
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComparative study of the solvolytic deconstruction of corn stover lignin in batch and flow-through reactors†
Daniel
Vincent Sahayaraj‡
 ab,
Lusi
A‡
c,
Emma M.
Mitchell
a,
Xianglan
Bai
*c and
Jean-Philippe
Tessonnier
*ab
ab,
Lusi
A‡
c,
Emma M.
Mitchell
a,
Xianglan
Bai
*c and
Jean-Philippe
Tessonnier
*ab
aDepartment of Chemical and Biological Engineering, Iowa State University, Ames, Iowa 50011, USA. E-mail: tesso@iastate.edu
bCenter for Biorenewable Chemicals (CBiRC), Ames, Iowa 50011, USA
cDepartment of Mechanical Engineering, Iowa State University, Ames, Iowa 50011, USA. E-mail: bxl9801@iastate.edu
First published on 2nd September 2021
Abstract
Solvolysis of industrial corn stover lignin was carried out in methanol and ethanol to deconstruct lignin into monomers and small oligomers using various reactor configurations. We demonstrate that it is possible to overcome the limitations inherent to batch reactors by carrying out the solvolytic deconstruction in flow-through reactor systems as they allow for the continuous removal and rapid cooling of the products. Monomers and solvent-soluble product yields achieved in flow-through reactors were higher under all tested reaction conditions. We also investigated the effects of various reaction parameters and phase of solvent (sub- and supercritical) on the distribution and yield of lignin-derived products using complementary characterization techniques. p-Coumaric acid and ferulic acid were obtained as primary monomers with very high selectivity and a maximum monomer yield of 14.45 wt% was achieved at 250 °C and 85 bar in ethanol. Subsequent upgrading of the deconstructed fractions through fast pyrolysis was also studied. In addition to the increased yield of phenolic monomers (23.86 wt% vs. 10.29 wt%), char formation during pyrolysis was substantially decreased (26.36 wt% vs. 40.95 wt%) under optimized conditions. The beneficial impact of the solvolytic pretreatment was also reflected in an enhanced aromatic hydrocarbon yield during catalytic fast pyrolysis.
Introduction
Lignin is an abundant, cheap, and underutilized source of carbon for the production of biobased fuels and chemicals. It accounts for 15–30 wt% of all lignocellulosic biomass and is primarily composed of three backbone units: guaiacyl, sinapyl, and p-hydroxyphenyl linked nonlinearly and randomly by aryl-ether and C–C linkages.1–3 Despite its enormous potential, practical valorization is still lacking because of its intrinsic heterogeneity and recalcitrant nature.4,5 For this reason, conventional biorefinery schemes such as the paper and pulp industry focus on extracting ‘valuable’ carbohydrate fractions (cellulose and hemicellulose) for further downstream applications while discarding lignin (i.e., delignification).6 Due to the harsh reaction conditions applied to cleave the lignin-carbohydrate linkages, the already complex lignin polymer is transformed into a more condensed residue.7 This complicates any further upgrading or recovery of products, and, as a result, lignin and lignin residues are mainly used as low-value boiler fuels.8 In order to improve the economic viability of biorefineries, sustainable technologies to transform lignin into value-added products are critically needed.9,10The selective conversion of lignin remains an elusive challenge for researchers due to the starting material's heterogeneous nature. Therefore, the growing focus is on transforming target structural units in lignin to value-added products and chemicals. Herbaceous biomass such as corn stover comprises extensive amounts of hydroxycinnamic acid pendant units, which are primarily associated with lignin through both ester and ether linkages (∼18% p-coumaric acid on a lignin basis).11–13 The extraction of hydroxycinnamic acids is of considerable interest because they play a vital role in the chemical industry as antioxidants, antimicrobials, precursors for value-added chemicals, and synthesis of functional polymers.12,14–17 Additionally, about 67 million tons of corn stover are generated in Iowa alone each year as a by-product of corn production. Farmers use only a fraction of the harvest as bedding and feed for cattle. However, there is increasing evidence that excess stover is a source of pathogens and allelopathy that are detrimental to crop yields.18–20 As the production far exceeds the current needs, new valorization technologies are desperately needed to utilize this agricultural residue.
Solvolytic deconstruction (or solvolysis) of lignin has shown promise in breaking down lignin into monomeric phenols.21 This approach combines the synergistic effect of thermal cracking and the solvent-induced cleavage of bonds in the biopolymer. However, the low monomer yields and inevitable formation of char due to the condensation and repolymerization of reactive intermediates generated during solvolysis represent significant roadblocks toward the scaling of these processes. In this regard, common batch reactors are effective towards disassembling lignin but cannot prevent these parasitic secondary transformations as the reactive intermediates remain in the hot reactor during the entire course of the reaction. More recently, flow-through reactors were introduced as an alternative configuration for lignin depolymerization.22 In these reactors, lignin is semi-continuously depolymerized into monomeric phenols and the depolymerized fragments are immediately removed from the reaction zone to be collected or further upgraded through catalytic treatment in a downstream reactor. Studies using flow-through reactors have until now focused on the kinetics of lignin solvolysis and, when applicable, the stability of the applied catalyst.22–25 However, these studies have been mainly exploratory and further research is needed to determine the relative merits of deconstructing lignin in a flow-through configuration over batch reactor configuration. The prospect of continuous removal and rapid cooling of the depolymerized product in a flow-through reactor could be beneficial towards curtailing secondary reactions and enhancing monomer and depolymerized product yields.
In addition to these technical advantages, fundamental insights into the solvolytic deconstruction could be drawn using flow-through systems by obtaining time-resolved product distributions and yields for various reaction conditions. The temperature and solvent properties are already known to influence the dominant reaction pathway for lignin deconstruction in batch reactors under sub- and supercritical conditions.26,27 Methanol and ethanol were chosen as solvents in these studies owing to their low environmental impact and their ability to dissolve lignin effectively under reaction conditions.28–30 Kim et al. studied the depolymerization of organosolv lignin in subcritical and supercritical ethanol and found that the yield of phenolic monomers increased with temperature, while the yield of the oil (liquid) phase decreased.31 A comprehensive study conducted for lignin depolymerization using supercritical ethanol and formic acid by Riaz et al. led to higher lignin-derived monomers and curtailed the solvent decomposition reactions.32 Other alcohols were also evaluated in this work, but ethanol afforded the best compromise between lignin conversion and phenolic monomer yield. However, in all these studies, batch reactors limited the insights gained into the deconstruction process. Flow-through systems could play a pivotal role in understanding the differences in the solvent's role and contribution under sub- and supercritical conditions.
In the present work, we studied the deconstruction of lignin in both batch and flow-through systems and drew normalized comparisons between the two reactor configurations. We first show that the repolymerization of reactive intermediates lowers the product yield but that this parasitic reaction can be curtailed by minimizing the residence time of the depolymerized fragments in the hot zone of the reactor. Next, we obtained mechanistic insights into the solvolytic depolymerization of corn stover lignin in subcritical and supercritical alcohols using the flow-through reactor system. By combining GPC, GC-MS, and NMR, we gained an understanding of the depolymerization process and parameters that govern the size distribution and yield of lignin-derived products. The subsequent upgrading of the deconstructed lignin to aromatic monomers using non-catalytic and catalytic pyrolysis was also evaluated to assess the potential benefits of the solvolytic depolymerization on further upgrading. The results obtained in the present study are significant as they provide fundamental insights into the solvolytic deconstruction of lignin and enable the rational design of catalysts for the subsequent upgrading of the deconstructed lignin monomers and oligomers.
Results and discussion
Efficiency of lignin deconstruction in batch and flow-through systems
The deconstruction experiments were carried out under both subcritical (170 °C, 30 bar) and supercritical conditions (250 °C, 85 bar) of MeOH and EtOH, in both the batch and flow-through systems. For consistency, the same reaction temperatures and pressures were selected for MeOH and EtOH, although the critical points of the two solvents are different (MeOH: Tc = 240 °C and Pc = 81 bar; EtOH: Tc = 241 °C and Pc = 63 bar). In addition, the lignin mass to solvent volume ratio was kept constant at 5 mg mL−1 to draw normalized comparisons between the two reaction systems.The phenolic monomers detected were p-coumaric acid, ferulic acid, vanillin, and 4-hydroxy-3,5-dimethoxybenzaldehyde under all tested conditions. p-Coumaric and ferulic acids were obtained with high selectivity (total selectivity >95%) compared to the other monomers, which were detected in small concentrations, independently of the reaction system. The product distributions were identical for both solvents, although variations were observed for the monomer yields. As shown in Fig. 1, an initial increase followed by a decrease in monomer yield was observed with time for the batch reactor, which can be attributed to secondary reactions where monomers recondense to form larger oligomers. In contrast, the deconstructed fragments were removed continuously in the flow-through system and, as a result, the extent of secondary reactions was attenuated. The maximum cumulative monomer yield was lower in the batch reactor than in the flow-through reactor for most reaction conditions. Notably, the maximum monomer yield reached ∼4 wt% in the batch system vs. ∼10 wt% in the flow-through reactor at 170 °C and 30 bar. However, the maximum monomer yields were comparable in both systems at 250 °C, 85 bar. The difference between the monomer yields can be partially attributed to enhanced lignin dissolution in flow-through reactors.33–35 In flow-through systems, fresh solvent continuously contacts the packed bed, thereby granting the maximum diffusive flux.
 | ||
| Fig. 1 Cumulative monomer yields obtained in batch (orange) and flow-through (black) systems for MeOH and EtOH at (a) 170 °C, 30 bar, (b) 250 °C, 85 bar. | ||
To further characterize the differences between batch and flow-through deconstruction processes, the total mass of solvent-soluble products was measured at the end of the reaction for both systems. Since char and very high molecular weight fragments are insoluble in MeOH and EtOH at room temperature, this measure indicates the extent of lignin depolymerization and/or repolymerization reactions. As shown in Fig. 2a, the fraction of solvent-soluble products obtained with the flow-through system was much higher than with the batch reactor for all reaction conditions and identical lignin concentrations. The difference between soluble product yields was more prominent under supercritical conditions due to a higher degree of repolymerization in the batch reactor (∼75 wt% solvent-soluble products for flow-through vs. ∼38 wt% for the batch system).
 | ||
| Fig. 2 Fraction of solvent-soluble products (wt%) obtained after treating lignin in batch and flow-through systems for 2 hours. (a) Effect of reaction conditions on product yield at 5 mg of lignin per mL of solvent processed. (b) Effect of lignin concentration at 250 °C, 85 bar, EtOH. | ||
As previously mentioned, this result cannot be wholly attributed to alleviating repolymerization reactions since a batch reactor is inherently mass transfer limited.23,34 As shown in Fig. 2b, additional batch reactor experiments were performed at 250 °C, 85 bar, EtOH, and this time, the concentration was varied between 2.5 and 10 mg of lignin per mL of solvent. The amount of solvent-soluble products increased to nearly ∼53 wt% when doubling the amount of solvent to reach a lignin concentration of 2.5 mg mL−1 but was still lower than that of the flow-through system. In contrast, when the lignin concentration was increased to 10 mg mL−1, only 20.9 wt% of products were recovered. Furthermore, time-resolved mass balance at these reaction conditions revealed that the mass of solvent-soluble products decreased continuously with time for the batch reactor (Fig. S1†). Although the dissolution of lignin is enhanced under supercritical conditions, the undesirable effect of secondary reactions and repolymerization drastically reduces the yield of lignin-derived products obtained in the batch reactor.
To gain further insights into secondary reactions, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w) mixture of p-coumaric acid and ferulic acid, the main monomeric products generated during solvolysis, was heated in MeOH and EtOH and the obtained solutions were subsequently analyzed by GPC (Fig. 3 and Fig. S2†). These molecules are said to be reactive due to the C
:1 (w/w) mixture of p-coumaric acid and ferulic acid, the main monomeric products generated during solvolysis, was heated in MeOH and EtOH and the obtained solutions were subsequently analyzed by GPC (Fig. 3 and Fig. S2†). These molecules are said to be reactive due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C functionality on their side chain.36–38 Both higher molecular fragments due to repolymerization and lower molecular weight fragments due to cracking were observed within only 30 minutes. At 250 °C, the formation of both higher and lower molecular weight species was more pronounced than at 170 °C for both solvents. Peaks corresponding to dimers and trimers (∼700 Da) were observed at 170 °C, while oligomers (>2000 Da) were observed at 250 °C for both solvents. Hence, higher temperatures lead to higher molecular weight products due to repolymerization reactions in addition to promoting cracking reactions.39,40 These results are significant as lignin oligomers have a greater propensity to repolymerize than monomers, as crosslinking is facilitated due to proximity and orientation effects.41 This was observed from the molecular weight distribution profile of products in the batch reactor, where the percentage of larger molecular weight species decreased with increasing time due to secondary reactions to form char/insoluble products (Fig. S3†). The solvent-soluble product yield and molecular weight decreased as reaction time progressed in the batch reactor, indicating enhanced secondary reactions, which convert lignin into insoluble char and gases. In summary, the higher yields of monomers and solvent-soluble products in the flow-through reactor can be attributed to higher dissolution rates and fractionation of products, which mitigate secondary reactions.23,35
C functionality on their side chain.36–38 Both higher molecular fragments due to repolymerization and lower molecular weight fragments due to cracking were observed within only 30 minutes. At 250 °C, the formation of both higher and lower molecular weight species was more pronounced than at 170 °C for both solvents. Peaks corresponding to dimers and trimers (∼700 Da) were observed at 170 °C, while oligomers (>2000 Da) were observed at 250 °C for both solvents. Hence, higher temperatures lead to higher molecular weight products due to repolymerization reactions in addition to promoting cracking reactions.39,40 These results are significant as lignin oligomers have a greater propensity to repolymerize than monomers, as crosslinking is facilitated due to proximity and orientation effects.41 This was observed from the molecular weight distribution profile of products in the batch reactor, where the percentage of larger molecular weight species decreased with increasing time due to secondary reactions to form char/insoluble products (Fig. S3†). The solvent-soluble product yield and molecular weight decreased as reaction time progressed in the batch reactor, indicating enhanced secondary reactions, which convert lignin into insoluble char and gases. In summary, the higher yields of monomers and solvent-soluble products in the flow-through reactor can be attributed to higher dissolution rates and fractionation of products, which mitigate secondary reactions.23,35
 | ||
| Fig. 3 Molecular weight distributions of the products obtained after 30 min using a 1:1 w/w mixture of p-coumaric and ferulic acids (total mass of 10 mg) in 2 mL of solvent at various conditions in (a) MeOH and (b) EtOH. The result obtained for p-coumaric acid (pCA) is provided as a reference. | ||
Products and product distributions after lignin deconstruction in flow-through reactors
As mentioned in the previous section, the monomeric products generated were p-coumaric acid, ferulic acid, vanillin, and 4-hydroxy-3,5-dimethoxybenzaldehyde. In terms of cumulative monomeric yield obtained, MeOH at 170 °C and 30 bar performed better than EtOH under identical conditions (11.41 wt% vs. 9.65 wt%) while EtOH at 250 °C and 85 bar gave the highest yield (Fig. 4) among the reaction conditions tested (14.45 wt%).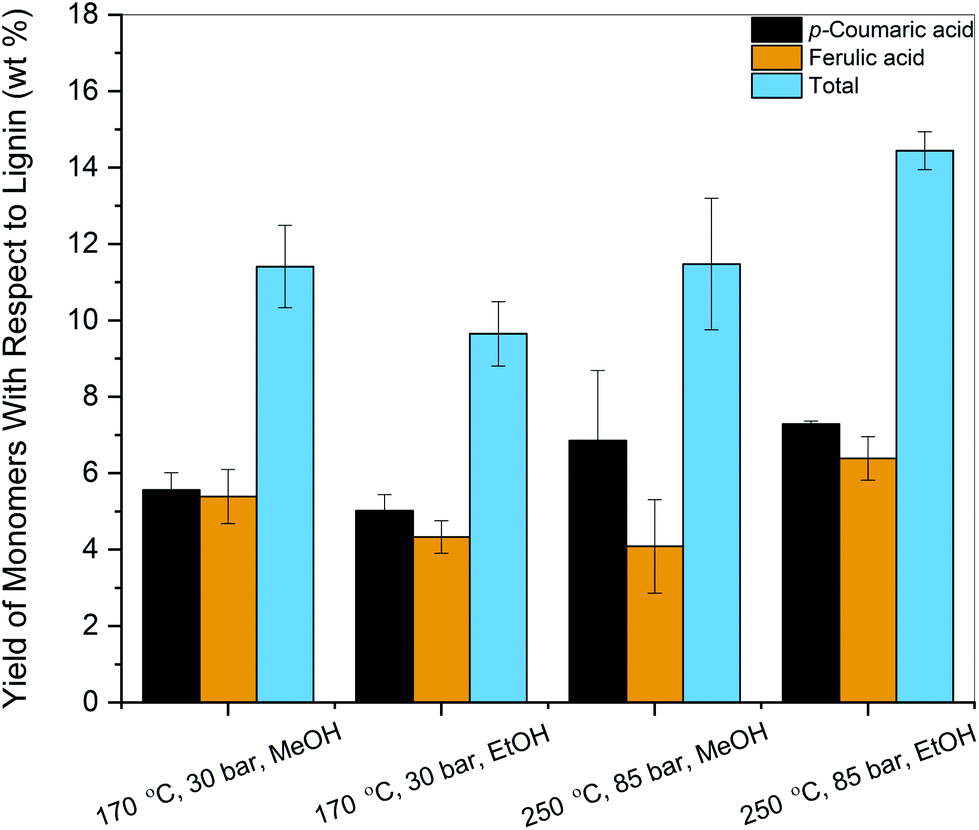 | ||
| Fig. 4 Cumulative yields of phenolic monomers obtained for various operating conditions using a flow-through reactor system. | ||
The difference between yields in supercritical MeOH and EtOH could be due to the lower critical pressure of ethanol than methanol. Therefore, depolymerization in ethanol operates at a much higher supercritical state than methanol. In addition, as seen in Fig. 3, results from model compound reactivity studies also indicated that secondary reactions were more prevalent in MeOH than EtOH at 250 °C, 85 bar. Although studies using similar feedstocks have observed the presence of decarboxylation products, such as 4-vinyl phenol and 4-vinyl guaiacol, these monomers were not observed under our reaction conditions.42
Although there are numerous reports on lignin depolymerization in the literature, only a few studies produced monomers with high selectivity. The formation of 4-ethyl phenolics via mild hydrogenolysis from corn stover lignin was reported using a Ru/C catalyst. A maximum yield of 3.1 wt% of 4-ethylphenol and 1.3 wt% of 4-ethyl guaiacol was produced with 65 vol% ethanol/water as the solvent.43 Nandiwale et al. demonstrated that the thermo-solvolytic treatment of corn stover lignin in acetic acid followed by depolymerization over Zr-KIT-5 catalyst yielded 28 wt% of identified phenolic monomers and 37% selectivity towards acetylated monomers.44 The high monomeric yields were attributed to better accessibility to the mesopores by the smaller lignin fragments formed during the thermal pretreatment. The selective conversion of lignin from corncob residue to monophenols was achieved via a two-step process.45 The lignin component in corncob residue was first selectively degraded to oligomers at 200 °C in a H2O–THF (3:7, v/v) co-solvent system and deconstructed further to monophenols at 300 °C. The yield of total monophenols reached 24.3 wt% and 4-ethylphenol, 2,6-dimethoxyphenol, and 4-ethyl guaiacol were the predominant products, representing 86.8% of the identified monomers. To date, the monomer yields achieved by lignin solvolysis without the addition of a catalyst have generally been below 10 wt%. Higher yields have sometimes been reported, albeit with low product selectivities.21 In essence, we demonstrated a strategy to selectively generate hydroxycinnamic acids in high yields in the absence of an heterogeneous catalyst using a flow-through solvolysis system.
The yield of monomers and mass of solvent-soluble products obtained under various operating conditions as a function of time are reported in Fig. 5. Phenolic monomers and lignin-derived products were observed only in the first three samples, which indicates that their extraction from lignin was complete within the corresponding reaction time (∼70 min). As can be seen in Fig. 5a, the sample collected at 30 min contained the highest amount of monomers, and the yield decreased as a function of time. No monomers were observed after 70 min. A similar trend was observed for the mass of the deconstructed sample collected, where the amount leveled to less than 5 wt% in each sample after 70 min (Fig. 5b). Nearly ∼70–75 wt% of the lignin loaded in the bed was collected by the end of the reaction. The monomer yields were normalized with the mass of solvent-soluble products for each sample point to further understand the effect of reaction conditions on product distribution. EtOH at 250 °C and 85 bar had the highest monomer/solubilized products ratio (Fig. 5c), meaning that these conditions were the most effective to deconstruct and solubilize the lignin polymer. Fig. 5c also revealed that flow-through reactors could deconstruct and fractionate lignin to produce highly concentrated hydroxycinnamic acids fractions within a short reaction time. Less than 15% of the initially loaded lignin was observed as char left as a residue in the reactor bed, regardless of the reaction conditions. The mass balance closure was about 80–88 wt% at all tested reaction conditions.
 | ||
| Fig. 5 (a) Yield of monomers (wt%), (b) mass of solvent-soluble products collected (wt%), and (c) monomer/sample ratio (wt/wt%) as a function of time for various operating conditions. | ||
The collected samples were further characterized by GPC to gain insights into this deconstruction process. The products obtained through solvolysis were significantly deconstructed compared to the initial lignin (5368 Da), and the extent of the deconstruction increased with increasing temperature (Table 1). Since the sample collected at 30 min contained nearly 60–70% of the total depolymerized lignin, this sample was used to gauge the extent of deconstruction as a function of reaction conditions. Lignin depolymerized in supercritical ethanol was the most deconstructed sample, with the smallest average molecular weight (∼1249 Da) of all tested reaction conditions. As shown in Table 1, the products’ molecular weight increased significantly after 70 min (>2000 Da) for all reaction conditions. This indicates that the smaller fragments were eluted first, followed by larger fragments, which could be due to recondensation reactions or preferential dissolution of smaller fragments.46 Nevertheless, the fractions obtained after 70 min only account for 15% of the total amount of the depolymerized product collected. It is worth mentioning that the molecular weight of the solvent-soluble products obtained from the flow-through system was higher than those from the batch reactor (Table S1†). This difference can be ascribed to the reduced secondary transformations and improved mass transfer in flow-through reactors. In batch reactor, the secondary reactions and mass transfer limitations promote both repolymerization and cracking. Although the molecular weights of the solvent-soluble products were smaller in the batch reactor, the yields of solvent-soluble products decreased as lignin was further converted to char and gases as the reaction progressed.
| Reaction conditions | ||||
|---|---|---|---|---|
| Time (min) | 170° C, 30 bar, MeOH | 250° C, 85 bar, MeOH | 170° C, 30 bar, EtOH | 250° C, 85 bar, EtOH |
| 30 | 1548 | 1338 | 1310 | 1249 |
| 50 | 1547 | 1383 | 1571 | 1378 |
| 70 | 2045 | 2028 | 1909 | 1777 |
| 90 | 2704 | 2138 | 2158 | 1923 |
| 110 | 2781 | 2355 | 2268 | 2268 |
| 130 | 2627 | 2366 | 2372 | 2499 |
The structural properties of the deconstructed lignin products obtained under different reaction conditions were analyzed by 2D HSQC NMR. An example of these spectra is shown in Fig. 6. Peaks from methoxy and β-O-4 substructures were prominent in the side chain region of the spectra (side chain region: δC/δH = 50–90/3.0–5.5 ppm). The peak corresponding to γ-p-coumaroylated β-ether units (β-O-4 (pCA)) is also observed in this region. However, contributions from phenylcoumaran, dibenzodioxocin, and resinol substructures were not within detection limits. Intense p-coumaric and ferulic acid peaks, which are characteristic for herbaceous lignin, were observed in the aromatic region (aromatic region: δC/δH = 90–155/6.0–8.0). Syringyl (S) correlations dominate the guaiacyl (G) correlations. The p-hydroxyphenyl (H) correlations were not present, in good agreement with other studies.47,48
 | ||
| Fig. 6 2D HSQC NMR spectra of (a) corn stover lignin and (b) the product obtained at 250 °C, 85 bar, EtOH along with their relative integrals. | ||
For a semi-quantitative comparison, the amount of inter-unit linkages and lignin (H, G, and S) units from untreated lignin and deconstructed samples obtained at different reaction conditions were estimated through the integration of the contours in the HSQC spectra, according to the literature (Table 2, Table S2 and eqn (S1)†).49 Upon depolymerization, the intensity of β-O-4 (pCA) ether linkages greatly decreased in all samples. The cleavage of the ether-linked hydroxycinnamic acid units occurred at around 170 °C while the ester-linked units were released under milder conditions.12,50 Therefore, the formation of p-coumaric and ferulic acid is through the cleavage of these linkages under the examined reaction conditions, while the other ether linkages present in lignin require higher temperatures to be cleaved.50 However, only minor variations were observed in the aromatic region. This indicated no significant occurrence of demethoxylation reactions or selective solubilization of the S or G units present in lignin.
| Lignin | 170° C, 30 bar, EtOH | 250° C, 85 bar, EtOH | 170° C, 30 bar, MeOH | 250° C, 85 bar, MeOH | |
|---|---|---|---|---|---|
| a Expressed per 100 aromatic units based on the relative integrals of characteristic peaks in the 2D HSQC spectra using the formula in ESI.† | |||||
| β-O-4a (%) | 29.48 | 22.03 | 20.24 | 25.21 | 26.6 |
| β-O-4(pCA)a (%) | 49.11 | 32.64 | 28.73 | 27.38 | 31.02 |
| β-O-4(pCA)/β-O-4 (%) | 59.17 | 40.02 | 34.52 | 36.51 | 37.02 |
| S/G | 2.09 | 2.11 | 1.91 | 2.02 | 1.75 |
FTIR analysis of the solvent-soluble products revealed that –OH (3370 cm−1), aliphatic –CH3 and –CH2 (2925 and 2851 cm−1), and carbonyl (1705 cm−1) functionalities decreased until 70 min but leveled off at longer time intervals (Fig. S4†). Based on the above results, the abundancy of the functionalities corresponds to the hydroxycinnamic acid concentrations in the solvent-soluble products. Compared to those produced at early reaction times, the solvent-soluble products obtained at longer reaction times show more aromatic condensed structures. Therefore, the flow-through reaction could reactively fractionate lignin into solvent-solubles with tailored compositions and properties.
Scheme 1 shows the overall reaction pathway for the solvolytic depolymerization of corn stover organosolv lignin. Lignin is solubilized in the reaction solvent, and the fragments undergo solvolysis to release the ester and ether-linked hydroxycinnamates. The deconstructed lignin and the monomers, which remain solubilized, are transferred to the sample collection unit. Simultaneously secondary reactions such as repolymerization and cracking reactions can also take place, leading to the formation of char and gases. However, the extent of secondary reactions was significantly lower in comparison to the batch system. Note that not all the loaded lignin was dissolved in the reaction solvent. The insoluble lignin with higher molecular weights was retained in the reactor bed for a longer time and was converted to char or gaseous products in the flow-through system.
 | ||
| Scheme 1 Proposed overall reaction network for the depolymerization of corn stover lignin in batch and flow-through reactors. | ||
Pyrolysis and catalytic fast pyrolysis of solvent-depolymerized lignin
Fast pyrolysis uses fast heating rates and high temperatures (450–600 °C) to break down organic material in the absence of oxygen to produce a bio-oil mainly comprised of phenolic monomers and oligomers. The monomers are desired as they can serve as intermediates to commodity chemicals, whereas the oligomers generated are difficult to upgrade and valorize.38 During lignin pyrolysis, the loss of carbon in the form of char (35–50 C %) leads to a reduction in the yield of volatile species generated and presents a roadblock towards scaling up this process.51 Pretreatment of lignin prior to pyrolysis has been shown to improve liquid product yield as well as the quality of the bio-oil.52 Thus, lignin and the solvent-soluble samples obtained at various reaction conditions for 30 min were pyrolyzed at a temperature of 500 °C, and the yield of phenolic monomers is represented in Fig. 7. Among the phenolic monomers, 4-vinyl phenol (VP) and 4-vinyl guaiacol (VG) were the primary products. Vinyl phenolics can be produced both from the degradation of hydroxycinnamic acids and through the cleavage of β-aryl ether linkages in lignin.53 Experiments with p-coumaric acid as the starting compound generated nearly 90 wt% of VP (Fig. S5†). Similarly, VG arises from ferulic acid after decarboxylation during pyrolysis. The other phenolic monomers included compounds such as phenol, guaiacol, 4-ethyl guaiacol, vanillin.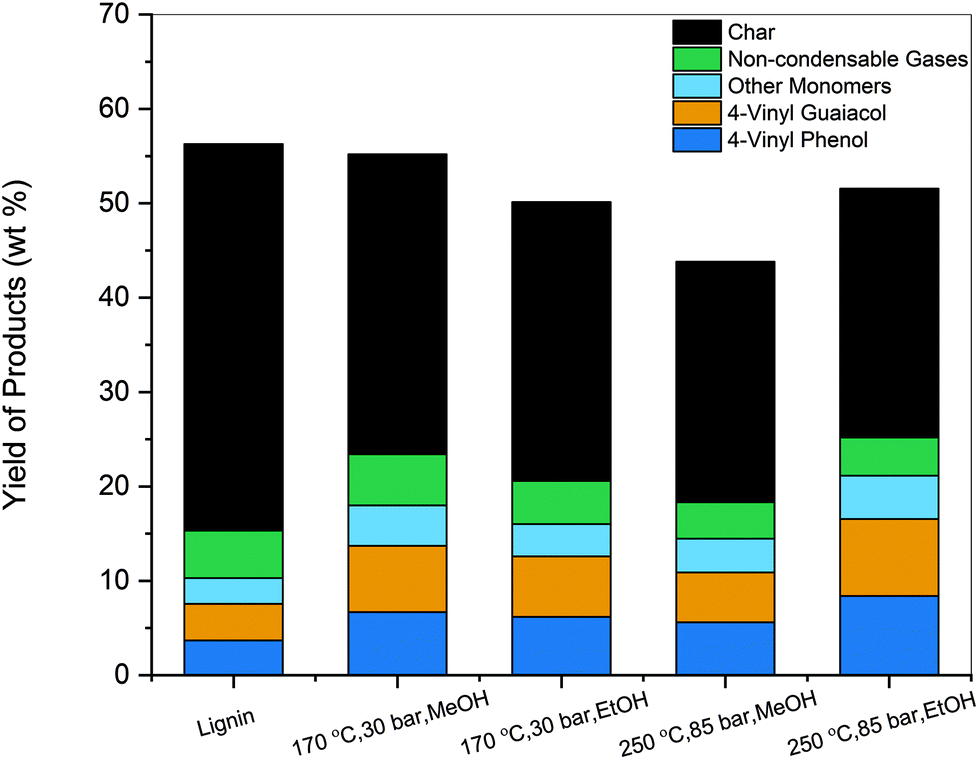 | ||
| Fig. 7 Yields of products obtained from the fast pyrolysis at 500 °C of untreated corn stover lignin and depolymerized lignin products (30 min sample). | ||
The total monomeric yield was 10.3 wt% for untreated organosolv corn stover, which was in agreement with prior experimental studies.51,54 The combined yield of VP and VG was 7.57 wt%. In contrast, the pretreated samples showed an increase in the total amount of monomers generated. The sample deconstructed at 250 °C, 85 bar, EtOH showed the highest increase with a total of 23.86 wt% of phenolic monomers produced. The high selectivity towards VP and VG was maintained, with a combined yield of 19.06 wt%. Vinyl phenolics are valuable products as they are used as food additives, antioxidants and are industrially relevant raw materials.55 Increase in yields of compounds such as vanillin, phenol, 4-ethyl phenol, 4-ethyl guaiacol were also observed. The samples depolymerized using EtOH showed a more significant increase in yields compared to MeOH. The solvolytic deconstruction of lignin caused the solvent-soluble samples to contain partially depolymerized lignin units with a less condensed structure than that of raw lignin. Therefore, more volatile species were formed, and the yield of phenolic products was enhanced compared to raw lignin itself.
The formation of char during lignin pyrolysis retained nearly 40–55 wt% of the initial mass, allowing only a portion of the feed to volatilize and be converted into products.54 In contrast, the increase in monomeric yields in the solvent-pretreated samples was accompanied by the reduction in char content. Samples treated using subcritical conditions of the solvent showed a 10 wt% reduction in char, while supercritical conditions decreased the amount even further by 15 wt%. Thus, the partial deconstruction and cleavage of bonds in the lignin polymer prior to pyrolysis could potentially reduce the formation of reactive species susceptible to repolymerization and char formation.56
The sample obtained at 30 min for 250 °C, 85 bar, EtOH generated the highest amount of phenolic monomers. Therefore, these conditions were selected for further investigation. The samples collected between 30 and 130 min were pyrolyzed, and the results are presented in Fig. 8. In general, higher yields compared to lignin were obtained for samples depolymerized and collected within the first 90 min. These results were in accordance with the GPC results, where larger weight fragments were extracted at longer times. It is imperative to account for the loss of mass/carbon in every stage of treatment to enable accurate comparisons relative to untreated lignin.56 As shown in Fig. 8, the normalized yield considers the mass of all samples obtained after flow-through solvolytic treatment (eqn (S2)†). The normalized yield was higher than that of lignin (14.73 wt% vs. 10.3 wt%), and the combined yield of VP and VG was 11.19 wt%.
 | ||
| Fig. 8 Yields of phenolic monomers obtained from fast pyrolysis of lignin and sample depolymerized at 250 °C, 85 bar, EtOH as a function of time. Left Y-axis: Yield of phenolic monomers (wt%), Right Y-axis: Mass of the solvent-soluble products obtained at corresponding time point from flow-through solvolytic treatment. | ||
Catalytic fast pyrolysis (CFP) involves direct deoxygenation of pyrolytic vapors, primarily using solid acid catalysts, to produce a product stream composed of aromatic hydrocarbons. Although studies have shown that it is feasible to convert lignin pyrolytic vapors/bio-oil into aromatics and olefins through CFP, the yields are much lower than whole biomass/cellulose-derived bio-oil.57 To test our hypothesis, i.e., the pretreatment could enhance the yield of aromatic products, catalytic fast pyrolysis experiments were performed at 500 °C with ZSM-5 zeolite. The yield of aromatics obtained from the sample pretreated at 250 °C, 85 bar, EtOH (30 min) was significantly improved compared to that of lignin (14.03 C % vs. 5.79 C %) (Table 3). Research efforts are underway to further study the effect of flow-through pretreatment on catalytic fast pyrolysis and hydrodeoxygenation reactions.
| Lignin | 250 °C, 85 bar, EtOH (30 min) | |
|---|---|---|
| Total aromatic yield (C %) | 5.79 | 14.03 |
| Distribution of aromatic product yield (C%) | ||
| Benzene | 0.54 | 1.23 |
| Toluene | 1.2 | 3.24 |
| Xylene | 1.01 | 2.61 |
| C9 | 0.23 | 0.57 |
| C10 | 1.46 | 3.39 |
| C10+ | 1.32 | 2.98 |
Conclusion
In this study, corn stover lignin was depolymerized in methanol and ethanol under both subcritical and supercritical conditions using batch and flow-through reactor systems. p-Coumaric acid and ferulic acid were obtained as primary monomers with very high selectivity (>95%) at all tested reaction conditions. The yield of monomeric products obtained in a flow-through reactor was typically higher than in a batch reactor. A higher yield of solvent-soluble products was also obtained from the flow-through reactor due to a decrease in repolymerization reactions and improved mass transfer. In contrast, repolymerization reactions that lead to the undesirable formation of char were observed in the batch reactor. The occurrence of secondary reactions was further confirmed using p-coumaric acid and ferulic acid as lignin model compounds. The extent of these reactions was more pronounced at higher temperatures. Therefore, the higher yields of monomers and deconstructed products in the flow-through reactor were attributed to higher dissolution rates and separation of products, which mitigates secondary reactions.Mechanistic insights into the solvolytic deconstruction process were obtained using the flow-through reactor. Ethanol at supercritical conditions (250 °C, 85 bar, EtOH) was found to be the most effective towards deconstructing lignin among all operating conditions evaluated, with a maximum monomeric yield of 14.45 wt%. The investigation of the reaction mechanism using HSQC NMR revealed that p-coumaric and ferulic acid formation occur through the cleavage of ester and ether-linked hydroxycinnamates. GPC analysis revealed that the lignin was significantly deconstructed due to solvolytic treatment from ∼5000 Da to <2500 Da at all reaction conditions.
The solvent-soluble products were then subjected to fast pyrolysis and catalytic fast pyrolysis. The formation of char during pyrolysis was also substantially decreased from 40 wt% to about 25 wt% for the sample treated using 250 °C, 85 bar, EtOH. When subjected to catalytic fast pyrolysis using a ZSM-5 catalyst, this deconstructed product showed an increase in aromatic product yield from 5.79 C% to 14.03 C%.
Experimental
Chemicals and materials
Corn stover-derived organosolv lignin was obtained from Archer Daniels Midland (ADM). HPLC grade methanol (MeOH, Sigma Aldrich) and ethanol (EtOH, Decon Labs) were used as solvents for all solvolysis experiments. Both alcohols are recommended green solvents in the CHEM21 solvent guide due to their favorable safety score (MeOH: 4; EtOH: 4), health score (MeOH: 7; EtOH: 3), and environmental score (MeOH: 5; EtOH: 3).28p-Coumaric acid (99%) and ferulic acid (99%) were obtained from MP Biomedicals Inc. NMR solvents namely, dimethylsulfoxide (DMSO)-d6 (99.9%) and pyridine-d5 (99.9%) were purchased from Cambridge Isotopes Inc. Tetrahydrofuran (Certified), contains about 0.025% butylated hydroxytoluene as a preservative was purchased from Fisher Scientific. ZSM-5 was purchased from Zeolyst International in its ammonium form (CBV 3024E with SiO2/Al2O3 = 30). The purchased NH4-ZSM-5 was calcined in air at 550 °C for 10 hours using a 5 °C min−1 ramp to obtain the acidic H-ZSM-5 form of the zeolite before catalytic testing. The derivatizing agent for GC analysis, N,O-bis(trimethylsilyl)trifluoroacetamide with trimethylchlorosilane (BSTFA + TMCS), was purchased from Sigma Aldrich. Additional chemicals, including a list of the calibration standards used for fast pyrolysis and catalytic fast pyrolysis experiments, are provided in the ESI (Table S3†). Polystyrene standards purchased from Agilent (Agilent Technologies, Inc., Santa Clara, CA) were used to calibrate molecular weights as a function of retention times for the GPC analysis.Flow-through lignin depolymerization
Flow-through lignin depolymerization studies were performed using a fixed-bed flow reactor setup in an up-flow configuration. Each experiment was performed using a new 316 stainless-steel reactor (0.25 inch in outer diameter and 7.33 inch in length) loaded with 150 mg of lignin. The detailed schematics of the packed reactor and setup are presented in Fig. S6 and S7.† The system was purged three times with N2, pressurized to the desired pressure at room temperature, and maintained at the set point using a back-pressure regulator. The solvent was fed into the reactor at a constant flow rate of 0.25 mL min−1 (Series II HPLC Pump, SSI). The benchtop heating controller (Omega) was programmed to the set temperature prior to starting the tests. The reactor took around 4–10 min (heating rate: 33–35 °C min−1) to reach the set temperature. The temperature was maintained using aluminum heat transfer blocks wrapped in heating tape and an insulating blanket. Liquid products were collected using a gas–liquid separator kept at 5 °C. The first sample was collected after 30 min by emptying the separator, and then subsequent samples were taken every 20 min using the same procedure. The volume of each sample collected was measured and checked for consistency with the solvent flow rate. The samples were then filtered using a 0.22 μm Nylon syringe filter and subjected to GC-MS-FID analysis. The yield of the monomers generated was calculated relative to the mass of initial lignin loaded as follows: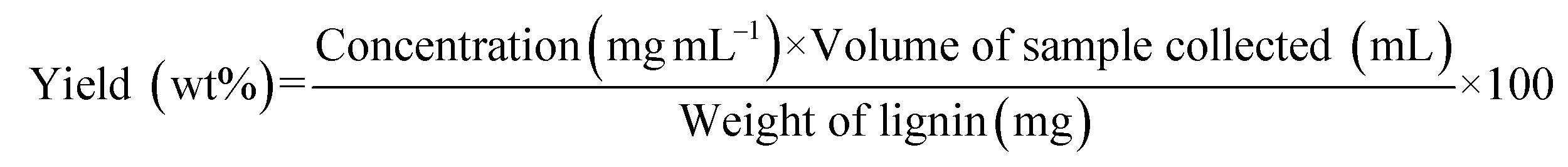 | (1) |
It is important to note that the concentrations measured by GC and the calculated yield correspond to averages over the time interval considered. Independent experiments were carried out also to evaluate the mass of the non-volatile products collected at every time point. Once a sample was collected, the vial was dried under flowing air for a day and placed in a vacuum oven overnight at 40 °C to evaporate the solvent. All reactions were run in triplicates.
Batch reactor lignin depolymerization
Batch reactor experiments were performed by loading 750 mg of lignin and 200 mL of the desired solvent in a 300 mL autoclave reactor made of 316 stainless-steel (Parker-Hannifin, high temperature, bolted closure laboratory batch reactor system with URC II reactor controller). Lignin mass and solvent volumes were adjusted to match the mass/volume ratio used for the flow-through experiments. The reactor was initially purged three times and then filled with 30 bar N2 at room temperature. It took 30 min to 1 h for the reaction mixture to reach the target temperature. The time at which the system reached the set temperature was noted as time zero. Approximately 5 mL samples were collected every 30 min, and the reaction was carried out for a total time of 2 hours. The samples were filtered using a 0.22 μm Nylon syringe filter and were then analyzed by GC-MS-FID. The yield of the monomers generated was calculated relative to the mass of lignin initially loaded as follows: | (2) |
The mass of non-volatile solid products was determined by bringing the reactor back to room temperature and recovering its content. Following this, the reactor was washed with an excess amount of solvent, and the solutions were combined in the same beaker. The contents were then filtered to remove solid residues, and the filtrate was dried. The mass of solvent-soluble products was measured after evaporating the solvent overnight inside a fume hood.
Model compound reactivity study
The experiments with model compounds were conducted in a mini 316 stainless-steel Swagelok reactor consisting of two 0.5 inch plugs and a 0.5-inch port connector (Fig. S8†). The total inner volume of the reactor was 3.6 mL. For each experiment, 5 mg of p-coumaric acid and 5 mg of ferulic acid were added to 2.0 mL of solvent. An industrial fluidized sand bed was heated to the set temperature, and the reactor was dropped into the bed for heating. After the desired reaction time was reached, the reactor was quickly taken out of the bed and immediately quenched in a water bath at room temperature.GC-MS-FID analysis
The filtered reaction products were analyzed using an Agilent 7890B GC-MS-FID, equipped with two identical Phenomenex ZB 1701 capillary columns connected to MS and FID detectors. The following temperature program conditions were used for the analysis: the temperature was held at 40 °C for 3 min, then ramped to 280 °C at 4 °C min−1, and held at 280 °C for 4 min. A five-point calibration was performed for each compound to achieve a regression coefficient higher than 0.99. A representative chromatogram and an example calculation of monomer yield are provided in Fig. S9.†Derivatization followed by GC-MS-FID was also used to unambiguously identify products such as p-coumaric acid and ferulic acid using the procedure described in the following reference.24 To perform this analysis, the solvent was first evaporated and the dried products were redissolved in tetrahydrofuran (THF).
GPC analysis
Gel permeation chromatography (GPC) was conducted using a Dionex/Thermo Scientific Ultimate 3000 Binary Semipreparative LC System (Sunnyvale, CA) equipped with two Agilent PLgel 3 μm 100 Å 300 × 7.5 mm columns, one Mesopore 300 × 7.5 mm column and a diode array detector (DAD). THF was used as the mobile phase at a flow rate of 0.3 mL min−1. Dried reaction products were redissolved in THF for analysis. The wavelengths used for analysis were 254 nm, 263 nm, and 280 nm. The GPC analysis of the raw lignin was carried out with pre-acetylated lignin to increase its solubility in THF.58HSQC NMR
Measurements were performed using 70–80 mg of raw lignin or deconstructed reaction product dissolved in 650 μL of a 4:1 v/v mixture of DMSO-d6 and pyridine-d5. The mixture was sonicated for over 2 hours to obtain a homogenous sample. The spectra were collected with a Bruker Avance III 600 MHz spectrometer using the pulse sequence ‘hsqcetgpsisp2.2’ at 25 °C, with the following parameters: interscan relaxation delay of 0.5 s, 32 scans with the total acquisition time of 3 hours, a spectral width of 220 ppm in f1 and 12 ppm in f2, with centers around 90 ppm and 5 ppm, respectively. The DMSO solvent peak was used as an internal reference (δH/δc: 2.49/39.5). All data processing was carried out using MestReNova v 12.0.1 software. The signals were assigned based on ref. 48,59 and 60.
FTIR
A Thermo Scientific Nicolet iS10 (Thermo Fisher Scientific Inc., Waltham, MA) equipped with a Smart iTR accessory was used to conduct the Fourier transform infrared (FTIR) analysis and determine the functional groups in the samples. Each sample was scanned 32 times from 750 cm−1 to 4000 cm−1 at a resolution of 4 cm−1 and an interval of 1 cm−1. Peaks were assigned using the following references.61,62Fast pyrolysis and catalytic fast pyrolysis tests
Lignin and the samples obtained from solvolytic depolymerization were pyrolyzed in a Frontier tandem micropyrolyzer system (Frontier laboratory, Japan) equipped with an auto-shot sampler (Rx-3050 TR, Frontier Laboratories, Japan) connected to an Agilent 7890B GC-FID-TCD-MS (Fig. S10†). During pyrolysis, a deactivated stainless-steel cup containing approximately 500 μg of the sample was dropped into a preheated furnace where the sample was pyrolyzed within a second. Helium gas was used as both the sweep and carrier gas. The vapors exiting the pyrolyzer were directly carried to a GC for analysis. For catalytic fast pyrolysis (CFP), a fixed bed reactor containing approximately 10 mg of the zeolite catalyst (50 and 70 mesh) was packed in the second reactor. The temperatures of the pyrolysis reactor and the fixed bed reactor were both set at 500 °C.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation under the grant number CBET-1706046. The authors thank Dr Sarah Cady and Dr Shu Xu for assistance with NMR characterization, Dr Patrick Johnston for help with GPC and other analyses.References
- P. McKendry, Bioresour. Technol., 2002, 83, 37–46 CrossRef CAS PubMed.
- M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29–41 CrossRef CAS.
- Y. Zeng, S. Zhao, S. Yang and S.-Y. Ding, Curr. Opin. Biotechnol., 2014, 27, 38–45 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695 CrossRef CAS PubMed.
- T. Renders, S. Van den Bosch, S. F. Koelewijn, W. Schutyser and B. F. Sels, Energy Environ. Sci., 2017, 10, 1551–1557 RSC.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. d. Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- J. E. Holladay, J. F. White, J. J. Bozell and D. Johnson, Top value-added chemicals from biomass-Volume II—Results of screening for potential candidates from biorefinery lignin, Pacific Northwest National Lab.(PNNL), Richland, WA (United States), 2007 Search PubMed.
- Z. Strassberger, S. Tanase and G. Rothenberg, RSC Adv., 2014, 4, 25310–25318 RSC.
- L. Cao, I. K. M. Yu, Y. Liu, X. Ruan, D. C. W. Tsang, A. J. Hunt, Y. S. Ok, H. Song and S. Zhang, Bioresour. Technol., 2018, 269, 465–475 CrossRef CAS PubMed.
- E. M. Anderson, R. Katahira, M. Reed, M. G. Resch, E. M. Karp, G. T. Beckham and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2016, 4, 6940–6950 CrossRef CAS.
- T. N. Linh, H. Fujita and A. Sakoda, Bioresour. Technol., 2017, 232, 192–203 CrossRef CAS PubMed.
- A. U. Buranov and G. Mazza, Food Chem., 2009, 115, 1542–1548 CrossRef CAS.
- P. A. Kroon and G. Williamson, J. Sci. Food Agric., 1999, 79, 355–361 CrossRef CAS.
- B. M. Upton and A. M. Kasko, Biomacromolecules, 2019, 20, 758–766 CrossRef CAS PubMed.
- S. D. Karlen, P. Fasahati, M. Mazaheri, J. Serate, R. A. Smith, S. Sirobhushanam, M. Chen, V. I. Tymokhin, C. L. Cass, S. Liu, D. Padmakshan, D. Xie, Y. Zhang, M. A. McGee, J. D. Russell, J. J. Coon, H. F. Kaeppler, N. de Leon, C. T. Maravelias, T. M. Runge, S. M. Kaeppler, J. C. Sedbrook and J. Ralph, ChemSusChem, 2020, 13, 2012–2024 CrossRef CAS PubMed.
- P. A. Johnston, H. Zhou, A. Aui, M. M. Wright, Z. Wen and R. C. Brown, Biomass Bioenergy, 2020, 138, 105579 CrossRef CAS.
- D. Ertl, Sustainable Corn Stover Harvest, Iowa Corn Promotion Board, 2013, https://www.iowacorn.org/media/cms/IowaCornResearchBrochure_Final_IFT_F4B608A12ED16.pdf Search PubMed.
- C. A. Seifert, M. J. Roberts and D. B. Lobell, Agron. J., 2017, 109, 541–548 CrossRef.
- H. P. Singh, D. R. Batish and R. K. Kohli, Crit. Rev. Plant Sci., 1999, 18, 757–772 CrossRef CAS.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- E. M. Anderson, M. L. Stone, R. Katahira, M. Reed, G. T. Beckham and Y. Román-Leshkov, Joule, 2017, 1, 613–622 CrossRef CAS.
- E. M. Anderson, M. L. Stone, M. J. Hülsey, G. T. Beckham and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2018, 6, 7951–7959 CrossRef CAS.
- I. Kumaniaev, E. Subbotina, J. Sävmarker, M. Larhed, M. V. Galkin and J. S. M. Samec, Green Chem., 2017, 19, 5767–5771 RSC.
- S. M. G. Lama, J. Pampel, T.-P. Fellinger, V. P. Beškoski, L. Slavković-Beškoski, M. Antonietti and V. Molinari, ACS Sustainable Chem. Eng., 2017, 5, 2415–2420 CrossRef CAS.
- T. L.-K. Yong and Y. Matsumura, Ind. Eng. Chem. Res., 2012, 51, 11975–11988 CrossRef CAS.
- T. L.-K. Yong and Y. Matsumura, Ind. Eng. Chem. Res., 2013, 52, 5626–5639 CrossRef CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. R. McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- L. Shuai and J. Luterbacher, ChemSusChem, 2016, 9, 133–155 CrossRef CAS PubMed.
- J. Y. Kim, S. Oh, H. Hwang, T. S. Cho, I. G. Choi and J. W. Choi, Chemosphere, 2013, 93, 1755–1764 CrossRef CAS PubMed.
- A. Riaz, D. Verma, H. Zeb, J. H. Lee, J. C. Kim, S. K. Kwak and J. Kim, Green Chem., 2018, 20, 4957–4974 RSC.
- D. S. Zijlstra, C. A. Analbers, J. Korte, E. Wilbers and P. J. Deuss, Polymers, 2019, 11, 1913 CrossRef CAS PubMed.
- B. Yang and C. E. Wyman, Biotechnol. Bioeng., 2004, 86, 88–95 CrossRef CAS PubMed.
- C. Liu and C. E. Wyman, Ind. Eng. Chem. Res., 2003, 42, 5409–5416 CrossRef CAS.
- X. Huang, T. I. Koranyi, M. D. Boot and E. J. Hensen, ChemSusChem, 2014, 7, 2276–2288 CrossRef CAS PubMed.
- K. H. Kim, R. C. Brown, M. Kieffer and X. Bai, Energy Fuels, 2014, 28, 6429–6437 CrossRef CAS.
- X. Bai, K. H. Kim, R. C. Brown, E. Dalluge, C. Hutchinson, Y. J. Lee and D. Dalluge, Fuel, 2014, 128, 170–179 CrossRef CAS.
- H. Pińkowska, P. Wolak and A. Złocińska, Chem. Eng. J., 2012, 187, 410–414 CrossRef.
- Wahyudiono, M. Sasaki and M. Goto, Chem. Eng. Process., 2008, 47, 1609–1619 CrossRef CAS.
- E. I. Kozliak, A. Kubátová, A. A. Artemyeva, E. Nagel, C. Zhang, R. B. Rajappagowda and A. L. Smirnova, ACS Sustainable Chem. Eng., 2016, 4, 5106–5122 CrossRef CAS.
- Y. Ye, Y. Zhang, J. Fan and J. Chang, Ind. Eng. Chem. Res., 2011, 51, 103–110 CrossRef.
- Y. Ye, Y. Zhang, J. Fan and J. Chang, Bioresour. Technol., 2012, 118, 648–651 CrossRef CAS PubMed.
- K. Y. Nandiwale, A. M. Danby, A. Ramanathan, R. V. Chaudhari and B. Subramaniam, ACS Sustainable Chem. Eng., 2018, 7, 1362–1371 CrossRef.
- Z. Jiang, T. He, J. Li and C. Hu, Green Chem., 2014, 16, 4257–4265 RSC.
- D. T. Balogh, A. A. d. S. Curvelo and R. A. M. C. De Groote, Holzforschung, 1992, 46, 343–348 CrossRef CAS.
- X. Qiao, C. Zhao, Q. Shao and M. Hassan, Energy Fuels, 2018, 32, 6022–6030 CrossRef CAS.
- H. Kim and J. Ralph, Org. Biomol. Chem., 2010, 8, 576–591 RSC.
- H. Zhou, L. Tan, Y. Fu, H. Zhang, N. Liu, M. Qin and Z. Wang, ChemSusChem, 2019, 12, 1213–1221 CrossRef PubMed.
- S. Wang, W. Gao, H. Li, L. P. Xiao, R. C. Sun and G. Song, ChemSusChem, 2018, 11, 2114–2123 CrossRef CAS PubMed.
- S. Zhou, Y. Xue, A. Sharma and X. Bai, ACS Sustainable Chem. Eng., 2016, 4, 6608–6617 CrossRef CAS.
- L. Fan, Y. Zhang, S. Liu, N. Zhou, P. Chen, Y. Cheng, M. Addy, Q. Lu, M. M. Omar, Y. Liu, Y. Wang, L. Dai, E. Anderson, P. Peng, H. Lei and R. Ruan, Bioresour. Technol., 2017, 241, 1118–1126 CrossRef CAS PubMed.
- Y.-C. Qu, Z. Wang, Q. Lu and Y. Zhang, Ind. Eng. Chem. Res., 2013, 52, 12771–12776 CrossRef CAS.
- P. R. Patwardhan, R. C. Brown and B. H. Shanks, ChemSusChem, 2011, 4, 1629–1636 CrossRef CAS PubMed.
- P. Terpinc, T. Polak, N. Segatin, A. Hanzlowsky, N. P. Ulrih and H. Abramovic, Food Chem., 2011, 128, 62–69 CrossRef CAS PubMed.
- A. Saraeian, M. W. Nolte and B. H. Shanks, Renewable Sustainable Energy Rev., 2019, 104, 262–280 CrossRef CAS.
- G. Zhou, P. A. Jensen, D. M. Le, N. O. Knudsen and A. D. Jensen, Green Chem., 2016, 18, 1965–1975 RSC.
- P. Buono, A. Duval, P. Verge, L. Averous and Y. Habibi, ACS Sustainable Chem. Eng., 2016, 4, 5212–5222 CrossRef CAS.
- S. D. Mansfield, H. Kim, F. Lu and J. Ralph, Nat. Protoc., 2012, 7, 1579–1589 CrossRef CAS PubMed.
- J. C. del Rio, J. Rencoret, P. Prinsen, A. T. Martinez, J. Ralph and A. Gutierrez, J. Agric. Food Chem., 2012, 60, 5922–5935 CrossRef CAS PubMed.
- J. Hu, D. Shen, S. Wu, H. Zhang and R. Xiao, Energy Fuels, 2014, 28, 4260–4266 CrossRef CAS.
- R. J. Sammons, D. P. Harper, N. Labbé, J. J. Bozell, T. Elder and T. G. Rials, BioResources, 2013, 8(2), 2752–2767 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Schematic of reactor setups, HSQC NMR quantification, example monomer yield quantification, additional GPC and FTIR results. See DOI: 10.1039/d1gc02420e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |