
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regioselective and water-assisted surface esterification of never-dried cellulose: nanofibers with adjustable surface energy†
Marco
Beaumont‡
 *ab,
Caio G.
Otoni‡
c,
Bruno D.
Mattos‡
b,
Tetyana V.
Koso
d,
Roozbeh
Abidnejad
b,
Bin
Zhao
b,
Anett
Kondor
e,
Alistair W. T.
King
d and
Orlando J.
Rojas
*bf
*ab,
Caio G.
Otoni‡
c,
Bruno D.
Mattos‡
b,
Tetyana V.
Koso
d,
Roozbeh
Abidnejad
b,
Bin
Zhao
b,
Anett
Kondor
e,
Alistair W. T.
King
d and
Orlando J.
Rojas
*bf
aDepartment of Chemistry, Institute of Chemistry for Renewable Resources, University of Natural Resources and Life Sciences Vienna (BOKU), Konrad-Lorenz-Straße 24, A-3430 Tulln, Austria
bDepartment of Bioproducts and Biosystems, School of Chemical Engineering, Aalto University, P.O. Box 16300, Espoo FI-00076, Finland. E-mail: orlando.rojas@ubc.ca; marcobeaumont1@gmail.com
cDepartment of Materials Engineering (DEMa), Federal University of São Carlos (UFSCar), Rod. Washington Luís, km 235, São Carlos, SP 13565-905, Brazil
dMaterials Chemistry Division, Department of Chemistry, University of Helsinki, AI Virtasen aukio 1, FI-00560 Helsinki, Finland
eSurface Measurement Systems Ltd., Rosemont Rd, Wembley, London HA0 4PE, UK
fDepartments of Chemical & Biological Engineering, 2360 East Mall; Chemistry, 2036 Main Mall, and Wood Science, 2424 Main Mall, The University of British Columbia, Vancouver, BC V6T 1Z3, Canada
First published on 2nd August 2021
Abstract
A new regioselective route is introduced for surface modification of biological colloids in the presence of water. Taking the case of cellulose nanofibers (CNFs), we demonstrate a site-specific (93% selective) reaction between the primary surface hydroxyl groups (C6-OH) of cellulose and acyl imidazoles. CNFs bearing C6-acetyl and C6-isobutyryl groups, with a degree of substitution of up to 1 mmol g−1 are obtained upon surface esterification, affording CNFs of adjustable surface energy. The morphological and structural features of the nanofibers remain largely unaffected, but the regioselective surface reactions enable tailoring of their interfacial interactions, as demonstrated in oil/water Pickering emulsions. Our method precludes the need for drying or exchange with organic solvents for surface esterification, otherwise needed in the synthesis of esterified colloids and polysaccharides. Moreover, the method is well suited for application at high-solid content, opening the possibility for implementation in reactive extrusion and compounding. The proposed acylation is introduced as a sustainable approach that benefits from the presence of water and affords a high chemical substitution selectivity.
Introduction
The run towards renewable nanomaterials is increasing in pace, and efforts are aimed at the isolation and modification of nanoparticles derived from structured biomass. Biological colloids, especially cellulose nanofibers (CNFs), are promising examples in this regard given the access to a wide variety of sources (e.g., forestry and agricultural side-streams). In contrast to nanofibers from synthetic polymers, the cellulosic counterparts are obtained according to a top-down approach. The latter takes advantage of the hierarchical fibrous structure of plants with primary structural building blocks formed by the cellulose (nano)fibers. These entities offer unique intrinsic properties and can be assembled into materials1 with outstanding mechanical properties2–4 and specific surface area (SSA).5,6 The 2,2,6,6-tetramethylpiperidine-1-oxyl radical (TEMPO)-mediated oxidation has been introduced to install carboxylate groups onto cellulose in a regioselective manner.7,8 This reaction has enabled effortless fibrillation of fibers into individual TEMPO-oxidized CNFs. Unfortunately, several challenges still remain for a wide adoption of TEMPO oxidation, including costs, toxicity, and the chemical degradation that can occur during the oxidation, resulting in a functional cellulose with a significantly lower molar mass than the starting material.9 Such effects ultimately decrease the mechanical performance of cellulosic materials,10 setting a limit for TEMPO-mediated oxidation and, generally, a restriction of the introduced functionalities to hydrophilic moieties.Chemically modified, hydrophobic CNFs are usually prepared by esterification to aliphatic esters preferably confined to the CNF surface. Related processes are in high demand given the need to endow CNFs with water resistance.11,12 So far, these modifications have been conducted either with dried cellulosic materials12–14 or in organic solvents, after solvent exchange.11 The required extensive water removal from cellulose in general leads to near irreversible formation of inter-fibrillar bonds, significant reduction of SSA and aspect ratio and reactivity – a phenomenon termed as “hornification”.15,16 This effect ultimately reduces dispersibility, water retention capacity,15 and the quality and performance of the obtained nanofibers in application.17,18 Moreover, full removal of water from cellulose is an enormous challenge, given the high hygroscopicity of cellulose and its strong interactions with water.19 As an alternative, solvent exchange of water-swollen fibers with an organic solvent avoids drying of cellulose but requires large solvent volumes and time- and energy consuming processing steps.11
While a plethora of chemical modification routes have been reported for (nano)celluloses, surface modifications are distinctive given that they do not affect the bulk of the material, i.e., the crystallinity of the sample is preserved, and its molar mass is not reduced. This is not the case for the frequently used acid-catalyzed modifications with acetic anhydride in toluene/acetic acid to prepare acetylated nanocelluloses,11,20 as well as similar approaches that are based on the industrial heterogeneous acid-catalyzed acetylation.21 In addition, surface esterification of dried cellulose and subsequent fibrillation into CNFs requires considerable energy, e.g., extensive ball-milling coupled with ultra-sonication.12,14 This also leads to a reduction in the cellulose molecular weight and crystallinity,12,14 also compromising its polymeric chain integrity.22
The surface-confined esterifications herein take place only at the accessible hydroxyl groups of the fibers, and do not influence the hierarchical structure of native cellulose and its crystalline domains. These form the basis of its inherently astonishing mechanical properties.23,24 Taking in consideration the challenges of surface modification, it is clear that the current methods to obtain, for example, surface-acetylated cellulose, are still far from meeting the need for a straightforward and sustainable process. In terms of sustainability, the main obstacles of the current methods relate to the high energy demand to completely remove water from cellulose, and the need for large volumes of organic solvents used in the solvent exchange of water. In addition, most esterification approaches are based on hazardous and non-sustainable solvents, mostly toluene,25 and require considerable energy input during the reaction, as heating11,20 or milling.12,14
We recently introduced a method for surface esterification of CNFs that tolerates water and occurs under mild conditions.16 This approach is based on the use of N-acetylimidazole as acetylation agent, which has also been shown to enable regioselective modification of dried cellulose fibers.26 However, this recent method lacks versatility since it only enables introduction of acetyl groups and it has not been demonstrated so far that it allows also the regioselective esterification of never-dried cellulose fibers, which is preferable to produce high-performance CNFs. Herein, we introduce a facile and versatile approach for the regioselective introduction of hydrophobic ester moieties onto the primary surface hydroxyl groups (C6-OH) of cellulose (Fig. 1a–d). The modification is sustainable and tackles most issues of current esterification methods, as no drying of cellulose or solvent-exchanges are necessary. In addition, the reaction is energy-efficient proceeding at room temperature; and uses an acetone/water mixture as solvent, which has less hazard and environmental impact than traditionally used organic solvents.27 We introduced successfully isobutyryl and acetyl moieties and, in an effort to fully describe the role of the substituent group, we extensively characterized the native and modified CNFs, for surface energy and interactions with water, highlighting the interfacial activity using model multiphase systems.
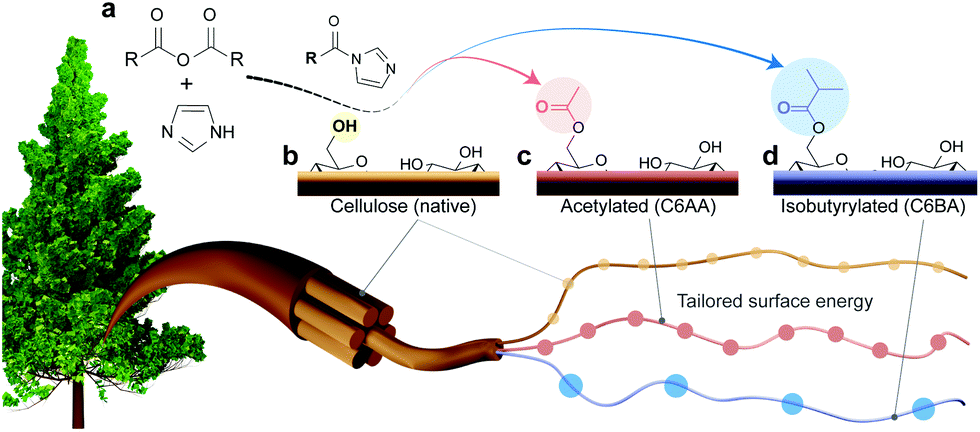 | ||
| Fig. 1 (a) C6-Regioselective esterification of cellulose in the presence of water. (b) Cellulose with imidazole-catalyzed reaction using (c) acetic or (d) isobutyric anhydrides to yield cellulose nanofibers with tailored surface energy. | ||
Results and discussion
We introduce a method that enables direct esterification of never-dried cellulose through reaction of N-acylimidazoles, formed in situ, with the accessible hydroxyl groups at the surface of cellulose. This route is offered as an alternative to those that require either (a) removal of water from wet cellulose pulp, e.g. through spray/evaporative/freeze-drying, which can impair the mechanical performance of the produced nanocelluloses,17,18 or (b) solvent exchange, which in turn requires a high volume of organic solvents and centrifugation steps. The present approach expands our previous wet acetylation method that used a solution of N-acetylimidazole in DMSO.16 We replaced DMSO, an environmentally problematic solvent, with a greener alternative, acetone.25 This brought also the advantage during work-up, namely, the lower fiber swelling in acetone/water enabled more efficient washing for solvent removal as in case of prior DMSO/water system.28 Herein, most notably, and compared to previous efforts, we demonstrate that the wet esterification approach can be extended to different N-acylimidazoles, which are produced in situ through reaction of carboxylic acid anhydrides with imidazole (Fig. 1a).Never-dried cellulose fibers (50 wt% solid content and 50 wt% water) were mixed with an imidazole solution in acetone before addition of either acetic anhydride or isobutyric anhydride to induce the formation of the respective N-acylimidazole. We chose the branched isobutyryl group to introduce higher hydrophobicity than the acetyl one, while exhibiting high colloidal stability in aqueous dispersions. The latter was not the case for longer alkyl chains, such as hexanoyl group (data not shown). The wet esterification proceeded spontaneously in the absence of heating to yield acetylated (C6AA) and isobutyrylated (C6BA) cellulose fibers, respectively. The modified fibers were further processed into CNFs through microfluidization. The infrared spectra in Fig. 2a indicate the successful introduction of the ester moieties, while other analyses (gel permeation chromatography and wide-angle X-ray diffraction) confirmed the success of the reaction under mild conditions (Fig. 2b and c). The crystalline structure of cellulose was not affected by the modification, while the weight-averaged degree of polymerization was only slightly reduced (Table 1). The latter is in contrast to regioselective modification via TEMPO-oxidation, which is known to cause significant polymer degradation, reducing the degree of polymerization by ca. 30–40%, even if performed under acidic or neutral conditions.29,30 As determined by nuclear magnetic resonance (NMR), the C6AA and C6BA samples featured degrees of substitution (DS) of 0.17 and 0.10, respectively, accounting to functionalization degrees between 1.0 and 0.6 mmol g−1. The regioselectivity of the esterification with N-acetylimidazole was confirmed by NMR (Fig. 2d), which showed that 93% of the C6-OH were modified (based on hydroxyl reactivity, the remaining 7% substitution mostly occurs at C2-OH). Usually, regioselective acetylation of cellulose requires the use of protecting groups and multi-step protocols.31 The high regioselectivity of acylation, with in situ N-acylimidazole, is rationalized by the decreased accessibility of the secondary hydroxyl groups, in presence of water.26
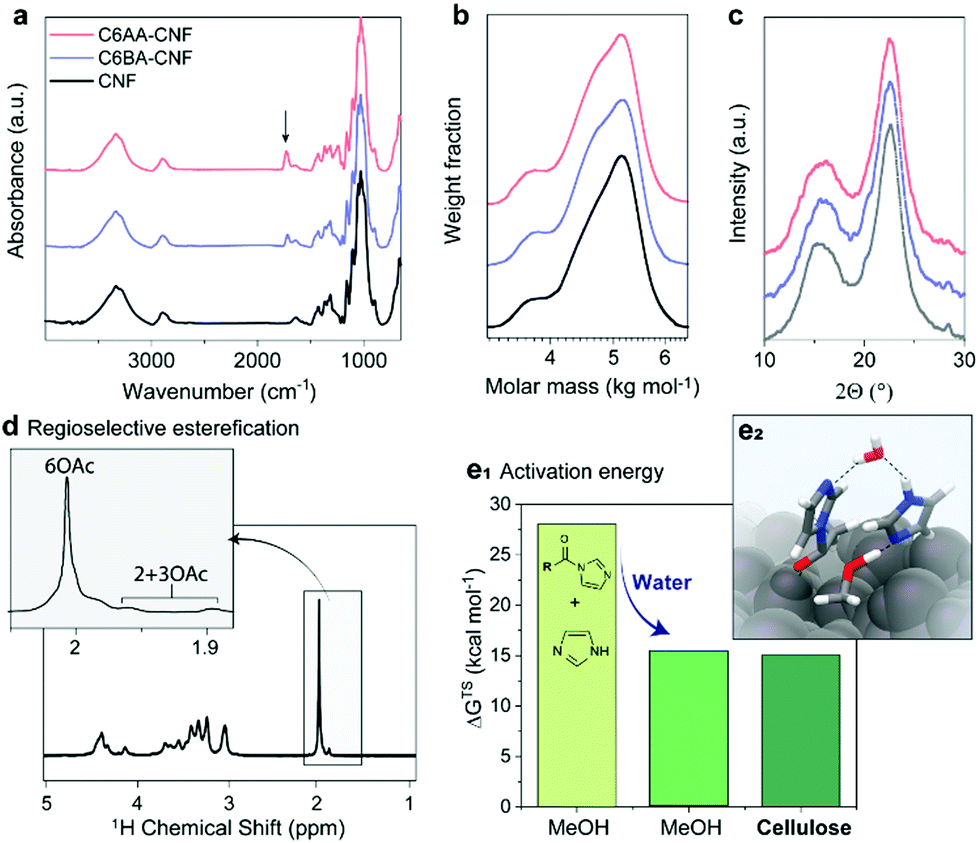 | ||
| Fig. 2 Chemical properties of acetylated (C6AA-CNF), isobutyrylated (C6BA-CNF), and native (CNF) cellulose nanofibers. The successful introduction of the ester groups is demonstrated through the presence of the IR carbonyl band (at 1727 cm−1) (a) and the diffusion-edited 1H-nuclear magnetic resonance spectrum (d). The procedure is mild and does not significantly impact the polymer chain integrity (b) and crystalline structure (c), as determined by gel permeation chromatography and wide-angle X-ray diffraction, respectively. The esterification is largely regioselective (d) and can be carried out in the presence of water (e1). In fact, the Gibb's free energy (ΔGTS) activation barrier was previously calculated for acetylation of methanol (MeOH) and cellulose with N-acetylimidazole and we show that water decreases the activation barrier and is further decreased through specific interactions with the cellulose surface (e2). Data for panels e1 and e2 were reproduced from the literature.26 | ||
| Feature | CNF | C6AA-CNF | C6BA-CNF |
|---|---|---|---|
| a Median surface energies are reported: γd, dispersive; γab, specific (acid–base); γt, total surface energies and γab/γt ratio. b Weight-averaged value. c Determined by DVS (water vapor) according to the Brunauer–Emmett–Teller model. | |||
| γ d (mJ m−2) | 48.6 | 30.4 | 37.5 |
| γ ab (mJ m−2) | 23.3 | 7.9 | 16.1 |
| γ t (mJ m−2) | 71.8 | 38.8 | 54.0 |
| γ ab/γta | 0.32 | 0.20 | 0.30 |
| k acid/kbase | 0.29 | 0.25 | 0.41 |
| Specific surface areac (m2 g−1) | 158 | 168 | 164 |
| Degree of polymerizationb | 969 | 895 | 899 |
| Degree of crystallinity (%) | 52 | 52 | 52 |
| Tensile strength (MPa) | 165 ± 20 | 186 ± 11 | 179 ± 12 |
| Elastic modulus (GPa) | 6.5 ± 0.3 | 7.5 ± 0.3 | 7.1 ± 0.3 |
Apart from enabling regioselective modification, the presence of water also promotes the reaction (Fig. S10†). The acetylation of never-dried (wet) fibers proceeds at significantly higher reaction rate if compared to the acetylation of dried fibers. This indicates a direct involvement of water in the reaction mechanism.26 As further confirmation, we calculated the Gibb's free energy of activation (gas-phase acetylation in methanol) and found that it was significantly reduced, by 56%, in the presence of water (Fig. 2e1 and e2). Additional modelling of the surface acetylation of the cellulose Iβ allomorph revealed a further decrease in activation energy due to interactions of N-acetylimidazole with the cellulose surface.
Earlier reports showed that partial esterification of cellulose in the presence of protic organic solvents increased the defibrillation tendency.11,13,16 In our solvent system (acetone/water) this effect was not significant due to the lower swelling of the cellulose structure, which restricts the modification to its surface, also maintaining the integrity of the isolated colloids. The CNFs were ca. 20 nm in diameter (Fig. 3a–f) and their length spanned the range of several hundreds of nanometers (>600 nm) to few microns (ca. 5 μm). Note: The diameters of the nanofibers were obtained from negative contrast SEM images following the procedures described by Mattos–Tardy et al.32 Nanofibers of this size are especially attractive to induce cohesion in particle composites.3 Tailoring CNF surface energy, for example, to achieve lower hydrophilicity, is expected to widen the use of CNFs as adhesives, and in multiphase materials. Overall, given the nanofiber dimensions, our regioselective modification enables a systematic evaluation of the effect of interfacial interactions of cellulose.
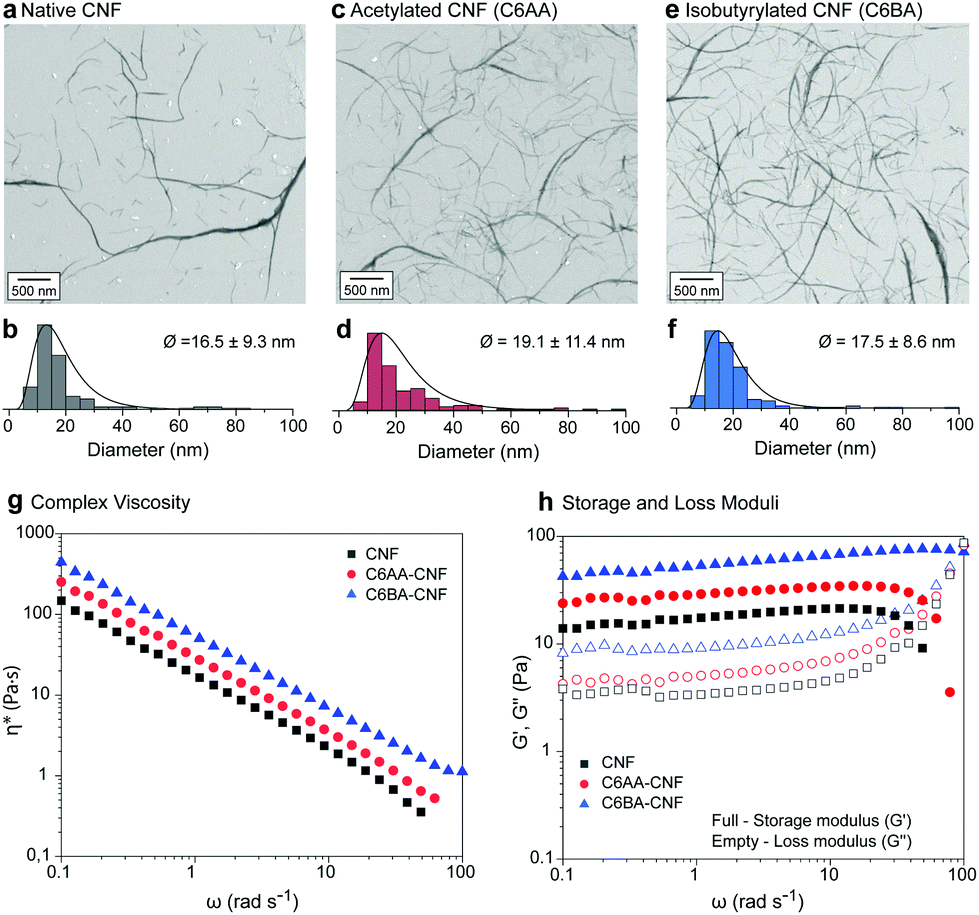 | ||
| Fig. 3 Negative contrast SEM images and diameter distribution (lognormal) of native CNF (a and b) and its esterified counterparts, namely C6AA-CNF (c and d) and C6BA-CNF (e and f). Oscillatory rheological profiles of aqueous suspensions (1.5 wt%) of CNF, C6AA-CNF, and C6BA-CNF: (g) flow profiles, and (h) storage and loss moduli. | ||
The morphological similarities of native CNF, C6AA-CNF, and C6BA-CNF were further confirmed by studying their rheological behavior (Fig. 3g and h). All nanofibers underwent pronounced shear thinning with apparent and complex viscosity profiles within the same range of values (Fig. S5†). However, the C6-OH-esterified CNF displayed slightly higher viscosity than their native analogues. Moreover, the complex viscosity indicated a dominant viscoelastic behavior for all nanofiber suspensions, with the yield stress point (G′ = G′′) shifting to higher shear stress values in the order C6BA-CNF > C6AA-CNF > CNF. The results cannot be explained by the nanofiber dimensions alone but their distinct self-interactions as well as interactions with water, both of which led to a ‘viscosifying’ effect. In parallel, C6AA-CNF and C6BA-CNF displayed remarkably higher water exclusion feature (Fig. S3a†) when compared native CNF, also favoring fiber-fiber interactions centered on hydrophobic effects. The formation of an excluded water shell around nanocelluloses is an additional factor explaining the high viscosity of the C6BA-CNF and C6AA-CNF suspensions, as shown even at low mass fraction.33
Compared to C6AA-CNF and C6BA-CNF, a higher water retention, and therefore higher swelling, was observed for native CNF (Fig. S3a†). C6AA-CNF and C6BA-CNF, in turn, retained water (Fig. S3a†), but we speculate that the higher molecular steric hindrance at their nanofiber–nanofiber interface created steric exclusion in the nanofiber network and consequently increasing water excluded volume and leading to higher viscosity. Altogether, our proposed C6-regioselective hydrophobization can be taken as a toolbox for fine tuning the colloidal behavior of CNF, for instance to manipulate associative interactions (Fig. S3b†) and the rheological behavior (Fig. 3g and h), both of which are central to several technological efforts (e.g., filament spinning34 and 3D printing35). We demonstrate the effect of C6-esterfication over the colloidal behavior of CNFs by its induced precipitation in the presence of various fractions of ethanol at increase G-forces. We show that C6-esterfied CNFs are more stable in conditions that would normally coagulate native CNF (Fig. S4†).
C6-OH-esterification allows for the tailoring of CNF for uses in multiphase systems. This is achieved by adjusting the surface energy and, thus, interfacial behavior at the air, liquid, and solid interfaces. As such, we used inverse gas chromatography (iGC) to elucidate the interactions of the unmodified CNF and the esterified CNFs with gases, namely acetone, acetonitrile, chloroform, dichloromethane, ethanol and ethyl acetate. The introduced ester groups reduced the total surface energy (γt) of the CNF (Table 1). As expected, the specific (acid–base) term (γab) corresponding to polar, short-range interactions (mostly hydrogen bonds)36,37 was reduced, as the primary hydroxyl groups – now esterified – would be otherwise involved in polar gas interactions (Table 1 and Fig. 4a). In comparison to native CNF with a γab of ca. 23 mJ m−2, C6AA-CNF featured the lowest γab, ca. 8 mJ m−2, followed by C6BA-CNF, ca. 16 mJ m−2. The lower specific surface energy of the former is attributed to the higher surface coverage by acetyl groups, as indicated by the higher DS (0.17 amounting to a modification of 73% of the maximum theoretical C6-OH, calculated from the cellulose crystallite size).16,38 The bulkier isobutyryl group was introduced to 43% of the available C6-OH groups. In a similar vein, the dispersive term γd, referring to nonpolar, long-range interactions,39 influenced more significantly the total surface energy of C6AA-CNF, suggesting that the degree of surface coverage by the substituent moiety was more critical than its size. The surface chemistry of the samples was assessed using the Gutmann acid (kacid) and base (kbase) numbers. Consistent with the literature,39,40 the kbase for C6AA-CNF and C6BA-CNF was consistently higher than kacid, indicating that the samples possessed preferential electron-donating (basic) abilities. Dependent on changes in charge density and relative abundance of the functional group, kacid/kbase can be shifted to slightly lower values, in case of C6AA-CNF due to the negative charge density of the carbonyl group. In case of longer alkyl chains, the positive charge of the alkyl chains dominates increasing the electron-withdrawing interactions (Fig. S1†). The observations discussed above for modified CNF in gaseous aprotic solvents differed from that in water, the protic analogue, both as vapor and as a liquid. Firstly, the capacity of CNF to adsorb water vapor was retained given the abundant density of hydroxyls on C2 and C3 (plus residual unmodified C6-OH) – note the similar equilibrium moisture contents (EMC) among the samples in dynamic vapor sorption (DVS; Fig. S7a†). Several factors are known to play a role in water interactions: while on one hand the reduced occurrence of hydroxyl groups is expected to lessen the EMC in C6-OH-esterified samples, the bulkier, less polar substituents might sterically impair interparticle cohesiveness and open the otherwise hidden sorption sites, accommodating more water molecules.41 Along these lines, we note that films made from C6-OH-esterified CNF displayed a higher SSA (Table 1). Importantly, the difference in moisture content at a given relative humidity, between adsorption (MCad) and desorption (MCdes) cycles, shows the hysteresis typical of cellulose with MCdes > MCad.19 The higher hysteresis of native CNF (Fig. S6b†) was expected and may reflect the interplay between the increased occurrence of hydroxyls taking part in the (ad/de)sorption process, the larger moisture-induced structural changes, and the more pronounced hygroexpansion, all of which increase the hydrophilicity.42,43 Related features are confirmed by using liquid water as a probe, for instance, by measuring the water contact angle (WCA) of the films (Fig. 4c1): the introduction of acetyl groups doubled the WCA of unmodified CNF (Fig. 4c2), and it was further increased in the case of C6BA-CNF films (Fig. 4c3). Although the surface hydrophilicity of the CNF film was markedly reduced upon esterification, as one would expect, the size of the substituent ester group also affects the behavior: the higher WCA of C6BA-CNF, which is less substituted compared to C6AA-CNF, is explained by the fact that BA has more carbon atoms and is a bulkier moiety. This relationship between alkyl chain length and hydrophobization has been demonstrated with CNF butyrate and palmitate esters.44 The progressive variation of WCA with time gives further insight into the hydrophilic/hydrophobic character:45 during the first minute, the WCA of native CNF varied by ca. 17% while that of C6-OH-esterified CNF was more consistent, ca. 4% (Fig. S6†), pointing to the more limited influence of capillarity and polarity in the latter system.
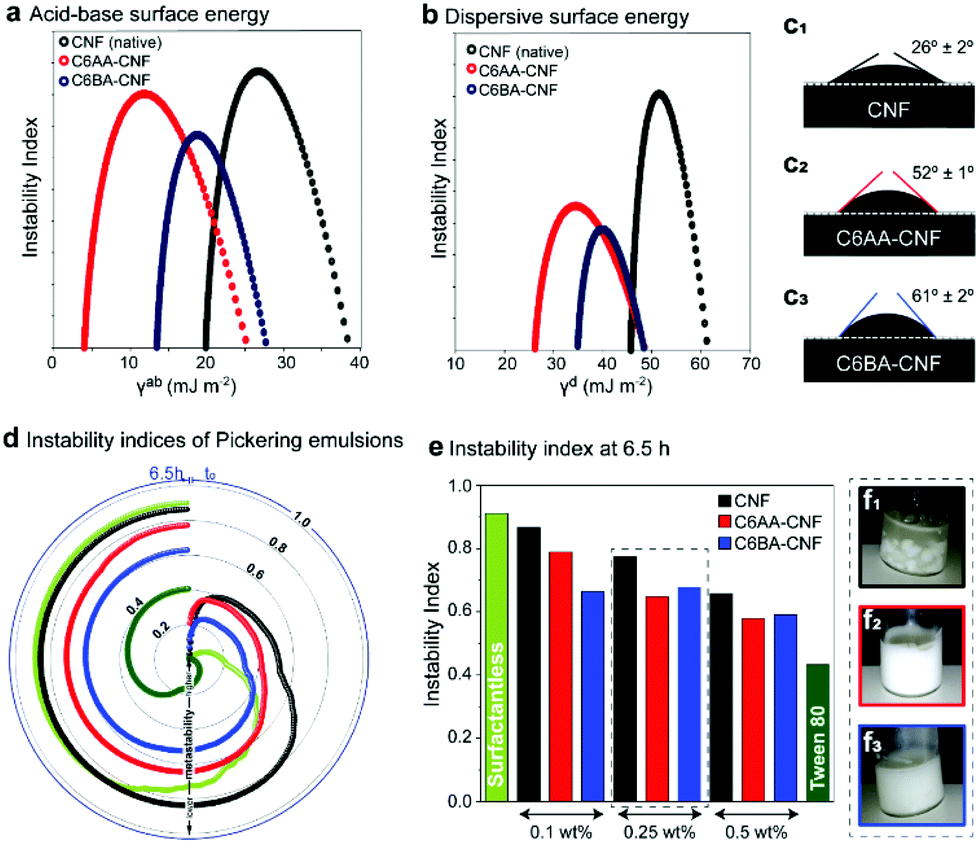 | ||
| Fig. 4 The acid–base and dispersive surface energy of unmodified cellulose nanofibers (CNF) is shifted to lower values upon modification due to the introduction of alkyl ester groups (C6AA-CNF in red and C6BA-CNF in blue) (a and b). Due to the higher surface coverage this effect is more pronounced for C6AA-CNF. The water contact angle depends on the alkyl chain length (c1–c3) and it is significantly higher for the isobutyrylated sample. This agrees with the instability indices of the Pickering emulsions prepared with sunflower oil (d and e), and the visual appearance of the respective Pickering emulsions from native CNF (f1), C6AA-CNF (f2), and C6BA-CNF (f3). | ||
By changing the surface energy of CNF, it is possible to tune the behavior at the oil/water interface, for instance in particle-stabilized colloidal multiphase liquid systems, the so-called Pickering emulsions. While the surface activities of all particles were all negligible, as demonstrated by the surface tension values of aqueous CNF suspensions (71 ± 1 mN m−1, determined by pendant drop in air – this value was constant during 30 min for all CNFs), the interfacial tension between a drop of water with suspended nanofibers and sunflower oil was reduced from the original value of 24 ± 1 mN m−1 by ca. 15% (CNF), 22% (C6AA-CNF), and 20% (C6BA-CNF). The small but clear reduction in interfacial tension of C6-OH-esterified CNF points to a better performance as a Pickering stabilizer. The macroscopic phase separation behavior within a centrifugal field was monitored via light transmittance (Fig. S8†). These profiles were used in the calculation of the emulsion instability index (EI), ranging from 0 to 1 and with lower values indicating higher kinetic stability. The evolution of the EI with time is depicted in Fig. 4d and e. As expected, O/W systems in the absence of any stabilizer presented the highest EI over the whole range. The emulsions stabilized with a nonionic surfactant, Tween 80, gave the lowest EI (Fig. 4e). The emulsions stabilized by C6-OH-esterification gave an EI that was lower than the other systems. After equilibration, the EI of all formulations corroborated the higher Pickering stabilization capacity of the C6-OH-esterified particles, which showed a higher tendency to adsorb at the interface given the presence of the hydrophobic moieties (Fig. 4f1–3).
Robust films were assembled from all the CNFs. The tensile strength and elastic moduli were higher for the C6-esterified CNF samples (Table 1 and Fig. S9†). It has been shown that the mechanical performance of cellulosic constructs can be improved by mild acetylation, i.e. without affecting their molar mass, and a subsequent fibrillation.46,47 Since fiber acetylation has been also shown to diminish the mechanical properties,48 the results of these nanofibrillated systems could be reasonably assumed to result from a more extensive fibrillation in the case of surface acetylated fibers. However, since our protocol did not significantly influence the dimensions of the prepared CNF, the increased mechanical properties can only be explained to originate from the regioselective introduction of functional groups. We speculate that the presence of alternating hydrophobic functional groups enables the formation of stronger cohesive interactions upon consolidation of the respective nanofiber suspension.
The SSA of the materials displayed similar values for all CNFs, confirming a similar nanofibrillar assembly for all of them. Entanglement and interconnectivity of the nanonetwork plays a fundamental role in transferring cohesion from single nanofibers to their macroscaled constructs. Given the higher mechanical properties of the C6-esterifeid samples, one can infer that nanofiber–nanofiber interactions played a determining role to enable efficient stress transfer mechanisms across the material. Steric hindrance and interfibrillar repulsion are expected to take place in the C6-OH-esterified nanofibers, which could enable a better densification of the system, for example, by drying after consolidation. The effects of interfibrillar repulsion as far as the formation of a well-organized nanofiber network have been previously discussed in terms of electrostatic interactions (as applicable to TEMPO-oxidized CNF);49 however, the present case of esterified CNF opens further opportunities for CNF deployment.
Conclusions
We report for the first time a versatile and simple preparation of esterified CNF by using a highly regioselective modification (hydrophobization) of cellulose. Acetyl and isobutyryl units were successfully introduced (functionalization degree of 0.6 to 1.0 mmol g−1) following in situ formation of N-acetylimidazoles and N-isobutyrylimidazole from the corresponding anhydrides. The modification was confirmed to be selective to the surface of cellulose and to preserve the integrity of the crystalline structure and polymer chains. The regioselective introduction of acetyl and isobutyryl groups facilitated the production of nanofibers with tunable surface energy, as demonstrated by the measured interactions with aprotic gases, water vapor, and oil and water. Our results indicated that the regioselective modification increased the metastability of Pickering emulsions and enabled specific interactions with various fluids, depending on the surface coverage and size of the ester alkyl group. The increased suspension viscosity and storage moduli as well as the mechanical properties of films produced from the nanofibers indicated the prominent role of cohesive inter-chain interactions. In this regard, the regioselective modification of CNF offers an opportunity beyond the state-of-the-art. Overall, we introduce a chemical pathway for the controlled hydrophobization of nanocelluloses in the presence of water. The introduced method allows installing a wide range of functionalities and expands the application prospects of cellulose-based colloids.Experimental
Materials
Cellulose fibers of high purity (93% cellulose, 4% hemicellulose) were provided as never-dried bleached beech sulfite dissolving pulp (50 wt% solid content) by Lenzing AG (Lenzing, Austria) and used in the production of the esterified pulp samples (C6AA-Cell and C6BA-Cell). Acetic anhydride (≥99%), isobutyric anhydride (97%), imidazole (ACS reagent, ≥99%), and acetone (DMSO, ≥99%) were purchased from Sigma-Aldrich (Merck Life Science OY, Finland). The never-dried pulp was pretreated before modification with a coffee blender to increase its SSA. Sunflower seed oil from Helianthus (CAS no. 8001-21-6) was used as provided by Sigma-Aldrich (Saint Louis, MO, USA) to fabricate emulsions.Preparation of esterified cellulose nanofibers
The pre-treated never-dried cellulose fibers (5 g dry cellulose content, 50 wt% solid content, 31 mmol) were added into 21.5 mL of 3 M imidazole solution in acetone and mixed with a glass rod for 5 min. After 30 min of equilibration in a closed container, 31 mL of 1 M carboxylic acid anhydride* in acetone (* either with acetic anhydride to yield C6AA-Cell or isobutyric anhydride to yield C6BA-Cell) was added and the reaction mixture was stirred for 24 h at room temperature to complete the esterification. Afterwards, the suspensions were filtered to remove the solvent, washed twice with acetone (2 × 50 mL) and 1 M aqueous solution of NaHCO3 (2 × 50 mL) and then four times with DI water (4 × 50 mL). Successful removal of solid reactants by washing was confirmed by IR (Fig. S13†) and NMR was used to confirm complete removal of solvent residues on basis of reported NMR chemical shifts.50The esterified fibers (quantitative mass yield) were diluted to 0.5 wt%, mixed in a kitchen blender and fibrillated by six passes in a microfluidizer (200 and 100 μm chambers at 2000 bar). The processed nanofibers, either acetylated (C6AA-CNF) or isobutyrylated (C6BA-CNF), were compared to native CNF, which was fibrillated at the same conditions.
Structural and morphological characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMSO-d6 (20:80 wt%) electrolyte solution.51,52 The samples were magnetically stirred at room temperature until they went clear, which typically took approx. 1 hour. If the samples did not go clear during that period, the temperature was increased to 60 °C. All NMR runs were recorded on a Bruker AVANCE NEO 600 MHz spectrometer equipped with a 5 mm SmartProbe™ set to 65 °C.
:DMSO-d6 (20:80 wt%) electrolyte solution.51,52 The samples were magnetically stirred at room temperature until they went clear, which typically took approx. 1 hour. If the samples did not go clear during that period, the temperature was increased to 60 °C. All NMR runs were recorded on a Bruker AVANCE NEO 600 MHz spectrometer equipped with a 5 mm SmartProbe™ set to 65 °C.
The diffusion-edited 1H experiment used a 1D bipolar-pulse pair with stimulated echo (BPPSTE)53 diffusion-ordered spectroscopy (DOSY) pulse sequence (Bruker pulse program ‘ledbpgp2s1d’), with 1 s relaxation delay (d1), 0.5 s acquisition time (aq), 16 dummy scans (ds), 128 transient scans (ns), a sweep-width (sw) of 20 ppm with the transmitter offset on 6.1 ppm (o1p), diffusion time (d20) of 200 ms, gradient recovery delay (d16) of 0.2 ms, eddy current delay (d21) of 5 ms, diffusion gradient pulse duration (p30) of 2.5 ms, and z-gradient strength (gpz6) of 90% at 50 G cm−1 (probe z-gradient strength). These conditions are specific to the NMR apparatus above and may need reoptimization for other systems. The diffusion-edited 1H spectra for the samples C6AA- and C6BA-cellulose fibers are shown in Fig. S11a and S12a.† The degree of substitution was determined from 1H-NMR diffusion-edited spectra of C6AA- and C6BA-Cell relating the protons from the glucose monomer unit to the protons of the acetyl or isobutyryl group, respectively. The acetyl peak in the spectrum of C6BA-Cell (Fig. S12a†) is as impurity neglected for the calculation of the degree of substitution.
The HSQC experiments used a multiplicity-edited phase sensitive HSQC sequence with echo/antiecho-TPPI gradient selection (Bruker pulse program ‘hsqcedetgp’).54 The parameters were as follows: spectral widths were 13 ppm and 165 ppm, with transmitter offsets (o1p) of 6.18 and 75 ppm, for 1H and 13C dimensions, respectively. The time-domain size (td1) in the indirectly detected 13C-dimension (f1) was 512, corresponding to 256 t1-increments for the real spectrum. There were 16 dummy scans and 64 scans, an acquisition time of 0.065 s for f2 and a relaxation delay of 1.5 s. Spectral resolution was increased by zero-filling, by setting the size of the spectra in both dimensions to 1024 Hz. Sine squared (90°) window functions were used in f1 and f2. Respective HSQC spectra are shown in Fig. S11b and S12b.†
The regioselectivity of the acetylation in presence of imidazole and acetic anhydride was determined by peak fitting of the 1H-NMR spectrum (Fig. S13†), according to a previously published work.26
Surface energy and activity
The samples (about 5 mg) were packed into individual 4 mm ID silanized glass columns and run at a series of surface coverage with alkanes and polar probe molecules in order to determine the dispersive surface energy (γDs) as well as the acid–base free energy of adsorption (ΔGSP). The sample column was pre-conditioned for 2 h at 30 °C and 0% relative humidity with 10 mL min−1 helium carrier gas. The experiment was conducted at 30 °C at a 10 mL min−1 total flow rate of helium and methane was used to determine the dead volume of the system.
The dispersive surface energy (γDs) analysis was performed by measuring the net retention volume VN (measured retention volume minus dead volume) for a series of n-alkane eluents (in this case: hexane, heptane, octane, and nonane). The Dorris and Gray method was applied during the analysis: a plot of RTln(VN) versus the alkane carbon number at a certain surface coverage produces a linear correlation. The dispersive surface energy of the solid sample can be determined from the slope of the line according to the following equation:
| Slope = 2(γCH2γDs)½ × NAaCH2, |
The specific contribution to the total surface energy (γABs) was obtained by first measuring the specific free energies of desorption for different polar probe molecules, ΔGSP. These values were determined by measuring the net retention volume of polar probe molecules (acetone, chloroform, ethyl acetate, toluene, and dichloromethane) on the samples. Polar probe points are located above the alkane straight line on the RTln(VN) versus PD plot. The vertical distance to the straight line is equal to the specific component of the free energy of desorption, ΔGSP. From the ΔGSP values based on the van Oss approach, the specific surface energy (γABs) of the samples were calculated. The specific contribution is subdivided into an acid γs+ and a base γs− parameter of the surface energy. In this approach, the Della Volpe scale is employed, with a pair of mono-functional acidic and basic probe molecules (dichloromethane, γs+ = 124.58 mJ m−2 and ethyl acetate, γs− = 475.67 mJ m−2). The surface chemistry of the samples was assessed using the Gutmann acid (KA) and base (KB) numbers, determined based on the Gutmann approach using the following polar probes: dichloromethane, ethyl acetate, acetone, and chloroform. The KA and KB values of the samples were calculated using the ΔGSP values of polar probes at a particular surface coverage and listed in Table 1.
To represent the heterogeneity of the samples in a more illustrative manner, the surface energy distributions are obtained by a point-by-point integration of the surface energy profiles, resulting in plots of γDs, γABs, and γTs surface energies versus percentage of surface (area increment), as shown in Fig. 4a and b. The medians γD,50s, γAB,40s, and γT,50s are defined as the surface energies at which half of the population lies below this value listed in Table 1. The ratio of the γABs and γTs indicates the surface polarity and it is also listed in Table 1.
The total work of cohesion was determined according to geometric mean method using the surface energy components of the samples:
| WtotalCohesion = 2[(gDs × gDs)½ + (gs+ × gs−)½ + (gs− × gs+)½]. |
Emulsion formation and stability
:1. Water and sunflower oil were also mixed at the same ratio, but in the absence of CNF or any surface-active molecules (surfactantless; herein taken as negative control) or added with 0.25 wt% Tween 80 (positive control). The systems were tip-sonicated (500 W at 20% amplitude) for 3 min (7 s on/3 s off steps) in an ice bath to avoid excessive heating.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Prof. Watson Loh and the São Paulo Research Foundation (FAPESP; grant no. 2016/13926-7) are acknowledged for granting access to LUMiSizer, and Caroline Silva for experimental support. This research was funded in part by the Austrian Science Fund (FWF) (J4356), FAPESP (grant no. 2019/00370-9), the Academy of Finland (Project #311255, ‘WTF-Click-Nano’) and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 788489).Notes and references
- T. Li, C. Chen, A. H. Brozena, J. Y. Zhu, L. Xu, C. Driemeier, J. Dai, O. J. Rojas, A. Isogai, L. Wågberg and L. Hu, Nature, 2021, 590, 47–56 CrossRef CAS PubMed.
- N. Mittal, F. Ansari, K. Gowda, V. C. Brouzet, P. Chen, P. T. Larsson, S. V. Roth, F. Lundell, L. Wågberg, N. A. Kotov and L. D. Söderberg, ACS Nano, 2018, 12, 6378–6388 CrossRef CAS PubMed.
- B. D. Mattos, B. L. Tardy, L. G. Greca, T. Kämäräinen, W. Xiang, O. Cusola, W. L. E. Magalhães and O. J. Rojas, Sci. Adv., 2020, 6, eaaz7328 CrossRef CAS PubMed.
- B. D. Mattos, L. G. Greca, B. L. Tardy, W. L. E. Magalhães and O. J. Rojas, Small, 2018, 14, 1801256 CrossRef.
- Y. Kobayashi, T. Saito and A. Isogai, Angew. Chem., Int. Ed., 2014, 10394–10397 CrossRef CAS.
- S. F. Plappert, J.-M. Nedelec, H. Rennhofer, H. C. Lichtenegger and F. W. Liebner, Chem. Mater., 2017, 29, 6630–6641 CrossRef CAS.
- A. Isogai and Y. Zhou, Curr. Opin. Solid State Mater. Sci., 2019, 23, 101–106 CrossRef CAS.
- A. Isogai and L. Bergström, Curr. Opin. Green Sustainable Chem., 2018, 12, 15–21 CrossRef.
- A. Isogai, T. Saito and H. Fukuzumi, Nanoscale, 2011, 3, 71–85 RSC.
- Z. Fang, B. Li, Y. Liu, J. Zhu, G. Li, G. Hou, J. Zhou and X. Qiu, Matter, 2020, 2, 1000–1014 CrossRef.
- A. Tripathi, M. Ago, S. A. Khan and O. J. Rojas, ACS Appl. Mater. Interfaces, 2018, 10, 44776–44786 CrossRef CAS PubMed.
- X. Kang, P. Sun, S. Kuga, C. Wang, Y. Zhao, M. Wu and Y. Huang, ACS Sustainable Chem. Eng., 2017, 5, 2529–2534 CrossRef CAS.
- M. Jonoobi, J. Harun, A. P. Mathew, M. Z. B. Hussein and K. Oksman, Cellulose, 2010, 17, 299–307 CrossRef CAS.
- P. Huang, M. Wu, S. Kuga, D. Wang, D. Wu and Y. Huang, ChemSusChem, 2012, 5, 2319–2322 CrossRef CAS PubMed.
- U. Weise, T. Maloney and H. Paulapuro, Cellulose, 1996, 189–202 CrossRef.
- M. Beaumont, S. Winklehner, S. Veigel, N. Mundigler, W. Gindl-Altmutter, A. Potthast and T. Rosenau, Green Chem., 2020, 22, 5605–5609 RSC.
- T. Saito, Y. Nishiyama, J.-L. Putaux, M. Vignon and A. Isogai, Biomacromolecules, 2006, 7, 1687–1691 CrossRef CAS PubMed.
- K. Abe, S. Iwamoto and H. Yano, Biomacromolecules, 2007, 8, 3276–3278 CrossRef CAS PubMed.
- M. Chen, B. Coasne, R. Guyer, D. Derome and J. Carmeliet, Nat. Commun., 2018, 9, 3507 CrossRef PubMed.
- S. Ifuku, M. Nogi, K. Abe, K. Handa, F. Nakatsubo and H. Yano, Biomacromolecules, 2007, 8, 1973–1978 CrossRef CAS PubMed.
- H. Steinmeier, Macromol. Symp., 2004, 208, 49–60 CrossRef CAS.
- Z. Ling, T. Wang, M. Makarem, M. Santiago Cintrón, H. N. Cheng, X. Kang, M. Bacher, A. Potthast, T. Rosenau, H. King, C. D. Delhom, S. Nam, J. V. Edwards, S. H. Kim, F. Xu and A. D. French, Cellulose, 2019, 26, 305–328 CrossRef CAS.
- I. Sakurada, Y. Nukushina and T. Ito, J. Polym. Sci., 1962, 57, 651–660 CrossRef CAS.
- I. Diddens, B. Murphy, M. Krisch and M. Müller, Macromolecules, 2008, 41, 9755–9759 CrossRef CAS.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. R. McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- M. Beaumont, P. Jusner, N. Gierlinger, A. W. T. King, A. Potthast, O. J. Rojas and T. Rosenau, Nat. Commun., 2021, 12, 2513 CrossRef CAS PubMed.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, R. C. McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- T. Saito, M. Hirota, N. Tamura and A. Isogai, J. Wood Sci., 2010, 56, 227–232 CrossRef CAS.
- T. Saito, M. Hirota, N. Tamura, S. Kimura, H. Fukuzumi, L. Heux and A. Isogai, Biomacromolecules, 2009, 10, 1992–1996 CrossRef CAS PubMed.
- S. C. Fox, B. Li, D. Xu and K. J. Edgar, Biomacromolecules, 2011, 12, 1956–1972 CrossRef CAS PubMed.
- B. D. Mattos, B. L. Tardy and O. J. Rojas, Biomacromolecules, 2019, 20, 2657–2665 CrossRef CAS PubMed.
- M. A. Hubbe, P. Tayeb, M. Joyce, P. Tyagi, M. Kehoe, K. Dimic-Misic and L. Pal, BioResources, 2017, 12, 9556–9661 CAS.
- M. J. Lundahl, V. Klar, L. Wang, M. Ago and O. J. Rojas, Ind. Eng. Chem. Res., 2017, 56, 8–19 CrossRef CAS.
- R. Ajdary, B. L. Tardy, B. D. Mattos, L. Bai and O. J. Rojas, Adv. Mater., 2020, 2001085 Search PubMed.
- N. Rjiba, M. Nardin, J.-Y. Dréan and R. Frydrych, J. Colloid Interface Sci., 2007, 314, 373–380 CrossRef CAS PubMed.
- M. Rückriem, A. Inayat, D. Enke, R. Gläser, W.-D. Einicke and R. Rockmann, Colloids Surf., A, 2010, 357, 21–26 CrossRef.
- Y. Okita, T. Saito and A. Isogai, Biomacromolecules, 2010, 11, 1696–1700 CrossRef CAS PubMed.
- M. Beaumont, A. Kondor, S. Plappert, C. Mitterer, M. Opietnik, A. Potthast and T. Rosenau, Cellulose, 2017, 24, 435–440 CrossRef CAS.
- Y. Peng, D. J. Gardner, Y. Han, Z. Cai and M. A. Tshabalala, J. Controlled Release, 2013, 405, 85–95 CAS.
- W. Yu, B. Xu, B. Fan, Z. Yaoli and X. Wang, BioResources, 2020, 15, 159–171 CAS.
- T. Yang, E. Ma and J. Cao, Holzforschung, 2018, 72, 943–950 CrossRef CAS.
- M. Cordin, U. J. Griesser and T. Bechtold, Cellulose, 2017, 24, 1837–1847 CrossRef CAS.
- S. Vuoti, R. Talja, L.-S. Johansson, H. Heikkinen and T. Tammelin, Cellulose, 2013, 20, 2359–2370 CrossRef CAS.
- C. S. R. Freire, A. J. D. Silvestre, C. P. Neto, M. N. Belgacem and A. Gandini, J. Appl. Polym. Sci., 2006, 100, 1093–1102 CrossRef CAS.
- J. C. Bletzinger, Ind. Eng. Chem., 1943, 35, 474–480 CrossRef CAS.
- W. H. Aiken, Ind. Eng. Chem., 1943, 35, 1206–1210 CrossRef CAS.
- S. Yang, Q. Xie, X. Liu, M. Wu, S. Wang and X. Song, RSC Adv., 2018, 8, 3619–3625 RSC.
- F. Jiang and Y.-L. Hsieh, ACS Sustainable Chem. Eng., 2016, 4, 1041–1049 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- A. W. T. King, V. Mäkelä, S. A. Kedzior, T. Laaksonen, G. J. Partl, S. Heikkinen, H. Koskela, H. A. Heikkinen, A. J. Holding, E. D. Cranston and I. Kilpeläinen, Biomacromolecules, 2018, 19, 2708–2720 CrossRef CAS PubMed.
- T. Koso, D. Rico del Cerro, S. Heikkinen, T. Nypelö, J. Buffiere, J. E. Perea-Buceta, A. Potthast, T. Rosenau, H. Heikkinen, H. Maaheimo, A. Isogai, I. Kilpeläinen and A. W. T. King, Cellulose, 2020, 27, 7929–7953 CrossRef CAS.
- D. H. Wu, A. D. Chen and C. S. Johnson, J. Magn. Reson., Ser. A, 1995, 115, 260–264 CrossRef CAS.
- W. Willker, D. Leibfritz, R. Kerssebaum and W. Bermel, Magn. Reson. Chem., 1993, 31, 287–292 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc02292j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |