6b of the ESI,† respectively.
The FTIR spectra of raw CSW and the solid residues after extraction showed absorption bands with main peaks at 1026 cm−1 (C–O stretching of the C–O–C group of the anhydroglucose ring of cellulose) and at 1232 cm−1 (C–O–C stretching in phenol–ether bonds of lignin, C–O stretching in syringyl rings, C–O–C symmetric stretching in cellulose/hemicellulose, and OH plane deformation), commonly associated with the overlapping of characteristics bands of cellulose, hemicellulose and lignin in lignocellulosic materials.77,78 The peak at 1542 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching in the aromatic ring likely associated with polyphenols81) in the spectrum of raw CSW was not longer observed in the spectra of the solid residues after the MWDA extraction (Fig. 6b and Fig. S6 of the ESI†). The intensity of the peaks at 1733 cm−1 (CO stretching in ester groups mainly associated with hemicellulose), 1618 cm−1 (CC stretching and COOH group stretching in the aromatic ring due to the overlapping of characteristics bands of cellulose, hemicellulose and lignin), 1367 cm−1 (CH3 symmetrical angular vibration of cellulose/hemicellulose), 1315 cm−1 (CH2 wagging of cellulose), 1202 cm−1 (O–H bending of cellulose/hemicellulose), 1157 cm−1 (C–O–C asymmetric-stretching of cellulose/hemicellulose), 1103 cm−1 (C–O–H stretching in secondary and tertiary alcohols in cellulose/lignin), 833 cm−1 (C–H out-of-plane deformations in aromatic rings associated with the syringyl nuclei in lignin) increased in the solid residues after the MWDA extraction, indicating their enrichment in cellulose/hemicellulose. The solid residues of CSW after the extraction processes showed the characteristics of hardwoods due to their characteristic peaks at 833 cm−1, 1157 cm−1, 1733 cm−1.59,79
C stretching in the aromatic ring likely associated with polyphenols81) in the spectrum of raw CSW was not longer observed in the spectra of the solid residues after the MWDA extraction (Fig. 6b and Fig. S6 of the ESI†). The intensity of the peaks at 1733 cm−1 (CO stretching in ester groups mainly associated with hemicellulose), 1618 cm−1 (CC stretching and COOH group stretching in the aromatic ring due to the overlapping of characteristics bands of cellulose, hemicellulose and lignin), 1367 cm−1 (CH3 symmetrical angular vibration of cellulose/hemicellulose), 1315 cm−1 (CH2 wagging of cellulose), 1202 cm−1 (O–H bending of cellulose/hemicellulose), 1157 cm−1 (C–O–C asymmetric-stretching of cellulose/hemicellulose), 1103 cm−1 (C–O–H stretching in secondary and tertiary alcohols in cellulose/lignin), 833 cm−1 (C–H out-of-plane deformations in aromatic rings associated with the syringyl nuclei in lignin) increased in the solid residues after the MWDA extraction, indicating their enrichment in cellulose/hemicellulose. The solid residues of CSW after the extraction processes showed the characteristics of hardwoods due to their characteristic peaks at 833 cm−1, 1157 cm−1, 1733 cm−1.59,79
The content of cellulose, hemicellulose and lignin in the solid residues, obtained after polyphenol extraction using ChCl–oxalic acid dihydrate, was predicted using a partial least squares (PLS) model previously reported for the determination of the chemical composition of hard and softwood samples.3,59 The PLS model was reconstructed using the FTIR spectra of the wood samples in the region of 1900–800 cm−1 (i.e. the fingerprint of wood components). The first derivative of the normalized FTIR spectra was used as the X matrix and the chemical composition of lignin, cellulose and hemicellulose (determined by Van Soest analysis) as the Y matrix. The model was developed using JMP software and seven principal components that account for 98% of the variance in the X matrix and 98.8% of the variance in the Y matrix.
The compositional prediction of the raw CSW and solid residue samples was then performed by loading their first derivative of the normalized FTIR spectra (Fig. 7c) into the X matrix of the model and by applying the cross-validation technique.
Fig. 7d shows the modelled percentage of cellulose, hemicellulose and lignin in the dry solid residues and raw CSW (labeled at time = 0 min). Using this approach, the prediction of the raw CSW constituents was: 24.1 ± 1.2% hemicellulose, 37.4 ± 1.9% cellulose and 21.1 ± 1.1% lignin. The composition of the solid residues after extraction depends on the extraction conditions: hemicellulose was about 18.3 ± 0.6–24.7 ± 1.0% and slightly decreased when the temperature and extraction time increased. The cellulose content ranged from 37.4 ± 1.5 to 51.3 ± 1.8% and was enhanced by time and temperature increments. Cellulose was actually the main component that reached 51 ± 1.8% after extraction at 85 °C and 30 min (Fig. 7d). Lignin showed the opposite behavior: the proportion of lignin (21.1 ± 1.1–10.5 ± 0.3%) significantly decreased with increasing extraction temperatures. The lowest amount of lignin (10.5 ± 0.3%) was obtained at 85 °C and 60 min of MW irradiation time (Fig. 7d). This suggests a simultaneous beneficial effect of both the acid-based DES used and MWs on the extraction of lignin. The high yield of polyphenols and TPC enhancements observed at high temperatures and extraction times are likely promoted by the extraction of lignin that is further hydrolyzed.
The PLS analysis of the solid residues obtained after extraction in water for 30 min at 65 °C (23.1 ± 0.9% hemicellulose, 38.5 ± 1.9% cellulose, 19.3 ± 0.8% lignin) or ChCl–oxalic acid (23.8 ± 1.1%, hemicellulose, 39.0 ± 1.9% cellulose, 19.2 ± 1.1% lignin) highlighted that these solvents are less efficient for the delignification of biomasses, given that these values are very close to those determined in raw CSW. An exception is represented by the slight decrease in hemicellulose and lignin composition (about 2%). On the other hand, the use of 25% water in the ChCl–oxalic acid–H2O system promoted the lignin removal, yielding a solid residue after 30 min at 65 °C characterized by 23.0 ± 0.9% hemicellulose, 41.2 ± 1.6% cellulose and 17.0 ± 0.7% lignin. Delignification in the ChCl–oxalic acid–H2O system was lower than that obtained with ChCl–oxalic acid dihydrate under the same extraction conditions.
Thus, ChCl–oxalic acid dihydrate is effective in the delignification pretreatment of lignocellulosic biomass for the production of cellulose-enriched materials. The combination of microwaves and acid-based DESs in delignification processes has been reported before.49,50,58,82 Usually, a short irradiation time is combined with a rather high working temperature (>100 °C). It is worth noting that the same ChCl–oxalic acid dihydrate DES here used was proven to be effective in the delignification of poplar wood flour after a very short irradiation time (3 minutes) and at mild temperatures (80 °C),58 thus in agreement with our findings.
Conclusions
The yield of polyphenols isolated from CSW by the MWDA extraction process was substantially improved using a ChCl–oxalic acid dihydrate DES in comparison to other acid-based ChCl DESs. The nature of the water present in this system affected the thermal stability and the dipolarity/polarizability solvatochromic parameter π*, which is in turn related to the MW absorption properties of the system. For the first time, the nature of the same amount of water in a DES, namely crystallization water derived from the HBD component or water added to the formed DES, has been shown to play a crucial, positive role in the extraction of added-value compounds from CSW.The ChCl–oxalic acid dihydrate DES was able to promote CSW decomposition probably through the lignin-hemicellulose complex break-down. Quantitative HPLC analysis highlighted how the polyphenol composition of the extracts varied as a function of the MWDA extraction parameters and thus the possibility of tailoring polyphenol extracts to a target composition by selecting specific extraction conditions. The residues obtained through MWDA extraction were characterized by FTIR, SEM and TGA and their relative lignin/hemicellulose/cellulose composition was assessed by partial least squares regression (PLS) of their FTIR spectra, which confirmed the enhanced lignin removal at the most forced MWDA extraction conditions.
Overall, the approach disclosed in this work highlights the benefits and potential of combining MW and acid-based DESs for the valorization of CSW. The key features of this novel process are (a) the effective extraction of polyphenols from a biomass waste exploiting the unique ‘designer solvent’ potential of DESs and (b) the obtainment of cellulose-enriched materials for further upgrading. Concerning the first aspect, the nature of the water present in a DES was shown to be a crucial parameter for the fine tuning of the DES properties, as important as the type of the HBA and HBD components, their molar ratio and the amount of water added to the DES.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the BIHO (Bando Incentivi di Ateneo Horizon e Oltre) project of the University of Pisa and Era-Net Cofund SUSFOOD2 Call 2017 and MIUR for financial support (ImPrOVE project) and Ortofrutticola del Mugello s.r.l. (Italy) for providing the CSW. The authors also would like to thank A. Barbini and F. Pardini (INO-CNR) for their valuable technical support.References
- P. Lidström, J. Tierney, B. Wathey and J. Westman, Tetrahedron, 2001, 57, 9225–9283 CrossRef.
- J. González-Rivera, A. Spepi, C. Ferrari, C. Duce, I. Longo, D. Falconieri, A. Piras and M. R. Tiné, Green Chem., 2016, 18, 6482–6492 RSC.
- J. González-Rivera, C. Duce, B. Campanella, L. Bernazzani, C. Ferrari, E. Tanzini, M. Onor, I. Longo, J. Cabrera Ruiz, M. R. Tiné and E. Bramanti, Ind. Crops Prod., 2021, 161, 113203 CrossRef.
- S. Sinnwell and H. Ritter, Aust. J. Chem., 2007, 60, 729–743 CrossRef CAS.
- J. González-Rivera, C. Duce, V. Ierardi, I. Longo, A. Spepi, M. R. Tiné and C. Ferrari, ChemistrySelect, 2017, 2, 2131–2138 CrossRef.
- A. Spepi, C. Duce, C. Ferrari, J. González-Rivera, Z. Jagličić, V. Domenici, F. Pineider and M. R. Tiné, RSC Adv., 2016, 6, 104366–104374 RSC.
- J. Asomaning, S. Haupt, M. Chae and D. C. Bressler, Renewable Sustainable Energy Rev., 2018, 92, 642–657 CrossRef CAS.
- D. Dallinger and C. O. Kappe, Chem. Rev., 2007, 107, 2563–2591 CrossRef CAS PubMed.
- C. O. Kappe and A. Stadler, Microwaves in Organic and Medicinal Chemistry, Wiley-VCH, 2005 Search PubMed.
- G. Squillaci, F. Apone, L. M. Sena, A. Carola, A. Tito, M. Bimonte, A. De Lucia, G. Colucci, F. La Cara and A. Morana, Process Biochem., 2018, 64, 228–236, DOI:10.1016/j.procbio.2017.09.017.
- A. Morana, G. Squillaci, S. M. Paixão, L. Alves, F. La Cara and P. Moura, Energies, 2017, 10, 1–14 CrossRef.
- F. Rodrigues, J. Santos, F. B. Pimentel, N. Braga, A. Palmeira-de-Oliveira and M. B. P. P. Oliveira, Food Funct., 2015, 6, 2854–2860 RSC.
- U. Y. Youn, M. S. Shon, G. N. Kim, R. Katagiri, K. Harata, Y. Ishida and S. C. Lee, Food Sci. Biotechnol., 2016, 25, 1169–1174 CrossRef CAS PubMed.
- B. S. Jung, N. Lee, D. S. Na, H. H. Yu and H.-D. Paik, J. Sci. Food Agric., 2016, 96, 2097–2102 CrossRef CAS PubMed.
- A. Aires, R. Carvalho and M. J. Saavedra, Waste Manag., 2016, 48, 457–464 CrossRef CAS PubMed.
- A. Sorice, F. Siano, F. Capone, E. Guerriero, G. Picariello, A. Budillon, G. Ciliberto, M. Paolucci, S. Costantini and M. G. Volpe, Molecules, 2016, 21, 1–16 Search PubMed.
- P. Tuyen, T. Xuan, D. Khang, A. Ahmad, N. Quan, T. T. Tu Anh, L. Anh and T. Minh, Antioxidants, 2017, 6, 1–14 CrossRef PubMed.
- B. Gullón, G. Eibes, I. Dávila, M. T. Moreira, J. Labidi and P. Gullón, Carbohydr. Polym., 2018, 192, 75–83 CrossRef PubMed.
- F. M. Vella, B. Laratta, F. La Cara and A. Morana, Nat. Prod. Res., 2018, 32, 1022–1032 CrossRef CAS PubMed.
- K. H. Hong, Cellulose, 2018, 25, 2745–2753 CrossRef CAS.
- D. Pinto, M. d. l. L. Cádiz-Gurrea, S. Sut, A. S. Ferreira, F. J. Leyva-Jimenez, S. Dall’acqua, A. Segura-Carretero, C. Delerue-Matos and F. Rodrigues, J. CO2 Util., 2020, 40, 101194 CrossRef CAS.
- J. C. M. Barreira, I. C. F. R. Ferreira, M. B. P. P. Oliveira and J. A. Pereira, Food Chem., 2008, 107, 1106–1113 CrossRef CAS.
- A. Cerulli, A. Napolitano, M. Masullo, J. Hošek, C. Pizza and S. Piacente, Food Res. Int., 2020, 129, 108787 CrossRef CAS PubMed.
- F. Lameirão, D. Pinto, E. F. Vieira, A. F. Peixoto, C. Freire, S. Sut, S. Dall'acqua, P. Costa, C. Delerue-Matos and F. Rodrigues, Antioxidants, 2020, 9, 267 CrossRef PubMed.
- D. Pinto, A. M. Silva, V. Freitas, A. Vallverdú-Queralt, C. Delerue-Matos and F. Rodrigues, ACS Food Sci. Technol., 2021, 1, 229–241 CrossRef CAS.
- D. Pinto, E. F. Vieira, A. F. Peixoto, C. Freire, V. Freitas, P. Costa, C. Delerue-Matos and F. Rodrigues, Food Chem., 2021, 334, 127521 CrossRef CAS PubMed.
- G. Vazquez, E. Fontenla, J. Santos, M. S. Freire, J. González-Álvarez and G. Antorrena, Ind. Crops Prod., 2008, 28, 279–285 CrossRef CAS.
- J. Ziivkovic, Z. Zekovic, I. Mujic, V. Tumbas, D. Cvetkovic and I. Spasojevic, Food Technol. Biotechnol., 2009, 47, 421–427 Search PubMed.
- G. Vázquez, J. González-Alvarez, J. Santos, M. S. Freire and G. Antorrena, Ind. Crops Prod., 2009, 29, 364–370 CrossRef.
- M. do C. B. M. de Vasconcelos, R. N. Bennett, S. Quideau, R. Jacquet, E. A. S. Rosa and J. V. Ferreira-Cardoso, Ind. Crops Prod., 2010, 31, 301–311 CrossRef CAS.
- M. Nazzaro, M. V. Mottola, F. La Cara, G. Del Monaco, R. P. Aquino and M. G. Volpe, Chem. Eng. Trans., 2012, 27, 331–336 Search PubMed.
- A. Fernández-agulló, M. S. Freire, G. Antorrena, J. A. Pereira and J. González-álvarez, Sep. Sci. Technol., 2014, 2, 267–277 CrossRef.
- J. S. Ham, H. Y. Kim and S. T. Lim, Ind. Crops Prod., 2015, 73, 99–105 CrossRef CAS.
- M. research Report, Polyphenols Market Size, Share & Trends Analysis Report By Product (Grape Seed, Green Tea, Cocoa), By Application (Beverages, Food, Feed, Dietary Supplements, Cosmetics), And Segment Forecasts, 2019–2025.
- Y. Chen and T. Mu, Green Energy Environ., 2019, 4, 95–115 CrossRef.
- E. Husanu, A. Mero, J. Gonzalez Rivera, A. Mezzetta, J. Cabrera Ruiz, F. D’Andrea, C. S. Pomelli and L. Guazzelli, ACS Sustainable Chem. Eng., 2020, 8, 18386–18399 CrossRef.
- J. Y. An, L. T. Wang, M. J. Lv, J. D. Wang, Z. H. Cai, Y. Q. Wang, S. Zhang, Q. Yang and Y. J. Fu, Microchem. J., 2021, 160, 105616 CrossRef CAS.
- P. Gullón, B. Gullón, A. Romaní, G. Rocchetti and J. M. Lorenzo, Trends Food Sci. Technol., 2020, 101, 182–197 CrossRef.
- J. L. K. Mamilla, U. Novak, M. Grilc and B. Likozar, Biomass Bioenergy, 2019, 120, 417–425 CrossRef CAS.
- A. Bjelić, B. Hočevar, M. Grilc, U. Novak and B. Likozar, Rev. Chem. Eng., 2020, 20190077 CrossRef.
- B. Bradić, U. Novak and B. Likozar, Green Process. Synth., 2019, 9, 13–25 Search PubMed.
- C. L. Yiin, K. L. Yap, A. Z. E. Ku, B. L. F. Chin, S. S. M. Lock, K. W. Cheah, A. C. M. Loy and Y. H. Chan, Bioresour. Technol., 2021, 333, 125195 CrossRef CAS PubMed.
- P. Bhagwat, A. Amobonye, S. Singh and S. Pillai, Biomass Convers. Biorefin., 2021 DOI:10.1007/s13399-021-01745-x.
- A. Mišan, J. Nađpal, A. Stupar, M. Pojić, A. Mandić, R. Verpoorte and Y. H. Choi, Crit. Rev. Food Sci. Nutr., 2020, 60, 2564–2592 CrossRef PubMed.
- M. A. R. Martins, S. P. Pinho and J. A. P. Coutinho, J. Solution Chem., 2019, 48, 962–982 CrossRef CAS.
- K. Shahbaz, F. S. Mjalli, G. Vakili-nezhaad, I. M. Alnashef, A. Asadov and M. M. Farid, J. Mol. Liq., 2016, 222, 61–66 CrossRef CAS.
- J. González-Rivera, E. Husanu, A. Mero, C. Ferrari, C. Duce, M. R. Tinè, F. D'Andrea, C. S. Pomelli and L. Guazzelli, J. Mol. Liq., 2020, 300, 112357 CrossRef.
- J. Huang, X. Guo, T. Xu, L. Fan, X. Zhou and S. Wu, J. Chromatogr. A, 2019, 1–3 CrossRef PubMed.
- P. D. Muley, J. K. Mobley, X. Tong, B. Novak, J. Stevens, D. Moldovan, J. Shi and D. Boldor, Energy Convers. Manage., 2019, 196, 1080–1088 CrossRef CAS.
- Z. Chen and C. Wan, Bioresour. Technol., 2018, 250, 532–537 CrossRef CAS PubMed.
- L. Hladnik, F. A. Vicente, U. Novak, M. Grilc and B. Likozar, Ind. Crops Prod., 2021, 164, 113359 CrossRef CAS.
- M. Panic, V. Gunjevic, G. Cravotto and I. Radojcic Redovnikovic, Food Chem., 2019, 300, 125185 CrossRef CAS PubMed.
- M. Panić, M. Radić Stojković, K. Kraljić, D. Škevin, I. Radojčić Redovniković, V. Gaurina Srček and K. Radošević, Food Chem., 2019, 283, 628–636 CrossRef PubMed.
- C. B. T. Pal and G. C. Jadeja, J. Food Sci. Technol., 2019, 56, 4211–4223 CrossRef CAS PubMed.
- C. B. T. Pal and G. C. Jadeja, Food Sci. Technol. Int., 2019, 1–15 Search PubMed.
- S. Chanioti and C. Tzia, Innovative Food Sci. Emerging Technol., 2018, 48, 228–239 CrossRef CAS.
- T. Wang, J. Jiao, Q. Gai, P. Wang, N. Guo, L. Niu and Y. Fu, J. Pharm. Biomed. Anal., 2017, 145, 339–345 CrossRef CAS PubMed.
- Y. Liu, W. Chen, Q. Xia, B. Guo, Q. Wang, S. Liu, Y. Lui, J. Li and H. Yu, ChemSusChem, 2017, 10, 1692–1700 CrossRef CAS PubMed.
- H. Chen, C. Ferrari, M. Angiuli, J. Yao, C. Raspi and E. Bramanti, Carbohydr. Polym., 2010, 82, 772–778 CrossRef CAS.
- Y. Nakamura, S. Tsuji and Y. Tonogai, J. Health Sci., 2003, 49, 45–54 Search PubMed.
- Y. Dai, G. J. Witkamp, R. Verpoorte and Y. H. Choi, Anal. Chem., 2013, 85, 6272–6278 CrossRef CAS PubMed.
- A. S. D. Ferreira, R. Craveiro, A. R. Duarte, S. Barreiros, E. J. Cabrita and A. Paiva, J. Mol. Liq., 2021, 342, 117463 CrossRef CAS.
- M. E. Di Pietro, M. Tortora, C. Bottari, G. Colombo Dugoni, R. V. Pivato, B. Rossi, M. Paolantoni and A. Mele, ACS Sustainable Chem. Eng., 2021, 9(36), 12262–12273 CrossRef CAS.
- M. E. Di Pietro, O. Hammond, A. Van Den Bruinhorst, A. Mannu, A. Padua, A. Mele and M. Costa Gomes, Phys. Chem. Chem. Phys., 2021, 23, 107–111 RSC.
- P. G. Jessop, D. A. Jessop, D. Fu and L. Phan, Green Chem., 2012, 14, 1245–1259 RSC.
- Y. Ma, Q. Xia, Y. Liu, W. Chen, S. Liu, Q. Wang, Y. Liu, J. Li and H. Yu, ACS Omega, 2019, 4, 8539–8547 CrossRef CAS PubMed.
- S. Hong, X.-J. Shen, Z. Xue, Z. Sun and T.-Q. Yuan, Green Chem., 2020, 22, 7219–7232 RSC.
- X. Yue, T. Suopajärvi, O. Mankinen, M. Mikola, A. Mikkelson, J. Ahola, S. Hiltunen, S. Komulainen, A. M. Kantola, V.-V. Telkki and H. Liimatainen, J. Agric. Food Chem., 2020, 68, 15074–15084 CrossRef CAS PubMed.
- G. Colombo Dugoni, A. Mezzetta, L. Guazzelli, C. Chiappe, M. Ferro and A. Mele, Green Chem., 2020, 22, 8680–8691 RSC.
- X.-D. Hou, G.-J. Feng, M. Ye, C.-M. Huang and Y. Zhang, Bioresour. Technol., 2017, 238, 139–146 CrossRef CAS PubMed.
- C. Li, C. Huang, Y. Zhao, C. Zheng, H. Su, L. Zhang, W. Luo, H. Zhao, S. Wang and L.-J. Huang, Processes, 2021, 9, 384 CrossRef CAS.
- M. Damle and N. Dalavi, Int. J. Appl. Sci. Biotechnol., 2015, 3, 434–438 CrossRef CAS.
- J. S. Boles, D. A. Crerar, G. Grissom and T. C. Key, Geochim. Cosmochim. Acta, 1988, 52, 341–344 CrossRef CAS.
- J. F. Yuan, T. T. Wang, D. H. Wang, G. H. Zhou, G. X. Zou, Y. Wang, M. G. Gong and B. Zhang, Food Bioprocess Technol., 2020, 13, 1246–1254 CrossRef CAS.
- M. Nuruddin, M. Hosur, M. J. Uddin, D. Baah and S. Jeelani, J. Appl. Polym. Sci., 2016 DOI:10.1002/app.42990.
- E. S. Morais, A. M. Da Costa Lopes, M. G. Freire, C. S. R. Freire and A. J. D. Silvestre, ChemSusChem, 2021, 14, 686–698 CrossRef CAS PubMed.
- F. Xu, J. Yu, T. Tesso, F. Dowell and D. Wang, Appl. Energy, 2013, 104, 801–809 CrossRef CAS.
- X. Li, Y. Wei, J. Xu, N. Xu and Y. He, Biotechnol. Biofuels, 2018, 11, 1–16 CrossRef.
- X. Colom and F. Carrillo, J. Wood Chem. Technol., 2005, 25, 1–11 CrossRef CAS.
- J. Lehto, J. Louhelainen, T. Kłosińska, M. Drożdżek and R. Alén, Biomass Convers. Biorefin., 2018, 8, 847–855 CrossRef.
- R. Ragupathi Raja Kannan, R. Arumugam and P. Anantharaman, Curr. Bioact. Compd., 2011, 7, 118–125 CrossRef.
- G. Grillo, E. Calcio Gaudino, R. Rosa, C. Leonelli, A. Timonina, S. Grygiškis, S. Tabasso and G. Cravotto, Molecules, 2021, 26, 798 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc03450b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |