
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing the activity of gold supported catalysts by oxide coating: towards efficient oxidations†
Camila P.
Ferraz
 ab,
Sara
Navarro-Jaén
a,
Liane M.
Rossi
b,
Franck
Dumeignil
a,
Mohamed N.
Ghazzal
c and
Robert
Wojcieszak
*a
ab,
Sara
Navarro-Jaén
a,
Liane M.
Rossi
b,
Franck
Dumeignil
a,
Mohamed N.
Ghazzal
c and
Robert
Wojcieszak
*a
aUniv. Lille, CNRS, Centrale Lille, Univ. Artois, UMR 8181 – UCCS – Unité de Catalyse et Chimie du Solide, F-59000 Lille, France. E-mail: robert.wojcieszak@univ-lille.fr
bDepartamento de Química Fundamental, Instituto de Química, Universidade de São Paulo, 05508-000 São Paulo, Brazil
cInstitute de Chimie Physique, UMR 8000 CNRS, Université Paris-Saclay, 91405 Orsay, France
First published on 13th October 2021
Abstract
Core–shell SiO2@Au@TiO2 materials have shown excellent catalytic performances in the base-free oxidation of furfural (FF) to furoic acid (FA). The enhanced catalytic behaviour with respect to that observed over supported SiO2@TiO2@Au catalysts suggests a key role of the TiO2 shell, which could prevent Au deactivation by irreversible FA adsorption, improve metal–oxide interaction and presumably, give rise to the formation of new active sites located at the perimeter of the Au–TiO2 interface.
With anthropogenic climate change and depletion of fossil fuels being major concerns in our days, a shift in the energy paradigm towards a “green” future has become imperative.1–3 In this context, the use of lignocellulosic biomass as a feedstock to produce biofuels, energy, and renewable chemicals appears as a promising alternative for reducing greenhouse gases (GHGs) emissions and for mitigating climate change.4–7
Among the lignocellulosic biomass derivatives, furfural (FF) belongs to the top 10 platform molecules, which can be further valorised into various key chemicals.8,9 In particular, oxidation of FF to produce furoic acid (FA) (Fig. 1) is of great interest due to the application of the latter as a starting material in the agrochemical, pharmaceutical, flavour, and fragrances industries.7,10
 | ||
| Fig. 1 Principle of synthesis of furoic acid (FA) from furfural (FF). | ||
FA is industrially produced from FF via a Cannizzaro reaction in the presence of a NaOH aqueous solution and strong oxidant agents, followed by an acidification step using H2SO4.11 However, in the envisaged “green” context, the corrosive and toxic nature of the employed reagents makes this process undesirable.7,12 Thereby, research efforts in the last decades have aimed at accomplishing such an oxidation process in a sustainable way, avoiding the use of a base and employing “green” oxidants such as H2O2, O2, or air.13–15
Heterogeneous catalysts consisting of gold nanoparticles (AuNPs) dispersed on inorganic oxides have shown very promising results for the partial oxidation of FF under green oxidation conditions4,16–18 (Table S1†). These materials’ activity, selectivity, and stability are governed by different parameters such as the metal particle size, their acid–base properties, and metal–support interaction.19,20 Thus, careful control of the catalyst structure constitutes a key aspect for obtaining stable catalysts while boosting the catalytic performance. Usually, higher oxidation activity is achieved only in the presence of an additional base, in aqueous or organic media. A proposed alternative to facilitate the workup procedure for product purification (neutralization of the additional homogeneous base by an acid, conventionally H2SO4 as aforementioned) is to bring the necessary basicity through the use of a basic support such as MgO, CaO, or hydrotalcite, which thus involves the design of new catalysts (Table S1†). Still, catalyst stability in aqueous media can be compromised due to the leaching of the support.21,22 Another drawback when working in homogeneous base-free conditions is the deactivation of the catalysts by irreversible adsorption of the formed acid on the metal surface.23,24 Taking this into account, we hypothesised that encapsulating Au nanoparticles within an oxide shell could solve the issue of catalyst deactivation due to FA adsorption. Since this strategy could hinder the access of the reactants to the active site, controlling the thickness of the oxide layer becomes essential. We selected TiO2 as the encapsulating oxide phase. Its non-basic character25 could avoid the support leaching during the reaction, whereas its combination with Au nanoparticles have shown promising results in oxidation reactions.26 This type of solids in which an oxide layer coats the metal particles has been proposed as a potential catalytic material due to enhanced control of interfacial sites which participate in the catalytic processes.27 However, their catalytic applications have been scarcely studied28,29 and to the best of our knowledge, they were never tested in the catalytic valorisation of biomass-derived products.
Herein, we report a highly active, selective, and stable “core–shell” material for base-free oxidation of furfural in water using air as an oxidant. The catalyst consists of SiO2-loaded AuNPs covered by an ultra-thin TiO2 oxide shell. We found that using a fully controlled synthesis of core–shell SiO2@Au@TiO2 materials (Fig. 2c), remarkable efficient heterogeneous catalysts can be prepared. The performance of the core–shell material was compared with a “conventional” system with AuNPs grown at the surface of a pre-formed SiO2@TiO2 core–shell support material (SiO2@TiO2@Au, Fig. 2a and Fig. S3–S7†) and that of a system with AuNPs partially embedded in the SiO2@TiO2 core–shell (SiO2@Auem@TiO2, Fig. 2b), meaning different types of metal–oxide interactions. The result is an enhancement of the catalytic performances in the oxidation of FF when a strong metal–support interaction (SMSI) is achieved, pointing out a clear correlation between the catalyst architecture and the catalyst performance.
 | ||
| Fig. 2 Schematic representation and TEM micrographs of the prepared catalysts. (a) SiO2@TiO2@Au, (b) SiO2@Auem@TiO2, (c) SiO2@Au@TiO2. | ||
The studied catalysts with a 2 wt% Au nominal content were prepared by a soft chemistry method previously described by a part of the present authors.30,31 To synthesize the “conventional” SiO2@TiO2@Au catalyst, AuNPs were loaded on pre-formed SiO2@TiO2 core–shell nanoparticles. Well-dispersed AuNPs deposited on the TiO2 shell were obtained, as revealed by TEM analysis (Fig. 2a and Fig. S3–S7†). Then, two different solids in which the extent of the metal–support interaction was increased were prepared by a modified synthesis method, using SiO2-loaded AuNPs as a starting material. In the SiO2@Auem@TiO2 catalyst, titanium isopropoxide (TTIP) was deposited on the surface of the SiO2@Au catalyst without specific control of the hydrolysis conditions (namely of pH and time). In this case, a “buried-like” structure consisting of partially embedded AuNPs was obtained.
The TiO2 shell was homogeneously deposited on the SiO2 surface via the intermediate titanium hydroxide (Ti(OH)4) formation, and Au nanoparticles were partially embedded into TiO2 (Fig. 2b). The same procedure was carried out to prepare the SiO2@Au@TiO2 catalyst, except that we used a slow, fully controlled TTIP hydrolysis.30,31 This method led to the formation of core–shell particles in which a homogeneous TiO2 shell with an average thickness of 3 nm encapsulated the AuNPs (Fig. 2c). Mean Au nanoparticle size calculated from TEM micrographs was ca. 5 nm in each catalyst.
The conversion achieved in the base-free oxidation of FF in water using air as an oxidant with the three Au catalysts under similar conditions is presented in Fig. 3. In addition, the blank experiment with the SiO2@TiO2 catalyst support is shown for comparison.
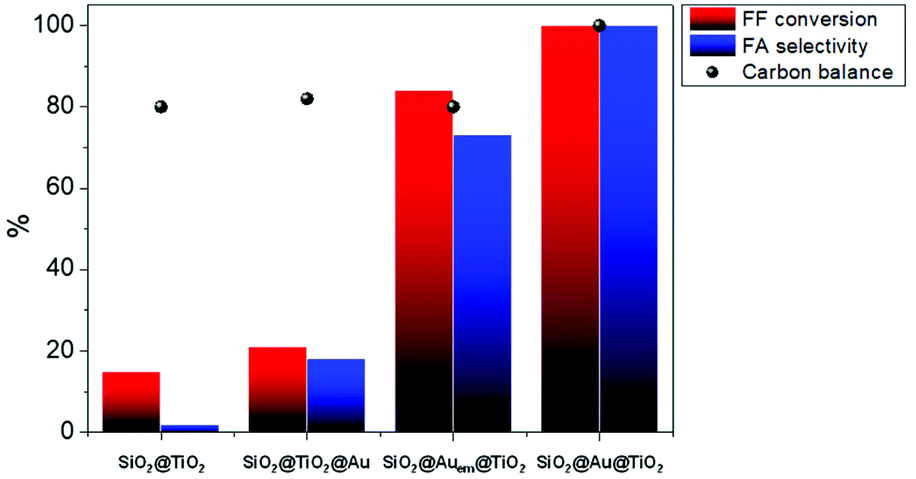 | ||
Fig. 3 FF conversion, selectivity to FA, and carbon balance over SiO2@TiO2, supported SiO2@TiO2@Au, partially embedded SiO2@Auem@TiO2 and core–shell SiO2@Au@TiO2 catalysts (FF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au ratio = 100, 2 h, 24 bar of air, 600 rpm). :Au ratio = 100, 2 h, 24 bar of air, 600 rpm). | ||
The SiO2@TiO2 catalyst support presents itself 15% conversion and yields mainly condensation products, confirming that Au is indispensable for the oxidation reaction. However, the sole presence of well-dispersed Au nanoparticles is not sufficient to boost the catalytic performance, according to the low conversion (21%) and selectivity (18%) values observed for the SiO2@TiO2@Au supported catalyst. The significant enhancement of the catalytic activity observed for the SiO2@Auem@TiO2 catalyst, where the TiO2 support partially embeds AuNPs, already suggests an essential role of the Au–TiO2 interface. The performance shown by the SiO2@Au@TiO2 core–shell catalyst, which quantitatively converted furfural into furoic acid, corroborates this hypothesis. The material with TiO2-encapsulated AuNPs thus presented remarkable catalytic activity compared to its supported and embedded counterparts, with even no by-products originating from side reactions detected during the catalytic cycle. In light of the promising results with 100% carbon balance, we also studied the influence of the Au loading on the catalytic behaviour of the SiO2@Au@TiO2 core–shell catalyst. The results are given in Table 1. Unlike the supported catalysts (Table 2), results using the core–shell materials demonstrate that the total and selective conversion of FF to FA can be achieved even at Au loadings as low as 0.25 wt%.
| Catalyst | Au content (wt%) | FF conversion (%) | FA selectivity (%) | Carbon balance (%) |
|---|---|---|---|---|
| SiO2@TiO2 | 0 | 15 | 2 | 80 |
| 0.25% SiO2@Au@TiO2 | 0.13 | 100 | >99 | >99 |
| 0.5% SiO2@Au@TiO2 | 0.53 | 100 | >99 | >99 |
| 1% SiO2@Au@TiO2 | 1.13 | 100 | >99 | >99 |
| 2% SiO2@Au@TiO2 | 2.02 | 100 | >99 | >99 |
| Catalyst | Au content (wt%) | FF conversion (%) | FA selectivity (%) | Carbon balance (%) |
|---|---|---|---|---|
| SiO2@TiO2 | 0 | 15 | 2 | 80 |
| 0.25% SiO2@TiO2@Au | 0.28 | 70 | 12 | 38 |
| 0.5% SiO2@TiO2@Au | 0.56 | 53 | 79 | 88 |
| 1% SiO2@TiO2@Au | 1.26 | 34 | 56 | 85 |
| 2% SiO2@TiO2@Au | 1.37 | 21 | 18 | 82 |
This finding could indicate that the new active species formed on the Au–TiO2 interface are much more active than gold itself in the oxidation of aldehydes. However, standard characterization by XRD (Fig. S8†) and XPS (Fig. S1 and S2†) did not permit to observe differences in the TiO2 structure. Indeed, the TiO2 layer deposited on gold is very thin (1–2 nm) and XPS analysis depth is estimated of about 8 nm.
Various interpretation concerning the way Au-based catalysts activity are documented, and commented below in the light of our new results presented herein:
(i) It has been proposed for Au-supported catalysts that the accessible contact zone between the nanoparticles and the support at the bottom edge of the nanoparticles plays an important role in the catalytic performance, with the active sites located at this specific rim position at the junction between the metallic nanoparticles and the oxidic carrier.27 Through this concept, optimizing the quantity of “rim sites” or “rim-like sites” could be a mean of accessing better catalytic performance.
