
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bio-energy conversion with carbon capture and utilization (BECCU): integrated biomass fermentation and chemo-catalytic CO2 hydrogenation for bioethanol and formic acid co-production†
Nils
Guntermann‡
a,
Hendrik G.
Mengers‡
 b,
Giancarlo
Franciò
a,
Lars M.
Blank
*b and
Walter
Leitner
*ac
b,
Giancarlo
Franciò
a,
Lars M.
Blank
*b and
Walter
Leitner
*ac
aInstitute of Technical and Macromolecular Chemistry, RWTH Aachen University, Aachen, Germany. E-mail: walter.leitner@cec.mpg.de
bInstitute of Applied Microbiology – iAMB, Aachen Biology and Biotechnology – ABBt, RWTH Aachen University, Aachen, Germany
cMax Planck Institute for Chemical Energy Conversion, Mülheim an der Ruhr, Germany
First published on 15th November 2021
Abstract
We present an integrated process for in situ CO2 hydrogenation during bioethanol production by combining bio- and chemocatalysis. The biphasic catalytic system used comprises an aqueous whole-cell fermentation broth and a tailored organometallic catalyst in the organic phase. Glucose is converted to ethanol, and the by-product CO2 is simultaneously upgraded to formic acid in a single reactor unit. Under the optimized conditions, 26% of the generated CO2 was hydrogenated directly.
Decoupling the production of chemical energy carriers and products from the exploitation of fossil resources is an essential pillar in the development towards a more sustainable future (“de-fossilisation”).1 Biomass and carbon dioxide are considered the most important alternative carbon resources in post-fossil scenarios. Technologies for their utilization are typically developed separately, but their complementarity is recognized as an important factor to reach the ambitious goal of a closed anthropogenic carbon cycle. Using sunlight as the energy source, biomass effectively accumulates CO2 from the atmosphere mainly in the form of carbohydrates. Their biochemical conversion into products of higher energy density leads, however, to a partial yet unavoidable release of carbon dioxide as the coupled by-product. These biogenic CO2 streams may be envisaged as carbon feedstocks for valuable products, e.g. via “power-to-X” technologies using green hydrogen. Such concepts of bio- and chemo-catalytic co-processing can help in maximizing the carbon balance of biomass utilization while minimizing the energy requirement for atmospheric CO2 sequestration and conversion (Fig. 1).
 | ||
| Fig. 1 Utilization of renewable resources for biofuel production through fermentation and additional catalytic CO2-reduction to value-added products. | ||
Here, we present a fully integrated bio- and chemo-catalytic process where carbohydrate fermentation and CO2 hydrogenation occur simultaneously in a single reactor. The fermentation of the C6 sugar glucose (C6H12O6) using the yeast Saccharomyces cerevisiae converts two-thirds of the carbon to two equivalents of ethanol (C2H5OH) with the release of two equivalents of CO2, which is hydrogenated in situ to give formic acid (HCOOH) using a tailor-made ruthenium catalyst (Fig. 2).
 | ||
| Fig. 2 Overall reaction pathways of the integrated process for combined carbohydrate fermentation to ethanol and CO2 hydrogenation to formic acid. | ||
Chemo- and bio-catalysis are broadly used in industry and academia for distinct processes. Their integration may result in fruitful synergies where one of the two is not sufficient in the chemical value chain. Thus, combining both catalytic disciplines is a longstanding research objective and a topic of several reviews.2 However, the combination of bio-catalytic transformations using living or even proliferating cells with transition metal catalysed reactions is particularly challenging.2 Integrated approaches comprising both catalyst types in a single reactor are often hampered by diverging demands of the two very different catalytic systems like temperature, solvent, an oxygen atmosphere, additives, and complex growth media. A rare example is the deprotection of allyloxycarbonyl-p-aminobenzoic acid mediated by hyperstoichiometric amounts of a ruthenium complex, where the free acid is used as a nutrient for the growth of auxotrophic E. coli.3 The synthesis of phenyl cyclopropane in vitro was also reported, where glucose is first converted by engineered E. coli to styrene, which then reacts further via metallocarbene transfer using an iron(III) phthalocyanine catalyst.4
The anaerobic fermentation of carbohydrates with S. cerevisiae is arguably the most established biotechnological process.5 While the released CO2 is used partially for carbonation in the beverage industry, there is currently no usage of it as a chemical feedstock. One possibility to convert CO2 to a valuable product is its hydrogenation to formic acid, which is used today as an agrochemical and also discussed as a hydrogen storage material.6–8 Molecular ruthenium complexes are known to be particularly effective and robust catalysts for this reaction, are able to operate under mild conditions and can tolerate aqueous media.9 Therefore, a direct combination of the two catalytic transformations was envisaged comprising a biphasic system organic/water to separate the organometallic catalyst from the fermentation broth. Effective compartmentalization of the ruthenium catalyst in the organic phase was hoped to prevent any negative interference between the chemo- and bio-catalyst and potentially allow for recycling of the noble metal catalyst. The designed reactive system is shown in Fig. 3.
