
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A solvent-reagent selection guide for Steglich-type esterification of carboxylic acids†
Andrew
Jordan
 *a,
Kyran D.
Whymark‡
b,
Jack
Sydenham‡
b and
Helen F.
Sneddon
*b
*a,
Kyran D.
Whymark‡
b,
Jack
Sydenham‡
b and
Helen F.
Sneddon
*b
aGlaxoSmithKline Carbon Neutral Laboratory for Sustainable Chemistry, Jubilee Campus, University of Nottingham, 6 Triumph Rd, Nottingham NG7 2GA, UK. E-mail: andrew.jordan@nottingham.ac.uk
bGSK, Medicines Research Centre, Gunnels Wood Road, Stevenage, Herts SG1 2NY, UK. E-mail: helen.f.sneddon@gsk.com; Fax: +44 (0)1438768302; Tel: +44 (0)1438 763645
First published on 29th July 2021
Abstract
The Steglich esterification is a widely employed method for the formation of esters under mild conditions. A number of issues regarding the sustainability of this transformation have been identified, chiefly the use of hazardous carbodiimide coupling reagents in conjunction with solvents with considerable issues such as dichloromethane (DCM) and N,N-dimethylformamide (DMF). To overcome these issues, we have developed a solvent-reagent selection guide for the formation of esters via Steglich-type reactions with the aim of providing safer, more sustainable conditions. Optimum reaction conditions have been identified after high-throughput screening of solvent-reagent combinations, namely the use of Mukaiyama's reagent (Muk) in conjunction with solvent dimethyl carbonate (DMC). The new reaction conditions were also exemplified through the synthesis of a small selection of building-block like molecules and includes the formation of t-butyl esters.
Introduction
Esterification is widely regarded as an essential transformation within organic and medicinal chemistry as the formation of ester functional groups is critical for the synthesis of many commercially available drugs and building blocks.1 Furthermore, esters find extensive use within the fragrance,2,3 flavour,4 and functional materials industries,5 as well as having a large prevalence within natural products, lipids, pheromones and other biologically active compounds.6 Thus, synthetic methodology affording ester functional groups has received a considerable amount of research interest (Fig. 1).7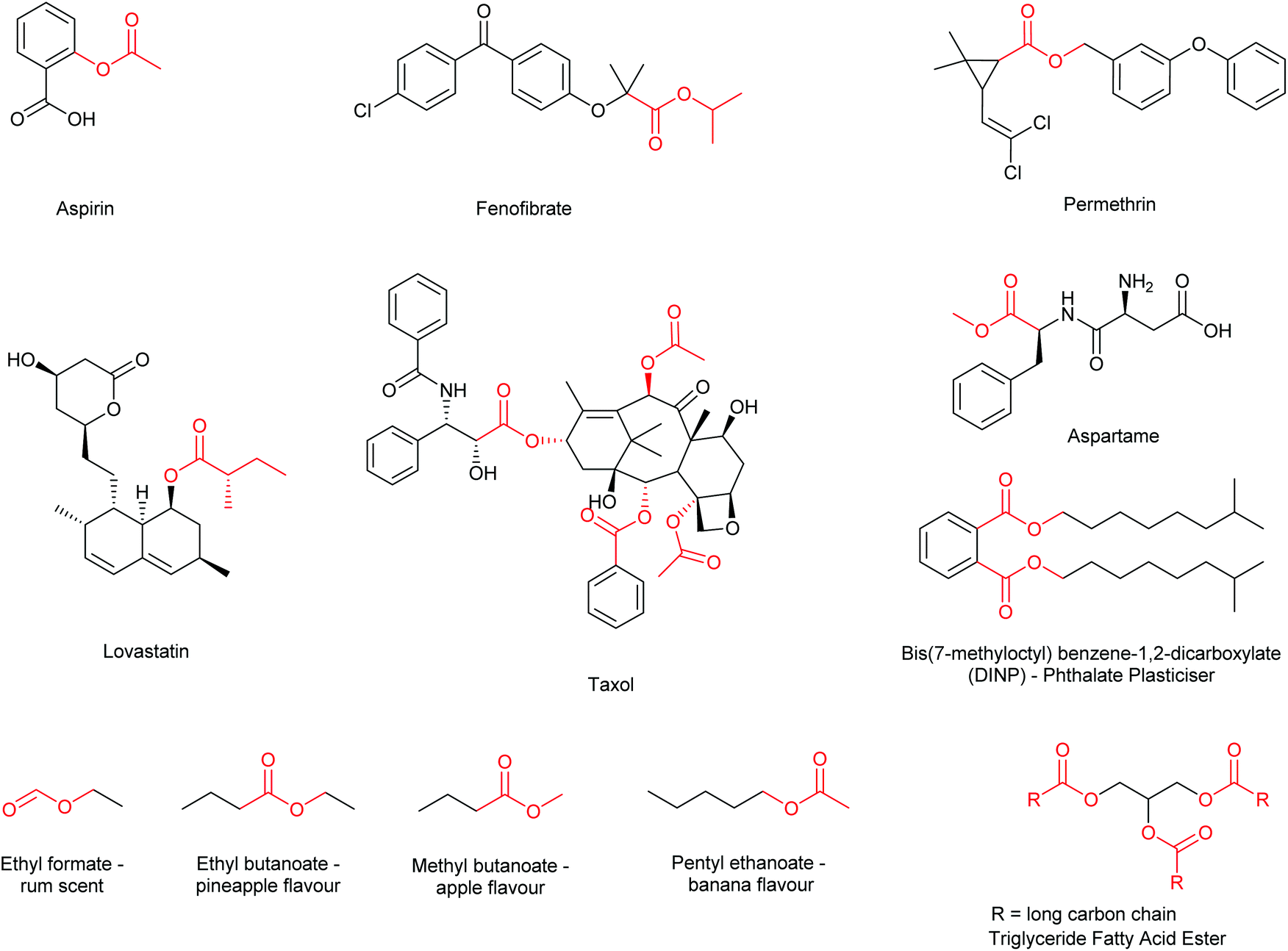 | ||
| Fig. 1 A selection of ester containing molecules including pharmaceuticals (aspirin, fenofibrate, taxol, lovastatin), insecticide (permethrin), flavours (ethyl formate) and flavour enhancers (aspartame), scents (ethyl butanoate, methyl butanoate, pentyl ethanoate), plasticisers (DINP), and natural molecules (triglycerides). | ||
Methodology often employed in the synthesis of esters includes the traditional acid-catalysed Fischer–Speier method (catalytic HCl, H2SO4etc.); first reported in 1895 and one of the most commonly employed methods today.8 Variants of the Fischer–Speier esterification have also been reported in the literature using an array of Lewis acid catalysts, such as scandium(III) triflate in acetic acid9 and ferric chloride in mesitylene,10 (and other non-polar solvents such as toluene, xylene and heptane). One potential downside to the simple utility of the Fischer–Speier methodology is the requirement to carry out the reaction under acidic conditions, thus rendering acid sensitive moieties/molecules incompatible with this methodology.
An alternative method for the formation of esters under mild and neutral conditions is the Steglich esterification.11,12 In 1978, Wolfgang Steglich and Bernhard Neises first reported an adapted esterification procedure utilising the amide coupling agent N,N′-dicyclohexylcarbodiimide (DCC) and organocatalyst 4-dimethylaminopyridine (DMAP), Scheme 1.11,12 This method was derived from amide coupling procedures that reported DCC as an effective coupling agent. The Steglich esterification proceeds under mild conditions, ambient temperatures, often neutral pH, and can afford a wide range of esters containing challenging substituents such as the acid liable, sterically hindered tert-butyl group, which under Fischer–Speier esterification conditions would undergo elimination. Catalytic amounts of DMAP are required for this esterification due to DMAP possessing a much greater nucleophilicity than that of the alcohol. DMAP accelerates the reaction rate by reacting with the O-acylisourea intermediate to form a highly activated electrophilic acylated pyridinium intermediate. This additionally prevents the possibility of an intramolecular 1,3-rearrangment of the O-acylisourea intermediate to an N-acylurea species,13 which is unable to react with alcohols.
 | ||
| Scheme 1 A typical DCC + DMAP mediated Steglich esterification. | ||
Although DCC is widely employed as the coupling agent for Steglich esterification, due to the effectiveness of affording the activated ester intermediate, there are many concerns associated with its use. As DCC is an irritant, can cause organ damage and is classified as an allergen, many precautions must be taken during its use.14 Another drawback associated with DCC is that it produces a by-product, N,N′-dicyclohexylurea (DCU) which is insoluble in water and only partially soluble in many organic solvents.15,16 Whilst the poor solubility of the DCU by-product results in its easy removal by filtration from reaction mixtures, it can prove difficult to remove remaining trace amounts, even via column chromatography, often making purification laborious.16,17 Organic solvent soluble carbodiimide derivatives such as N,N′-diisopropylcarbodiimide (DIC) can overcome some of the aforementioned issues. Although many other variants of the Steglich esterification exist (such as the Yamaguchi esterification utilising the coupling agent 2,4,6-trichlorobenzoyl chloride),18,19 the Steglich esterification remains one of the most widely applied synthetic methodologies for synthesis of esters.20
Steglich esterification typically employs solvents and reagents which are less than ideal from an environmental, health and safety perspective (EHS).21,22 A recent analysis of the literature associated with the synthesis of esters via Steglich type methods showed that the most commonly employed reaction solvents were DCM, DMF, THF, and CH3CN.20 Furthermore, commonly employed coupling reagents such as DCC and DIC are considered problematic by the GlaxoSmithKline (GSK) reagent selection guides due to EHS concerns e.g. DIC is toxic by inhalation.21 Therefore, the development of a solvent-reagent selection guide for the formation of esters highlighting safer, more sustainable alternatives, would be of importance and great utility to both academia and industry. Inspired by previously developed solvent-reagent guides developed in collaboration with GSK, the authors have undertaken this investigation with the aim of discovering safer, more sustainable alternatives in which to conduct coupling reagent promoted esterification reactions in the same vein as the Steglich esterification.
Method
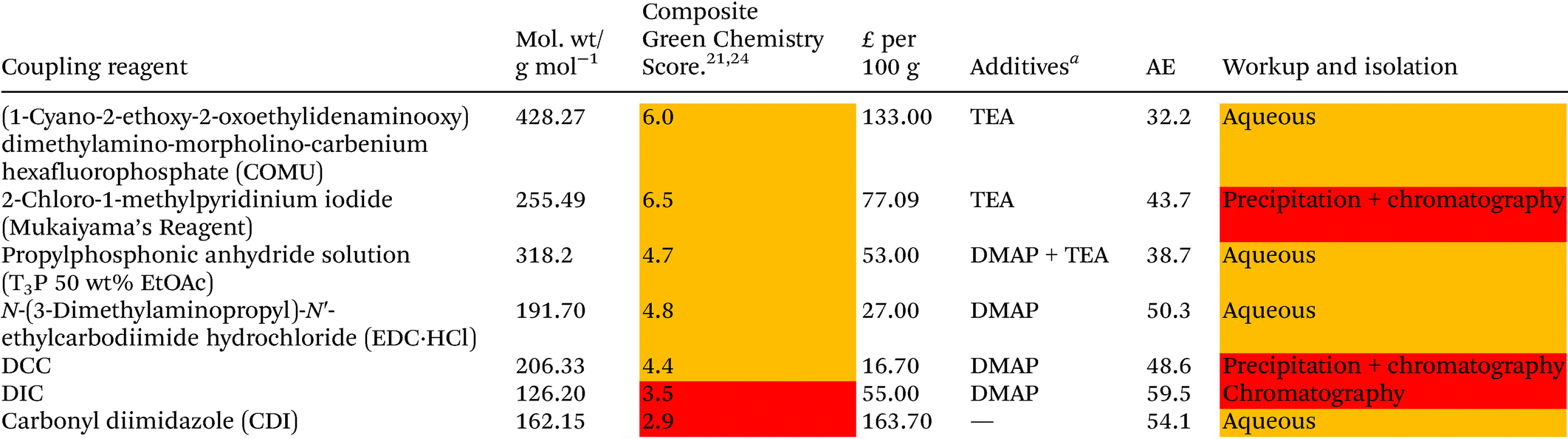
Solvent selection criteria for potentially safer, more sustainable Steglich esterification reactions were ones that performed just as well, if not better than, commonly utilised solvents such as DCM. A short list of potential solvent replacements were chosen from a GSK solvent sustainability guide.22 Solvents with favourable EHS scores encompassing a broad variety of physicochemical properties and functional groups were chosen, Table 1. DCM was also chosen as a solvent against which to benchmark potential replacements. Solvent price was also examined as recent editorial direction provided by ACS Sustainable Chemistry and Engineering has outlined the need to not only consider the environmental, health, safety and sustainability of a solvent, but also its economic value/cost i.e. a “triple bottom line”.23 Potential coupling reagents were similarly identified from a reagent selection guide, Table 2, using DIC as a benchmark reagent.21,24 DCC was not chosen due to the propensity for large amounts of urea precipitate to form during the reactions making them incompatible with high throughput HPLC analysis, vide infra. A theoretical AE was calculated for each coupling reagent in advance of them being utilised to assess the potential wastefulness of each reaction, Table 2. Thus, sustainability criteria can be considered in advance of exploratory chemistry even being conducted, highlighting the potential power of utilising green chemistry metrics from an early stage, Scheme 2. From the outset, Mukaiyama's reagent (2-chloro-1-methylpyridinium iodide a.k.a Muk) and EDC-HCl appeared to be favourable contenders as a potential DIC/DCC replacement from an EHS and AE perspective. | ||
| Scheme 2 Some green and sustainable chemistry considerations. | ||
| a 2.5 kg. |
|---|

|
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 Coupling reagent: acid:alcohol, 5 vol solvent
:1:1 Coupling reagent: acid:alcohol, 5 vol solvent
Four simple test reactions comprising aryl and alkyl acids and alcohols were chosen with which to carry out solvent-reagent screening, Scheme 3. It was envisioned that solvent-reagent-substrate reactivity trends across the four classes of test reactions could be identified to assist in applying solvent-reagent selection criteria to more complicated molecules. Esterification reactions were conducted for 24 hours at room temperature and percentage yields were determined by HPLC, see SI for details. Reactions were conducted at 0.2 M concentration to allow for rapid analysis by HPLC without the need for further dilution. Ideal solvents were considered to be those that could effectively replace DCM as a “drop in” solvent and be broadly applicable across all four reaction classes. Ideal coupling reagents were those that could effectively promote esterification in good yields, produce the least waste, and score favourably according to the criteria outlined in Table 2. Lastly, it was desirable to be able to conduct reactions at room temperature rather than heating if possible as reactions conducted at ambient temperature represent a saving in terms of energy cost.25,26 In total, 192 initial screening reactions comprising eight solvents and six coupling reagents were conducted using model reactions 1–4.
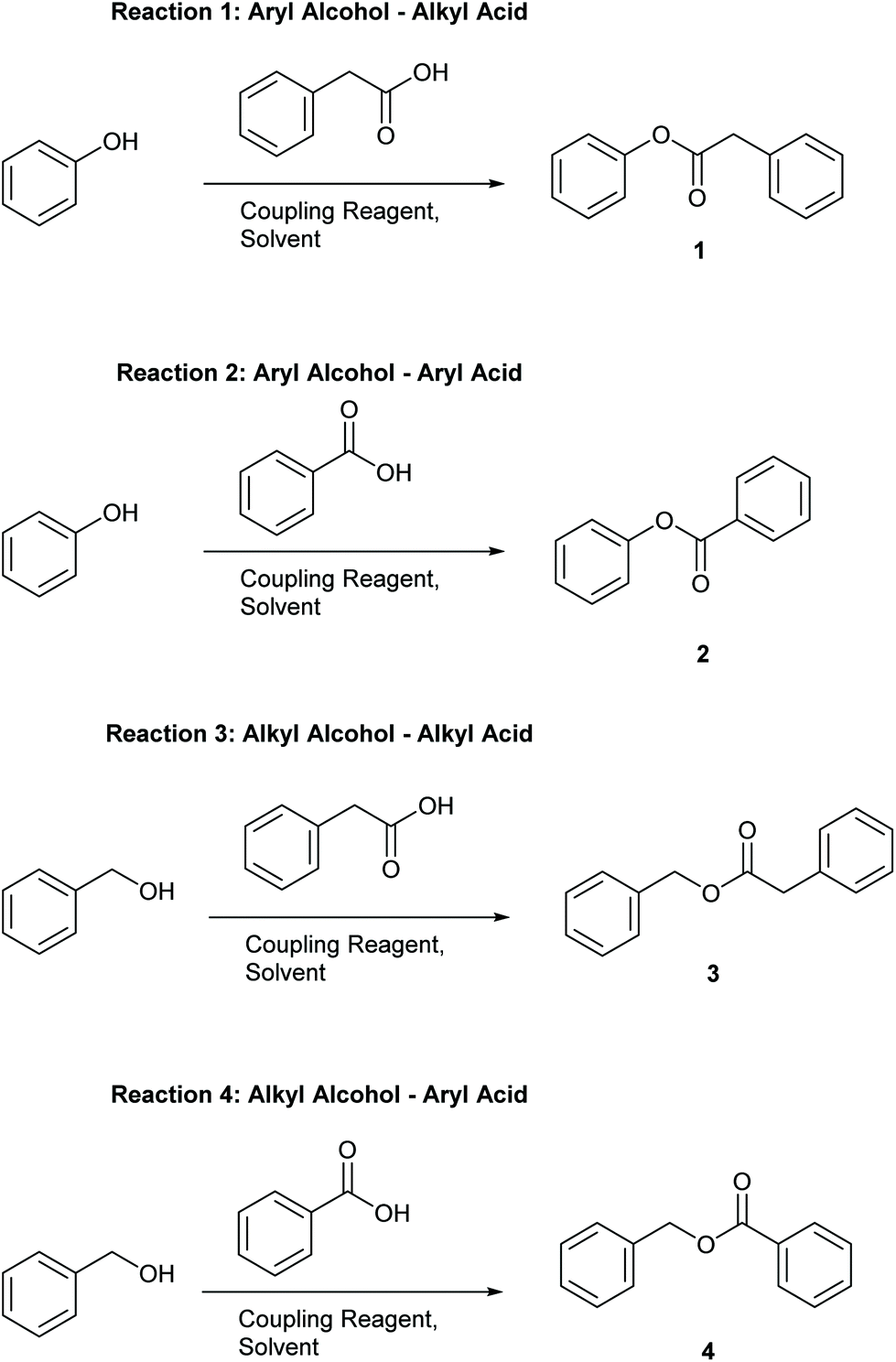 | ||
| Scheme 3 Test reactions chosen for solvent-reagent condition screening. Reaction conditions: 0.2 mmol acid, 0.6 mmol alcohol, 1 mL solvent, 1.0 equivalent of coupling reagent, 3.0 equivalents of TEA if required, 5 mol% DMAP if required. Stir at room temperature for 24 hours. | ||
Results
Coupling reagents
As can be seen from Table 3, the “classic” combination of DIC with 5 mol% DMAP consistently performed well in all four reaction classes. However, DIC is one of the worst scoring reagents in terms of composite EHS score, Table 2, second only to CDI. Due to the poor EHS profile of DIC it was decided not to investigate its use in conjunction with more sustainable solvents any further. Utilising aromatic acids with DIC also lead to a greater formation of N-acyl urea byproduct,13 an occurrence that is not particularly solvent dependant.13 When the reaction of O-acyl urea with alcohol is slow, there is a greater chance of an O–N-acyl shift occurring.13 One area in which DIC excelled was when aryl alcohols were utilised i.e. reactions 1 and 2, Table 3, giving consistently higher yields than any other coupling reagent initially screened.
T3P-DMAP showed comparable performance to DIC in reaction 3 only, when utilising DMC, iPrOAc or CPME as solvents. Very poor conversion was observed when T3P was used in conjunction with aryl acids, reaction 4 (16–41% yield), or with aryl alcohols reactions 1–2. No further investigation using T3P was conducted.
EDC-HCl gave moderate yields overall in all four reactions, performing well in reaction 3 but only when anisole was used as solvent (73% yield). A gummy residue was observed on the bottom of the reaction vials for all of the EDC-HCl reactions and may indicate poor solubility of the EDC-HCl in the test solvents at 0.2 M concentration. Examples of EDC-HCl mediated Steglich esterification reactions demonstrated in the literature by Lutjen et al. are conducted at 0.075 M in CH3CN. Esterification of (E)-cinnamic acid with benzyl alcohol was reported to give an isolated yield of 76%.30 Note: the work of Lutjen et al. employed an excess of DMAP and reactions were conducted at 45° C,30 furthermore CH3CN, whilst one of the more favourable dipolar aprotic solvents, still has a number of sustainability concerns, including its miscibility with water leading to biotreatment and incineration issues.22
CDI performance was moderate for reactions 1, 3, 4 but was very poor in reaction 2, a trend which was similarly observed in the authors’ earlier work employing thiol substrates with carboxylic acids.31
Reactions 2–4, conducted using COMU, generally showed moderate to good conversion, though yields for reaction 1 were lower. COMU was however shown to be a superior reagent when more dilute conditions were employed, with less anhydride side-product observed compared to other reagents at concentrations of 0.05 M, see ESI Table S1† for details.
Muk stood out as an excellent choice of coupling reagents in reactions 3 and 4 in conjunction with most solvents screened, e.g.Table 3, reaction 3, iPrOAc 89%, reaction 4 DMC 80% Furthermore, the reaction could be effectively conducted at room temperature for 24 hours, or heated to 60 °C for 3 hours, giving almost identical results, Table 3 reactions 3, 4 using DMC as solvent. Muk does however require the presence of a tertiary amine base which may preclude it from use with base sensitive moieties.
Overall Muk and EDC-HCl stood out as potential replacements for DIC when EHS profile, yield and potential AE were considered. COMU was not considered for any further investigation as its AE was too poor (32.2, Table 2) to justify its further use. CDI was not broadly compatible, e.g. little to no reactivity in reaction 2. T3P-DMAP showed moderate utility though was considerably poorer in reactions 1, 2 and 4 than EDC-HCl. Investigation into the use of EDC-HCl was later halted due to solubility concerns during optimisation, see ESI Table S3,† leaving Muk as the coupling reagent of choice.
Solvents
Regarding general solvent performance, from the results depicted in Table 3, it can be seen that both DMC, iPrOAc and anisole all performed just as well as, or outperformed DCM across all four reaction classes and were considered potential candidates as replacement solvents. 2-MeTHF and CPME were not as broadly compatible (e.g. reaction 3, Table 3) and cyclopentanone was not as high yielding. A final decision to utilise DMC and iPrOAc was made as they performed the best across all four reaction classes in terms of yield and both have highly favourable EHS green chemistry composite score, Table 2. Anisole was discounted due to its elevated boiling point of 154 °C. iPrOAc was later shown to give lower yields on scale-up to 5 mmol (28% yield reaction 3), see ESI Table S3† and was discounted from further investigation leaving DMC as the leading alternative solvent.Aqueous systems were also briefly investigated using water, water-surfactant mixtures, and biphasic systems though none appeared to give satisfactory results. See ESI Table S1† for more detail.
Safer, more sustainable bases
Having already screened a number of alternative tertiary amine bases, Mukaiyama et al. demonstrated that a number of bases other than TEA could be utilised including tributylamine and 2,6-lutidine.33 It was also demonstrated that as the basicity of the tertiary amine decreased, so too did yield; α-picoline, pyridine and N,N-diethylaniline gave lower yields, descending in that order.33 When considering the EHS profile of the organic bases screened by Mukaiyama et al.,33 2,6-lutidine is considered preferable to TEA as it is less flammable and less toxic.34 Thus, we investigated whether 2,6-lutidine could be utilised as a drop-in replacement for TEA in conjunction with Muk and DMC as reaction solvent. Gratifyingly, yield was not affected by this simple replacement leading to the use of a safer, more sustainable amine base alternative. We also investigated the use of aqueous NaHCO3 mixed with DMC in a 1:9 ratio as an alternative to an organic base. This combination proved to be incompatible with Muk and was not investigated further.
Optimised conditions
Taking yield, general synthetic utility, and EHS profile into account, the reagent-solvent-base combination of Muk–DMC-2,6-lutidine were considered as the optimum for further esterification substrate screening. Reactions could be conducted at room temperature for 24 hours or at 60 °C for 3–8 hours (reaction concentration dependant) with negligible difference observed in terms of yield. Reaction concentrations between 0.5 and 1.0 M are also considered important. See ESI Table S3 and Fig. S1 and 2† for further reaction optimisations conducted around reaction concentration, time, and workup/isolation.Workup and isolation
Workup for esterification reactions utilising Muk have traditionally involved filtration of precipitated 1-methylpyridin-2(1H)-one, compound 6 (Scheme 4), followed by purification by silica gel chromatography.32 Calculating the logP of the pyridone waste product using MarvinSketch predicts a value of 0.24 suggesting that the molecule is water soluble. Similarly protonated 2,6-lutidine should also be water soluble, thus the potential for purifying reaction mixtures using an aqueous organic extraction should be possible. To test this theory the 2 mmol scale synthesis of isopropyl ester 9 was conducted in DMC. The reaction mixture was first diluted with EtOAc to a final volume of 15 mL followed by gravity filtration to remove any precipitated pyridone 6. The organic filtrate was then washed with water (1 × 10 mL) followed by 2 mL brine to break the resulting emulsion. The organic layer was then washed with 1 M aqueous HCl (1 × 10 mL) to remove the 2,6-lutidine, followed by saturated aqueous NaHCO3 (1 × 10 mL) to attempt remove any unreacted carboxylic acid. TLC analysis of the organic layer showed complete removal of 2,6-lutidine and a faint spot remained for unreacted phenylacetic acid, however a strong baseline impurity still remained and the organic layer had a yellow colour (desired product was a colourless oil). The reaction mixture was then concentrated in vacuo and adsorbed directly onto a 3 cm plug of silica gel in a 4 cm diameter sinter funnel. The product was then eluted using a single portion of 50 mL 10% EtOAc:cyclohexane under vacuum filter conditions. This simple silica plug filtration allowed for elution of the desired product in one portion directly into a round bottomed flask. Removal of solvent in vacuo gave an excellent purity profile (see ESI Fig. S1†). For reactions that utilise alcohol starting materials that possess poor aqueous solubility, e.g. phenol for the synthesis of 1, then silica gel chromatography was utilised for purification. The E-factor and process mass intensities of reactions utilising silica gel chromatography were heavily affected due to the larger volumes of solvent utilised.
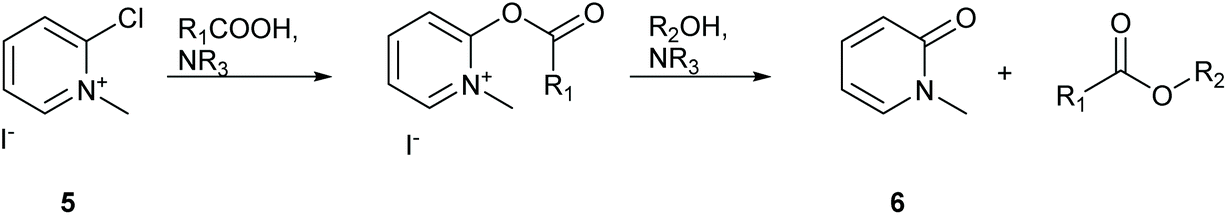 | ||
| Scheme 4 Formation of carboxylic esters utilising Muk 5.32 | ||
Substrate screening
To further evaluate the utility and robustness of the optimised conditions identified, a brief substrate scope was conducted utilising a number of acids and alcohols. Reactions were conducted using equimolar amounts of acid and alcohol unless specified otherwise.From the results depicted in Fig. 2, it can be generally said that the optimised conditions allowed easy access to numerous functionalised esters from primary, secondary, tertiary, and phenolic alcohols, in good to excellent yields. Gratifyingly, sterically hindered t-butyl phenylacetate was isolated in 83% yield. It was also demonstrated that reactions could be driven to completion by using a slight excess of either alcohol or acid, depending on which was considered more precious or more laborious to separate during product isolation (e.g.9: 92% – 2.0 equiv. alcohol, 13: 99% – 2.0 equiv. acid). Phenolic esters were also shown to be accessible through this methodology as well giving a much higher yield of 1 (92%) when compared to the original screening result of just 28%, Table 3. One potential reason for the poorer result observed during the screening in Table 3 may be that the excess of phenol originally used may have solidified in the reaction mixture leading to poor stirring.
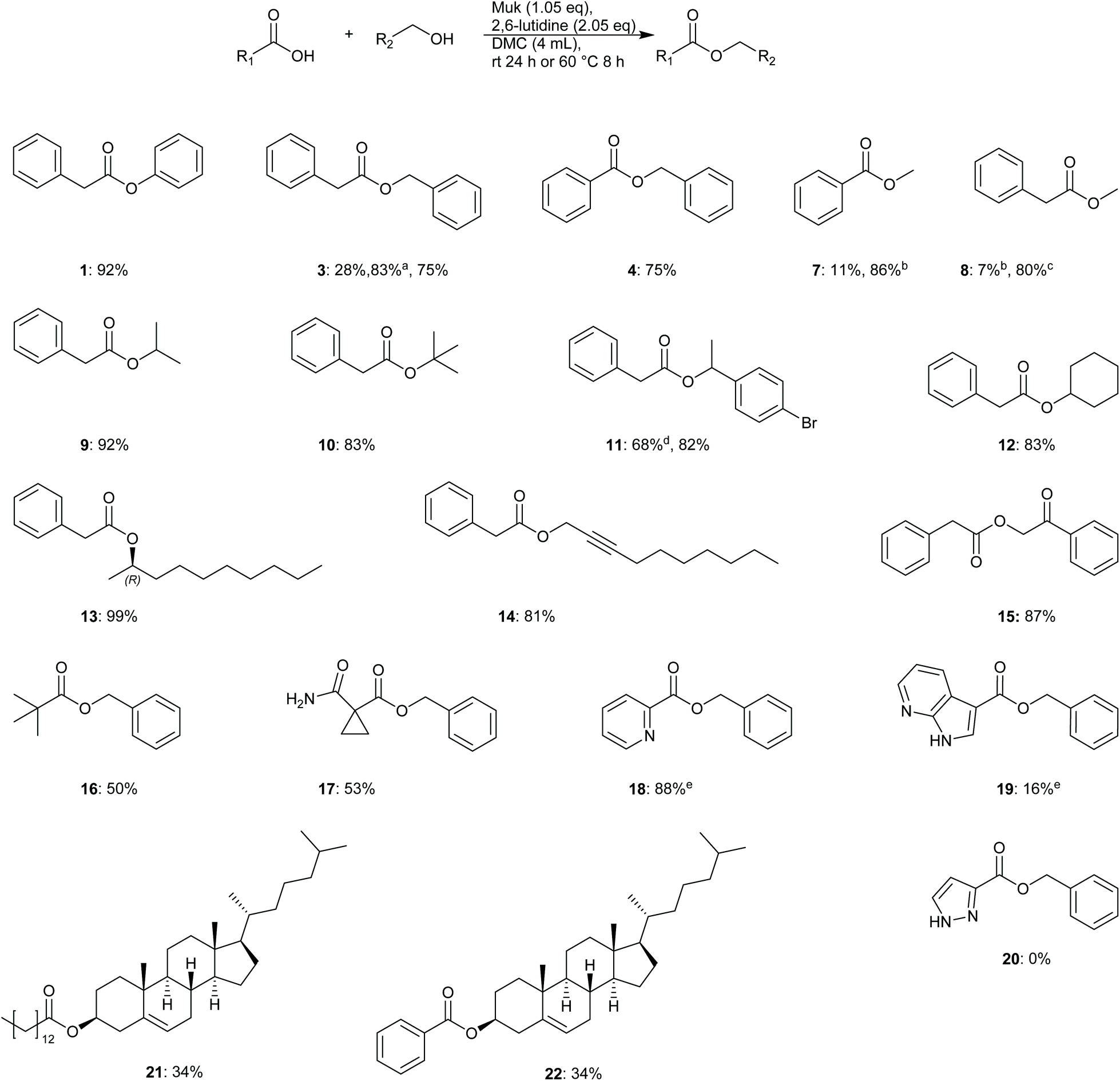 | ||
| Fig. 2 Substrates evaluated for esterification using optimised conditions. Reported yields are isolated unless stated otherwise. aYield determined by HPLC-UV. b5 mol% DMAP added. cReaction conducted in 4:1 DMC:MeOH and 1-methylimidazole (3.0 equiv.) was utilised as base. dReaction conducted in 2 mL solvent. eTEA (3.0 equiv.) used as base. | ||
Acid compatibility was also assessed; picolinic acid was converted to benzyl picolinate 18 in excellent yield (88%), though the stronger base TEA was utilised in this reaction to avoid protonation of the product. More sterically hindered acids such as pivalic acid 16 and cyclopropyl derivative 17 gave poorer yields of 50 and 53% respectively. None of product 20 was observed and only unreacted benzyl alcohol was recovered from this reaction mixture. Steroidal substrates 21–22 were also synthesised but in moderate yields.
Troubleshooting – methyl esters
Difficulties were initially encountered synthesising esters from primary alcohols with poor isolated yields obtained e.g. methyl benzoate 7 (11%) and methyl phenylacetate 8 (9%). HPLC analysis of methyl benzoate reactions showed the presence of predominantly benzoic anhydride. It was discovered that for reactions utilising aryl acids that the addition of 5 mol% DMAP allowed for smooth conversion of benzoic acid to the desired ester in a much higher yield of 86% when 3–5 equivalents of methanol were used. The reason for the improvement may be that reactions utilising benzoic acids can also form benzoic anhydride as noted by Mukaiyama et al. in their original publication.32 The presence of DMAP may serve to catalyse alcohol acetylation via in situ formed benzoic anhydride as reported by Xu et al.35 Unfortunately, the same strategy did not work for reactions utilising phenylacetic acid; no phenylacetic anhydride was detected by HPLC. Instead HRMS analysis of reaction mixtures showed that Muk may be reacting directly with methanol under the reaction conditions to give a 2-methoxy ether derivative, see compound 23, ESI Fig. S3 and 4,† for details. To overcome the poor yields obtained, the base 2,6-lutidine was switched for 1-methylimidazole, as evinced by Zhao et al.,36 and the solvent composition was altered to a 4:1 DMC:methanol mixture. Gratifyingly, a much-improved isolated yield of 8 (80%) was obtained using this method. One potential reason for this improvement may be due to generation of an activated ester via an imidazole species, similar to the reported mechanism for carbonyldiimidazole coupling reactions,37 or potentially a rate enhancement via imidazole·HCl catalysis.37 The effect is most likely not pKa driven as both 1-methylimidazole and 2,6-lutidine possess similar pKa values (H2O: 7.06 vs. 6.72)38,39 and decreasing pKa has previously shown to correlate with decreasing yields.33 Further investigation was not conducted as it was considered outside the main aims and scope of this study.
Green chemistry metrics
For the substrates synthesised, depicted in Fig. 2, a green chemistry metrics evaluation was conducted using the Chem21 workbook.28 Metrics include AE, reaction mass efficiency (RME) and PMI. For the AE calculations, Muk has been considered as a reactant as per the calculations used in Table 2. As can be seen from the metrics calculated in Table 4, the AE of each of the reactions is between 33 and 68, with the average being 46.6. The RME's calculated show unsurprisingly that reactions with poorer yields such as 19 (16%) gave a much poorer efficiency score (7.6). This is simply due to the small mass of product when compared to the total mass of reactants. Average RME was 30.0. Most insightful are the PMI metrics which show a high process mass intensity for the synthesis of all products, ranging from 173 up to 2043. The reason for the high PMI metric across the board is due to the quantities of solvent (both aqueous and organic) that are required for workup and isolation of products. There was also a difference in PMI observed between workup methods e.g. for reactions 1, 3, 4, 7 which were isolated by silica gel chromatography, the PMI was in the range of 304–508. For reactions isolated by aqueous workup and silica plug filtration 9–12 the PMI values ranged from 173–282. For clarity, aqueous workup solvents such as 1 M HCl and saturated aqueous bicarbonate solutions have been included in the PMI calculation. Overall, due to the quantities of solvent required for workup and isolation, this methodology will suffer from poorer PMI unless solvent recycling is implemented, or isolation by other means such as distillation/precipitation/recrystallisation can be achieved.| Product | AE | RME | PMI |
|---|---|---|---|
| a Product purified by silica gel chromatography. b Purified by aqueous workup and silica plug filtration. See workup and isolation section for details. c No aqueous workup was used. | |||
| 1a | 43.7 | 39.0 | 304.6 |
| 3a | 45.3 | 33.0 | 350.5 |
| 4a | 43.7 | 31.8 | 373.6 |
| 7a | 33.2 | 24.0 | 507.6 |
| 8a | 35.4 | 23.9 | 494.5 |
| 9b | 39.5 | 31.0 | 276.2 |
| 10b | 41.3 | 33.2 | 281.9 |
| 11b | 53.9 | 42.8 | 173.4 |
| 12b | 44.4 | 35.7 | 249.3 |
| 13b,c | 50.3 | 29.2 | 301.2 |
| 14b | 49.9 | 39.5 | 203.4 |
| 15a | 48.2 | 40.7 | 269.6 |
| 16a | 41.3 | 20.1 | 615.3 |
| 17a | 44.5 | 23.0 | 692.1 |
| 18a | 43.8 | 37.3 | 322.9 |
| 19a | 48.0 | 7.6 | 2043.3 |
| 21b | 68.6 | 23.1 | 376.2 |
| 22a | 64.2 | 21.2 | 464.4 |
Conclusion
In conclusion, we have successfully identified safer, greener, more sustainable solvent-reagent conditions with which to conduct Steglich-type esterification reactions via solvent-reagent condition screening. The results of the solvent-reagent screen have been compiled, Table 3, to provide a visual solvent-reagent selection guide for the formation of esters and will provide a useful go-to reference point for organic and medicinal chemists seeking to conduct this type of transformation. The optimised conditions utilised equimolar quantities of both alcohol and acid, were conducted in safer, more sustainable solvent dimethyl carbonate, and used Muk instead of DIC/DCC. The use of Muk allows for safer esterification reactions to be conducted free from the hazards associated with the use of DIC/DCC. Furthermore, alternative base 2,6-lutidine was utilised to great effect, effectively replacing more harmful bases such as TEA with a safer alternative.34 The optimised reaction conditions were shown to proceed at either room temperature over a 24 hours period or at 60 °C for 8 hours, though some hindered substrates required longer reaction times. The general utility of the new reaction conditions were exemplified using a set of alcohols and carboxylic acids to form building-block like molecules. In general, good to excellent yields were obtained, even for hindered alcohols such as t-butyl alcohol (83%), though hindered acids such as pivalic acid appeared to fare less well giving moderate yields (50%). One drawback remains however and that is the method of isolation. Though purification could be achieved in rapid time (30 min), large PMI values were observed for several products due to isolation by aqueous workup followed by silica plug filtration or by silica gel chromatography. The PMI values will only be reduced though the use of other isolation methods such as distillation or countered by solvent recycling. In summary, the “classic” Steglich esterification solvent-reagent combination of a carbodiimide coupling reagent, DMAP and DCM can effectively be replaced with the greener, safer, more sustainable combination of Mukaiyama's reagent, 2,6-lutidine, and dimethylcarbonate.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the EPSRC for funding via Prosperity Partnership EP/S035990/1 and GlaxoSmithKline for postdoctoral funding. We would like to kindly thank Nicholas Bennett for helpful discussions in preparation of this manuscript. We would also like to kindly thank Heather Barnett and Ross Hryczanek for their support of Kyran and Jack during their student placements to GSK during 2020.References
- P. Ertl, E. Altmann and J. M. McKenna, J. Med. Chem., 2020, 63, 8408–8418 CrossRef CAS PubMed.
- G. D. Yadav and M. S. M. Mujeebur Rahuman, Clean Technol. Environ. Policy, 2003, 5, 128–135 CrossRef CAS.
- O. D. Sparkman, Z. E. Penton and F. G. Kitson, in Gas Chromatography and Mass Spectrometry, ed. O. D. Sparkman, Z. E. Penton and F. G. Kitson, Academic Press, Amsterdam, 2nd edn, 2011, DOI:10.1016/B978-0-12-373628-4.00015-0, pp. 285–292.
- R. J. Ouellette and J. D. Rawn, in Organic Chemistry, ed. R. J. Ouellette and J. D. Rawn, Elsevier, Boston, 2014, DOI:10.1016/B978-0-12-800780-8.00021-8, pp. 699–745.
- A. D. Godwin, in Applied Polymer Science: 21st Century, ed. C. D. Craver and C. E. Carraher, Pergamon, Oxford, 2000, DOI:10.1016/B978-008043417-9/50011-8, pp. 157–175.
- M. Tsakos, E. S. Schaffert, L. L. Clement, N. L. Villadsen and T. B. Poulsen, Nat. Prod. Rep., 2015, 32, 605–632 RSC.
- M. Kouichi, Y. Rina and O. Yohei, Recent Advances in the Synthesis of Carboxylic Acid Esters, Carboxylic Acid – Key Role in Life Sciences, ed. G. I. Badea and G. L. Radu, IntechOpen, 2018, DOI:10.5772/intechopen.74543.
- E. Fischer and A. Speier, Ber. Dtsch. Chem. Ges., 1895, 28, 3252–3258 CrossRef CAS.
- A. G. M. Barrett and D. Christopher Braddock, Chem. Commun., 1997, 351–352, 10.1039/a606484a.
- Y. Sugi, K. Komura, A. Ozaki and N. Ieda, Synthesis, 2008, 3407–3410 CrossRef.
- G. Höfle, W. Steglich and H. Vorbrüggen, Angew. Chem., Int. Ed. Engl., 1978, 17, 569–583 CrossRef.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed. Engl., 1978, 17, 522–524 CrossRef.
- A. H. M. Schotman, Recl. Trav. Chim. Pays-Bas, 1991, 110, 319–324 CrossRef CAS.
- S.-Y. Han and Y.-A. Kim, Tetrahedron, 2004, 60, 2447–2467 CrossRef CAS.
- A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS PubMed.
- F. S. Gibson, M. S. Park and H. Rapoport, J. Org. Chem., 1994, 59, 7503–7507 CrossRef CAS.
- B. Neises and W. Steglich, Org. Synth., 1985, 63, 183 CrossRef CAS.
- J. Inanaga, K. Hirata, H. Saeki, T. Katsuki and M. Yamaguchi, Bull. Chem. Soc. Jpn., 1979, 52, 1989–1993 CrossRef CAS.
- I. Dhimitruka and J. SantaLucia, Org. Lett., 2006, 8, 47–50 CrossRef CAS PubMed.
- A. Jordan, P. Stoy and H. F. Sneddon, Chem. Rev., 2021, 121, 1582–1622 CrossRef CAS PubMed.
- A. Jordan and H. F. Sneddon, Green Chem., 2019, 21, 1900–1906 RSC.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- D. T. Allen, N. Gathergood, P. Licence and B. Subramaniam, ACS Sustainable Chem. Eng., 2020, 8, 14627–14629 CrossRef CAS.
- J. P. Adams, C. M. Alder, I. Andrews, A. M. Bullion, M. Campbell-Crawford, M. G. Darcy, J. D. Hayler, R. K. Henderson, C. A. Oare, I. Pendrak, A. M. Redman, L. E. Shuster, H. F. Sneddon and M. D. Walker, Green Chem., 2013, 15, 1542–1549 RSC.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, 1998, p. 30 Search PubMed.
- Price for 2.5 L solvent according to PSDS, https://chase-home-offsite.psds.ac.uk/, (accessed 17/06/2021).
- C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–3121 RSC.
- Reagent pricing per 100 g according to PSDS, https://chase-home-offsite.psds.ac.uk/, (accessed 17/06/2021).
- A. B. Lutjen, M. A. Quirk, A. M. Barbera and E. M. Kolonko, Bioorg. Med. Chem., 2018, 26, 5291–5298 CrossRef CAS PubMed.
- A. Jordan and H. F. Sneddon, Green Chem., 2019, 21, 1900–1906 RSC.
- T. Mukaiyama, M. Usui, E. Shimada and K. Saigo, Chem. Lett., 1975, 4, 1045–1048 CrossRef.
- K. Saigo, M. Usui, K. Kikuchi, E. Shimada and T. Mukaiyama, Bull. Chem. Soc. Jpn., 1977, 50, 1863–1866 CrossRef CAS.
- R. K. Henderson, A. P. Hill, A. M. Redman and H. F. Sneddon, Green Chem., 2015, 17, 945–949 RSC.
- S. Xu, I. Held, B. Kempf, H. Mayr, W. Steglich and H. Zipse, Chem. – Eur. J., 2005, 11, 4751–4757 CrossRef CAS PubMed.
- H. Zhao, Z. Song, J. V. Cowins and O. Olubajo, Int. J. Mol. Sci., 2008, 9, 33–44 CrossRef CAS PubMed.
- E. K. Woodman, J. G. K. Chaffey, P. A. Hopes, D. R. J. Hose and J. P. Gilday, Org. Process Res. Dev., 2009, 13, 106–113 CrossRef CAS.
- 1-Methylimidazole Product Information, https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma/Product_Information_Sheet/1/m8878pis.pdf, (accessed 01/06/2021).
- S. Shimizu, N. Watanabe, T. Kataoka, T. Shoji, N. Abe, S. Morishita and H. Ichimura, in Ullmann's Encyclopedia of Industrial Chemistry, 2000, DOI:10.1002/14356007.a22_399.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc02251b |
| ‡ These authors contributed equally to this publication. |
| This journal is © The Royal Society of Chemistry 2021 |