
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Incorporation of catechyl monomers into lignins: lignification from the non-phenolic end via Diels–Alder cycloaddition?†
Daisuke
Ando‡
 a,
Fachuang
Lu
ab,
Hoon
Kim
a,
Alexis
Eugene
a,
Yuki
Tobimatsu
c,
Ruben
Vanholme
de,
Thomas J.
Elder
f,
Wout
Boerjan
de and
John
Ralph
*agh
a,
Fachuang
Lu
ab,
Hoon
Kim
a,
Alexis
Eugene
a,
Yuki
Tobimatsu
c,
Ruben
Vanholme
de,
Thomas J.
Elder
f,
Wout
Boerjan
de and
John
Ralph
*agh
aDepartment of Energy, Great Lakes Bioenergy Research Center, Wisconsin Energy Institute, Madison, Wisconsin 53726, USA. E-mail: jralph@wisc.edu; Tel: (+1) 608-890-2429
bState Key Laboratory of Pulp and Paper Engineering, South China University of Technology, 381 Wushan Rd, Tianhe District, Guangzhou, 510640, P.R. China
cResearch Institute for Sustainable Humanosphere, Kyoto University, Gokasho, Uji 611-0011, Japan
dDepartment of Plant Biotechnology and Bioinformatics, Ghent University, B-9052 Gent, Belgium
eCenter for Plant Systems Biology, VIB, B-9052 Gent, Belgium
fUSDA-Forest Service, Southern Research Station, 521 Devall Drive, Auburn, AL 36849, USA
gDepartment of Biochemistry, University of Wisconsin-Madison, Madison, Wisconsin 53706, USA
hDepartment of Biological System Engineering, University of Wisconsin-Madison, Madison, Wisconsin 53726, USA
First published on 26th October 2021
Abstract
Canonical lignification occurs via the coupling of phenolic radicals, in which chain extension can occur only from phenolic ends of growing polymer chains. Radical coupling of catechyl monomers, including caffeyl and 5-hydroxyconiferyl alcohols, gives rise to benzodioxane units in the polymer. Anticipating that a catechol could oxidize to its o-benzoquinone analog under the dehydrogenative (oxidative) conditions of lignification, we examined the possibility that an o-benzoquinone, as the diene component, could also incorporate into lignin via another mechanism, the Diels–Alder cycloaddition reaction. The o-benzoquinone derived from methyl 5-hydroxyvanillate and 4-O-methylconiferyl alcohol served as models for the diene and dienophile, respectively, and produced Diels–Alder products in vitro. Two types of Diels–Alder products were found: (i) when the 1,2-diketone of the quinone acts as the diene in a hetero-Diels–Alder reaction, a benzodioxane structure was produced with a different regiochemistry than the benzodioxane isomer produced via radical coupling; (ii) when the quinone's diene participated in the Diels–Alder reaction, a distinctive oxatricyclo structure was produced. Both features may be used as markers for the occurrence of Diels–Alder reactions in lignification. Examination of natural lignins derived from catechyl monomers, however, did not reveal evidence for such products. The conclusion is that the only significant reactions in lignification are combinatorial radical coupling reactions of the single-electron-oxidized phenolics and that polymer chain extension therefore occurs only from the phenolic end-units even in the special case of plants that utilize catechyl monomers for lignification.
Introduction
Lignification is the process by which phenolic monomers polymerize, in planta, to form lignin polymers.1–4 The primary canonical monomers, termed monolignols, are p-coumaryl, coniferyl, and sinapyl alcohols, hydroxycinnamyl alcohols bearing 0, 1, or 2 methoxyls ortho to the phenol (and, consequently, meta to the 3-carbon sidechain). After polymerization, these result in the p-hydroxyphenyl (H), guaiacyl (G), and syringyl (S) aromatic units in the lignin polymer. The polymerization requires the formation of monolignol, oligolignol, and/or polymer radicals, created by 1-electron oxidation (dehydrogenation) using H2O2-requiring peroxidases or O2-requiring laccases, as shown in Fig. 1A for guaiacyl lignins from coniferyl alcohol 1. The enzymes do not necessarily need direct contact with the oligomeric/polymeric phenolic substrate, as demonstrated in a manganese peroxidase system in which phenolic radicals were generated via a redox shuttle such as Mn(II)/Mn(III) oxalate.5 Phenolic intermediaries that are more easily oxidized such as p-coumarate, or monomer radicals themselves, may also oxidize the polymer by radical transfer.3,4,6–12 | ||
| Fig. 1 (A) Generation of the radical 1˙ from coniferyl alcohol 1, a monolignol, and the radical 2˙ from an oligolignol or lignin chain 2. Dimerization of 1 (via its radical 1˙) produces, following rearomatization reactions, one of 3 primary dimers 3a–c; cross-coupling of a monolignol 1 with an oligomer 2 (endwise coupling), the main reaction in lignification, (again via the radicals 1˙ and 2˙ of each) lengthens the chain by one unit and produces new end-units 4a–b; cross-coupling can also join two growing chains 2 to produce 5-5- and 4-O-5-coupled units 5a–b. The bonds formed during the radical coupling step are highlighted by being bolded. Additional coupling to continue the lignification can occur at the positions noted by the small arrows; obviously, these new products also have the general structure represented by 2. (B) An attempt to cross-couple a monolignol 1 (via its radical 1˙) with an etherified (phenol-protected) monolignol such as 4-O-methylconiferyl alcohol (3,4-dimethoxycinnamyl alcohol) 6 results in quantitative recovery of starting material 6 along with the usual array of products from the monolignol 1, i.e., 3a, 3b, 3c as shown in Fig. 2. N.R.: no reaction. (C) What happens to cinnamyl alcohol endgroups in lignification? Top: The usual case is illustrated using a β-ether dimer 3a. The phenolic radical 3a˙ can be generated and, as usual, we can understand the coupling pathways available by drawing resonance forms. The resonance form shown illustrates how the single-electron radical density can reach as far as C1, but there is no mechanism by which it can extend into the sidechain. The available coupling possibilities are therefore at 4-O, 5, or 1 (as indicated by the dotted arrows on 3a). Bottom: A special case in which lignification can occur on an etherified unit.19 In the phenylcoumaran dimer 3b, the generated radical 3b˙ can, via an actual reaction, ring-open and undergo radical coupling at its β-position with a phenolic unit radical 2˙ to produce, e.g., the intermediate 7. Following normal rearomatization, the product is 8 that appears as though it derives from coupling with the etherified B-unit in 3b, but it is actually not derived that way (see text). | ||
The subsequent coupling mechanism has some striking features that often surprise organic chemists. First, the polymerization is not a series of radical addition reactions in a chain reaction typified by the polymerization of styrene to polystyrene. Instead, every chain-extension event is a radical coupling (quenching) reaction between two radicals to generate a non-radical neutral species (Fig. 1A); forming a new phenolic radical is required again for the next chain-extension. The apparently energetically profligate process of lignification requires that the newly produced phenolic end-group is again re-oxidized to form a new phenolic radical for further polymerization. A likely advantage of such an apparently energy-inefficient process is that it is controllable, and not subject to runaway radical reactions that are the bane of living systems.
The initial reaction, as illustrated in Fig. 1A for coniferyl alcohol, may be either the (dehydro)dimerization of two monolignol radicals (via β-β-, β-5-, or β-O-4-coupling) to produce dehydrodimers 3, or by the cross-coupling of a monolignol radical with any of a variety of initiating or nucleating phenolic monomer radicals (not shown); examples include ferulate (on arabinoxylans, in monocots),8 and tricin (a flavone, mainly in monocots).13,14 Here, and in subsequent polymerization, a second striking feature of the lignification mechanism is encountered. The process is a non-enzymatic combinatorial chemical one in the sense that the monomer may couple with another monomer or the growing polymer to form several different products, even if polymerization is from a single monomer type (such as coniferyl alcohol, Fig. 1A); more possibilities arise if the supplied monomer can vary. Ramifications of this combinatorial, chemically-controlled process are noted below.
The major polymerization reaction involves coupling of a monolignol radical with a radical from the phenolic end of a growing polymer 2 (via 4-O-β- or 5-β-coupling) to produce the single-unit-extended polymer 4. From coupling reactions involving a monolignol, which invariably couples at its β-position, the resulting intermediate products are reactive quinone methides that undergo rearomatization via nucleophilic addition at the α-position by, e.g., water or an internal aliphatic or phenolic hydroxyl group, as typically observed after β-O-4, β-β, or β-5 coupling reactions, respectively, to dimers 3a–c or oligomers 4a–b. Two lignin chains 2 may also couple at their phenolic ends, via 5-5- or 4-O-5-coupling, linking the two chains together and producing units 5 in the polymer (Fig. 1A; the minor β-1 coupling pathway is not shown). By convention, the new bond formed during the coupling reaction (β-O-4, β-5, β-β, β-1, 5-5, and 4-O-5) is used to designate that coupling pathway and the structure in the lignin dimer, or the dimeric unit in an oligomer/polymer, that results following the intermediate's rearomatization.
Under physiological conditions, the radical coupling reactions are considered to be irreversible, as are the following rearomatization reactions, such that the distribution of isomers or of different products reflects kinetic control and not thermodynamics.3 For example, if allowed to equilibrate under acidic conditions, both β-guaiacyl and β-syringyl ethers equilibrate to ∼50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 erythro:threo (anti:syn, RS/SR:RR/SS) thermodynamic ratios, but β-syringyl ethers are produced in approximately 75:25 ratio in vivo and under biomimetic in vitro conditions, and remain that way in the lignin polymer, as dictated by the kinetics of rearomatization.3,15
:50 erythro:threo (anti:syn, RS/SR:RR/SS) thermodynamic ratios, but β-syringyl ethers are produced in approximately 75:25 ratio in vivo and under biomimetic in vitro conditions, and remain that way in the lignin polymer, as dictated by the kinetics of rearomatization.3,15
A key feature of these polymerization reactions is that they are purely chemical and not enzyme- or protein-directed.1,16,17 The resulting polymer gains two new optical centers each time a monomer couples with another phenolic; as a result of the combinatorial coupling, even from a single type of monolignol monomer, individual polymer chains are structurally and/or stereochemically highly diverse. The complexity is such that even a simple β-ether 20-mer derived from solely β-O-4-coupling reactions to produce a homopolymer has 38 optical centers and consequently 238 optical isomers, or half that many, 237 (over 137 billion), possible physically distinct isomers. Clearly lignin is a very different polymer than, for example, a protein in which every molecule has the same sequence and structure, and is only a single isomer;18 this is not to say that proteins may not end up becoming different when post-translationally modified, for example. Another corollary of this combinatorial and purely chemical process is that there is no prescribed order of assembly of monomers into the polymer, nor is there any prescribed sequence of the types of units produced by the various coupling regiochemistries available. Because certain couplings are chemically favored over others, the coupling is not, however, statistically random (as sometimes suggested) but reflects coupling and cross-coupling propensities in a statistically weighted manner. As complex as the resulting lignin structure is, however, the lignification process itself is delightfully simple, as only a single chemical mechanism, that of radical coupling, is involved. In many ways lignin biosynthesis is therefore simpler than, for example, the biosynthesis of hemicellulosic polysaccharides.
Even after this mechanism of lignification is accepted, it is still frequently not appreciated that the reaction is exclusively via the coupling (quenching) of two radicals (Fig. 1A). Why is it not possible to take a single radical and couple it with a neutral molecule possessing a double bond as in, for example, polystyrene polymerization? Is it possible, for example, to take an etherified monolignol, i.e., one that is phenol-protected, and react it with a monolignol radical (Fig. 1B)? Such reactions are of course known in chemistry, but they involve oxidizing reagents that are considerably stronger than those available in the plant world with its peroxidases and laccases for the process. Lignin chemists have long known that this type of radical cross-reaction (Fig. 1B) will not occur during lignification, but we offer further evidence herein.
A valid question remains, however. Are there viable ways to extend the polymer from an etherified hydroxycinnamyl alcohol at the non-phenolic end of the chain? Traditionally within the lignin literature, the answer is a very strong ‘No!’ – the double bond of a hydroxycinnamyl alcohol in which its phenol is etherified is not reactive in a lignification sense. The simple reasoning is that it is not possible for a phenolic radical, generated in a structure containing an etherified hydroxycinnamyl alcohol unit, to propagate any single-electron density to that sidechain double-bond. For example, Fig. 1C, in dimer 3a, the ring B cinnamyl alcohol is etherified, and therefore cannot enter into any coupling reactions even after generating the ring A phenolic radical 3a˙. In chemical terms, it is not possible to use the mechanistic rules of ‘electron-pushing’ to transfer single-electron density to the β-carbon in that B-ring double-bond. Recently, however, an apparent exception to this rule in the rather special case of phenylcoumaran dimer 3b was revealed, Fig. 1C.19 Because of an actual chemical reaction pathway (not simply resonance), the phenylcoumaran radical 3b˙ can ring-open and, in a concerted reaction, undergo radical coupling via its B-ring moiety with a phenolic radical 2˙. The result for the example of Bβ-O-4 coupling to give the bis-(quinone methide) intermediate 7, following the usual water addition and re-cyclization of the phenylcoumaran in the rearomatization step, is the apparent extension of the chain via the double-bond of a phenol-etherified unit in products such as 8 (Fig. 1C). Preliminary density functional theory (DFT) calculations [at the B3LYP/6-31+G(d) level] indicate that the reaction from 3b˙ to 7 is exothermic (at −8.65 kcal mol−1) indicating that the reaction should occur; the rearomatization reaction to 8 is also obviously energetically downhill. This mechanism had escaped even conjecture until the appearance of the paper revealing this possibility.19 The evidence from experiments using strategically labeled compounds provides an apparent exception to the rule that phenol-etherified units cannot participate in lignification. It must be stressed, however, that this process involves a true chemical reaction as shown in Fig. 1C. As such, it clearly represents a mechanism by which a phenol-etherified cinnamyl alcohol unit can be involved in a radical coupling reaction and, in addition to occurring in a biomimetic in vitro system, almost certainly also occurs during lignification. This eye-opening exception aside, it can still be asserted that, in general, phenol-etherified units cannot inherently generate radicals that can undergo radical coupling, and certainly not by resonance from a distant phenolic radical, i.e., in general, etherified phenolic units cannot participate in the radical coupling reactions of lignification.
Are there other ways in which polymer chain extension might occur on etherified hydroxycinnamyl alcohol end-units in lignins? Interesting candidates may be found in plants deficient in O-methyltransferases. Truncated monolignol biosynthesis in such plants produces catechol-containing alternative monomers caffeyl alcohol and 5-hydroxyconiferyl alcohol, resulting in catechyl and 5-hydroxyguaiacyl units (C- and 5H-lignin units) in the polymer. Considerations of the reactivity and polymerization mechanisms of catechyl monomers relate not solely to genetically engineered plants deficient in O-methyltransferases, as natural mutants with the same characteristics are well-known in agriculture. The brown-midrib mutants of maize (bm3 or bmr3) and sorghum (bm12 or bmr12) are the classic examples that demonstrate the plants’ utilization of 5-hydroxyconiferyl alcohol monomers in lignification.20–22 More recently, C-lignin and 5H-lignin polymers derived partially or entirely from caffeyl alcohol or 5-hydroxyconiferyl alcohol have been discovered in the seedcoats of various plants.23–27 Additionally, the stilbene piceatannol, a catechol, has recently been discovered as a lignin monomer in various fruit endocarp tissues and bark.28–31 We reasoned that such catechyl monomers could produce ortho-benzoquinones under the dehydrogenative (oxidative) conditions of lignification; quinones, including o-quinones, are known plant metabolites.32 Indeed, early on it was contended that such catechyl monomers, if produced, would spontaneously oxidize or disproportionate to o-benzoquinones that could not then undergo radical coupling reactions and therefore lignification in a normal sense.33,34 The incorporation of the catechyl monomers into the single-electron oxidative processes of lignification has, however, been decisively confirmed, as reviewed.3,4,12,35 Nevertheless, we hypothesized that o-benzoquinones, if formed, could readily act as the diene component of classical Diels–Alder reactions, with cinnamyl alcohol units, even those present in etherified structures, being the required dienophile, analogously to reported reactions of galloyl quinones.36 Aqueous intermolecular Diels–Alder reactions are known.37 We report here a demonstration that, in the special case of catechyl monomers reacting with etherified hydroxycinnamyl alcohols, in vitro Diels–Alder reactions can indeed occur and produce diagnostic marker units for such reactions. We then explore whether such reactions can be found to occur during in planta lignification.
Results and discussion
The major goal of this research was to establish whether there were plausible mechanisms by which, under special circumstances, etherified hydroxycinnamyl alcohol units in lignins might participate in lignification. We were particularly interested in determining whether lignification with catechyl monomers could, via their o-benzoquinones, open up the possibility of Diels–Alder reactions in addition to the prototypical radical coupling reactions. We first needed to reaffirm that etherified hydroxycinnamyl alcohols cannot and do not participate in radical coupling reactions under lignification conditions.Establishing that etherified monolignols do not normally participate in lignification
Here we attempt to show a feature that is well-known in the lignin field, namely that cinnamyl alcohols without the (conjugated) phenolic-OH group cannot form radicals under lignification conditions and nor can they somehow cross-couple with a (phenolic) radical, i.e., that etherified hydroxycinnamyl alcohol moieties cannot participate in lignification. We took coniferyl alcohol 1 and 4-O-methylconiferyl alcohol 6 (=3,4-dimethoxycinnamyl alcohol) in a biomimetic peroxidase/H2O2in vitro system (Fig. 1B) wherein, as shown in Fig. 2, the coniferyl alcohol 1 undergoes dehydrodimerization to produce four dimers 3, the two isomers of the β-ether 3a, and the single isomers each of the phenylcoumaran 3b and the resinol 3c. Coniferyl alcohol's phenol-methylated analog, 4-O-methylconiferyl alcohol 6, remains totally intact; quantification of 6 before and after reaction (in triplicate) reveals that it has not reacted, and no cross-products from it were observed. We hope that this demonstration will be sufficiently convincing to confirm the contention that a free phenol is required for radical coupling, and perhaps avert further notions of cross-coupling reactions with etherified components from being entertained in the lignin literature.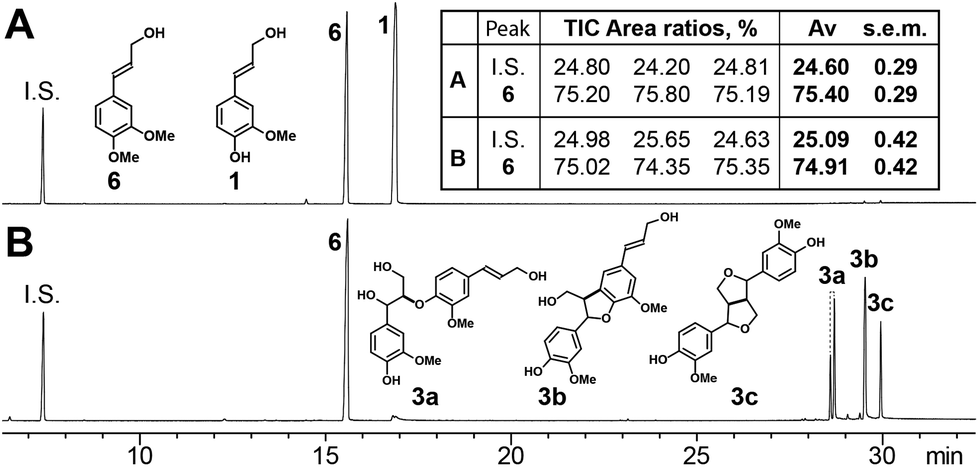 | ||
| Fig. 2 Total-ion chromatogram of (A) starting mixture and (B) product mixture, showing that 4-O-methylconiferyl alcohol 6 does not participate in radical coupling reactions whereas coniferyl alcohol 1 is converted by the biomimetic peroxidase-H2O2 system to dimers by coupling β-O-4 (3a, two isomers, syn or threo, anti or erythro), β-5 (3b, one isomer, trans), and β-β (3c, one isomer). I.S.: 3,4-dimethoxybenzaldehyde. Average, s.e.m. (standard error of the mean) from 3 replicates. | ||
Possibility of Diels–Alder reactions between the double-bond of a cinnamyl alcohol as the dienophile and an o-benzoquinone as the diene
Catechyl structures can potentially be oxidized to form o-benzoquinones under the dehydrogenative (oxidative) conditions of lignification (Fig. 3). Horseradish peroxidase oxidized catechol to o-benzoquinone in vitro, for example.38 A Diels–Alder reaction is a concerted [4 + 2] addition reaction between a conjugated diene and an alkene, the latter of which is termed a dienophile. Ideally, one of the pair is electron-deficient (substituted with electron-withdrawing groups) and the other is electron-rich (substituted with electron-donating groups). In the case of o-benzoquinones derived from catechyl monomers in lignification, obviously the quinone is electron-deficient, whereas the monolignol is electron-rich. An o-benzoquinone has two modes by which it may act as a diene – the actual diene itself in the traditional Diels–Alder reaction, or the 1,2-diketone in a so-called hetero-Diels–Alder reaction.39,40 Such cycloaddition reactions have even been noted in a somewhat lignin-related sense.41 After initially discovering a Diels–Alder product in an attempted in vitro radical coupling reaction between coniferyl alcohol 1 and methyl 5-hydroxyvanillate 11, as will be described below, we undertook a more careful evaluation of the products from a simple etherified analog of coniferyl alcohol, 4-O-methylconiferyl alcohol 6 (Fig. 3), and the o-benzoquinone 9 derived explicitly from methyl 5-hydroxyvanillate 11 under oxidative conditions and isolated before the reaction. | ||
| Fig. 3 Diels–Alder reaction between a quinone and a cinnamyl alcohol. (A) In the top line (black reaction arrows) are the reaction products from reacting 4-O-methylconiferyl alcohol 6 with the quinone 9 from catechol 11 (methyl 5-hydroxyvanillate). Notably, two Diels–Alder products are formed: a benzodioxane 12 and an oxatricyclo compound 13. Percentages are relative to the total yield of the four products (which totals 100%); actual yields are provided in the Experimental section. (B) The bottom scheme (red reaction arrows) is from the attempted reaction of coniferyl alcohol 1 with catechol 11 to produce the β-O-4-coupling product, benzodioxane model 12′a, from which was isolated Diels–Alder product 15; compound 15 could be produced via first producing either 14, the β-5 dimer of coniferyl alcohol, followed by Diels–Alder coupling with quinone 9, or via Diels–Alder product 13a followed by radical coupling with further coniferyl alcohol 1. Methylation of benzodioxane 12′a produced the benzodioxane 12′, which is an isomer of 12. In other words, the Diels–Alder reaction product, benzodioxane 12, that resembles a radical coupling product is not the same product 12′ that is produced by radical coupling (followed by simple phenol-methylation). Compounds are colored green to show the units sourced from the cinnamyl alcohol 1 (but dark blue for the second coniferyl alcohol 1 moiety added) and magenta from the quinone 9 or catechol 11; black bonds are those created in the [4+2]-cycloaddition reaction or from the subsequent internal trapping of the diketone by the γ-OH; the bold black bond in 12′a, 12′, 14 and 15 is the bond formed during radical coupling of coniferyl alcohol 1, and the gray bonds are from the internal cyclization (rearomatization) of the quinone methide intermediate. | ||
Initial discovery of a Diels–Alder product from an attempted in vitro radical cross-coupling reaction between coniferyl alcohol and methyl 5-hydroxyvanillate
Early in the process of characterizing lignins in COMT-deficient plants we attempted to make compounds to authenticate the new benzodioxane units being discovered.22–24,26,29,42–49 We sought the benzodioxane product 12′a (Fig. 3) as an intermediate compound to synthesize such a benzodioxane lignin model compound.42 By cross-coupling coniferyl alcohol 1 with methyl 5-hydroxyvanillate 11, we also isolated (unreported at the time) a modest (5%) yield of a product that had as many as nine HMBC NMR correlations to some of its protons – see later in Fig. 5 and 7. We deduced this product to be the trimer 15 (Fig. 3), as reported here (below) for the first time, a Diels–Alder [4+2]-cycloaddition product. Two possible reaction pathways, which cannot be distinguished here, could lead to this product. Coniferyl alcohol 1 could directly undergo a Diels–Alder reaction with quinone 9 (formed by in situ oxidation of catechol 11 under the dehydrogenation conditions) to form compound 13a which could then subsequently chain-elongate to 15via radical coupling as usual between the phenolic radical of 13a and a further radical from coniferyl alcohol 1. Alternatively, two coniferyl alcohol 1 monomers could first dimerize via radical coupling to the β-5 (phenylcoumaran) dehydrodimer 14, which could then subsequently act as a dienophile in a Diels–Alder reaction with diene 9 to produce 15. Compound 13a was not isolated from the mixture but that doesn't discount its possible intermediacy. We assume, but did not verify, that higher oligomers of coniferyl alcohol that, like compound 14, retain the cinnamyl alcohol end-unit, could also undergo the Diels–Alder reaction with diene 9 to form higher-molecular-mass products. We discuss the nature of the trimer 15 and the production of benzodioxanes from either radical coupling or Diels–Alder cyclization below after first examining the simpler reaction depicted in the top line of Fig. 3. | ||
| Fig. 4 COSY and NOESY spectra of compound 13 (500 MHz, in CDCl3). (A) COSY spectrum highlighting correlations between protons 2′ (red) and β (cyan). (B) NOESY spectrum highlighting correlations between protons 2′ and 6 and/or 2 (red), and between protons α and γ1 (cyan); negative contours, mainly associated with the diagonal and devoid of useful information, are in a light gray to deemphasize them. | ||
 | ||
| Fig. 5 1H–13C correlation spectra of compound 13 (500 MHz, in CDCl3). HSQC (red contours, and with assignments) and HMBC spectra (black) showing 3-bond correlations of protons 6′ (yellow highlighting) and α (green highlighting) to carbon 2′ that are useful for defining the stereochemistry. Bicyclo[2.2.2]octanes (including 13 here) from Diels–Alder reactions have HMBC correlations between a proton and carbons 2- or 3-bonds separated that initially seem to be too numerous to be physically possible, but are in fact beautiful signatures of such structures. For example, as shown by the green and yellow shading, proton α correlates with nine carbons: 6′, β, γ, 3′, 2, 6, 1, 2′, and 4′ (in chemical shift order); the correlation to 4′ is too distant to be shown in this plot region (but is present). Similarly, proton β correlates with five carbons: 6′, α, 3′, 1′, and 1; the 2′ proton correlates with 5 carbons: 6′, 1′, 5′, 7′, and 4′; and proton 6′ correlates with 7 carbons: β, α, 1′, 5′, 2′, 7′, and 4′. Contours colored light gray are from residual 1-bond correlations (split by the 1-bond 1H–13C coupling constant) from some peaks due to deviation from the set average value (145 Hz); darker gray contours are from minor impurities. The structure below shows the complex color-coded HMBC correlations from protons in the oxatricyclo ring to carbons within 3-bonds, also confirming its regiochemical assignment of compound 13. | ||
 | ||
| Fig. 6 Diels–Alder reactions and products from 4-O-methylconiferyl alcohol 6 and the quinone 9 from methyl 5-hydroxyvanillate 11. Two so-termed ‘inverse electron-demand’ (because the diene is electron-deficient and the dienophile is electron-rich) Diels–Alder reactions are possible. (A) The conventional Diels–Alder reaction in which the diene itself acts as the diene component. Because of the electronic requirements (see text), there is a strongly favored regiochemical possibility, producing 13 (and its optical isomer). (B) The ‘hetero-Diels-Alder’ reaction in which the o-benzoquinone acts as the diene. To satisfy the electronic requirements, the regiochemistry, if it were described as resulting from radical coupling, appears as a β-O-5 product 12; because of the concerted nature of the reaction, strictly trans-benzodioxane 12 is produced. The regiochemistry and the trans-only nature of the benzodioxane ring are therefore significantly different from the outcome with radical coupling, in which only the β-O-4 coupling product 12′ (Fig. 3, and see later in Fig. 8) is produced, and in which the ring-closure from the quinone methide intermediate allows both trans- and cis-benzodioxane rings to form, admittedly with the cis-ring being minor (∼5%). Modeling doesn't allow a prediction of a favored benzodioxane as the Gibbs free energy (GFE) levels are similar (Fig. S1†); lower GFEs are predicted for both benzodioxanes 12 and 12′ than for the conventional Diels–Alder product 13, also proving to be of limited value given that the latter is the major product. Note: In the structures on the left (and also in the orientational figures in the middle), the magenta arrows show the conventional concerted Diels–Alder electron-pushing with the anticipated (and observed) regiochemistry from electronic considerations, whereas the dashed gray arrows indicate (part of) the ionic rendition – a useful tool for predicting the reaction regiochemistry [see, for example, https://en.wikipedia.org/wiki/Diels–Alder_reaction]. The full range of potential products is shown in Fig. S1.† | ||
 | ||
| Fig. 7 1H–13C correlation spectra of compound 15 (700 MHz, in acetone-d6). HSQC (red contours, and with assignments) and HMBC spectra (black) showing (with green highlighting) correlations of protons 6′ and Bα to carbon 2′ that are useful for defining the stereochemistry. Bicyclo[2.2.2]octanes (including 15 here) from Diels–Alder reactions have HMBC correlations between a proton and carbons 2- or 3-bonds separated that initially seem to be too numerous to be physically possible, but are in fact a beautiful signature of such structures. For example, as shown by the green shading, proton Bα correlates with nine carbons: 6′, Bβ, Bγ, 3′, B2, B6, B1, 2′, and 4′ (in chemical shift order); the correlation to 4′ is too distant to be shown in this plot region but is clearly seen in the data. Similarly, proton Bβ correlates with six carbons: 6′, Bα, Bγ (weak, and below the level drawn in the figure), 3′, 5′, and B1; the 2′ proton correlates with 5 carbons: 6′, 5′ (weak, and below the level drawn in the figure), 1′, 7′, and 4′; and proton 6′ correlates with 7 carbons Bβ, Bα, 5′, 1′, 2′, 7′, and 4′. Contours colored light gray are from residual 1-bond correlations (split by the 1-bond 1H–13C coupling constant) from some peaks due to deviation from the set average value (145 Hz); darker gray contours are from minor impurities. The structure below shows the complex color-coded HMBC correlations from protons in the oxatricyclo ring to carbons within 3-bonds, also confirming its regiochemical assignment of compound 15. | ||
Diels–Alder products from the coupling of 4-O-methylconiferyl alcohol and the quinone from methyl 5-hydroxyvanillate
After the observation that the Diels–Alder product 15 was produced in our attempts to cross-couple coniferyl alcohol 1 and methyl 5-hydroxyvanillate 11, we more carefully examined the pathway and products from the reactions between a simpler phenol-protected monolignol, 4-O-methylconiferyl alcohol 6 (that could not be involved in radical coupling reactions because it lacks the requisite phenol), and the quinone 9 (Fig. 3). The quinone 9 was therefore generated explicitly from methyl 5-hydroxyvanillate 11 and isolated before the reaction.Reaction of quinone 9 with 6 at room temperature produced a mixture of four main products (Fig. 3, top row) for which structures could readily be determined by NMR analysis. In addition to the starting material 6, one product was 4-O-methylconiferaldehyde (3,4-dimethoxycinnamaldehyde) 10, with its diagnostic aldehyde peak at 9.65 ppm in the 1H NMR spectrum. This compound derived from the oxidation of compound 6 by the quinone 9, as corroborated by also finding the reduction product, catechol 11. The third product was the 1,4-benzodioxane 12. In the 1H NMR spectrum, the α and β proton peaks were observed at 5.01 and 4.07 ppm (Fig. 8), similarly to those found in some 1,4-benzodioxane structures in lignin and neolignans, and analogously to benzodioxanes prevalent in the recently discovered C-lignin and 5H-lignin in certain seedcoats.23–27 This compound 12 was determined to be a trans-stereoisomer by the coupling constant between protons α and β (J = 8.17 Hz); the cis-isomer of 12 cannot be formed via a concerted Diels–Alder reaction with trans-4-O-methylconiferyl alcohol 6 as the dienophile. Without the presence of a phenolic-OH on compound 6, it was concluded that this benzodioxane structure 12 had to have been formed by the Diels–Alder reaction between the diketone of the o-benzoquinone 9 and the alkene in 6, i.e., that quinone 9 is capable of reacting as a diene in two senses; in this case it is the 1,2-diketo system that is the diene in a ‘hetero-Diels–Alder’ reaction.39,40 However, benzodioxane 12 was a regioisomer of the analogous product formed by radical coupling – see below.
 | ||
| Fig. 8 1H–13C correlation spectra of the benzodioxane 12 from the Diels–Alder reaction between (A) 4-O-methylconiferyl alcohol 6 and the quinone 9 from methyl 5-hydroxyvanillate 11, and (B) of the benzodioxane 12′ from the radical coupling reaction between coniferyl alcohol 1 and methyl 5-hydroxyvanillate 11 followed by simple phenolic methylation to produce the compound analogous to 12 and allow the determination that these are isomers and are not the same products. HSQC (red contours and assignments) spectra are overlaid (from separate spectrum/experiments). HMBC spectra (black correlations) show (with green highlighting) (A) correlations of proton β to carbon 5′ and proton α to carbon 4′ that define the regiochemistry, and (B) correlations of proton α to carbon 5′ and proton β to carbon 4′ that define its different regiochemistry. Some correlations required vertical expansion (by the factor indicated beside the box) to be seen on the current plot. Other correlations aid in assignments. Of major significance are the following points. (A) This benzodioxane 12 is the product of apparent β-O-5-coupling, but the mechanism is in fact [4+2]-cycloaddition. As shown by the green shading, proton α correlates with six carbons: γ, β, 2, 6, 1, and, importantly, 4′ (in chemical shift order). Similarly, proton β correlates with three carbons: α, 1, and, importantly, 5′. (B) The radical coupling benzodioxane 12′ is the product of β-O-4-coupling, as is usual for coupling of a monolignol with a phenolic. As shown by the green shading, proton α correlates with six carbons: γ, β, 2, 6, 1, and, importantly, 5′ (in chemical shift order). Similarly, proton β correlates with four carbons: γ, α, 1, and, importantly, 4′, establishing the β-O-4-coupling. Note that the cis-isomer in the benzodioxane ring system, cis-12′, is readily seen, in the 1D proton and carbon projections, and as highlighted for the proton α and β correlations by the dark blue contours; the cis-ring isomer is typically seen at about the 5% level in such radical coupling products. For both A and B, contours colored light gray are from residual 1-bond correlations (split by the 1-bond 13C–1H coupling constant) from some peaks due to deviation from the set average value (145 Hz) or are from minor impurities. | ||
The final product was the expected oxatricyclo Diels–Alder product 13 (Fig. 3, 6, and S2†). In this case, it is the conjugated diene itself that has acted as the diene in the Diels–Alder reaction, quite analogously to reported reactions of galloyl quinones.36 The structure was determined by NMR analysis using normal assignment principles from the typical array of 1D and 2D NMR experiments – 1H (Fig. S2†), 13C (Fig. S4A†), COSY (Fig. 4A), NOESY (Fig. 4B), HSQC (Fig. 5, red contours), and HMBC (Fig. 5, black contours). Confirmation was from reported 1H NMR data of a compound that has the common oxatricyclo structure.50 In the COSY spectrum (Fig. 4A), proton 2′ correlated with protons α and 6′, but not with the β proton. Proton β correlated with protons α, 6′, and γ, but not with the 2′ proton. These data strongly suggest that protons 2′ and β are the most separated in the proton coupling network. In the NOESY spectrum (revealing protons that are spatially close, Fig. 4B), proton 2′ correlated with protons 2 and 6 (as well as the 3′-OMe). Proton α correlated with proton γ1 but not with proton γ2 (Fig. 4B), observations that are also consistent with the structure 13 drawn. Further confirmation was provided by the carbon 2′ correlations in the HMBC spectrum that are generally limited to 3-bonds (Fig. 5); Carbon 2′ correlated with protons 6′ and α, but not with proton β. HMBC spectra are particularly diagnostic of these types of Diels–Alder products (such as 13) because of the incredible number of correlations possible between protons and carbons that are within 3-bonds of each other; proton α, for example, correlates with nine carbons, 6′, β, γ, 3′, 2, 6, 1, 2′, and 4′ – all but the 4′ correlation (that was nevertheless observed, as indicated) are shown in Fig. 5.
Rationalization of Diels–Alder coupling to produce a single isomer of product 13
The Diels–Alder possibilities for reaction of the diene 9 with dienophile 6 are referred to as being ‘inverse electron-demand’ Diels–Alder reactions because the diene is electron-deficient and the dienophile is electron-rich, well known reactions that are opposite to the originally reported ‘conventional’ or ‘normal electron-demand’ reaction.51,52 In the case of the Diels–Alder reaction in which the conjugated double-bonds (and not the quinone system) in o-benzoquinone 9 act as the diene, four products from the cycloaddition with dienophile 6, two bicyclo isomers and two oxatricyclo isomers, can be drawn (Fig. S1†). However, the only Diels–Alder product is that anticipated and rationalized if the reaction is considered to be ionic (as most readily visualized via the gray arrows and resonance forms drawn in Fig. 6A), i.e., the oxatricyclo product 13. The concerted Diels–Alder cycloaddition reaction is shown by the magenta arrows; the alternative orientations (Fig. S1B–D†) simply don't comport with the electronics. NMR confirms this structure 13 as the main/sole Diels–Alder product of this type obtained, clarifying that the Diels–Alder reaction can occur using the diene in the quinone structure 9 and the double-bond in the 4-O-methylconiferyl alcohol 6 as the dienophile. As noted above, the Diels–Alder reaction in which the diketone in 9 can act as the diene is also produced, as discussed below.Molecular modeling can also be used to help rationalize the products 12 and 13 observed from the Diels–Alder coupling of 4-O-methylconiferyl alcohol 6 and quinone 9, Fig. 3 and 6. Firstly, the proposed inverse electron-demand nature of this Diels–Alder system in which the LUMO of the diene and the HOMO of the dienophile is confirmed by a much smaller gap between these frontier molecular orbitals. Next, per the experimental results, compound 13 is the main Diels–Alder product, with the benzodioxane 12 also detected. Computationally, 12 is the more stable by 8.1 kcal mol−1 (Fig. 6). This relatively large thermodynamic difference is probably due to the retention of two aromatic rings in the benzodioxane. Among the bicyclic and tricyclic Diels–Alder adducts the tricyclic compounds are the most stable, and compound 13 is the most stable among these. Despite the energy calculations suggesting that the benzodioxane 12 is significantly more stable than the oxatricyclo product 13, both products were observed in comparable amounts (with 13 predominating) implying that both of these Diels–Alder reactions can occur between these two substrates, and with roughly similar propensities.
Diels–Alder coupling reactions produce trimer 15
As seen from the NMR data, the product 13 (Fig. 4, 5 and Fig. S2, S4A†) from reaction of 4-O-methylconiferyl alcohol 6 and quinone 9 (Fig. 3) and the product 15 (Fig. 7 and Fig. S3, S4B†) isolated from the reaction of coniferyl alcohol 1 with catechol 11 (via its quinone 9 and involving at least one coniferyl alcohol radical to produce the β-5 moiety), share significant structural similarities, particularly with the oxatricyclo moiety. In each case, the regiochemistry of the reaction can be defined from particularly the HMBC correlations observed, Fig. 5 and 7, and are supported by NOESY correlations. Particularly striking again are the HMBC correlations between proton α in 13 and proton Bα in 15 with all nine carbons within 3 bonds of it in each case (Fig. 5 and 7).It is perhaps evident in the 1D projections on Fig. 7, but is more clearly revealed in the 1D spectra in Fig. S3 and S4B,† that most of the proton and carbon multiplets in the spectra of 15 are split into two resolved sets each. This is because there are two isomers in essentially equal proportion, but it might not be obvious how these arise. There are clearly two stereocenters (at Aα and Aβ) in the phenylcoumaran moiety, and five in the oxatricyclo system. However, because these are all constrained relative to each other in each moiety, it is equivalent to having just one stereocenter in the phenylcoumaran and one in the oxatricyclo system, but these are independent of each other, so they could be like RR/SS and RS/SR isomers (as are syn and anti, or threo and erythro isomers, for example). As shown in Fig. 7, it appears that the phenylcoumaran moiety in 15 is in the trans-configuration (because there is only one real isomer of the dimer 14, Fig. 3) such that αβ is either RS or SR. The stereochemistry of the oxatricyclo system of 15 in the order 3′, Bα, Bβ, 6′ appears to be SRRR/RSSS. The two isomers combined would be the two pairs of optical isomers RSSRRR/SRRSSS and SRSRRR/RSRSSS. Again, it is easiest to equate this compound to having a single optical center in each moiety, as with typical threo/erythro or syn/anti isomer pairs. Incidentally, we have previously seen this NMR separation of isomers in which the two optical centers, even when separated by many bonds within the molecule, are in separate moieties, as will be revealed separately (unpublished).53
Benzodioxanes resulting from hetero-Diels–Alder vs. radical coupling
Since their original discovery in lignins,42–44,54 benzodioxane units are now well-known to result from lignification in which catechyl lignin monomers such as caffeyl alcohol or 5-hydroxyconiferyl alcohol are involved; as noted above, certain seedcoat lignins are derived solely from such monomers resulting in lignins that are almost exclusively composed of benzodioxane units.23–27 In lignins and in model reactions, they have always been described to be the benzodioxanes derived from 4-O-β-coupling of the catechol with a monomer. Although theoretically possible, the 5-O-β-coupled benzodioxane isomers have not been described to occur. The rationalization is that the product derives from the more conjugated (and therefore more stable) 4-O-radical, even though radical transfer to produce the presumably more reactive 5-O-radical would be energetically trivial.The benzodioxane 12 produced by the Diels–Alder reaction between 4-O-methylconiferyl alcohol 6 and the o-benzoquinone 9 from methyl 5-hydroxyvanillate 11 can be described as a 5-O-β or β-O-5 product (were it to derive from radical coupling, Fig. 3). This structure was particularly evidenced by the 3-bond HMBC correlations between proton α and carbon 4′, and between proton β and carbon 5′ (Fig. 8A, the carbon assignments of which can be quite unambiguously assigned by first principles, i.e., without resorting to chemical shift arguments, by analysis of the combined correlation data from the various experiments). To confirm the structure of the radical coupling product 12′a (Fig. 3) between coniferyl alcohol 1 and methyl 5-hydroxyvanillate 11, we methylated its phenolic-OH using methyl iodide, thereby creating the isomer 12′ (Fig. 3). In contrast to its Diels–Alder-produced counterpart 12, the HMBC correlations in 12′ were between proton α and carbon 5′, and between proton β and carbon 4′ (Fig. 8B), establishing 12′a firmly as the 4-O-β-coupling product.
Clearly, the Diels–Alder reaction and the radical coupling pathway produced opposite geometric isomers. Another observation was that radical coupling produced some 5% of the cis-benzodioxane ring (Fig. 8B) whereas we noted (and can fully rationalize) only the trans-benzodioxane from the Diels–Alder reaction. Although we cannot unambiguously claim that there was no cis-isomer of 12, it is logical that there was none as the stereochemistry of the benzodioxane ring is fully defined by the concerted [4+2]-cycloaddition reaction. The exciting realization is that we now have two product structures (the β-O-5-benzodioxane as in 12, and the oxa-tricyclo unit as in 13) that, if found in lignin, would be diagnostic for the occurrence of Diels–Alder reactions during lignification, and perhaps the cis-(4-O-β)-benzodioxane is a ‘marker’ for radical coupling. That said, because of the similarity of the spectra and the closely matched correlations in the HSQC spectra, Fig. 8, we don't expect to be able to resolve and distinguish the two possibilities in the broader HSQC spectra from actual lignins.
Are Diels–Alder reactions produced in lignification that involves catechyl monomers?
Plants’ utilization of catechyl monomers in lignification has been noted in mutants or transgenics deficient in O-methyltransferases,20,43 as we've reported in a number of reviews.3,4,11,12,17,18,55,56 In two independent studies, Arabidopsis was engineered to derive its lignin from 5-hydroxyconiferyl alcohol.57,58 Whereas the levels of 5-hydroxyguaiacyl (5H) units in the lignin of wild-type Arabidopsis plants remained below the detection limit, lignin of engineered lines contained up to ∼70% 5H units. More recently, specialized C-lignins derived from 100% caffeyl alcohol,23–27 or 5H-lignins from 100% 5-hydroxyconiferyl alcohol,24 have been discovered. These lignins are the most likely to involve the production of quinones by oxidation/dehydrogenation and therefore have the most chance of being produced via Diels–Alder reactions. We don't have the exact models to provide comparative data for the products that would be expected in these lignins and predicting shifts from our model to the C- or 5H-lignin does not seem to be particularly reliable. The most diagnostic way for the oxatricyclo products (as in compounds 13 and 15) is to look for the diagnostic set of nine HMBC correlations from proton α (as in Fig. 5 and 7) along with the five correlations from proton β, but HMBC experiments, even on clean lignins, usually have only weak correlations at best for minor structures. The more sensitive way is to attempt to find the set of HSQC correlations consistent with these Diels–Alder structures (from the best predicted shifts). In searching through all of the NMR data from our publications on C- and 5H-lignins noted above, we could not find any convincing evidence for the presence of Diels–Alder products in any of them.Can we leverage the observation that the benzodioxane regiochemistry is different to provide further evidence for radical coupling vs Diels–Alder mechanisms in lignification? Again, the HMBC approach is not very sensitive, and the α- and β-H/C correlations are not so different that they would be expected to be well-separated in HSQC spectra, particularly when considering that the peaks in lignin spectra are significantly broader than in the models. Clearly, better spectra, and HMBCs are going to be necessary to seek benzodioxane regioisomers and the other diagnostic Diels–Alder products, or we need to look for such products by more sensitive methods, such as by HPLC-MS (although it is not obvious how to release them efficiently from the polymer; we suspect that hydrogenolysis may be the best way as it fully cleaves benzodioxane structures26), but so far we simply do not see any evidence for either. What is clearly evident is the presence of the cis-benzodioxane rings for C- and 5H-lignins,23–27 as well as in piceatannol lignins,28 an observation again consistent with radical coupling but not Diels–Alder reactions.
We therefore have to accommodate the current lack of evidence for Diels–Alder coupling products produced between a hydroxycinnamyl alcohol, whether etherified or not, and an o-benzoquinone that might logically be produced from the catechyl units in such systems. This was particularly sobering after identifying Diels–Alder products in our model reactions, and the tantalizing prospect that it could reveal a new mechanism in lignification – a mechanism that would allow chain-extension at the phenolic and even at the non-phenolic ends of the lignin polymer chain. However, this absence of verifiable structures derived via Diels–Alder coupling in lignins has particularly important ramifications regarding lignification. First, lignification, the process of growing lignin polymer chains from hydroxycinnamyl alcohol (or other) monomers, needs to be recognized even more strongly now as being constrained to, rather strictly, radical coupling reactions. Second, coupling is exclusively via the radical produced from oxidative ‘radicalization’ of the 4-OH (even if that may be in simple equilibrium with other phenolic radicals); only products from the 4-O radical are observed from the 3,4-dihydroxyphenyl monomers such as caffeyl alcohol and 5-hydroxyconiferyl alcohol. Third, after radical coupling, rearomatization can occur via internal trapping by the 3-OH for catechyl units and the 5-OH for 5-hydroxyguaiacyl units, whereby the benzodioxane rings are primarily trans (E) with minor cis (Z) ring-isomers. Additionally, although catechyl units might be expected to produce quinones under the oxidative (dehydrogenative) conditions present in the cell wall during lignification, all lignin units identified to date comport with single-electron oxidation processes and radical coupling and not from these two-electron-oxidized species and their possible [4+2]-cycloaddition (Diels–Alder) reactions.
The latter point, that quinones don't appear to be involved to any significant extent in lignification, is important because the conversion of catechols to quinones has at times been strongly advocated in the literature.33,34 The notion that catechols are problematic for lignification has been mitigated by the repeated observations of lignins from such catechols in a range of viable natural, mutant, and transgenic plants, in which the monomers clearly incorporate into the polymer by the canonical radical coupling processes, as we have reviewed.3,4,12,35 Here we find that quinones can form, at least from strongly conjugated species like methyl 5-hydroxyvanillate 11, in simulated lignification processes (Fig. 3), from which they may react to produce dimers and higher oligomers. The lack of evidence for Diels–Alder and other quinone reactions complicating the polymer structure from a variety of lignins from plants utilizing high levels of catechyl monomers, however, suggests that processes other than radical coupling are at the very least minor. We remain open to the possibility that trace quantities of such Diels–Alder products may be detected in lignins at some point, but there is currently no evidence that cycloaddition reactions represent a significant pathway; small unassigned NMR 1H–13C correlation peaks in some lignins certainly remain to be elucidated. Additional evidence has come recently from the manner in which piceatannol, a catechyl stilbene from outside the monolignol biosynthetic pathway, incorporates into lignins; again, the product array noted is entirely compatible with radical-coupling arguments.28–31,59,60 A credible explanation for an absence of Diels–Alder products and mechanisms is likely premature, but it may be because of the exact nature of the substrates themselves, the caffeyl alcohol and the 5-hydroxyconiferyl alcohol that, even as quinones, are not as electron deficient as the methyl 5-hydroxyvanillate 11 examined here, or perhaps simply that radical coupling reactions simply out-compete the disproportionation reaction required to generate the quinone (for which two catechyl radicals need to encounter each other) in the cell wall. Regardless, and as evidence has already shown (above), catechols do undergo (one-electron oxidation and) efficient radical coupling to produce either copolymeric or homopolymeric lignins.
Conclusions
Two sets of reactions are shown here to operate if catechyl structures can be oxidized to their o-benzoquinones, (Fig. 3, and as summarized in Fig. 9). First, the quinone can oxidize a cinnamyl alcohol to a cinnamaldehyde, itself being reduced (back) to the catechol (Fig. 3). More importantly in the context of this paper, the quinone can function with either the 1,2-diketone or the conjugated diene as the diene component of a Diels–Alder reaction, with any cinnamyl alcohol unit, including those that are not free-phenolic, functioning as the dienophile (Fig. 9). At least with the components used here (Fig. 3), quinone 9 and dienophile 6, the major Diels–Alder products were single isomers (although certainly as enantiomeric pairs) of the benzodioxane 12 and the oxatricyclo product 13 (Fig. 3). The key implication is that catechols such as caffeyl alcohol and 5-hydroxyconiferyl alcohol can first oxidize to o-quinones and subsequently may undergo either/both of two Diels–Alder reactions with general cinnamyl alcohols, including the monolignols, to potentially produce benzodioxane and oxatricyclo products. Importantly, a free-phenolic cinnamyl alcohol unit is not required, so the Diels–Alder mechanism would provide a way of extending the lignin polymer from the non-phenolic ‘starting end’ of a lignin chain, a pathway that is not typically possible via the usual radical-coupling pathways of lignification. As we can find no evidence of Diels–Alder products in natural lignins, even in lignins derived solely from catechyl monomers, we are forced to contend that they are not significant in the lignification of plants examined to date. As it stands, all product units seen in lignins of diverse types therefore comport with their being derived solely from radical coupling in single-electron oxidation processes. No further modification is therefore required at this point to the existing theory of lignification as a process of combinatorial radical coupling of phenols, with lignins deriving from a variety of phenolic monomers including the many examples now of non-canonical monomers.3,6,8,11–14,17,28–31,49,56,58–81 | ||
| Fig. 9 Summary of radical coupling vs in vitro Diels–Alder pathways and products. (A) Radical coupling of a monolignol with a general catechyl lignin end-unit; (B) Diels–Alder reaction of a general cinnamyl alcohol unit (usually on the starting end of a polymer chain) with a quinone. Note that the radical coupling pathway produces benzodioxanes 12′ from β-O-4 coupling, whereas the Diels–Alder coupling produces benzodioxanes 12 that would be described as β-O-5-coupled were they derived from radical coupling. | ||
Experimental
Materials
ortho-Chloranil, methyl gallate, sodium borohydride were purchased from Acros Organics. Acetone, acetone-d6, chloroform-d, 3,4-dimethoxycinnamic acid, ethyl chloroformate, toluene, and THF were purchased from Sigma-Aldrich (St Louis, MO, USA).Methods
The mass spectra were acquired in the range of 50–1000 m/z units on a Bruker Daltonics quadrupole time-of-flight (QTOF) Impact II instrument (Bruker, Billerica, MA, USA), operating in negative-ion mode [ESI−]. The nebulizing gas pressure was set at 4 bar, the drying gas flow at 8 L min−1, the drying gas temperature was at 210 °C, and the capillary voltage was set at 3.50 kV. Sodium formate was used as a calibration standard.
COSY (homonuclear correlation spectroscopy) at 500 MHz: cosygpmfqf pulse program, acquired from 10 to 0 ppm in F2 (1H) with 2k data points (acquisition time, 205 ms) and 10 to 0 ppm in F1 (1H) with 512 increments (F1 acquisition time, 102 ms) of 2 scans with a 1.5 s interscan delay, 30.5 min total acquisition time, processed to 2k × 1k using sine-squared apodization in each dimension.
NOESY (nuclear Overhauser effect spectroscopy) at 500 MHz: noesyph pulse program, acquired from 10 to 0 ppm in F2 (1H) with 2k data points (acquisition time, 205 ms) and 10 to 0 ppm in F1 (1H) with 256 increments (F1 acquisition time, 25.6 ms) of 8 scans with a NOESY mixing time of 300 ms, and a 2 s interscan delay, 1 h 26 min total acquisition time, processed to 2k × 1k using Gaussian apodization (GB = 0.001, LB = −0.1) in F2 and cosine-squared F1.
HSQC (Heteronuclear single-quantum coherence) for compounds 12 and 13 at 500 MHz: hsqcetgpsi2.2 pulse program, acquisition from 10 to 0 ppm in F2 (1H) with 2k data points (acquisition time, 200 ms) and 200 to 0 ppm in F1 (13C) with 400 increments (F1 acquisition time, 8 ms) of 2 scans with a 1 s interscan delay; the d24 delay was set to 0.89 ms (1/8J, J = 140 Hz) optimized for all multiplicities, 17 min total acquisition time, processed to 2k × 1k using Gaussian apodization (GB = 0.001, LB = −0.1) in F2 and cosine-squared in F1.
HSQC for compounds 12′ and 15 at 700 MHz: hsqcetgpsi2.2 pulse program, acquisition from 11.65 to −0.65 ppm in F2 (1H) with 3448 data points (acquisition time, 200 ms) and 215 to −5 ppm in F1 (13C) with 618 increments (F1 acquisition time, 8 ms) of 4 scans with a 1 s interscan delay, the d24 delay was set to 0.89 ms (1/8J, J = 140 Hz) optimized for all multiplicities, 51 min total acquisition time, processed to 4k × 1k using Gaussian apodization (GB = 0.001, LB = −0.1) in F2 and cosine-squared in F1.
HMBC (heteronuclear multiple-bond correlation) for compounds 12 and 13 at 500 MHz: hmbcgplpndqf pulse program, acquisition from 10 to 0 ppm in F2 (1H) with 4k data points (acquisition time, 410 ms) and 200 to 0 ppm in F1 (13C) with 400 increments (F1 acquisition time, 16 ms) of 4 scans with a 1 s interscan delay, long-range J-coupling evolution time d6 of 80 ms (JLR = 6.25 Hz), 40.5 min total acquisition time, processed to 4k × 1k using Gaussian apodization [GB = 0.195 (=d6/AQ), LB = −20)] in F2 and sine in F1.
HMBC for compounds 12′ and 15 at 700 MHz: hmbcgplpndqf pulse program, 11.65 to −0.65 ppm in F2 (1H) with 4096 data points (acquisition time, 238 ms) and 215 to −5 ppm in F1 (13C) using non-linear sampling (NUS, 25%) with 1160 increments (F1 acquisition time, 30 ms) of 64 scans with a 1 s interscan delay, long-range J-coupling evolution time d6 of 80 ms (JLR = 6.25 Hz), 5 h total acquisition time, NUS processed to 4k × 2k using Gaussian apodization [GB = 0.337 (=d6/AQ), LB = −10)] in F2 and sine in F1.
The rest (90 mL) of the starting mixture was used for the typical peroxidase-mediated free-radical coupling reaction. Thus, 13 mg of urea-H2O2 was added while stirring, followed by adding 0.5 mg (dissolved in 1 mL water) of horseradish peroxidase (EC 1.11.1.7, 181 purpurogallin units per mg solid, type II, Sigma-Aldrich). The mixture was stirred for 20 min, and then was extracted with an equal volume of EtOAc (90 mL, 2×), the combined EtOAc solution was washed with saturated NH4Cl solution and processed as above. About 6 mL of the EtOAc were evaporated using rotary evaporation under reduced pressure. The resulting product mixture was silylated using N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) and subjected to GC-MS analysis.
GC conditions: injector temperature 250 °C; initial temperature 100 °C, hold for 1 min; ramp at 5 °C min−1 to 180 °C, hold for 1 min; then ramp at 10 °C to 300 °C, and hold for 8 min. MS detector: ion source temperature 300 °C; EI mode. GC column: ZB-5HT, length 15 m; thickness 0.25 μm; diameter 0.25 mm.
:hexane = 1:2) to obtain 4-O-methylconiferyl alcohol 6 (1.639 g, 8.44 mmol). NMR (500 MHz, CDCl3) δH: 3.88 (s, 3H, 3-OMe), 3.90 (s, 3H, 4-OMe), 4.31 (d, J = 5.86 Hz, 2H, γ), 6.25 (dt, J = 5.98, 15.79 Hz, 1H, β), 6.55 (d, J = 15.86 Hz, 1H, α), 6.82 (d, J = 8.17 Hz, 1H, 5), 6.92 (dd, J = 1.90, 8.17 Hz, 1H, 6), 6.94 (d, J = 1.90 Hz, 1H, 2); δC: 55.8 (OMe), 55.9 (OMe), 63.8 (γ), 108.7 (2), 111.0 (5), 119.7 (6), 126.4 (β), 129.6 (1), 131.1 (α), 148.8 (3), 148.9 (4).
Diels–Alder benzodioxane product trans-12. NMR (500 MHz, CDCl3) δH: 3.55 (bm, Hz, 1H, γ1), 3.81 (m, 1H, γ2), 3.897 (s, 3H, OMe), 3.899 (s, 3H, OMe), 3.902 (s, 3H, OMe), 3.904 (s, 3H, OMe), 4.07 (ddd, J = 8.15, 3.9, 2.8 Hz, 1H, β), 5.01 (dd, J = 8.15 Hz, 1H, α), 6.90 (d, J = 8.25 Hz, 1H, 5), 6.94 (d, J = 2.0 Hz, 1H, 2), 7.01 (dd, J = 2.0, 8.25 Hz, 1H, 6), 7.24 (d, J = 1.9 Hz, 1H, 2′), 7.38 (d, J = 1.9 Hz, 1H, 6′); δC: 52.2 (7′-OMe), 55.9 (3′-OMe), 56.0 (3/4-OMe), 56.2 (4/3-OMe), 61.6 (γ), 76.7 (α), 77.9 (β), 105.5 (2′), 110.2 (2), 111.2 (5), 111.8 (6′), 120.4 (6), 122.3 (1′), 127.9 (1), 137.7 (4′), 143.2 (5′), 148.8 (3′), 149.3 (4*), 149.8 (3*), 166.7 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); * implies assignments may be reversed. LC-QTOF-MS: m/z [M − H]− calculated for C20H21O8: 389.1236; found: 389.1180.
O); * implies assignments may be reversed. LC-QTOF-MS: m/z [M − H]− calculated for C20H21O8: 389.1236; found: 389.1180.
Diels–Alder product 13. NMR (500 MHz, CDCl3) δH: 7.06 (dd, J = 1.13, 2.15 Hz, 1H, 2′), 6.77 (d, J = 8.20 Hz, 1H, 5), 6.58 (dd, J = 2.20, 8.20 Hz, 1H, 6), 6.56 (d, J = 2.20 Hz, 1H, 2), 4.38 (dd, J = 3.25, 8.30 Hz, 1H, γ2), 4.05 (dd, J = 2.15, 4.45 Hz, 1H, 6′), 4.02 (d, J = 8.30 Hz, 1H, γ1), 3.85 (s, 6H, 7′-OMe + 4-OMe), 3.81 (s, 3H, 3-OMe), 3.57 (s, 3H, 3′-OMe), 3.43 (t, J = 1.50 Hz, 1H, α), 2.85 (m, J = 1.6 Hz, 1H, β); δC: 200.47 (4′), 163.89 (7′), 148.44 (3), 148.41 (4), 138.45 (2′), 132.25 (1′), 131.44 (1), 121.17 (6), 112.42 (2), 110.74 (5), 95.65 (5′), 86.78 (3′), 74.39 (γ), 55.81 (4-OMe), 55.70 (3-OMe), 54.30 (3′-OMe), 52.56 (7′-OMe), 49.67 (α), 47.21 (β), 44.90 (6′). LC-QTOF-MS: m/z [M − H]− calculated for C20H21O8: 389.1236; found: 389.1233.
Benzodioxane product trans-12′. NMR (500 MHz, CDCl3) δH: 3.56 (bm, 1H, γ1), 3.87 (s, 3H, 7′-OMe), 3.89 (s, 6H, 3,4-OMe), 3.91 (bm, 1H, γ2), 3.93 (s, 3H, 3′-OMe), 4.06 (ddd, 1H, J = 2.6, 3.6, 8.2 Hz, β), 4.99 (d, J = 8.2 Hz, 1H, α), 6.89 (d, J = 8.3 Hz, 1H, 5), 6.93 (d, J = 2.0 Hz, 1H, 2), 7.00 (dd, J = 2.0, 8.3 Hz, 1H, 6), 7.23 (d, J = 1.9 Hz, 1H, 2′), 7.37 (d, J = 1.9 Hz, 1H, 6′); δC: 52.1 (7′-OMe), 55.90 (3/4-OMe), 55.92 (4/3-OMe), 56.2 (3′-OMe), 61.3 (γ), 75.9 (α), 78.8 (β), 105.3 (2′), 109.9 (2), 111.1 (5), 112.2 (6′), 120.0 (6), 122.3 (1′), 128.1 (1), 137.1 (4′), 143.9 (5′), 148.4 (3′), 149.3 (3), 149.6 (4), 166.6 (7′). LC-QTOF-MS: m/z [M + Na]+ calculated for C20H22O8Na+: 413.1207; found: 413.1168.
Diels–Alder product 15. [There are two isomers (see Results and Discussion), so most peaks have two entries; for the 13C NMR data, the two shifts are separated by a comma before the assignment; note that we use 3 significant figures only to show that the two peaks were resolved, and the difference between them]. NMR (700 MHz, acetone-d6) δH: 7.59 (s, 0.5H, A4-OH), 7.58 (s, 0.5H, A4-OH), 7.03 (d, J = 2.20 Hz, 0.5H, A2), 7.02 (d, J = 2.20 Hz, 0.5H, A2), 6.95 (m, 0.5H, 2′), 6.94 (m, 0.5H, 2′), 6.87 (d, J = 8.10 Hz, 1H, A6), 6.80 (d, J = 8.10 Hz, 1H, A5), 6.72 (m, 0.5H, B6), 6.66 (m, 0.5H, B6), 6.62 (bd, J = 1.6 Hz, 1H, B2), 6.23 (s, 0.5H, 5′-OH), 6.22 (s, 0.5H, 5′-OH), 5.52 (d, J = 6.9 Hz, 0.5H, Aα), 5.51 (d, J = 6.9 Hz, 0.5H, Aα), 4.33 (dd, J = 3.5, 8.1 Hz, 0.5H, Bγ2), 4.31 (dd, J = 3.5, 8.1 Hz, 0.5H, Bγ2), 4.03 (d, J = 8.10 Hz, 0.5H, Bγ1), 4.02 (d, J = 8.10 Hz, 0.5H, Bγ1), 3.87 (m, 1H, 6′), 3.83 (m, 1H, Aγ2), 3.815 (s, 1.5H, A3-OMe), 3.811 (s, 1.5H, A3-OMe), 3.809 (s, 1.5H, 7′-OMe), 3.788 (s, 1.5H, 7′-OMe), 3.785 (s, 3H, B3-OMe), 3.77 (m, 1H, Aγ1), 3.549 (s, 1.5H, 3′-OMe) 3.547 (s, 1.5H, 3′-OMe), 3.546 (m, 0.5H, Bα), 3.538 (m, 0.5H, Bα), 3.489 (dd, J = 6.3, 6.5 Hz, 0.5H, Aβ), 3.486 (dd, J = 6.3, 6.5 Hz, 0.5H, Aβ), 2.87 (m, 1H, Bβ); δC: 201.20, 201.08 (4′), 164.72, 164.65 (7′), 148.35, 148.34 (A3), 148.29, 148.28 (B4), 147.27, 147.25 (A4), 144.43, 144.36 (B3), 139.49, 139.48 (2′), 134.36, 134.27 (A1), 134.07, 133.93 (B1), 133.51, 133.47 (1′); 129.86, 129.74 (B5), 119.53 (A6), 118.89, 118.81 (B6), 115.63 (A5), 114.95, 114.81 (B2), 110.45, 110.43 (A2), 97.43, 97.37 (5′), 88.49, 88.45 (Aα), 88.20, 88.10 (3′), 74.67, 74.59 (Bγ), 64.77, 64.60 (Aγ), 56.272, 56.265 (B3-OMe), 56.259, 56.251 (A3-OMe), 54.88, 54.80 (Aβ), 54.01, 53.92 (3′-OMe), 52.50, 52.49 (7′-OMe), 50.12, 50.11 (Bα), 48.45, 48.27 (Bβ), 47.01 (6′). LC/Q-TOF MS: m/z [M + Na]+ calculated for C29H30O11Na+: 577.1680; found: 577.1609.
Computational details
All calculations have been performed using Gaussian 16, Revision C.01. Optimizations were completed using the default criteria, with the M06-2X density functional method and the 6-311++G(d,p) basis set with the GD3 empirical dispersion correction. Frequency calculations were also performed to verify the identification of a minimum, as evidenced by the absence of imaginary frequencies and for the determination of thermal corrections for thermodynamic quantities.Author contributions
D. A., F. L., and J. R. originated the project on Diels–Alder reactions of o-benzoquinones; D. A. and F. L. performed the reactions, isolated and purified products and, with J. R., identified and structurally validated all products; R. V., W. B., T. J. E., J. R. and F. L. worked on clarifying misconceptions of lignification; T. J. E. performed the molecular modeling and DFT calculations; Y. T., J. R., and H. K. reanalyzed NMR data of lignins from various prior studies seeking evidence for Diels–Alder products; D. A. produced the original draft manuscript and figures; J. R. finalized the text, produced final figures and ESI;† A. E. and H. K. provided structural analysis data; all authors edited the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. R., F. L., H. K., A. E., and the laboratory operation supporting D. A. were funded by the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science DE-SC0018409). D. A. was supported by JSPS Overseas Research Fellowships. Y. T. acknowledges support from JSPS KAKENHI (#JP20H03044). We thank Dr Steven Karlen for assistance with QTOF-MS analysis. W. B. and R. V. acknowledge support by SBO-FISCH through the ARBOREF project (grant no. 140894). For T. J. E., this work was also made possible in part by a grant of high-performance computing resources and technical support from the Alabama Supercomputer Authority.References
- K. Freudenberg and A. C. Neish, Constitution and Biosynthesis of Lignin, Springer-Verlag, Berlin-Heidelberg-New York, 1968 Search PubMed.
- K. V. Sarkanen and C. H. Ludwig, Lignins: Occurrence, Formation, Structure and Reactions, Wiley-Interscience, New York, 1971 Search PubMed.
- J. Ralph, K. Lundquist, G. Brunow, F. Lu, H. Kim, P. F. Schatz, J. M. Marita, R. D. Hatfield, S. A. Ralph, J. H. Christensen and W. Boerjan, Phytochem. Rev., 2004, 3, 29–60 CrossRef CAS.
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519–546 CrossRef CAS PubMed.
- H. Önnerud, L. Zhang, G. Gellerstedt and G. Henriksson, Plant Cell, 2002, 14, 1953–1962 CrossRef PubMed.
- J. Ralph, M. Bunzel, J. M. Marita, R. D. Hatfield, F. Lu, H. Kim, P. F. Schatz, J. H. Grabber and H. Steinhart, Phytochem. Rev., 2004, 3, 79–96 CrossRef CAS.
- R. D. Hatfield, J. Ralph and J. H. Grabber, Planta, 2008, 228, 919–928 CrossRef CAS PubMed.
- J. Ralph, Phytochem. Rev., 2010, 9, 65–83 CrossRef CAS.
- U. Takahama, Physiol. Plant., 1995, 93, 61–68 CrossRef CAS.
- S. Sasaki, T. Nishida, Y. Tsutsumi and R. Kondo, FEBS Lett., 2004, 562, 197–201 CrossRef CAS PubMed.
- R. Vanholme, K. Morreel, C. Darrah, P. Oyarce, J. H. Grabber, J. Ralph and W. Boerjan, New Phytol., 2012, 196, 978–1000 CrossRef CAS PubMed.
- J. Ralph, C. Lapierre and W. Boerjan, Curr. Opin. Biotechnol., 2019, 56, 240–249 CrossRef CAS PubMed.
- J. C. del Río, J. Rencoret, P. Prinsen, Á. T. Martínez, J. Ralph and A. Gutiérrez, J. Agric. Food Chem., 2012, 60, 5922–5935 CrossRef PubMed.
- W. Lan, J. Rencoret, F. Lu, S. D. Karlen, B. G. Smith, P. J. Harris, J. C. del Río and J. Ralph, Plant J., 2016, 88, 1046–1057 CrossRef CAS PubMed.
- G. Brunow, O. Karlsson, K. Lundquist and J. Sipilä, Wood Sci. Technol., 1993, 27, 281–286 CAS.
- J. Ralph, J. Peng, F. Lu, R. D. Hatfield and R. F. Helm, J. Agric. Food Chem., 1999, 47, 2991–2996 CrossRef CAS PubMed.
- J. Ralph, G. Brunow, P. J. Harris, R. A. Dixon, P. F. Schatz and W. Boerjan, in Recent Advances in Polyphenol Research, ed. F. Daayf, A. El Hadrami, L. Adam and G. M. Ballance, Wiley-Blackwell Publishing, Oxford, UK, 2008, vol. 1, ch. 2, pp. 36–66 Search PubMed.
- J. Ralph and L. L. Landucci, in Lignin and Lignans; Advances in Chemistry, ed. C. Heitner, D. R. Dimmel and J. A. Schmidt, CRC Press (Taylor & Francis Group), Boca Raton, FL, 2010, ch. 5, pp. 137–234, DOI:10.1201/EBK1574444865.
- Y. Matsushita, Y. Oyabu, D. Aoki and K. Fukushima, R. Soc. Open Sci., 2019, 65, 29, DOI:10.1186/s10086-019-1809-1.
- C. Lapierre, M. T. Tollier and B. Monties, C. R. Acad. Sci., Ser. III, 1988, 307, 723–728 CAS.
- Y. Barrière, J. Ralph, V. Méchin, S. Guillaumie, J. H. Grabber, O. Argillier, B. Chabbert and C. Lapierre, C. R. Biol., 2004, 327, 847–860 CrossRef PubMed.
- J. M. Marita, W. Vermerris, J. Ralph and R. D. Hatfield, J. Agric. Food Chem., 2003, 51, 1313–1321 CrossRef CAS PubMed.
- F. Chen, Y. Tobimatsu, D. Havkin-Frenkel, R. A. Dixon and J. Ralph, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1772–1777 CrossRef CAS PubMed.
- F. Chen, Y. Tobimatsu, L. Jackson, J. Nakashima, J. Ralph and R. A. Dixon, Plant J., 2013, 73, 201–211 CrossRef CAS PubMed.
- Y. Tobimatsu, F. Chen, J. Nakashima, L. Jackson, L. L. Escamilla-Treviño, R. A. Dixon and J. Ralph, Plant Cell, 2013, 25, 2587–2600 CrossRef CAS PubMed.
- Y. Li, L. Shuai, H. Kim, A. H. Motagamwala, J. K. Mobley, F. Yue, Y. Tobimatsu, D. Havkin-Frenkel, F. Chen, R. A. Dixon, J. S. Luterbacher, J. A. Dumesic and J. Ralph, Sci. Adv., 2018, 4, eaau2968 CrossRef PubMed.
- X. Wang, C. Zhuo, X. Xiao, X. Wang, M. L. Docampo-Palacios, F. Chen and R. A. Dixon, Plant Cell, 2020, 32, 3825–3845 CrossRef CAS PubMed.
- J. C. del Río, J. Rencoret, A. Gutiérrez, H. Kim and J. Ralph, Plant Physiol., 2017, 174, 2072–2082 CrossRef PubMed.
- J. Rencoret, H. Kim, A. B. Evaristo, A. Gutiérrez, J. Ralph and J. C. del Río, J. Agric. Food Chem., 2018, 66, 138–153 CrossRef CAS PubMed.
- J. Rencoret, D. Neiva, G. Marques, A. Gutiérrez, H. Kim, J. Gominho, H. Pereira, J. Ralph and J. C. del Río, Plant Physiol., 2019, 180, 1310–1321 CrossRef CAS PubMed.
- J. C. del Río, J. Rencoret, A. Gutiérrez, T. Elder, H. Kim and J. Ralph, ACS Sustainable Chem. Eng., 2020, 8, 4997–5012 CrossRef.
- K. O. Eyong, V. Kuete and T. Efferth, in Medicinal Plant Research in Africa, 2013, ch. 10, pp. 351–391, DOI:10.1016/b978-0-12-405927-6.00010-2.
- A. M. Anterola and N. G. Lewis, Phytochemistry, 2002, 61, 221–294 CrossRef CAS PubMed.
- L. B. Davin, M. Jourdes, A. M. Patten, K.-W. Kim, D. G. Vassao and N. G. Lewis, Nat. Prod. Rep., 2008, 25, 1015–1090 RSC.
- R. Vanholme, B. Demedts, K. Morreel, J. Ralph and W. Boerjan, Plant Physiol., 2010, 153, 895–905 CrossRef CAS PubMed.
- K. S. Feldman, S. Quideau and H. M. Appel, J. Org. Chem., 1996, 61, 6656–6665 CrossRef CAS PubMed.
- P. A. Grieco, K. Yoshida and P. Garner, J. Org. Chem., 1983, 48, 3137–3139 CrossRef CAS.
- A. Sadler, V. V. Subrahmanyam and D. Ross, Toxicol. Appl. Pharmacol., 1988, 93, 62–71 CrossRef CAS PubMed.
- V. Nair and S. Kumar, J. Chem. Soc., Chem. Commun., 1994, 1341–1342 RSC.
- V. Nair, B. Mathew, K. V. Radhakrishnan and N. P. Rath, Tetrahedron, 1999, 55, 11017–11026 CrossRef CAS.
- J. C. Wozniak, D. R. Dimmel and E. W. Malcolm, J. Wood Chem. Technol., 1989, 9, 513–534 CrossRef CAS.
- J. Ralph, C. Lapierre, F. Lu, J. M. Marita, G. Pilate, J. Van Doorsselaere, W. Boerjan and L. Jouanin, J. Agric. Food Chem., 2001, 49, 86–91 CrossRef CAS PubMed.
- J. M. Marita, J. Ralph, C. Lapierre, L. Jouanin and W. Boerjan, J. Chem. Soc., Perkin Trans. 1, 2001, 2939–2945 RSC.
- J. Ralph, C. Lapierre, J. Marita, H. Kim, F. Lu, R. D. Hatfield, S. A. Ralph, C. Chapple, R. Franke, M. R. Hemm, J. Van Doorsselaere, R. R. Sederoff, D. M. O'Malley, J. T. Scott, J. J. MacKay, N. Yahiaoui, A.-M. Boudet, M. Pean, G. Pilate, L. Jouanin and W. Boerjan, Phytochemistry, 2001, 57, 993–1003 CrossRef CAS PubMed.
- J. M. Marita, J. Ralph, R. D. Hatfield, D. Guo, F. Chen and R. A. Dixon, Phytochemistry, 2003, 62, 53–65 CrossRef CAS PubMed.
- K. Morreel, J. Ralph, F. Lu, G. Goeminne, R. Busson, P. Herdewijn, J. L. Goeman, J. Van der Eycken, W. Boerjan and E. Messens, Plant Physiol., 2004, 136, 4023–4036 CrossRef CAS PubMed.
- F. Lu, J. M. Marita, C. Lapierre, L. Jouanin, K. Morreel, W. Boerjan and J. Ralph, Plant Physiol., 2010, 153, 569–579 CrossRef CAS PubMed.
- K. Morreel, H. Kim, F. Lu, O. Dima, T. Akiyama, R. Vanholme, C. Niculaes, G. Goeminne, D. Inzé, E. Messens, J. Ralph and W. Boerjan, Anal. Chem., 2010, 82, 8095–8105 CrossRef CAS PubMed.
- Y. Tobimatsu, S. Elumalai, J. H. Grabber, C. L. Davidson, X. Pan and J. Ralph, ChemSusChem, 2012, 5, 676–686 CrossRef CAS PubMed.
- C. S. Chu, T. H. Lee, P. D. Rao, L. D. Song and C. C. Liao, J. Org. Chem., 1999, 64, 4111–4118 CrossRef CAS.
- G. J. Bodwell and Z. Pi, Tetrahedron Lett., 1997, 38, 309–312 CrossRef CAS.
- G. J. Bodwell, Z. Pi and I. R. Pottie, Synlett, 1999, 477–479 CrossRef CAS.
- R. Gao, F. Lu, G. Goeminne, K. Morreel, W. Boerjan and J. Ralph, TBD, 2021 Search PubMed , unpublished.
- L. Jouanin, T. Goujon, V. de Nadaï, M.-T. Martin, I. Mila, C. Vallet, B. Pollet, A. Yoshinaga, B. Chabbert, M. Petit-Conil and C. Lapierre, Plant Physiol., 2000, 123, 1363–1373 CrossRef CAS PubMed.
- B. A. Simmons, D. Loqué and J. Ralph, Curr. Opin. Plant Biol., 2010, 13, 313–320 CrossRef CAS PubMed.
- R. Vanholme, B. De Meester, J. Ralph and W. Boerjan, Curr. Opin. Biotechnol., 2019, 56, 230–239 CrossRef CAS PubMed.
- J.-K. Weng, H. Mo and C. Chapple, Plant J., 2010, 64, 898–911 CrossRef CAS PubMed.
- R. Vanholme, J. Ralph, T. Akiyama, F. Lu, J. Rencoret Pazo, J. Christensen, A. Rohde, K. Morreel, R. De Rycke, H. Kim, B. Van Reusel and W. Boerjan, Plant J., 2010, 64, 885–897 CrossRef CAS PubMed.
- T. Elder, J. C. del Río, J. Ralph, J. Rencoret, H. Kim and G. T. Beckham, Phytochemistry, 2019, 164, 12–23 CrossRef CAS PubMed.
- J. C. del Río, J. Rencoret, A. Gutiérrez, W. Lan, H. Kim and J. Ralph, in Recent Advances in Polyphenol Research, ed. J. Reed, V. de Freitas and S. Quideau, Wiley-Blackwell, 2021, vol. 7, ch. 7, pp. 177–206 Search PubMed.
- J. Ralph, in The Science and Lore of the Plant Cell Wall Biosynthesis, Structure and Function, ed. T. Hayashi, Universal Publishers (BrownWalker Press), Boca Raton, FL, 2006, pp. 285–293 Search PubMed.
- R. Vanholme, K. Morreel, J. Ralph and W. Boerjan, Curr. Opin. Plant Biol., 2008, 11, 278–285 CrossRef CAS PubMed.
- J. H. Grabber, P. F. Schatz, H. Kim, F. Lu and J. Ralph, BMC Plant Biol., 2010, 10, 1–13 CrossRef PubMed.
- J. H. Grabber, D. Ress and J. Ralph, J. Agric. Food Chem., 2012, 60, 5152–5160 CrossRef CAS PubMed.
- S. Elumalai, Y. Tobimatsu, J. H. Grabber, X. Pan and J. Ralph, Biotechnol. Biofuels, 2012, 5, 1–13 CrossRef PubMed.
- J. H. Grabber, N. Santoro, C. E. Foster, S. Elumalai, J. Ralph and X. Pan, BioEnergy Res., 2015, 8, 1391–1400 CrossRef CAS.
- W. Lan, K. Morreel, F. Lu, J. Rencoret, J. C. del Río, W. Voorend, W. Vermerris, W. Boerjan and J. Ralph, Plant Physiol., 2016, 171, 810–820 CAS.
- W. Lan, F. Lu, M. Regner, Y. Zhu, J. Rencoret, S. A. Ralph, U. I. Zakai, K. Morreel, W. Boerjan and J. Ralph, Plant Physiol., 2015, 167, 1284–1295 CrossRef CAS PubMed.
- Y. Mottiar, R. Vanholme, W. Boerjan, J. Ralph and S. D. Mansfield, Curr. Opin. Biotechnol., 2016, 37, 190–200 CrossRef CAS PubMed.
- R. Rinaldi, R. Jastrzebshi, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- N. B. Eloy, W. Voorend, W. Lan, M. de Lyra Soriano Saleme, I. Cesarino, R. Vanholme, R. A. Smith, G. Goeminne, A. Pallidis, K. Morreel, J. Nicomedes Jr., J. Ralph and W. Boerjan, Plant Physiol., 2017, 173, 998–1016 CrossRef CAS PubMed.
- W. Lan, F. Yue, J. Rencoret, J. C. del Río, W. Boerjan, F. Lu and J. Ralph, Polymers, 2018, 10, 916 CrossRef PubMed.
- J. C. del Río, J. Rencoret, A. Gutierrez, H. Kim and J. Ralph, J. Agric. Food Chem., 2018, 66, 4402–4413 CrossRef PubMed.
- P. Y. Lam, Y. Tobimatsu, N. Matsumoto, S. Suzuki, W. Lan, Y. Takeda, M. Yamamura, M. Sakamoto, J. Ralph, C. Lo and T. Umezawa, Nat. Sci. Rep., 2019, 9, 11597 CrossRef PubMed.
- P. Y. Lam, A. C. Lui, M. Yamamura, L. Wang, Y. Takeda, S. Suzuki, H. Liu, F. Y. Zhu, M. X. Chen, J. Zhang, T. Umezawa, Y. Tobimatsu and C. Lo, New Phytol., 2019, 223, 204–219 CrossRef CAS PubMed.
- W. Lan, J. Rencoret, J. C. del Río and J. Ralph, in Lignin: Biosynthesis, Functions, and Economic Significance, ed. F. Lu and F. Yue, Nova Science Publisher, Inc., Hauppauge, NY, USA, 2019, ch. 3, pp. 51–78 Search PubMed.
- J. H. Grabber, C. L. Davidson, Y. Tobimatsu, H. Kim, F. Lu, Y. Zhu, M. Opietnik, N. Santoro, C. E. Foster, F. Yue, D. Ress, X. Pan and J. Ralph, Plant Sci., 2019, 287, 110070 CrossRef CAS PubMed.
- P. Oyarce, B. De Meester, F. Fonseca, L. de Vries, G. Goeminne, A. Pallidis, R. De Rycke, Y. Tsuji, Y. Li, S. Van den Bosch, B. Sels, J. Ralph, R. Vanholme and W. Boerjan, Nat. Plants, 2019, 5, 225–237 CrossRef CAS PubMed.
- A. C. W. Lui, P. Y. Lam, K. H. Chan, L. Wang, Y. Tobimatsu and C. Lo, New Phytol., 2020, 228, 269–284 CrossRef CAS PubMed.
- T. Elder, J. C. del Río, J. Ralph, J. Rencoret, H. Kim, G. T. Beckham and M. F. Crowley, ACS Sustainable Chem. Eng., 2020, 8, 11033–11045 CrossRef CAS.
- T. Elder, J. Rencoret, J. C. del Río, H. Kim and J. Ralph, Front. Plant Sci., 2021, 12, 642848 CrossRef PubMed.
- R. R. Scheline, Acta Chem. Scand., 1966, 20, 1182 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1 provides data relating to all possible Diels–Alder products, and Fig. S2–S8 show assigned 1D and 2D NMR spectra establishing the veracity of crucial compound 12, 12′, 13, and 15 assignments. See DOI: 10.1039/d1gc03022a |
| ‡ Current address: Institute of Wood Technology, Akita Prefectural University, 11-1 Aza Kaieizaka, Noshiro, Akita, Japan, 016-0876. |
| This journal is © The Royal Society of Chemistry 2021 |