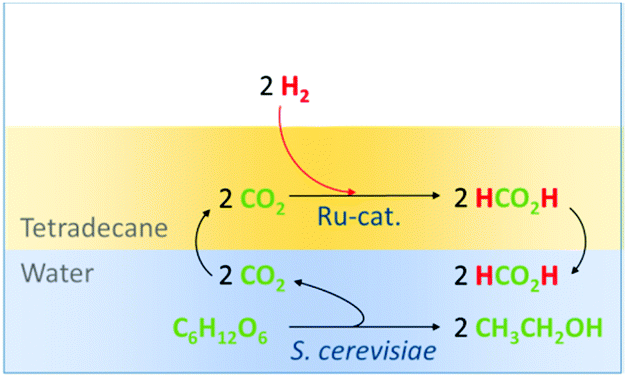 | ||
| Fig. 3 Schematic depiction of the envisaged multiphasic combination of both transformations. | ||
The proliferation of yeast cells in standard minimal growth medium (Verduyn for anaerobic growth, VfA)10 was used to assess the effect of mechanical, physical, and chemical stress on the fermentation process (Fig. 4). While formate had no effect on the cells (see Fig. S8, ESI†), the use of magnetic stirring bars of different geometries was found to exhibit a considerable effect on cell growth as compared to shaking, with a cross-shaped stirring bar having the least damaging impact. In standard Hungate tubes, cell growth was reduced by 23% under stirring as compared to shaking (see Fig. 4). The same growth behaviour was observed when conducting the fermentation in a stirred high pressure reactor with a glass inlet as required for the combined process. This value was set as the reference for 100% cell proliferation activity. In the closed reactor, a final pressure of 4.0 bar was detected due to the CO2 generation from fermentation after 24 h. Pressurizing the reactor initially with hydrogen at 60 bar caused a decrease in the cell growth to 76%, independent of the pressurization rate (see Fig. S2, ESI†). Still, a seven-fold growth over the inoculation time (t0) was observed under high pressure.
 | ||
| Fig. 4 Comparison of the relative cell count after 24 h incubation in a shaken Hungate tube (Hungate), stirred Hungate tube (Hungate stirred), stirred reactor (Reactor, set as 100%), stirred reactor with 60 bar H2 (+H2), stirred reactor with tetradecane (+TD), stirred reactor with tetradecane and 60 bar H2 (+TD +H2). Cells were counted with the Amphasys Z32 impedance flow cytometer.11 | ||
The effect of the additional organic phase on the growth of S. cerevisiae was investigated first in shaken Hungate tubes to select a solvent with minimal impact. While slightly polar methyl isobutyl carbinol (MIBC) and, to some extent, even the non-polar decane showed toxic effects, a negligible impact was found for the long-chain alkanes dodecane, tetradecane, and hexadecane (see Fig. S3, ESI†). Dodecane and tetradecane resulted in a certain decrease of cell growth in the high pressure reactor upon stirring (1000 rpm), probably due to increased mixing. In the presence of hydrogen, tetradecane resulted in the retention of 44% of the unperturbed cell proliferation, making this biphasic system the most promising combination.
The selection of tetradecane as the catalyst phase imposes a specific solubility profile for the catalyst. Recently, some of us reported the ruthenium catalyst cis-[RuCl2(C12-dppm)2] bearing the tailored bidentate phosphine C12-dppm (bis(bis(4-dodecylphenyl)phosphanyl)methane) as a ligand (Fig. 2), which fulfils the requirement of partitioning almost exclusively into the non-polar organic phase in alkane/water systems.12 In agreement with this design concept, the addition of the complex to tetradecane had no effect on cell proliferation compared to the organic solvent alone.
Having established the principle compatibility of the individual components, the combined process was carried out in the biphasic system containing S. cerevisiae (OD600 nm = 1) in VfA (6 mL) with glucose (20 wt%) and cis-[RuCl2(C12-dppm)2] (4 μmol) in tetradecane (1 mL) under H2 atmosphere (60 bar). Besides the main products formate and ethanol, the concentrations of the residual glucose and the metabolites glycerol and acetate were determined by HPLC as well (see ESI, Tables 2–6†). After 24 h, an ethanol concentration of 725 mM and a cell growth of 56% with respect to the positive control were observed together with a formic acid concentration of 12.6 mM in the aqueous phase. No formic acid was detected in the organic phase. In sharp contrast, no significant amounts of formic acid were detected using the established water soluble catalyst for the hydrogenation of CO2 [RuCl2(PTA)4] (PTA = 1,3,5-triaza-7-phosphaadamantane)13 directly in the fermentation broth under otherwise identical conditions (see Fig. S4, ESI†). In pure water as well as in VfA, the PTA-based catalyst achieved a formic acid concentration of 3.0 mM and 2.7 mM, respectively, within 24 h at 5 bar CO2 and 60 bar H2. This demonstrates the inhibiting effect of the growing cells in the medium on the metal catalyst. The superior catalytic activity of the tailor-made ruthenium catalyst cis-[RuCl2(C12-dppm)2] confirms the importance of an effective compartmentalization.
Although the application of H2 pressure has a negative impact on cell growth, the extent of this effect is not strongly dependent on the final hydrogen pressure (see Fig. S2, ESI†). In contrast, the hydrogen pressure influence on formate production was significant as shown in Fig. S5.† Increasing the H2 pressure from 30 bar to 120 bar gave three times more formic acid after 24 h. No significant benefit of a further pressure increase was noted and thus, a partial pressure of 120 bar H2 was chosen for further studies.
The formation of formic acid from CO2 and H2 is an equilibrium reaction that is known to be greatly facilitated by the presence of amine bases.8 Various alkylamines were tested but resulted in no live cells after incubation in agreement with their known toxicity to microbes.14 Therefore, basic amino acids were considered as biologically benign alternatives.15
The addition of arginine and lysine was lethal for the cells, probably due to the pH shift or high osmolarity. Histidine, however, was tolerated to a significant extent. In 24 h incubation experiments, 50 mM concentration of histidine caused a growth reduction of 20%, which increased to 50% on rising the concentration of histidine to 100 mM. An increase of histidine concentration to up to 200 mM had no further impact on the cell growth (see Fig. S6, ESI†).
A series of experiments were conducted to find the optimum balance of histidine concentration between the negative effect on cell growth and the positive effect on the hydrogenation equilibrium (Fig. 5). Conducting the reaction for 24 h at different histidine loadings showed an overall positive effect, reaching a maximum production of formate of ca. 60 mM at an amino acid concentration of 100–150 mM. Extending the reaction time to 48 h resulted in the maximum formate concentration of 140 mM corresponding to the turnover number (TON) per ruthenium center of 200 using a histidine concentration of 150 mM. No histidine was found in the tetradecane phase by NMR analysis.
 | ||
| Fig. 5 Produced formic acid (c(FA)) at different histidine concentrations and reaction times. Conditions: S. cerevisiae (OD600 nm = 1) in VfA (6 mL) with glucose (20 wt%), cis-[RuCl2(C12-dppm)2] (4 μmol) in tetradecane (1 mL) and H2 (60 bar). The histidine concentration did not vary over the course of fermentation, so we do not expect any direct influence on the yeast metabolism (see Fig. S7, ESI†). | ||
Since the thermodynamically limited equilibrium of an equimolar amount of HCOOH to histidine was approached under the optimized conditions, a second series of experiments was carried out at a reduced catalyst loading (2 μmol instead of 4 μmol) and increased H2 pressure (120 bar). An overview including experiments without histidine under the same conditions can be found in Table 1, while the concentrations of the metabolites are listed in the ESI Tables S2–S5.†
| Entry | Time | Live cellsb [%] | Dead cellsb [%] | c(FA)c [mM] | TON | c(EtOH)c [mM] |
|---|---|---|---|---|---|---|
| a All experiments were conducted in fivefold determination; reactor volume = 12.5 mL, H2 = 120 bar, cis-[RuCl2(C12-dppm)2] = 2 μmol, tetradecane (1 mL), VfA (6 mL) with glucose (20 wt%), 1000 rpm, 30 °C. b Cell count determination using Amphasys Z32 relative to the PC in the reactor for 24 h and 48 h (each set as 100%). c Determined via HPLC calibrated by dilution series of known concentration. d Additional 150 mM histidine. | ||||||
| 1 | 24 h | 40.4 ± 4.6 | 20.9 ± 5.5 | 10 ± 2 | 31 ± 6 | 415 ± 62 |
| 2 | 24 h + Hisd | 20.1 ± 4.2 | 26.1 ± 8.2 | 25 ± 2 | 82 ± 9 | 154 ± 16 |
| 3 | 48 ha | 24.7 ± 11.5 | 32.0 ± 12.8 | 25 ± 3 | 83 ± 4 | 651 ± 93 |
| 4 | 48 h + Hisd | 31.4 ± 3.8 | 20.6 ± 5.5 | 128 ± 9 | 406 ± 8 | 484 ± 45 |
Overall, the total cell count after 24 and 48 h reaction times was comparable (see Table 1) and the pressure curves showed a much slower, if any, CO2 production after 24 h (see Fig. S12, ESI†), indicating dormant cells at this stage. The consumed amounts of glucose and the resulting ethanol concentrations match very well with the observed cell growth. The formic acid concentration in turn correlates mainly with the amount of histidine and the reaction time. Without a base, the concentration of formic acid reached 10 mM after 24 h and 25 mM after 48 h. In the presence of 150 mM histidine, 25 mM formic acid was obtained after 24 h that increased further to 133 mM after 48 h, corresponding to 89% of the maximum equilibrium conversion.
Under the reaction conditions in Table 1, the ruthenium catalyst produces 406 mol of HCOOH per mol of the charged complex in a single run (TON = 406). The catalyst is still active at the end of the reaction as demonstrated by re-using the organic phase. After removal of the product containing aqueous fermentation broth, fresh growth medium with new cells was added to the reactor and the fermentation/hydrogenation was repeated with the same catalyst phase. In the second run, a formic acid concentration of 99 mM was achieved, retaining 75% of the initial performance. Further recycling was hampered by foaming, preventing clean phase separation on a small laboratory scale, but a total turnover number of 1089 based on the initially charged ruthenium complex could be produced over five consecutive runs (see Table S6 and Fig. S19, ESI†). The ICP-MS analysis of the aqueous phase showed a total Ru loss of 4.7%, with the majority being lost after the first run (2.3%). The reduced activity therefore cannot be explained solely on the basis of catalyst leaching.
The mass balance of the fermentation products could be closed regarding the above mentioned metabolites between 88% and 96% (Tables S2–S5, ESI†), allowing the estimation of the carbon loss as CO2 from the stoichiometry of glucose conversion (Fig. 2). As the chosen standard laboratory strain S. cerevisiae S288C does neither show anaerobic formate production nor consumption (see Fig. S8, ESI†), the experimentally determined concentration of HCOOH corresponds directly to the upgraded by-product. Without histidine, 4% of the released carbon dioxide was converted after 48 h. The addition of the stabilizing base increased the reconversion rate to remarkable 26% within the same time even under the non-optimized small-scale conditions.
In conclusion, we have demonstrated the first example of an integrated process combining biomass fermentation with chemo-catalytic CO2 hydrogenation. The judicious choice of the individual components and the systematic investigation of the reaction system enabled us to upgrade 26% of the waste CO2 from fermentation into formic acid. While the downstream processing of the product containing the fermentation broth was not investigated under the laboratory conditions, the isolation of bioethanol can be expected to be achieved by following standard techniques.16 The formic acid product might be extracted and isolated subsequently also by established methods.17 Alternatively, the resulting aqueous phase containing the amino acid–formic acid mixture could even be considered directly for agrochemical applications such as cattle feed, similar to aqueous formic acid solutions which are already in use today.18 Co-feeding of the formate enriched broth to enhance microbial growth or product formation in other biotechnological processes could also be envisaged.19
While bio-energy conversion with carbon capture and storage (BECCS) is attracting significant interest as a potential large-scale carbon negative technology,20 the upgrading of waste CO2 to valuable products offers an attractive complementary approach to improve the carbon balance with additional economic potential (Fig. 1).7 The combination of both concepts is thus described by the term “bioenergy conversion with carbon capture and utilization (BECCU)”. Currently reported BECCU strategies comprise either biocatalytic approaches like algae-based biotransformations and biological Sabatier processes or focus on chemical transformations.21 The example presented here demonstrates that even fully integrated co-production of chemo- and biocatalysis can be achieved in certain cases. The highly dynamic progress in the fields of biomass conversion and catalytic CO2 chemistry offers a promising basis for further development of the general concept as contribution towards an increasingly closed anthropogenic carbon cycle.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – Exzellenzcluster 2186 “The Fuel Science Center” ID: 390919832 and by the Bundesministerium für Bildung und Forschung (BMFB, Federal Ministry of Education and Research, 031B0850A). The authors are responsible for the content of this publication. Open Access funding provided by the Max Planck Society.Notes and references
- (a) J. Rockström, O. Gaffney, J. Rogelj, M. Meinshausen, N. Nakicenovic and H. J. Schellnhuber, Science, 2017, 355, 1269–1271 CrossRef PubMed; (b) J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Science, 2020, 367, 397–400 CrossRef PubMed; (c) Climate change 2013. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, ed. T. F. Stocker, D. Qin, G.-K. Plattner, M. Tignor, S. K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex and P. M. Midgley, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2013 Search PubMed.
- (a) P. K. Sasmal, C. N. Streu and E. Meggers, Chem. Commun., 2013, 49, 1581–1587 RSC; (b) H. Gröger and W. Hummel, Curr. Opin. Chem. Biol., 2014, 19, 171–179 CrossRef; (c) S. Wallace, E. E. Schultz and E. P. Balskus, Curr. Opin. Chem. Biol., 2015, 25, 71–79 CrossRef PubMed; (d) F. Rudroff, M. D. Mihovilovic, H. Gröger, R. Snajdrova, H. Iding and U. T. Bornscheuer, Nat. Catal., 2018, 1, 12–22 CrossRef; (e) X. Huang, M. Cao and H. Zhao, Curr. Opin. Chem. Biol., 2020, 55, 161–170 CrossRef PubMed.
- Y. Lee, A. Umeano and E. P. Balskus, Angew. Chem., Int. Ed., 2013, 52, 11800–11803 CrossRef PubMed.
- S. Wallace and E. P. Balskus, Angew. Chem., Int. Ed., 2015, 54, 7106–7109 CrossRef PubMed.
- Global Bioenergy Statistics, World Bioenergy Association, 2019, https://www.worldbioenergy.org/uploads/201210%20WBA%20GBS%202020.pdf Search PubMed.
- (a) W. Leitner, Angew. Chem., Int. Ed. Engl., 1995, 34, 2207–2221 CrossRef; (b) P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259–272 CrossRef; (c) C. Federsel, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 6254–6257 CrossRef; (d) P. G. Jessop, F. Joó and C.-C. Tai, Coord. Chem. Rev., 2004, 248, 2425–2442 CrossRef CAS; (e) W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 CrossRef CAS.
- (a) B. M. Bhanage, Y. Ikushima, M. Shiraia and M. Arai, Chem. Commun., 1999, 1277–1278 RSC; (b) S. Moret, P. J. Dyson and G. Laurenczy, Nat. Commun., 2014, 5, 4017 CrossRef CAS; (c) M. Scott, B. Blas Molinos, C. Westhues, G. Franciò and W. Leitner, ChemSusChem, 2017, 10, 1085–1093 CrossRef CAS PubMed.
- C. Verduyn, E. Postma, W. A. Scheffers and J. P. van Dijken, Yeast, 1992, 8, 501–517 CrossRef CAS PubMed.
- I. Heidmann, G. Schade-Kampmann, J. Lambalk, M. Ottiger and M. Di Berardino, PLoS One, 2016, 11, e0165531 CrossRef.
- (a) M. Scott, C. G. Westhues, T. Kaiser, J. C. Baums, A. Jupke, G. Franciò and W. Leitner, Green Chem., 2019, 21, 6307–6317 RSC; (b) C. M. Jens, M. Scott, B. Liebergesell, C. G. Westhues, P. Schäfer, G. Franciò, K. Leonhard, W. Leitner and A. Bardow, Adv. Synth. Catal., 2019, 361, 307–316 CrossRef CAS.
- G. Laurenczy, F. Joó and L. Nádasdi, Inorg. Chem., 2000, 39, 5083–5088 CrossRef CAS PubMed.
- T. Cai, L. Chen, Q. Ren, S. Cai and J. Zhang, J. Hazard. Mater., 2011, 186, 59–66 CrossRef CAS.
- D. WEI, H. Junge and M. Beller, Chem. Sci., 2021, 12, 6020–6624 RSC.
- A. A. Kiss and C. S. Bîldea, in Intensification of Biobased Processes, ed. A. Górak and A. Stankiewicz, Royal Society of Chemistry, Cambridge, 2018, pp. 62–85 Search PubMed.
- (a) T. Schaub and R. A. Paciello, Angew. Chem., Int. Ed., 2011, 50, 7278–7282 CrossRef CAS; (b) T. Brouwer, M. Blahusiak, K. Babic and B. Schuur, Sep. Purif. Technol., 2017, 185, 186–195 CrossRef CAS; (c) S. Şahin, Ş. S. Bayazit, M. Bilgin and İ. İnci, J. Chem. Eng. Data, 2010, 55, 1519–1522 CrossRef.
- R. E. Muck, E. M. G. Nadeau, T. A. McAllister, F. E. Contreras-Govea, M. C. Santos and L. Kung, J. Dairy Sci., 2018, 101, 3980–4000 CrossRef CAS PubMed.
- (a) N. J. Claassens, I. Sánchez-Andrea, D. Z. Sousa and A. Bar-Even, Curr. Opin. Biotechnol., 2018, 50, 195–205 CrossRef CAS; (b) J. La Gonzalez de Cruz, F. Machens, K. Messerschmidt and A. Bar-Even, ACS Synth. Biol., 2019, 8, 911–917 CrossRef; (c) S. Zobel, J. Kuepper, B. Ebert, N. Wierckx and L. M. Blank, Eng. Life Sci., 2017, 17, 47–57 CrossRef CAS PubMed; (d) O. Yishai, S. N. Lindner, J. La Gonzalez de Cruz, H. Tenenboim and A. Bar-Even, Curr. Opin. Chem. Biol., 2016, 35, 1–9 CrossRef CAS.
- (a) S. Kar, A. Goeppert and G. K. S. Prakash, Acc. Chem. Res., 2019, 52, 2892–2903 CrossRef CAS PubMed; (b) G. Lee, Y. C. Li, J.-Y. Kim, T. Peng, D.-H. Nam, A. Sedighian Rasouli, F. Li, M. Luo, A. H. Ip, Y.-C. Joo and E. H. Sargent, Nat. Energy, 2021, 6, 46–53 CrossRef CAS; (c) V. Stavrakas, N.-A. Spyridaki and A. Flamos, Sustainability, 2018, 10, 2206 CrossRef CAS.
- (a) Department of Energy Office of Energy Efficiency and Renewable Energy, Algae Production CO2 Absorber with Immobilized Carbonic Anhydrase. DE-EE0007092, 2020; (b) IAE Bioenergy, Deployment of BECCS/U value chains. Technological pathways, policy options and business models, 2020; (c) M. Nevander, J. Kärki and J. Lehtonen, Bioplastics, 2021, 16, 42–43 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc02915k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |