
Furan platform chemicals beyond fuels and plastics
Roman
Bielski
 *ab and
Grzegorz
Grynkiewicz
*c
*ab and
Grzegorz
Grynkiewicz
*c
aWilkes University, Dept Pharmaceutical Sciences, Wilkes-Barre, PA 18766, USA
bChemventive, LLC, Chadds Ford, PA 19317, USA. E-mail: bielski1@verizon.net
cProfessor Emeritus (Pharmaceutical Research Institute), Rydygiera 8, 01-793, Warsaw, Poland. E-mail: grynicz@gmail.com
First published on 25th August 2021
Abstract
Herein, we offer some perspective on the metamorphosis that has already begun to take place in substantial parts of the chemical industry, i.e., a switch from traditional resources such as crude oil to biomass. This change requires the workhorse of chemical reactants and petroleum refineries to be replaced with biorefineries. The present perspective offers a brief look at the manufacture and uses of furan platform chemicals (FPCs) directly available from biomass (furfural and 5-hydroxy-methylfurfural). Next, we discuss the difficulties encountered when a secondary FPC, 2,5-furandicarboxylic acid, moves from the lab to large-scale manufacture. The main purpose of the present article is to show the spectacular range of compounds that can be economically synthesized from biomass via FPCs. The fate of selected FPCs and their potential are shown herein as an example only, where many more simple or complex chemicals can be obtained. We believe that there are excellent applications of bio-based materials besides the broadly promoted manufacture of fuels and monomers. This perspective looks at the types of reactions applicable to FPCs and a variety of methods for the synthesis of chiral furans.
 Roman Bielski | Roman Bielski was born in Katowice, Poland. He holds a Master's Degree in Chemical Engineering from Warsaw University of Technology (1970) and a Ph.D. in Organic Chemistry from the Institute of Organic Chemistry, Polish Academy of Sciences (1974). He was a Post-Doctoral Fellow at Imperial College, London with Sir Derek Barton (1976–1978). He worked in academia at Warsaw Agricultural University (1978–1987), Lehigh University (1987–8) and Cornell University (1988–1992). He co-founded several small companies (Petramec, Petrotraces, Attochrom/Biotraces, Cheminnolab, and Biosynthon). At present, he is one of two partners in Chemventive, LLC, one of two partners in Biosynthon, Inc. and an Adjunct Professor at Wilkes University. He is a member of the Chemical Consultants Network (Chair 2015–2016). |
 Grzegorz Grynkiewicz | Grzegorz Grynkiewicz was born in Czestochowa, Poland. He holds a Ph.D. in Chemistry {Chemistry Department, University of Warsaw (1968)} and D.Sc. (habilitation) {Institute of Organic Chemistry, Polish Academy of Sciences (IChO PAN, 1979)}. He became a Titular Professor in 1992. He was affiliated with IChO PAN (1962–1982) and the Pharmaceutical Research Institute in Warsaw since 1984. He retired recently. He co-authored over 200 publications, over 3 dozen review articles and more than 60 patents. His awards include the T. Sendzimir Memorial Medal (2000), S. Biniecki Memorial Medal (2006), Officer Cross of Merite de L'Invention at Eureka Exhibition in Brussels (2009), and Kostanecki Medal of the Polish Chemical Society for Achievements in Organic Chemistry (2014). His research interests include organic synthesis, natural products chemistry, medicinal chemistry, and analytical chemistry. |
1. Introduction
The first humans to generate heat used biomass. Burning dry wood and cellulose-based materials was perhaps not very effective, but as far as we know, this was not an issue. Thousands of years later, better sources of heat such as coal, crude oil and natural gas were discovered. Crude oil and natural gas have been utilized as (a) sources of fuels and (b) chemical industrial substrates for about 150 years during the booming chemical industry period. However, the use of these materials for combustion, and even household and construction materials is becoming less acceptable. Thus, currently, a potential alternative for these applications is biomass.1,2During the 20th century, chemical and related industries relied almost exclusively on fossil carbon feedstock. This has created an impressive collection of materials designed as fuels, solvents, commodity chemicals, polymers, etc., manufactured at multimillion ton quantities annually.3,4 The excessive emissions of carbon dioxide and discharge of enormous amounts of plastic and chemical waste into the environment are some consequences of this rather irresponsible exploitation of natural resources. Thus, this practice has proven to be catastrophic for the planetary equilibrium, endangering the biodiversity and future of mankind.
Consequently, it is generally agreed that a change in attitude is necessary. One of the integral parts of the change is accepting the postulates of the circular economy (CE). Utilizing plant-derived or waste-derived biomass fits perfectly into this concept, leading to a current technological revolution. This revolution is the consequence of a transition from thermo-chemical refineries based on the recovery of energy to novel solutions designed for manufacturing of chemicals. It must be emphasized that new solutions must follow the rules of atom economy and green chemistry, also warranting biodegradability and/or recirculation.5,6
Recently, novel methods for the treatment of lignocellulosic biomass (LCB) have been developed, allowing the synthesis of excellent, multipurpose fuels. These fuels contain oxygen atoms in already combusted molecules, offer the optimal composition of exhaust gases, which can be custom designed. It seems that biomass-derived fuels will replace petrochemicals, at least for some transition period. However, any practical heat generating combustion (excluding that of hydrogen) produces CO2. Thus, despite the fact that biomass is renewable, relevant bio-fuels will eventually be terminated, with the future relying on green energy sources.7–9
In the last two decades, LCB has become a formidable but sustainable alternative to natural gas and crude oil as a chemical industry starting material. This applies particularly to carbohydrate feedstocks suitable for further biotechnological or chemical transformations. Considering that the annual growth of biomass (globally amounting to 1011 tons) can supply the needs of the carbon-based chemical industry, the paramount problem of fossil carbon depletion may be addressed.10–12
It is clear that the transfer from refinery to bio-refinery (the term commonly referring to LCB stock, rather than to applied processes) has already begun to happen. However, the question is which bio-refinery will be with us, for example, in the next 30 years. We believe that a very small part of bio-refineries will produce customized fuels. Significantly, a larger part will produce monomers for the manufacture of biodegradable polymers. Furthermore, an even larger part of bio-refineries will react biomass (polysaccharides, monosaccharides, lignocelluloses, etc.) to produce 5-hydroxymethylfurfural (HMF), furfural (FUR), levulinic acid, etc., and perhaps, chiral, enantiopure entities much less complex than pentoses or hexoses. Many new, spectacular applications of biotechnology and chemical innovations based on chemocatalysis and molecular devices are implicated in energy transfer coupling to chemical cycles,13,14 suggesting that both chemo and bio-catalytic approaches can be economically viable in specific cases.
Biomass polysaccharides consist (after the hydrolysis of macromolecules) of only C-5 (pentoses) and C-6 (hexoses) monosaccharides. Consequently, relevant products directly derived from bio-refineries have either 5 (FUR) or 6 (HMF) carbon atoms. Of course, the initial bio-refinery products can be reacted further to produce compounds possessing any number of carbon atoms. Biomass-derived materials are single products or very simple mixtures. This is also true when non-sugar lignocellulosic materials are used as reactants. Isolating single components from bio-refinery products is relatively easy. It should be noted that while only a few chemicals are derived directly from biomass (each can start a new cascade of derivatives), crude oil consists of a plethora of compounds. The fact that most petrochemical products are mixtures is not important for materials to be combusted but essential for the reactants of chemical processes. Another advantage of biomass-derived compounds is that many of the formed compounds belong to furans. They are more functionalized than crude oil-derived hydrocarbons but significantly less functionalized than carbohydrates, and thus they are excellent chemical industry reactants.15–17
Many experts believe that the most effective use of biomass is the synthesis of monomers for the manufacture of biodegradable polymers. 2,5-Furandicarboxylic acid (FDCA) and related diols {2,5-dihydroxymethylfuran (DHMF), 2,5-dihydroxymethyltetrahydrofuran (DHMTHF), furan-derived aliphatic C4–C6 diols and diamines} are exceptionally promising monomers. However, an issue is that the manufacture of some of these compounds is more difficult than expected. Considering that furan-derived polymers including FDCA offer several advantages such as biodegradability over terephthalic acid (and other benzene-containing monomers)-based polymers, one can hope that effective chemical or bio-catalysts will be found. At present, FDCA is not yet a viable replacement for terephthalic acid (TPA).18
It seems that there is a third option (other than fuels and polymers) allowing the economical utilization of biomass-derived compounds. While this option has been proposed in some reviews,19–22 we believe that it has not been sufficiently promoted. Many of the biomass-derived products are wonderful substrates for the synthesis of a multitude of valuable materials. Their annual demand may be as small as a few kilograms to as large as thousands of tons. The value of the relevant markets varies dramatically and can be measured in billions of dollars. Potential products include pharmaceuticals, fragrances, nutraceuticals, alkaloids, amino acids, pheromones, specialty chemicals, etc. However, although the technical feasibility of manufacturing chemicals from LCB has been validated, some recently elaborated processes (both chemo and bio-catalytic) experience serious scaling up and commercialization problems.23,24
The main purpose of this perspective is to convince readers that taking advantage of biorefinery-derived chemicals as synthetic substrates is a highly economic possibility and a good alternative not only to fuels and plastic monomers from bio-refineries but also hydrocarbon refineries. Reviewing all possible products derived from bio-refineries would make this perspective very long. Thus, to make it more manageable, we selected furan platform chemicals (FPCs) as examples. Note that many more valuable chemicals can be obtained. In the following text, we briefly discuss the manufacture of primary furan platform chemicals (FUR and HMF) and the manufacture of, arguably, the most important secondary FPC, FDCA, as an example of difficulties encountered when scaling up the monomer production. Next, we look at procedures that can be applied directly to biomass carbohydrates or derived compounds (such as HMF and FUR), leading to enantiopure, chiral furan derivatives. Finally, we briefly list selected reactions applicable to FPC-based substrates.
2. Primary furan platform chemicals, their manufacture, and synthetic potential
2.1. Primary FPC – furfural (FUR)
Remarkably, furfural (FUR) was discovered as an individual oily entity in 1821 by J.W. Döbereiner during his experiments on ants and the oxidation of sugar substrates to formic acid and described in 1832.25 Döbereiner's observations on “artificial oil of ants”, as it was first called, were further investigated by a number of followers. An account of these historical developments can be found in Dunlop's monograph and some later reviews.26–28 The industrial production of furfural started in Cedar Rapids, Iowa, exactly 100 years later (in 1921), as a consequence of a coincidental excess of oat hulls and availability of redundant pressure cookers in the Quaker Oats Company plant manufacturing rolled oats.29 The batch sulfuric acid-catalyzed process was conducted in a pressure vessel at an elevated temperature. This process, after certain modifications, was introduced in China (Westpro – Huaxia technology)30,31 and is still used in FUR-producing countries (USA, Dominican Republic, China, South Africa, France, and Germany). It is based on a biomass feedstock such as corn cobs, oats, cottonseeds, rice hulls, rye, barley, and wheat straw; sugarcane bagasse or birch wood residues.32–36 An alternative, the Rosenlew continuous process, which does not use catalysts, is currently of minor economic significance but evokes interest in Brazil, where the large sugar manufacturing industry leaves over 75 million tons of bagasse annually.37 The requirement for raw material is simple, namely, it should have a considerably high content of hydrolysable hemicelluloses, such as branched xylans with a relatively low polymerization degree, which makes it more susceptible to hydrolysis. The remaining constituents of common lignocellulosic biomass (cellulose and lignin materials) should not be affected by the applied reaction conditions, even for a long period of time.38–40 Initially, FUR found application as a solvent for various organic materials and resins, for the production of sand binders in the foundry industry, and as a fungicide, insecticide and nematicide. Gradually its significance increased with the development of the petroleum industry, where it became indispensable as a selective solvent for cracking and reforming processes. Presently, ca. 400 thousand tons per annum of FUR is produced globally, mainly in China, with current prices fluctuating between 800 and 1600 € per ton. The current FUR market value is estimated to be $1.8 billion and is growing steadily at 5% annually. Approximately 70% of current production is consumed by the chemical industry for conversion into furfuryl alcohol (FA), other solvents {tetrahydrofuran (THF) and 2-methyltetrahydrofuran (MTHF)}, fuel additives and monomeric components for the manufacture of polymeric materials and chemical intermediates for various synthetic applications (Scheme 1).41–43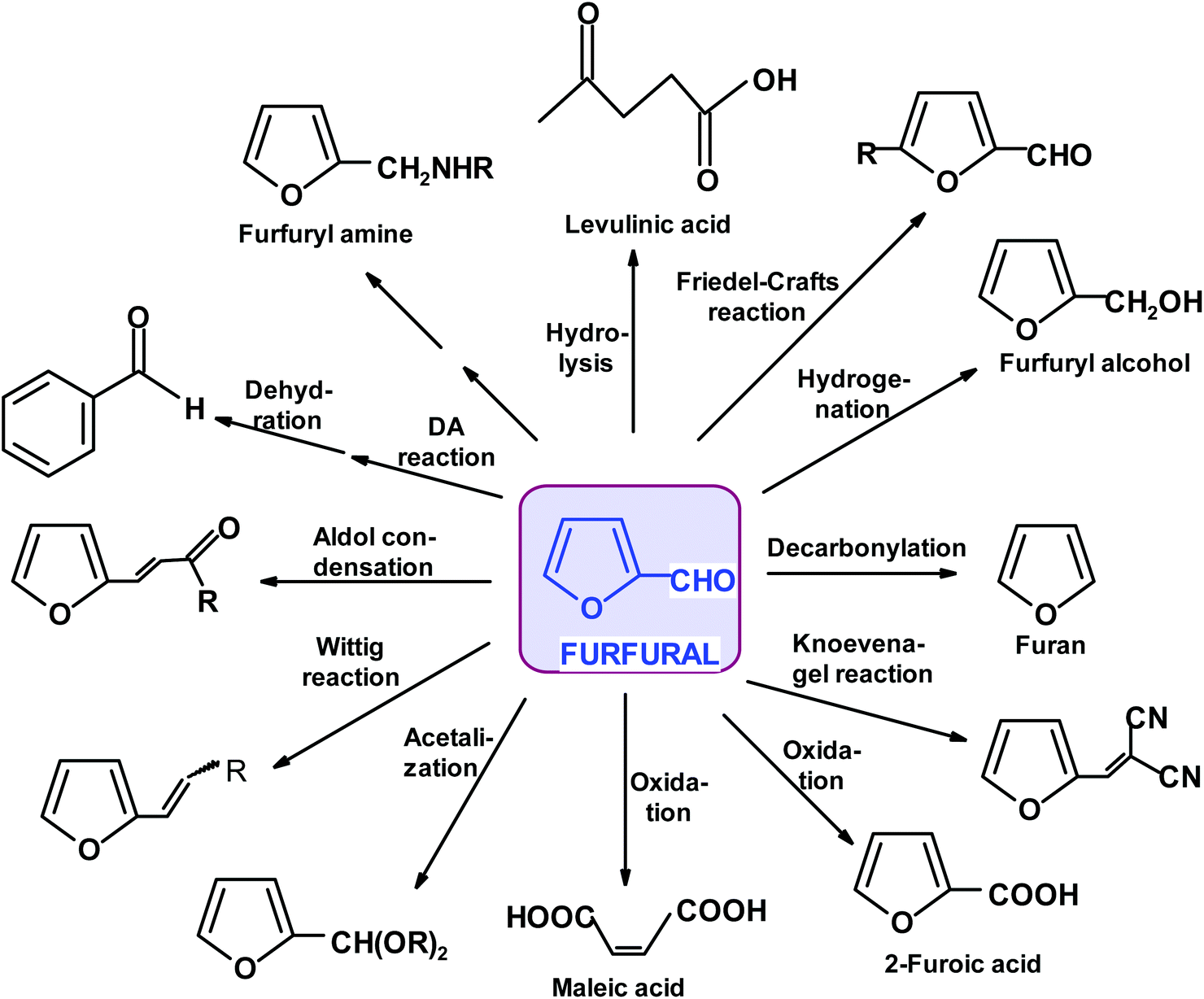 | ||
| Scheme 1 Major products formed directly (or almost directly) from furfural. | ||
Various R&D (research and development) studies reported all types of improvements in the manufacture of FUR resulting from detailed examinations of the chemistry, kinetics, catalytic effects, energy, mass transfer, etc. involved in the reaction.44–46 Presently, the yields achievable in industry are around 50% based on the monomeric pentose content in the hemicellulose raw material. However, it should be considered that ca. 30 t of steam for 1 t of product is needed to strip it from the reactor before it is lost to side reactions. The pentosane hydrolysis to D-xylose step is relatively slow in comparison to the isomerization and dehydration reactions of pentose. It must be emphasized that at any time FUR is present in a reactor, it can be lost to acetal formation via its reaction with hydroxyl group-rich sugar substrates. These condensates are susceptible to further polymerization, leading to insoluble humins. Consequently, the transformation of carbohydrates to furans still remains an open research field ripe for radical innovations, which can take advantage of new catalysts or process design, aiming at the effective separation of the desired reaction products from the unreacted biomass components.47,48 Two distinct lines of process research exist, starting from entirely different feedstocks. The first one, traditionally uses agricultural feedstocks, which require pretreatment to liberate hemicellulosic pentosanes before the application of homogeneous catalysts in the form of mineral or organic acids, metal salts solutions or ionic liquids. A recent study48 recommended simultaneous LCB fractionation and conversion as a one-pot operation using a biphasic solvent system composed of choline chloride and methyl isobutyl ketone at 170 °C (0.6% sulfuric acid; 60 min for 10.7% feedstock weight). The process solubilizes xylans and converts them to FUR (84% yield). FUR is distilled off, while the solvents extract lignans and the resultant separable pulp is highly enriched with cellulose, which can be further biocatalytically converted to glucose. Alternatively, the dehydration process starts from monomeric xylose and is carried out either under homogeneous or heterogenous conditions, with the aid of various Brønsted solid acid catalysts, ionic liquids, supported catalysts, or other constructs such as MOFs (metal–organic frameworks) and nano-composites.49–52 It should be noted that thermochemical and chemo-catalytic processing of FUR and FA can afford a much wider selection of products than the above-mentioned industrial chemicals. These include the exploitation of furan-specific chemistry as a Diels–Alder reaction for the manufacture of benzene (and other aromatic) derivatives53,54 and polymers,55,56 a variety of oligomerization reactions producing, for example, macrocyclic (calixarene type) ethers, linear constructs of interest to opto-electronic/electronic applications26,34,57,58 and ring-opening reactions (RORs). RORs of furan derivatives based on hydrogenation/hydrogenolysis reactions59,60 can provide assorted multifunctional derivatives including alcohols, ketones, acids, anhydrides, and lactones. The other category of relevant processes is skeletal rearrangements (Piancatelli; Achmatowicz; and aza-Achmatowicz),26,35,36,61–63 resulting in modified cyclic compounds such as pentanones, pyranes and pyrimidines.64–68 More examples of simple furanics (FPC) as useful synthons for the preparation of functionalized aliphatic, aromatic and heterocyclic compounds will be presented in the next paragraphs. In principle, each of the mentioned possibilities can be applied for starting a new value-added chain of products. In the case of FUR, we decided to mention its connection to pharmaceutical chemistry and clinical medicine, which are seldomly associated with renewable resources and platform chemicals. Furfural derivatives, particularly 5-nitrosubstituted, have found wide applications in pharmacology and medicine for the treatment of bacterial and parasitic infections. The synthesis of active pharmaceutical ingredients is illustrated in Scheme 2. A furan substrate was obtained via the treatment of furfural (protected as acylal) with nitric acid in a solution of acetic anhydride and acetic acid.69,70
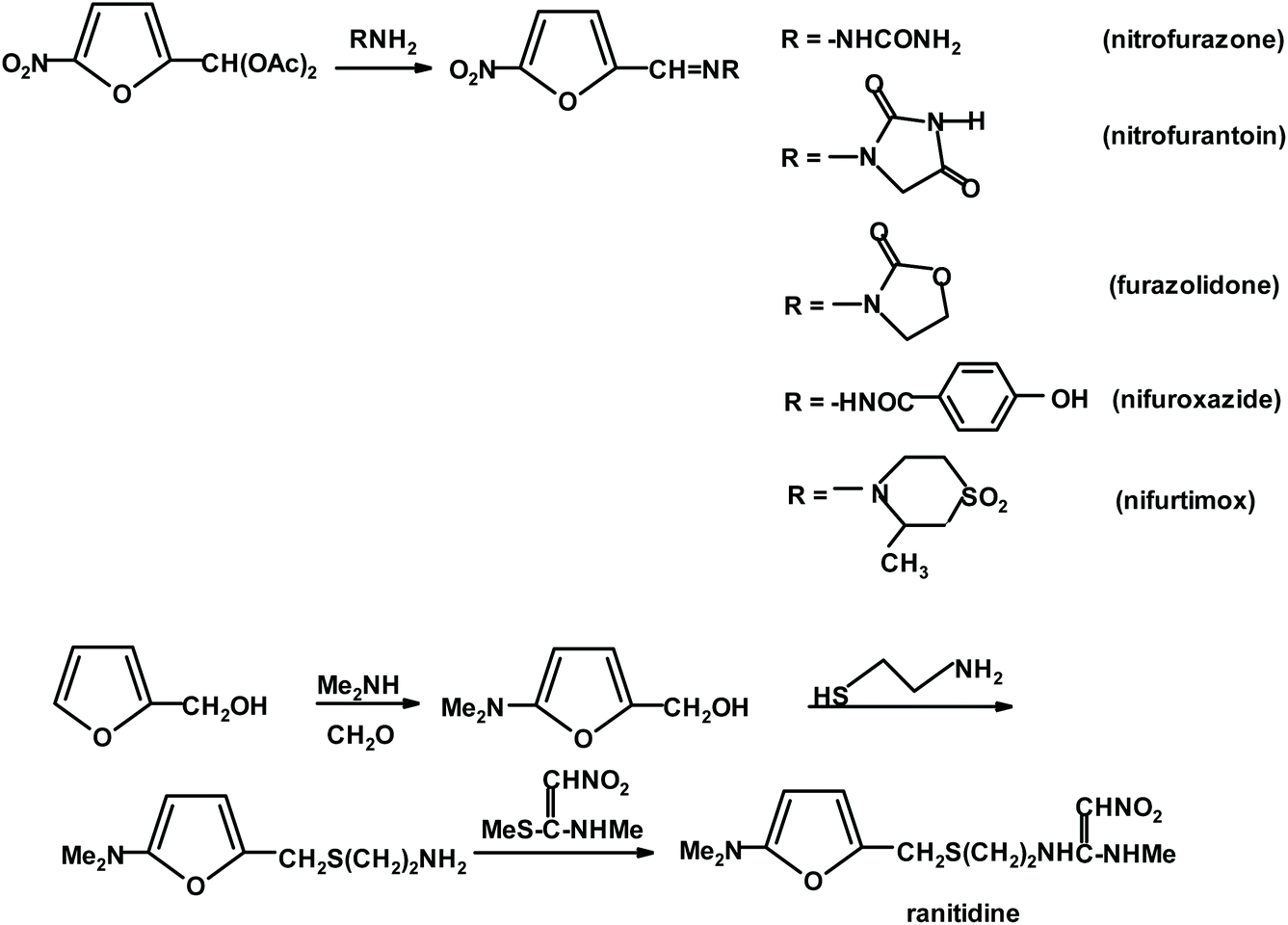 | ||
| Scheme 2 Derivatives of 5-nitrofurfural and furfuryl alcohol applied as clinical therapeutics. | ||
Additional examples of furan ring-containing synthetic medicines include antibiotics such as cefuroxime; antihypertensive such as prazosin, corticosteroid such as fluticasone furoate, and chemo-therapeutic such as lapatinib.71 Furthermore, some furan-2-carbohydrazides have demonstrated promising activity as orally active glucagon receptor antagonists.72 Interestingly, it is also possible to apply simple furan derivatives for entry into the nucleoside area, which is yet to attract vivid interest for the exploration of new antiviral medicines. Thus, furan 2-carboxylic esters are easily converted into C-glycosides when treated with tetra-O-acetyl-D-ribofuranose in the presence of tin tetrachloride. This approach offers an entry to new synthetic C-nucleoside analogs.66 Conversely, furan itself can be converted into nucleoside analogs via the 1,4-oxidative addition of carboxylic acid residues (e.g., by the action of lead tetrabenzoate), which can be replaced in the presence of Pd(0) catalysts by purine or pyrimidine basic components. This methodology also offers an option for catalytic desymmetrization via chiral catalysis to secure entry to both D- and L-analogs of natural nucleic acid constituents.73
As a platform chemical with a long industrial tradition but no petrochemical manufacturing process, FUR is an excellent example of a chemical intermediate that is technically and economically compatible with recently formulated sustainable development goals.74–76 However, although its markets seem fairly stable, the network of FUR follow-up intermediates and products is quite complex. Therefore, new factors such as the implementation and commercialization of furan-derived monomers for biodegradable plastics may significantly increase its demand. Research on LCB (and its hemicellulose components) conversion to FUR remains very vigorous since more selective technologies for the separation of the pentosane fraction from cellulose and lignin have become available, and more region-specific bio-waste materials are discovered.77–80
Remarkably, the accumulation of new knowledge on FUR chemistry and new processes for its manufacturing recently resulted in a new format, which includes a detailed discussion of its life cycle analysis (LCA). This format allows the realistic assessment of its technical applicability, environmental compliance, and commercialization potential.81–86
2.2 Primary FPC – 5-hydroxymethylfurfural (HMF)
Studies on the dehydration of carbohydrates, which commenced in 1840, are very well-documented in the literature. A critical review reporting the historical achievements in this area appeared in 1951 in Advances in Carbohydrate Chemistry.87 HMF was first obtained in 1895 by the action of oxalic acid on inulin. Interestingly, its structure was not elucidated until 1910. The interest in the chemistry and application of HMF are slowly evolving.87–89 Although the basic physicochemical properties and reactivity of HMF were established relatively early, only about one thousand papers have accumulated in the scientific literature towards the end of the 20th century.A radical change in the perception of the potential of HMF and appreciation for its diverse reactivity occurred when a switch from unrenewable chemical industry feedstock became unavoidable. At about the same time, it was realized that biomass-derived HMF is almost totally green and can replace petrochemical substrates for commodity and specialty chemicals.90–106 Thus, an examination of the research on HMF and its development is particularly inspiring for many reasons. Specifically, this compound is easily available from renewable feedstock, and it has a low molecular weight and is bi-functional and multi-reactive. Thus, it appears to be an ideal substrate and intermediate for industrial synthesis, particularly for the formation of polymeric materials. It can be easily converted to di-alcohol or dicarboxylic acid. Consequently, it is a good substrate for synthesizing monomers for the manufacture of polymers (and copolymers) such as polyesters and polyamides.12,107,108 Additionally, its similar structure to benzaldehyde and benzylic alcohol opens a plethora of additional functionalization abilities, many of which have already been experimentally confirmed.53,109,110
Conversely, HMF is relatively unstable. It easily undergoes re-hydration, a reaction that leads to the acyclic products levulinic acid and formic acid, which are both of interest as industrial chemicals.13,92,111,112 It is evident that the more easily isolable and more stable functional analogs of HMF, such as 5-halo-, 5-acyloxy and 5-alkoxy-methyl compounds, can be very useful when coupled to a suitable end-use application.113–115 The two main approaches for the transformation of HMF into more stable and valuable chemicals are oxygenation and hydrogenation/hydrogenolysis.116–118 There are many other processes for the formation of more stable products, which will be discussed later (Scheme 3). However, despite the fact that there has been over a century of research (recently very vigorous), HMF has not become a commodity chemical with dozens of applications, as earlier expected.119–121
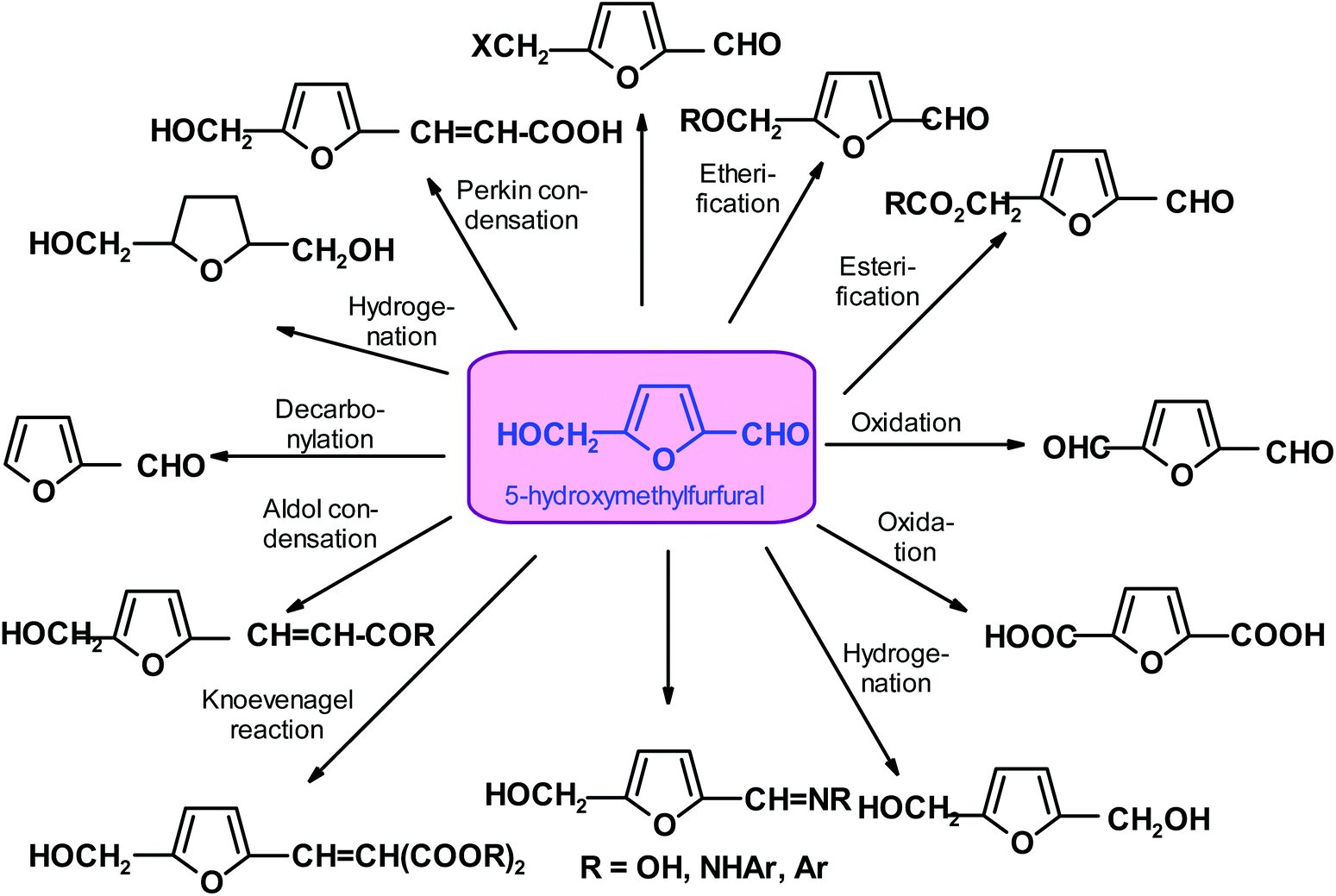 | ||
| Scheme 3 Selected products directly (or almost directly) available from HMF. | ||
In comparison to the R & D on furfural, the HMF story can be presented from different angles, exposing particular problems. The handling of HMF during its formation inevitably involves rather recalcitrant cellulosic feedstock under harsh chemical conditions. Thus, is clear that a successful process design is challenging. Routinely, the issues related to the feedstock, reaction environment, energy and mass transfer, catalysis, process design, isolation method and purification have been researched and discussed separately. Recently, the hydrothermal processing of LCB sugar components in an aqueous environment has been investigated theoretically and experimentally.122–124 An unavoidable conclusion is that after depolymerization, monosaccharide retro-aldol condensation reactions leading to glyceraldehyde, hydroxyacetone and pyruvaldehyde tend to dominate.125–127 Hence, hexose monosaccharides require catalysts for their efficient dehydrative conversion to HMF. This conclusion led to the development of many families of catalysts, starting from simple Brønsted or Lewis acids, but eventually comprising ion-exchange resins, zeolites, heteropolyacids, inorganic salts, ionic liquids, acid-functionalized mesoporous metals, carbon- and metal oxide-supported metals, metal oxides, etc.89,128–131
The instability of HMF under the conditions of its formation led to the search for solvents suitable for its extractive removal from the reaction media. A recent systematic overview of the reaction phases (RP) versus extraction phases (EP) applied to HMF formation processes allowed the formulation of guidelines for the rational selection of solvents. These guidelines satisfy technical requirements, together with environmental health and safety demands formulated by the ACS Green Chemistry – Pharmaceutical Roundtable.132,133 The screening of a pool of 177 solvents has recommended 15 candidates specially suitable for ternary equilibria (water–HMF–solvent), which combined with a large array of possible catalysts, illustrate the amount of data involved in the LCB conversion process design.134 The matrix of parameters is even further extended when HMF derivatives such as esters, ethers, acetals are considered, which requires the help of advanced chemoinformatics tools.135 A model hexose dehydration process136 involves the treatment of D-fructose with an aqueous tetraethylammonium bromide solution in the presence of Amberlyst-15 at 100 °C for 15 min. The product is isolated after ammonium salt precipitation with ethanol followed by filtration. Alternatively, the product is extracted with ethyl acetate, the solvent is evaporated and flash chromatography on silica gel gives the pure final product. Consequently, HMF is obtained in 79% yield at 97% purity. However, the product is unstable under ambient storage conditions, as evidenced by the prompt color development.
Together with this validated lab-scale preparation procedure, there are parallel efforts reporting the R&D on a prospective industrial process137 operated at a relatively very low temperature (70 °C). The conversion of fructose to HMF is based on a study of a non-aqueous two-phase system and 1,3-dimethylimidazolium chloride catalysis, which may be problematic from an economic viewpoint. The literature discussing the choice of catalyst for homogeneous or heterogeneous reaction conditions and product separation is extensive.102,105,106 It indicates the formation of competing side products and purification problems. Therefore, in situ derivatization methods are of particular interest, which convert HMF into more stable compounds. They have to retain the chemical versatility of HMF and must make its isolation, purification, storage and transport easier. Examples of these innovations138 have already been discussed.
A recent example of the employment of acetone in the hexose dehydrogenation process has highlighted the easy isolation of the double acetone condensation product (HMF–Acetone–HMF = HAH), which can be efficiently carried out by filtering the solid products that are insoluble in water.106
Perhaps surprisingly, it seems unlikely that chemo-catalytic HMF manufacturing technologies139 will be promptly replaced by new solutions emerging from organocatalysis and biotechnology. The combination term “biorefinery” was coined up to accommodate the concept of the sustainable processing of biomass into marketable products, with focus on renewable resources as the feedstock.140 At present, biomass conversion is quite demanding in terms of energy (relatively high temperatures and pressures), the need for complex catalysts and special equipment, and the environmental impact (e.g., generated chemical waste).45,141 Furthermore, although it is generally accepted that biotechnology, biocatalysis in particular, is a sustainable alternative to traditional chemical industries, making biorefineries greener by switch from chemical catalysts to industrial enzymes still faces many technical hurdles, which are typical in the implementation of bioengineering.142
The massive accumulation of literature on HMF research in recent years should translate to its vigorous commercialization. Unfortunately, the reality looks quite different. An excellent review143 on the technical development and scale-up of HMF processes in industry based on various sources including patents and company press releases concluded that presently there is only one commercial enterprise in operation (AVA Biochem, Muttenz, CH), which has a manufacturing capacity in the range of 500–1000 t a−1.144 The patented process was first developed by Food Chemical and Research Laboratories, Inc. as early as 1956. It used saccharose, which was autoclaved in the presence of protic acids in a butanol–water mixture at 150 °C, to afford HMF in 68% yield.
Developments that followed expanded the feedstock variety, catalyst range and reaction conditions, including the use of biphasic solvents. More recent (patented in 2013–2014) process improvements, which were conducted at BASF SE, focused on the operation modes (semi-batch, continuous, and pipe reactor) and the use of ionic liquid (1-ethyl-3-methylimidazolium methylsulfonate) as the solvent without additional catalysts.143 Meanwhile, the demand for down-the-line HMF conversion products such as FDCA (multipurpose monomer) and/or 2,5-dimethylfuran (DMF; fuel) seems to be increasing sharply. It has been predicted that the 2025 market value for FDCA, which is a functional analog of terephthalic acid, will reach $ 850 million.145
Well-documented chemical transformations of HMF are plentiful and extend far beyond the customarily mentioned hydrogenation and oxidation reactions. They open access to several categories of industrial materials such as fuels, solvents, monomers and plastic components, functional polymers for packaging, photovoltaics and optoelectronics.58,146,147 It should be stressed that FPCs (“furanics”) can be chemocatalytically (and in many instances also biocatalytically) converted to aliphatic and functionalized aliphatic compounds, benzene derivatives (via Diels–Alder cycloadditions), and a variety of carbocyclic and O- and N-heterocyclic compounds.24,62,148–150 Reactions producing new carbon–carbon bonds have been extensively applied to HMF and its derivatives, generating new structures. The resulting synthons enable the formation of various natural products containing HMF-related structures (rhemanones; pichiafuran C; sessiline, etc.),23,151,152 where some of these compounds exhibit interesting biological activities.
Although it has nothing to do with the manufacture of HMF, it is interesting to note that HMF is a common constituent of processed food. It results from thermal treatment or natural ageing, which involves non-enzymatic sugar browning and sugar-amino acid-initiated Maillard reactions.153 The high chemical reactivity of HMF is a subject of some toxicological concerns due to its supposed role in reactions parallel to non-enzymatic hemoglobin glycation, condensation with the endogenous antioxidant glutathione, etc.154 However, at the average estimated daily intake of below 100 mg, this compound is considered safe.155 HMF exhibits a wide range of interesting biological activities observed in vitro.156 For example, it has been selected by FDA as a drug candidate to undergo clinical trials for the treatment of the sickle cell anemia.157,158 HMF 5-citrate ester, a natural product known under the name Mumefural, improves human blood fluidity and also exhibits inhibitory effects towards the H1N1-type influenza virus.159 It seems likely that more compounds with a 2,5-disubstituted furan structure exist in a continuously expanding natural product space, which warrants interest in their metabolomics as a possible inspiration for future biotechnology and medicine. In the last two decades, there has been an avalanche of publications (approaching 900 per annum towards the end of this period) on all aspects of HMF chemistry, ranging from its preparation to various ways for its valorization and application. The most extensive critical review on this subject appeared in Green Chemistry recently.160
3. An example of an extraordinarily important secondary furan platform chemical: furan 2,5-dicarboxylic acid (FDCA)
As has been already pointed out, primary furan platform chemicals (FPC), FUR and HMF, have no direct application as widely marketable products. HMF, equipped with two reactive functional groups, is the most promising feedstock for both oxidative and reductive transformations. Both processes afford more stable chemicals, and thus are capable of entering many growing value-product chains. Thus, the exhaustive reduction of both 2,5-functional ring substituents can result in the formation of DMF. This compound has excellent characteristic as fuel for internal combustion engines and can replace gasoline. A change in the reaction conditions results in the formation of dihydroxymethyl furan (DHMF), which can replace diols (or relevant diamines) of petrochemical origin in many polymeric products.161,162HMF oxidation can be a stepwise and multidirectional process, engaging both substituents and the heterocyclic ring itself, affording C4 or C6 dicarboxylic acids or their derivatives.163,164 In this paragraph, we focus our attention on FDCA as an exemplary FPC chemical. It has highly desirable properties including stability, crystalline solid, safe storage and transportation, and nontoxicity, with a firmly fixed destination as a replacement for monomers currently derived from petrochemicals.
Much of the contemporary polymer industry relies on aromatic dicarboxylic acids such as terephthalic acid (TPA), which have an estimated annual production of 100 million tons. The market of the major product polyethylene terephthalate (PET) is valued at $60 billion. However, the future of PET is rather bleak due to its non-renewable origin, alleged estrogenic effects, and poor biodegradability. Consequently, many new polymeric materials, i.e., functional analogs of commercial plastic materials, based on petrochemical monomers (e.g., PET) have been obtained by switching from TPA to FDCA. The properties of these new materials have indeed proven to be superior in many respects to the petrochemical macromolecular composites of the previous century.
The chemistry of the conversion of HMF to FDCA seems rather simple as a laboratory exercise via stepwise oxidation (Scheme 4). However, the work on prospective chemocatalytic processes that meet Green Chemistry criteria and are economically viable has taken decades to develop in the form of an experimental pilot plant manufacture.165–168
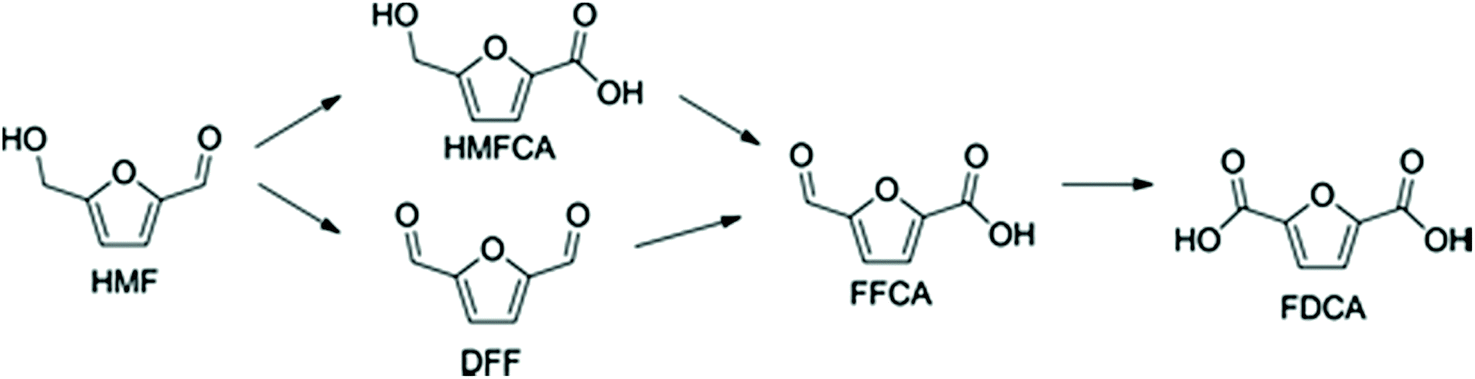 | ||
| Scheme 4 Main intermediates in the stepwise oxidation of HMF to FDCA. | ||
FDCA was first reported in 1876 as a product of mucic (galactaric) acid dehydration reaction by fuming hydrobromic acid under pressure.169 There are three main synthetic approaches to FDCA (and its analogs), i.e., oxidation of 2,5-disubstituted furans, such as HMF, dehydration of aldaric acids, and condensation of diglycolic acid derivatives with α-dicarbonyl compounds such as glyoxal. The former are of interest from the standpoint of LCB conversion and valorization.170–172 It should be noted that FPCs are inter-related along the C5–C6 axis through C1 transfer reactions, such as the reaction of FUR with formaldehyde and carboxylation/decarboxylation or carbonylation/decarbonylation processes. Thus, it is clear that commercially available furfural can also (together with HMF) be considered a viable substrate for FDCA.
The facile oxidation of FUR using nitric acid, followed by esterification with methanol affords methyl furoate, which readily reacts with formaldehyde in the presence HCl and zinc chloride to give a good yield of 5-chloromethylated derivative. The latter is conveniently transformed into FDCA by the action of nitric acid. FUR (and also FA) can be easily carboxylated, affording FFCA, the common intermediate on the pathway of HMF chemical oxygenation to the desired furanyl monomer.173–175 Since the beginning of the new century, the presumed availability of HMF as a principal FPC intermediate of the green polymer industry has channelled unprecedented research toward its transformation into FDCA, which is expected to fill the TPA market niche.
Standard stoichiometric oxidants, such as dichromate or permanganate, perform well on HMF in alkaline solution at ambient temperature, affording the desired FDCA salts within minutes. Unfortunately, employing this textbook chemistry leaves toxic metal waste, which is unacceptable in contemporary industrial enterprises. The industrial oxidation of hydrocarbons has been repeatedly developed as an aerobic catalytic process. Due to the structural similarity of FDCA to TPA, this process seems particularly relevant to the FPC feedstock. As already mentioned, TPA is the principal constituent of the major polyester (and polyamide) materials used for packaging in many industries, but mainly for food products.176
The research on the oxidation of p-xylene (PX) to TPA initially provided a hazardous commercial process based on the high temperature liquid phase-diluted nitric acid oxidation. Later, a much better system utilizing a cobalt/manganese/bromide catalyst was introduced. It operates in acetic acid medium at about 200 °C with air (15–30 bar) as the oxygen source and is known as the Amoco MC process. Under the harsh conditions, some of the substrate starts burning to CO2. Nevertheless, the reaction mixture still contains some unreacted intermediate, i.e., 4-carboxybenzaldehyde (or 4-formylbenzoic acid), which is highly undesirable in subsequent poly-condensations as a polymer chain growth breaker.177–179 The initial applications of the Amoco process to HMF have confirmed that rapid and selective substrate oxidation are achievable and the product can be separated by filtration, allowing the efficient recovery of the dissolved catalyst composition.
The results from the oxidation of xylene offered the much-desired proof-of-principle and strongly suggested that HMF oxidation is also achievable. However, it was soon realized that the situation of the reactive bifunctional HMF under the Amoco process conditions is considerably different from that of p-xylene containing no oxygen atoms in the molecule. HMF has an easily oxidizable primary hydroxyl group (its conversion to the primary oxidation product leads to the formation of diformylfuran (DFF)) and an aldehyde group of only slightly lower susceptibility towards oxidants (its oxidation leads to 5-hydroxymethyl-2-furancarboxylic acid (HMFCA)). Each of these intermediates can undergo following oxidation step, which is firmly determined by their functionality. Also, both afford the same common intermediate, 5-formyl-2-furancarboxylic acid (FFCA), which is one step away from the final product, FDCA. The substrate can undergo three types of side reactions, namely, overoxidation to carbon oxides; furan ring opening to maleic or fumaric acids, and finally condensations to polymeric materials, including insoluble humins. Thus, many more variables have to be considered for efficient optimization of the reaction conditions. The initially reported FDCA yield of 60.9%180 was further improved to ca. 90% with the total conversion of HMF requiring less than 20 min.181
Parallel research (hundreds of studies have been reported in the last couple of decades) on the heterogeneous catalytic aerobic oxidation of HMF is developing vigorously. It offers possible operational advantages such as the use of fixed bed flow reactors or porous and nanoscale catalysts. The prevailing trend is to turn from the traditionally applied metal oxides to noble metals (mono- and bimetallic systems on a variety of mineral supports) or composites containing them, such as Pt–C/AgO/CuO.182,183 It is generally accepted that both the physical form of the metallic catalyst and its support considerably influence its efficiency. This is particularly obvious for gold, which was added to the noble triade Pt, Pd, and Ru recently, and has proven to be most effective when applied as nanoparticles. The research on supports for newly designed nano-formulated catalysts is a quickly expanding field of R&D. Electrochemical oxidation of HMF is a very interesting option, particularly when coupled with a solar energy conversion unit or water splitting cell, which generates residual hydrogen, but there have been relatively few reports on this research thus far.184,185
At present, the main catalysts used in LCB transformations are chemical catalysts. Nevertheless, biocatalysis plays a continuously growing role. In the previous century, saccharification (breaking down cellulose into fermentable sugar subunits) has been considered the main initial target of LCB biotechnology. It turned out that chemical degradation methods may be rendered acceptable, at least as a temporary solution.186 From the perspective of circular bioeconomy dominance, access to effective biocatalytic transformations of cellulose as the main constituent of LCB may prove indispensable for generating added value from various platform chemicals.165,187–190 For the further conversion of de-polymerized saccharides and FPCs, a long tradition of industrial biocatalytic oxidation already spans from the use of whole cell microorganisms, through single enzyme preparations, to genetically engineered organisms.191 Moreover, even more powerful tools such as chemically evolved proteins with enzymatic function have been added recently.192 Naturally, all these developments strongly influence R&D aimed at highly valued products such as FDCA.
Single-enzyme catalysis research has somewhat limited perspective considering substrate specificity, which is poorly compatible with HMF bifunctionality (most enzymes oxidize either the alcohol or aldehyde functional group). Surprisingly, an oxidoreductase protein that can to convert HMF to FDCA (HMF/furfural oxidoreductase; HmfH) has been identified,193 but attempts of its expression in E. coli have failed.194 The next candidate for HMF oxidase was isolated from the Methylovorus sp. MP688 strain, and it was found that this alcohol oxidase can also oxidize the aldehyde group, albeit only in its hydrated gem-diol form. Crystal structure studies on HMFO (HMF oxidase) helped to guide the protein engineering investigation, which led to a mutant enzyme with over a 1000-fold increase in activity compared to wild-type oxidase.195,196
Expectedly, whole-cell catalysis has attracted considerably more attention, despite the alleged toxicity of HMF. Firstly, P. putida S 12 expressing HmfH was chosen due to its resistance to chemical stressors, which demonstrated successful batch fermentation of HMF on a 30 g L−1 scale, and later more strains capable of growth on HMF as a carbon source were identified.197,198 It has been suggested that enzyme cascade reactions (oxidases in tandem) may offer a viable solution for the more efficient conversion of HMF to FDCA.199 The cascade biocatalytic oxidation of HMF was realized by co-expression in E. coli vanillin dehydrogenase (VDH1) and HmfH, affording FDCA on a gram scale with 0.6 g L−1 productivity.200 There is also some interest in exploiting a thermophilic FDCA decarboxylase (PtHmfF) for the biocatalytic carboxylation of FA, but only low yields have been obtained thus far.201 In general, the chemocatalytic methods for the preparation of FDCA seem more ready for process development and commercialization than that based on biotechnology.165,202
4. Synthesis of chiral furan synthons from biomass (or its sugar components)
For the effective use of biomass-derived furans to synthesis high-value organic compounds, it is essential that chiral furan compounds are available in the enantiopure form. Here, we present enantiopure compounds that can be directly or indirectly manufactured from biomass. For brevity, we will not address all chiral compounds available from biomass but limit our discussion to chiral compounds comprising a furan moiety. Also, to the best of our knowledge, there is no review on the synthesis of chiral furans from biomass-related materials, which must be mentioned given that it justifies quoting relevant but relatively old publications.Before discussing the available methods, here we digress. Abundant monosaccharides such as glucose, fructose, and xylose contain a few chiral centers but are often too complex as starting materials for various synthons. Usually, there are too many OH groups and some must be removed. Sugar chemists have learnt a variety of tricks to selectively remove OH groups but carbohydrates including biomass sugar components as reactants still pose a challenge. Conversely, HMF and FUR, which are direct and non-expensive products from biomass, are reactive species but contain no chiral centers. Thus, we must either produce chiral compounds directly from biomass or transform non-chiral biomass-based derivatives into chiral compounds. Incidentally, one can argue that the addition of one carbon atom to C-6 in the glucose units of cellulose followed by the standard treatment of cellulose may produce chiral compounds.
Renewable biomass (LCB) consists mainly of well-known carbohydrate polymers. From the standpoint of its technical applications, LCB is “overoxidized”. Therefore, to enter a growing value chain of conversions to industrial chemicals, it needs to be de-polymerized and, at least partly, de-functionalized. In the main body of this review, we discuss mainly chemical means for the conversion of LCB to FPCs, which are products of monosaccharide dehydration. Nevertheless, we are perfectly aware of the intrinsic value of multiple chirality centers being lost in the process. Subsequently, we review the synthesis of chiral furans from biomass or FPCs given that we want to mention another possibility.
It is likely that apart from the mainstream biorefinery transformations, new chemistry will be developed, in which carbohydrate materials will be selectively de-functionalized and will have preselected chirality units preserved. This idea is best illustrated by the conversion of D-glucose to its bicyclic 1,6-dehydration product, levoglucosan, and further to levoglucosenone and Cyrene. They retain only half of their initial oxygen atoms and two chirality centers (Scheme 5).
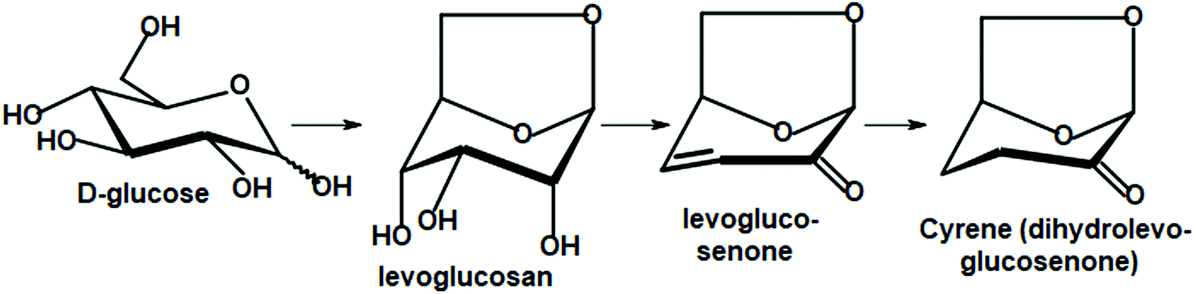 | ||
| Scheme 5 Derivatives containing two chiral centers available from glucose or cellulose. | ||
Although levoglucosenone is traditionally prepared in moderate yield via the laboratory-scale pyrolysis of certain cellulosic materials, new research on the chemocatalytic deoxydehydration of sugars seem to offer more efficient methods of controlled oxygen removal.203 Current advances in carbon–oxygen bond hydrogenolysis and hydrodeoxygenation take advantage of supported metal catalysts modified with various metal oxides. The utilized metals include Re, Mo, and W, and silica, alumina, carbon, titania, zirconia are used as supports. Rhenium, with its unusual valency range, is a particularly effective component of relevant catalytic systems.204–210
For the purpose of this perspective, we divide the methods for the synthesis of chiral furans from biomass into three categories. However, as is often the case with this type of artificial classification, some synthetic methods are difficult to qualify.
(A) Synthesis directly from biomass (or unprotected mono- or disaccharides),
(B) Synthesis via (modified) mono- or disaccharides, and
(C) Direct synthesis of non-chiral furans followed by enantioselective reactions.
In the following text, we briefly review the existing methods for the synthesis of chiral furan derivatives. By no means this review is exhaustive. Instead, we wish to show a plethora of methods allowing the direct or indirect transformation of biomass into more or less enantiopure chiral compounds containing a furan unit. Many of these compounds can serve as substrates for a variety of syntheses including total syntheses. To keep this article relatively short, where there are many articles describing the same process, we often quote only selected papers.
A1. The Garcia Gonzalez (GG) and related reactions
Discovered about 80 years ago, the Garcia Gonzalez reaction involves the reaction of unprotected monosaccharides with 1,3-dicarbonyl compounds in the presence of a Lewis acid. The conditions are usually mild, and the yields are often good. The products of this reaction are polyhydroxyalkylated furans or/and C-glycosylfurans (Fig. 1).
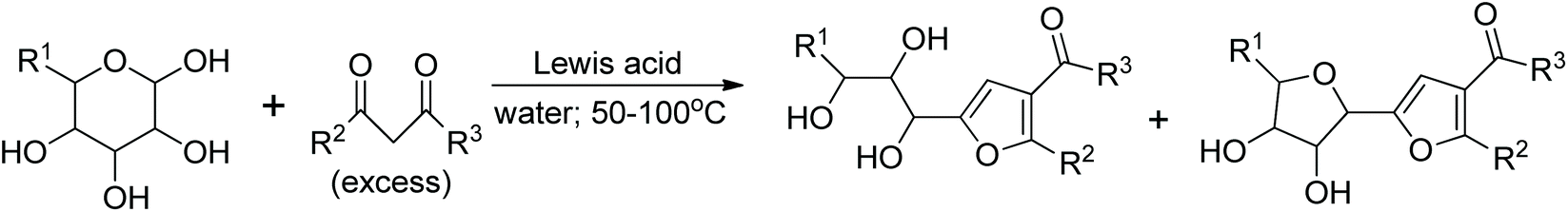 | ||
| Fig. 1 Garcia Gonzalez reaction products. | ||
During the last two decades, several new catalysts have been developed, resulting in substantially better yields, often lower reaction temperatures, highly improved reproducibility and regio- and stereoselectivity of reactions. The list of catalysts used to accomplish the GG reaction includes:
• Cerium chloride heptahydrate (CeCl3·7H2O) in water.211 More than a decade later, Misra et al. used the same catalyst to synthesize GG products, which were transformed into various anti-cancer drugs.212 Interestingly, three of these products exhibited a significant cytotoxic effect.
• Zinc chloride (ZnCl2).213 This catalyst is not new (it was used in the first GG reactions). It is worth noting that the D-arabinose-derived product was transformed into analogues of D- and L-serine.
• Scandium triflate {Sc(OTf)3}.214
• Indium chloride trihydrate (InCl3·3H2O).215
• Silica-supported cerium chloride heptahydrate (CeCl3·7H2O) and sodium iodide (NaI) as a Lewis acid promoter.216
• Iron(III) chloride (FeCl3) in ethanol/water.217
• Plant-waste derived, recyclable polymetallic chlorides.218
• Iron(III) triflate {Fe(OTf)3}.219 It is worth adding that satisfactory yields of GG products were obtained when potatoes (starch) were treated with HCl and directly reacted with 2,4-dipentanone.
• Zirconium chloride (ZrCl4) (Fig. 2).220
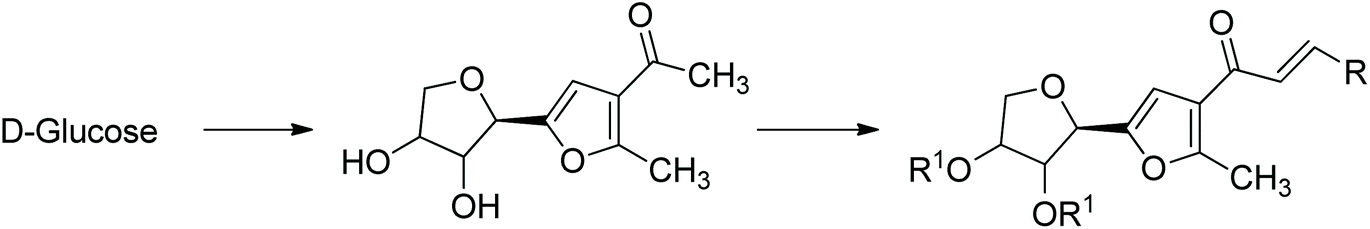 | ||
| Fig. 2 Jones, France et al. synthesis of chiral furans from glucose using ZrCl4.220 | ||
A similar method of synthesizing chiral furans starting from unprotected monosaccharides was developed recently by Lambu and Judeh.221 Instead of using 1,3-dicarbonyl compounds, they utilized malononitrile and the reaction was performed in the presence of an aqueous base such as triethylamine. The products are 2-amino-3-cyano-furans equipped with a chiral polyhydroxyalkyl group in the 5-position and the yields are very good.
Another very close relative to the GG reaction was developed by Zhuang et al.222 The activated methylene reactant is also malononitrile and an enzyme serves as the catalyst (Fig. 3).
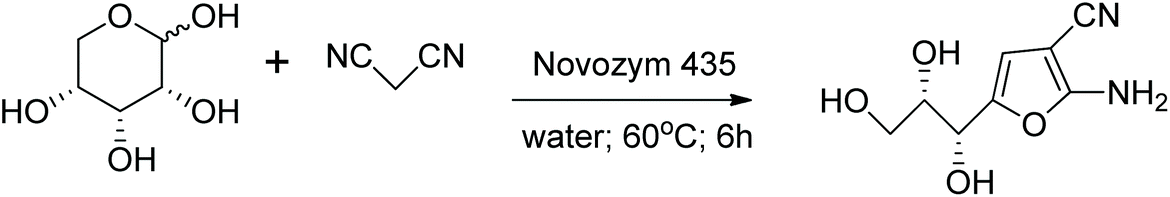 | ||
| Fig. 3 Chiral furans synthesized by Zhuang et al.222 | ||
The GG reaction and its relatives require the use of active methylene compounds. Note that all or most of these compounds are available from biomass. However, their synthesis is not part of this paper.
Boto and Hernández described a mild and effective method for the production of chiral furans (furyl carbinols) from monosaccharides.223 The method is different from the GG reaction, but the products are often similar. It requires the transformation of carbohydrates into polyhydroxylated ketones. Next, they are dehydrated and reacted under basic conditions to form furans. The scheme below shows the obtained yields and typical products (Fig. 4).
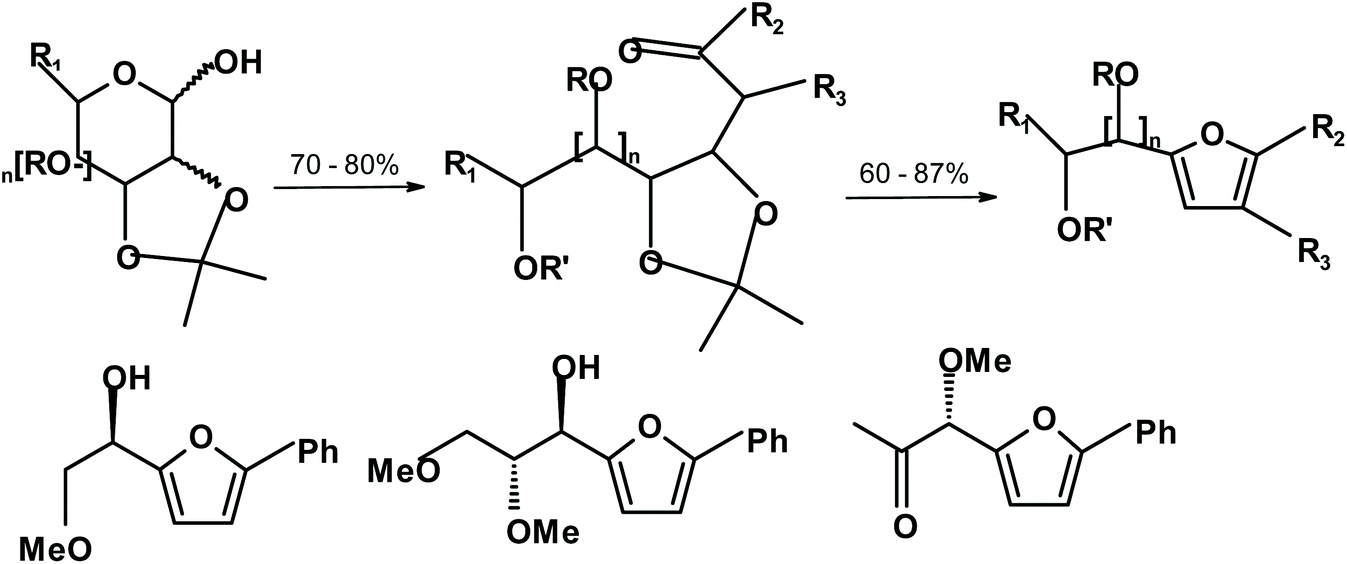 | ||
| Fig. 4 Chiral furans synthesized by Boto and Hernández.223 | ||
A2. Synthesis from disaccharides
Lichtenthaler's group published a series of very interesting papers describing the formation of chiral furans (such as glucosylmethylfurfural {GMF}) from unprotected disaccharides such as isomaltulose (glucosyl-α(1 → 6)-fructose).224 Isomaltulose is produced on a large scale from sucrose via Protaminobacter rubrum-induced O2 → O6-glucosyl transfer, and hence is clearly a biomass component. The free aldehyde group of the furyl unit of GMF has been transformed into a plethora of products.225,226 Also, glucose in GMFs may be replaced with other monosaccharides (Fig. 5).
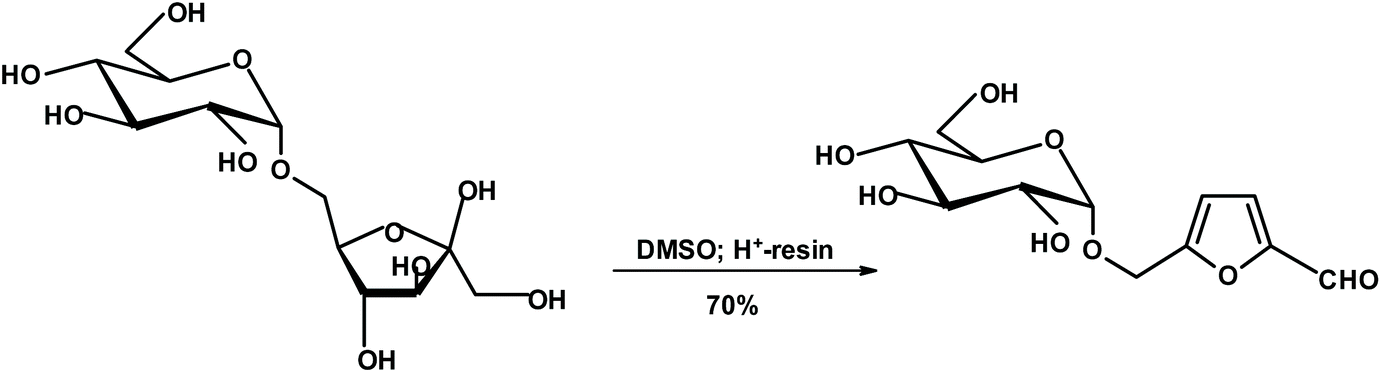 | ||
| Fig. 5 Lichtenthaler's monosaccharide-furan constructs.224 | ||
It should be emphasized that the Garcia Gonzalez products are very different from Lichtenthaler's GMFs. In the GG products, the chiral unit is directly connected to the furan ring. Thus, one can expect very high diastereoselectivities in reactions of the carbonyl group forming chiral center(s). The dr ratio for similar reactions performed on GMFs will not be very good. However, the products will still be diastereoisomers and separable by chromatography or crystallization.
A3. Synthesis from heptose
Almost 40 years ago, Fayet and Gelas reported that heating of a natural sugar (sedoheptulosan) for 60 min at 110–120 °C in the presence of anhydrous pyridinium chloride allowed the isolation of a chiral furan derivative, 5-(D-glycero-1,2-dihydroxyethyl)-2-furaldehyde.227 However, the yield of the isolated product was rather low (20–25%). Considering that the authors reported that they did not optimize the conditions, a significantly better yield is probably achievable. Also, it is the only example we are aware of that a sugar was directly dehydrated to a chiral furan without using any other reactants. Thus, the atom economy is good. Surprisingly, there have been no attempts to produce chiral furans from heptoses or modify sugars in biomass to relevant heptoses.
B1. Synthesis via sugar derivatives, glycals
Another methodology for the synthesis of chiral furans takes advantage of glycals. Thus, monosaccharides or biomass are first reacted to form glycals, which in turn are transformed into substituted or non-substituted (depending on the substrate structure) chiral compounds such as 2-(D-glycero-1,2-dihydroxyethyl)furan. The first practical method was developed almost 50 years ago when it was observed that glycals could be treated with a mercury salt in an aqueous acidic solution or in dioxan to produce furan diols.228–230 This procedure offers excellent yields (a diol with a protected primary hydroxyl group was synthesized from glucal in 92% yield).231 Moreover, inversion of the configuration at the chiral atom can be easily accomplished.232
Japanese reaserchers232 treated D-glucal with a catalytic amount of Sm(OTf)3 or RuCl2(PPh3)3 in the presence of 1 equiv. of H2O to afford an optically active furan diol in good yields (up to 70%).
Another effective catalyst for this reaction is indium chloride.233,234 For example, Babu and Balasubramanian233 achieved very good yields (82%) of furan diols starting from glucal and galactal.
Vankar and coworkers discovered some interesting uses of bismuth triflate. An attempt to form furan diols from glycals such as glucal or galactal using bismuth triflate in acetonitrile gave yields of the expected product of 59% and 46%, respectively (Fig. 6).235
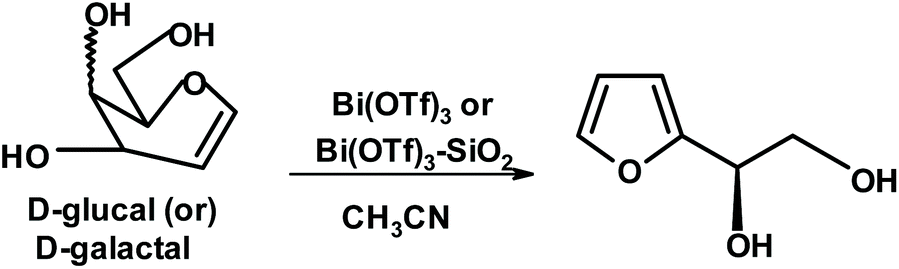 | ||
| Fig. 6 Synthesis of 2-(D-glycero-1,2-dihydroxyethyl)furan from glycals.235 | ||
J. S. Yadav and coworkers236 found a simple, efficient and environmentally benign protocol for this transformation utilizing Montmorillonite KSF as a catalyst. Protic acids can serve as successful catalysts also. HClO4-SiO2 has been shown to transform glycals into furan diols with yields as high as 89%.237
M. Teijeira et al.238 described the effective transformation of glucal into furan diol in the presence of InCl3·3H2O and an ionic liquid.
Shaw and collaborators transformed 3,4,6-tri-O-acetyl-D-glucal into various 2- and 2,3-substituted enantiopure furans. A mixture of Lewis acids (ZrCl4/ZnI2) acts as a catalyst (synergistic effect) and the yields are between 75% and 89%. Interesting products include 2-substituted 3-iodo-furans and (1-R)-1-(2′-furyl)-1,2-ethanediol with a primary hydroxyl group protected as acetate (Fig. 7).239
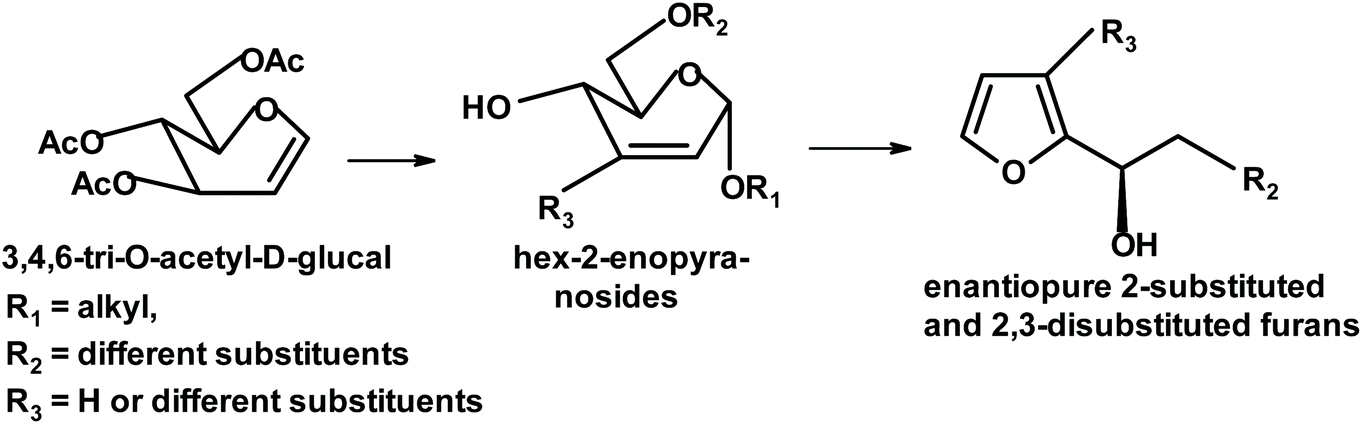 | ||
| Fig. 7 Shaw et al. synthesis of 3-substituted 2-furylethylenediol.239 | ||
B2. Synthesis via substituted glycals
There are a few methodologies for the manufacture of chiral furans from monosaccharides via (sugar derived) substituted glycals such as haloglycals, ketoglycals and nitroglycals. For example, polysubstituted chiral furans can be prepared via the tandem borylation/cross-coupling/cyclization of glycals described by Cossy et al.240 The following scheme explains the methodology and shows a typical product (trimethoxyalcohol), with yields up to 90% (Fig. 8).
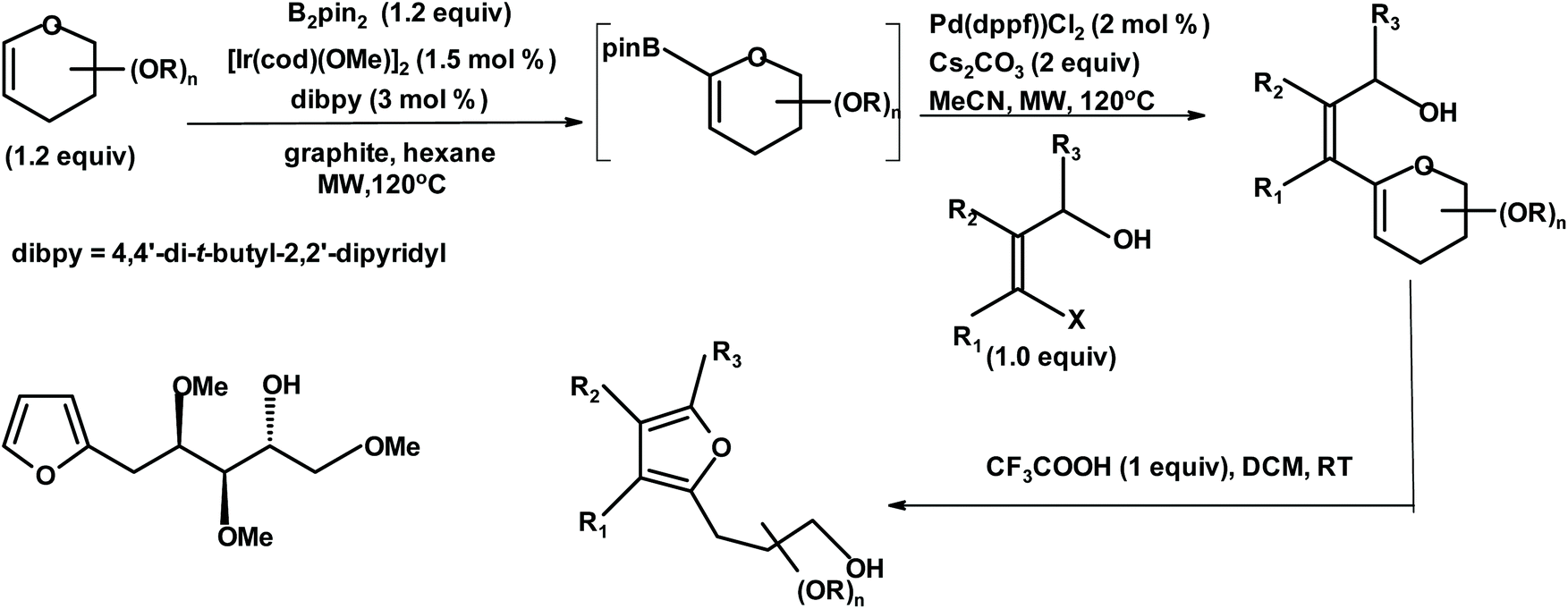 | ||
| Fig. 8 Cossy et al.240 methodology for the synthesis of chiral furans {B2pin2 = bis(pinacolato)diboron; [Ir(cod)(OMe)]2 = bis(1,5-cyclooctadiene)di-μ-methoxydiiridium(I); Pd(dppf)Cl2 = 1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II); MW = microwave; MeCN = acetonitrile; and DCM = dichloromethane}. | ||
Mukherjee et al. reacted acetylated iodoglycals with ArB(OH)2 in the presence of Pd(II) and HCOOH to form 2-acyl glycals, which were treated with TfOH (triflic acid) to produce 2,4-disubstituted furans with a chiral unit in the 2-position.241
Mal, Sharma and Das transformed glycals into iodoglycals, and then synthesized TMS-protected 2-alkynylated glycals.242 The following reaction is gold(III) chloride-catalyzed and produces various 5-aryl(alkyl)-3-formyl-2-polihydroxyalkylfurans. The yields are moderate. Das's group synthesized similar chiral compounds from glycal-derived 2-substituted 3-ketoglucals (Fig. 9).243
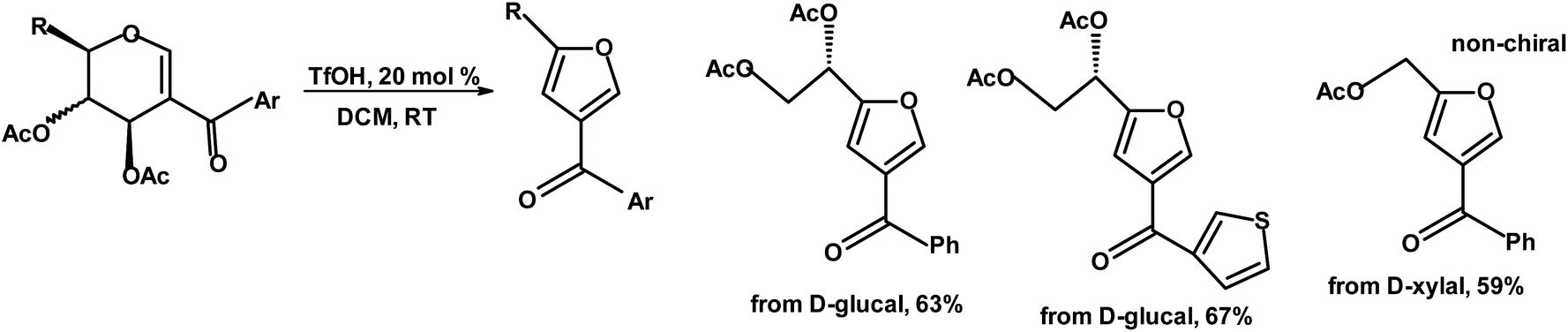 | ||
| Fig. 9 Mukherjee et al.241 synthesis of chiral 2,4-disubstituted furans. | ||
Mal and Das begin their synthesis of highly substituted chiral furans with haloglycals.244 The reaction was performed in DMSO in the presence of potassium carbonate, as shown in Fig. 10.
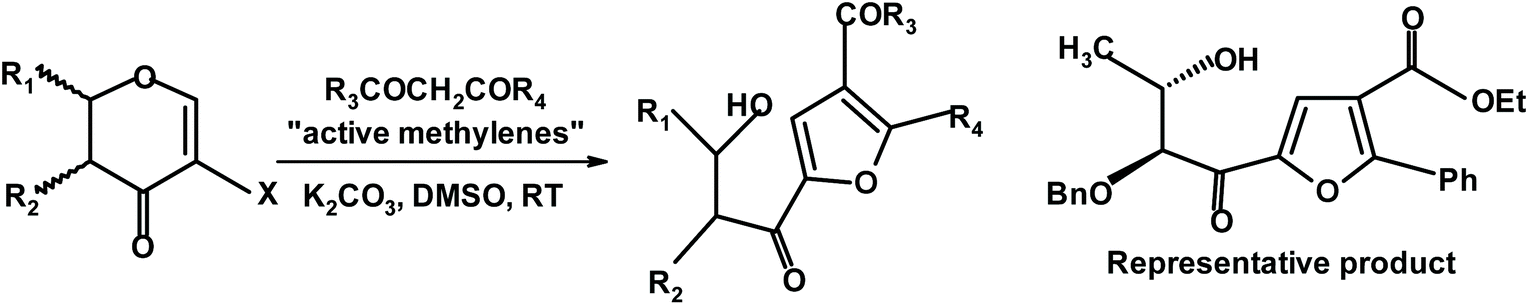 | ||
| Fig. 10 Mal and Das synthesis of chiral furans via haloglycals244 (DMSO = dimethyl sulfoxide). | ||
Gil and coworkers245 transformed glycals into nitroglycals, and, next performed reactions “on water” between nitroglycal and furan, 2-methylfuran and a furfural derivative. The yields of the interesting chiral furans (see Fig. 11) were very good.
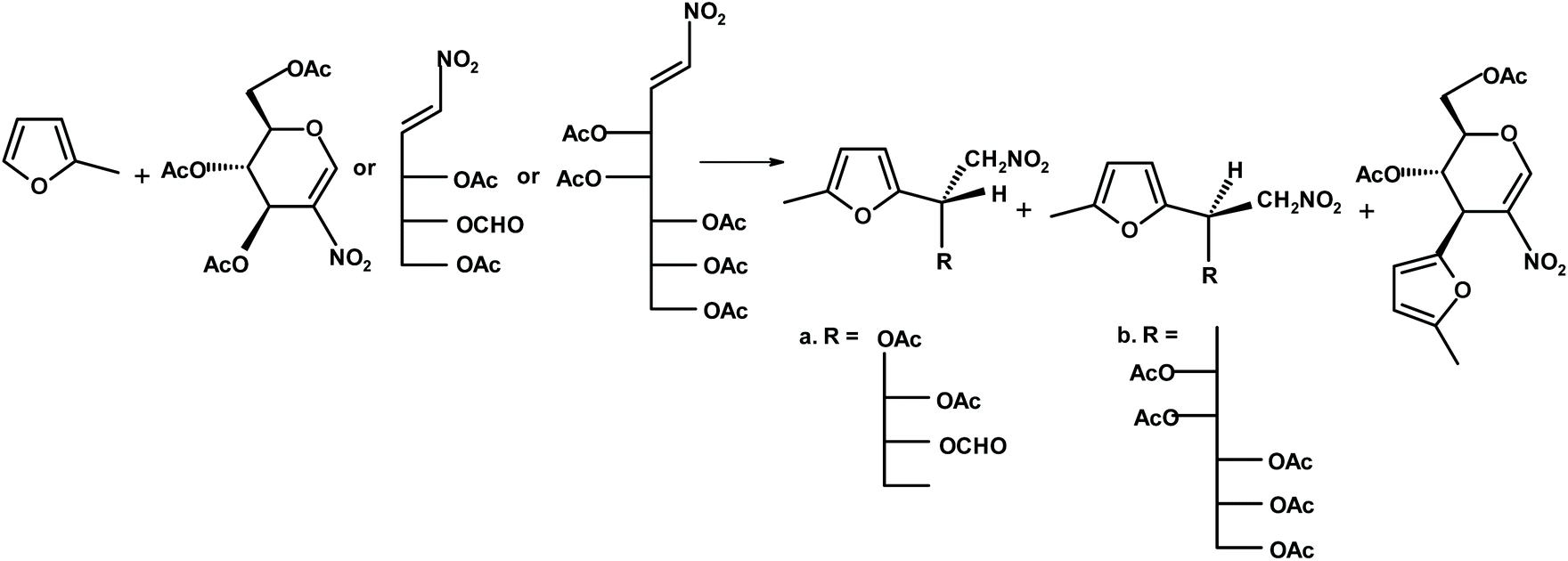 | ||
| Fig. 11 Gil et al.245 synthesis of chiral furans via nitroglycals. | ||
C1. Sharpless enantioselective dihydroxylation or aminohydroxylation of vinylfuran (VF)
This is one of the most elegant ways for the formation of enantiopure 1-(2-furanyl)-1,2-ethanediol. It requires substituted or not-substituted 2-vinylfuran to be available. One possibility is to manufacture vinylfuran directly from sugars using deoxydehydration catalysts.206,246 Scientists in the Netherlands246 used CpttReO3 as a new catalyst (Cptt = 1,3-di-tert-butylcyclopentadienyl) and produced a mixture of vinylfuran and furan at a ratio of 2.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (yield from D-mannose = 39%). Another method for the synthesis of vinylfuran begins with furfural, which can be transformed to vinylfuran via Wittig247–249 or Peterson olefination.250,251 A different method starting from furfural (HMF can be used also) requires Knoevenagel condensation with malonic acid followed by decarboxylation.252 A similar approach to vinylfurans includes an the aldol condensation of furfural with acetaldehyde (formed in situ) to form 3-(2′-furyl)-2-propenal,253 followed by oxidation and decarboxylation. The subsequent dihydroxylation or aminohydroxylation can be performed both on vinylfuran and on vinylfuran still having an additional carbon atom from the aldol condensation. Furthermore, another approach has been recently described by Lobo et al.,254 who reduced biomass-derived (substituted) acetylfuran, and then dehydrated the product to VF using ZSM-5 (Zeolite Socony Mobil-5) as an effective catalyst (Fig. 12). German and Cuban researchers249 described the direct transformation of 2-hydroxymethyl-5-acetyl furan into a VF derivative (Fig. 12).
:1 (yield from D-mannose = 39%). Another method for the synthesis of vinylfuran begins with furfural, which can be transformed to vinylfuran via Wittig247–249 or Peterson olefination.250,251 A different method starting from furfural (HMF can be used also) requires Knoevenagel condensation with malonic acid followed by decarboxylation.252 A similar approach to vinylfurans includes an the aldol condensation of furfural with acetaldehyde (formed in situ) to form 3-(2′-furyl)-2-propenal,253 followed by oxidation and decarboxylation. The subsequent dihydroxylation or aminohydroxylation can be performed both on vinylfuran and on vinylfuran still having an additional carbon atom from the aldol condensation. Furthermore, another approach has been recently described by Lobo et al.,254 who reduced biomass-derived (substituted) acetylfuran, and then dehydrated the product to VF using ZSM-5 (Zeolite Socony Mobil-5) as an effective catalyst (Fig. 12). German and Cuban researchers249 described the direct transformation of 2-hydroxymethyl-5-acetyl furan into a VF derivative (Fig. 12).
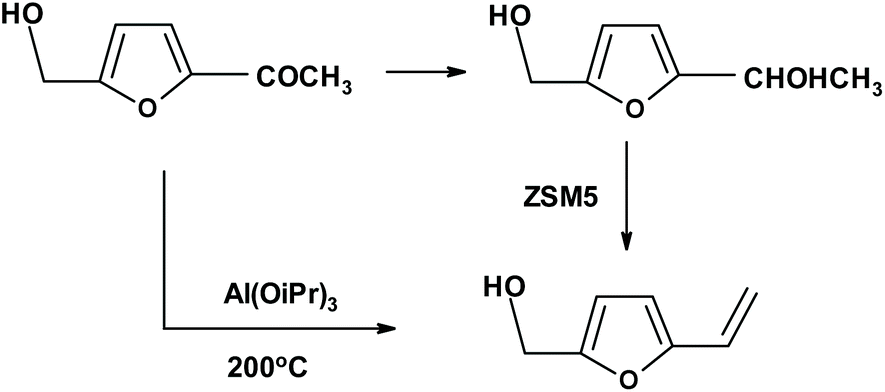 | ||
| Fig. 12 Synthesis of 5-hydroxymethyl-2-vinylfuran, an excellent substrate for enantioselective transformations. | ||
VF has been subjected successfully to the Sharpless asymmetric hydroxylation255,256 and aminohydroxylation.257,258
C2. Formation of 2-acetylfuran followed by enantioselective reduction of the carbonyl group
There are several methods for the synthesis of 2-acetylfyran but the Friedel–Crafts process is by far the most effective. The most popular method for the enantioselective reduction of aromatic (including furyl) ketones is the spectacular Noyori approach.259,260
Wong, Ciufolini and coworkers261 developed practical procedures for the enantioselective reduction of 2-acetylfuran to the corresponding carbinols with 88–90% ee using Thermoanaerobium brockii alcohol dehydrogenase coupled with an NADPH regeneration system.
Excellent reduction of acetylfuran results were also presented by Deska and coworkers,262 who transformed acetylfuran in a two-step, one-pot process to the Achmatowicz rearrangement product with almost perfect ee (Fig. 13).
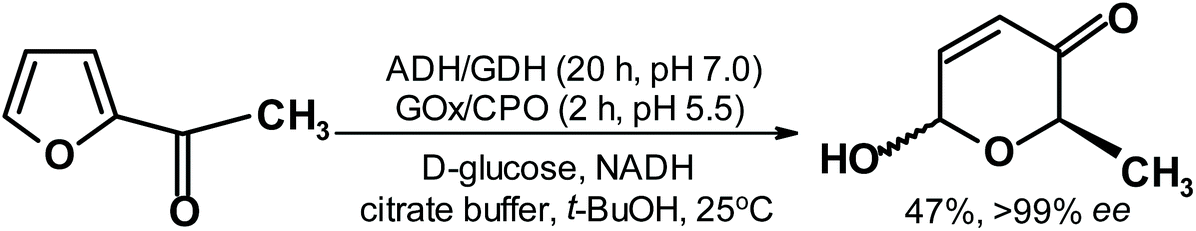 | ||
| Fig. 13 Deska et al.262 reduction of acetylfuran followed by the Achmatowicz reaction. | ||
A few years ago, Deska et al.263 described the elegant synthesis of both enantiomers of cis-osmundalactone. During the synthesis, they obtained both enantiomers of alcohol derived from the enzymatic reduction of acetylfuran, as shown in the following scheme (Fig. 14).
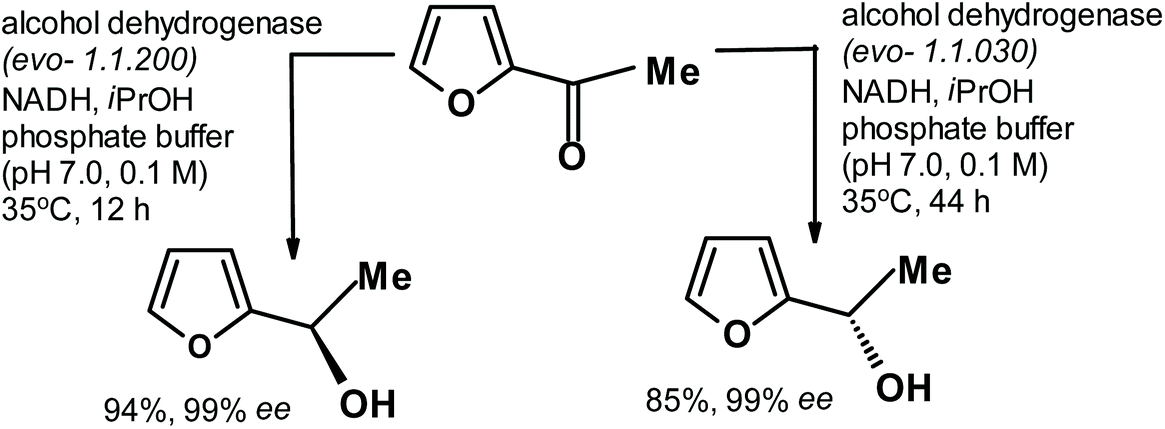 | ||
| Fig. 14 Enzymatic reduction of acetylfuran to produce both alcohol enantiomers.263 | ||
P.-F. Koh and T.-P. Loh264 performed an aldol condensation on protected HMF to form racemic alcohol. It was oxidized to furyl ketone, and subsequently reduced in the presence of a chiral catalyst to give the reduction product with impressive results (yield = 99% and ee = 98%) (see Fig. 15).
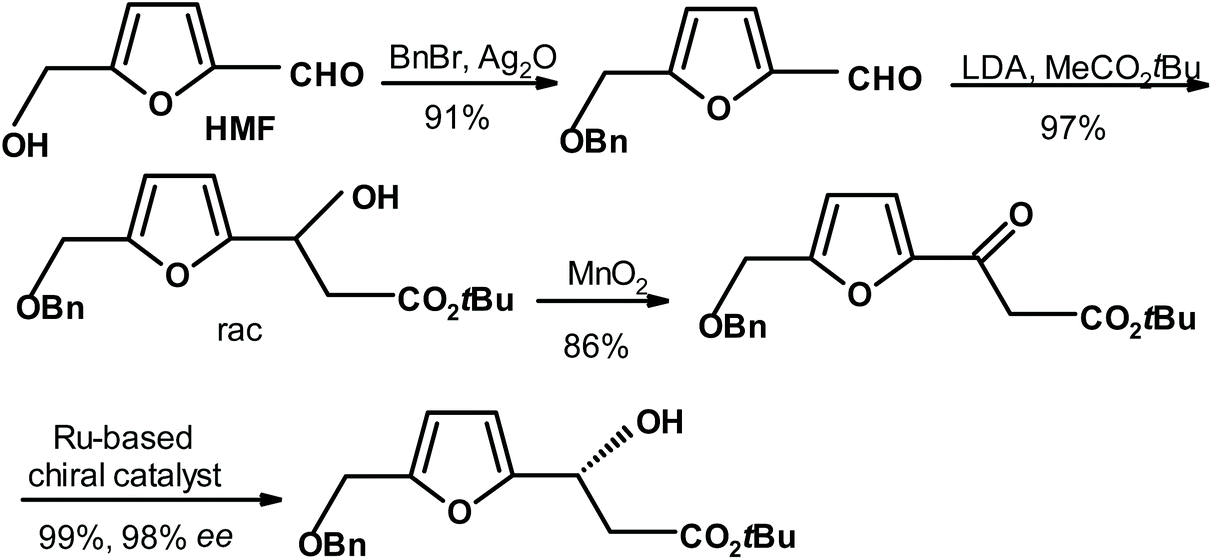 | ||
| Fig. 15 Koh and Loh264 synthesis of chiral alcohol via the enantioselective reduction of ketone. | ||
C3. Reactions on furan using chiral catalysts or with enantiopure compounds
Jurczak et al.265 performed the enantioselective Friedel–Crafts reaction of (1R)-8-phenylmenthyl glyoxylate with various substituted furans in the presence of Lewis acids such as SnCl4 and MgBr2. The following scheme shows the process, obtained yields and de (Fig. 16).
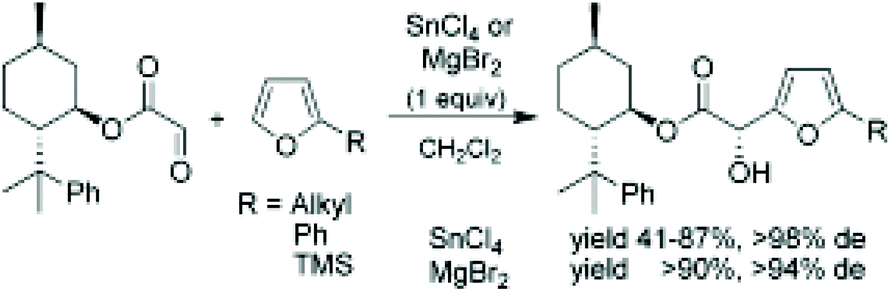 | ||
| Fig. 16 Jurczak et al.265 Friedel–Crafts reaction of chiral glyoxylate with furans. | ||
The same group achieved equally impressive results when performing the Friedel–Crafts reaction of achiral alkyl glyoxylates with furans in the presence of a 6,6′-dibromo-BINOL/Ti(IV) complex.266 The following scheme shows the results (Fig. 17).
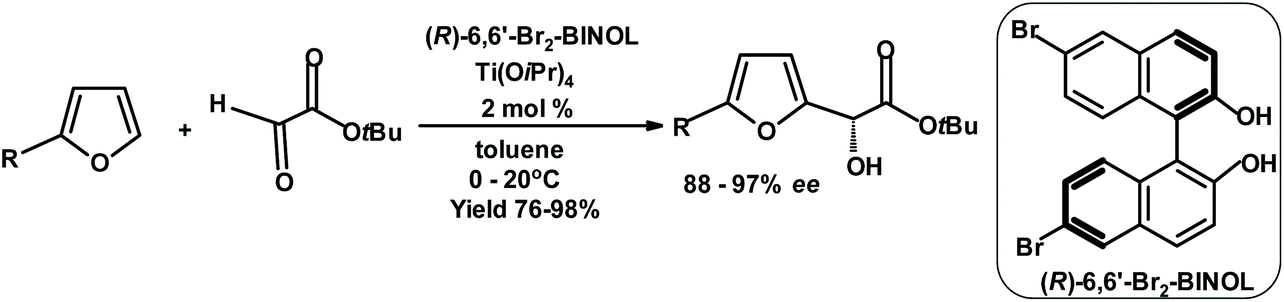 | ||
| Fig. 17 Jurczak et al.266 Friedel–Crafts reaction of glyoxylate with furans using a chiral catalyst. | ||
Yamazaki et al.267 studied the Friedel–Crafts reaction of ethenetricarboxylate with substituted furans in the presence of a chiral bisoxazoline–copper(II) complex (Fig. 18).
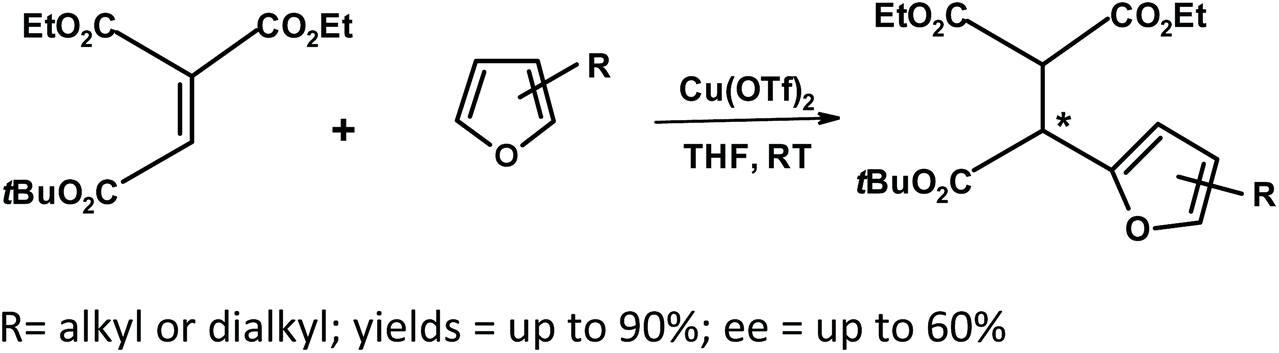 | ||
| Fig. 18 Yamazaki et al.267 Friedel–Crafts reaction of ethenetricarboxylate with furans using a chiral catalyst (Tf = triflate). | ||
Another group of Japanese researchers performed the aza-Friedel–Crafts reaction starting from 2-methoxyfuran (synthesizable from furan) using a chiral catalyst.268 The process is depicted in the scheme below. The yields were up to 95% and ee up to 97% (Fig. 19).
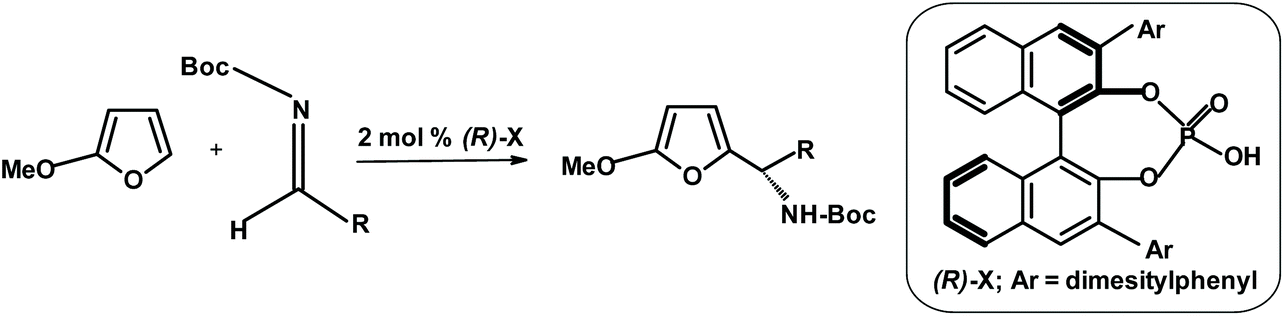 | ||
| Fig. 19 Uraguchi et al.268 aza-Friedel–Crafts reaction in the presence of a chiral catalyst. | ||
Recently, Schäfer et al.269 described the effective, enantioselective Suzuki–Miyaura coupling of heterocycles via the rhodium-catalysed allylic arylation of racemates. The examples include the formation of enantioenriched 2- and 3-substituted furan derivatives. Fig. 20 shows one of these examples.
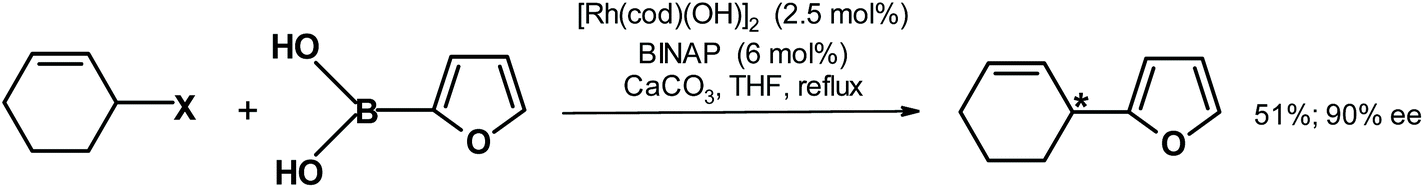 | ||
| Fig. 20 Suzuki–Miyaura coupling described by Schäfer et al.269 {[Rh(cod)(OH)]2 = cyclooctadiene rhodium chloride dimer; BINAP = ([1,1′-binaphthalene]-2,2′-diyl)bis(diphenyl-phosphane)}. | ||
C4. Reactions of the aldehyde group producing chiral products
Martin et al.270 reacted 4,5-disubstituted 2-furaldehyde with the boron enolate derived from the chiral oxazolidinone (shown in the rounded rectangle in Fig. 21). The condensation proceeded with a high degree (>98%) of diastereoselectivity to give the expected erythro-β-hydroxy adduct (90%).
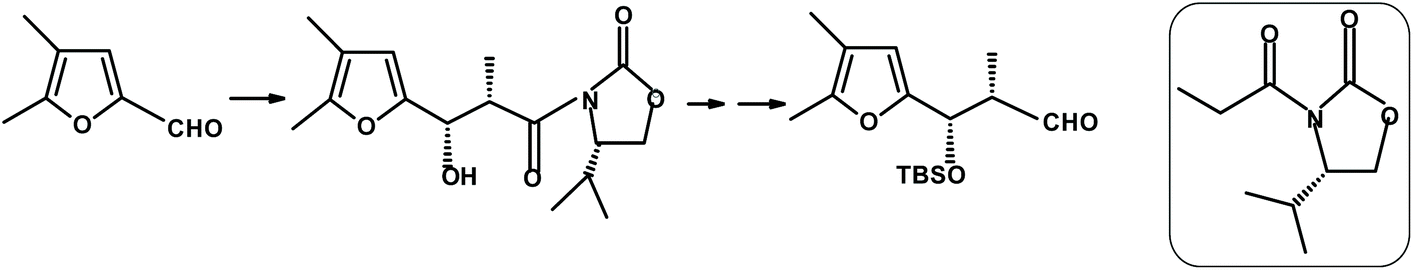 | ||
| Fig. 21 Martin et al.270 furfural condensation producing the erythro-β-hydroxy adduct. | ||
During the total synthesis of phorbol, Wender et al.271 performed a chiral oxazolidinone-based asymmetric aldol condensation of the HMF derivative with N-propionyl oxazolidinone (Fig. 22). The condensation occurred with a very high isolated yield (96% of single diastereoisomer after chromatography) and diastereoselectivity (98% de).
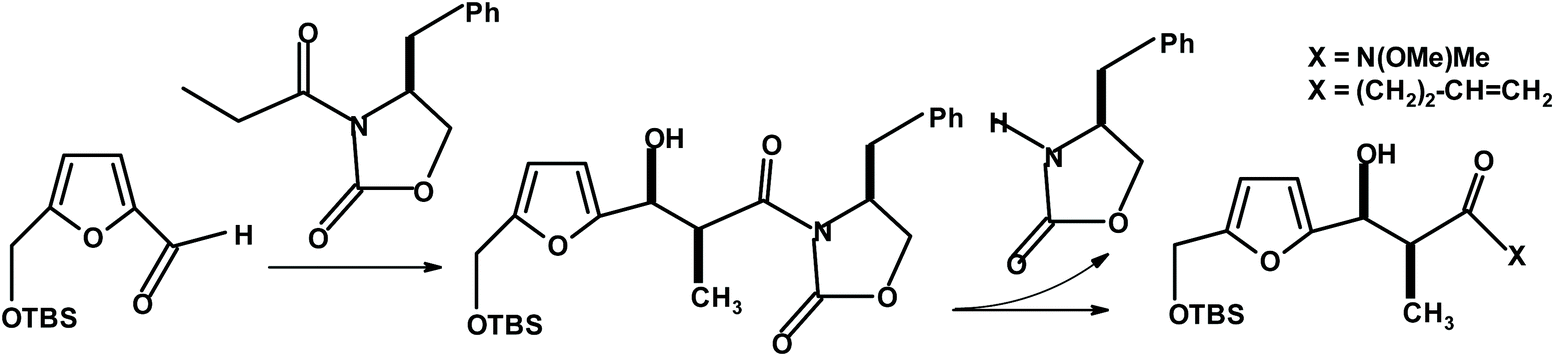 | ||
| Fig. 22 Wender et al.271 aldol condensation of HMF. | ||
Carreira and Krüger studied enantioselective dienolate additions to aldehydes, including furfural, mediated by tol-BINAP Cu(II) fluoride complexes.272 For furfural, they achieved the yield of 91% and ee of 94%. The product was further reacted to produce a chiral dihydroxy ester (protected as the isopropylidene derivative), as depicted in Fig. 23.
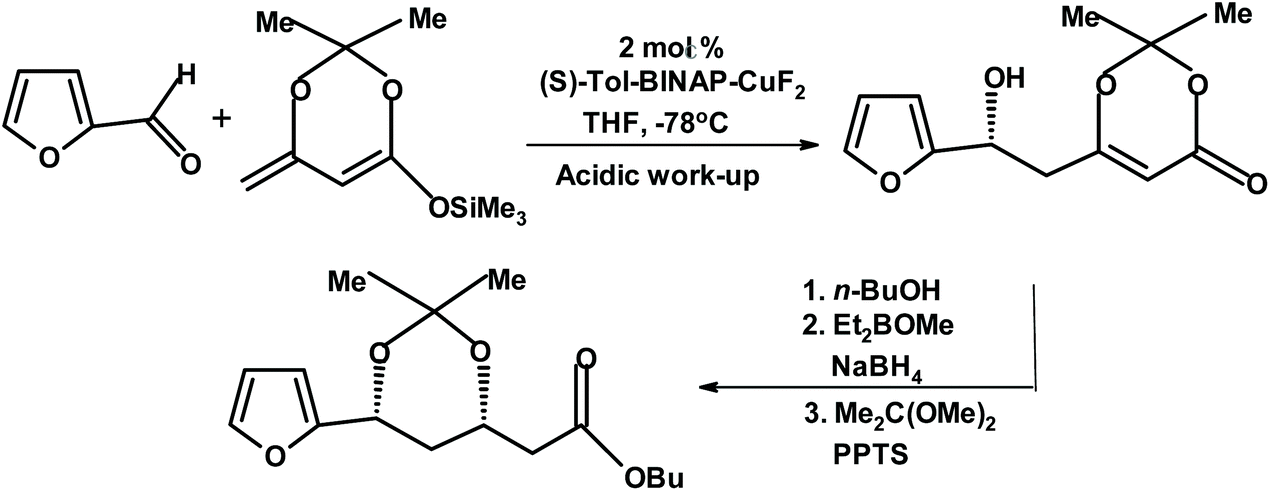 | ||
| Fig. 23 Carreira and Krüger dienolate addition to furfural.272 {(S)-Tol-BINAP = (S)-(−)-2,2′-bis(di-p-tolylphosphino)-1,1′-binaphthyl; and PPTS = pyridine p-toluenesulfonate.} | ||
Pedrosa and coworkers273 reacted aromatic aldehydes including 5-methyl-2-furfural, with chiral oxazolidine. In the case of the furan derivative, the yield was 69% and the de was 81% (Fig. 24).
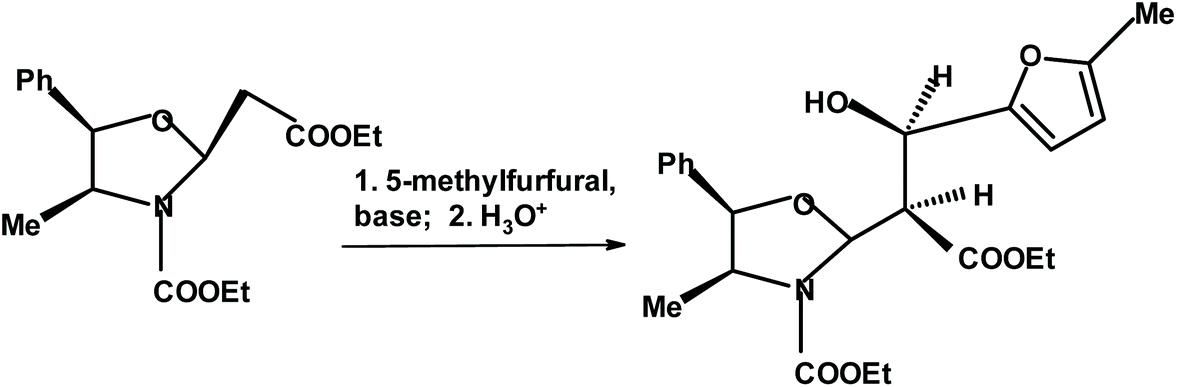 | ||
| Fig. 24 Pedrosa et al.273 condensation of chiral oxazolidine with 5-methylfurfural. | ||
Turkish chemists274 described a successful enantioselective Henry reaction. The reactants were various aromatic aldehydes including furfural. The scheme below depicts the ligand (chiral oxazoline) and its synthesis. The dominant enantiomer depends on the absolute configuration of the chiral centers in oxazoline. For furfural, the yields and ee approached 80% (Fig. 25).
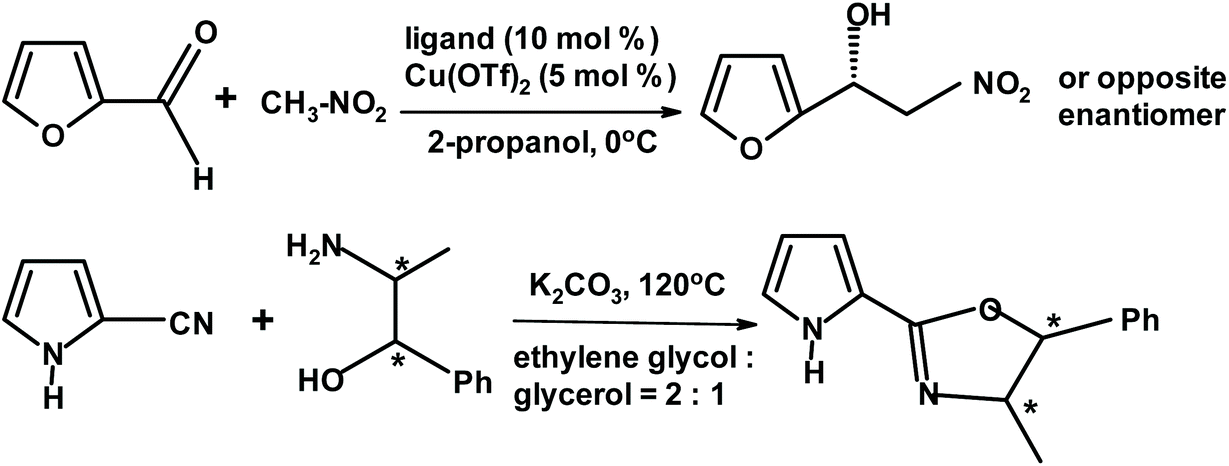 | ||
| Fig. 25 Henry reaction of furfural with nitromethane described by Aydin and Yuksekdanaci.274 | ||
Witczak, Bielski et al.275 reacted various 5-membered aromatic aldehydes (including HMF and 5-methyl furfural) with dihydrolevoglucosenone using piperidine as a base. The yields of the condensation products (shown in Fig. 26) were above 80%.
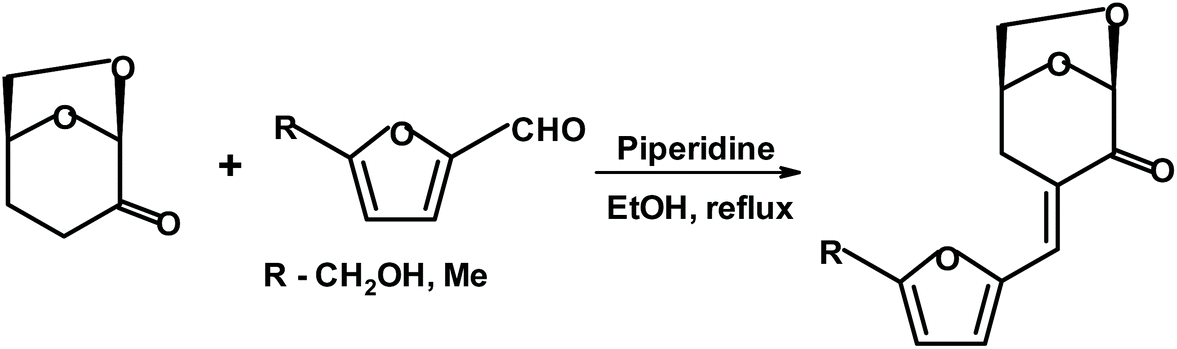 | ||
| Fig. 26 Aldol condensation of substituted furfurals with dihydrolevoglucosenone.275 | ||
Another approach developed by Grushin, van Leeuven and collaborators276 used furfural as a starting material. The furoin reaction produced a racemic product, furoin, which was hydrogenated using a chiral Ru or Ir catalyst to give hydrofuroin. The enantiomeric excess and diasteroisomeric ratios were very impressive (Fig. 27).
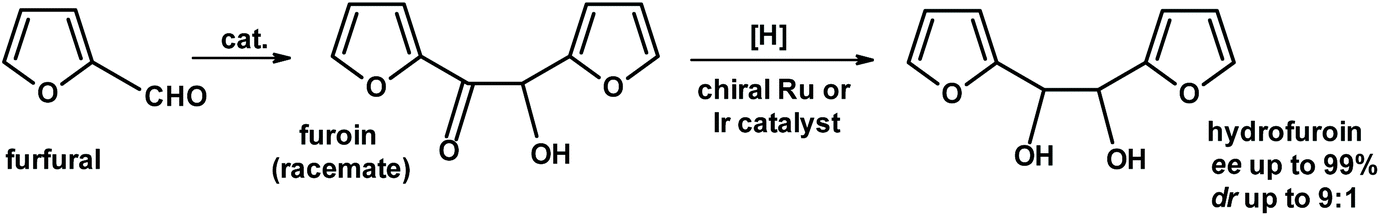 | ||
| Fig. 27 Enantioselective synthesis of hydrofuroin from furfural.276 | ||
Gong et al.277 developed an asymmetric allylation reaction at the benzylic position of benzylfurfurals, as depicted in Fig. 28.
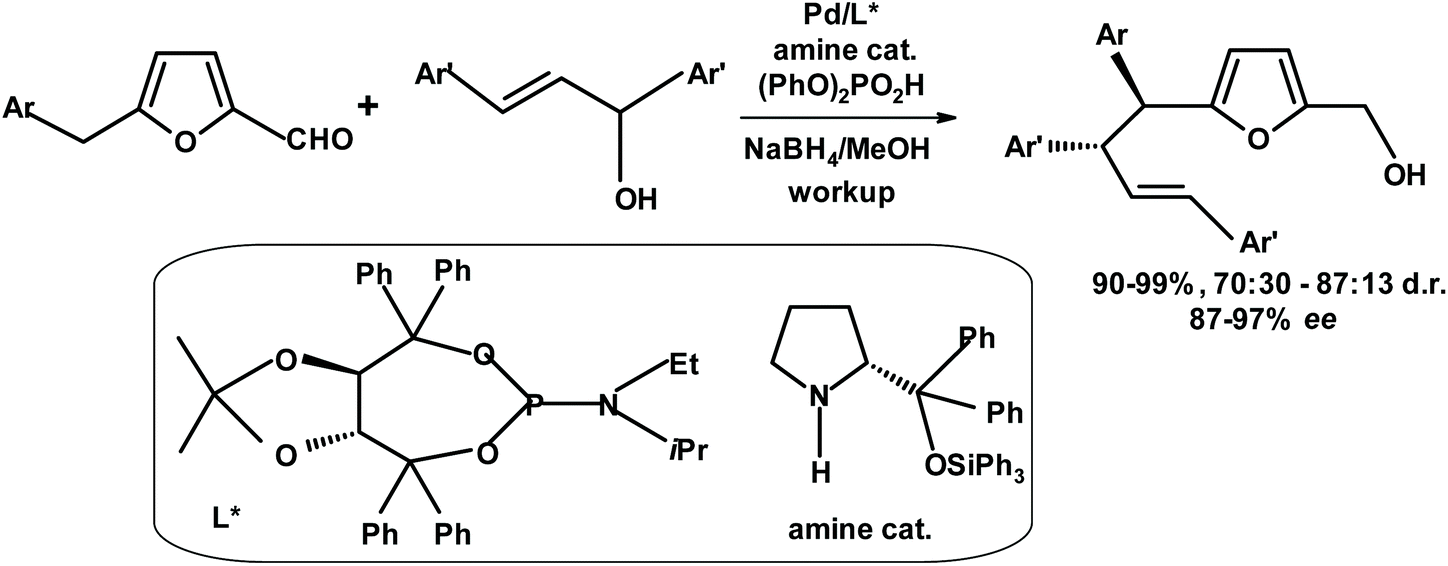 | ||
| Fig. 28 Asymmetric allylation developed by Gong et al.277 | ||
Brazilian scientists278 used fungi to accomplish the enantioselective reduction of a furfural-derived product, as shown in Fig. 29. The results were very impressive. For example, the use of the fungus Penicillium citrinum gave 98% yield of the S-reduced product with an enantiomeric ratio of 99%. Another fungus, Aspergillus sydowii, offered 92% isolated yield of the R product and the enantiomeric ratio was 97%.
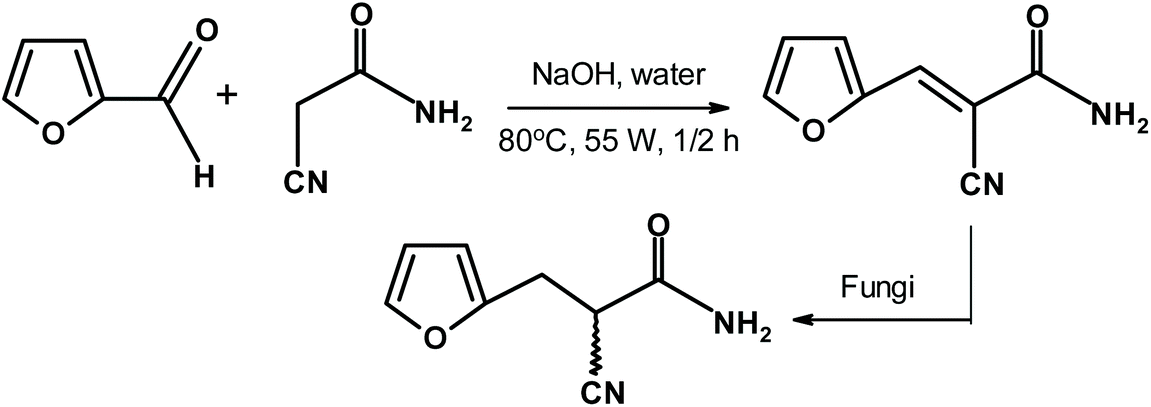 | ||
| Fig. 29 Chiral furans from enantioselective reduction using fungi.278 | ||
C5 Other reactions transforming non-chiral furans to chiral furans
Hailes and coworkers279 synthesized various furylamines starting from carbonyl compounds using transaminases and amines. One of the reactions was that of acetylfuran with Mycobacterium vanbaalenii (Mv-Tam) and an amine donor, benzylmethylamine. The yield was 54% and ee of the product was 78%.
Loh and coworkers280 synthesized furylamine with very high enantiopurity by taking advantage of the rhodium-catalyzed asymmetric arylation methodology developed by Hayashi et al. (Fig. 30)281
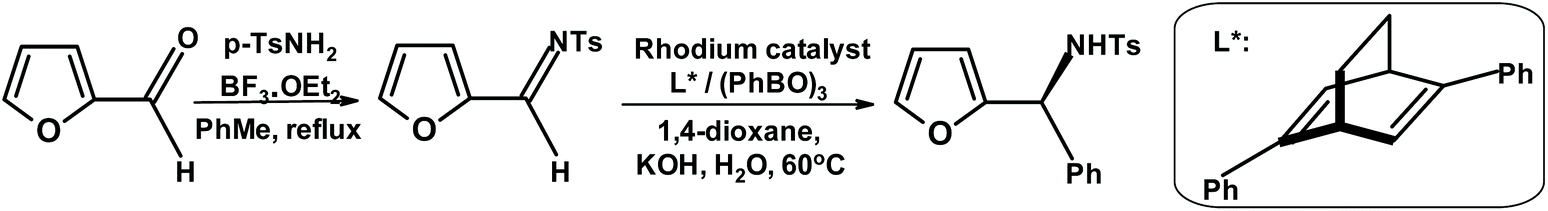 | ||
| Fig. 30 Asymmetric arylation described by Loh et al.280 | ||
Some reactions of sugars with achiral furan compounds may also lead to chiral furans. For example, Grynkiewicz and BeMiller,282 Grynkiewicz and Zamojski,283 and P. Sinaÿ et al.284 synthesized these constructs as a result of a Ferrier-type reaction directly between glycals and furans, with moderate yields.
Zhang and co-workers285 synthesized compounds similar to Lichtenthaler products but their starting materials were fructose-derived HMF and various glycals (Fig. 31).
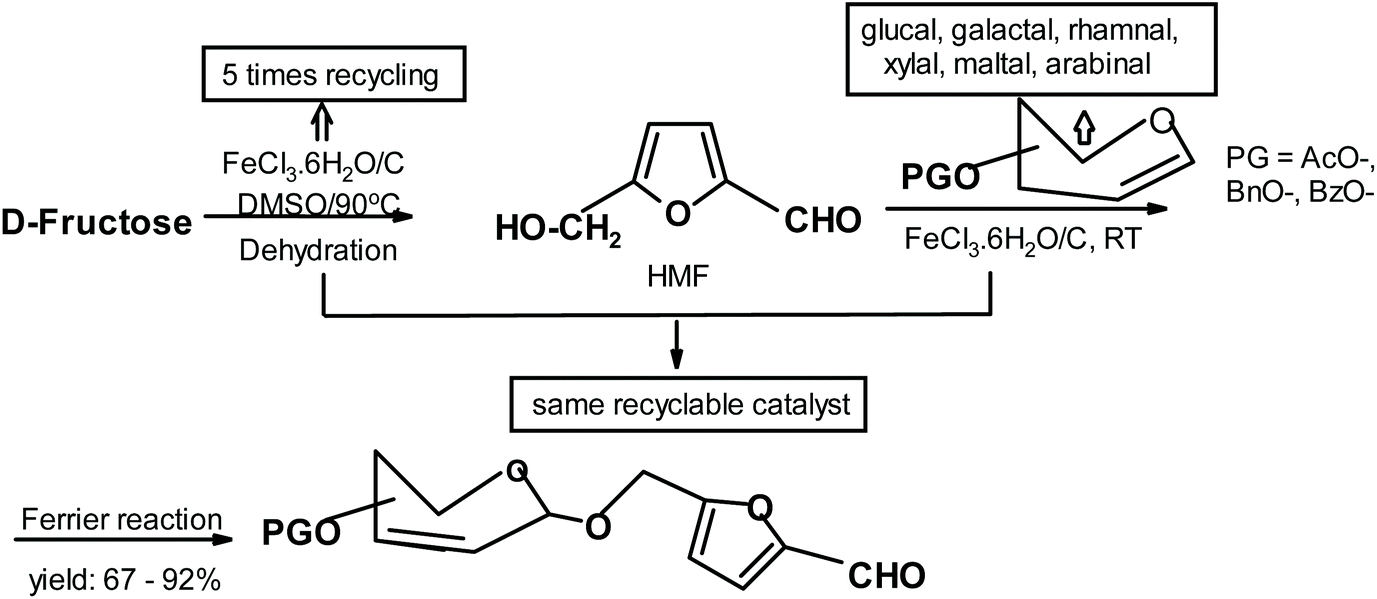 | ||
| Fig. 31 Zhang et al.285 synthesis of HMF-glycal adducts. | ||
Another, simple and effective method for the synthesis of chiral furans takes advantage of the direct reaction of methyl furoate with peracetylated ribofuranose in the presence of a Lewis acid.286 The product, methyl 5-(2′,3′,5′-tri-O-acetyl-β-D-ribofuranosyl)-2-furoate, was isolated in 60% yield and the β-anomer was the only product (Fig. 32).
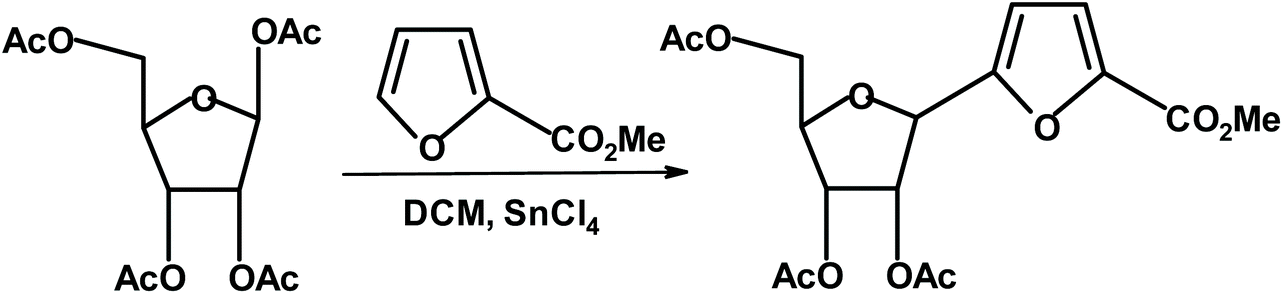 | ||
| Fig. 32 Cermola et al.286 synthesis of monosaccharide-methyl furoate construct. | ||
An entirely different strategy for synthesizing constructs containing furans and sugars was developed by Jarosz and coworkers.287 The synthesis starts with D-gluconolactone and can produce furans with the sugar unit either in the 2- or 3-position of the furan ring. The yields are moderate (Fig. 33).
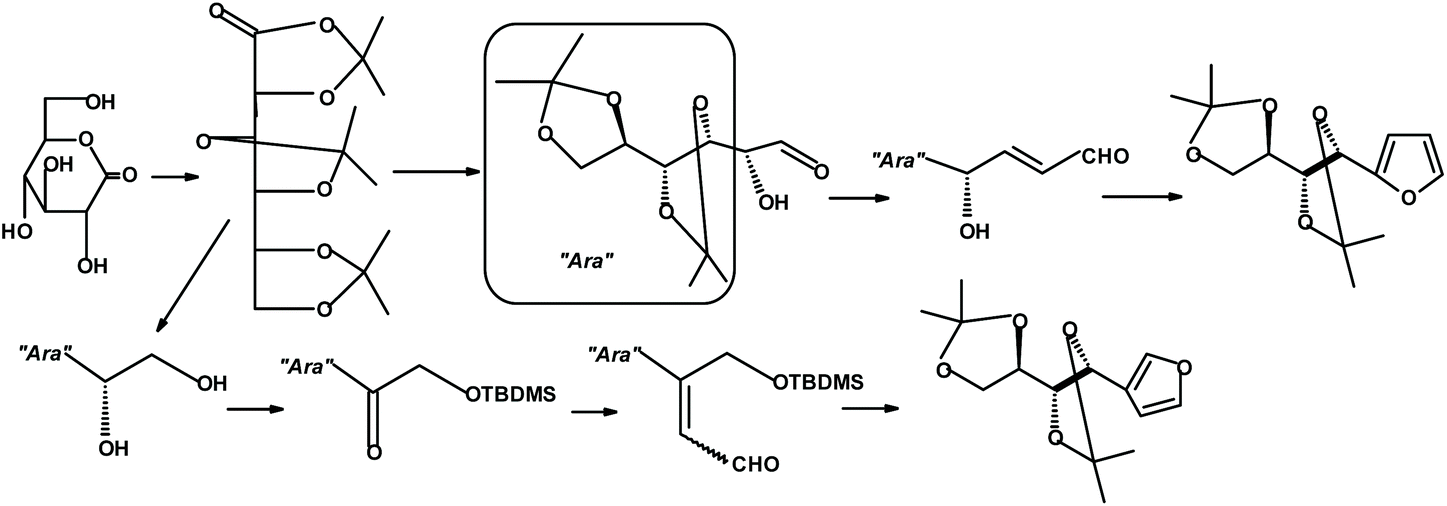 | ||
| Fig. 33 Jarosz et al.287 synthesis of monosaccharide-furan constructs. | ||
Lan, Xiao and coworkers288 synthesized highly complex chiral furan derivatives starting from biomass-based 2,5-dimethylfuran using a chiral phosphoric acid as the catalyst (Fig. 34). The yield and enantiomeric excess values were very good.
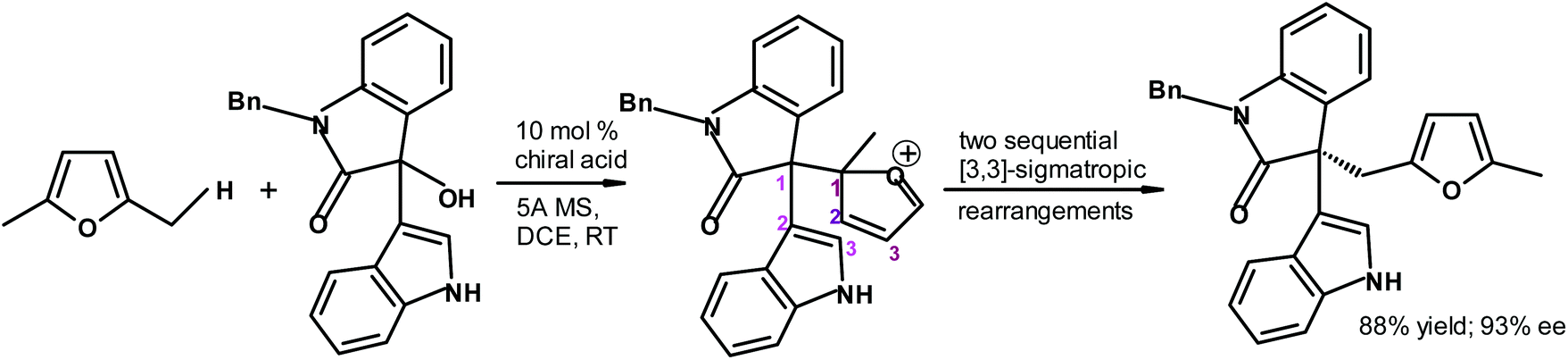 | ||
| Fig. 34 Lan, Xiao et al.288 chiral furan synthesis taking advantage of two sequential [3,3]-sigmatropic rearrangements {MS = molecular sieves and DCE = dichloroethane}. | ||
J. L. Marco289 described the synthesis of several chiral furans utilizing another monosaccharide derivative, 1,2:5,6-di-O-isopropylidene-α-D-glucofuranose. The products are convenient chiral precursors for the synthesis of furanoterpenes.
Of course, enantiopure furans can be formed from racemates as a result of chiral separations. For example, Irimie and coworkers290 used regio- and stereoselective lipases for the efficient kinetic resolution of racemic 1-(5-phenylfuran-2-yl)ethane-1,2-diols. All the yields were between 46% and 49% and the ee between 92% and 97%.
5. Very brief look at FPC reactions
Excluding the furan ring itself, major direct FPCs contain three highly reactive moieties, namely, an aldehyde group, hydroxymethyl group and hydrogen atom in the 2-position of the furan aromatic ring. These structural features make furanics potential objects of a large variety of reactions. This is the main reason why FPCs are formidable substrates for the chemical and related industries. Many of the processes listed here were already mentioned in this text. Nevertheless, it seems useful to very briefly look at the possibilities.5.1. Coupling at the furan C-2 position
Coupling allows for the introduction of various substituents to the carbon atom adjacent to the ring oxygen. The relevant processes include Friedel–Crafts alkylation and acylation,265,246,291,292 Heck reaction,293–296 Suzuki reaction,269,297,298 borylation,299–301 and carboxylation.173 Often the same compound can be synthesized using alternative coupling reactions. It is very difficult to compare various approaches given that the outcome depends on factors such as availability and cost of the reactants and catalysts, reaction conditions, and ease of isolating the product. Scheme 6 visualizes the most important reactions at C-2 position of furan.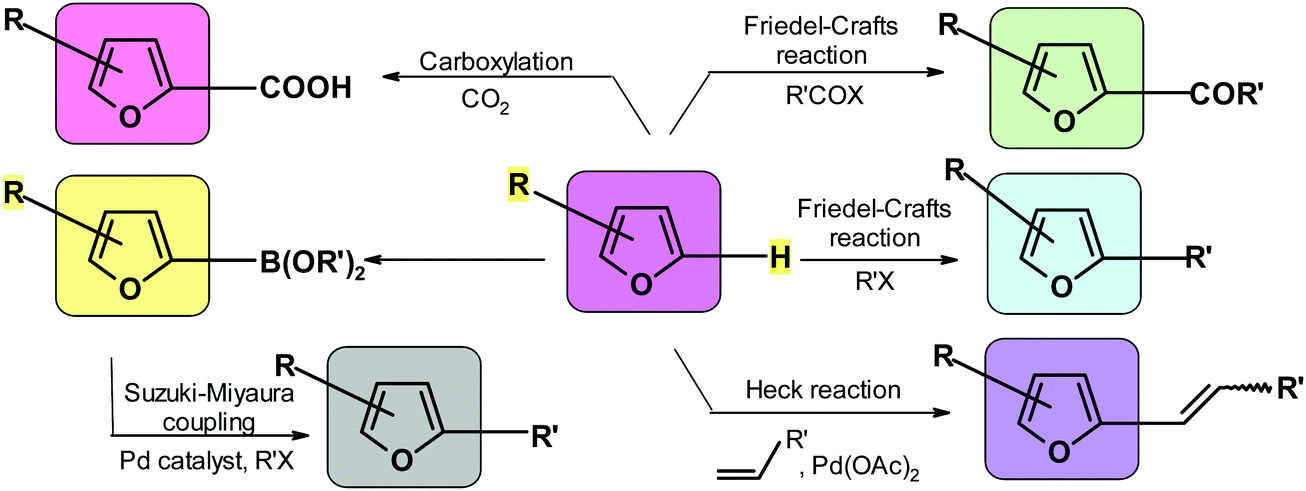 | ||
| Scheme 6 Reactions in which a furan hydrogen atom is replaced. | ||
5.2. Reactions of the aldehyde unit in HMF and FUR
The specific processes include aldol and related condensations,91,122,270–273,275,302 furoin condensation,276,303 acetal formation with alcohols,304–306 Henry reaction with nitro-compounds,274,307 formation of adducts (followed by dehydration) with amines, hydrazines, hydroxylamines and related compounds,91,308–310 decarbonylation,311 reduction to alcohol91 and oxidation to acid.91 The most important reactions of the furfural aldehyde group are depicted in Scheme 7.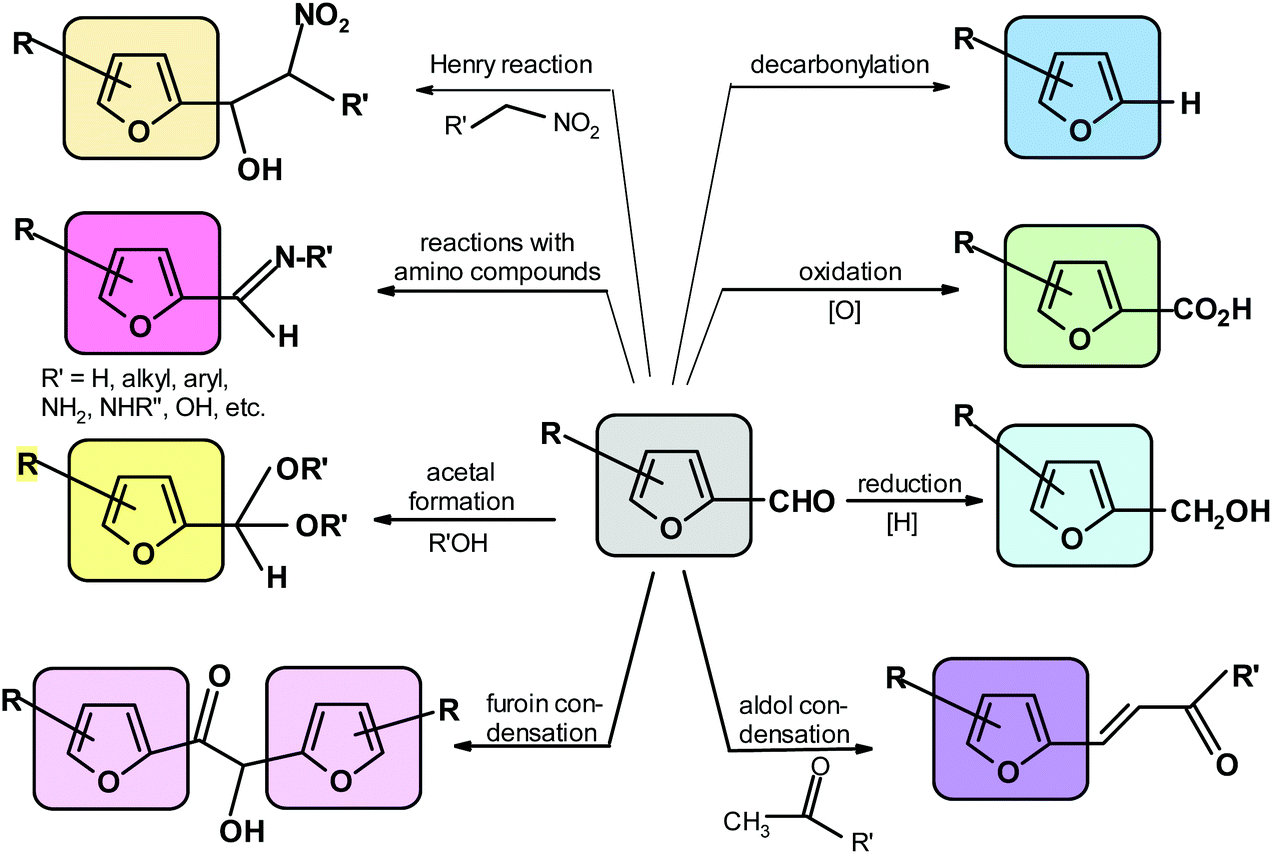 | ||
| Scheme 7 Major reactions of the FUR and HMF aldehyde group. | ||
5.3. Reactions of the hydroxymethyl group in HMF
They include oxidation to aldehyde or acid,91,312 nucleophilic substitution of the modified OH group to form halides,313 esterification,314 and etherification315,316 (Scheme 8).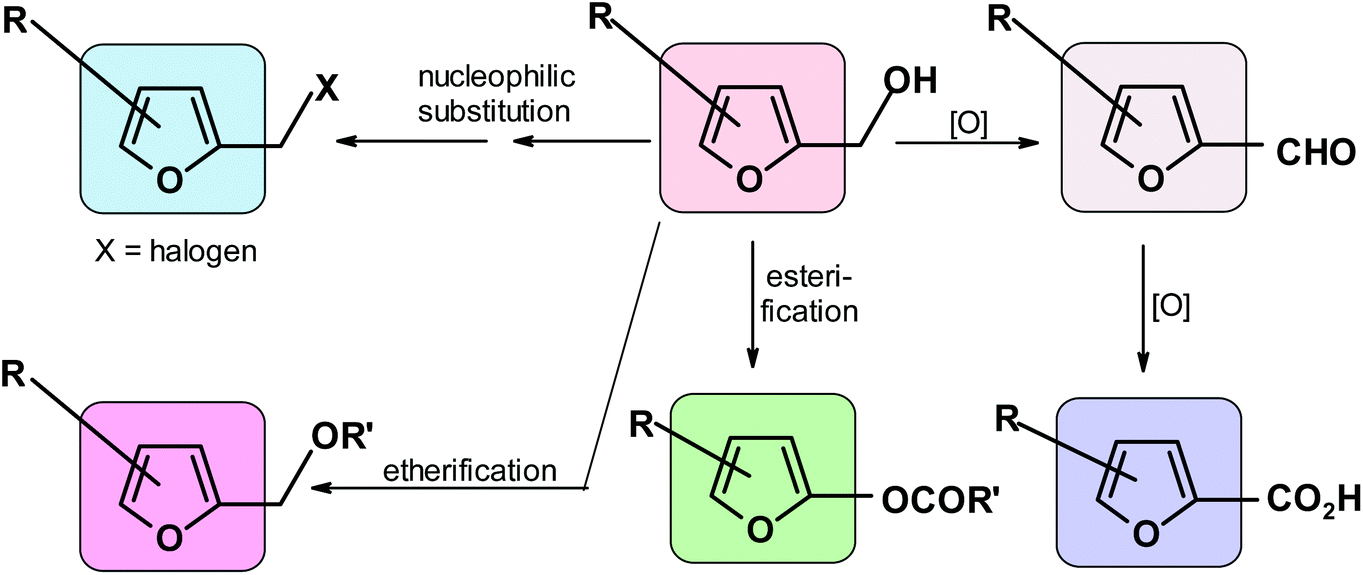 | ||
| Scheme 8 Main reactions of the HMF hydroxymethyl group. | ||
5.4. Reactions of the furan ring producing moieties containing no furan ring
Examples of relevant processes are the Diels–Alder reaction (it may offer benzene derivatives),68,317,318 pyrrole and other heterocycle formation via the Butin reaction,319 the oxa- and aza-Achmatowicz reaction forming 6-membered rings with O or N, respectively,64 the Piancatelli reaction forming 4-hydroxycyclopentenones,320–322 singlet oxygen oxidations producing cyclopentanones and hydrindanes323 and formation of maleic acid324 (Scheme 9).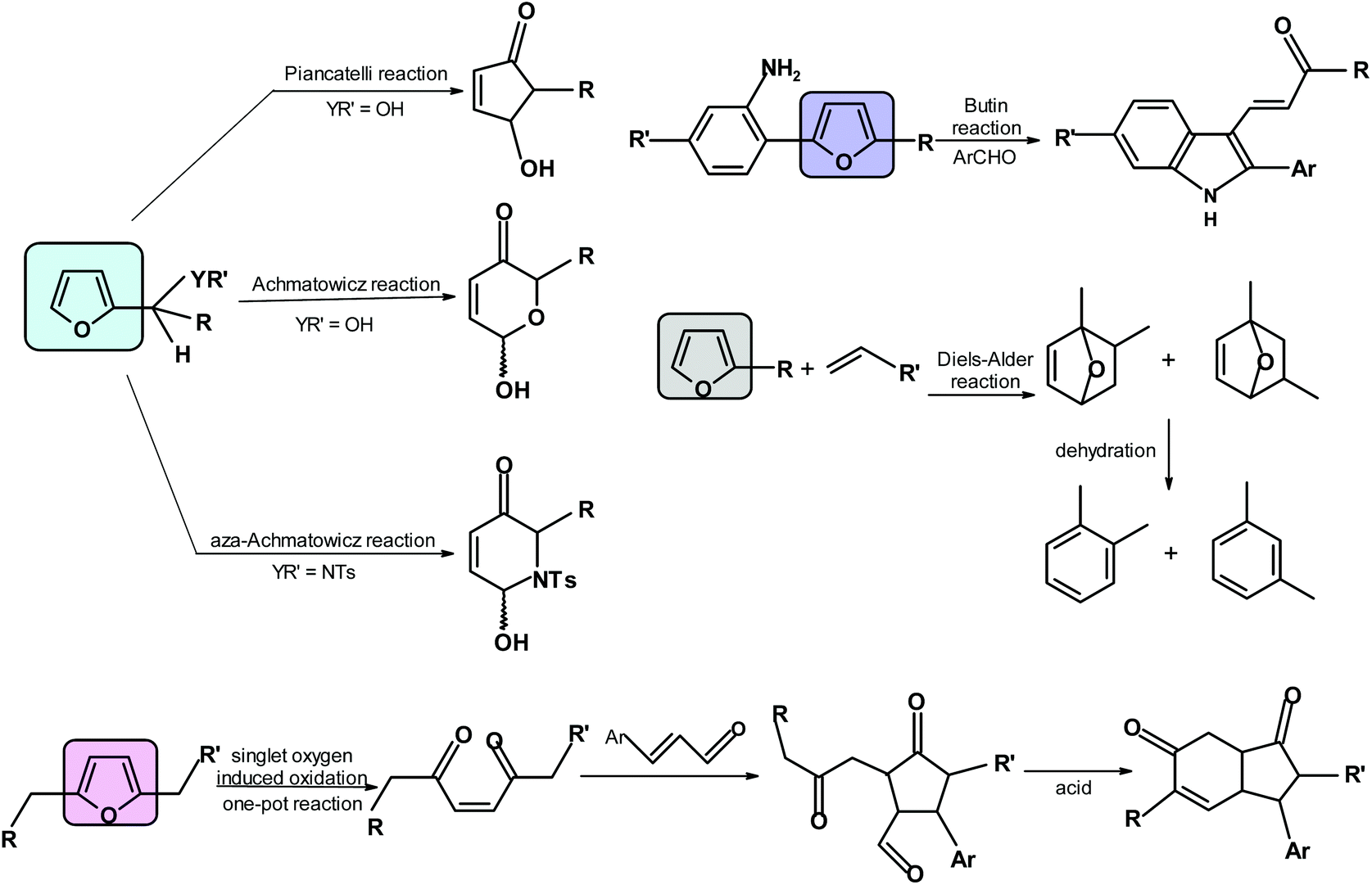 | ||
| Scheme 9 More important reactions of furans resulting in products with no furan ring. | ||
6. Conclusions/future perspectives
Around fifty years ago, most chemists believed that developing catalysts only slightly worse than natural catalysts (enzymes) in producing enantiopure compounds from non-chiral or racemic substrates would require an exorbitant R&D effort. Today, there are dozens of methodologies for the synthesis of chiral compounds with ee approaching 100%. Actually, chemists learned to use catalysts much less complex and wasteful than enzymes to produce results almost comparable to that in Nature.Thus, there is a good reason to expect that coupling financial resources with an improved intellectual effort will solve other equally difficult problems. Perhaps learning from Nature and adjusting its methods to our abilities will give direction. Incidentally, we understand how plants utilize CO2 to form a plethora of organic compounds. However, taking advantage of modified chlorophyll seems to be less popular than expected.
Nature can utilize carbohydrates in the body (or generally, in cells) to synthesize almost any chemical. Can we do this also? Is it too arrogant to try? It seems that transforming biomass into compounds such as FPCs, and next, into a variety of organics is one of the most promising and economic options. This approach takes advantage of Nature's work (carbon dioxide has already been transformed into organic compounds) but takes it further.
The present perspective describes some aspects of the biotechnical revolution occurring presently. One of the main symptoms of this process is the replacement of refineries with biorefineries. Biorefineries, using biomass as feedstock, offer various advantages over refineries, which include:
1. The feedstock of biorefineries is renewable.
2. It is significantly less complicated than that of refineries (highly complex mixture of hydrocarbons), which allows for a relatively easy access to FPCs and other chemicals.
3. The biorefinery feedstock consists mainly of monosaccharides and polysaccharides composed (after hydrolysis) of pentoses and hexoses. Consequently, the direct products are partially oxygenated C5 and C6 hydrocarbons. Their level of functionalization is ideal for further processes, leading to a plethora of valuable chemicals. As a result of various degradations and condensations, the resulting products may have almost any number of carbon atoms. They all are easily separable by process design. Note that petrochemical refineries start from a huge variety of hydrocarbons featuring similar physicochemical properties.
The biorefinery is a novel concept and technology, and thus it is experiencing learning difficulties. Time and effort are required to develop optimal solvents, conditions, reactants, catalysts or biocatalysts, etc. for specific processes. It is likely that the present difficulties such as problems in effectively forming FDCA from FUR or HMF will soon be overcome.
Two main areas of applications of biorefinery-derived furan-containing products have been proposed, i.e., custom-made fuels for combustion engines and monomers for polymerization. Particularly interesting are polycondensation products to replace polyethylene terephthalate and related condensation polymerization products. Note that biorefinery products not only can offer diacids but also various symmetric diols, diamines, dihalides, etc. Thus, interesting targets include polyesters, polyamides, polyurethanes, polyureas, etc. Of course, important monomers such as acrylic and hydroxypropionic acids are also available from biorefineries.
The main purpose of this perspective is to promote another area of applications for biorefinery products, i.e., synthetic industrial compounds belonging to specialty chemicals, fragrances, pharmaceuticals, nutraceuticals, mono- and disaccharides, alkaloids, etc. In this perspective, we looked at the possible applications of selected biorefinery-derived chemicals. We chose to briefly examine the most important FPC-derived (FUR and HMF) products. Herein, we attempted to highlight the commendable achievements and wonderful potential of the discussed materials. It was also shown that many enantiopure compounds can be formed directly from biomass or via intermediates such as modified sugars or HMF and FUR. FPCs are highly versatile starting materials that can enter reactions via the aldehyde group, hydroxymethyl group, C-2 of furan, and processes that transform furan into other entities such as other heterocycles and Diels Alder products.
Herein, we uncovered a clear need for accelerated research and development in this field. Although the formation of direct and indirect FPCs in the lab is reasonably well established, their industrial-scale production is considerably less advanced. The necessary R&D will have to focus on improved chemo- and bio-catalysts, isolation of individual products from the reaction mixture without any destruction of unreacted components such as cellulose, use and reuse of better solvents, etc. A successful implementation of effective biorefinery processes will require creativity, original thinking, time and, perhaps, some level of insanity.
If the biorefinery does not become a common reality, it will not be due to a lack of novel ideas. Recently Deska and coworkers325 incorporated “abiotic transformations into a recombinant bacterial host, which allowed the production of complex lactone building blocks. This whole cell system streamlines the synthetic cascade, eliminates isolation and purification steps, and provides a high degree stereoselectivity that has thus far been elusive in chemical methodology”. In other words, factory processes can take place in the cell.
Another potential methodology capable of dramatically improving the reaction kinetics is the use of strong, oriented electric fields.326–328 A catalyst such as an enzyme takes one molecule of one reactant and orients it against a molecule of another reactant. After completion of the reaction, the product is released, and the enzyme molecule is ready for the repetition of the cycle. A strong electric field can enforce mutual orientation of a significant part of all reactant molecules. The number of molecules oriented along the lines of the electric field depends on the dipole moment of the molecules, the process medium and strength of the electric field.
Flow chemistry has already become a common term in pharmaceutical R&D and manufacture, but to the best of our knowledge, to date, it has not been applied to large-scale processes.329,330 It is very likely it will become practical also in biorefinery-derived large-scale products.
Additionally, it is impossible to overstate the spectacular potential of the directed engineering of enzymes in modern industrial chemistry. The Nobel lecture of F. Arnold offers an excellent perspective on this issue.331
Of course, not only furan compounds are derived from biomass. There are benzene derivatives (from lignin), a plethora of non-cyclic compounds and others. Many bio-based products are presently more expensive from biomass than refineries. This is an issue of scale and some economic bias. When these materials are produced on a larger scale, they will become substantially less expensive and likely to be less costly than petroleum-derived equivalents.
The primary conclusion is very important, i.e., FPCs (which serve as an example here) are a marvelous alternative to (a) petrochemical refinery-derived fuels and other reactants and (b) thermochemical biorefinery-based fuels. This means that when carbon combustion is eliminated from energy-generating methods, we will have an excellent source of comparatively sophisticated chemicals directly and indirectly produced from renewable, safe, and green materials.
One can ask about priorities. Which biorefinery-related problems are the most urgent? The answer is obviously complex, but a few issues can be highlighted. As was already mentioned, the large-scale production of FDCA is essential. There are several reasons for the importance of FDCA production including the existing problems with PET polymers and the fact that LCB-derived polymers seem to be easier to collect, store and recycle than petrochemical plastics.
Also, we have shown that there are many good methods for the synthesis of enantiopure products from biomass or its components. However, it seems that there is a need for synthons containing one or two chiral centers that can be directly manufactured from biomass. This will dramatically facilitate the synthesis of various complex chemicals.
