
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organocatalytic epoxidation and allylic oxidation of alkenes by molecular oxygen†
Marina
Petsi
,
Maria
Orfanidou
and
Alexandros L.
Zografos
 *
*
Department of Chemistry, Aristotle University of Thessaloniki, University Campus, Old Chemistry Building, 54124, Thessaloniki, Greece. E-mail: alzograf@chem.auth.gr
First published on 14th October 2021
Abstract
Pyrrole-proline diketopiperazine (DKP) acts as an efficient mediator for the reduction of dioxygen by Hantzsch ester under mild conditions to allow the aerobic metal-free epoxidation of electron-rich alkenes. Mechanistic crossovers are underlined, explaining the dual role of Hantzsch ester as a reductant/promoter of the DKP catalyst and a simultaneous competitor for the epoxidation of alkenes when HFIP is used as a solvent. Expansion of this protocol to the synthesis of allylic alcohols was achieved by adding a catalytic amount of selenium dioxide as an additive, revealing a superior method to the classical application of t-BuOOH as a selenium dioxide oxidant.
Introduction
Epoxides and allylic alcohols are versatile intermediates that are commonly used to induce complexity in modern organic synthesis.1 Discoveries in the asymmetric epoxidation and allylic oxidation of alkenes coincide, not accidentally, with major breakthroughs in total synthesis, pharmaceutical industry and materials science. Pioneering methods for asymmetric epoxidation such as Mukaiyama,2 Sharpless3 and Jacobsen–Katsuki,4 despite their undeniable success are still not the methods of choice for the pharmaceutical industry, due to the strict requirements for metal impurities to be in the ppm scale.5 Considerable efforts towards the development of organocatalytic asymmetric epoxidation resulted in the powerful methods of Shi,6 List,7 and Hayashi8 among others,9 that utilize hydrogen peroxide, oxone and alkyl peroxides as the terminal oxidants. Greener variants utilizing dioxygen are rare especially when metal catalysts are absent (Scheme 1).10 | ||
| Scheme 1 Reported methods for the aerobic epoxidation and allylic hydroxylation of alkenes and the current work employing DKP as an organocatalyst for the epoxidation and allylic hydroxylation of electron-rich alkenes. | ||
Employment of methylhydrazine or isopropylbenzene under basic conditions or photochemical initiated use of tetramethylguanidine provide a reliable protocol only for the aerobic epoxidation of electron poor alkenes (Scheme 1).11 Complementary organocatalytic epoxidation of electron rich alkenes can be achieved by the Minisci reaction,12 where the epoxidation of terminal positions is favoured. The mild conditions employed by these protocols usually ensure their chemo- and regio-selectivity in complex systems but at the expense of precious (for the pharmaceutical industry) reaction time. Modern efforts to employ flow chemistry in Minisci or Mukaiyama reactions are surely of special practicality.13
In turn, monooxygenases are nature's choice to produce the desired oxidation levels of complexity in secondary metabolite structures.14 Commonly monooxygenases, apart from the active metallic center, contain flavin mononucleotide (FMN) or flavin adenine dinucleotide (FAD) cofactors that initiate the transfer of electrons from NADH or NADPH to the terminal dioxygen oxidant. Flexibility of their oxidation potentials is achieved by different bimetallic coordination patterns and by ligands attached to the metal center allowing either the epoxidation or allylic oxidation of alkenes by typically similar enzymes, while the more challenging CH-oxidation of unactivated hydrocarbons is achieved by high-valent metal oxo species.15
Considerable efforts have been made to achieve the biosynthetic levels of selectivity between epoxidation and allylic oxidation of alkenes by metal oxo complexes without success (Scheme 1).14a This is mainly attributed to the sensitive factors that rule the oxidative ability of high-valent metal oxo species.16 To this end, aerobic protocols managing to deliver chemoselective allylic oxidation are scarce and synthetically favoured protocols rely either on the use of selenium dioxide with t-BuOOH or palladium chemistry.17 Still, the pursuit for a green, aerobic protocol that can ideally have access to epoxides and allylic alcohols with a high degree of chemocontrol is in high demand.
Results and discussion
Recently, our group reported a green and efficient catalytic system for the oxidation of sulfides to sulfoxides by taking advantage of the ability of proline dipeptides to reduce dioxygen under essential neutral conditions.18 According to our studies,18b pyrrole-proline diketopiperazine (DKP) was found optimal as a green, neutral activator of dioxygen in polar solvents, to produce electrophilic hydroperoxy species 1 in its contact with air (Scheme 2). The isolable 1 is able to readily oxidize electron rich substrates when involved in the reaction, producing hemiaminal 2. A catalytic cycle was developed allowing the reduction of 2 back to DKP with the aid of Hantzsch ester (3) in HFIP. During our studies, we were aware that the same catalytic system was able to epoxidize alkenes but no studies were available regarding the chemoselectivity of this process nor its ability to be expanded to the allylic oxidation of alkenes. The mildness and greenness of this protocol which avoids toxic metals and hazardous oxidants, and also the obvious mechanistic similarities of our catalytic prototype to well-studied artificial riboflavin catalysts render DKP unique and prone for further investigation, as riboflavin epoxidation has not been reported.19 | ||
| Scheme 2 Merging pathways for the aerobic epoxidation of alkenes in HFIP. | ||
Screening of optimized epoxidation conditions was performed with Z-cyclooctene 5 as a model substrate (Table 1). As in the case of sulfide oxidation, HFIP was found to be the optimal solvent for the process providing cyclooctene oxide (6) in 93% yield (entry 4). Polar aprotic solvents (DMSO and DMF; entries 2 and 3) were able to initiate the epoxidation, but failed to retain the performance of the catalytic cycle due to their enhanced basic profile that shutters the hemiaminal reduction. The same was apparent even when a mixture of DMF with HFIP was used (entry 6). On the other hand, a mixture of HFIP and DCM (entry 7) was found effective to retain DKP catalysis, a result that is highly beneficial in the cases where substrates are practically insoluble in HFIP. Although protic solvents were known from our previous work that instantly kill the catalytic cycle by interfering with the reduction step, water mixtures were also tested in the presence of HFIP as a medium to introduce buffer salts in order to prevent the decomposition of labile epoxides. Unfortunately, these attempts (entry 5) failed to produce useful yields of 6. Further screening by additional equivalents of Hantzsch ester (3), an open vial setting or by heating the reaction mixture (entries 8–10, middle part of Table 1) resulted in diminished yields of 6, whereas addition of hydrogen peroxide as a terminal oxidant led only to low yields of 6. Blank reactions (lower part of Table 1) in the absence of (a) DKP and Hantzsch ester (entry 12), (b) Hantzsch ester (entry 13) and (c) DKP (entry 14) produced no product, 8% and 5–30% yields of 6, respectively. The range of epoxide yields witnessed when Hantzsch ester was used in HFIP in the presence of dioxygen was highly dependent on its purity and the existence of metal-traces and light.20 Based on these results, we initiated a parallel survey to establish the crucial factors allowing its oxidation and how this can result in the subsequent epoxidation of alkenes. It was known from our previous work18 that pyridine 4 produced from Hantzsch ester within the catalytic cycle promotes the ability of DKP to activate dioxygen. Thus, blank reactions were performed in the presence of pyridine 4 and water and found that 0.25 eq. of pyridine 4 and 25 eq. of water were essential to practically inhibit the epoxidation reaction induced by Hantzsch ester (entry 15). To our delight, the same additives allowed the epoxidation of cyclooctene in the presence of DKP (entry 16) without affecting the optimized yield. To this end, it is proposed that two reaction pathways are present during the epoxidation reaction (Scheme 2). The first involves the direct oxidation of Hantzsch ester by dioxygen in the presence of HFIP, resulting in the indirect epoxidation of the substrate, while the other implicates DKP as the mediator to achieve the aerobic epoxidation of alkenes. The Hantzsch ester path appears to halt in the presence of a substoichiometric amount of pyridine 4 and excess of water, in favour of DKP catalysis which is accelerated by the presence of pyridine 4. The inhibition of the Hantzsch ester path to activate dioxygen is particularly valuable in the cases where an enantioselective oxidation is pursued. In preliminary asymmetric experiments, it is indeed true that the absence of pyridine 4 and water in reaction media led to racemates, while their addition resulted in the enantioenrichment of the final products.21
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Solvent | Deviation from conditions | Conv.b | Yield of 6b (%) | |
| a Reaction conditions: all reactions ran in 4 mL vials at 0.20 mmol scale of substrate. b were determined by GC using 1,3,5-trimethoxybenzene as an internal standard. | |||||
| Solvent screening | 1 | DCM | — | 20 | 12 |
| 2 | DMSO | — | 10 | 6 | |
| 3 | DMF | — | 35 | 10 | |
| 4 | HFIP | — | 100 | 93 | |
| 5 | HFIP/H2O 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
— | 16 | 8 | |
| 6 | HFIP/DMF 3:1 |
— | 42 | 15 | |
| 7 | HFIP/DCM 3:1 |
— | 93 | 56 | |
| Condition screening | 8 | HFIP | 3 (2 eq.) | 75 | 73 |
| 9 | HFIP | Open vial | 55 | 50 | |
| 10 | HFIP | Heat 45 °C | 55 | 54 | |
| 11 | HFIP | No. 1, H2O2 (1 eq.) instead of O2 | 30 | 28 | |
| Blank reactions | 12 | HFIP | No. 3; No DKP | 0 | 0 |
| 13 | HFIP | No. 3 | 12 | 8 | |
| 14 | HFIP | No DKP | 10–44 | 5–35 | |
| 15 | HFIP | +4 (0.25 eq.); +H2O (25 eq.); No DKP | 10 | 5 | |
| 16 | HFIP | +4 (0.25 eq.); +H2O (25 eq.) | 100 | 93 | |

With these results in hand, we next moved to test our optimized protocol in different alkene substrates (Scheme 3). Various alkenes bearing terminal, internal, allylic and conjugated double bonds were tested. Alkenes bearing electron-rich double bonds were found optimal for fast oxidations in moderate to good yields (compounds 6–13; Scheme 3). Aromatic alkenes react slower compared to alkenes bearing an alkyl substitution, providing moderate yields of epoxides (compounds 15–20), while aromatic compounds bearing electron-withdrawing groups fail to epoxidise (compound 14). Allylic alcohols were surprisingly unreactive in most cases (compounds 23, 30, and 35), in contrast to classical methods that favor their epoxidation. The latter can be potentially attributed to the formation of a hydrogen bond between HFIP and allylic alcohol that sufficiently removes the electron density from the allylic p-bond. Thus, reactants bearing hydrogen bonding donor groups are able to be epoxidized with the lack of protection, while their alkene moieties remain unreactive. The lack of reactivity is also apparent for terminal monosubstituted alkenes, even after prolonged reaction times (compounds 36–38). The chemoselective characteristics described above allow the method to be amenable for the late-stage epoxidation of complex substrates bearing polyunsaturation (compounds 27–35, Scheme 3). The protocol is tolerant to multiple functionalities such as aromatics, esters, aldehydes, amides, silyloxy groups, ketals etc. Epoxidation can also be scaled up to several mmol scale of the substrate even using 2 mmol% of the catalyst without evidencing decrease on the yields of delivered products. In these cases, HFIP and DKP can be easily recycled and reused underlining the greenness and efficacy of the current protocol.
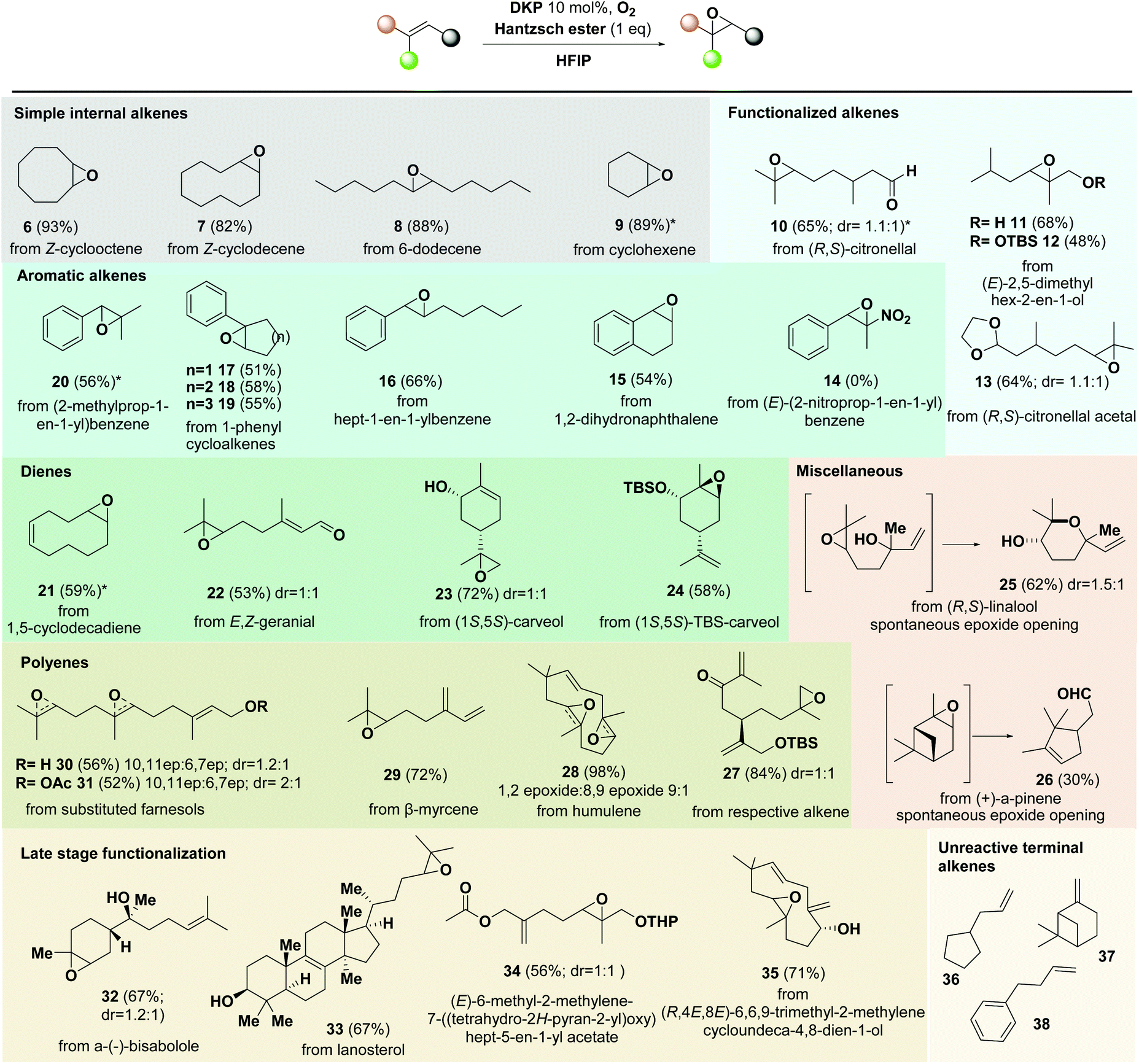 | ||
| Scheme 3 Reaction scope. Reaction conditions: the optimized conditions of entry 4 of Table 1 were used unless otherwise noted. Reactions in 0.2 mmol scale unless otherwise noted. Numbers in parentheses correspond to isolated yields. Asterisk symbol denotes yields on 2 mmol scale with 2 mol% DKP. | ||
For developing the idea of a biomimetic oxidative protocol that would ideally have the ability to access both epoxides and allylic positions, we sought to diverge from the prototype epoxide pathway discussed above towards the discovery of its allylic variation. During our screening on potential additives, only selenium dioxide was found appropriate to achieve the transformation allowing the allylic oxidation of alkenes (Scheme 4), while the typical oxygen active Lewis acids of CuI, Cu(OTf)2, FeCl3, Fe(TPP)Cl, etc. led only to decomposition or mixtures of allylic oxidation and epoxidation products. To this end, a catalytic amount of selenium dioxide allows the allylic oxidation of alkenes in good yields, in the cases where the epoxidation reaction is competent (internal electron rich alkene compounds 39, 40, and 43), whereas unreactive to epoxidation alkenes by our method provide excellent yields of allylic alcohol (terminal alkene compounds 41, 42, 44, and 45) (Scheme 4). It is interesting to note that comparison of the developed method with the classical Sharpless catalytic allylic oxidation, employing SeO2 with t-BuOOH in DCM shows superiority in isolated yields of the current method in every compound tested. The latter can be attributed to the beneficial effects of hydrogen bonding in HFIP, which inhibit the overoxidation of allylic alcohols prohibiting the production of undesired ketones or aldehydes, usual byproducts in the selenium dioxide reaction. Further screening of reaction conditions shows that diminished selenium dioxide loadings of 1–5% provide cleaner reaction profiles due to the avoidance of overoxidized products and produced selenoxides.
 | ||
| Scheme 4 Allylic oxidation of alkenes, protocol and proposed mechanism. Comparison of aerobic DKP-SeO2 allylic hydroxylation with the well-established allylic hydroxylation by SeO2 and t-BuOOH. (Remaining percentage corresponds to starting alkene). Reported yields refer to isolated products. | ||
The preferential ability of this protocol for allylic oxidation over epoxidation can be credited to the enhanced electrophilic profile of the SeO2-1 hybrid (intermediate I, Scheme 4, proposed mechanism) that is proposed to initiate the allylic oxidation reaction, over 1 which is responsible for the epoxidation reaction. The electrophilic enhancement is evident from the terminal alkene ability to react under these conditions, in contrast to their epoxidation. An ene-reaction of intermediate I with alkenes followed by a [2,3]-sigmatropic rearrangement produce selenoxide II with subsequent cleavage of aminal 2 (DKP-OH) (Scheme 4). Finally, regeneration of DKP-OOH (1) according to the oxygen-Hantzsch mediated catalytic cycle (Scheme 2) allows the cleavage of selenoxide II to provide allylic alcohol and regenerate the active allylic oxidant I.
Conclusions
In conclusion, an efficient organocatalytic aerobic protocol for the epoxidation of electron-rich alkenes is described which relies on the mediacy of pyrrole-proline DKP as an activator of dioxygen. The described protocol allows the chemoselective reaction of the most electron rich alkenes over the terminal, allylic and homoallylic positions, avoiding the use of toxic metals and hazardous peroxides as oxidants. The method under the same principles can also be expanded to provide allylic alcohols in good to excellent yields, if catalytic selenium dioxide is introduced as an additive. The latter is to the best of our knowledge the first catalytic aerobic allylic oxidation based on selenium dioxide.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Dr Michalis Kallitsakis, research associate at Aristotle University of Thessaloniki for helpful discussions and support regarding the GC analyses. We are also grateful to the collaborators Prof. Alexandros Koumbis and Prof. Christos Stathakis from Aristotle University of Thessaloniki for the generous donation of substrates for the current study. This work was supported by the project ‘OPENSCREEN-GR’ (MIS 5002691) which is implemented under the Action ‘Reinforcement of the Research and Innovation Infrastructure’, funded by the Operational Program ‘Competitiveness, Entrepreneurship and Innovation’ (NSRF 2014–2020) and co-financed by Greece and the European Union (European Regional Development Fund). Funding was also provided the by Research-Create-Innovate program (project code T1EDK-01161) which is implemented in collaboration with Pharmathen S.A. and co-financed by Greece and the European Union.Notes and references
- (a) Aziridines and Epoxides in Organic Synthesis, ed. A. K. Yudin, Wiley-VCH, 2006, ch. 7–8, pp. 229–313 Search PubMed; (b) Aziridines and Epoxides in Organic Synthesis, ed. A. K. Yudin, Wiley-VCH, 2006, ch. 7, pp. 229–269 Search PubMed; (c) C. Schneider, Synthesis, 2006, 3919–3944 CrossRef CAS; (d) C. M. Marson, Tetrahedron, 2000, 56, 8779–8794 CrossRef CAS; (e) J. Gorzynski Smith, Synthesis, 1984, 629–656 CrossRef.
- (a) T. Yamada, K. Imagawa, T. Nagata and T. Mukaiyama, Chem. Lett., 1992, 21, 2231–2234 CrossRef; (b) T. Nagata, K. Imagawa, T. Yamada and T. Mukaiyama, Chem. Lett., 1994, 23, 1259–1262 CrossRef; (c) T. Yamada, K. Imagawa and T. Mukaiyama, Chem. Lett., 1992, 21, 2109–2112 CrossRef; (d) T. Nagata, K. Imagawa, T. Yamada and T. Mukaiyama, Chem. Lett., 1995, 68, 1455–1465 CAS; (e) S. Inoki, T. Takai, T. Yamada and T. Mukaiyama, Chem. Lett., 1991, 20, 941–944 CrossRef; (f) K. Imagawa, T. Nagata, T. Yamada and T. Mukaiyama, Chem. Lett., 1994, 23, 527–530 CrossRef.
- (a) R. A. Johnson and K. B. Sharpless, Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Elsevier Science Ltd., 1991, ch. 3.2, vol. 7, pp. 389–436 Search PubMed; (b) A. Pfenninger, Synthesis, 1986, 89–116 CrossRef CAS; (c) M. G. Finn and B. Sharpless, J. Am. Chem. Soc., 1991, 113, 113–126 CrossRef CAS; (d) T. Katsuki and K. B. Sharpless, J. Am. Chem. Soc., 1980, 102, 5974–5978 CrossRef CAS; (e) Y. Gao, R. M. Hanson, J. M. Klunder, S. Y. Ko, H. Masamune and K. B. Sharpless, J. Am. Chem. Soc., 1987, 109, 5765–5780 CrossRef CAS.
- (a) W. Zhang, J. L. Loebach, S. R. Wilson and E. N. Jacobsen, J. Am. Chem. Soc., 1990, 112, 2801–2803 CrossRef CAS; (b) R. Irie, K. Noda, Y. Ito, N. Matsumoto and T. Katsuki, Tetrahedron Lett., 1990, 31, 7345–7348 CrossRef CAS; (c) B. D. Brandes and E. N. Jacobsen, J. Org. Chem., 1994, 59, 4378–4380 CrossRef CAS; (d) T. Linker, Angew. Chem., Int. Ed. Engl., 1997, 36, 2060–2062 CrossRef CAS; (e) E. N. Jacobsen, W. Zhang, A. R. Muci, J. R. Ecker and L. Deng, J. Am. Chem. Soc., 1991, 113, 7063–7064 CrossRef CAS; (f) T. Katsuki, Synlett, 2003, 281–297 CrossRef CAS.
- ICH Q3D(R1) Guidelines for elemental impuriries.
- (a) O. A. Wong and Y. Shi, Chem. Rev., 2008, 108, 3958–3987 CrossRef CAS PubMed; (b) Y. Shi, Acc. Chem. Res., 2004, 37, 488–496 CrossRef CAS PubMed; (c) O. B. Wang, A. Wong, M. X. Zhao and Y. Shi, J. Org. Chem., 2008, 73, 9539–9543 CrossRef PubMed; (d) H. Tian, X. She, L. Shu, H. Yu and Y. Shi, J. Am. Chem. Soc., 2000, 122, 11551–11552 CrossRef CAS; (e) Z. X. Wang, Y. Tu, M. Frohn, J. R. Zhang and Y. Shi, J. Am. Chem. Soc., 1997, 119, 11224–11235 CrossRef CAS; (f) Y. Tu, Z.-X. Wang and Y. Shi, J. Am. Chem. Soc., 1996, 118, 9806–9807 CrossRef CAS.
- (a) O. Lifchits, C. M. Reisinger and B. List, J. Am. Chem. Soc., 2010, 132, 10227–10229 CrossRef CAS PubMed; (b) X. Wang, C. M. Reisinger and B. List, J. Am. Chem. Soc., 2008, 130, 6070–6071 CrossRef CAS PubMed; (c) A. Lee, C. M. Reisinger and B. List, Adv. Synth. Catal., 2012, 354, 1701–1706 CrossRef CAS; (d) O. Lifchits, M. Mahlau, C. M. Reisinger, A. Lee, C. Fares, I. Polyak, G. Gopakumar, W. Thiel and B. List, J. Am. Chem. Soc., 2013, 135, 6677–6693 CrossRef CAS PubMed; (e) X. Wang and B. List, Angew. Chem., Int. Ed., 2008, 47, 1119–1122 CrossRef CAS PubMed.
- (a) B. P. Bondzic, T. Urushima, H. Ishikawa and Y. Hayashi, Org. Lett., 2010, 12, 5434–5437 CrossRef CAS PubMed; (b) T. Mukaiyama, K. Ogata, T. Ono and Y. Hayashi, ChemCatChem, 2015, 7, 155–159 CrossRef CAS.
- I. Triandafillidi, D. I. Tzaras and C. G. Kokotos, ChemCatChem, 2018, 10, 2521–2535 CrossRef CAS.
- For selected metal-catalyzed aerobic epoxidation please see: (a) M. D. Hughes, Y.-J. Xu and P. Jenkins, et al. , Nature, 2005, 437, 1132–1135 CrossRef CAS PubMed; (b) M. M. Montemore, M. A. van Spronsen and R. J. Madix, et al. , Chem. Rev., 2018, 118, 2816–2862 CrossRef CAS PubMed; (c) S. E. Allen, R. R. Walvoord and R. Padilla-Salinas, et al. , Chem. Rev., 2013, 113, 6234–6458 CrossRef CAS PubMed; (d) M. Hajimohammadi and N. J. Safari, Porphyrins Phthalocyanines, 2010, 14, 639–645 CrossRef CAS; (e) Y. Sun, Z. Xi and G. J. Cao, Mol. Catal. A: Chem., 2001, 166, 219–224 CrossRef CAS; (f) T. S. Symeonidis, A. Athanasoulis, R. Ishii, Y. Uozumi, Y. M. A. Yamada and I. N. Lykakis, ChemPhotoChem, 2017, 1, 479–484 CrossRef CAS.
- For selected organocatalytic aerobic epoxidations please see: (a) H. Kawai, S. Okusu and Z. Yuan, et al. , Angew. Chem., Int. Ed., 2013, 52, 2221–2225 CrossRef CAS PubMed; (b) Y. Wu, G. Zhou and Q. Meng, et al. , J. Org. Chem., 2018, 83, 13051–13062 CrossRef CAS PubMed.
- For selected references on Minisci epoxidation: (a) F. Minisci, C. Gambarotti, M. Pierini, O. Porta, C. Punta, F. Recupero, M. Lucarinib and V. Mugnaini, Tetrahedron Lett., 2006, 47, 1421–1424 CrossRef CAS; (b) C. Einhorn, J. Einhorn, C. Marcadal and J. L. Pierre, Chem. Commun., 1997, 447–448 RSC; (c) L. Melone, C. Gambarotti, S. Prosperini, N. Pastori, F. Recupero and C. Punta, Adv. Synth. Catal., 2011, 353, 147–154 CrossRef CAS; (d) T. Iwahama, S. Sakaguchi and Y. Ishii, Chem. Commun., 1999, 727–728 RSC.
- (a) R. Spaccini, L. Liguori, C. Punta and H. R. Bjørsvik, ChemSusChem, 2012, 5, 261–265 CrossRef CAS PubMed; (b) L. Vanoye, J. Wang, M. Pablos and C. de Bullefon, Catal. Sci. Technol., 2016, 6, 4724–4732 RSC.
- (a) M. Petsi and A. L. Zografos, Synthesis, 2018, 50, 4715–4745 CrossRef CAS; (b) D. E. T. Pazmino, M. Winkler, A. Glieder and M. W. Fraaijea, J. Biotechnol., 2010, 146, 9–24 CrossRef PubMed; (c) W. J. H. van Berkel, N. M. Kamerbeek and M. W. Fraaije, J. Biotechnol., 2006, 124, 670–689 CrossRef CAS PubMed; (d) M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841–2887 CrossRef CAS PubMed; (e) I. G. Denisov, T. M. Makris, S. G. Sligar and I. Schlichting, Chem. Rev., 2005, 105, 2253–2277 CrossRef CAS PubMed.
- (a) B. J. Wallar and J. D. Lipscomb, Chem. Rev., 1996, 96, 2625–2657 CrossRef CAS PubMed; (b) C. Krebs, D. G. Fujimori, C. T. Walsh and J. M. Bollinger Jr., Acc. Chem. Res., 2007, 40, 484–492 CrossRef CAS PubMed; (c) J. C. Price, E. W. Barr, B. Tirupati, J. M. Bollinger Jr. and C. Krebs, Biochemistry, 2003, 42, 7497–7508 CrossRef CAS PubMed.
- T. Terencio, E. Andrios, I. Gamba, M. Smec, M. Costas and J. Roithova, J. Am. Soc. Mass Spectrom., 2019, 30, 1923–1933 CrossRef CAS PubMed.
- (a) M. A. Warpehoski, B. Chabaud and K. B. Sharpless, J. Org. Chem., 1982, 47, 2897–2900 CrossRef CAS; (b) M. A. Umbreit and K. B. Sharpless, J. Am. Chem. Soc., 1977, 99, 5526–5528 CrossRef CAS; (c) M. S. Chen and M. C. White, J. Am., Chem. Soc., 2004, 126, 1346–1347 CrossRef CAS PubMed; (d) C. C. Patillo, I. I. Strambeanu, P. Calleja, N. A. Vermeulen, T. Mizuno and M. C. White, J. Am. Chem. Soc., 2016, 138, 1265–1272 CrossRef PubMed; (e) D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Chem. Rev., 2018, 118, 2636–2679 CrossRef CAS PubMed; (f) C. V. Kozack, J. A. Sowin, J. N. Jaworski, A. V. Iosub and S. S. Stahl, ChemSusChem, 2019, 12, 3003–3007 CrossRef CAS PubMed.
- (a) M. Petsi and A. L. Zografos, ACS Catal., 2020, 10(13), 7093–7099 CrossRef CAS; (b) For mechanistic study on the presented mechanism please see the supporting information of the parent reference pp. 33–60.
- R. Cibulka, Eur. J. Org. Chem., 2015, 915–932 CrossRef CAS.
- For oxidation of Hantzsch ester under different conditions please see: (a) X. Xu and H. Xu, Asian J. Chem., 2009, 21, 4599–4602 CAS; (b) D. H. Wang, Q. Liu, B. Chen, L. P. Zhang, C. H. Tung and L. Z. WU, Chin. Sci. Bull., 2010, 55, 2855–2858 CrossRef CAS; (c) S. Chen, M. S. Hossain Jr. and F. W. Foss, ACS Sustainable Chem. Eng., 2013, 1(8), 1045–1051 CrossRef CAS; (d) S. H. Mashraqui and M. A. Kamik, Tetrahedron Lett., 1998, 39, 4895–54898 CrossRef CAS; (e) N. Nakamichi, Y. Kawashita and M. Hayashi, Org. Lett., 2002, 4, 3955–3957 CrossRef CAS PubMed; (f) A. Saini, S. Kumar and J. r S. Sandhu, Synth. Commun., 2007, 37, 2317–2324 CrossRef CAS.
- Preliminary results of asymmetric epoxidation with the aid of asymmetric DKPs show low enantioselectivities (ee 5–9%) only for reactions bearing pyridine and water as additives when enantiopure DKPs were used.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, copies of NMR spectra and optimization procedures for the described protocols. See DOI: 10.1039/d1gc03029a |
| This journal is © The Royal Society of Chemistry 2021 |