
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Active role of lignin in anchoring wood-based stabilizers to the emulsion interface†
Danila M. de
Carvalho
 *a,
Maarit H.
Lahtinen
a,
Mamata
Bhattarai
a,
Martin
Lawoko
b and
Kirsi S.
Mikkonen
ac
*a,
Maarit H.
Lahtinen
a,
Mamata
Bhattarai
a,
Martin
Lawoko
b and
Kirsi S.
Mikkonen
ac
aFaculty of Agriculture and Forestry, Department of Food and Nutrition, FI-00014 University of Helsinki, P.O. Box 66, Finland. E-mail: danila.moraisdecarvalho@helsinki.fi; Tel: +358 29 4158295
bWallenberg Wood Science Center, Department of Fiber and Polymer Technology, Royal Institute of Technology, KTH, Teknikringen 56, 100 44, Stockholm, Sweden
cHelsinki Institute of Sustainability Science (HELSUS), FI-00014 University of Helsinki, P.O. Box 65, Finland
First published on 29th October 2021
Abstract
Hemicellulose-rich wood extracts show efficient capacity to adsorb at emulsion interfaces and stabilize them. Their functionality is enhanced by lignin moieties accompanying the hemicellulose structures, in the form of lignin-carbohydrate complexes (LCCs) and, potentially, other non-covalent associations. The formation and stability of emulsions is determined by their interfacial regions. These are largely unexplored assemblies when formed from natural stabilizers with a complex chemical composition. Understanding the structure of the interfacial region could facilitate both designing the extraction processes of abundant biomasses and unraveling a valuable industrial application potential for the extracts. Herein, we characterized the LCCs from the interface of oil-in-water emulsions stabilized by galactoglucomannan (GGM) or glucuronoxylan (GX)-rich wood extracts, using two-dimensional nuclear magnetic resonance (NMR) spectroscopy analysis. The type of covalent linkage between residual lignin and hemicelluloses determined their partitioning between the continuous and interfacial emulsion phases. Benzylether structures, only found in the interface, were suggested to participate in the physical stabilization of the emulsion droplets. In turn, the phenylglycosides, preferentially observed in the continuous phase, were suggested to interact with adsorbed stabilizers by electrostatic interaction. More hydrophobic lignin structures, such as guaiacyl lignin type, dibenzodioxocin substructures, and certain end groups also contributed to droplet stabilization. The elucidation of such attributes is of paramount importance for the biorefinery industry, enabling the optimization of extraction processes for the preparation of wood-based stabilizers and designed interfaces for novel and sustainable emulsion systems.
Introduction
Emulsions are dispersions formed by the combination of, at least, two immiscible-liquids. Due to the unique functionalities and potentialities of emulsions in modern industrial applications, emulsion science is rapidly evolving.1–3 In an oil-in-water emulsion, three distinguished regions are identified: the continuous water phase, the dispersed oil droplet, and the oil–water (o/w) interface containing the stabilizer agent.4 As water and oil do not form spontaneous dispersions, it is evident that the stabilizer plays a vital role in emulsion formation, influencing droplet size and emulsion stability and, eventually, inhibiting emulsified lipid oxidation.5,6 Consequently, the interfacial layer formed in the presence of stabilizer stands as a key region to the structural support of the emulsion. For this very reason, it is the interface that is the main region subjected to tensions, that, eventually, lead to emulsion break down.7 Therefore, improving stability of emulsion systems demands a deep understanding on the structure, behavior, and functionality of stabilizers and, consequently, the resultant interfacial layer.Industrial emulsions can be prepared using synthetic or natural stabilizers. In the production of synthetic stabilizers both, functional and structural attributes are controlled to enhance their performance in applications. This includes, for example, the engineering of highly pure molecules containing specific hydrophilic-lipophilic balance. In turn, natural stabilizers, including those from lignocellulosic sources, have a more complex composition and certain imbalance in hydrophilic-lipophilic domains, for which extensive fractionation, derivatization, and purification are applied prior to their use in emulsion systems. Despite that, natural stabilizers incorporate better into modern production platforms, supporting more sustainable formulations and clean-label applications.1,8,9 Recently, wood hemicelluloses were identified as efficient and multifunctional stabilizers for dispersion systems.10–14 Interestingly, studies have also proved that crude extracts were more active in emulsion stabilization than the purified ones, putting into question the need for intensive purification of wood-extracts. In fact, the heterogeneous composition of such extracts has been linked to their enhanced functionality as stabilizers, suggesting the occurrence of a natural combination of beneficial and complementary attributes of their various compounds.4,10,13,15 Hemicelluloses, including galactoglucomannan (GGM) and glucuronoxylan (GX), promote good steric stabilization of emulsion in varied conditions of pH and ionic strength.16 However, large amounts of hemicelluloses might be necessary to efficiently cover oil droplets since their structures typically lack hydrophobic sites.17,18 Opportunely, in crude wood-extracts the deficiency of intrinsic hydrophobic domains in hemicelluloses is compensated by the co-preservation of lignin moiety. The lignin, extracted from biomass in a relatively native form by certain processes, contribute to the steric stabilization of emulsions and effective protection of emulsified oil against oxidation.12,13 Lignin is a naturally hydrophobic web shaped aromatic structure that is found in close physiological association with hemicelluloses in wood tissues, including through covalent interactions.19–22 In line with this, a systematic study recently confirmed that part of the lignin-carbohydrates association in GGM extracts used as stabilizers has a covalent nature, identified as the so-called lignin-carbohydrate complex (LCC).15 These findings corroborated with the hypothesis that wood-based stabilizers are naturally amphiphilic, in which the gradient of polarity in the same molecule favors their anchoring to the oil droplet.1,12 Additionally, specific to the functionality of hemicelluloses as stabilizer agents, recent studies have demonstrated their suitable sensory profile and biocompatibility for application in life science applications, such as food and biomedical products.14,23
Despite advances in the chemical, structural, and sensory attributes of wood-based stabilizers, there are crucial aspects about their stabilization action that remain unresolved. This includes, for example, the understanding on the behavior and mechanisms, through which such complex wood-derivatives form the o/w interface. It is already known that only a certain amount of such stabilizer goes to the interface of the emulsion, whilst part of the stabilizer supplemented remains unabsorbed in the continuous phase.12,24 Likely, chemical and structural attributes of the different populations present in the composition of GGM and GX stabilizers are contributing to the selective distribution of such stabilizers. Moreover, the different function of such populations in each emulsion phase might be explained by differences in their chemical structure. Although crucial for enhancing the understanding on stabilization mechanisms, detailed chemical characterization studies on the interface of emulsions stabilized by GGM and GX are non-existent. The main reason for this is the limited efficiency of methods to recover stabilizers from emulsion phases. Herein, for the first time, the interface of emulsions stabilized by GGM and GX were studied in terms of active compounds in the droplet stabilization. GGM and GX samples with varied carbohydrates and lignin compositions were systematically applied in emulsification. A simple and efficient method based on liquid-extraction was developed for recovering pure stabilizers from emulsion phases and to enable further structural characterization. Moreover, the nature of lignin-carbohydrate interaction in GX stabilizers (fractionated by antisolvent process) was investigated following a similar approach used previously for GGM stabilizer,15 in support of the interface study. Results indicated specific structural attributes in wood-based stabilizers associated to the oil droplet stabilization and endorse the value and functionality of using heterogeneous assemblies, supplemented by various lignin structures, rather than purified carbohydrates. This new understanding also opens possibility in biorefineries for using their side-streams of crude hemicelluloses-rich extracts, naturally associated with lignin, in novel and sustainable emulsion systems.
Experimental
Materials
Rich hemicellulose extracts containing galactoglucomannan (GGM) and glucuronoxylan (GX) were extracted from spruce and birch sawdust, respectively, through a pressurized hot water flow-through extraction (PHWE) process according to Kilpeläinen et al.25 Spray-dried galactoglucomannan (sdGGM) and glucuronoxylan (sdGX) were recovered from the resultant water-extract by spray-drying. Chemical and structural compositions of sdGGM and their fractions, i.e., ethanol-precipitated (ep-) GGM and ethanol-soluble (es-) GGM, were previously published,15 and the fractionation of sdGX was performed as described below.Antisolvent fractionation and characterization of GX samples
The antisolvent fractionation of sdGX in ethanol was performed according to the described literature for sdGGM,15 with only minor modifications. First, sdGX was suspended in MilliQ-water to a ratio of 24% solids at room temperature overnight under constant agitating. Next, the sdGX suspension was added to absolute ethanol (suspension![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethanol ratio of 1:8), stirred for 10 minutes, and cooled at 4 °C overnight.10 The epGX was recovered by centrifugation (20 °C, 10000 rpm, and 5 min) (Eppendorf Centrifuge 5810 R), washed with absolute ethanol, and dried in a vacuum oven (40 °C, 48 h). The esGX was obtained after concentration of the supernatant using rotary evaporator (45–55 °C) and lyophilization. The fractionation yields were estimated gravimetrically. Chemical and structural characterization of GX samples, including carbohydrate and lignin compositions, acetyl groups content and molar mass were assessed according to previous publications.15,26
:ethanol ratio of 1:8), stirred for 10 minutes, and cooled at 4 °C overnight.10 The epGX was recovered by centrifugation (20 °C, 10000 rpm, and 5 min) (Eppendorf Centrifuge 5810 R), washed with absolute ethanol, and dried in a vacuum oven (40 °C, 48 h). The esGX was obtained after concentration of the supernatant using rotary evaporator (45–55 °C) and lyophilization. The fractionation yields were estimated gravimetrically. Chemical and structural characterization of GX samples, including carbohydrate and lignin compositions, acetyl groups content and molar mass were assessed according to previous publications.15,26
Preparation of oil-in-water emulsion using wood-based stabilizers
All GGM and GX samples were used to stabilize a suspension of water and hexadecane. Hexadecane, a hydrocarbon, was used as the oil phase herein instead of vegetable oil (mono- or polyunsaturated fatty acid) alternatives due to its minimal effect on the subsequent analytical method used for the assessment of the composition of stabilizers. A 1% (w/w) GGM (or GX) was suspended in water overnight at room temperature and constant agitation. Next, 5% (w/w) hexadecane was added and the suspension was mixed using an Ultra-Turrax (T-18 basic, IKA, Staufen, Germany) equipped with a disperser-type stirrer at 22000 rpm for 2 min to form a coarse emulsion. To obtain a fine dispersion, the coarse emulsion was subjected to microfluidizer (Microfluidizer 110Y, Microfluidics, Westwood, MA, USA) configured with 75 μm (Y-type F20Y) and 200 μm (Z-type H30Z) chambers in a series at a pressure of 800 bar under a continuous flow for a total of three passes. Emulsion was collected during the fourth pass. Samples used for emulsion characterization were supplemented with potassium sorbate (0.05% w/w) to prevent microbial growth and stored at room temperature.
Phase partitioning and stabilizer recovering from emulsion phases
472 rpm, 4 °C, and 15 min) (Thermo Scientific, Sorvall LYNX 6000 Centrifuge, rotor F14-6x250y) (Scheme 1). The emulsion separated into a cream (hexadecane droplets + stabilizers) and a clear serum phase (water + stabilizers), which was assisted by the solidification of hexadecane oil droplets at 4 °C. To recover stabilizer, the serum phase was collected and lyophilized. The stabilizer from the cream phase was recovered through liquid-extraction. Both fractions (i.e., supernatant and cream) were weighed for assessing the gravimetric yield of phase partitioning.
 | ||
| Scheme 1 Phase partitioning of emulsions by centrifugation and liquid-extraction process for recovering GGM and GX stabilizers from cream. | ||
:10 (w/v) was used for all extractions. Two sequential steps with intermediate centrifugation was applied for extraction using 1% Tween-20 (w/w): first at room temperature overnight and then at 40 °C for 2 h. For extractions using the solvents, the suspension was initially vortexed after solvent addition and then phase separation occurred overnight without agitation. Next, centrifugation (12472 rpm, 4 °C, and 15 min) was performed to obtain stabilizers in a hexadecane-free supernatant (Thermo Scientific, Sorvall LYNX 6000 Centrifuge, rotor F14-6x250y). Supernatant from extraction using Tween-20 was direct lyophilized for stabilizer recovery. Supernatant from extraction using solvents were first completely evaporated by flushing nitrogen, then resuspended in MilliQ-water, and lyophilized. Acetonitrile extraction provided the most efficient and selective stabilizer recovering process and was used in the remainder of the study (Scheme 1) (see attributes that qualified acetonitrile as the most suitable solvent to recover wood-based stabilizers from cream in ESI, Fig. S1†). GGM and GX stabilizers recovered from both, continuous phase and cream were structurally characterized by NMR spectroscopy. The efficient removal of hexadecane from GGM and GX recovered from continuous phase and cream was confirmed by Fourier transform infrared (FTIR) spectroscopy (ESI, Fig. S2 and Table S1†).
Emulsions characterization
Results and discussion
Aspects driving the anchoring of wood-based stabilizers in emulsion interface
The capacity of galactoglucomannan- and glucuronoxylan-rich extracts (from spruce and birch, respectively) to stabilize emulsions although well demonstrated and revised in the literature,1,10–13,24 still have some unresolved aspects. This includes, for example, the understanding of what chemical and structural attributes drive the distribution and define the performance of such stabilizers at the interface. A previous characterization of spruce-extract confirmed that sdGGM, esGGM, and epGGM have chemical and structural particularities, including a varied chemical composition and different types and abundance of LCC bonds.15 Herein, fundamental differences among sdGX, esGX, and epGX compositions were also demonstrated (ESI, Tables S4 and S5†). These two studies together provided a robust basis of data on softwood- and hardwood-based stabilizers. Moreover, the diversity of structures covered by the various GGM and GX samples, obtained by PHWE process, supported a systematic investigation on stabilizer migration towards interface and stabilization aspects. Such aspects are discussed in the subsequent sections.| Stabilizer | Phases | Distribution, % | Lignina, % | Carbohydratesa, % | S/G ratioa | LCC structuresb |
|---|---|---|---|---|---|---|
| a Obtained from NMR spectra by semi-quantitative analysis using the aromatic G2 (for GGM samples) and S2/6 (for GX samples) peaks reference. b PG: phenylglycoside; BE: benzylether, and GE: γ-ester. n.d. Not determined: only contours in the area of S lignin were identified. | ||||||
| sdGGM | Interface | 16.6 | 63.7 | 36.3 | — | PG BE1 GE |
| Continuous phase | 83.4 | 1.7 | 98.3 | — | PG GE | |
| esGGM | Interface | 28.0 | 95.0 | 5.0 | — | BE1 GE |
| Continuous phase | 72.0 | 16.6 | 83.4 | — | PG GE | |
| epGGM | Interface | 12.7 | 27.0 | 73.0 | — | GE |
| Continuous phase | 87.3 | 0.2 | 99.8 | — | PG GE | |
| sdGX | Interface | 12.9 | 43.6 | 56.4 | 7.6 | PG BE2 GE |
| Continuous phase | 87.1 | 6.1 | 93.9 | 10.3 | PG GE | |
| esGX | Interface | 28.8 | 87.4 | 12.6 | 5.1 | BE2 GE |
| Continuous phase | 71.2 | 15.2 | 84.8 | 6.8 | PG GE | |
| epGX | Interface | 17.1 | 15.5 | 84.5 | 9.8 | GE |
| Continuous phase | 82.9 | 0.9 | 99.1 | >9.8 n.d | GE | |
 | ||
| Fig. 1 Structure of the main lignin-carbohydrates bonds (A), lignin end groups (B), lignin interlinkages (C), and lignin subunits (D) found in wood-extracts. | ||
 | ||
| Fig. 2 Distribution of phenylglycoside (PG), benzylether (BE), and γ-ester (GE) LCC structures between emulsion phases in emulsions stabilized by sdGGM (A and B) and sdGX (C and D). For emulsions stabilized by the other GGM and GX samples see the ESI (Fig. S3†). The main assignments for lignin and carbohydrate structures in stabilized by GGM and GX samples are shown in ESI (Tables S2 and S3,† respectively). | ||
Benzylether structures were identified exclusively in the interface of emulsions stabilized by sdGGM, esGGM, sdGX, and esGX contours at δC/δH 80.2/4.50 ppm for BE1 structures in GGM samples and at δC/δH 82.3/5.20 ppm for BE2 structures in GX samples,29,30 suggesting a possible role of molecules containing benzylether structure in the physical stabilization of oil droplet. The absence of benzylether structures in the interface of emulsion stabilized by epGGM and epGX is explained by the fact that during the ethanol fractionation used to prepare such samples, the structures containing benzylether were preferentially solubilized in ethanol and concentrated in es- samples, rather than precipitated in the ep- samples. Phenylglycoside structures (δC/δH 102.8–99.8/5.18–4.75 ppm),29,31,33,34 although present in the interface of emulsions stabilized by sdGGM and sdGX, were more frequently observed in the continuous phase of emulsions stabilized by both GGM and GX samples. The role of molecules containing phenylglycoside structures in the emulsion stabilization seemed to be considerably more related to electrostatic interaction with adsorbed stabilizers (via hydrogen-bonding and hydrophobic interactions) than to their direct participation in the physical stabilization of oil droplet. The participation of molecules containing γ-ester structures (δC/δH 64.0–62.0/4.50–4.00 ppm)29,32 in the emulsion stabilization was not clear since they described a random and even distribution between emulsion phases, likely driven by other aspects of their chemical structure, as the composition and proportion of lignin and carbohydrate moieties, for example. It is noteworthy to mention that the present study did not investigate the origin of LCC's (wood or its processing) or whether the extract composition, extraction process, or further processing treatments can affect the type and amount of LCC's formed.
Lignin structure itself also contains a diversity of substructures (e.g., side and end groups), interlinkages (e.g., β-O-4, β-5, 5-5/4-O-β, β-1, and β-β), and subunits (i.e., H, G, and S) (Fig. 1B, C, and D, respectively).27,34–36 Such structural differences in stabilizers found adsorbed and unadsorbed were also investigated. A semi-quantitative analysis revealed a substantial increase in the abundance of syringyl (S) lignin type (δC/δH 103.9/6.68 ppm) for GX samples recovered from interface to continuous phase (Table 1). This result indicated that molecules containing guaiacyl (G) lignin type had a more clear function in oil droplet stabilization than the S lignin. Indeed, higher lignin hydrophobicity has being associated with the occurrence of condensation reactions,29 in which G lignin units have more sites for coupling. Although this finding might suggest a certain advantage of softwood-based over hardwood-based stabilizers in emulsion stabilization, due to the greater abundance of G lignin in the former, long-term stability of GX has being reported to be much better compared to GGM,13 indicating that other structural attributes contribute to the stabilization mechanisms. Strong signals for G lignin (δC/δH 110.9/6.94 ppm) were observed in all GGM samples recovered from emulsion phases (except for epGGM recovered from the continuous phase in which only a weak signal was observed) and weak signals for p-hydroxyfenyl (H) lignin (δC/δH 127.5/7.21 ppm) were only observed in the GGM samples more concentrated in lignin (i.e., sdGGM, esGGM and epGGM recovered from interface and esGGM recovered from continuous phase) (ESI, Table S2†). It is noteworthy to be aware that values obtained for the relative amount of H, S, and G lignin by semi-quantitative analysis did not represent the absolute values, being used only for comparison between similar samples. Particularly herein, this analysis was exclusively used to track the fractionation of the sample between the emulsion phases.
The variation in polarity of lignin caused by the presence of specific side/end groups seemed to play a role on anchoring certain lignin structures in the interface (Fig. 1). Lignin oxidized side chain (δC/δH 106.4/7.20–7.06 ppm)29 and dehydroconiferyl alcohol structures (δC/δH 34.4/1.67 ppm)37 were predominantly identified in GGM and GX stabilizers recovered from the interface (i.e., sdGGM, esGGM, epGGM, sdGX, and esGX). These findings connected such structures with an active participation in the oil droplet stabilization.
The type of lignin interlinkage varied between emulsifiers recovered from interface and continuous phase (ESI, Table S2†). The β-aryl ether (β-O-4) was identified in all GGM and GX samples recovered from continuous phase and interface (δC/δH 71.4/4.71 ppm),37 with exception of the lignin-depleted epGGM sample from continuous phases (Table 1). Similarly, GGM and GX recovered from samples more purified from lignin did not exhibit cross-signals due to the presence of phenylcoumaran (β-5) (δC/δH 87.2/5.43 ppm).37 Dibenzodioxocin (5-5/4-O-β) structures (δC/δH 86.2/3.91 ppm),37,38 which had been described as end-wise structures originated at the last stage of lignin supramolecular structure biosyntehsis,38,39 were only identified in sdGGM, esGGM and esGX stabilizers recovered from interface. This interesting result evidenced the efficiency of dibenzodioxocin to anchor lignin to the oil droplet, likely explained by the high hydrophobicity of such condensed structure. Cross-signals due to the presence of β-β (δC/δH 85.1/4.61 ppm and 42.3/1.84 ppm for resinol and secoisolariciresinol, respectively) and β-1 (δC/δH 81.2/5.00 ppm) were more abundant in GGM than in GX recovered samples,27 in accordance with results for their presence (or absence) in original GX samples (ESI, Table S5†). Moreover, recovered GGM and GX samples richer in lignin moiety, including G lignin units, presented a higher diversity of lignin interlinkage patterns.
Although the structures of hemicelluloses can differ in carbohydrate composition, chain sizes, pattern of substitutions, and acetylation,40–42 no clear distribution of various populations of hemicelluloses between emulsion phases was confirmed from the results. Only a small effect of galactose moiety was observed due to the absence of its cross-signal in esGGM from interface (δC/δH 105.0/4.26 ppm), but its preservation in the esGGM from continuous phase. It is also possible that low galactose content in esGGM from interface could have affected its identification by making the typical cross-signal not visible. Galactosyl side chains are typical structural feature in GGM from softwood, contrasting with the glucomannan from hardwood in which galactosyl substitutions are not present.42 Interestingly, the selective concentration of more linear carbohydrate in the interface suggested that this conformation could favor oil droplet covering by hemicelluloses. No or less substitution results in more insoluble packed polysaccharide morphology that are more compact,43 and could act as soft-Pickering type particles assisting interfacial stabilization by adsorbing at the droplet and forming a protective layer that prevents coalescence.1,44 However, the effect of carbohydrates linearity in the stabilization of emulsions still requires further investigation. Apart from this, other aspects of the carbohydrate structure were quite similar in stabilizers recovered from interface and continuous phase. Acetylation at C-2 and C-3 of mannosyl units and in C-2, C-3, and C-2,3 of xylosyl units was observed for the recovered GGM and GX, respectively. The structure of the main carbohydrate in GGM and GX were also confirmed.
Therefore, the overall results indicated that the carbohydrates described a more passive drive between emulsion phases. However, the lignin transported carbohydrates (chemically or physically associated) and actively anchored them at the interface according to specific structural attributes, including benzylether and phenylglycoside bonds, lignin type (especially G lignin moiety), lignin interlinkages (especially dibenzodioxocin – 5-5/4-O-β), and end groups (oxidized side chain and dehydroconiferyl alcohol structures) (Table 2).
| Adsorbed in the interface | Unadsorbed in the continuous phase | ||
|---|---|---|---|

|
Benzylether lignin-carbohydrate bond |

|
Phenylglycoside lignin-carbohydrate bond |
| Lignin: guaiacyl lignin type, dibenzodioxocin, oxidized side chain, and dehydroconiferyl alcohol | Lignin: syringyl lignin type | ||
| Hemicelluloses: more linear molecules | Hemicelluloses: more branched molecules | ||
Role of lignin in the stabilization on emulsion
 | ||
| Fig. 3 Global Turbiscan Stability Index (TSI) of the emulsions prepared using sdGGM, esGGM, epGGM, sdGX, esGX, and epGX as stabilizers (A). Measurements were performed at days 0, 7, and 30. Morphology of aggregates collected from the top of emulsions stabilized by esGGM and esGX at day 0 (B). | ||
Notably high heterogeneity in composition is not always an advantage. Assessing the other side of the spectrum, more heterogeneous wood-based stabilizers, i.e., esGGM (52.5% purity) and esGX (63.9% purity) also produced unstable emulsions (33.4 and 34.0 TSI, respectively). In both esGGM and esGX, the residual lignin accounted for about 30–33% of dry content (ESI, Table S4†).15 Moreover, larger flocs, likely originated from lignin, were visualized on the surface of emulsions stabilized by es- samples (Fig. 3B) after the microfuidization step. This observation indicated that the process might have resulted in morphological modification, possibly unfolding the hydrophobic parts, leading to aggregation. Interestingly, the role of lignin structures in stabilization seems to behave as Poles of a continuum, in which it deficiency caused reduction of stabilizer capacity and resulted in less stable emulsion, and its excess, not used for emulsion stabilization, tended to flocculate each other.
The emulsion droplets were predominantly small in diameter (median of about 0.1 μm), although larger droplets were also confirmed by the identification of bi- or tri-modal distribution patterns (Fig. 4). The occurrence of flocculation and coalescence was also confirmed by optical measurements, supporting the increasing droplet size during storage for certain emulsions (ESI, Fig. S4†). The variation in D[3,2] was investigated to assess the small-sized droplets, revealing changes in their size (Fig. 5). In general, emulsions stabilized by sdGGM, sdGX, and epGX pursued lower droplet sizes (0.06–0.07 μm) and only minimum increase in droplet size during storage (4.5–11.4% droplet size increment), corroborating previous discussion on stability. In turn, the droplet size in emulsions stabilized by esGGM (0.08 μm) and esGX (0.09 μm) were higher than that for sdGGM, sdGX, and epGX at day 0, had a lower increment rate or even decreased in size during storage, confirming that despite lignin flocculation (Fig. 3B), on regards to small droplets the emulsion stabilized by es- samples presented a satisfactory stability. In addition to the presence of flocs, physical attributes of esGGM and esGX, as their particle-type, also contributed to the high droplet size of their correspondent emulsions. Although the final droplet size of emulsions stabilized by particles is larger than that stabilized by molecules, the greater adsorption energy required for the formation of emulsions stabilized by particles results in a rigid interfacial layer. This enhances resistance against desorption and, consequently, minimizes changes in the droplet size during storage.49 The higher droplet size of emulsion stabilized by epGGM (D[3,2] higher than 0.09 μm) is also consistent with its lower stability during storage (higher global TSI value). Both results were likely caused by the lower lignin content of epGGM, which harmed its capacity of perform long-term stabilization.
 | ||
| Fig. 4 Droplet size distribution in days 0, 1, 7, and 30 of emulsions stabilized by sdGGM (A), esGGM (B), epGGM (C), sdGX (D), esGX (E), and epGX (F) n = 3. | ||
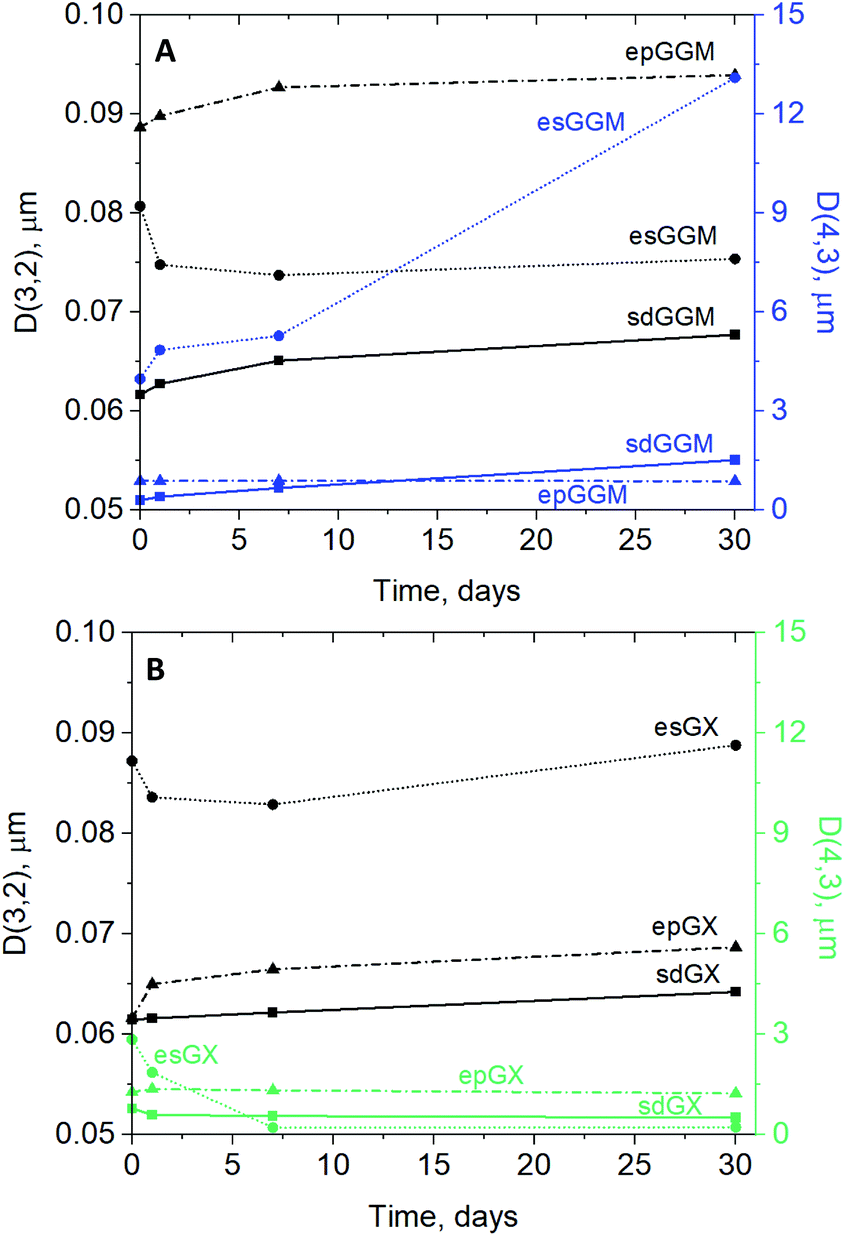 | ||
| Fig. 5 Surface-average diameter D[3,2] and volume-average diameter [4,3] at days 0, 1, 7, and 30 of emulsions stabilized by GGM (i.e., sdGGM, esGGM, epGGM) (A) and GX (i.e., sdGX, esGX, and epGX) (B). | ||
The D[4,3] assessment, which is sensitive to the size variation of large droplets, revealed a similar droplet size for sd- and ep- samples during 30 days storage for GGM and GX samples (0.3–1.5 μm) (Fig. 5). The large-sized droplets in emulsions prepared using esGGM, which was already higher than its counterparts at day 0 (4.0 μm), substantially increased during storage (day 1 = 4.9 μm; day 7 = 5.3 μm; and day 30 = 13.1 μm) as a result of flocculation and coalescence. The unexpected decreasing of large-sized droplets for esGX during storage (from 2.9 μm at day 0 to 0.3 μm at day 30) indicated a possible problem in sampling large particles flocculated in the upper part of emulsion (samples were collected from half height of the emulsion volume), resulting in the assessment of small droplets only.
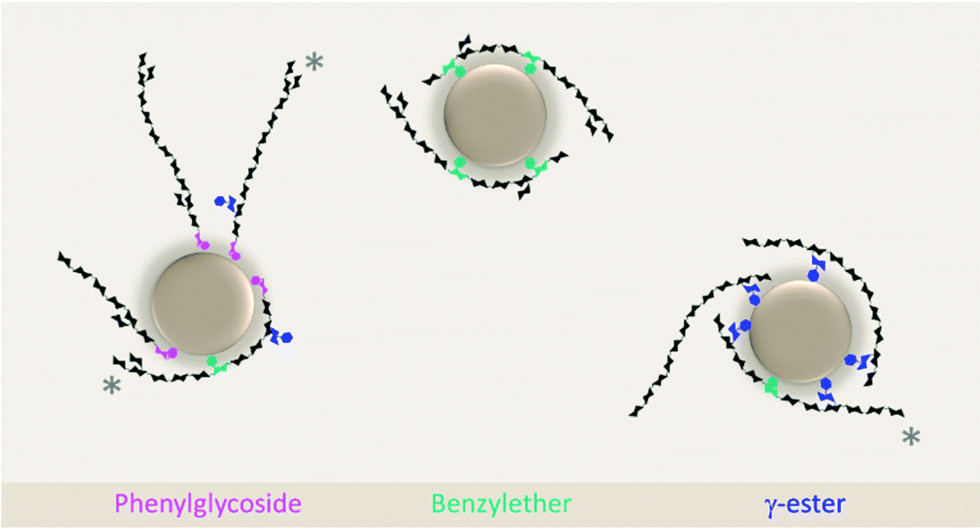 | ||
| Fig. 6 Hypothetical anchoring patterns of phenylglycoside (pink), benzylether (green), and γ-ester (blue) structures in the interface of the oil droplet in an oil-in-water emulsion. Gray asterisks (*) mark structures containing simultaneously more then one type of lignin-carbohydrate linkage. | ||
On the other hand, molecules containing phenylglycoside depend on other structural attributes to perform efficient physical stabilization of oil droplet. In phenylglycoside structures, the lignin-carbohydrate bond occurs only at the reducing end of the hemicellulose chain and is limited to one bond per molecule, placing the hydrophobic sites horizontally aligned to the hemicelluloses.12 Thus, after lignin moiety anchoring to the oil droplet, the hemicellulose tail is likely oriented tangentially to the oil droplet, where only a weaker physical interfacial stabilization is performed. It might be possible that other hydrophobic sites along the hemicelluloses tail (e.g., benzylether and γ-ester bonds) could create additional anchoring points in phenylglycoside-containing stabilizers. However, due to the tangential alignment of the hemicellulose tail towards the oil droplet a certain spacing between the phenylglycoside and the subsequent hydrophobic site might be necessary to allow the bending of the molecule to establish new anchoring points.
Besides lignin involved in LCC bonds, it is also reasonable to assume that a certain amount of free-lignin from samples could be involved in the stabilization of emulsion as amphiphilic surfactant or by Pickering-mechanism, as previously reported.1,51,52 This was corroborated by the concentration of lignin structures in the interface of the studied emulsions (Table 1), where they performed active stabilization. Although typically associated to hydrophobic nature, the lignin also contains hydrophilic moieties (due to certain functional groups) that, when preserved during processing conditions, have the capacity to provide an amphiphilic behavior to the lignin and,53 consequently, favors it function as stabilizer.54 However, the use of lignin as emulsion stabilizer is still limited by its complex and heterogeneous structure.55 Therefore, to allow novel applications, the preparation of more homogeneous lignin nanoparticles, with tuned shape, size, and surface activity have been recently investigated.54–58 Having spherical, homogeneous, and smaller sizes, the lignin nanoparticles are stable in a wide range of pH and ionic strength and performs better stabilization of Pickering emulsions.53,54,56 Such emulsion systems possess various possibilities of application still to be explored, including in life science products.
It may also be possible that certain types of nanostructured assemblies/aggregates (from hemicelluloses and lignin) that behave as particles and form Pickering type emulsions are involved in the stabilization mechanism of the emulsions herein investigated. The tendency of lignin to form aggregates in certain conditions is already know.59,60 In regards to hemicelluloses, this possibility is supported by recent findings that demonstrated that GGM is only partially soluble and the existence of colloidal entities, i.e. aggregates and assemblies, is quite frequent.61–63 Similarly, the occurrence of aggregates and assemblies from GX in colloidal state is also possible. The chemical heterogeneity of GGM and GX is likely behind their heterogeneous conformational structure (i.e., molecules and particle-like morphology), in which the association at the interface depends on the LCC bond type.
Elucidation of chemical and structural attributes of birch-wood stabilizers (for supporting the findings obtained for emulsion stabilization)
 | ||
| Fig. 7 Mass balance, chemical and structural aspects of sdGX, esGX, and epGX obtained from birch-wood extract. Chemical composition of samples includes glucuronoxylan (GX), glucomannan (GM), pectin, and lignin contents. Structural aspects as the average value for the degree of acetylation (DSAc), molar mass (Mw) and the dispersity index (DI) of molar mass distribution are also presented. | ||
The sdGX presented a heterogeneous chemical composition (68.9% glucuronoxylan, 6.3% glucomannan, 4.0% pectin, and 19.9% lignin) (Fig. 7). Its lower molar mass value observed for sdGX (4.2 kDa) in comparison to larger values reported for glucuronoxylan elsewhere (16.5–23.7 kDa)26,64 confirmed the occurrence of a partial hydrolysis of carbohydrates during PHWE. This was also evidenced by the broader molar mass distribution observed for sdGX (DI = 3.5) (ESI, Fig. S5†), that contrasted to the relatively monodisperse nature of purer glucuronoxylans (1.1–1.7).26,64 During the PHWE, part of the acetyl groups from hemicelluloses were utilized as chemical source for the hydrolysis.65 As consequence, the residual degree of acetylation (DSAc) of sdGX was only 0.46, a lower value compared to that attributed to native glucuronoxylan (0.7).47 After such deacetylation, acetylation distribution of sdGX (0.5:1.0:0.2, at C-2, C-3, C-2,3 respectively) was still consistent with that reported for other hardwood hemicelluloses, predominanlty observed at the C-3 position.66,67 The S/G ratio of sdGX (6.8) was substantially higher than the value typically reported for birch-wood (3.25),68 suggesting a higher susceptibility of S lignin units to be hydrolyzed/removed by PHWE. However, the value was still within the range reported for birch-wood when various tissues are systematically assessed (1.0–7.0).69 Again, although the values obtained for S/G ratio by semi-quantitative analysis did not represent the absolute values, an interesting variation in the S and G content caused by the ethanol-fractionation was herein observed and discussed for the samples obtained from the sdGX.
The antisolvent fractionation concentrated lignin moities in esGX (63.9% glucuronoxylan, 2.8% glucomannan, 2.9% pectin, and 30.4% lignin), but only a slight variation in the S/G ratio (6.1) compared to the sdGX was noticed. Low molar mass carbohydrates populations (1.2 kDa) were preferentially solubilized by the ethanol. Such populations were also more homogeneous (dispersity index of 1.9) than those identified in sdGX and epGX, likely due to the reduced presence of carbohydrates other than glucuronoxylan (less than 6%). Comparatively, the glucuronoxylan in esGX was slightly less subtituted by acetyl groups (DSAc of 0.40) than that in sdGX and epGX and less substitutes at C-3 position resulted (0.7:1.0:0.3, at C-2, C-3, C-2,3 respectively).
The epGX, although predominantly formed by glucuronoxylan (75.2%), was also richer in glucomannan (7.6%) and pectin (4.5%) than the sdGX. Moreover, the antisolvent fractionation proved to be molar mass dependent, precipiting preferentially higher molar mass (4.9 kDa) carbohydrates. The high dispersity index of epGX (3.1) corroborated with the diversity of carbohydrates in the sample. However, the DSAc (0.48) and acetylation distribution (0.5:1.0:0.2, at C-2, C-3, C-2,3 respectively) were similar to that observed in sdGX sample (ESI, Table S4†). Interestingly, epGX was substantially purified from lignin (12.7%) and especially depleted from lignin guaiacyl units (S/G ratio of 11.6). These two aspects in combination contributed to the increased polarity of the epGX sample.
 | ||
| Fig. 8 Cross-signal for phenylglycoside (PG), benzylether (BE), and γ-ester (GE) bonds identified in sdGX (A), esGX (B), and epGX (C). | ||
The presence of phenylglycoside and γ-ester was identified in all GX samples (i.e., sdGX, esGX, and epGX), indicating a nonspecific effect of the antisolvent fractionation of these LCC bonds. Multiple contours at the region assigned for phenylglycoside (δC/δH 104.5–99.8/5.18–4.75 ppm) supported the participation of various types of carbohydrates in this bond,29,31,33,34 possibly including the residual glucomannan moieties. The abundance of acidic sites in all GX, e.g., glucuronic acid branches and acetyl groups substitutions, likely favored the formation of phenylglycoside (via acid-catalyzed hydrolysis)34,70 and stable γ-ester bonds.29,32
Unlike with the β-O-4, β-5, and (resinol) β-β interlinkages that were identified in all GX samples (i.e., sdGX, esGX, and epGX), 5-5/β-O-4 interlinkage was only identified in sdGX and esGX. Likely the higher abundance of G lignin moiety in sdGX and esGX provided more available sites for 5-5 interlinkage than in epGX. This finding also confirmed the increased hydrophobicity of lignin moiety connected to the presence of condensed structures.29 The main assignments for lignin structure in GX samples are shown in ESI (Table S5†).
Conclusions
Preserving certain lignin structures in wood-based stabilizers favored their anchoring to the interface and enhanced their emulsion stabilization capacity. Benzylether structures were found exclusively in the interface of oil-in-water emulsions, suggesting their participation in the physical stabilization of oil droplets. Phenylglycoside structures, although observed in the interface of emulsions prepared by some of the wood-based stabilizers, were more frequent in the continuous phase, suggesting that such structures are less active in physical droplet stabilization. Moreover, the interface was substantially concentrated with hydrophobic lignin substructures, such as guaiacyl lignin type, dibenzodioxocin interlinkage (5-5/4-O-β), and certain end groups (oxidized side chain and dehydroconiferyl alcohol structures). They likely compensated for the lack of a hydrophobic domain in hemicelluloses, creating a suitable balance of hydrophobic-lipophilic domains in the wood-based stabilizers (either by chemical bonding or electrostatic interaction). Finally, the carbohydrate moiety in wood-based stabilizers seemed to perform a passive drive between emulsion phases, in which the lignin (including all aspects herein discussed) enabled carbohydrate-delivery to the interface. Recognizing that the chemical complexity of natural stabilizers benefits their stabilization capacity and makes it possible to design simple and mild extraction methods in biorefinery industries. This would not only avoid unnecessary purification steps but also prioritize the preservation of those attributes active in the stabilization of interfaces. The new understanding herein achieved is expected to enable further studies including exploring the use of vegetable oil alternatives in the design of optimized interfaces, which would enable novel and sustainable applications for wood-based stabilizers, including in food, pharmaceuticals, and cosmetics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Troy Faithfull for his help editing the manuscript. We also thank Petri Kilpeläinen from Natural Resources Institute Finland (Luke) for providing the GGM and GX spray-dried samples and for fruitful discussions. The facilities and expertise of the HiLIFE NMR unit at the University of Helsinki, a member of Instruct-ERIC Centre Finland, FINStruct, and Biocenter Finland are gratefully acknowledged. DMdC acknowledges Tandem Forest Values for funding (TFV 2018-0016). We thank Prof. Maija Tenkanen for her support and encouragement for developing this project.Notes and references
- K. S. Mikkonen, Green Chem., 2020, 22, 1019–1037 RSC.
- D. J. McClements, Food emulsions: principles, practices, and techniques, CRC press, 2015 Search PubMed.
- L. Bai, S. Huan, O. J. Rojas and D. J. McClements, J. Agric. Food Chem., 2021, 69, 8944–8963 CrossRef CAS PubMed.
- S. Sivapratha and P. Sarkar, Crit. Rev. Food Sci. Nutr., 2018, 58, 877–892 CrossRef CAS PubMed.
- S. Petersen and J. Ulrich, Chem. Eng. Technol., 2013, 36, 398–402 CrossRef CAS.
- D. J. McClements and E. Decker, J. Agric. Food Chem., 2018, 66, 20–35 CrossRef CAS PubMed.
- A.-J. Choi, C.-J. Kim, Y.-J. Cho, J.-K. Hwang and C.-T. Kim, Food Bioprocess Technol., 2011, 4, 1119–1126 CrossRef CAS.
- B. Ozturk and D. J. McClements, Curr. Opin. Food Sci., 2016, 7, 1–6 CrossRef.
- D. J. McClements, L. Bai and C. Chung, Annu. Rev. Food Sci. Technol., 2017, 8, 205–236 CrossRef PubMed.
- K. S. Mikkonen, D. Merger, P. Kilpeläinen, L. Murtomäki, U. S. Schmidt and M. Wilhelm, Soft Matter, 2016, 12, 8690–8700 RSC.
- K. S. Mikkonen, C. Xu, C. Berton-Carabin and K. Schroën, Food Hydrocolloids, 2016, 52, 615–624 CrossRef CAS.
- M. Lehtonen, M. Merinen, P. O. Kilpeläinen, C. Xu, S. M. Willför and K. S. Mikkonen, J. Colloid Interface Sci., 2018, 512, 536–547 CrossRef CAS PubMed.
- M. H. Lahtinen, F. Valoppi, V. Juntti, S. Heikkinen, P. O. Kilpeläinen, N. H. Maina and K. S. Mikkonen, Front. Chem., 2019, 7, 1–18 CrossRef.
- P. Naidjonoka, M. Fornasier, D. Pålsson, G. Rudolph, B. Al-Rudainy, S. Murgia and T. Nylander, Colloids Surf., B, 2021, 203, 111753 CrossRef CAS PubMed.
- D. M. d. Carvalho, M. H. Lahtinen, M. Lawoko and K. S. Mikkonen, ACS Sustainable Chem. Eng., 2020, 8, 11795–11804 CrossRef.
- E. Dickinson, Soft Matter, 2008, 4, 932–942 RSC.
- A. S. Lim and Y. H. Roos, J. Food Eng., 2016, 171, 174–184 CrossRef CAS.
- A. Gharsallaoui, K. Yamauchi, O. Chambin, E. Cases and R. Saurel, Carbohydr. Polym., 2010, 80, 817–827 CrossRef CAS.
- P. Oinonen, L. Zhang, M. Lawoko and G. Henriksson, Phytochemistry, 2015, 111, 177–184 CrossRef CAS PubMed.
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519–546 CrossRef CAS PubMed.
- R. Vanholme, B. Demedts, K. Morreel, J. Ralph and W. Boerjan, Plant Physiol., 2010, 153, 895–905 CrossRef CAS PubMed.
- O. M. Terrett and P. Dupree, Curr. Opin. Biotechnol., 2019, 56, 97–104 CrossRef CAS PubMed.
- S. Kirjoranta, A. Knaapila, P. Kilpeläinen and K. S. Mikkonen, Cellulose, 2020, 27, 7607–7620 CrossRef CAS.
- M. Bhattarai, L. Pitkänen, V. Kitunen, R. Korpinen, H. Ilvesniemi, P. O. Kilpeläinen, M. Lehtonen and K. S. Mikkonen, Food Hydrocolloids, 2019, 86, 154–161 CrossRef CAS.
- P. O. Kilpeläinen, S. S. Hautala, O. O. Byman, L. J. Tanner, R. I. Korpinen, M. K. J. Lillandt, A. V. Pranovich, V. H. Kitunen, S. M. Willför and H. S. Ilvesniemi, Green Chem., 2014, 16, 3186–3194 RSC.
- D. M. de Carvalho, J. Berglund, C. Marchand, M. E. Lindström, F. Vilaplana and O. Sevastyanova, Carbohydr. Polym., 2019, 220, 132–140 CrossRef PubMed.
- H. Kim, J. Ralph and T. Akiyama, BioEnergy Res., 2008, 1, 56–66 CrossRef.
- M. Balakshin, E. Capanema and A. Berlin, in Studies in Natural Products Chemistry, ed. R. Atta ur, Elsevier, Amsterdam, 2014, vol. 42, pp. 83–115 Search PubMed.
- N. Giummarella and M. Lawoko, ACS Sustainable Chem. Eng., 2017, 5, 5156–5165 CrossRef CAS.
- H. Nishimura, A. Kamiya, T. Nagata, M. Katahira and T. Watanabe, Sci. Rep., 2018, 8, 1–11 CAS.
- A. Martínez-Abad, N. Giummarella, M. Lawoko and F. Vilaplana, Green Chem., 2018, 20, 2534–2546 RSC.
- M. Balakshin, E. Capanema, H. Gracz, H.-m. Chang and H. Jameel, Planta, 2011, 233, 1097–1110 CrossRef CAS PubMed.
- X. Du, M. Pérez-Boada, C. Fernández, J. Rencoret, J. C. del Río, J. Jiménez-Barbero, J. Li, A. Gutiérrez and A. T. Martínez, Planta, 2014, 239, 1079–1090 CrossRef CAS.
- N. Giummarella, M. Balakshin, S. Koutaniemi, A. Kärkönen and M. Lawoko, J. Exp. Bot., 2019, 70, 5591–5601 CrossRef CAS PubMed.
- J. Ralph, C. Lapierre and W. Boerjan, Curr. Opin. Biotechnol., 2019, 56, 240–249 CrossRef CAS.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- C. S. Lancefield, H. L. J. Wienk, R. Boelens, B. M. Weckhuysen and P. C. A. Bruijnincx, Chem. Sci., 2018, 9, 6348–6360 RSC.
- E. M. Kukkola, S. Koutaniemi, E. Pöllänen, M. Gustafsson, P. Karhunen, T. K. Lundell, P. Saranpää, I. Kilpeläinen, T. H. Teeri and K. V. Fagerstedt, Planta, 2004, 218, 497–500 CrossRef CAS PubMed.
- N. Terashima, M. Yoshida, J. Hafrén, K. Fukushima and U. Westermark, Holzforschung, 2012, 66, 907–915 CAS.
- D. M. de Carvalho, A. Martínez-Abad, D. V. Evtuguin, J. L. Colodette, M. E. Lindström, F. Vilaplana and O. Sevastyanova, Carbohydr. Polym., 2017, 156, 223–234 CrossRef.
- A. Teleman, M. Nordström, M. Tenkanen, A. Jacobs and O. Dahlman, Carbohydr. Res., 2003, 338, 525–534 CrossRef CAS PubMed.
- A. Ebringerová, Z. Hromádková and T. Heinze, in Polysaccharides I: Structure, Characterization and Use, ed. T. Heinze, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, pp. 1–67, DOI:10.1007/b136816.
- S. Dumitriu, Polysaccharides: Structural Diversity and Functional Versatility, CRC Press, 2004 Search PubMed.
- E. Dickinson, Food Hydrocolloids, 2017, 68, 219–231 CrossRef CAS.
- M. H. Lahtinen, F. Valoppi, V. Juntti, S. Heikkinen, P. O. Kilpeläinen, N. H. Maina and K. S. Mikkonen, Front. Chem., 2019, 7, 1–18 CrossRef.
- Y.-F. Li, P.-P. Yue, X. Hao, J. Bian, J.-L. Ren, F. Peng and R.-C. Sun, RSC Adv., 2020, 10, 4657–4663 RSC.
- E. Sjöström, in Wood Chemistry (Second Edition), ed. E. Sjöström, Academic Press, San Diego, 1993, pp. 51–70 Search PubMed.
- K. Alba, M. Dimopoulou and V. Kontogiorgos, LWT, 2021, 144, 111235 CrossRef CAS.
- D. Gonzalez Ortiz, C. Pochat-Bohatier, J. Cambedouzou, M. Bechelany and P. Miele, Engineering, 2020, 6, 468–482 CrossRef.
- V. Mikulcová, R. Bordes, A. Minařík and V. Kašpárková, Food Hydrocolloids, 2018, 80, 60–67 CrossRef.
- Z. Wei, Y. Yang, R. Yang and C. Wang, Green Chem., 2012, 14, 3230–3236 RSC.
- L. B. Brenelli, L. R. B. Mariutti, R. Villares Portugal, M. A. de Farias, N. Bragagnolo, A. Z. Mercadante, T. T. Franco, S. C. Rabelo and F. M. Squina, Ind. Crops Prod., 2021, 167, 113532 CrossRef CAS.
- M. Österberg, M. H. Sipponen, B. D. Mattos and O. J. Rojas, Green Chem., 2020, 22, 2712–2733 RSC.
- M. H. Sipponen, M. Smyth, T. Leskinen, L.-S. Johansson and M. Österberg, Green Chem., 2017, 19, 5831–5840 RSC.
- W. Zhao, B. Simmons, S. Singh, A. Ragauskas and G. Cheng, Green Chem., 2016, 18, 5693–5700 RSC.
- P. Figueiredo, M. H. Lahtinen, M. B. Agustin, D. M. de Carvalho, S.-P. Hirvonen, P. A. Penttilä and K. S. Mikkonen, ChemSusChem, 2021, 14, 1–14 CrossRef.
- M. B. Agustin, P. A. Penttilä, M. Lahtinen and K. S. Mikkonen, ACS Sustainable Chem. Eng., 2019, 7, 19925–19934 CrossRef CAS.
- M. R. V. Bertolo, L. B. Brenelli de Paiva, V. M. Nascimento, C. A. Gandin, M. O. Neto, C. E. Driemeier and S. C. Rabelo, Ind. Crops Prod., 2019, 140, 111591 CrossRef CAS.
- M. H. Sipponen, H. Lange, M. Ago and C. Crestini, ACS Sustainable Chem. Eng., 2018, 6, 9342–9351 CrossRef CAS PubMed.
- W. Zhao, L.-P. Xiao, G. Song, R.-C. Sun, L. He, S. Singh, B. A. Simmons and G. Cheng, Green Chem., 2017, 19, 3272–3281 RSC.
- M. Bhattarai, I. Sulaeva, L. Pitkänen, I. Kontro, M. Tenkanen, A. Potthast and K. S. Mikkonen, Carbohydr. Polym., 2020, 241, 116368 CrossRef CAS PubMed.
- M. Bhattarai, F. Valoppi, S.-P. Hirvonen, S. Hietala, P. Kilpeläinen, V. Aseyev and K. S. Mikkonen, Food Hydrocolloids, 2020, 102, 105607 CrossRef CAS.
- S. Kishani, F. Vilaplana, W. Xu, C. Xu and L. Wågberg, Biomacromolecules, 2018, 19, 1245–1255 CrossRef CAS PubMed.
- A. Jacobs and O. Dahlman, Biomacromolecules, 2001, 2, 894–905 CrossRef CAS PubMed.
- T. E. Amidon and S. Liu, Biotechnol. Adv., 2009, 27, 542–550 CrossRef CAS PubMed.
- D. Evtuguin, J. Tomás, A. S. Silva and C. Neto, Carbohydr. Res., 2003, 338, 597–604 CrossRef CAS PubMed.
- A. Teleman, M. Tenkanen, A. Jacobs and O. Dahlman, Carbohydr. Res., 2002, 337, 373–377 CrossRef CAS PubMed.
- Z. Wang, S. Winestrand, T. Gillgren and L. J. Jönsson, Biomass Bioenergy, 2018, 109, 125–134 CrossRef CAS.
- K. V. Fagerstedt, P. Saranpää, T. Tapanila, J. Immanen, J. A. A. Serra and K. Nieminen, Plants, 2015, 4, 183–195 CrossRef PubMed.
- D. M. Clode, Chem. Rev., 1979, 79, 491–513 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc02891j |
| This journal is © The Royal Society of Chemistry 2021 |