Arguably, another acute issue is the lack of operationally reliable and economically sound information in the literature on integrated validated processes comprising the effective separation of LCB components and channeling them into selective reaction pathways. Only recently things have begun to change. For example, Won et al.332 announced the GVL (gamma valerolactone) solvent-assisted chemo-catalytic treatment of LCB into an insoluble cellulose fraction, soluble C-5 sugar fraction and lignin utilized for heat and/or power generation. Interestingly, through the application of assorted unit operations such as dehydration, hydrogenation, oxidation, and hydration, the main LCB constituents, cellulose and hemicellulose, were effectively converted through multistep pathways into C6 monomer synthon (FDCA) and C5 aliphatic component (1,5-pentanediol). The authors included techno-economic analysis and life-cycle assessments for the products and their intermediates, discussing present market prices and predictable trends.
Conflicts of interest
There are no conflicts to declare.References
- https://op.europa.eu/en/publication-detail/-/publication/8c4de15d-a17d-11eb-b85c-01aa75ed71a1/language-en/format-PDF/source-201342421 .
- P. Morseletto, J. Ind. Ecol., 2020, 24, 763 CrossRef.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- J. Garcia-Martinez, Angew. Chem., Int. Ed., 2021, 60, 4956 CrossRef CAS PubMed.
- T. A. Bender, J. A. Dabrovski and M. R. Gagné, Nat. Rev. Chem., 2018, 2, 35 CrossRef CAS.
- R. A. Sheldon and M. Norton, Green Chem., 2020, 22, 6310 RSC.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484 CrossRef CAS PubMed.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS PubMed.
- A. T. Ubando, C. B. Felix and W.-H. Chen, Bioresour. Technol., 2020, 299, 122585 CrossRef CAS PubMed.
- A. Kelloway and P. Daoutidis, Ind. Eng. Chem. Res., 2013, 53, 5261 CrossRef.
- S. Farzad, M. A. Mandegari, M. Guo, K. F. Haigh, N. Shah and J. F. Görgens, Biotechnol. Biofuels, 2017, 10, 87 CrossRef PubMed.
- A. Duque, C. Alvarez, P. Domenech, P. Manzanares and A. D. Moreno, Processes, 2021, 9, 206 CrossRef.
- J.-M. Choi, S.-S. Han and H.-S. Kim, Biotechnol. Adv., 2015, 33, 1443 CrossRef CAS PubMed.
- C. K. Winkler, J. H. Schrittwieser and W. Kroutil, ACS Cent. Sci., 2021, 7, 55 CrossRef CAS PubMed.
- J. C. Serrano-Ruiz, R. Luque and A. Sepulveda-Escribano, Chem. Soc. Rev., 2011, 40, 5266 RSC.
- A. Tursi, Biofuel Res. J., 2019, 22, 962 CrossRef.
- K. Kohli, R. Prajapati and B. K. Sharma, Energies, 2019, 12, 233 CrossRef CAS.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540 CrossRef CAS PubMed.
- C. H. Zhou, X. Xia, C. X. Lin, D. S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588 RSC.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538 RSC.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497 RSC.
- C. Chatterjee, F. Pong and A. Sen, Green Chem., 2015, 17, 40 RSC.
- J. van Haveren, E. L. Scott and J. Sanders, Biofuels, Bioprod. Biorefin., 2008, 2, 41 CrossRef CAS.
- G. Fiorentino and M. Ripa, Biofuels, Bioprod. Biorefin., 2017, 11, 195 CrossRef CAS.
- J. W. Döbereiner, Ann. Pharm., 1832, 3, 141 CrossRef.
- H. J. Brownlee and C. S. Miner, Ind. Eng. Chem., 1948, 40, 2001 CrossRef.
- S. Dutta, S. De, B. Saha and M. Imteyaz Alam, Catal. Sci. Technol., 2012, 2, 2025 RSC.
- G. Machado, S. Leon, F. Santos, R. Lourega, J. Dullius, M. E. Mollmann and P. Eichler, Nat. Resour. J., 2016, 7, 115 CAS.
- K. Yan, G. Wu, T. Lafleur and C. Jarvis, Renewable Sustainable Energy Rev., 2014, 38, 663 CrossRef CAS.
- D. T. Win, Aust. J. Technol., 2005, 8, 185 Search PubMed.
- S. Rivas, C. Vila, V. Santos and J. C. Parajó, Holzforschung, 2016, 70, 901 CAS.
- C. Aellig, D. Scholz, P. Y. Dapsens, C. Mondelli and J. Pérez-Ramírez, Catal. Sci. Technol., 2015, 5, 142 RSC.
- X. Li, P. Jia and T. Wang, ACS Catal., 2016, 6, 7621 CrossRef CAS.
- L. Ricardi, W. Verboom, J. P. Lange and J. Huskens, ChemSusChem, 2020, 13, 3589 CrossRef PubMed.
- A. Mazar, O. Ajao, M. Benali, N. Jemaa, W. W. Al-Dajani and M. Paleologou, ACS Sustainable Chem. Eng., 2020, 8, 17345 CrossRef CAS.
- D. Weidener, W. Leitner, P. Domínguez de María, H. Klose and P. M. Grande, ChemSusChem, 2021, 14, 909 CrossRef CAS PubMed.
- J. F. L. Silva, M. A. Selicani, T. L. Junqueira, B. C. Klein, S. Vaz Júnior and A. Bonomi, Brazilian J. Chem. Eng., 2017, 34, 623 CrossRef CAS.
- K. J. Zeitsch, The Chemistry and Technology of Furfural and Its Many By-products, Elsevier Science, Amsterdam NL, 2000 Search PubMed.
- C. M. Cai, T. Zhang, R. Kumar and C. E. Wyman, J. Chem. Technol. Biotechnol., 2014, 89, 2 CrossRef CAS.
- L. C. Nhien, N. V. D. Long, S. Kim and M. Lee, Biochem. Eng. J., 2016, 116, 166 CrossRef CAS.
- J.-P. Lange, E. van der Heide, J. van Buijtenen and R. Price, ChemSusChem, 2012, 5, 150 CrossRef CAS PubMed.
- A. E. Eseyin and P. H. Steele, Int. J. Adv. Chem., 2015, 3, 42 CrossRef.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. López Granados, Energy Environ. Sci., 2016, 9, 1144 RSC.
- A. Llevot, P.-K. Dannecker, M. von Czapiewski, L. C. Over, Z. Söyler and M. A. R. Meier, Chem. – Eur. J., 2016, 22, 11510 CrossRef CAS.
- S. Chen, R. Wojcieszak, F. Dumeignil, E. Marceau and S. Royer, Chem. Rev., 2018, 118, 11023 CrossRef CAS PubMed.
- R. Gérardy, D. P. Debecker, J. Estager, P. Luis and J.-C. M. Monbaliu, Chem. Rev., 2020, 120, 7219 CrossRef PubMed.
- V. Krzelj, J. van Kampen, J. van der Schaaf and M. F. Neira d'Angelo, Ind. Eng. Chem. Res., 2019, 58, 16126 CrossRef CAS.
- Z. Chen, X. Bai, A. Lusi, W. A. Jacoby and C. Wan, Bioresour. Technol., 2019, 289, 121708 CrossRef PubMed.
- A. Mittal, S. K. Black, T. B. Vinzant, M. O'Brien, M. P. Tucker and D. K. Johnson, ACS Sustainable Chem. Eng., 2017, 5, 5694 CrossRef CAS.
- B. M. Matsagar, S. A. Hossain, T. Islam, H. R. Alamri, Z. A. Alothman, Y. Yamauchi, P. L. Dhepe and K. C.-W. Wu, Sci. Rep., 2017, 7, 13508 CrossRef PubMed.
- P. Sudarsanam, E. Peeters, E. V. Makshina, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2019, 48, 2366 RSC.
- R. Fang, A. Dhakshinamoorthy, Y. Li and H. Garcia, Chem. Soc. Rev., 2020, 49, 3638 RSC.
- G. Shen, B. Andrioletti and Y. Queneau, Curr. Opin. Green Sustain. Chem., 2020, 26, 100384 CrossRef.
- R. C. Cioc, M. Lutz, E. A. Pidko, M. Crockatt, J. C. van der Waal and P. C. A. Bruijnincx, Green Chem., 2021, 23, 367 RSC.
- A. Gandini, D. Coelho, M. Gomes, B. Rei and A. Silvestre, J. Mater. Chem., 2009, 19, 8656 RSC.
- A. Gandini and T. M. Lacerda, Prog. Polym. Sci., 2015, 48, 1 CrossRef CAS.
- W. Sliwa, Chem. Heterocycl. Compd., 2004, 40, 683 CrossRef CAS.
- A. R. Chaudhry, B. U. Haq, S. Muhammad, A. Laref and A. Irfan, Comput. Theor. Chem., 2019, 1147, 20 CrossRef CAS.
- Z. J. Brentzel, K. J. Barnett, K. Huang, C. T. Maravelias, J. A. Dumesic and G. W. Huber, ChemSusChem, 2017, 10, 1351 CrossRef CAS PubMed.
- C. Xu, E. Paone, D. Rodríguez-Padrón, R. Luque and F. Mauriello, Renewable Sustainable Energy Rev., 2020, 127, 109852 CrossRef CAS.
- J. P. M. Nunes, C. A. M. Afonso and S. Caddick, RSC Adv., 2013, 3, 14975 RSC.
- C. Piutti and F. Quartieri, Molecules, 2013, 18, 12290 CrossRef CAS PubMed.
- R. Ramos, A. Grigoropoulos, N. Perret, M. Zanella, A. P. Katsoulidis, T. D. Manning, J. B. Claridgea and M. J. Rosseinsky, Green Chem., 2017, 19, 1701 RSC.
- R. Bielski and G. Grynkiewicz, Tetrahedron, 2021, 85, 132058 CrossRef CAS.
- P.-F. Koh, P. Wang, J.-M. Huang and T.-P. Loh, Chem. Commun., 2014, 50, 8324 RSC.
- F. van Der Pijl, F. L. van Delft and F. P. J. T. Rutjes, Eur. J. Org. Chem., 2015, 4811 CrossRef CAS.
- I. V. Trushkov, M. G. Uchuskin and A. V. Butin, Eur. J. Org. Chem., 2015, 2999 CrossRef CAS.
- F. A. Kucherov, K. I. Galkin, E. G. Gordeev and V. P. Ananikov, Green Chem., 2017, 19, 4858 RSC.
- D. Olender, J. Zwawiak and L. Zaprutko, Pharmaceuticals, 2018, 11, 54 CrossRef PubMed.
- M. D. Delost, D. T. Smith, B. J. Anderson and J. T. Njardson, J. Med. Chem., 2018, 61, 10996 CrossRef CAS PubMed.
- F. Hasegawa, K. Niidome, C. Migihashi, M. Murata, T. Negoro, T. Matsumoto, K. Kato and A. Fujii, Bioorg. Med. Chem. Lett., 2014, 24, 4266 CrossRef CAS PubMed.
- A. Astarita, F. Cermola, M. R. Iesce and L. Previtera, Tetrahedron, 2008, 64, 6744 CrossRef CAS.
- B. M. Trost and Z. Shi, J. Am. Chem. Soc., 1996, 118, 3037 CrossRef CAS.
- M. Stafford-Smith, D. Griggs, O. Gaffney, F. Ullah, B. Reyers, N. Kanie, B. Stigson, P. Shrivastava, M. Leach and D. O'Connell, Sustain. Sci., 2017, 12, 911 CrossRef PubMed.
- P. T. Anastas and J. B. Zimmerman, Curr. Opin. Green Sustain. Chem., 2018, 13, 150 CrossRef.
- A. M. Noce, Chem. Eng. News, 2018, 96(22), 43 Search PubMed.
- J. B. Binder, J. J. Blank, A. V. Cefali and R. T. Raines, ChemSusChem, 2010, 3, 1268 CrossRef CAS PubMed.
- X. Hu, R. J. M. Westerhof, D. Dang, L. Wu and C. Z. Li, ACS Sustainable Chem. Eng., 2014, 2, 2562 CrossRef CAS.
- J. S. Luterbacher, D. Martin-Alonso and J. A. Dumesic, Green Chem., 2014, 16, 4816 RSC.
- G. Zang, A. Shah and C. Wan, J. Cleaner Prod., 2020, 260, 120837 CrossRef CAS.
- Y. Luo, Z. Li, X. Li, X. Liu, J. Fan, J. H. Clark and C. Hu, Catal. Today, 2019, 319, 14 CrossRef CAS.
- P. Rachamontree, T. Donzou, K. Cheeenkachorn, M. Sriyariyanun and K. Rattanaporn, Appl. Sci. Eng. Prog., 2020, 13, 3 Search PubMed.
- G. Guillén-Gosálbez, F. You, Á. Galán-Martín, C. Pozo and I. E. Grossmann, Curr. Opin. Chem. Eng., 2019, 26, 170 CrossRef.
- I. Ioannou, S. C. D'Angelo, Á. Galán-Martín, C. Pozo, J. Pérez-Ramírez and G. Guillén-Gosálbez, React. Chem. Eng., 2021, 6, 1179 RSC.
- B. R. Bakshi, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 265 CrossRef PubMed.
- J. Kleinekorte, L. Fleitmann, M. Bachmann, A. Kätelhön, A. Barbosa-Póvoa, N. von der Assen and A. Bardow, Annu. Rev. Chem. Biomol. Eng., 2020, 11, 203 CrossRef PubMed.
- F. H. Newth, Adv. Carbohydr. Chem., 1951, 6, 83 CAS.
- J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979 CrossRef CAS PubMed.
- S. P. Teong, G. Yi and Y. Zhang, Green Chem., 2014, 16, 215 RSC.
- B. M. F. Kuster, Starch, 1990, 42, 314 CrossRef CAS.
- J. Lewkowski, ARKIVOC, 2001, 2, 17 Search PubMed.
- Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933 CrossRef.
- A. A. Rosatella, S. P. Simeonov, R. F. M. Frade and C. A. M. Afonso, Green Chem., 2011, 13, 754 RSC.
- R. J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499 CrossRef CAS PubMed.
- C.-H. Zhou, X. Xia, C.-X. Lin, D.-S. Tong and J. Beltramini, Chem. Soc. Rev., 2011, 40, 5588 RSC.
- B. Liu and Z. H. Zhang, ChemSusChem, 2016, 9, 2015 CrossRef CAS PubMed.
- P. Bhaumik and P. L. Dhepe, Catal. Rev. Sci. Eng., 2016, 58, 36 CrossRef CAS.
- A. C. G. Vries, Chem. Rec., 2016, 16, 2787 CrossRef PubMed.
- P. K. Rout, A. D. Nannaware, O. Prakash, A. Kalra and R. Rajasekharan, Chem. Eng. Sci., 2016, 142, 318 CrossRef CAS.
- H. Zhang, H. Pan and S. Yang, Curr. Nanosci., 2017, 13, 513 CAS.
- V. M. Chernyshev, O. A. Kravchenko and V. P. Ananikov, Russ. Chem. Rev., 2017, 86, 357 CrossRef CAS.
- F. A. Kucherov, L. V. Romashov, K. I. Galkin and V. P. Ananikov, ACS Sustainable Chem. Eng., 2018, 6, 8064 CrossRef CAS.
- G. G. Millán, S. Hellsten, J. Llorca, R. Luque, H. Sixta and A. M. Balu, ChemCatChem, 2019, 11, 2022 CrossRef.
- J. Esteban, A. J. Vorholt and W. Leitner, Green Chem., 2020, 22, 2097 RSC.
- L. Hu, Y. Jiang, Z. Wu, X. Wang, A. He, J. Xu and J. Xu, J. Cleaner Prod., 2020, 276, 124219 CrossRef CAS.
- H. Chang, I. Bajaj, G. Huber, C. T. Maravelias and J. Dumesic, Green Chem., 2020, 22, 5285 RSC.
- A. Gandini, Green Chem., 2011, 13, 1061 RSC.
- R. De Clerq, M. Dusselier and B. F. Sels, Green Chem., 2017, 19, 5012 RSC.
- W. Fan, C. Verrier, Y. Queneau and F. Popowycz, Curr. Org. Synth., 2019, 4, 583 CrossRef PubMed.
- K. I. Galkin and V. P. Ananikov, ChemistryOpen, 2020, 9, 1135 CrossRef CAS PubMed.
- J. Mitra, X. Zhou and T. Rauchfuss, Green Chem., 2015, 17, 307 RSC.
- W. Deng, Q. Zhang and Y. Wang, Sci. China: Chem., 2015, 58, 29 CrossRef CAS.
- S. Kang, J. Fu and G. Zhang, Renewable Sustainable Energy Rev., 2018, 94, 340 CrossRef CAS.
- M. Mascal, ChemSusChem, 2015, 8, 3391 CrossRef CAS PubMed.
- H. Wang, T. Deng, Y. Wang, Y. Qi, X. Hou and Y. Zhu, Bioresour. Technol., 2013, 136, 394 CrossRef CAS PubMed.
- P. P. Upare, Y. K. Hwang and D. W. Hwang, Green Chem., 2018, 20, 879 RSC.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164 CrossRef CAS PubMed.
- C. Chatterjee, F. Pong and A. Sen, Green Chem., 2015, 17, 40 RSC.
- L. L. Adduci, T. A. Bender, J. A. Dabrowski and M. R. Gagné, Nat. Chem., 2015, 7, 576 CrossRef CAS PubMed; L. L. Adduci, T. A. Bender, J. A. Dabrowski and M. R. Gagné, Nat. Chem., 2015, 7, 759 CrossRef PubMed.
- R. L. De Souza, H. Yu, F. Rataboul and N. Essayem, Challenges, 2012, 3, 212 CrossRef.
- G. Yi, S. P. Teong, X. Li and Y. Zhang, ChemSusChem, 2014, 7, 2131 CrossRef CAS PubMed.
- K. I. Galkin and V. P. Ananikov, ChemSusChem, 2019, 12, 2976 CrossRef CAS PubMed.
- T. M. Aida, K. Tajima, M. Watanabe, Y. Saito, K. Kuroda, T. Nonaki, H. Hattori, R. L. Smith, Jr. and K. Arai, J. Supercrit. Fluids, 2007, 42, 110 CrossRef CAS.
- A. S. Amarasekara, L. T. D. Williams and C. C. Ebede, Carbohydr. Res., 2008, 343, 3021 CrossRef CAS PubMed.
- L.-K. Ren, L.-F. Zhu, T. Qi, J.-Q. Tang, H.-Q. Yang and C.-W. Hu, ACS Catal., 2017, 7, 2199 CrossRef CAS.
- M. Bicker, D. Kaiser, L. Ott and H. Vogel, J. Supercrit. Fluids, 2005, 36, 118 CrossRef CAS.
- A. Ranoux, K. Djanashvili, I. W. C. E. Arends and U. Hanefeld, ACS Catal., 2013, 3, 760 CrossRef CAS.
- G. S. Svenningsen, R. Kumar, C. E. Wyman and P. Christopher, ACS Catal., 2018, 8, 5591 CrossRef CAS.
- B. Saha and M. M. Abu-Omar, Green Chem., 2014, 16, 24 RSC.
- S. Siankevich, Z. Fei, R. Scopoletti, G. Laurenczy, S. Katsyuba, N. Yan and P. J. Dyson, ChemSusChem, 2014, 7, 1647 CrossRef CAS PubMed.
- S. Eminov, J. D. E. T. Wilton-Ely and J. P. Hallett, ACS Sustainable Chem. Eng., 2014, 2, 978 CrossRef CAS.
- R. Fang, A. Dhakshinamoorthy, Y. Li and H. Garcia, Chem. Soc. Rev., 2020, 49, 3638 RSC.
- V. G. Zuin, I. Eilks, M. Elschami and K. Kümmerer, Green Chem., 2021, 23, 1594 RSC.
- J. A. Haack and J. E. Hutchison, ACS Sustainable Chem. Eng., 2016, 4, 5889 CrossRef CAS.
- J. Esteban, A. J. Vorholt and W. Leitner, Green Chem., 2020, 22, 2097 RSC.
- T. W. Walker, A. K. Chew, R. C. Van Lehn, J. A. Dumesic and G. W. Huber, Top. Catal., 2020, 63, 649 CrossRef CAS.
- S. P. Simeonov, J. A. S. Coelho and C. A. M. Afonso, Org. Synth., 2016, 93, 29 CrossRef CAS.
- M. Knierbein, M. Voges and C. Held, Org. Process Res. Dev., 2020, 24, 1052 CrossRef CAS.
- S. L. Barbosa, M. de S. Freitas, W. T. P. dos Santos, D. L. Nelson, S. I. Klein, G. C. Clososki, F. J. Caires, A. C. M. Baroni and A. P. Wentz, Sci. Rep., 2021, 11, 1919 CrossRef CAS PubMed.
- A. T. Ubando, C. B. Felix and W.-H. Chen, Bioresour. Technol., 2020, 299, 122585 CrossRef CAS PubMed.
- S. D. Tilley, Adv. Energy Mater., 2019, 9, 1802877 CrossRef.
- J. B. Heo, Y.-S. Lee and C.-H. Chung, Biotechnol. Adv., 2019, 37, 107422 CrossRef CAS PubMed.
- C. Thoma, J. Konnerth, W. Sailer-Kronlachner, P. Solt, T. Rosenau and H. W. G. van Herwijnen, ChemSusChem, 2020, 13, 3544 CrossRef CAS PubMed.
- S. Krawielitzki, Chimia, 2020, 74, 776 CrossRef CAS PubMed.
- https://www.globenewswire.com/news-release/2019/04/12/1803565/0/en/2-5-Furandicarboxylic-Acid-FDCA-Market-Size-Worth-Around-850-Million-By-2025-Acumen-Research-and-Consulting.html .
- O. Gidron, A. Dadvand, Y. Sheynin, M. Bendikov and D. F. Perepichka, Chem. Commun., 2011, 47, 1976 RSC.
- A. Islam, Z. Liu, R. Peng, W. Jiang, T. Lei, W. Li, L. Zhang, R. Yang, G. Qian and Z. Ge, Chin. J. Polym. Sci., 2017, 35, 171 CrossRef CAS.
- L. I. Palmer and J. R. de Alaniz, Synlett, 2014, 25, 8 CAS.
- R. Chung, D. Yu, V. T. Thai, A. F. Jones, G. K. Veits, J. R. de Alaniz and J. E. Hein, ACS Catal., 2015, 5, 4579 CrossRef CAS.
- S. Sowmiah, L. F. Veiros, J. M. S. S. Esperanca, L. P. N. Rebelo and C. A. M. Afonso, Org. Lett., 2015, 17, 5244 CrossRef CAS PubMed.
- H. Quiroz-Florentino, R. I. Hernández-Benitez, J. A. Aviña, E. Burgueño-Tapia and J. Tamariz, Synthesis, 2011, 1106 CAS.
- V. Ilkei, K. Faragó, Z. Sánta, M. Dékány, L. Hazai, Jr., C. Szántay and G. Kalaus, Int. J. Org. Chem., 2014, 4, 309 CrossRef.
- M. Hellwig and T. Henle, Angew. Chem., Int. Ed., 2014, 53, 10316 CrossRef CAS PubMed.
- F. J. Tessier, Pathol. Biol., 2010, 58, 214 CrossRef CAS PubMed.
- L. A. Peterson, Chem. Res. Toxicol., 2013, 18, 6 Search PubMed.
- M. Banerjee, H. K. S. Kumar and M. Banerjee, Int. J. Res. Phytochem. Pharmacol., 2015, 5, 48 Search PubMed.
- E. Oder, M. K. Safo, O. Abdulmalik and G. J. Kato, Br. J. Hematol., 2016, 175, 24 CrossRef CAS PubMed.
- G. G. Xu, P. P. Pagare, M. S. Ghatge, R. P. Safo, A. Gazi, Q. Chen, T. David, A. B. Alabbas, F. N. Musayev, J. Venitz, Y. Zhang, M. K. Safo and O. Abdulmalik, Mol. Pharmaceutics, 2017, 14, 3499 CrossRef CAS PubMed.
- H. Sugimura, M. Kikuchi, S. Kato, W. Sekita and I. Sasaki, Tetrahedron, 2016, 72, 7638 CrossRef CAS.
- Q. Hou, X. Qi, M. Zhen, H. Qian, Y. Nie, C. Bai, S. Zhang, X. Bai and M. Ju, Green Chem., 2021, 23, 119 RSC.
- A. Gandini, T. M. Lacerda, A. J. Carvalho and E. Trovatti, Chem. Rev., 2016, 116, 1637 CrossRef CAS PubMed.
- G. Z. Papageorgiu, D. G. Papageorgiu, Z. Terzopoulou and D. N. Bikiaris, Eur. Polym. J., 2016, 83, 202 CrossRef.
- S. E. Davis, B. N. Zope and R. J. Davis, Green Chem., 2012, 14, 143 RSC.
- D. Zhang and M.-J. Dumont, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1478 CrossRef CAS.
- A. A. Ghatta, J. D. E. T. Wilton-Ely and J. P. Hallet, Green Chem., 2021, 23, 1716 RSC.
- G. Dedes, A. Karnaouri and E. Topakas, Catalysts, 2020, 10, 743 CrossRef CAS.
- M. Sajid, X. Zhao and D. Liu, Green Chem., 2018, 20, 5427 RSC.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G.-J. M. Gruter, J. F. J. Coelho and A. D. J. Silvestre, Polym. Chem., 2015, 6, 5961 RSC.
- R. Fittig and H. Heizelmann, Chem. Ber., 1876, 9, 1198 Search PubMed.
- S. Thiyagarajan, A. Pukin, J. van Haveren, M. Lutz and D. S. van Es, RSC Adv., 2013, 3, 15678 RSC.
- K. Kohli, R. Prajapati and B. K. Sharma, Energies, 2019, 12, 233 CrossRef CAS.
- X. Fei, J. Wang, J. Zhu, X. Wang and X. Liu, ACS Sustainable Chem. Eng., 2020, 8, 8471 CrossRef CAS.
- A. Banerjee, G. R. Dick, T. Yoshino and M. W. Kanan, Nature, 2016, 531, 215 CrossRef CAS PubMed.
- G. R. Dick, A. D. Frankhouser, A. Banerjee and M. W. Kanan, Green Chem., 2017, 19, 2966 RSC.
- Y. Shao, X. Hu, Z. Zhang, K. Sun, G. Gao, T. Wei, S. Zhang, S. Hu, J. Xing and Y. Wang, Green Energy Environ., 2019, 4, 400 CrossRef.
- J. Pang, M. Zheng, R. Sun, A. Wang, X. Wang and T. Zhang, Green Chem., 2016, 18, 342 RSC.
- X. Zuo, P. Venkitasubramanian, D. H. Busch and B. Subramaniam, ACS Sustainable Chem. Eng., 2016, 4, 3659 CrossRef CAS.
- H. Ban, Y. Zhang, S. Chen, Y. Cheng, T. Pan, L. Wang and X. Li, ACS Sustainable Chem. Eng., 2020, 8, 8011 CrossRef CAS.
- J. Zhang, Y. Liu, Z. Qi, L. He and L. Peng, BioResources, 2020, 15, 4502 Search PubMed.
- W. Partenheimer and V. V. Grushin, Adv. Synth. Catal., 2001, 343, 102 CrossRef CAS.
- S. Chen, Y. Cheng, H. Ban, Y. Zhang, L. Zheng, L. Wang and X. Li, Ind. Eng. Chem. Res., 2020, 8, 8011 Search PubMed.
- P. Sudarsanam, E. Peeters, E. V. Makshina, V. I. Parvulescu and B. F. Sels, Chem. Soc. Rev., 2019, 48, 2366 RSC.
- Y. Yamamoto, M. Ota, S. Kodama, K. Michimoto, A. Nomoto, A. Ogawa, M. Furuya and K. Kawakami, ACS Omega, 2021, 6, 2239 CrossRef CAS PubMed.
- D. J. Chadderdon, L. Xin, J. Qi, Y. Qiu, P. Krishna, K. L. More and W. Li, Green Chem., 2014, 16, 3778 RSC.
- J. Weidner, S. Barwe, K. Sliozberg, S. Piontek, J. Masa, U.-P. Apfel and W. Schuhmann, Beilstein J. Org. Chem., 2018, 14, 1436 CrossRef PubMed.
- K. K. Meier, S. M. Jones, T. Kaper, H. Hansson, M. J. Koetsier, S. Karkehabadi, E. I. Solomon, M. Sandgren and B. Kelemen, Chem. Rev., 2018, 118, 2593 CrossRef CAS PubMed.
- H. Guo, Y. Chang and D.-J. Lee, Bioresour. Technol., 2018, 252, 198 CrossRef CAS PubMed.
- H. Østby, L. D. Hansen, S. J. Horn, V. G. H. Eijsin and A. Várnai, J. Ind. Microbiol. Biotechnol., 2020, 47, 623 CrossRef PubMed.
- A. Dadwal, S. Sharma and T. Satyanarayana, Front. Microbiol., 2020, 11, 1387 CrossRef PubMed.
- Z. Zhou, X. Ju, J. Chen, R. Wang, Y. Zhong and L. Li, Bioresour. Technol., 2021, 319, 124159 CrossRef CAS PubMed.
- J.-J. Dong, E. Fernandez-Fueyo, F. Hollmann, C. E. Paul, M. Pesic, S. Schmidt, S. Younes and W. Zhang, Angew. Chem., Int. Ed., 2018, 57, 9238 CrossRef CAS PubMed.
- S. C. Hammer, A. M. Knight and F. H. Arnold, Curr. Opin. Green Sustain. Chem., 2017, 7, 23 CrossRef.
- F. Koopman, N. Wierckx, J. H. de Winde and H. J. Ruijssenaars, Bioresour. Technol., 2010, 101, 6291 CrossRef CAS PubMed.
- W. P. Dijkman, D. E. Groothuis and M. W. Fraaije, Angew. Chem., Int. Ed., 2014, 53, 6515 CrossRef CAS PubMed.
- W. P. Dijkman, C. Binda, M. W. Fraaije and A. Mattevi, ACS Catal., 2015, 5, 1833 CrossRef CAS.
- C.-T. Hsu, Y.-C. Kuo, Y.-C. Liu and S.-L. Tsai, Microbiol. Biotechnol., 2020, 13, 1094 CrossRef CAS PubMed.
- C.-F. Yang and C.-R. Huang, Bioresour. Technol., 2016, 2014, 311 CrossRef PubMed.
- C.-F. Yang and C.-R. Huang, J. Biosci. Bioeng., 2018, 125, 407 CrossRef CAS PubMed.
- S. M. McKenna, S. Leimkuehler, S. Herter, N. J. Turner and A. J. Carnell, Green Chem., 2015, 17, 3271 RSC.
- X. Wang, ACS Sustainable Chem. Eng., 2020, 8, 4341 CrossRef CAS.
- K. A. P. Payne, S. A. Marshall, K. Fisher, M. J. Cliff, D. M. Cannas, C. Yan, D. J. Heyes, D. A. Parker, I. Larrosa and D. Leys, ACS Catal., 2019, 9, 2854 CrossRef CAS PubMed.
- A. D. K. Deshan, L. Atanda, L. Moghaddam, D. W. Rackemann, J. Beltramini and W. O. S. Doherty, Front. Chem., 2020, 8, 659 CrossRef CAS PubMed.
- X.-n. Ye, Q. Lu, X. Wang, H.-q. Guo, M.-s. Cui, C.-q. Dong and Y.-p. Yang, ACS Sustainable Chem. Eng., 2017, 5, 10815 CrossRef CAS.
- K. Tomishige, Y. Nakagawa and M. Tamura, Green Chem., 2017, 19, 2876 RSC.
- K. Tomishige, Y. Nakagawa and M. Tamura, Curr. Opin. Green Sustain. Chem., 2020, 22, 13 CrossRef.
- M. Tamura, Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2020, 10, 3805 RSC.
- K. Tomishige, Y. Nakagawa and M. Tamura, Chin. Chem. Lett., 2020, 31, 1071 CrossRef CAS.
- G. K. Cook and M. A. Andrews, J. Am. Chem. Soc., 1996, 118, 9448 CrossRef CAS.
- M. Shiramizu and D. Toste, Angew. Chem., Int. Ed., 2012, 51, 8082 CrossRef CAS PubMed.
- L. L. Adduci, T. A. Bender, J. A. Dabrowski and M. R. Gagne, Nat. Chem., 2015, 7, 576 CrossRef CAS PubMed.
- A. K. Misra and G. Agnihotri, Carbohydr. Res., 2004, 339, 1381 CrossRef CAS PubMed.
- A. Dutta, D. Dhara, P. K. Parida, A. Si, R. Yesuvadian, K. Jana and A. K. Misra, RSC Adv., 2017, 7, 28853 RSC.
- L. Molina, A. J. Moreno-Vargas, A. T. Carmona and I. Robina, Synlett, 2006, 2006, 1327 CrossRef.
- S. Sato, Y. Naito and K. Aoki, Carbohydr. Res., 2007, 342, 913 CrossRef CAS PubMed.
- J. S. Yadav, B. V. S. Reddy, M. Sreenivas and G. Satheesh, Synthesis, 2007, 1712 CrossRef CAS.
- G. Bartoli, J. G. Fernández-Bolaños, G. Di Antonio, G. Foglia, S. Giuli, R. Gunnella, M. Mancinelli, E. Marcantoni and M. Paoletti, J. Org. Chem., 2007, 72, 6029 CrossRef CAS PubMed.
- L. Nagarapu, M. V. Chary, A. Satyender, B. Supriya and R. Bantu, Synthesis, 2009, 2278 CrossRef CAS.
- V. Escante, T. K. Olszewski, E. Petit and C. Grison, ChemSusChem, 2014, 7, 1915 CrossRef PubMed.
- A. D. Sutton, J. K. Kim, R. Wu, C. B. Hoyt, D. B. Kimball, L. A. Silks III and J. C. Gordon, ChemSusChem, 2016, 9, 2298 CrossRef CAS PubMed.
- N. Ronaghi, D. M. Fialho, C. W. Jones and S. France, J. Org. Chem., 2020, 85, 15337 CrossRef CAS PubMed.
- M. R. Lambu and Z. M. A. Judeh, Green Chem., 2019, 21, 821 RSC.
- J. Liu, F. Li, X. Zheng, J. Su, Y. Yu, L. Wang and H. Zhuang, Process Biochem., 2021, 101, 99 CrossRef CAS.
- A. Boto, D. Hernández and R. Hernández, Org. Lett., 2007, 9, 2021 CrossRef PubMed.
- F. W. Lichtenthaler, D. Martin, T. Weber and H. Schiweck, Liebigs Ann. Chem., 1993, 1993, 967 CrossRef.
- F. W. Lichtenthaler, Acc. Chem. Res., 2002, 35, 728 CrossRef CAS PubMed.
- D. Martin and F. W. Lichtenthaler, Tetrahedron: Asymmetry, 2006, 17, 756 CrossRef CAS.
- C. Fayet and J. Gelas, Heterocycles, 1983, 20, 1563 CrossRef CAS.
- F. Gonzalez, S. Lesage and A. S. Perlin, Carbohydr. Res., 1975, 42, 267 CrossRef CAS.
- F. M. Hauser, S. R. Ellenberger and W. P. Ellenberger, Tetrahedron Lett., 1988, 29, 4939 CrossRef CAS.
- M. Saeed, T. Ilg, M. Schick, M. Abbas and W. Voelter, Tetrahedron Lett., 2001, 42, 7401 CrossRef CAS.
- E. N. Tzanetou, K. M. Kasiotis, P. Magiatis and S. A. Haroutounian, Molecules, 2007, 12, 735 CrossRef CAS PubMed.
- M. Hayashi, H. Kawabata and K. Yamada, J. Chem. Soc., Chem. Commun., 1999, 1999, 965 RSC.
- B. S. Babu and K. K. Balasubramanian, J. Org. Chem., 2000, 65, 4198 CrossRef CAS PubMed.
- S. B. Boga and K. K. Balasubramanian, ARKIVOC, 2004,(viii), 87 Search PubMed.
- J. L. Babu, A. Khare and Y. D. Vankar, Molecules, 2005, 10, 884 Search PubMed.
- J. S. Yadav, B. V. S. Reddy and A. V. Madhavi, J. Mol. Catal. A: Chem., 2005, 226, 213 CrossRef CAS.
- A. Agarwal, S. Rani and Y. D. Vankar, J. Org. Chem., 2004, 69, 6137 CrossRef CAS PubMed.
- M. Teijeira, Y. Fall, F. Santamarta and E. Tojo, Tetrahedron Lett., 2007, 48, 7926 CrossRef CAS.
- M. Saquib, I. Husain, B. Kumar and A. K. Shaw, Chem. – Eur. J., 2009, 15, 6041 CrossRef CAS PubMed.
- A. N. Butkevich, L. Meerpoel, I. Stansfield, P. Angibaud, A. Corbu and J. Cossy, Org. Lett., 2013, 15, 3840 CrossRef CAS PubMed.
- N. Hussain, M. Bhardwaj, A. Ahmed and D. Mukherjee, Org. Lett., 2019, 21, 3034 CrossRef CAS PubMed.
- K. Mal, A. Sharma and I. Das, Chem. – Eur. J., 2014, 20, 11932 CrossRef CAS PubMed.
- K. Mal, A. Kaur and I. Das, Asian J. Org. Chem., 2015, 4, 1132 CrossRef CAS.
- K. Mal and I. Das, J. Org. Chem., 2016, 81, 932 CrossRef CAS PubMed.
- V. Luque-Agudo, M. V. Gil, E. Román and J. A. Serrano, Green Chem., 2016, 18, 3844 RSC.
- J. Li, M. Lutz, M. Otte and R. J. M. Klein Gebbink, ChemCatChem, 2018, 10, 4755 CrossRef CAS PubMed.
- J. Arekion, M. Delmas and A. Gasset, Biomass, 1983, 3, 59 CrossRef CAS.
- D. Balachari, N. Quinn and A. O'Doherty, Tetrahedron Lett., 1999, 40, 4769 CrossRef CAS.
- A. Mehner, A. L. Montero, R. Martinez and S. Spange, Molecules, 2007, 12, 634 CrossRef CAS PubMed.
- Z. Jian, L. Falivene, G. Boffa, S. O. Sánchez, L. Caporaso, A. Grassi and S. Mecking, Angew. Chem., Int. Ed., 2016, 55, 14378 CrossRef CAS PubMed.
- S. O. Sánchez, F. Marra, A. Dibenedetto, M. Aresta and A. Grassi, Macromolecules, 2014, 47, 7129 CrossRef.
- R. Skowronski, G. Grabowski, J. Lewkowski, G. Descotes, L. Cottier and C. Neyret, Org. Prep. Proced. Int., 1993, 25, 353 CrossRef CAS.
- G. Li, W. Jiao, Z. Sun, Y. Zhao, Z. Shi, Y. Yan, L. Feng, Y. Zhang and Y. Tang, ACS Sustainable Chem. Eng., 2018, 6, 4316 CrossRef CAS.
- S. Prodinger, M. F. K. Verstreken and R. F. Lobo, ACS Sustainable Chem. Eng., 2020, 8, 11930 CrossRef CAS.
- T. Taniguchi, H. Ohnishi and K. Ogasawara, Chem. Commun., 1996, 1996, 1477 RSC.
- L.-X. Liao, Z.-M. Wang, H.-X. Zhang and W.-S. Zhou, Tetrahedron: Asymmetry, 1999, 10, 3649 CrossRef CAS.
- M. A. Ciufolini, C. Y. W. Hermann, Q. Dong, T. Shimizu, S. Swaminathan and N. Xi, Synlett, 1998, 105 CrossRef CAS.
- M. H. Haukaas and G. A. O'Doherty, Org. Lett., 2001, 3, 401 CrossRef CAS PubMed.
- T. Ohkuma, M. Koizumi, M. Yoshida and R. Noyori, Org. Lett., 2000, 2, 1749 CrossRef CAS PubMed.
- R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40 CrossRef CAS PubMed.
- D. G. Drueckhammer, C. F. Barbas III, K. Nozaki, C.-H. Wong, C. Y. Wood and M. A. Ciufolini, J. Org. Chem., 1988, 53, 1607 CrossRef CAS.
- D. Thiel, D. Doknić and J. Deska, Nat. Commun., 2014, 5, 5278 CrossRef CAS PubMed.
- F. Blume, Y.-C. Liu, D. Thiel and J. Deska, J. Mol. Catal. B: Enzym., 2016, 134, 280 CrossRef CAS.
- P.-F. Koh and T.-P. Loh, Green Chem., 2015, 17, 3746 RSC.
- P. Kwiatkowski, J. Majer, W. Chaładaj and J. Jurczak, Org. Lett., 2006, 8, 5045 CrossRef CAS PubMed.
- J. Majer, P. Kwiatkowski and J. Jurczak, Org. Lett., 2008, 10, 2955 CrossRef CAS PubMed.
- S. Yamazaki, S. Kashima, T. Kuriyama, Y. Iwata, T. Morimoto and K. Kakiuchi, Tetrahedron: Asymmetry, 2009, 20, 1224 CrossRef CAS.
- D. Uraguchi, K. Sorimachi and M. Terada, J. Am. Chem. Soc., 2004, 126, 11804 CrossRef CAS PubMed.
- P. Schäfer, T. Palacin, M. Sidera and S. P. Fletcher, Nat. Commun., 2017, 8, 15762 CrossRef PubMed; P. Schäfer, T. Palacin, M. Sidera and S. P. Fletcher, Nat. Commun., 2018, 9, 16216 CrossRef PubMed.
- S. F. Martin, C. Gluchowski, C. L. Campbell and R. C. Chapman, J. Org. Chem., 1984, 49, 2512 CrossRef CAS.
- P. A. Wender, K. D. Rice and M. E. Schnute, J. Am. Chem. Soc., 1997, 119, 7897 CrossRef CAS.
- J. Krüger and E. M. Carreira, J. Am. Chem. Soc., 1998, 120, 837 CrossRef.
- M. Garcia-Valverde, J. Nieto, R. Pedrosa and M. Vicente, Tetrahedron, 1999, 55, 2755 CrossRef CAS.
- A. E. Aydin and S. Yuksekdanaci, Tetrahedron: Asymmetry, 2013, 24, 14 CrossRef.
- Z. J. Witczak, R. Bielski and D. M. Mencer, Tetrahedron Lett., 2017, 58, 4069 CrossRef CAS.
- A. Kabro, E. C. Escudero-Adán, V. V. Grushin and P. V. N. M. van Leeuven, Org. Lett., 2012, 14, 4014 CrossRef CAS PubMed.
- Y.-L. Su, Z.-Y. Han, Y.-H. Li and L.-Z. Gong, ACS Catal., 2017, 7, 7917 CrossRef CAS.
- D. E. Q. Jimenez, J. C. Barreiro, F. M. dos Santos Jr., S. P. de Vasconcellos, A. L. M. Porto and J. M. Batista Jr., Chirality, 2019, 31, 534 CrossRef CAS PubMed.
- A. Dunbabin, F. Subrizi, J. M. Ward, T. D. Sheppard and H. C. Hailes, Green Chem., 2017, 19, 397 RSC.
- P.-F. Koh, P. Wang, J.-M. Huang and T.-P. Loh, Chem. Commun., 2014, 50, 8324 RSC.
- N. Tokunaga, Y. Otomaru, K. Okamoto, K. Ueyama, R. Shintani and T. Hayashi, J. Am. Chem. Soc., 2004, 126, 13584 CrossRef CAS PubMed.
- G. Grynkiewicz and J. N. BeMiller, Carbohydr. Res., 1982, 108, C1 CrossRef CAS.
- G. Grynkiewicz and A. Zamojski, Z. Naturforsch., 1980, 35b, 1024 CrossRef CAS.
- M. Sollogoub, A. J. Pearce, A. Hérault and P. Sinaÿ, Tetrahedron: Asymmetry, 2000, 11, 283 CrossRef CAS.
- Z. Ding, X. Luo, Y. Ma, H. Chen, S. Qiu, G. Sun, W. Zhang, C. Yu, Z. Wu and J. Zhang, J. Carbohydr. Chem., 2018, 37, 81 CrossRef CAS.
- A. Astarita, F. Cermola, M. R. Iesce and L. Previtera, Tetrahedron, 2008, 64, 6744 CrossRef CAS.
- S. Jarosz, M. Mach, K. Szewczyk, S. Skóra and Z. Ciunik, Eur. J. Org. Chem., 2001, 2955 CrossRef CAS.
- L. Xu, H. Chen, J. Liu, L. Zhou, Q. Liu, Y. Lan and J. Xiao, Org. Chem. Front., 2019, 6, 1162 RSC.
- J. L. Marco, Tetrahedron, 1989, 45, 1475 CrossRef CAS.
- L. C. Bencze, C. Paizs, M. I. Tosa and F. D. Irimie, Tetrahedron: Asymmetry, 2011, 22, 675 CrossRef CAS.
- M. Opietnik, A. Jungbauer, K. Mereiter and T. Rosenau, Curr. Org. Chem., 2012, 16, 2739 CrossRef CAS.
- D. S. Park, K. E. Joseph, M. Koehle, C. Krumm, L. Ren, J. N. Damen, M. H. Shete, H. S. Lee, X. Zuo, B. Lee, W. Fan, D. G. Vlachos, R. F. Lobo, M. Tsapatsis and P. J. Dauenhauer, ACS Cent. Sci., 2016, 2, 820 CrossRef CAS PubMed.
- A. Vasseur, J. Muzart and J. Le Bras, Chem. – Eur. J., 2011, 17, 12556 CrossRef CAS PubMed.
- L. Chen, C. Bruneau, P. Dixneuf and H. Doucet, Tetrahedron, 2013, 69, 4381 CrossRef CAS.
- S. Gao, Z. Wu, F. Wu, A. Lin and H. Yao, Adv. Synth. Catal., 2016, 358, 4129 CrossRef CAS.
- J. Le Bras and J. Muzart, Catalysts, 2020, 10, 571 CrossRef CAS.
- A. A. Zen, J. W. Aylott and W. C. Chan, Tetrahedron Lett., 2014, 55, 5521 CrossRef.
- D. Bhattacherjee, M. Rahman, S. Ghosh, A. K. Bagdi, G. V. Zyryanov, O. N. Chupakhin, P. Das and A. Hajrab, Adv. Synth. Catal., 2021, 363, 1597 CrossRef CAS.
- J. V. Obligacion, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 4133 CrossRef CAS PubMed.
- M.-A. Légaré, M.-A. Courtemanche, É. Rochette and F.-G. Fontaine, Science, 2015, 349, 513 CrossRef PubMed.
- L. Britton, J. H. Docherty, A. P. Dominey and S. P. Thomas, Molecules, 2020, 25, 90 CrossRef PubMed.
- J. Cueto, L. Faba, E. Díaz and S. Ordóñez, ChemCatChem, 2017, 9, 1765 CrossRef CAS.
- L. Wang and E. Y.-X. Chen, ACS Catal., 2015, 5, 6907 CrossRef CAS.
- M. Y. Chernyak, V. E. Tarabanko, A. A. Morosov and A. A. Kondrasenko, J. Siberian Federal Univ., Chem. 2, 2016, 9, 146 CrossRef.
- K. S. Arias Carrascal, A. Garcia-Ortiz, M. J. Climent Olmedo, A. Corma Canós and S. Iborra Chornet, ACS Sustainable Chem. Eng., 2018, 6, 4239 CrossRef.
- Y. Du, Y.-L. Feng, C.-L. Zou, X. Wu and W. Huang, Prog. React. Kinet. Mech., 2018, 43, 21 CrossRef CAS.
- M. Phukan, K. J. Borah and R. Borah, Green Chem. Lett. Rev., 2009, 2, 249 CrossRef CAS.
- A. Cukalovic and C. V. Stevens, Green Chem., 2010, 12, 1201 RSC.
- W. Fan, Y. Queneau and F. Popowycz, RSC Adv., 2018, 8, 31496 RSC.
- X. Wang, W. Chen, Z. Li, X. Zeng, X. Tang, Y. Sun, T. Lei and L. Lin, J. Energy Chem., 2018, 27, 209 CrossRef.
- J. Mitra, X. Zhou and T. Rauchfuss, Green Chem., 2015, 17, 307 RSC.
- V. Singh and V. Singh, Synth. Commun., 2010, 40, 1280 CrossRef CAS.
- R. Rajmohan, S. Gayathri and P. Vairaprakash, RSC Adv., 2015, 5, 10040 RSC.
- A. Sengupta, T. Dey, M. Ghosh, J. Ghosh and S. Ghosh, J. Inst. Eng. (India): Ser. E, 2012, 93, 31 Search PubMed.
- D. R. Chaffey, C. Alamillo-Ferrer, T. E. Davies, S. H. Taylor, N. C. O. Tomkinson and A. E. Graham, ACS Omega, 2019, 4, 15985 CrossRef CAS PubMed.
- J. Luo, J. Yu, R. J. Gorte, E. Mahmoud, D. G. Vlachos and M. A. Smith, Catal. Sci. Technol., 2014, 4, 3074 RSC.
- J. J. Pacheco and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8363 CrossRef CAS PubMed.
- I. Scodeller, K. De Oliveira Vigier, E. Muller, C. Ma, F. Guégan, R. Wischert and F. Jérôme, ChemSusChem, 2021, 14, 313 CrossRef PubMed.
- V. T. Abaev, I. V. Trushkov and M. G. Uchuskin, Chem. Heterocycl. Compd., 2016, 52, 973 CrossRef CAS.
- D. Fisher, L. I. Palmer, J. E. Cook, J. E. Davis and J. R. de Alaniz, Tetrahedron, 2014, 70, 4105 CrossRef CAS.
- D. Lebœuf, E. Schulz and V. Gandon, Org. Lett., 2014, 16, 6464 CrossRef PubMed.
- C. Verrier, S. Moebs-Sanchez, Y. Queneau and F. Popowycz, Org. Biomol. Chem., 2018, 16, 676 RSC.
- D. Kalaitzakis, M. Sofiadis, V. Tsopanakis, T. Montagnon and G. Vassilikogiannakis, Org. Biomol. Chem., 2020, 18, 2817 RSC.
- Y. Lou, S. Marinkovic, B. Estrine, W. Qiang and G. Enderlin, ACS Omega, 2020, 5, 2561 CrossRef CAS PubMed.
- Y.-C. Liu, Z.-L. Wu and J. Deska, ChemRxiv, 2021, 5, 11.
- C. D. Fu, Y. He and J. Pfaendtner, J. Phys. Chem. A, 2019, 123, 3080 CrossRef CAS PubMed.
- V. Vaissier Welborn, L. R. Pestana and T. Head-Gordon, Nat. Catal., 2018, 1, 649 CrossRef.
- S. Shaik, D. Mandal and R. Ramanan, Nat. Chem., 2016, 8, 1091 CrossRef CAS PubMed.
- R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2 CrossRef CAS.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796 CrossRef CAS PubMed.
- F. H. Arnold, Angew. Chem., Int. Ed., 2019, 58, 14420 CrossRef CAS PubMed.
- H. Kim, S. Lee, Y. Ahn, J. Lee and W. Won, ACS Sustainable Chem. Eng., 2020, 8, 12419 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |