
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultrasound-assisted extraction of metals from Lithium-ion batteries using natural organic acids†
Xiong
Xiao
 a,
Billy W.
Hoogendoorn
a,
Yiqian
Ma
b,
Suchithra
Ashoka Sahadevan
c,
James M.
Gardner
c,
Kerstin
Forsberg
b and
Richard T.
Olsson
*a
a,
Billy W.
Hoogendoorn
a,
Yiqian
Ma
b,
Suchithra
Ashoka Sahadevan
c,
James M.
Gardner
c,
Kerstin
Forsberg
b and
Richard T.
Olsson
*a
aDepartment of Fiber and Polymer Technology, KTH Royal Institute of Technology, Teknikringen 56, 11428 Stockholm, Sweden. E-mail: rols@kth.se
bDepartment of Chemical Engineering, KTH Royal Institute of Technology, Teknikringen 42, 11428 Stockholm, Sweden
cDepartment of Chemistry, KTH Royal Institute of Technology, Teknikringen 30, 11428 Stockholm, Sweden
First published on 29th September 2021
Abstract
An ultrasound-assisted extraction (leaching) method of valuable metals from discarded lithium-ion batteries (LiBs) is reported. Mild organic citric or acetic acids were used as leaching agents for a more environmentally-friendly recovery of the lithium, nickel, cobalt, and manganese from the discharged and crushed lithium nickel-manganese-cobalt oxide (NMC) LiBs. The extraction was performed with the presence/absence of continuous ultrasound (US) energy supplied by a 110 W ultrasonic bath. The effect of temperature (30–70 °C), reducing agent concentration (H2O2: 0–2.0 vol%), as well as choice of specific acid on the metal dissolution were investigated. The US leaching decreased the leaching time by more than 50% and improved the leached percentage of Li, Mn, Co, and Ni due to the local heat and improved mass transfer between solid and liquid interfaces in the process. The X-ray diffraction results of residues from the US leaching further confirmed an improved dissolution of the crushed layered NMC structure, resulting in the significant improvement of the leached amounts of the valuable metals. Furthermore, it is demonstrated that using citric acid eliminated the need of additional reducing agents and suppressed the dissolution of copper (Cu) due to its inhibitor effect on the Cu surface, i.e. compared with using acetic acid as leaching reagent. Overall, it is shown that recovery of the battery metals can be facilitated and carried out in a more energy-efficient manner at low temperatures (50 °C) using ultrasound to improve metal ions mass transportation in the residue layers of the NMC during the organic acid leaching.
1 Introduction
Lithium-ion batteries (LiBs) are increasing in demand due to their use in electronic vehicles (EVs). LiBs have been responsible for more than half of the energy output from all batteries since 2017 and the use of LiBs is expected to increase to 75% of the market by 2025.1 At the same time, the most frequently used functional metal elements, i.e. lithium, nickel, and cobalt, are limited in supply and global geological exploration predicts that a shortage of the elements may occur within 25 years.2 This calls for intense efforts to develop strategies to reuse, recover, and recycle different battery types to avoid the build-up of valuable materials in landfills.3 Resource efficient metals recycling, allowing for continuous raw material supply, is here a cornerstone in a future sustainable battery market. The recycling of valuable metals requires facile processing with high selectivity and demands as low energy input as possible, with minimal environmental impact for the extraction of the metal ions from their host materials. This ultimately demands that the energy and emissions (volatile compounds and toxic greenhouse gases) associated with the strategies to recover the metals are overviewed in complete lifecycle analysis (LCA) studies.1,4 Hydrometallurgy represents a promising recycling method since it requires lower energy input and generates lower levels of toxic emissions than pyrometallurgy.5,6 In hydrometallurgy recycling, battery shredding is frequently used to obtain the process raw material consisting of a mixture of iron, aluminium, copper, in addition to the active battery metals, and graphite, liquid electrolytes, plastics, and binders.7 The polymers are sieved off in a flotation process,6 leaving behind a mixture of materials that are processed with strong and harsh acids to dissolve the metals for further separation by leaching, i.e. before precipitation,8 solvent extraction,1 and/or electrowinning of the valuable metals.9 The drawbacks of the processing include occasionally time-consuming steps including calcination (ca. 450–700 °C), organic solution treatment, and NaOH treatment for removal of battery binders and Al, with reported losses of valuable metals, e.g. lithium, etc. and emission of toxic greenhouse gases.10–14Previously, ultrasound was demonstrated as an efficient and green technology that can be used in the process of extraction from biological samples. Most ultrasonic baths send mechanical pressure waves with frequencies greater than 20 kHz through the liquid medium. By sending the ultrasonic waves through an aqueous medium, microbubbles are formed.15 The collapse of the bubbles generates local temperatures of 5000 K along with the production of highly reactive free radicals.16 The events will cause physical effects, such as turbulence, shear forces, shock waves, and microjets, resulting in increased mass transfer while degassing the solution of the dissolved gases. Ultrasound may however also allow for improving mass transfer in the extraction of valuable battery metals to an extent that more environment-friendly organic acids can be used in the recovery of the metals. So far, investigations on the extent of the improved efficiency of metal extraction from used LiBs using ultrasound are scarce.12,13,17,18 In these studies, the ultrasound extraction of metals from LiBs focuses on the dissolution of LiCoO2, LiNiO2, and LiMn2O4, and occasionally reports on the leaching kinetics of these materials with the complexity of added reducing agents. The more environment-friendly acids, so far reported, include formic acid,19 lactic acid,14 acetic acid,20 and citric acid.21 In the absence of ultrasound, these weaker organic acids would normally be regarded as insufficiently strong for efficient extractions and require the use of additional chemicals or pre-treatments steps before the actual extraction experiments (leaching process) to reach recovery levels >95%, see Table S1.† In LiBs recycling, low and high-power ultrasound vibration was used to assist the electrode delamination process during disassembly of batteries.22
In this work, the ultrasound-assisted extraction of the metals from lithium nickel–manganese–cobalt oxide (NMC) batteries (the most common lithium battery type) is reported for the first time, and the kinetics of the extraction is described in detail. The difference between leaching with, and without ultrasound, using weak acetic or citric acids is shown. Citric acid was used as a natural, mildly reducing (E0 = −0.18 V vs. NHE), weak triprotic acid that is commercially produced by fermentation.18,23 It is ‘green’, abundant, comparatively inexpensive (to other organic acids), and has been widely used by the food and beverages industry, and as a non-toxic household detergent for years.24 The citric acid was compared with acetic acid due to its inexpensive nature at high purity, and high availability, although not as acidic as the citric acid i.e. being the sole organic acid with a pKa value of 4.76![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 25 (see Table S2†). The batteries used were shredded 24 kW h Volvo C30 NMC batteries. The leaching processing was however carried out as a universal approach, making the demonstrated methods applicable to work with other battery types. It is shown that ultrasound significantly increases the leaching rate of the battery metals during the processing. The leaching time was reduced by more than 50% when using a standard laboratory ultrasonic bath, as opposed to when carrying out traditional extraction in a stirred reactor. Almost complete metal ion extraction is demonstrated, using the acids in combination with the ultrasound. The citric acid further improved the leached percentage of lithium, nickel, cobalt, and manganese while suppressing the dissolution of Cu, when compared with acetic acid. The results were confirmed with multiple measurements and characterizations of all battery residues before, and after, the leaching process.
25 (see Table S2†). The batteries used were shredded 24 kW h Volvo C30 NMC batteries. The leaching processing was however carried out as a universal approach, making the demonstrated methods applicable to work with other battery types. It is shown that ultrasound significantly increases the leaching rate of the battery metals during the processing. The leaching time was reduced by more than 50% when using a standard laboratory ultrasonic bath, as opposed to when carrying out traditional extraction in a stirred reactor. Almost complete metal ion extraction is demonstrated, using the acids in combination with the ultrasound. The citric acid further improved the leached percentage of lithium, nickel, cobalt, and manganese while suppressing the dissolution of Cu, when compared with acetic acid. The results were confirmed with multiple measurements and characterizations of all battery residues before, and after, the leaching process.
2 Experimental
2.1 Materials
Discarded NMC LiBs were delivered as a shredded ‘blackmass’ powder from Volvo Cars AB (Sweden), Fig. 1a. The ‘blackmass’ was prepared by crushing followed by removing a magnetic fraction via magnetic separation. Plastics, copper, aluminium foils were removed by air separation and sieving, and the binder was kept in contrast to previous works adapting ultrasound to remove the binder.22 The use of calcination at >600 °C was avoided to reduce the energy demands of the entire recycling/recovery of metals procedure, eliminating the unnecessary generation of greenhouse gases.26 The citric acid (C6H8O7, ≥99.5%), acetic acid (C2H4O2, ≥99.7%), 37% HCl (analytical grade), ≥65% HNO3 (analytical grade), and hydrogen peroxide (H2O2, 30 wt%) were purchased from Sigma-Aldrich AB, Sweden. Standard solutions of 1000 mg L−1 ICP-OES for Li, Ni, Co, Mn, Cu, Al, and Fe in 2–5 w% HNO3 or HCl solutions for ICP-OES calibration were purchased from Alfa Aesar, Sweden. MiliQ water (>18.2 M) was used in all the experiments. | ||
| Fig. 1 (a) Barrels of discharged and ground batteries, (b) SEM images of fine ground LiBs powder, and (c) the fine ground powder in high magnification, (d) size distributions of fine ground powder derived from >50 micrographs using min. 600 particles for accurate statistics, (e) metal elements content variation (ICP-OES measurements) from top to bottom of the Barrel 1 and 2, respectively, (f) EDS maps taken from (c). Triple samples tested on each location in the barrels confirmed the nonuniform powder composition from the natural fractionation that had occurred in the coarse ground powder, demonstrated by the error bars in Fig. 1e. | ||
2.2 Leaching/extraction methods
:HCl = 1:3, v/v) at 343 K for 5 h to determine the contents of metals in the ground LiBs powder. The experiment was repeated thrice to ensure that the contents of the metals in the reference the sample was accurately determined.
400 rpm for 2 min before the removal of the solid phase, dilution, and analysis by inductively coupled plasma-optical emission spectrometry (ICP-OES).
2.3 Characterization
2.4 Microscopy
The morphology and size distribution of the samples were observed in a field emission scanning electron microscope (FE-SEM, Hitachi S-4800) and SEM images were taken at an acceleration voltage of 15.0 kV, and a current of 10 mA. The size of the particles was obtained by manually measuring 600 particles using ImageJ (National Institute of Health, Maryland, USA). Sputtering of the samples was performed for 20 s using a Cressington 208HR, equipped with a Pt/Pd target. Energy-dispersive X-ray spectroscopy (EDS) was carried out at 15.0 kV to measure the distribution of the elements in the LiBs powder before and after leaching.2.5 Characterization of elemental compositions
All solutions were diluted into metal concentration ranges useful in the elemental analysis. The concentrations of metal ions in the solutions were determined by ICP-OES (Thermo Fisher iCAP 7400, USA), using at least 3 suitable wavelengths that did not interfere with the wavelengths of other metal ions in the solution. The calculation of the leached percentage is represented as follows: | (1) |
 | (2) |
2.6 Crystallography
The ground LiBs powder and all sample residues were characterized by X-ray diffraction (XRD), and the PANalytical X'pert3 software was used for peak identification. The XRD patterns were recorded on a powder diffractometer operating at 45 kV and 40 mA using Cu Kα radiation (λ = 0.15418 nm).2.7 Viscosity
Kinetic viscosities of the aqueous solutions were measured using an Ubbelohde capillary viscometer from Schott. The capillary used was a type II (Di = 1.13 mm, K ≈ 0.1) and a type 0c (capillary constant = 0.0031476 mm2 s−2). The capillary was placed in a Lauda iVisc (LAUDA, Germany), an automated infrared measuring device, which measures the time of the experiments. The iVisc with capillary was placed in a water bath (Lauda ET 15) at 298 K, and the temperature was regulated with an immersion heating thermostat (Lauda Eco Silver).3 Results and discussion
3.1 Analysis of the ground LiBs powder provided
Fig. 1a shows the two barrels containing fine and coarse ground LiBs ‘blackmass’ powder. The barrels were 100 × 60 cm (height × diameter), containing ca. 100 kg ground powder. Fig. 1b and c shows that even the finest ground battery powder displayed significant particle size differences. The size distribution of the fine ground powder is displayed in Fig. 1d, showing that the average particle size was in the range of ca. 2–8 μm (average: 5.9 ± 0.3 μm), although large sized flakes sized up to 2 mm could occasionally be observed Fig. 1b.Fig. 1e shows the elemental presence in the top and bottom powder fractions of the barrels for the fine and coarse ground powder, as a comparison. The coarse ground powder contained a markedly higher content of aluminium and copper compared to the more valuable metals Co, Ni, and Mn (right hand side columns, Fig. 1e). The coarse ground powder also demonstrated a wider particle size distribution (Fig. S2b†), which explained a more extensive fractionation that had occurred during the barrel transportation. It was thus evident from the elemental analysis and microscopy that only the fine ground powder, with almost identical metal composition on the top and bottom (left hand side columns, Fig. 1e) was useful as starting material for developing optimized extraction protocols, i.e. due to its more uniform composition. The fine ground powder was therefore further analysed and used in all the remaining experiments. The larger size particles in Fig. 1b (representing less than 5% of the particle fraction, although a considerable part of the metallic mass content) was however firstly filtered off and identified as composed of aluminium or copper (Fig. S2†). The larger sized particles were accordingly excluded from the extraction protocols focusing on the active battery materials.
Fig. 1c shows a magnified area in Fig. 1b, displaying the finer particles, with the dominant phase represented in particle sizes by the histogram shown in Fig. 1d. Fig. 1f shows the EDS mapping images of the same area displayed in Fig. 1c. The detected elements were Co, Ni, and Mn (top row), while the bottom row shows C, O, and F. The latter elements were remains from the conductive additives and binders used in the LiBs fabrication process. Lithium is not shown in Fig. 1f since it was not detected by the normal EDS probe analysis due to its low characteristic radiation energy.27 For comparison, Fig. S3† shows the EDS mapping of the residue of the same LiBs powder after aqua regia leaching digestion, demonstrating that the aqua regia leaching digestion could effectively be used for complete extraction (100%) of the Ni, Co, to obtain the reference values synonymous with complete extraction of all active battery metals from the remaining carbon residuals.
Fig. 2 shows the TGA curves and XRD patterns of the fine ground LiBs powder before and after aqua regia leaching digestion. TGA was carried out under O2 flow to ensure that all organic materials in the fine ground LiBs powder were removed, and its relative content could be determined. The results show that before aqua regia leaching digestion about 61.8 wt% residue remained after heating to 950 °C, which was associated with the metal fraction of the battery powder. After aqua regia leaching digestion, less than 1 wt% material remained although a small amount of ash existed. The results were consistent with an almost complete oxidation of the organic materials (Fig. 2a), at a temperature above 500 °C. Moreover, XRD characterization was used to investigate the crystalline phases in the fine ground powder before and after the aqua regia leaching digestion.
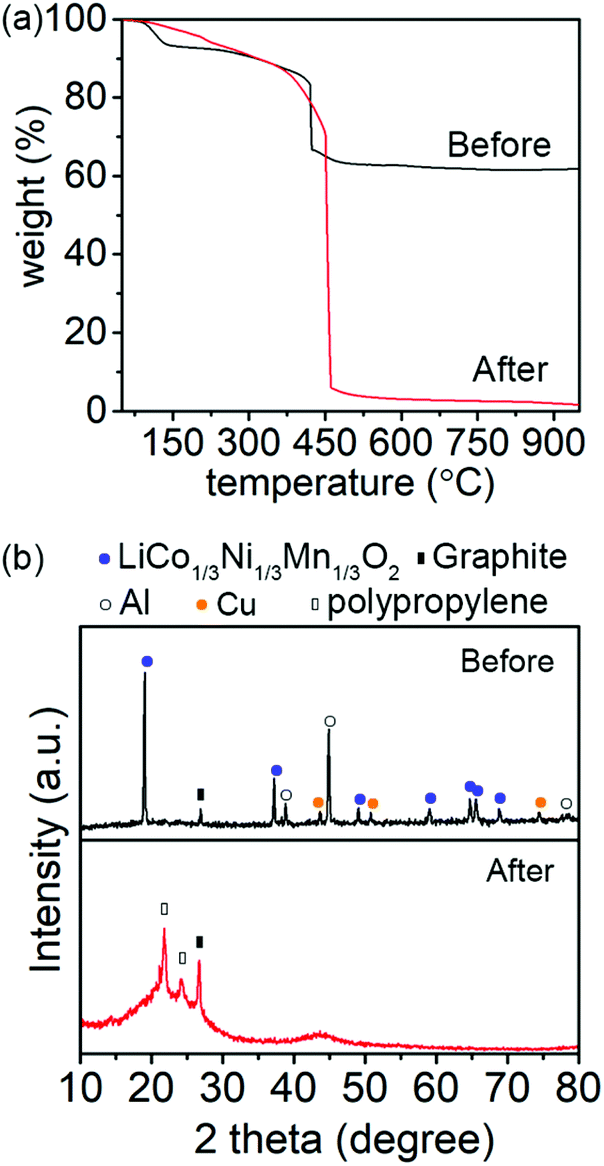 | ||
| Fig. 2 (a) TGA with O2 flow and (b) XRD of the fine ground LiBs powder before and after aqua regia leaching digestion. The peaks for related phases referred to ICDD database: (LiNi1/3Co1/3Mn1/3)O2 (NMC) (PDF 56-0147), graphite (PDF 08-0415), Al (PDF 01-1180), Cu (JCPDS 003-1018), and polypropylene (PDF 61-1416). | ||
The peaks in the diffractograms before leaching originated from the layered NMC phase (active material), Al, Cu, and graphite, respectively, Fig. 2b (top). After the aqua regia leaching digestion, the remaining material was graphite and polypropylene (PP). The PP came from the separator/battery casing, see Fig. 2b (bottom). The TGA and XRD of powders before and after aqua regia leaching agreed with the EDS mapping and confirmed that a full extraction of the metal elements had been carried out.
ICP-OES was used to carry out the quantitative analysis of the aqua regia leachate composition. The concentration (mmol g−1) and standard deviation of all metals are summarized and shown in Table 1. Fe was detected in the fine ground LiBs powder at a concentration of 0.048 mmol (pristine powder) g−1, which is not included in Table 1 due to its comparably small amount. The metal mole-to-mole ratio of the active material Li:Mn:Co:Ni was ca. 1.289:0.315:0.319:0.366, reflecting that the composition of active material (LiMn0.315Co0.319Ni0.366O2) and electrolyte in the hybrid EV battery was closed to LiNi1/3Co1/3Mn1/3O2. The phase composition was also in agreement with the diffraction pattern in Fig. 2b (top), although the additional 29% Li originated from the electrolyte (LiPF6) that was present in the sample. The Al and Cu in Fig. 2b originated from the current collector and were not shown in the EDS mapping (Fig. 1f) due to their large particle sizes and coincidentally were found in similar amounts as the targeted metal ions (Ni, Co, and Mn). In total, the weight of Al, Cu, and the NMC phase occupied ca. 65.7 wt% of the fine ground powder, which was in good agreement with the remaining residue percentage (61.8 wt%, Fig. 2a) obtained from the gravimetric measurements.
In summary, the characterization in terms of the natural fractionation and deviation of uniformity (coarse vs. fine ground powder), the phase characterization, and the chemical composition allowed only the fine ground powder to be identified as a useful reference material. The coarse ground powder was always displaying different powder characteristics at different locations in the barrel and was therefore discarded as a source for more experiments.
3.2 Effect of ultrasound on metal ion extraction using soft acids
Fig. 3 shows the effect of ultrasound on the metal ion extraction from the fine ground LiBs powder using 1.5 mol L−1 citric acid, in the absence of any H2O2 reducing agent, which commonly is used to improve leaching efficiency.1 The most extended leaching times were selected to 24 h (1440 min) on the basis of possibly reaching full extraction of the NMC material within a reasonable time frame. In the first 150 min, the leaching behaviour of the Li, Mn, Co, Ni, and Al in the US leaching system and the traditional oil bath were similar and the extracted amount per time was almost identical, see overlapping curves in Fig. 3. After 150 min, the leached percentage of Li, Mn, Co, and Ni in the US leaching system was higher than that in the oil bath, reaching on average ca. 97 ± 3% after 1440 min. Among Li, Mn, Co and Ni, the most effective leaching was found for the cobalt reaching 100% extraction, while Li and Mn were extracted to 94% and 96%, see Table 2. Also, the leached percentage of Al at 1440 min was significantly improved from 48% to 60% using the ultrasound in the leaching process. An increase in temperature from 323 to 343 K (Fig. S4†, Table 2) of the oil bath was insufficient to reach the same extraction efficiency as when solely using the ultrasound for the Li, Mn, Co, Ni metal ions, although Al leaching was sensitive to an increase temperature from 323 K to 343 K. More specifically, raising the temperature 20 K resulted in more aluminium extracted compared to when only ultrasound was used. | ||
| Fig. 3 Effect of leaching method (US vs. oil bath) on metal ions extraction from the fine ground LiBs powder. Error bars are standard errors from triplicate measurement of different batches. Conditions: citric acid, 1.5 mol L−1; S/L, 25 g L−1, H2O2, 0 vol%; ultrasonic bath temperature, 323 K; oil bath temperature, 323 K; stirring speed, 500 rpm. The lines between measurement points serve as the guide to the eye. | ||
| ICP-OES result | Metal elements (mmol (pristine powder) g−1) | |||||
|---|---|---|---|---|---|---|
| Li | Mn | Co | Ni | Al | Cu | |
| Comment: The data for each ion was acquired as repeated twice with separate ICP-OES measurements. The deviation was maximum ±3% with an exception of Cu (maximum ±7%). | ||||||
| Oil bath, 323 K | 89% | 89% | 87% | 91% | 48% | 83% |
| US, 323 K | 94% | 96% | 100% | 99% | 60% | 48% |
| Oil bath, 343 K | 90% | 91% | 93% | 92% | 78% | 54% |
Table 3 show over time that the active battery metals could be extracted much faster using ultrasound. The required times for reaching 20%, 50%, 80%, and 90% of the total amount of extractable metal ions (Li, Ni, Co, and Mn) are shown in Table 3.
| Times (t, min) | Li | Mn | Co | Ni |
|---|---|---|---|---|
| Comment: The data in the table with parentheses represent the required time in an oil bath. Data from Fig. 3, see dashed lines for 80 and 90%. | ||||
| t 20% | 20 (20) | 44 (40) | 43 (45) | 47 (41) |
| t 50% | 75 (75) | 140 (250) | 160 (290) | 150 (240) |
| t 80% | 270 (650) | 430 (975) | 420 (1125) | 420 (940) |
| t 90% | 600 (>1440) | 790 (>1440) | 685 (>1440) | 675 (1440) |
Among the metals, the fastest extraction was observed for the lithium, showing that 80% of the total amount of extractable Li (t80%) could be reached within 270 min. The same extent of extraction, using traditional stirring, required over 650 min to be completed. At the same time, 420 min was needed to extract 80% of the cobalt (t80%) using the ultrasound in comparison to 1125 min using the traditional extraction, see Table 3 and Fig. 3.
The X-ray diffraction patterns of residues, as dependent on the leaching time with/without ultrasound, are shown in Fig. S5† and Fig. 4. From 0 to 720 min, a shift of the 19.05° peak (representing the NMC interlayer direction), occurred from a higher to a lower angle (Δ 0.8°, see Fig. 4a). The shift was caused by the proton exchange accompanied with water molecules28 intercalated within the NMC layered space, resulting in an expansion of the interlayer distance, see Fig. 4b. With an additional increase in the leaching time to 960 min, the peak shifted back to a higher angle due to the destruction of the layered NMC phase and the formation of a spinel-type LiMn2O4. The spinel-type LiMn2O4 can retain its structure even when exposed to harsh HCl acid solutions.29 After 1440 min leaching, the diffractogram peaks associated with the LiMn2O4 spinel structure disappeared in the residues from the US leaching method while they remained in the residues from the oil bath leaching at 323 K. The results suggested that the ultrasound not only assisted in the diffusion of the valuable metal ions into the solution, but also had an impact on the dissolution of the more stable LiMn2O4 spinel structure.
 | ||
| Fig. 4 (a) X-ray diffraction patterns of residues in the leaching system with/without ultrasound depending on leaching time at 323 K. (b) Crystal structures of LiCo1/3Ni1/3Mn1/3O2 (NMC) and (c) spinel-type LiMn2O4 (draw by VESTA30), Li (green ball/octahedral), Ni/NiO6 (grey ball/octahedral), Co (blue ball/octahedral), and Mn (purple ball/octahedral), vacancy (white ball). | ||
The only exception from the general observation that ultrasound assisted leaching outperformed the traditional oil bath leaching was the Cu element. The leached percentage of Cu at 1440 min was supressed during the leaching, see Table 2. This was related to the depletion of dissolved O2 in the solution (caused by the intense ultrasound), which have been reported to interact with Cu and citric acid in conditions without any oxidizing agents.31 For this reason, ultrasound has traditionally also been used for degassing and release of gases from solutions.32,33 A lower O2 concentration consequently interfered with the Cu oxidation and dissolution mechanisms as previously reported.31 This would be in line with the results showing that extraction carried out at higher temperature in the oil bath, see Table 2 (343 K) also suppressed Cu leaching since a higher temperature results in a lower percentage of naturally dissolved O2.34
3.3 Kinetics study with/without ultrasound on metal ions extraction
To further understand how ultrasound influenced the leaching process, the shrinking core model of noncatalytic fluid-solid reactions20,35 was used to assign the contribution of different mass transfer phenomena to the observed leaching behaviour. The schematic diagram of the shrinking core model was shown in Scheme S1† and the kinetics of it is described by the following three equations representing different leaching rate contributions to the model:(1) Liquid boundary layer mass transfer contribution (eqn (3)):
 | (3) |
(2) Surface chemical reaction contribution (eqn (4)):
 | (4) |
(3) Residue layer diffusion contribution (eqn (5)):
 | (5) |
| X = k1t | (6) |

| 1 − (1 − X)1/3 = k2t | (7) |

| 1 − 3(1 − X)2/3 + 2(1 − X) = k3t | (8) |
where, k1, k2, and k3 (min−1) are the slopes of the fitted lines of X, (1 − X)1/3 and 1 − 3(1 − X)2/3 + 2(1 − X) vs. t (leaching time), respectively. The different contributions were accordingly fitted to the leaching data from the curves shown in Fig. 3. Fig. 5 shows the fitting of the data to the residue layer diffusion contribution factor (1 − 3(1 − X)2/3 + 2(1 − X) vs. t), whereas Fig. S6† shows the same data fitted to the contributions refer as liquid boundary layer mass transfer (X vs. t) and surface chemical reaction ((1 − X)1/3vs. t).

 | ||
| Fig. 5 Kinetics analysis during the leaching of the fine ground LiBs powder with/without ultrasound, adapting leaching time data to the residue layer diffusion contribution (eqn (5)) of the shrinking core model. Conditions: citric acid, 1.5 mol L−1; S/L, 25 g L−1, H2O2, 0 vol%; temperature, 323 K; stirring speed, 500 rpm. | ||
The results show that for both systems (with/without ultrasound), the fitting lines of Li, Mn, Co, Ni, and Al were in agreement with the residue layer diffusion contribution to the shrinking core model. A linear relationship (k3) with a higher regression coefficient (R2) value (≥0.9855) could be established, whereas no relationships could be established for any of the other contributions to the shrinking core model (Fig. S6†).
The specific R2 values are shown in Table 4 for the different ions. The only exception was the leaching of Cu that instead reasonably well correlated with the reaction control contribution, showing a R2 value of 0.989 when the extraction was done in absence of ultrasound (Fig. S6†). This result again suggested that having oxygen dissolved in the solution contributed to a uniformly progressing Cu dissolution reaction (see section 3.2). Overall, it was evident that the rate limiting contribution to the US leaching behavior could be assigned to the residue layer diffusion (eqn (5) and (8)), while the ultrasound favoring the residue layer diffusion may also be used to suppress the dissolution of copper, compare Table 2.
| Li (1st stage) | Li (2nd stage) | Mn | Co | Ni | Al | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| k 3 × 10−4 (min−1) | R 2 | k 3 × 10−4 (min−1) | R 2 | k 3 × 10−4 (min−1) | R 2 | k 3 × 10−4 (min−1) | R 2 | k 3 × 10−4 (min−1) | R 2 | k 3 × 10−4 (min−1) | R 2 | |
| Comment: Copper was excluded in the table because of its low fitting R2 value from the US leaching system, regardless of the control models described in the shrinking core model. Conditions: citric acid, 1.5 mol L−1; S/L, 25 g L−1, temperature, 323 K; stirring speed, 500 rpm. | ||||||||||||
| Without US | 15.6 | 0.9916 | 3.52 | 0.9917 | 4.41 | 0.9980 | 3.81 | 0.9981 | 4.36 | 0.9981 | 0.79 | 0.9855 |
| With US | 15.3 | 0.9814 | 7.28 | 0.9763 | 8.75 | 0.9944 | 9.30 | 0.9945 | 9.47 | 0.9956 | 0.99 | 0.9982 |
Table 4 shows the k3 values for Li, Mn, Co, Ni, and Al, respectively, as well as their related R2 values from the fitted slopes based on residue layer diffusion contribution (Fig. 5). The k3 values represent the release rate of the metal ions as the steepness of the linear slope in Fig. 5. It is shown that the Li extraction that required a two-stage fitting of the residue layer diffusion contribution in the 1st stage leaching demonstrated k3 values that were independent of the leaching method (oil bath or ultrasound), and ca. 2–4 times higher than other metal ions in the NMC. This was caused by some of the lithium originating from the electrolyte, making it easier to release to the acid solution. During the 2nd stage, the release of the lithium was dominantly from the solid phase and therefore occurred at a slower rate, with k3 value similar to the Mn, Co, and Ni metal ions.
Notably, although the k3 values for Mn, Co, and Ni were similar (Fig. 5 and Table 4), ultrasound always yielded higher release rates (regardless of the specific metal ion) suggesting that this release was caused by ultrasound impact on the solid phase. This observation was supported by the observation of almost identical k3 values for Li during the 1st stage leaching, compare 15.6 × 10−4 to 15.3 × 10−4 min−1 (without and with US).
Fig. 6 shows the SEM micrographs of the blackmass residue after the leaching had been carried out, with or without ultrasound, for the leaching times; 1.5 h, 3 h, and 6 h. The corresponding average values of extracted metal ions (Mn, Co and Ni) are included as marked ‘Ext. xx%’ in the upper righthand corners of the micrographs. The morphology of the NMC particles was initially made up of larger ca. 10 μm spherical secondary particles shown in Fig. 6a (white arrows), which consisted of smaller aggregated primary particles of irregular sizes (Fig. 6b). The primary particles ranged in size from ca. 0.5–3.0 μm in diameter, and the boundaries between these particles are distinguishable as fused surfaces (red arrows) in Fig. 6b. The surface of the primary particles was smooth although covered by significantly smaller particles with approximate size of 40–100 nm (Fig. 6b). With ultrasound treatment during the leaching, the secondary particles completely fell apart into the primary particles after 1.5 h and all the adhesion between primary particles were lost (Fig. S7c†). Without ultrasound, the secondary particles still held together for longer time than 3 h, i.e. the positioning of the primary particles remained almost the same in the larger secondary particles, see Fig. 6d, f and Fig. S7d.† Accordingly, a significant portion of the NMC surfaces were inaccessible to the acid during the early hours of the leaching when performed in absence of US. The extended particle size decrease in the US system consequently resulted in enhanced residue layer diffusion for the ultrasound system, see eqn (8). It is noteworthy that the smaller 40–100 nm particles disappeared completely in both cases. It is suggested that the disappearance of these particles followed a similar dissolution behavior as the aluminum, see Fig. 5, which showed the best fitting to a linear dissolution behavior from 0 to 1440 min (R2 = 0.9982, Table 4). Another observation was that the morphology of the particles exposed to ultrasound had developed more faceted edges, compare Fig. 6c and d. These facetted edges of the samples became most pronounced with longer extraction times (>6 h), while appearing only after 3 h with for the traditional extraction without ultrasound. Fig. 7a and b show high-resolution images of typical NMC primary particles with the layered grains after 3 h of extraction. The stacked sheet appearance was suggested to exist as a consequence of a more intense metal ion drainage in the surface of the particles, see arrow in Fig. 7b.
 | ||
| Fig. 6 Scanning electron micrographs of the pristine LiBs powder before the ultrasound extraction/leaching of battery metals (a and b), and after (c–h) citric acid leaching. The top row shows ultrasound assisted leaching while bottom row shows the oil bath leaching for different times, where c and d is 1.5 h, e and f 3 h, and g and h 6 h. The average values (displayed as inserts in each micrograph) of the extracted metal ions (excluding Li) were derived from Fig. 3. | ||
 | ||
| Fig. 7 Scanning electron micrographs of the primary particle of the layered NMC grains after 3 hours of extraction. Note the visible channel formations along different facets (a and b). | ||
It should be noted that lithium-proton ion exchange dominated in the earlier stages of extraction. Thus H+/H3O+ replaced Li+ sites, leaving primarily a Ni/Mn/CoO6 built layered structure (Fig. 7b) with less lithium present than represented by the crystal structure in Fig. 4b. This intercalation of H3O+ into the interlayers to some extent resulted in the shift of (003) to the lower 2 theta angle (Fig. 4a). It is suggested that although the morphological differences shown in Fig. 6 and 7 primarily related to initially breaking up the secondary particles, a more efficient metal ion drainage of the primary particles was always present when the ultrasound was used. This was evidenced by k3 values remaining constant for leaching times >3 h up to 24 h (1440 min) as shown in Fig. 3.
3.4 The effect of temperature on the release order of metal ions
Fig. 8 shows the effect of temperature on the metal ion release from the LiBs powder for times from 0 to 1440 min in absence of ultrasound. The curves in Fig. 8 show that the leaching rate of all metals steadily increased with higher temperature (from 303 K to 343 K) during the first ca. 300 min and thereafter changed more moderately for all systems. The increase in leaching rate with increasing temperature was a consequence of the increasing dissociation constant of the citric acid,36 since all metal ions showed the same behavior. The first dissociation constant of citric acid increased from 7.7 × 10−4 s−1 to 8.1 × 10−4 s−1 when the temperature was increased from 303 K to 323 K,36 leading to the formation of more available H+ ions and a faster chemical reaction. Moreover, a high temperature also increased the rate of mass diffusion for the released metal ions in the residue layer, which also contributed to the increased leaching rate, see section 3.3. | ||
| Fig. 8 Effect of temperature on leaching behaviour of metal ions (from the fine ground LiBs powder) depending on leaching time. The errors (error bars) were derived from triplicate leaching experiments. Conditions: citric acid, 1.5 mol L−1; S/L, 25 g L−1, stirring speed, 500 rpm. The lines between measurement points serve as the guide to the eye. | ||
Table 5 summarize the leached percentages of all the metals at 1440 min. At 303 K only 71% Li, 53% Mn, 52% Co, and 54% Ni were released, while at 333 K ca. 20% more Li were released and about 40% more of the divalent Mn, Co, and Ni.
| Leached percentage | Metal elements | |||||
|---|---|---|---|---|---|---|
| Li | Mn | Co | Ni | Al | Cu | |
| Comment: The data for each ion was acquired as repeated more than twice. The deviation was maximum ±3% with an exception of Cu (maximum ±7%). | ||||||
| 303 K | 71% | 53% | 52% | 54% | 28% | 54% |
| 313 K | 77% | 72% | 66% | 70% | 34% | 73% |
| 323 K | 89% | 89% | 87% | 91% | 48% | 83% |
| 333 K | 92% | 92% | 93% | 93% | 58% | 77% |
| 343 K | 90% | 91% | 93% | 92% | 78% | 54% |
A further increase in temperature to 343 K resulted in more leached Al at 1440 min, whereas that of Li, Mn, Co, and Ni remained essentially the same and showed no additional leached amount of metal ions. The only exception from the general behavior that an increase in temperature favored more dissolved metal ions was for the copper (Cu). Here the leached percentage reached a maximum of 83% at 323 K and then gradually decreased to 54% when temperature was increased to 343 K. The explanation was argued related to the depletion of the dissolved O2 causing a supressed dissolution of Cu as described in section 3.2.
The activation energies (Ea) were calculated according to eqn (9) to investigate if there was a correlation between the minimum energy required to release of Li, Ni, Co, and Mn and the total amount of leached metal ions, as well as rate of the release of metal ions in the NMC grain structure. The activation energies were derived for the oil bath system since the Ea values were required to be calculated for uniform temperature conditions, which could not be established for the US system.15,16
 | (9) |
K vs. 1/T, the Ea values for leaching of Li (1st stage), Li (2nd stage), Mn, Co, and Ni were determined from the slopes of the corresponding fitted lines in Fig. 9.
 | ||
| Fig. 9 Arrhenius plots for Li, Co, Ni, and Mn leaching using the k3 values obtained from fitting slopes of residue layer diffusion control model. Conditions: citric acid, 1.5 mol L−1; S/L, 25 g L−1; stirring speed, 500 rpm. | ||
Fig. 9 shows that the following Ea values could be established for the residue layer diffusion of the different ions: 67.9 (Li 1st stage), 39.6 (Li 2nd stage), 55.7 (Mn), 56.6 (Co), and 57.2 (Ni) KJ mol−1, respectively. The activation energy for the 1st stage Li leaching was in fact significantly higher than other metal elements implying that a slower leaching rate should have existed although the opposite was observed. It should be highlighted here on the basis of the Li ion location in the NMC phase that the proton exchange process likely has a different reaction/diffusion path as compared to the other metal ions due to its location within the interlayer space of the NMC. This is in agreement with the structural data, wherein direct bonding holds Co/Mn/Ni in an oxide-rich coordination sphere, as shown in Fig. 4b. The 1st stage Li leaching was also reflecting contributions from both the electrolyte Li ion release process and the proton exchange process within the Ni/Co/MnO6 layers (see Fig. 4b and Fig. 7). It was calculated that approximately 50% of the Li leaching connected to the 1st stage leaching was related to the electrolyte releasing process, see further section 3.1. It is noteworthy that while the electrolyte release process may have had a small contribution to the derived Ea value for the 1st Li leaching part, the residue layer diffusion for the first Ea value may have required an even higher activation energy due to the intense electrostatic attractions in the interlayer space of the NMC structure. On the contrary, during the 2nd stage of Li leaching, Li showed a diffusion rate similar to the Mn, Co and Ni metals ions (Table S3†), although the estimated Ea was lower, i.e. 39.6 kJ mol−1. A suggested explanation would be that the layered structure of the NMC started to dissociate at this stage as it was converted into the spinel structure (see Fig. 4c), thereby facilitating the release of Li. At this stage, the activation energy based on the surface residue layer diffusion was on the order of Ni > Co > Mn > Li (2nd stage), which shows that the binding between Ni-citrate is the strongest while that of Li-citrate is the weakest among all the metal ions in NMC. A prevailing composition of Ni-citrate may thus exist in the residue layer interface surrounding the dissolving NMC grains.
The R2 values for the Arrhenius plots of Cu and Al, shown in Fig. S10 and Table S4,† are significantly lower than the other elements. Since the Arrhenius equation assumes that the activation energy is indenpendent on temperature,37 this observation suggests that the leaching mechanisms of Al and Cu were temperature dependent to a larger degree than for the NMC-metals. The deviation from the Arrhenius equation can especially be expected for Cu, since the temperature directly affects the concentration of dissolved oxygen in the solution.
3.5 The impact of ultrasound depending on acid species
Table 6 summarize the total amounts of extracted metal ions for the two different acids over 24 h (1440 min).| Leached percentage | Metal elements | |||||
|---|---|---|---|---|---|---|
| Li | Ni | Co | Mn | Al | Cu | |
| 1.5 mol L−1 citric acid | 89% | 89% | 87% | 91% | 48% | 83% |
| 4.5 mol L−1 acetic acid | 83% | 75% | 72% | 73% | 53% | 86% |
The effect of the strength of the organic acid on the leaching behavior was investigated using a weaker organic acid, namely the monocarboxylic acetic acid (pKa = 4.76).25 4.5 mol L−1 acetic acid was used to normalize the concentration for the reduced number of carboxylic groups. Fig. 10 shows the leaching behavior when using 4.5 mol L−1 acetic acid and 1.5 mol L−1 citric acid at 323 K. During the first 150 min, the leaching behaviors of the acids were almost similar, thereafter the leached percentage of Li, and especially Mn, Co, and Ni showed to be significantly higher for the citric acid, although the higher number of carboxylic groups had been compensated for with 3 times higher concentration of acetic acid. Thus, without any additional reducing agent, 1.5 M citric acid (pH ≈ 1.14) showed better transition metal (Ni, Mn, Co) leaching performance compared to the 4.5 M acetic acid (pH ≈ 1.74). The leached percentage of Cu in 1.5 mol L−1 citric acid was, however, lower than in 4.5 mol L−1 acetic acid during the entire course of the reaction. This may be explained as a result from that citric acid has been reported as an efficient chelating agent to suppress the Cu dissolution in acid.38
 | ||
| Fig. 10 Effect of organic acid species on extraction of metal ions from the fine ground LiBs powder in oil bath at 323 K. Conditions: citric acid, 1.5 mol L−1; acetic acid, 4.5 mol L−1; 0 vol% H2O2; S/L, 25 g L−1; stirring speed, 500 rpm. The lines between measurement points serve as the guide to the eye. | ||
The leaching behavior with/without ultrasound using acetic acid was also investigated. As shown in Fig. S11,† the leached percentage of the targeted metal ions were significantly improved with ultrasound also for the acetic acid, reaching ca. 96% ± 2% (Table 7). The result further highlighted the impact of ultrasound in improving the extraction of metals from the blackmass, irrespectively of the acid species. This might be due to the formation of hydroxyl radicals in the US leaching system, possibly allowing continuous formation of hydrogen peroxides.39 Fig. S12† shows the overlapping nature of the citric and the acetic acid during the entire leaching cycles, suggesting that the primary difference between the acids was their leaching rate (speed) under the influence of the ultrasound.
| Leached percentage | Metal elements | |||||
|---|---|---|---|---|---|---|
| Li | Ni | Co | Mn | Al | Cu | |
| Oil bath, 323 K | 83% | 75% | 72% | 73% | 53% | 86% |
| US, 323 K | 94% | 97% | 97% | 95% | 60% | 73% |
The Ea values for leaching of Li (1st stage), Li (2nd stage), Mn, Co, and Ni were 70.1, 41.5, 44.4, 45.1, and 44.1 kJ mol−1, respectively (see Fig. S13, S14, S15, and Tables S5, S6† for their derivation). The activation energies for the citric and the acetic acid were thus similar for the Li (1st stage) and Li (2nd stage), while for Mn, Co, and Ni the Ea values were more than 10 kJ mol−1 lower for the acetic acid as compared to the citric acid. The reason to the lower activation energy for the divalent metal ions when using acetic acid is presently unknown. It was speculated being a consequence of a less restricted diffusion through the residue layers on the surface of the NMC grains. This was based on the presumption that the complex formation of the extracted divalent metal ions (Mn, Co, and Ni) and the citric acid likely formed stronger complexes by directing 2 oxygen rich carboxylic groups of the acid towards the positively charged metal ions, while leaving one carboxyl group facing outwards towards the bulk acid solution. This configuration stands in contrast to the nonpolar methyl group facing outwards in the case of the acetic acid when its carboxyl group in turned towards the metal ions.
The viscosities of the aqueous solutions of 4.5 mol L−1 acetic acid and 1.5 mol L−1 citric acid, before and after the 24 h leaching process, were taken as an indicative measure of this possible complex binding and coordination of the divalent metal ions (Table 8). The viscosity of the aqueous solution of the 1.5 mol L−1 citric acid was significantly higher than that of the 4.5 mol L−1 acetic acid. The differences in viscosity before and after (Δ) leaching of the 1.5 mol L−1 citric acid solution was: Δ = 0.33 mm2 s−1, which in turn also was higher than that of 4.5 mol L−1 acetic acid solution: Δ = 0.17 mm2 s−1. The higher viscosity of the citric acid system was suggested to reflect the stronger metal–organic ion complex binding. This would be in agreement with the higher activation energy required for the diffusion of transition metal ions through the residue layer in the 1.5 mol L−1 citric acid.
| Acid species | 1.5 mol L−1 citric acid | 4.5 mol L−1 acetic acid | ||
|---|---|---|---|---|
| Before | After | Before | After | |
| Comment: The data for each viscosity was acquired as repeated five times measurements at 298 K. The deviation was maximum ±0.03%. | ||||
| Viscosity (mm2 s−1) | 1.67 | 2.00 | 1.33 | 1.50 |
3.6 The effect of H2O2 concentration
Hydrogen peroxide is frequently used to reduce high-valence value metal ions (Co3+, Mn3+/4+) to lower valence values (Co2+, Mn2+), making them more soluble and facilitating their release from the NMC structure during the leaching.40 The H2O2 concentration effect was therefore evaluated. Fig. S16† shows the leached percentage of metal ions over the 1440 min leaching for different H2O2 concentrations from 0 to 2.0 vol%. The leaching rate of all metals were increased with an increase in the H2O2 concentration during the first 150 min, except for the Al ions. After 150 min, the leaching rate changed more moderately with increasing H2O2 concentration. Fig. S16† shows that no improvement in total amount of leached metals occurred when increasing the H2O2 concentration from 0 to 1.0 vol%. More than 1.0 vol% H2O2 resulted in a decreased in the leached percentages for all metals at 1440 min, with only ca. 80% being leached for 2.0 vol% of H2O2. This was caused by the precipitation of the copper ions, occurring with extensive addition of H2O2. The X-ray diffraction of the residues after 1440 min leaching with different H2O2 concentrations from 0 to 2.0 vol% confirmed that the peaks attributed to NMC disappeared when the H2O2 concentration reached 1.5 vol%, while the peaks attributed to Cu(OH)2 phase (ca. 22.8°) became apparent when the H2O2 concentration exceeded 1.5 vol%, see Fig. S17.† The explanation to the decrease in total amount of leached metal ions (for 2.0 vol% of H2O2) was therefore suggested to have occurred as a result of the Cu(OH)2 precipitating along with possible coprecipitates of Li, Mn, Ni, and Co. The formation of the Cu(OH)2 (s) might be due to the fact that H2O2 functions as an oxidizing agent, improving the dissolution of Cu in acid solutions.41 The quick dissolution of solid-phase Cu/CuOx to Cu2+ is accompanied by a large consumption of protons, resulting in an increased concentration of OH− at the solid/liquid interface. The sudden increase in pH at the interface eventually leads to Cu2+ precipitating in the form of Cu(OH)2. Cu(OH)2 has the lowest solubility constant (Ksp), log(Ksp) = −19.32,42 apart from the aluminium hydroxide with log(Ksp) = −33.5,42 however, citric acid has been reported to prevent precipitation of Al(OH)3 when the citric acid/aluminium ratio is higher than 0.843 (which was the case in our work).Fig. S18† shows the curves in Fig. S16† as fitted to the residue layer diffusion contribution of the shrinking core model. The leaching processes of all NMC metal elements could be divided into two stages in the presence of H2O2, with the corresponding k3 values being summarized in Table S7.† When 1.5 vol% of H2O2 was added, the k3 values of the transition metal elements (Ni, Mn, and Co) were ca. 2–3 times higher during the 1st stage (initial 150 min) than when no H2O2 was added. The increased rate constants were synonymous with a faster release of the structurally bonded metal ions. The increase of the k3 value in the 1st stage was attributed to:
• The H2O2 reducing Co and Mn from high valence values to low valence ones, which improved the ability of acid to solvate the metal ions,
• The decomposition of the H2O2 resulting in the formation of bubbles, which improves the mixing of the solution, contributing to a facilitated mass transfer and a higher leaching rate.
After 150 min (the 2nd stage), the k3 values decreased and became similar, regardless of H2O2 concentration. It was suggested that H2O2 mainly worked during the beginning of the leaching process due to its depletion. Overall, it was concluded that the addition of H2O2 enhanced the leaching rate of transition metal elements, whereas the total amount of metal ions decreased at excessively high H2O2 concentrations due to the formation of intermediate hydroxide phases (e.g., Cu(OH)2).
4 Conclusions
Extraction of lithium-ion battery metals from blackmass (leaching) was carried out using mild organic acids (citric and acetic) in the presence of ultrasound. The ultrasound significantly impacted the leaching, resulting in, on average 97% ± 3% metal ion recovery, which was a substantially higher amount of recovered metal ions than for the same conditions when only mechanical stirring was used (89% ± 2%). The highest recoveries were achieved for cobalt and nickel, reaching >99%, while lithium and manganese were recovered at 94–96% efficiency. In addition, a facilitated dissolution of the spinel LiMn2O4 (formed during the leaching) was observed. The effect of temperature and leaching time was investigated (correlated with ICP-OES measurements) for the Li, Mn, Co, and Ni metal ions. It was shown that when ultrasound was used only half of the leaching time (≤600 min) was required to reach the same percentage of target metals (ca. 90%) as the mechanically stirred leaching procedure reached in 1440 min (24 h). The time and temperature data for different metal ions were fitted to the shrinking core model. The results revealed that the leaching of Li, Ni, Mn, Co, and Al (from the fine ground LiBs powder) was controlled by residue layer diffusion, and could be associated with the residue layer diffusion contribution of the shrinking core model. The ultrasound contributing to an improved residue layer mass diffusion. The immediate effect of the ultrasound was to facilitate the liberation of primary NMC particles (within 90 min) from larger-sized secondary particles, as demonstrated by microscopy. However, although the ultrasound provided better acid access to the surface of the aggregated, smaller primary NMC particles, the leaching rate remained markedly higher over the entire 24 h (1440 min) extraction process. The only exception could be established for Cu, which was controlled by the surface chemical reaction contribution of the model. This was suggested to be caused by the limited amount of dissolved O2 in the solution, which resulted from the ultrasonic degassing of the acidic extraction solutions. Using citric acid, as compared to acetic acid, further suppressed the dissolution of Cu due to the chelating effect of citrate ions with the surface of Cu. The addition of H2O2 enhanced the leaching rate of Ni, Mn, Co, and Li, especially in the first 150 min, but showed less improvement or even decreased the maximum leached percentage of metal ions due to the co-precipitation with Cu2+ ions. The leached percentage using an even weaker organic acid (acetic acid, pKa: 4.76) demonstrated that also for this acid ultrasound had a marked effect with extraction values reaching 96% ± 2% with ultrasound, while the value was 77% ± 5% without ultrasound. It could also be concluded that the ultrasound had a greater impact on the leaching results than that of the use of either citric or acetic acid. Overall, the technique shortened leaching time, eliminated reducing agents, and omitted reported pre-treatment procedures (e.g., binder remover chemicals, and pre-calcination above 600 °C). It is therefore suggested that ultrasound provides a promising route towards energy-efficient and more environment-friendly extraction of targeted metals in discarded lithium-ion batteries. Finally, it was concluded that the blackmass composition strongly depended on the method of powder preparation, highlighting the importance of using fine ground blackmass to avoid challenges in the interpretation of leaching data due to natural fractionation from transport and storage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This article was carried out within the PERLI (Processes for Efficient Recycling of Lithium-ion Batteries) project 48228-1 granted by the Swedish Energy Agency. Assoc. Prof. M. Petranikova at Chalmers University of Technology is acknowledged for establishing the contact with Volvo Cars and providing the Volvo C30 blackmass powder under energy authority grant no: 48204-1.Notes and references
- T. Or, S. W. D. Gourley, K. Kaliyappan, A. Yu and Z. Chen, Carbon Energy, 2020, 2, 6–43 CrossRef CAS.
- J. C. Kelly, Q. Dai and M. Wang, Mitigation Adapt. Strategies Global Change, 2019, 371–396 Search PubMed.
- L. Dahllöf, M. Romare and A. Wu, Mapping of lithium-ion batteries for vehicles, Nordic Council of Ministers/Publication Unit, Denmark, 2019 Search PubMed.
- D. L. Thompson, J. M. Hartley, S. M. Lambert, M. Shiref, G. D. J. Harper, E. Kendrick, P. Anderson, K. S. Ryder, L. Gaines and A. P. Abbott, Green Chem., 2020, 22, 7585–7603 RSC.
- E. Melin, State-of-the-art in reuse and recycling of lithium-ion batteries – A research review, The Swedish Energy Agency, Eskilstuna, 2019 Search PubMed.
- X. Zheng, Z. Zhu, X. Lin, Y. Zhang, Y. He, H. Cao and Z. Sun, Engineering, 2018, 4, 361–370 CrossRef CAS.
- J. Diekmann, C. Hanisch, L. Froböse, G. Schälicke, T. Loellhoeffel, A.-S. Fölster and A. Kwade, J. Electrochem. Soc., 2017, 164, A6184–A6191 CrossRef CAS.
- Y. Ma, M. Svärd, X. Xiao, J. M. Gardner, R. T. Olsson and K. Forsberg, Metals, 2020, 10, 1–16 Search PubMed.
- C. Lupi and M. Pasquali, Miner. Eng., 2003, 16, 537–542 CrossRef CAS.
- J. Zhang, J. Hu, W. Zhang, Y. Chen and C. Wang, J. Cleaner Prod., 2018, 204, 437–446 CrossRef CAS.
- P. Liu, L. Xiao, Y. Chen, Y. Tang, J. Wu and H. Chen, J. Alloys Compd., 2019, 783, 743–752 CrossRef CAS.
- R. Golmohammadzadeh, F. Rashchi and E. Vahidi, Waste Manage., 2017, 64, 244–254 CrossRef CAS PubMed.
- L. Li, L. Zhai, X. Zhang, J. Lu, R. Chen, F. Wu and K. Amine, J. Power Sources, 2014, 262, 380–385 CrossRef CAS.
- L. Li, E. Fan, Y. Guan, X. Zhang, Q. Xue, L. Wei, F. Wu and R. Chen, ACS Sustainable Chem. Eng., 2017, 5, 5224–5233 CrossRef CAS.
- C. E. Brennen, Cavitation and bubble dynamics, 2013 Search PubMed.
- E. Ciawi, J. Rae, M. Ashokkumar and F. Grieser, J. Phys. Chem. B, 2006, 110, 13656–13660 CrossRef CAS PubMed.
- W. Y. Wang, C. H. Yen and J. K. Hsu, Sep. Sci. Technol., 2020, 55, 3028–3035 CrossRef CAS.
- Y. J. Shih, S. K. Chien, S. R. Jhang and Y. C. Lin, J. Taiwan Inst. Chem. Eng., 2019, 100, 151–159 CrossRef CAS.
- W. Gao, X. Zhang, X. Zheng, X. Lin, H. Cao, Y. Zhang and Z. Sun, Environ. Sci. Technol., 2017, 51, 1662–1669 CrossRef CAS PubMed.
- W. Gao, J. Song, H. Cao, X. Lin, X. Zhang, X. Zheng, Y. Zhang and Z. Sun, J. Cleaner Prod., 2018, 178, 833–845 CrossRef CAS.
- X. Chen, B. Fan, L. Xu, T. Zhou and J. Kong, J. Cleaner Prod., 2016, 112, 3562–3570 CrossRef CAS.
- C. Lei, I. Aldous, J. M. Hartley, D. L. Thompson, S. Scott, R. Hanson, P. A. Anderson, E. Kendrick, R. Sommerville, K. S. Ryder and A. P. Abbott, Green Chem., 2021, 4710–4715 RSC.
- A. E. Danks, S. R. Hall and Z. Schnepp, Mater. Horiz., 2016, 3, 91–112 RSC.
- H. Poerwono, K. Higashiyama, H. Kubo, A. T. Poernomo, S. Suharjono, I. K. Sudiana, G. Indrayanto and H. G. Brittain, Citric Acid, Elsevier Masson SAS, 2001, vol. 28 Search PubMed.
- D. D. Perrin, B. Dempsey and E. P. Serjeant, pKa Prediction for Organic Acids and Bases, 1981 Search PubMed.
- X. Chen, C. Luo, J. Zhang, J. Kong and T. Zhou, ACS Sustainable Chem. Eng., 2015, 3, 3104–3113 CrossRef CAS.
- W. J. Wolfgong, Handb. Mater. Fail. Anal. with Case Stud. from Aerosp. Automot. Ind, 2016, pp. 279–307 Search PubMed.
- X. Xiao, F. Hayashi, H. Shiiba, S. Selcuk, K. Ishihara, K. Namiki, L. Shao, H. Nishikiori, A. Selloni and K. Teshima, J. Phys. Chem. C, 2016, 120, 11984–11992 CrossRef CAS.
- Q. H. Zhang, S. P. Li, S. Y. Sun, X. S. Yin and J. G. Yu, Chem. Eng. Sci., 2010, 65, 169–173 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- H. Nord, Acta Chem. Scand., 1955, 9, 430–437 CrossRef CAS.
- D. G. Eskin, Ultrasonic degassing of liquids, Elsevier Ltd, Uxbridge, UK, 2015 Search PubMed.
- A. R. Kaiser, C. A. Cain, E. Y. Hwang, J. B. Fowlkes and R. J. Jeffers, J. Acoust. Soc. Am., 1996, 99, 3857–3859 CrossRef.
- D. Tromans, Hydrometallurgy, 1998, 48, 327–342 CrossRef CAS.
- P. R. Taylor, M. de Matos and G. P. Martins, Metall. Trans. B, 1983, 14, 49–53 CrossRef.
- R. G. Bates and G. D. Pinching, J. Am. Chem. Soc., 1949, 71, 1274–1283 CrossRef CAS.
- K. J. Laidler, J. Chem. Educ., 1984, 61, 494–498 CrossRef CAS.
- T. C. Hu, S. Y. Chiu, B. T. Dai, M. S. Tsai, I. C. Tung and M. S. Feng, Mater. Chem. Phys., 1999, 61, 169–171 CrossRef CAS.
- K. Makino, M. M. Mossoba and P. Riesz, J. Phys. Chem., 1983, 87, 1369–1377 CrossRef CAS.
- L. P. He, S. Y. Sun, X. F. Song and J. G. Yu, Waste Manage., 2017, 64, 171–181 CrossRef CAS PubMed.
- J. C. Chen and W. T. Tsai, Mater. Chem. Phys., 2004, 87, 387–393 CrossRef CAS.
- R. Smith and A. Martell, Critical Stability Constants Volume 4: Inorganic Complexes, 1976, vol. 4 Search PubMed.
- D. M. Dabbs, U. Ramachandran, S. Lu, J. Liu, L. Q. Wang and I. A. Aksay, Langmuir, 2005, 21, 11690–11695 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc02693c |
| This journal is © The Royal Society of Chemistry 2021 |