(ii) The observed differences in the catalytic behaviour when AuNPs are supported or encapsulated under a TiO2 shell point towards a possible change in the reaction mechanism. With AuNPs exposed on the catalyst surface and in the absence of a base, the furfural molecule should adsorb on the surface-exposed gold and then follow the oxidation to the acid. Recently, Megías-Sayago et al.32 studied the base-free oxidation of glucose to gluconic acid over Au/C catalysts and suggested that the oxidation reaction occurs preferably on gold and not on the metal–support interface or the support, in good agreement with previous studies.33 In this hypothesis, the reaction occurs through the formation of a carbonyl conjugated radical, which is subsequently oxidized. Since O2 dissociation is not favoured on the Au surface, the activation of O2 takes place via the formation and dissociation of peroxide (OOH*) and hydrogen peroxide (HOOH*) intermediates.34 Consequently, in our SiO2@TiO2@Au supported catalysts, furfural would adsorb on AuNPs to be subsequently oxidized through hydrogen peroxide formation. Since the reaction, in this case, occurs on the Au surface, the deactivation of the catalyst is probably taking place due to the irreversible adsorption of the FA formed during the reaction.
(iii) When a TiO2 layer encapsulates AuNPs, furfural adsorption on the surface of gold should be excluded, which suggests that the oxidation process occurs through a different reaction mechanism. Whereas the SiO2@TiO2 catalyst support favoured the formation of condensation products, our SiO2@Au@TiO2 core–shell catalysts showed excellent catalytic performance, which confirms that the presence of Au is essential. The controlled architecture of the SiO2@Au@TiO2 catalyst generates an enhanced gold-titania surface interaction, with the active sites presumably located in the periphery of the TiO2/Au interface. In fact, some research groups have recently proposed the Au-assisted Mars-van Krevelen mechanism as possibly occurring during CO oxidation over Au/TiO2 catalysts, which entails the participation of lattice oxygen atoms near the periphery of Au/TiO2 in the oxidation process. The desorption of the CO2 formed generates an oxygen vacancy, which is subsequently replenished by O.35–37 The presence of Au in close interaction with the TiO2 support plays a fundamental role, presumably facilitating the formation of oxygen lattice vacancies in the Au/TiO2 perimeter thanks to the ability of gold to accommodate the excess electrons generated during the oxygen vacancies formation. Thus, in our case, furfural could adsorb on TiO2 and react with the lattice oxygen atoms located at the periphery of the TiO2/Au interface. The oxidation of furfural to furoic acid could proceed through the Au-assisted Mars-van Krevelen mechanism, that is, via the cyclic formation and replenishment of oxygen vacancies. Another possible mechanism could imply the adsorption of furfural on the surface of TiO2 and its subsequent oxidation through the formation of hydrogen peroxide on gold. Such a mechanism could take place assuming that diffusion of O2 through the TiO2 layer is possible. Regardless of the reaction path, furfural adsorption does not occur on the surface of the metal particles, which prevents the accumulation of reaction products on the metal surface leading to catalyst deactivation.
According to these hypotheses, the catalytic behaviour of the SiO2@Auem@TiO2 catalyst could be explained by the simultaneous occurrence of both mechanisms since Au can be found both on the surface or partially embedded in the TiO2 layer.
The present results pave the way towards the design of highly efficient catalysts for oxidation reactions. We demonstrate using variable catalyst design that the TiO2/Au interface has a key role in the reported enhancement of FF oxidation. Nevertheless, how the furfural oxidation proceeds in such a complex catalytic system is an aspect needing to be clarified. Thus, DFT studies and deep characterization of these materials are in progress to elucidate the mechanism of this reaction and the real nature of the extremely efficient active site.
Conclusions
TiO2-encapsulated AuNPs, SiO2@Au@TiO2 core–shell catalysts, have proved excellent activity, selectivity, and stability in the base-free oxidation of furfural using air as an oxidant. The materials showed a hundredfold catalytic activity increase with respect to conventional supported Au catalysts (SiO2@TiO2@Au), even using very low gold loadings, with 100% selectivity. The activity enhancement process could be explained by a change in the reaction mechanism induced by an extended TiO2–Au interaction in the core–shell catalysts. In the supported catalysts, furfural adsorption would take place on AuNPs, and possibly the deactivation of the catalyst is due to furoic acid adsorption. In the case of the core–shell catalysts, furfural adsorption would take place on the very thin TiO2 layer just covering the metal particles, thus avoiding the irreversible adsorption of the acid on gold and the subsequent catalyst deactivation. Our results demonstrate how the increase in the extent of the interface between the support and metal significantly increases the overall catalytic activity via a synergic effect between both components. Indeed, once embedded, the stability of gold increases and the adsorption of the reaction products on the surface of gold is excluded. Consequently, this work provides new insights into the design of heterogeneous catalysts, suggesting that the core–shell SiO2@Au@TiO2 catalysts could be used for various oxidation processes, even under harsh conditions.Author contributions
CPF: Conceptualization, data curation, formal analysis, writing – original draft, SNJ: data curation, writing – original draft, LMR: funding acquisition, supervision, validation, writing – review & editing, FD: supervision, validation, writing – review & editing, MNG: conceptualization, formal analysis, methodology, validation, writing – review & editing, RW: conceptualization, project administration, supervision, writing – original draft, writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
LMR is grateful to FAPESP (2016/16738-7, 2018/26253-6) for financial support. LMR acknowledges CNPq (306024/2019-5). The financial support of this work was provided by the French National Research Agency (ANR), through the IngenCat project (ANR-20-CE43-0014).Notes and references
- R. Calvo-Serrano, M. Guo, C. Pozo, Á. Galán-Martín and G. Guillén-Gosálbez, ACS Sustainable Chem. Eng., 2019, 7, 10570–10582 CrossRef CAS.
- S. A. Montzka, E. J. Dlugokencky and J. H. Butler, Nature, 2011, 476, 43–50 CrossRef CAS PubMed.
- C. M. Liu, N. K. Sandhu, S. T. McCoy and J. A. Bergerson, Sustain. Energy Fuels, 2020, 4, 3129–3142 RSC.
- R. Wojcieszak, C. P. Ferraz, J. Sha, S. Houda, L. M. Rossi and S. Paul, Catalysts, 2017, 7, 352 CrossRef.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- P. L. Arias, J. A. Cecilia, I. Gandarias, J. Iglesias, M. López Granados, R. Mariscal, G. Morales, R. Moreno-Tost and P. Maireles-Torres, Catal. Sci. Technol., 2020, 10, 2721–2757 RSC.
- D. Y. Murzin, E. Bertrand, P. Tolvanen, S. Devyatkov, J. Rahkila, K. Eränen, J. Wärnå and T. Salmi, Ind. Eng. Chem. Res., 2020, 59, 13516–13527 CrossRef CAS.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539 RSC.
- A. E. Eseyin and P. H. Steele, Int. J. Adv. Chem., 2015, 3, 42 CrossRef.
- K. J. Zeitsch, in The chemistry and technology of furfural and its many by-products, Elsevier B. V., Amsterdam, 2000, vol. 13, ch. 19, pp. 159–163 Search PubMed.
- C. Xu, E. Paone, D. Rodriguez-Padron, R. Luque and F. Mauriello, Chem. Soc. Rev., 2020, 49, 4273–4306 RSC.
- C. P. Ferraz, N. J. S. Costa, E. Teixeira-Neto, Â. A. Teixeira-Neto, C. W. Liria, J. Thuriot-Roukos, M. T. Machini, R. Froidevaux, F. Dumeignil, L. M. Rossi and R. Wojcieszak, Catalysts, 2020, 10, 75 CrossRef CAS.
- F. Menegazzo, T. Fantinel, M. Signoretto, F. Pinna and M. Manzoli, J. Catal., 2014, 319, 61–70 CrossRef CAS.
- C. Megías-Sayago, S. Navarro-Jaén, R. Castillo and S. Ivanova, Curr. Opin. Green Sustain. Chem., 2020, 21, 50–55 CrossRef.
- E. Taarning, I. S. Nielsen, K. Egeblad, R. Madsen and C. H. Christensen, ChemSusChem, 2008, 1, 75–78 CrossRef CAS PubMed.
- F. Menegazzo, M. Manzoli, A. di Michele, E. Ghedini and M. Signoretto, Top. Catal., 2018, 61, 1877–1887 CrossRef CAS.
- M. Douthwaite, X. Huang, S. Iqbal, P. J. Miedziak, G. L. Brett, S. A. Kondrat, J. K. Edwards, M. Sankar, D. W. Knight, D. Bethell and G. J. Hutchings, Catal. Sci. Technol., 2017, 7, 5284–5293 RSC.
- C. Ampelli, K. Barbera, G. Centi, C. Genovese, G. Papanikolaou, S. Perathoner, K. J. Schouten and J. K. van der Waal, Catal. Today, 2016, 278, 56–65 CrossRef CAS.
- N. Dimitratos, C. Hammond and P. P. Wells, in Gold Catalysis: Preparation, Characterization, and Applications, ed. L. Prati and A. Villa, CRC Press, Boca Raton, 2015 Search PubMed.
- C. P. Ferraz, M. Zieliński, M. Pietrowski, S. Heyte, F. Dumeignil, L. M. Rossi and R. Wojcieszak, ACS Sustainable Chem. Eng., 2018, 6, 16332–16340 CrossRef CAS.
- A. Roselli, Y. Carvalho, F. Dumeignil, F. Cavani, S. Paul and R. Wojcieszak, Catalysts, 2020, 10, 73 CrossRef CAS.
- M. Besson and P. Gallezot, Catal. Today, 2000, 57, 127–141 CrossRef CAS.
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, Chem. Rev., 2018, 118, 11023–11117 CrossRef CAS PubMed.
- E. Farfan-Arribas and R. J. Madix, J. Phys. Chem. B, 2003, 107, 3225–3233 CrossRef CAS.
- Z. Duan and G. Henkelman, ACS Catal., 2015, 5, 1589–1595 CrossRef CAS.
- J. Zhang and J. Will Medlin, Surf. Sci. Rep., 2018, 73, 117–152 CrossRef CAS.
- C. Wu, L. Lin, J. Liu, J. Zhang, F. Zhang, T. Zhou, N. Rui, S. Yao, Y. Deng, F. Yang, W. Xu, J. Luo, Y. Zhao, B. Yan, X. D. Wen, J. A. Rodriguez and D. Ma, Nat. Commun., 2020, 11, 5767 CrossRef CAS PubMed.
- G. Wang, F. Luo and F. Zhao, React. Kinet., Mech. Catal., 2021, 132, 155–170 CrossRef CAS.
- G. D. Gesesse, C. Wang, B. K. Chang, S. H. Tai, P. Beaunier, R. Wojcieszak, H. Remita, C. Colbeau-Justin and M. N. Ghazzal, Nanoscale, 2020, 12, 7011–7023 RSC.
- G. D. Gesesse, T. Le Neel, Z. Cui, G. Bachelier, H. Remita, C. Colbeau-Justin and M. N. Ghazzal, Nanoscale, 2018, 10, 20140–20146 RSC.
- C. Megías-Sayago, L. F. Bobadilla, S. Ivanova, A. Penkova, M. A. Centeno and J. A. Odriozola, Catal. Today, 2018, 301, 72–77 CrossRef.
- T. Ishida, N. Kinoshita, H. Okatsu, T. Akita, T. Takei and M. Haruta, Angew. Chem., Int. Ed., 2008, 47, 9265–9268 CrossRef CAS PubMed.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74–78 CrossRef CAS PubMed.
- P. Mars and D. W. van Krevelen, Chem. Eng. Sci., 1954, 3, 41–59 CrossRef CAS.
- M. A. Saqlain, A. Hussain, M. Siddiq and A. A. Leitão, Appl. Catal., A, 2016, 519, 27–33 CrossRef CAS.
- P. Schlexer, D. Widmann, R. J. Behm and G. Pacchioni, ACS Catal., 2018, 8, 6513–6525 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc02889h |
| This journal is © The Royal Society of Chemistry 2021 |