
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical hydrogenation of bioprivileged cis,cis-muconic acid to trans-3-hexenedioic acid: from lab synthesis to bench-scale production and beyond†
Marco Nazareno
Dell'Anna
 ab,
Mathew
Laureano
ab,
Hamed
Bateni
ab,
John E.
Matthiesen
ab,
Ludovic
Zaza
ab,
Michael P.
Zembrzuski
a,
Thomas J.
Paskach
a and
Jean-Philippe
Tessonnier
*ab
ab,
Mathew
Laureano
ab,
Hamed
Bateni
ab,
John E.
Matthiesen
ab,
Ludovic
Zaza
ab,
Michael P.
Zembrzuski
a,
Thomas J.
Paskach
a and
Jean-Philippe
Tessonnier
*ab
aDepartment of Chemical and Biological Engineering, Iowa State University, Ames, IA 50011, USA. E-mail: tesso@iastate.edu
bCenter for Biorenewable Chemicals (CBiRC), Ames, IA 50011, USA
First published on 27th July 2021
Abstract
The integration of microbial and electrochemical conversions in hybrid processes broadens the portfolio of products accessible from biomass. For instance, sugars and lignin monomers can be biologically converted to cis,cis-muconic acid (ccMA), a bioprivileged intermediate, and further electrochemically upgraded to trans-3-hexenedioic acid (t3HDA). This novel monounsaturated monomer is gaining increasing attention as it can substitute adipic acid in Nylon 6,6 to introduce desired properties and yield polyamides with performance advantages. The implementation of t3HDA for advanced polymer production is, however, hampered by the low productivities achieved to date, in the order of milligrams per hour per cm2. Here, we report on new synergies between microbial and electrochemical conversions and present a simple strategy to enhance the productivity of t3HDA by over 50 times. Specifically, we show that the broth composition has a dramatic role on the subsequent electrochemical step. Broth with neutral pH and high ccMA titer obtained from bacteria was found to enhance the electrochemical hydrogenation while impeding the parasitic hydrogen evolution reaction. As a result, high productivities were achieved under industrially-relevant current densities (200–400 mA cm−2). The effect of other parameters that are key for scale up and continuous operation, namely reactor configuration, potentiostatic/galvanostatic operation mode, and cathode material are also discussed. The experimental results served as input parameters for a detailed technoeconomic analysis and the blueprint of a hybrid microbial electrosynthesis process for t3HDA production.
Introduction
Biomass represents an abundant source of renewable carbon that can be harnessed to curb the chemical industry's environmental footprint. However, the adoption of biomass as a feedstock for chemical manufacturing remains slow due to unfavorable economics. For instance, biobased adipic acid, terephthalic acid, acrylonitrile, and isoprene (i.e. direct replacements) currently suffer higher production costs than when these monomers are manufactured from petroleum.1–4 It becomes increasingly clear that a direct competition with the petrochemical industry for the production of commodities is futile in an era of low oil prices. Therefore, opportunities for the biorenewable chemical industry to grow and strive will initially come from chemicals that are not easily accessible from fossil feedstocks, specifically functional replacements and novel compounds. These bioproducts are extremely valuable as they give access to materials with performance advantages over their petrochemical counterparts.5,6 The most successful example to date is 2,5-furandicarboxylic acid (FDCA). This performance-advantaged monomer is central to polyethylene furanoate (PEF), a 100% biobased and recyclable polyester with superior mechanical and thermal properties than polyethylene terephthalate (PET) and 3–6 times better O2 and CO2 barrier properties, respectively.7,8The report published by the U.S. Department of Energy on the “Top 10” platform chemicals from biomass has guided much of the work on biorenewable chemicals over the past decade, including on FDCA.9,10 However, recent reaction network analysis has revealed new biobased platform molecules with similar or better potential for diversification.6,11 Among the discovered compounds, muconic acid (MA) and triacetic acid lactone (TAL) were shown to possess molecular structures with distinct reactivity patterns that give readily access to both direct replacements and novel species with broad end-use applications.12–15 The unique potential for diversification of these biopriviledged compounds offers significant advantages for derisking the associated technologies and pushing new molecules to market. MA and TAL are produced through synthetic biology, using metabolically-engineered yeasts and bacteria.15–17 As such, the compounds co-exist in fermentation broths with a wide range of inorganic salts, amino acids, and biogenic impurities that are susceptible to alter the heterogeneous catalysts used in downstream chemical transformations.16,18–21 For example, the Miller and Dumesic groups demonstrated that common amino acids like cysteine and methionine poison precious metal catalysts, sometimes irreversibly.20,21 These catalysts only tolerate minute amounts of impurities, typically in the ppm range. Therefore, costly separation and purification steps are required to recover the intermediate before it can be further reacted. These steps can account for up to 60% of the total production cost and,22 as a result, jeopardize the adoption and broad implementation of these chemicals despite an increasing demand for performance-advantaged chemicals and materials. Hence, alternative conversions that tolerate complex feedstocks are highly desired (Scheme 1).
 | ||
| Scheme 1 cis,cis-Muconic acid (ccMA) produced biologically from sugar or lignin can be electrochemically hydrogenated to trans-3-hexenedioic acid (t3HDA), a monounsaturated analogue of adipic acid (AA). In contrast to thermocatalytic conversions, the electrochemical route is not inhibited by the biogenic impurities. The electrochemical transformation can take place directly in the fermentation broth, under ambient conditions, using renewable electricity and the water molecules of the broth as sole reagents. | ||
Hybrid microbial electrosynthesis (HMES) emerges as a technology with unparalleled advantages for bioprocessing and bioprocess intensification due to the seamless integration of microbial fermentation and organic electrosynthesis, remarkable resistance of electrode materials to biogenic impurities, mild operating conditions, use of electrons as green reagent for redox transformations, and facile integration with renewable energy sources.16,23–33 Taking also into account that HMES promotes the use of the fermentation broth's inorganic salts as electrolyte and water solvent as hydrogen source for the electrochemical step, it complies with most of the 12 Principles of Green Chemistry,34 making this route particularly attractive from a sustainability perspective, especially if green electrons are supplied by wind or solar energy. With the rapid drop in renewable electricity costs, the electrification of chemical transformations currently presents the most promising opportunities to promote sustainability without compromising on economic competitivity.35–37
In addition to the aforementioned technical advantages, HMES also opens new reaction pathways distinct from thermochemical and thermocatalytic routes and that can give access to new products that are hardly obtainable through conventional transformations.16,18,19,38,39 Recently, our group explored the electrohydrogenation of cis,trans-muconic acid (ctMA) and demonstrated that the reaction can take place directly upon fermentation with metabolically-engineered S. cerevisiae using protons provided by the acidic (pH 4) broth.16,18,19 This electrohydrogenation produced the novel compound trans-3-hexenedioic acid (t3HDA) with near stoichiometric yields and close to 100% faradaic efficiency (FE) using a lead cathode.18 The value of this monounsaturated diacid was subsequently demonstrated by substituting adipic acid with t3HDA to introduce desired properties including hydrophobicity into conventional polyamides, thereby enabling the synthesis of performance-advantaged Nylon 6,6.40–42 This example highlights the opportunities arising from hybrid biomass conversions combining synthetic biology and electrochemistry. However, the road to kiloton-scale electrobiorefineries remains long and uncertain as this technology is in its infancy.
The present work investigates the electrohydrogenation of cis,cis-muconic acid (ccMA) to t3HDA in an electrochemical flow reactor and identifies parameters that are key to reach a kilogram per month bench-scale production. Combining H/D isotopic tracing, cyclic voltammetry, linear sweep voltammetry, and ccMA solubility measurements, it is shown that fermentation and electrohydrogenation are not independent steps. Instead, synergies exist that, if controlled, can enhance the t3HDA production rate by over 50 times. Other parameters related to technological aspects such as flow reactor configuration, up and downstream operation, operating voltage, and current density have been studied. We also show how to reduce the effect of cathodic corrosion and leaching to increase the longevity and durability of the electrochemical system. The data collected served as input parameters for a detailed technoeconomic analysis of the integrated process.
Results and discussion
Kinetic and mechanistic investigations have shown that the thermocatalytic hydrogenation of MA proceeds sequentially to produce adipic acid with near stoichiometric yields.43–47 Muconic acid first undergoes 2,3- and 2,5-hydrogenations to form 2- and 3-hexenedioic acid, respectively, which are further hydrogenated to the saturated diacid (Scheme 1). The 2,3-hydrogenation pathway is preferred when the reaction is performed in an organic solvent using a precious metal catalyst and H2 gas. In contrast, we have previously reported that the electrohydrogenation of MA in the fermentation broth preferentially produces t3HDA and that further hydrogenation to adipic acid is hardly ever observed, even with a Pd foil cathode.19 These differences may be due to solvent effects, the nature of the metal electrode, and/or site blocking by salts and biogenic impurities present in the broth. Any of these effects may alter the kinetics of parallel and sequential reactions as well as the reaction mechanism. Therefore, the thermochemical and electrochemical reactions were further investigated in order to identify strategies to increase t3HDA productivity and reach bench-scale production levels.Mechanisms of the thermochemical and electrochemical hydrogenations of cis,cis-muconic acid in aqueous media
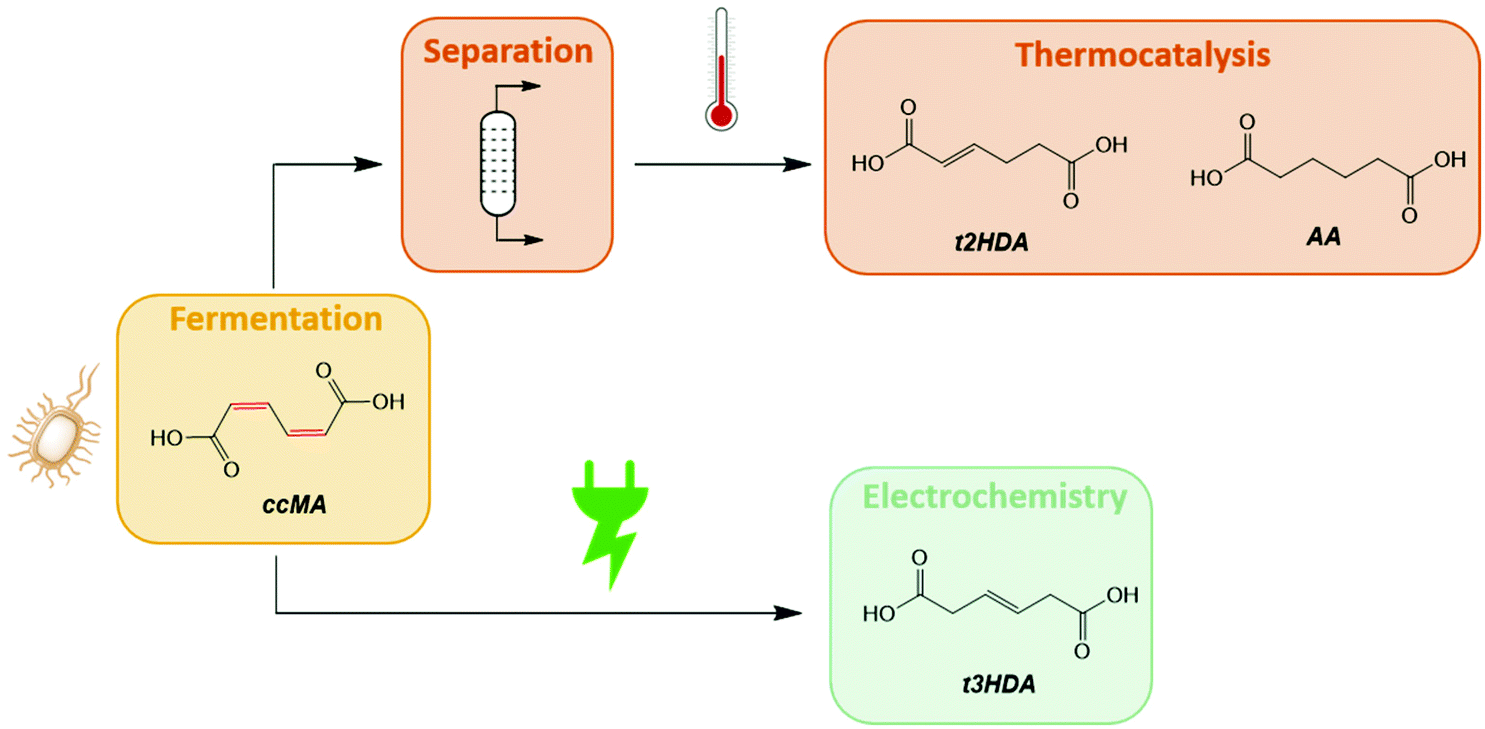
As prior kinetic studies were performed in organic media,47 we first investigated the effect of water and of the fermentation broth on the thermocatalytic hydrogenation of ccMA. The reactions were performed with Pd/C under flowing H2 at 70 °C using a conventional batch reactor system. No substantial change in the reaction pathway was observed with water as a solvent compared to prior studies (Fig. 1a). ccMA was converted to the monounsaturated diacids and further hydrogenated to adipic acid. It is to be noted that both the cis and trans isomers of 2HDA and 3HDA were detected, i.e. the thermocatalytic reaction was not stereoselective. In addition, the production of 3HDA was delayed relative to 2HDA, which could indicate that 3HDA was not a product of ccMA hydrogenation but, instead, was formed through isomerization of 2HDA.44 Additional experiments starting with 2HDA as a reactant would be required to test this hypothesis. However, these experiments are not currently possible as 2HDA is not commercially available.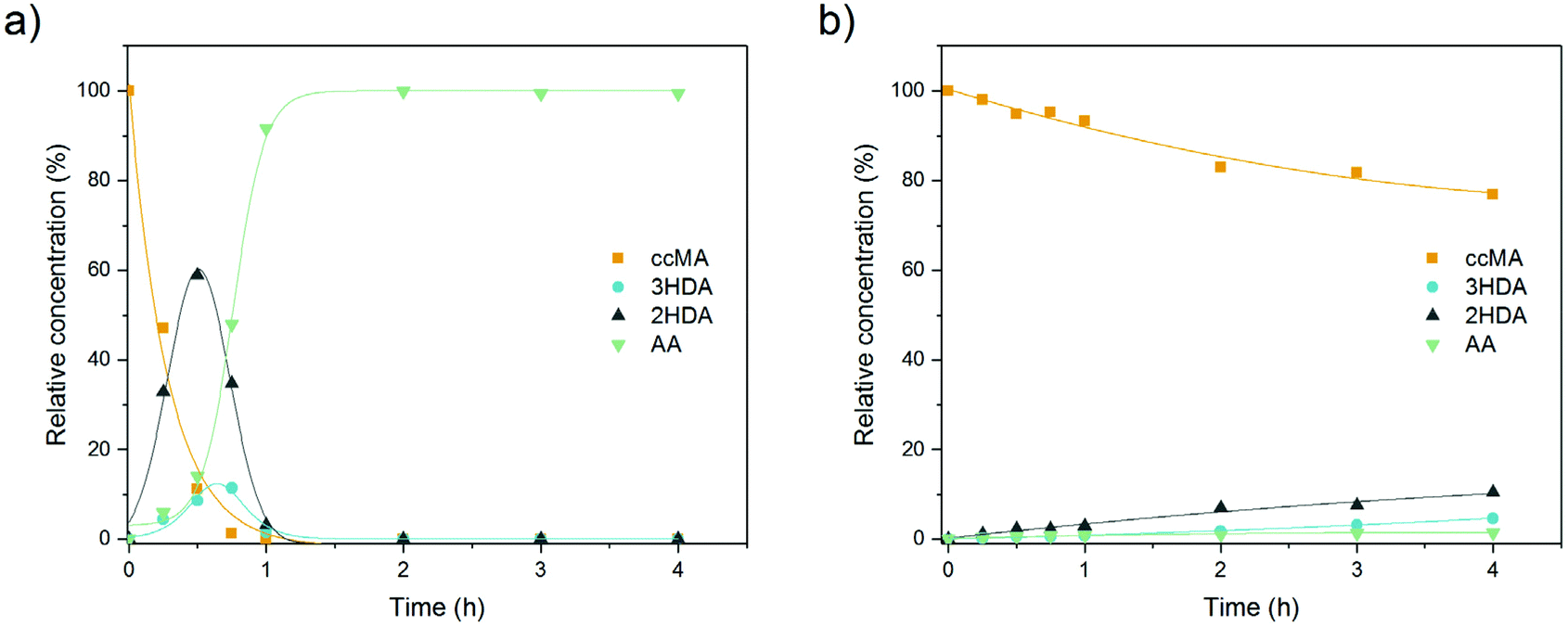 | ||
| Fig. 1 Relative concentrations of ccMA, 2HDA, 3HDA, and AA during the thermocatalytic hydrogenation of (a) a ccMA model solution and (b) ccMA in the fermentation broth. 2HDA and 3HDA indicate a mixture of cis and trans isomers where the trans isomer is the major product. | ||
Substantial differences were observed when replacing the model solution by fermentation broth (Fig. 1b). Although the concentration of ccMA was the same as for the model solution, the initial rate dropped by a factor 25 and the hydrogenation to adipic acid was nearly suppressed. The monounsaturated acids became the main reaction products with a 2HDA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3HDA ratio of nearly 1:1. Additional experiments using model solutions containing representative impurities found in the broth revealed that leftover proteins were the main cause of catalyst deactivation (Fig. S1†). In contrast to prior studies, amino acids (alanine, tyrosine, and glutamic acid) had only a minor role in the observed change in rate and selectivity. In any case, we can safely exclude solvent effects and selective site blocking as being responsible for the high rate and selectivity to t3HDA observed for the electrohydrogenation reaction.
:3HDA ratio of nearly 1:1. Additional experiments using model solutions containing representative impurities found in the broth revealed that leftover proteins were the main cause of catalyst deactivation (Fig. S1†). In contrast to prior studies, amino acids (alanine, tyrosine, and glutamic acid) had only a minor role in the observed change in rate and selectivity. In any case, we can safely exclude solvent effects and selective site blocking as being responsible for the high rate and selectivity to t3HDA observed for the electrohydrogenation reaction.
Considering the significant differences observed for the thermo- and electrohydrogenation reactions, H/D isotopic tracing experiments were performed to gain further insights into the mechanism of the electrochemical transformation. To this end, the electrohydrogenation was performed with model solutions under both acidic (pD 1 using 0.1 M D2SO4/D2O) and neutral conditions (pD 7 using 0.1 M K2SO4/D2O adjusted with a few drops of KOD) and the reaction products were analyzed by 1H NMR (Fig. 2 and S2†). The signals at 5.66 ppm and 3.10 ppm with a 1:1 integration revealed that deuterium was exclusively added to the carbon atoms in the 2 and 5 positions. This result, along with the fact that deuterated 2HDA was never observed in our experiments, excludes a pathway involving a 2,3-hydrogenation followed by isomerization of 2HDA to 3HDA. 2,5-Hydrogenations have been documented for unsaturated dienes and were shown to follow a radical-type mechanism.48 The formation of a radical anion is facilitated by the electron withdrawing effect of the carboxylic functional groups and the molecular conjugation that stabilizes the radical intermediate.48 Hence, the electrohydrogenation is likely to proceed through a sequential electron transfer-chemical protonation (ECEC) mechanism where the cathode first supplies an electron to ccMA to form a radical that accepts a proton either from the water solvent (at neutral pH) or from the electrolyte (for reactions at low pH). The molecule then reorganizes and undergoes a second electronation-protonation in the α position of the other carboxylic acid group (Scheme 2). Further H/D isotopic tracing experiments performed with Pb and Pd electrodes under acidic conditions revealed no effect from the pH (pD) and no effect from hydrogen chemisorbed on the precious metal catalyst (Fig. S2†). These experiments further support that both H2O and H+ (from the electrolyte) can serve as a source of hydrogen, and chemisorbed hydrogen from HER does not participate in the reaction. These observations are consistent with the literature on electrochemical hydrogenations in aqueous media via ECEC mechanism.33,49–53
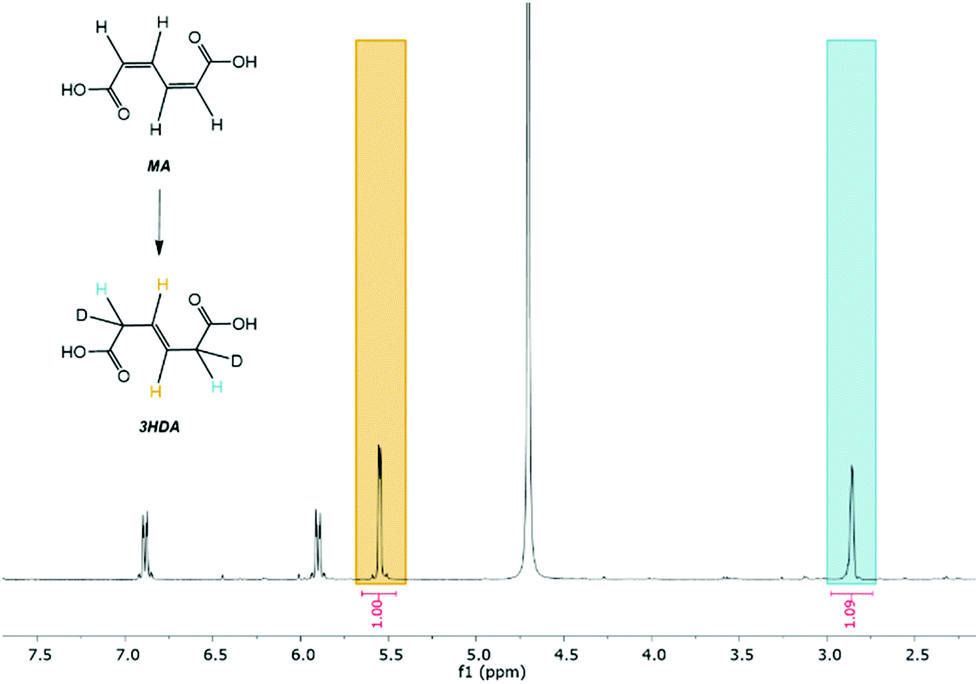 | ||
| Fig. 2
1H NMR spectrum collected after electrohydrogenation of 5 g L−1 of ccMA in 0.1 M K2SO4/D2O using a lead (Pb) electrode kept at a constant potential of −2.0 V vs. Ag/AgCl. The pD was adjusted to 7 with a few drops of KOD. H2 gas was bubbled throughout the reaction to investigate the possible contribution of H2 formed through the hydrogen evolution reaction (HER). The signals at 3.10 and 5.66 ppm correspond to the protons in α and β of the carboxylic functional groups. The ratio between the integrated areas of the peaks is 1:1, which indicates a 2,5-addition of deuterium during the electrohydrogenation reaction. | ||
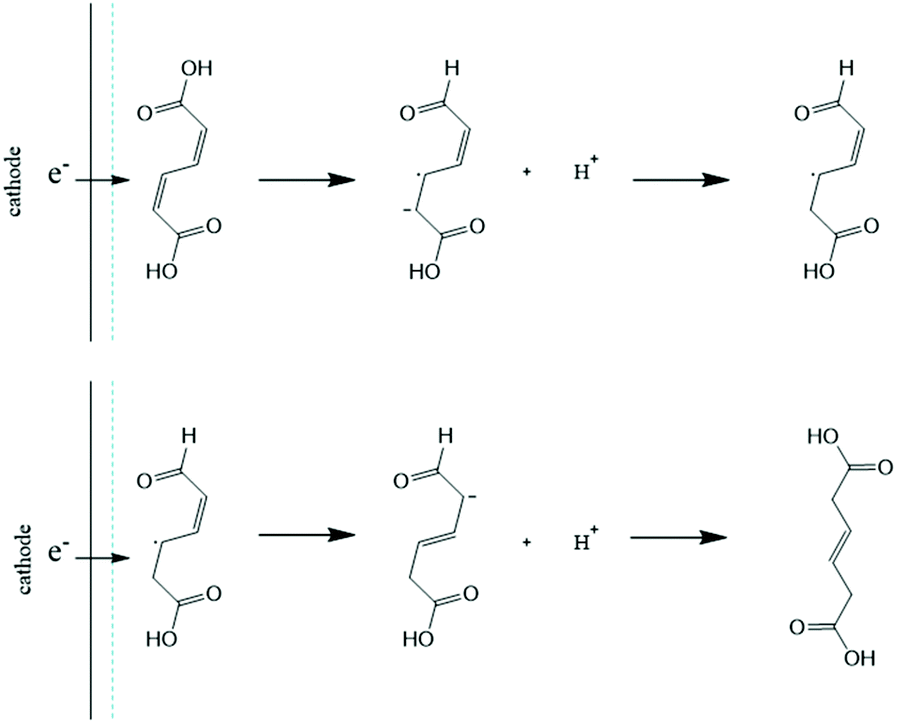 | ||
| Scheme 2 Proposed electronation-protonation mechanism for the electrohydrogenation of ccMA to t3HDA. | ||
Electronation–protonations are well-documented in organic electrosynthesis. They consist in an outer-sphere process where electrons tunnel through a thin layer of electrolyte to react with the substrate in solution.33,54 As such, this process does not require the substrate to adsorb and bind to the cathode's surface. This mechanism explains the high tolerance of the studied process to salts and biogenic impurities, i.e. species at the electrode surface do not disturb the outer-sphere electrohydrogenation reaction. This mechanism implies a relatively simple rate law where [ccMA] is the main factor governing the rate of the reaction. It also suggests that the ccMA radical anion may react with any hydrogen source, including water molecules under neutral pH conditions. This hypothesis, if confirmed, would provide interesting routes for process optimization and was, therefore, further explored.
Solubility of cis,cis-muconic acid under neutral pH conditions and implications on its electrohydrogenation
A wide range of organic compounds bearing amine or carboxylic functional groups present a solubility that depends on pH. The protonation/deprotonation of the molecule yields organic ions with significantly higher solubility in aqueous media than the neutral form of the molecule.55–57 In the case of carboxylic acids, the solubility is an exponential function of the pH. Fig. 3 shows the concentration of ccMA at saturation as a function of pD. The data points were fitted using the Henderson–Hasselbalch equation where the asymptotic value of the solubility at very low pD represents the solubility limit of the free acid in aqueous media, which for ccMA is 0.5 g L−1. Conversely, at pD above 5 the solubility reaches values >70 g L−1, which corresponds to the solubility limit of the muconate salt. This limit also depends on the nature of the counterion and, therefore, determining the exact value is beyond the scope of this study.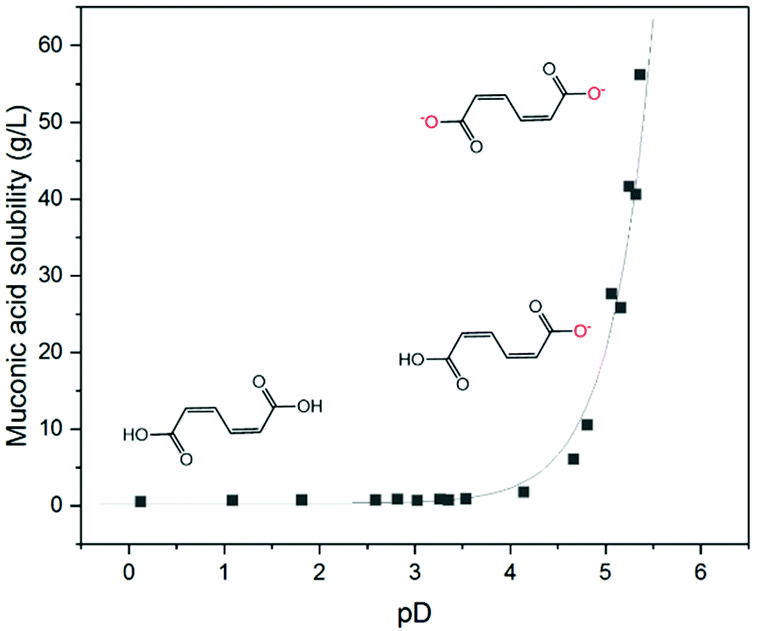 | ||
Fig. 3
ccMA solubility as a function of pD. The experimental data points were fitted using the Henderson–Hasselbalch equation 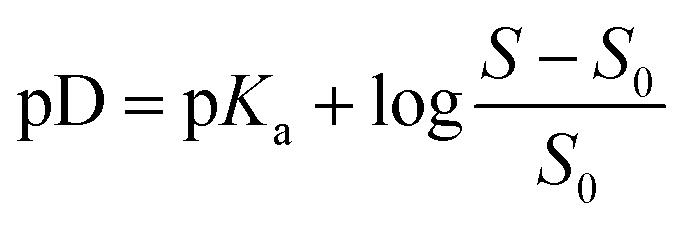 where S is the solubility of the ionic species (cis,cis-muconate) and S0 is the solubility of free acid (cis,cis-muconic acid). where S is the solubility of the ionic species (cis,cis-muconate) and S0 is the solubility of free acid (cis,cis-muconic acid). | ||
As [ccMA] is the main factor governing the rate of the reaction, Fig. 3 suggests high rates and high productivities can only be achieved near neutral pH. However, the species in solution under these conditions would be the muconate dianion (ccMA2−). ccMA2− may display a different reactivity than ccMA and, in addition, the repulsion of the dianion by the negatively charged cathode may ultimately hamper or even prevent the cathodic hydrogenation reaction. Therefore, we investigated next the reactivities of ccMA and ccMA2− by cyclic voltammetry (CV). Fig. 4 shows the voltammograms recorded for the blank electrolytes and 0.5 g L−1ccMA solutions at pH 1 and 7. In the presence of ccMA, the CVs exhibited an onset potential at −0.6 V for pH 1 and −1.5 V vs. Ag/AgCl for pH 7, followed by a mass transfer limiting current region corresponding to the electrochemical hydrogenation to t3HDA. The second onset potential (−1.0 V for pH 1 and −2.0 V for pH 7) present for both the blank and MA solutions corresponds to the parasitic HER. The sharp peaks at −0.45 V and −0.75 V vs. Ag/AgCl in the voltammograms correspond to the oxidation and reduction of the lead electrode with the typical Gaussian shape for surface reactions.58 It is also evident that the reverse reaction, i.e. t3HDA oxidation/dehydrogenation, did not take place in the scanned window of potential, which is consistent with an irreversible hydrogenation reaction.
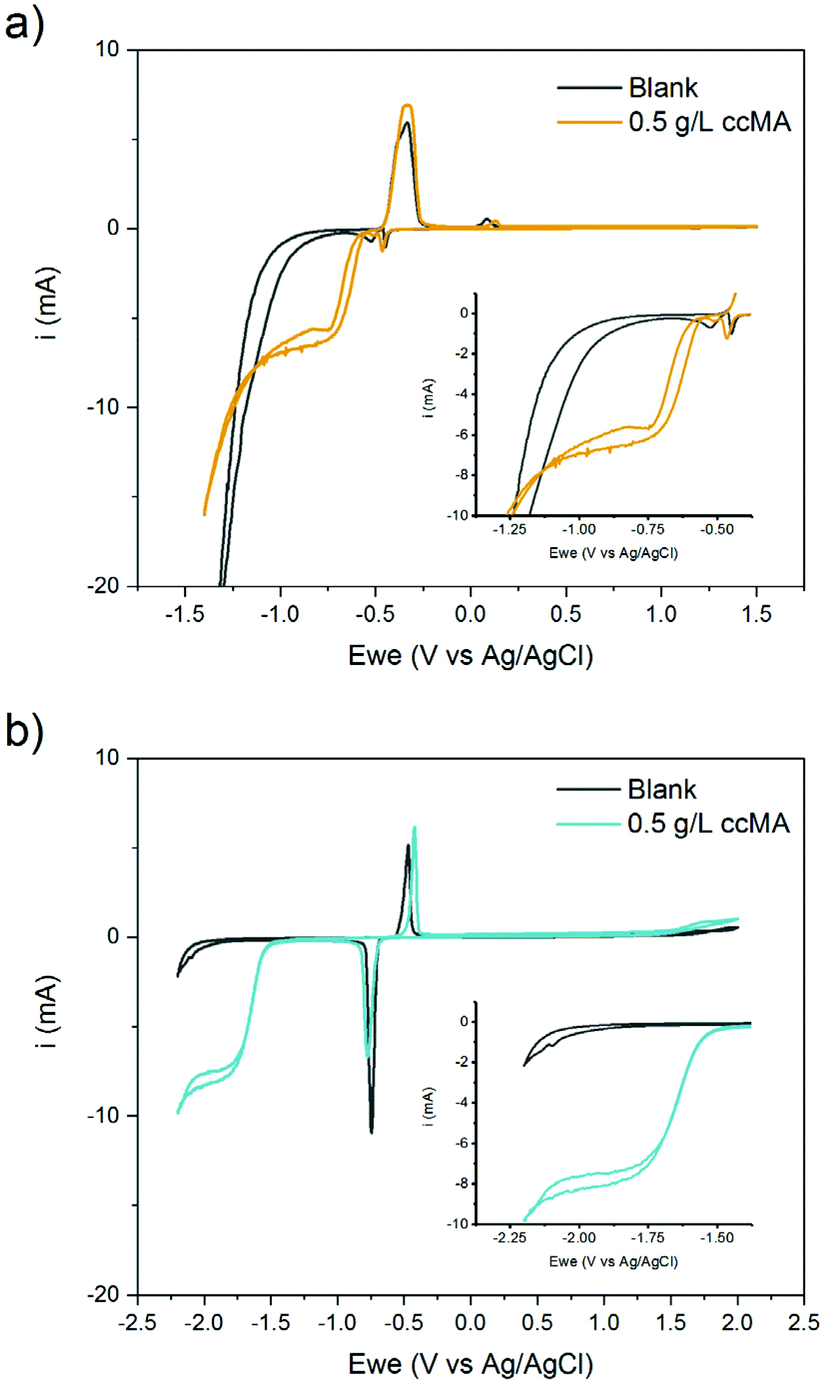 | ||
| Fig. 4 Cyclic voltammograms for blank and ccMA solutions at (a) pH 1 and (b) pH 7. The insets show magnified plots of the window of potentials corresponding to the electrohydrogenation and hydrogen evolution reactions. | ||
The ∼1 V shift of the onset potential towards more negative values when increasing the pH from 1 to 7 indicates an increase in the energetic barrier for the hydrogenation reaction. The higher energy needed for the reaction to proceed could be due to the protonation state of MA (ccMA2−), the change in hydrogen source (H2O instead of H3O+), the change in electrolyte composition (sulfuric acid for the solution at pH 1, potassium sulfate for pH 7), or to a combination of these factors (eqn (1)–(3)).
Overall reaction:
| ccMA2− + H2O → t3HDA2− + ½O2 | (1) |
Cathodic half-reaction:
| ccMA2− + 2H2O + 2e− → t3HDA2− + 2OH− | (2) |
Anodic half-reaction:
| 2OH− → H2O + ½O2 + 2e− | (3) |
This shift in onset potential can be partially balanced by increasing the MA concentration (Fig. 5). The onset potential reverted from −1.65 V for the 0.5 g L−1 solution to −1.4 V at 70 g L−1ccMA2− (Fig. 5 and S3†). Higher concentrations were not tested as 70 g L−1 represents the highest titer achieved to date with metabolically-engineered microbes. This shift in onset potential also translated into high currents and, therefore, in higher reaction rates and productivities. Surprisingly, the fermentation broth performed even better than the model solution although pH and ccMA concentrations were the same. The most likely explanation is that the other species in solution (salts, aminoacids etc.) destabilize ccMA2− and enhance its reactivity. It is also to be noted that the plateau corresponding to the mass transfer limiting current region vanished above 30 g L−1, indicating that high ccMA2− concentrations enhance both reaction kinetics and mass transfer.
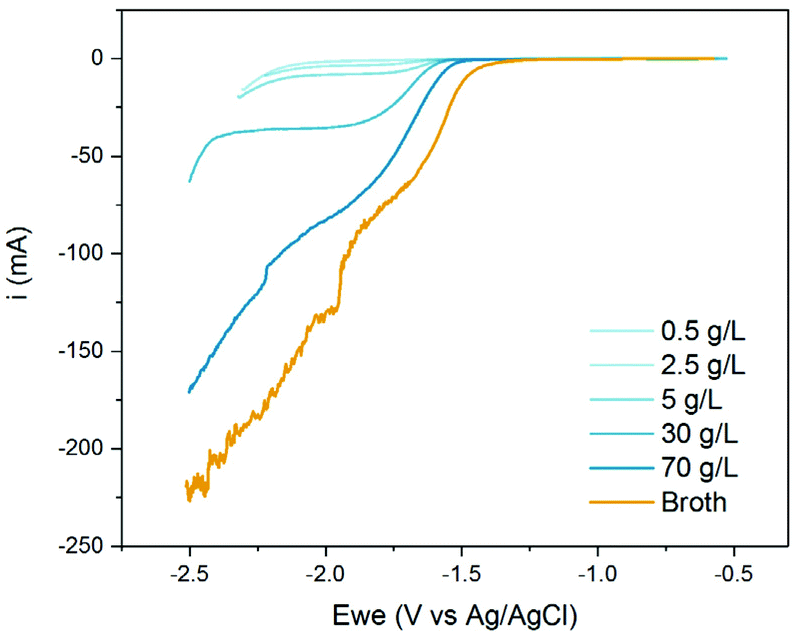 | ||
| Fig. 5 Linear sweep voltammograms for aqueous solutions at pH 7 with different concentrations of ccMA (blue). The voltammogram obtained with the fermentation broth is shown for comparison (orange). | ||
Bulk electrolysis in flow reactor systems
Although a higher pH comes with an increase in the energy cost, working under neutral conditions seems to be the only viable strategy to increase the concentration of ccMA in solution and achieve high reaction rates. Therefore, these conditions were selected to evaluate the scalability of the electrohydrogenation process under industrially relevant conditions, i.e. using galvanostatic (constant current) instead of potentiostatic (constant voltage) conditions.Various reactor configurations were investigated using both undivided and divided cells (with Nafion® membrane), in single- and double-compartment mode (Fig. S4†). Adding a membrane to decouple anodic and cathodic reactions did not offer any performance advantage as neither ccMA2− nor t3HDA2− were prone to oxidation under our reaction conditions (Fig. S5†). Similarly, the double-compartment mode offered no real advantage over the single-compartment undivided cell, indicating that rate and faradaic efficiency are governed by the current density. Therefore, most of the subsequent experiments were performed using an undivided single-compartment reactor system (Fig. S6†).
The main challenge when performing the electrohydrogenation reaction in a flow reactor under constant current is the formation of oxygen bubbles at the anode (confirmed by MS analysis of the gas phase, Fig. S7†). The bubbles can increase the resistance of the cell, hinder the desired reaction, and lower the faradaic efficiency. On the other hand, the gas evolution can contribute to the internal mixing of the reactor and improve the mass transfer.59–61 Phenomena associated to bubbles are more remarked at high current densities as gas formation is very rapid.59 It is possible to mitigate these issues by increasing the volumetric flow rate and by purging the solution with an inert gas. In a loop reactor, a high volumetric flow rate enhances the detachment of the bubbles from the electrode surface while purging the reservoir with inert gas prevents the recirculation of the bubbles and flushes them out of the solution. Looping an electrochemical reactor is already used in industry, for example for the electrochemical production of adiponitrile in Monsanto's process, to flush the hydrogen gas byproduct and prevent the degradation of the desired product.62,63
The productivity increased rapidly when increasing the current density from 50 to 400 mA cm−2, albeit the correlation between production rate and current density was not linear (Table S1†). At 400 mA cm−2 the time of electrolysis for the ccMA broth decreased by a factor ∼3.5 compared to the same reaction performed at 50 mA cm−2 (Fig. 6.). For all the current densities, the t3HDA yield remained close to 90% meaning that the higher current had little to no effect on the degradation of ccMA and t3HDA (Fig. S8†). However, above 200 mA cm−2 the faradaic efficiency dropped due to the higher currents favouring the parasitic HER (Table S1†).58 A good trade-off between productivity and faradaic efficiency was obtained for tests conducted using the single-compartment configuration under a constant current density of 100–200 mA cm−2 (Table S1†).
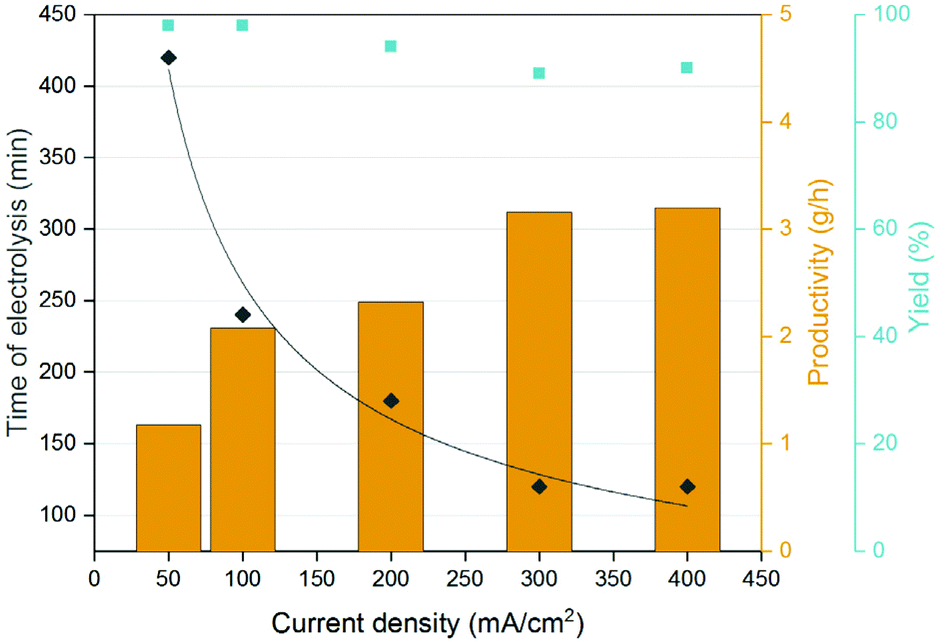 | ||
| Fig. 6 Time of electrolysis, yield, and productivity as a function of the current density used for the electrochemical hydrogenation of a fermentation broth containing ∼70 g L−1 of ccMA at pH 7. The trendline for the time of electrolysis is meant to guide the eye. | ||
To demonstrate the reusability and durability of the reactor and electrode system, 2 L of fermentation broth were converted in two batches of 1 L each. By keeping the current density at 200 mA cm−2, the two cycles showed a productivity consistent with the results obtained at a smaller scale using 100 mL of broth (Fig. S9†). After reaching the desired conversion, the reacted broth was filtered on activated carbon (Norit CN1) to remove organics and other soluble impurities. The filtrate was then acidified to pH 0 by using a small amount of 18.4 M sulfuric acid and crystallized at 4 °C overnight. The t3HDA crystals were then recovered by filtration and dried for 2 days in air. About 155 g of dried product was collected (Fig. 7a) and analyzed by 1H NMR (Fig. 7b) and GC-MS (Fig. S10†) for purity. Both techniques revealed a small amount of unreacted ccMA (5%) as the only impurity.
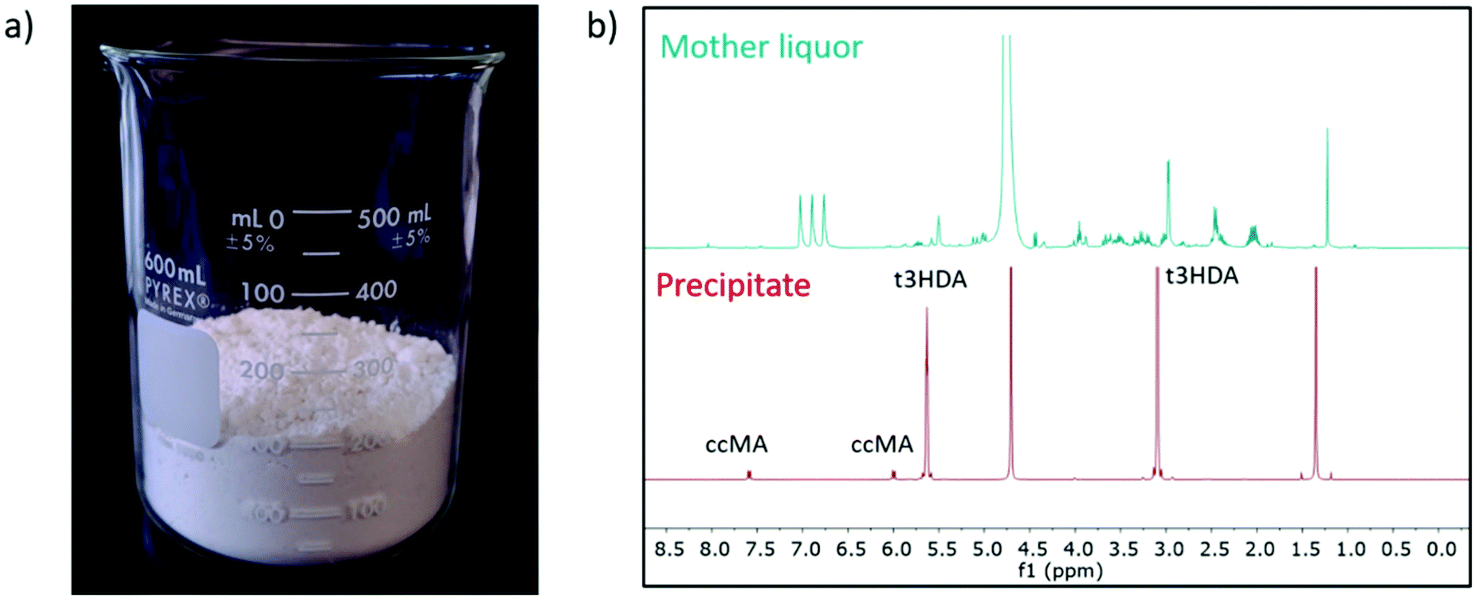 | ||
| Fig. 7 Purified product (a) recovered after electrochemical processing of 2 L of fermentation broth. The 1H NMR spectra (b) of the mother liquor and t3HDA precipitate highlight the efficiency of the separation-purification process. Moreover, unreacted ccMA (5%) is the sole impurity in the recovered product. | ||
Kinetic modelling
A simple model was derived using the flow reactor data to get further insights into the reaction kinetics and determine the rate law that would be needed for further scale up. The model consists of a two-parameters kinetic law with a mixed zero-first reaction order with respect to [ccMA]. The eqn (4)–(6) can be easily solved to determine the concentration of ccMA (eqn (4)), t3HDA (eqn (5)), and the faradaic efficiency (eqn (6)) as a function of time.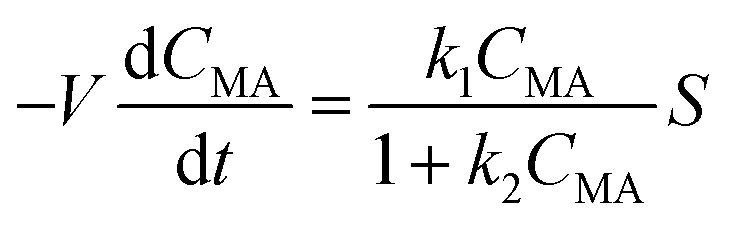 | (4) |
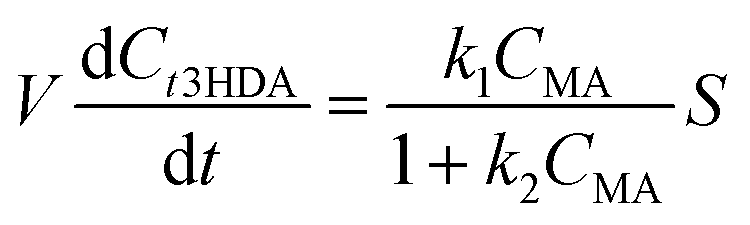 | (5) |
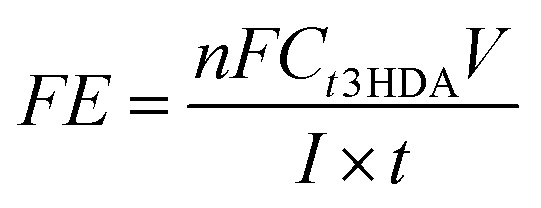 | (6) |
As can be seen from Fig. 8 and S11,† a good fit was obtained between the model and the experimental dataset. The reaction is initially of zeroth order and proceeds with a constant rate where the set current density determines the rate of the electrohydrogenation reaction. This can also be seen in eqn (4) and (5) as the term k2CMA ≫ 1 leading to a rate being equal to k1/k2. High reaction rates are obtained because the high concentration of ccMA abundantly satisfies the number of electrons provided by the cathode. In contrast to (electro)catalytic reactions that are governed by surface processes, the kinetics of the electrochemical hydrogenation are solely controlled by the current, in good agreement with the proposed outer-sphere electronation-protonation mechanism (Scheme 2). The model indicates that the reaction is not limited by mass transfer either, which is consistent with the LSV results (Fig. 5). However, Fig. 5 also predicts that mass transfer limitations could become relevant as [ccMA] drops. At low [ccMA], k2CMA ≪ 1 and the rate becomes equal to k1CMA. The reaction then turns to first order. These equations also indicate that the range of concentrations at which the reaction is free of mass transfer and follows zeroth order kinetics could potentially be extended by increasing the volumetric flow rate and/or improving the reactor design.
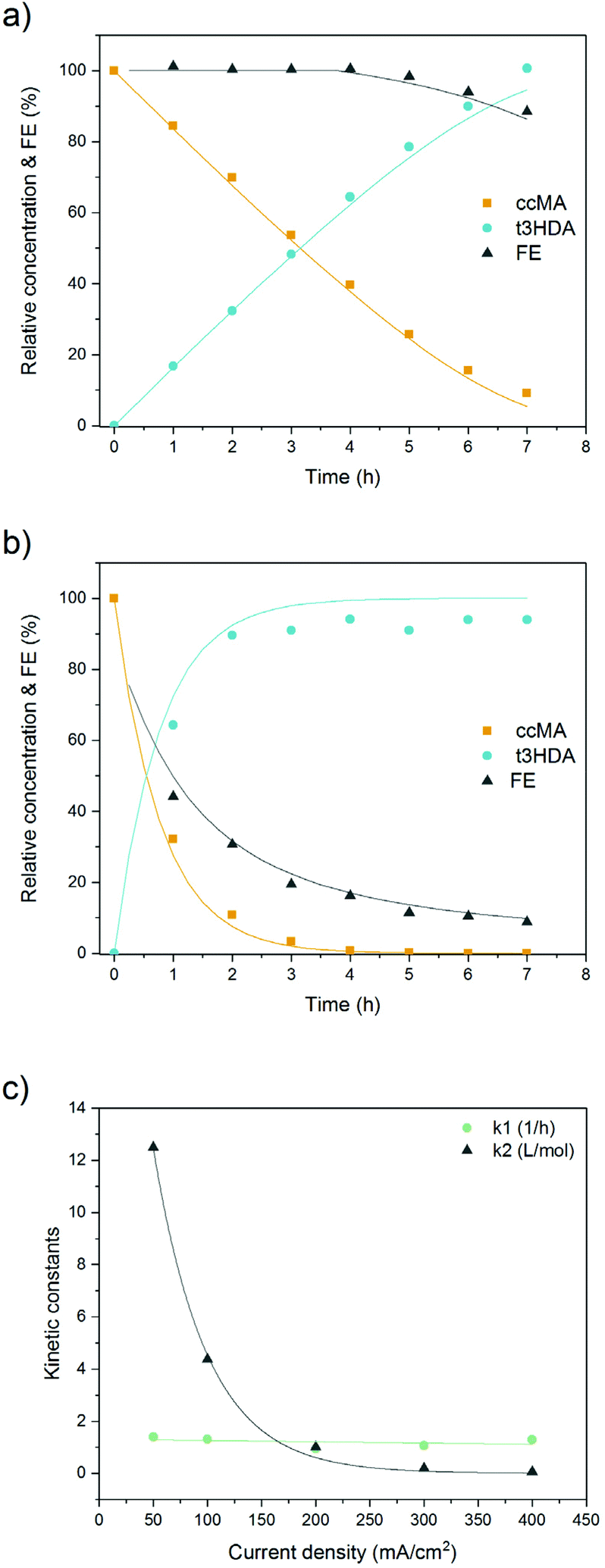 | ||
| Fig. 8 Relative concentrations of ccMA and t3HDA and evolution of the faradaic efficiency (FE) with reaction time. The reactions were performed using ccMA fermentation broth at pH 7 with a lead cathode at (a) 50 mA cm−2 and (b) 400 mA cm−2. (c) Dependence of the kinetic constants on current density. k1 is the mass transfer coefficient normalized to the volume and surface employed. | ||
The effect of the current density on the rate constants was also investigated. As can be seen from Fig. 8c, k1 presents a linear trend while k2 decreases exponentially with increasing currents. The dependence with the current density suggests that the empirical model proposed is consistent with theoretical models and equations present in the literature.64 At low current densities (50–100 mA cm−2), the reaction is initially kinetically controlled where the constant current determines a constant rate of reaction. Towards the end of the reaction, mass transfer limitations take place and the reaction slows down. The mass transfer limiting conditions result in a first order for the reaction rate.58 This is the case for 200, 300, and 400 mA cm−2 where the rate of reaction is only slightly enhanced by the mixing due to bubble formation at both electrodes.
Cathodic corrosion and suitable replacement for Pb
Cathodic corrosion is an issue that is often overlooked or underestimated in academic research but that is gaining increasing attention due to its importance during scale up and electrochemical manufacturing.65–68 ICP-OES analysis of the reacted broth revealed lead impurities with concentrations in the order of hundreds of ppb (Fig. S12†). Lead is a highly toxic metal that is known to dissolve into aqueous solutions. Although its Pourbaix diagram indicates that Pb should remain in its metallic state under negative potentials, Pb corrosion and leaching are unavoidable. The corrosion may be due to the mechanical stress caused by H2 formation and evolution at the surface or to chelation by the muconate dianions.38,65As lead is toxic even at very low concentrations (the limit in drinking water is 15 ppb),69 we explored alternative electrode materials that do not present this hazard. Suitable candidates were searched based on the known toxicity of the metal and of its ions, and the metal's Gibbs free energy of hydrogen binding, a key descriptor for HER activity.70,71 Bismuth was found to present a high hydrogen overpotential and among the lowest toxicity and ecotoxicity among heavy metals. Industrially, Bi is found in a large range of products including pharmaceuticals, cosmetics, and paints among others. However, Bi is a brittle metal that is difficult to manufacture in plates. Therefore, Bi electrodes were manufactured in house by melting 1 kg of high purity bismuth ingots at 280 °C, followed by cooling, and cutting to the desired shape (Fig. S13†). To our surprise, the performance of these Bi electrodes was almost identical to commercial Pb cathodes for a fraction of the cost (Fig. 9). Bismuth electrodes were further investigated using CV with different concentrations of muconic acid. The voltammograms exhibited very similar onset potentials and concentration dependence as Pb (Fig. S14 and S15†).
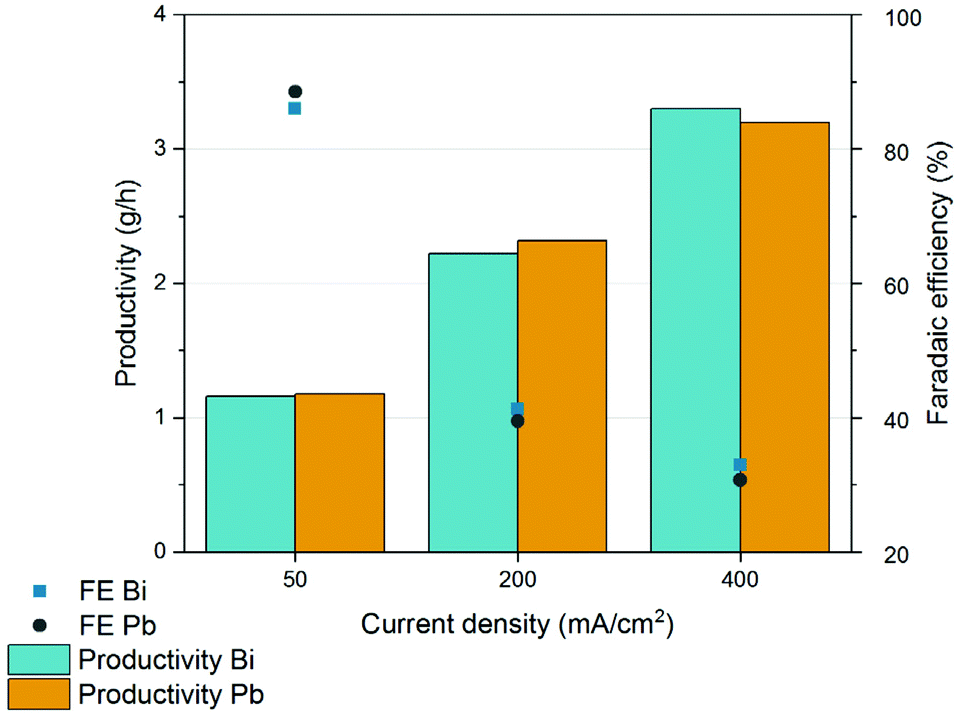 | ||
| Fig. 9 Chronopotentiometric results obtained when processing the fermentation broth in an undivided single-compartment electrochemical flow reactor equipped with lead and bismuth cathodes. | ||
Process design and technoeconomic analysis
The promising experimental results for the electrochemical conversion of ccMA to t3HDA has prompted further investigations into technoeconomic considerations for the scale up and commercialization of this process. Previously, an Early Stage Techno Economic Analysis software (ESTEA) was utilized to perform a simplified analysis and determine the feasibility of this process at an industrial level.18 However, the experiments performed in the present work were necessary to carry out a more detailed technoeconomic analysis using Aspen and outline an industrial design for the conversion of glucose into muconic acid through biological fermentation followed by electrochemical hydrogenation to t3HDA with the goal of minimizing operating costs.Non-conventional components and components outside of Aspen databanks were used in this simulation and required the use of several functional group estimation techniques. Specifically, t3HDA was not present in Aspen's database and required the use of both the UNIFAC and JOBACK functional group methods to estimate key parameters. Furthermore, while solubility data was provided, the crystallization of t3HDA required the addition of the MOFASA functional group method to estimate the solid heat capacities. The results of this design are dependent on these models and prompt further investigation to ensure their validity.
In the final design (Fig. 10 and S16†), glucose and water are fed into large fermentation reactors and allowed to ferment for 72 hours. Compressed air is fed into the reactors to promote growth of aerobic bacteria and assist in the agitation of the vessels. A recycle cooling loop is utilized to maintain the fermenter temperature at 45 °C and to promote further mixing. Following fermentation, the mixture is cooled to 25 °C prior to entering an intermediate storage tank meant to accommodate process overflows. The cooled fermentation broth enters a centrifuge where cells and nutrients are recycled to save on raw material costs. The clarified broth continues to the electrochemical reactor where ccMA is converted to t3HDA and concentrated via an anion adsorption unit. The concentrated t3HDA is desorbed and sent to agitated crystallization reactors where the product is allowed to crystallize. Finally, the stream enters a vacuum drum filter that removes residual moisture leaving dry t3HDA crystals. The separation and crystallization steps were designed based on work published previously.18
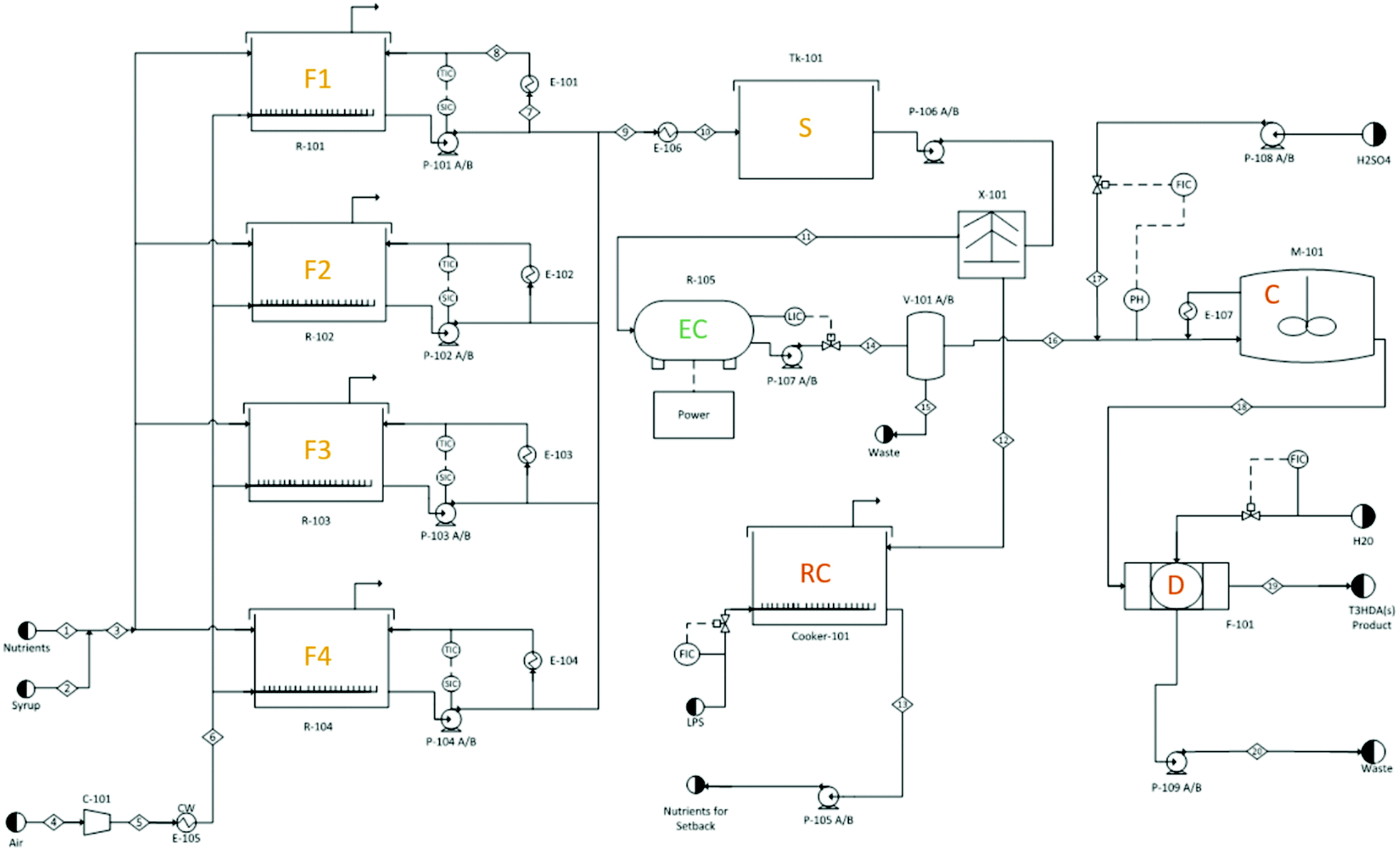 | ||
| Fig. 10 Aspen flow diagram for a glucose to t3HDA plant. F1, F2, F3, and F4 (fermentation reactors), S (intermediate storage), EC (electrochemical hydrogenation reactor), RC (recycle cooker), C (t3HDA crystallizer), D (belt filter/dryer). For further details, see Fig. S16.† | ||
Special consideration was given to the electrochemical reactor because it was found to represent the majority of both utility costs and capital costs. A parametric optimization was done investigating the impact of voltage vs. the equivalent annual operating costs on this unit operation. For the purpose of this analysis, a minimum cost operating point was discovered at approximately 5.7 volts with a current density of 200 mA cm−2 (Fig. S17†). However, Fig. S17† prompts further investigation to determine the true operating minimum that may occur between two sets of data points.
The final Aspen flowsheet design was sized to produce 83 ktonne per year of t3HDA. The cost of manufacturing not considering depreciation (COMd) was $131744371 and a product cost of $1.75 per kg was obtained for t3HDA (see ESI and Tables S2–S11†). A breakdown of the manufacturing costs is presented in Table S11† and Fig. 11. Fermentation and electrochemical conversion represent 9 and 12% of t3HDA's production costs, which corresponds to $0.17 and $0.22 per kg of high purity product, respectively. The raw materials (sugar feedstock) represent the main cost driver, like in most industrial bioprocesses.
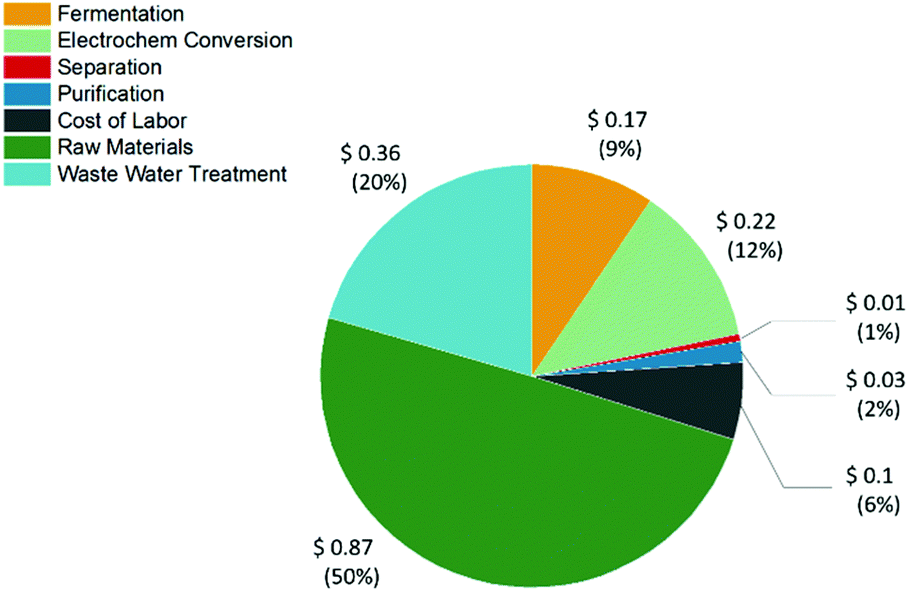 | ||
| Fig. 11 Distribution of the overall production cost of t3HDA ($1.75 per kg) between the major cost categories, namely raw materials, fermentation, electrochemical conversion, separation, purification, cost of labor, and waste water treatment. The cost of raw materials is the most dominant. | ||
Conclusion
Prior studies on the electrochemical hydrogenation of biologically-produced MA to t3HDA achieved a maximum productivity of 44 gt3HDA per month, leaving little hope for an industrial production of this novel monomer. New insights into the mechanism of the electrochemical hydrogenation gained through the present work changed these prospects and provided unprecedented opportunities for synergies between microbial and electrochemical conversions. Specifically, isotopic tracing revealed that the electrochemical hydrogenation of ccMA proceeds through sequential outer-sphere electronation-protonation. As adsorption on the electrode surface is not required, the reaction can accommodate a broad range of broth compositions, making it a robust and versatile process for the selective production of t3HDA. Isotopic tracing and cyclic voltammetry also showed that (i) water molecules may replace protons as a hydrogen source, and (ii) against all expectations the muconate dianion reacts with the negatively charged cathode and accepts electrons to form a radical dianion susceptible to electrohydrogenation.The flexibility of the process together with the reactivity of the muconate dianion made it possible to switch to bacteria for the microbial step and explore pH neutral broths. The higher pH compared to the broth obtained from yeasts allowed MA concentrations in solution in the order of 70 g L−1, which enhanced the reaction kinetics. Moreover, the higher pH also impeded the hydrogen evolution reaction due to proton reduction (HER), which had beneficial effects on the faradaic efficiency. These improvements allowed the electrochemical reaction to proceed at higher current densities under galvanostatic conditions, common in industrial processes. The increase in current density combined with the transition from batch to flow reactor enhanced the productivity by up to 52-fold from 0.044 kgt3HDA per month to 1.5–2.3 kgt3HDA per month.
The experimental data was used to model and optimize an industrial process for t3HDA production in terms of operating costs and energy consumption. Operating at a current density of 200 mA cm−2 offered a good compromise between productivity, faradaic efficiency, and operating costs, with an estimated production cost of $1.75 per kgt3HDA. Furthermore, replacing conventional Pb cathodes by Bi plates enhanced the sustainability of the process without compromising on performance.
This advancement in MA electrohydrogenation technology will enable the development of novel performance-advantaged polymers using biobased t3HDA and its derivatives. Moreover, the findings from this work are broadly applicable and provide guidance for the development of other HMES processes combining microbial and electrochemical transformations.
Experimental
Chemicals and model solutions
Chemicals were purchased as follows: cis–cis muconic acid (98%, Acros Organics), trans-3-hexenedioic acid (98%, Tokyo Chemical Industries), deuterium oxide (99.9% D, Sigma-Aldrich), dimethylmalonic acid (TraceCERT® grade for qNMR, Sigma-Aldrich), potassium deuteroxide (98% D, Sigma-Aldrich), sulfuric acid-d2 (99.5% D, Sigma-Aldrich), 5 wt% Pd on activated carbon (Sigma-Aldrich). A fermentation broth containing 70–80 g L−1 of ammonium muconate at pH 7 was provided by a third party.The electrolytes used for voltammetry consisted of 100 mL of either 0.1 M H2SO4 for experiments conducted at pH 1 or of a mixture of 0.5 M sodium phosphate buffer and 0.1 M K2SO4 for experiments at pH 7. The resistance of both solutions was similar. Model solutions were prepared by dissolving the amount of cis,cis-muconic acid required to reach the desired concentration. If needed, the pH was then corrected with a few drops of 1 M KOH.
The model solution used for the thermocatalytic test presented in Fig. 1a had a ccMA concentration of 75 g L−1 and its pH was brought to 7 with drops of 0.35 M NH4OH. NH4OH was preferred over KOH in this case to avoid the presence of any species that may poison the catalyst.
Electrochemical analysis and reactions
Cyclic and linear sweep voltammetry (CV and LSV, respectively) were conducted in a three-electrode cell with a lead (Pb) or bismuth (Bi) rotating disk electrode (RDE) as working electrode, Ag/AgCl as reference electrode, and a platinum wire as counter electrode (Pine Research Instrumentation, Durham, NC). The experiments were conducted with a BioLogic VSP-300 potentiostat (BioLogic, Claix, France). The solution was purged by bubbling argon gas for 15 minutes before the analysis as well as during the experiment. The RDE rotation speed was set at 1600 rpm (MSR rotator, Pine Research Instrumentation). The uncompensated resistance of the solution (Rs) was measured by potentiostatic electrochemical impedance spectroscopy (PEIS) prior to each experiment. The measured Rs value was then used to correct the potential on the working electrode. The scan rate for CV and LSV was set at 50 mV s−1 and the window of potential was adjusted depending on the pH of the solution to account for pH-dependent shifts of the peaks and onset potentials.The flow reactor experiments were conducted in a Micro Flow Cell® purchased from ElectroCell (Amherst, NY). The reactor was configured either as a single compartment cell with a PTFE frame separating the two electrodes or as a double compartment cell using a Nafion membrane (Fig. S4†). The electrode gap was 5 mm for both configurations. Pb and Bi plates were used as working electrodes and platinized titanium as counter electrode. Bismuth electrodes were prepared in house using commercially available 99.99% purity bismuth ingots (Rotometals, San Leandro, CA). The ingots were melted using a cast iron pan and a hot plate able to reach 300 °C. After cooling, the resulting plate was cut to reactor dimensions and sanded to remove any oxidized bismuth and obtain an evenly polished surface. The RDE was manufactured using a similar protocol. An Ag/AgCl reference electrode was inserted in the PTFE frame of the flow reactor. The exposed surface area of the working and counter electrodes was 10 cm2. Bulk electrolysis experiments were performed under constant current (galvanostatic mode) using 100 mL of model solution or fermentation broth that were looped through the reactor at 180 ml min−1 using a Fisherbrand™ GP1000 pump (ThermoFisher). Power was supplied by the BioLogic VSP-300 potentiostat for low current experiments and a Sorensen AMETEK XPH 35-5 power unit for experiments that required currents above 1 A.
Samples of 250 μL were withdrawn every hour. The aliquots were subsequently dried in air and the solid was redissolved in a deuterium oxide solution containing dimethylmalonic acid as an internal standard. The weights of the dried and deuterated samples were recorded before each analysis and all calculations were based on weight fractions. The samples were then analyzed by 1H NMR using a Bruker 600 MHz spectrometer to determine conversion and selectivity. The amount of product obtained and the current applied were used to calculate the faradaic efficiency. A more detailed explanation of the quantitative NMR (qNMR) method is available online.72
Large scale experiments were performed with the same flow reactor equipped with a 10 cm2 Bi working electrode and using two 1 L batches of fermentation broth. Upon completion of the first run, the reacted broth was quickly replaced by 1 L of fresh broth without stopping the reaction system. Conversion, selectivity, and rates of both runs were compared to confirm the stability of the reaction system during continuous operation for approximately 70 hours. Upon completion, both batches were combined and filtered on Norit CN1 activated carbon to adsorb soluble organic impurities including colorants. The filtrate was acidified to pH 0 by using a small amount of 18.4 M sulfuric acid and crystallized at 4 °C overnight. The crystals of t3HDA were then recovered using vacuum filtration with a Buchner funnel equipped with a Whatman 50 filter, then dried for 2 days in air.
Elemental analysis
Metal leaching into the electrolyte was quantified through inductively coupled plasma-optical emission spectrometry (ICP-OES) using a PerkinElmer Optima 8000 (Shelton, CT). In this case, we viewed the axial range for lead (wavelength of 220.357 nm) and bismuth (wavelength of 223.061 nm). Calibration curves were built from standard solutions of bismuth dissolved in 10% nitric acid (PerkinElmer) and lead dissolved in 0.5% nitric acid (Aristar, VWR). Samples were filtered through a 0.22 μm filter to remove any precipitation and then diluted in nitric acid solution to the desired ratio.Solubility measurements
Solubility measurements were directly conducted in 10 mL of deuterium oxide with a pD set based on the concentration of sulfuric acid-d2 or potassium deuteroxide. A specific amount of muconic acid was slowly added and the pD was constantly measured through a pH-meter. The recorded pH value was corrected to the pD value using pD = pH + 0.4.73 Solutions were stirred at 800 rpm until saturation was reached and pH measurement was constant. 980 μL of the solution was then filtered and diluted with 20 μL of D2O solution with internal standard. The samples were then analyzed by 1H NMR to determine the concentration of muconic acid or muconate in solution.Thermocatalytic hydrogenation
Thermocatalytic hydrogenation tests were performed in a three neck round-bottom flask with a continuous stream of H2 at atmospheric pressure. 50 mL of muconic acid model solution or fermentation broth were reacted at 70 °C in the presence of 300 mg of commercial 5 wt% Pd/C. Samples were collected at specific time intervals and analyzed by 1H NMR using the same method as for the electrochemical tests.Conflicts of interest
J. P. T. is a founder and scientific advisor of Sumatra Biorenewables, LLC, a start-up company that specializes in novel biobased monomers derived from muconic acid.Acknowledgements
This material is based upon work supported in part by the National Science Foundation under grant numbers CBET-1512126 and IIP-1820147. We also acknowledge the support of Iowa State University through the Regents Innovation Fund.References
- E. M. Karp, T. R. Eaton, V. Sànchez i Nogué, V. Vorotnikov, M. J. Biddy, E. C. D. Tan, D. G. Brandner, R. M. Cywar, R. Liu, L. P. Manker, W. E. Michener, M. Gilhespy, Z. Skoufa, M. J. Watson, O. S. Fruchey, D. R. Vardon, R. T. Gill, A. D. Bratis and G. T. Beckham, Science, 2017, 358, 1307 CrossRef CAS PubMed.
- O. A. Abdelrahman, D. S. Park, K. P. Vinter, C. S. Spanjers, L. Ren, H. J. Cho, K. Zhang, W. Fan, M. Tsapatsis and P. J. Dauenhauer, ACS Catal., 2017, 7, 1428–1431 CrossRef CAS.
- T. W. Lyons, D. Guironnet, M. Findlater and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 15708–15711 CrossRef CAS PubMed.
- C.-C. Chang, S. K. Green, C. L. Williams, P. J. Dauenhauer and W. Fan, Green Chem., 2014, 16, 585–588 RSC.
- N. Fitzgerald and A. Bailey, Moving Beyond Drop-In Replacements: Performance-Advantaged Biobased Chemicals, U.S. Department of Energy Office of Energy Efficiency & Renewable Energy, 2018 Search PubMed.
- B. H. Shanks and L. J. Broadbelt, ChemSusChem, 2019, 12, 2970–2975 CrossRef CAS.
- M. Sajid, X. Zhao and D. Liu, Green Chem., 2018, 20, 5427–5453 RSC.
- X. Fei, J. Wang, J. Zhu, X. Wang and X. Liu, ACS Sustainable Chem. Eng., 2020, 8, 8471–8485 CrossRef CAS.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass: Volume I – Results of Screening for Potential Candidates from Sugars and Synthesis Gas, Report DOE/GO-102004-1992; TRN: US200427%%671, National Renewable Energy Lab., Golden, CO (US), 2004 Search PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- X. Zhou, Z. J. Brentzel, G. A. Kraus, P. L. Keeling, J. A. Dumesic, B. H. Shanks and L. J. Broadbelt, ACS Sustainable Chem. Eng., 2018, 7, 2414–2428 CrossRef.
- J. Huo and B. H. Shanks, Annu. Rev. Chem. Biomol. Eng., 2020, 11, 63–85 CrossRef.
- B. H. Shanks and P. L. Keeling, Green Chem., 2017, 19, 3177–3185 RSC.
- I. Khalil, G. Quintens, T. Junkers and M. Dusselier, Green Chem., 2020, 22, 1517–1541 RSC.
- M. Chia, T. J. Schwartz, B. H. Shanks and J. A. Dumesic, Green Chem., 2012, 14, 1850–1853 RSC.
- M. Suastegui, J. E. Matthiesen, J. M. Carraher, N. Hernandez, N. Rodriguez Quiroz, A. Okerlund, E. W. Cochran, Z. Shao and J. P. Tessonnier, Angew. Chem., Int. Ed., 2016, 55, 2368–2373 CrossRef CAS PubMed.
- J. M. Leavitt, J. M. Wagner, C. C. Tu, A. Tong, Y. Liu and H. S. Alper, Biotechnol. J., 2017, 12, 1600687 CrossRef PubMed.
- J. E. Matthiesen, M. Suastegui, Y. T. Wu, M. Viswanathan, Y. Qu, M. F. Cao, N. Rodriguez-Quiroz, A. Okerlund, G. Kraus, D. R. Raman, Z. Y. Shao and J. P. Tessonnier, ACS Sustainable Chem. Eng., 2016, 4, 7098–7109 CrossRef CAS.
- J. E. Matthiesen, J. M. Carraher, M. Vasiliu, D. A. Dixon and J. P. Tessonnier, ACS Sustainable Chem. Eng., 2016, 4, 3575–3585 CrossRef CAS.
- T. J. Schwartz, R. L. Johnson, J. Cardenas, A. Okerlund, N. A. Da Silva, K. Schmidt-Rohr and J. A. Dumesic, Angew. Chem., Int. Ed., 2014, 53, 12718–12722 CrossRef CAS PubMed.
- Z. Zhang, J. E. Jackson and D. J. Miller, Bioresour. Technol., 2008, 99, 5873–5880 CrossRef CAS PubMed.
- I. Bechthold, K. Bretz, S. Kabasci, R. Kopitzky and A. Springer, Chem. Eng. Technol., 2008, 31, 647–654 CrossRef CAS.
- F. J. Holzhäuser, J. Artz, S. Palkovits, D. Kreyenschulte, J. Büchs and R. Palkovits, Green Chem., 2017, 19, 2390–2397 RSC.
- C. H. Lam, S. Das, N. C. Erickson, C. D. Hyzer, M. Garedew, J. E. Anderson, T. J. Wallington, M. A. Tamor, J. E. Jackson and C. M. Saffron, Sustainable Energy Fuels, 2017, 1, 258–266 RSC.
- Z. L. Li, S. Kelkar, L. Raycraft, M. Garedew, J. E. Jackson, D. J. Miller and C. M. Saffron, Green Chem., 2014, 16, 844–852 RSC.
- Y. Kwon, K. J. P. Schouten, J. C. van der Waal, E. de Jong and M. T. M. Koper, ACS Catal., 2016, 6, 6704–6717 CrossRef CAS.
- A. S. May and E. J. Biddinger, ACS Catal., 2020, 10, 3212–3221 CrossRef CAS.
- E. J. Biddinger and M. A. Modestino, Electrochem. Soc. Interface, 2020, 29, 43–47 CAS.
- Y. Qiu, L. Xin, D. J. Chadderdon, J. Qi, C. Liang and W. Li, Green Chem., 2014, 16, 1305–1315 RSC.
- J. A. Lopez-Ruiz, E. Andrews, S. A. Akhade, M.-S. Lee, K. Koh, U. Sanyal, S. F. Yuk, A. J. Karkamkar, M. A. Derewinski, J. Holladay, V.-A. Glezakou, R. Rousseau, O. Y. Gutiérrez and J. D. Holladay, ACS Catal., 2019, 9, 9964–9972 CrossRef CAS.
- Y. Song, U. Sanyal, D. Pangotra, J. D. Holladay, D. M. Camaioni, O. Y. Gutierrez and J. A. Lercher, J. Catal., 2018, 359, 68–75 CrossRef CAS.
- A. M. Román, J. C. Hasse, J. W. Medlin and A. Holewinski, ACS Catal., 2019, 9, 10305–10316 CrossRef.
- X. H. Chadderdon, D. J. Chadderdon, J. E. Matthiesen, Y. Qiu, J. M. Carraher, J. P. Tessonnier and W. Li, J. Am. Chem. Soc., 2017, 139, 14120–14128 CrossRef CAS PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- J. Seidler, J. Strugatchi, T. Gärtner and S. R. Waldvogel, MRS Energy Sustain., 2020, 7, E42 CrossRef.
- D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386–12400 RSC.
- S. Mohle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef.
- R. Palkovits and I. Delidovich, Philos. Trans. R. Soc., A, 2018, 376, 20170064 CrossRef PubMed.
- S. Abdolmohammadi, N. Hernández, J.-P. Tessonnier and E. W. Cochran, in Green Polymer Chemistry: New Products, Processes, and Applications, American Chemical Society, Washington, DC, 2018, vol. 1310, ch. 22, pp. 355–367 Search PubMed.
- N. Hernandez, M. Yan, E. W. Cochran, J. E. Matthiesen and J.-P. Tessonnier, Bioadvantaged Nylon: Polycondensation of 3-Hexenedioic Acid With Hexamethylenediamine, US10793673B2, 2020.
- J.-P. Tessonnier, J. E. Matthiesen, J. W. Nelson, E. W. Cochran, S. Abdolmohammadi and N. Hernandez, Functionalization of Trans-3-Hexenedioic Acid for the Production of Hydrophobic Polyamides and Chemical Resistance Thereof, US20190153156A1, 2018.
- S. Capelli, D. Motta, C. Evangelisti, N. Dimitratos, L. Prati, C. Pirola and A. Villa, ChemCatChem, 2019, 11, 3075–3084 CrossRef CAS.
- S. Capelli, A. Rosengart, A. Villa, A. Citterio, A. Di Michele, C. L. Bianchi, L. Prati and C. Pirola, Appl. Catal., B, 2017, 218, 220–229 CrossRef CAS.
- S. Scelfo, R. Pirone and N. Russo, Catal. Commun., 2016, 84, 98–102 CrossRef CAS.
- D. R. Vardon, M. A. Franden, C. W. Johnson, E. M. Karp, M. T. Guarnieri, J. G. Linger, M. J. Salm, T. J. Strathmann and G. T. Beckham, Energy Environ. Sci., 2015, 8, 617–628 RSC.
- D. R. Vardon, N. A. Rorrer, D. Salvachua, A. E. Settle, C. W. Johnson, M. J. Menart, N. S. Cleveland, P. N. Ciesielski, K. X. Steirer, J. R. Dorgan and G. T. Beckham, Green Chem., 2016, 18, 3397–3413 RSC.
- P. Neta and R. W. Fessenden, J. Phys. Chem., 1972, 76, 1957–1961 CrossRef CAS.
- J. Lessard, in Organic Electrochemistry, ed. O. Hammerich, B. Speiser, CRC Press, Taylor & Francis Group, 5th edn, 2016, ch. 44, pp. 1657–1672 Search PubMed.
- C. Sandford, M. A. Edwards, K. J. Klunder, D. P. Hickey, M. Li, K. Barman, M. S. Sigman, H. S. White and S. D. Minteer, Chem. Sci., 2019, 10, 6404–6422 RSC.
- S. W. Boettcher and Y. Surendranath, Nat. Catal., 2021, 4, 4–5 CrossRef CAS.
- J. Anibal and B. Xu, ACS Catal., 2020, 10, 11643–11653 CrossRef CAS.
- R. Chen, C. Yang, Z. Zhou, F. Haeffner, A. Dersjant, N. Dulock, Q. Dong, D. He, J. Jin, Y. Zhao, J. Niu and D. Wang, Angew. Chem., Int. Ed., 2021, 7534–7539 CrossRef CAS.
- D. E. Blanco, P. A. Prasad, K. Dunningan and M. A. Modestino, React. Chem. Eng., 2020, 5, 136–144 RSC.
- N. Jain, G. Yang, S. E. Tabibi and S. H. Yalkowsky, Int. J. Pharm., 2001, 225, 41–47 CrossRef CAS.
- S. H. Yalkowsky, R. B. Patel and D. Alantary, ADMET & DMPK, 2015, 3, 359–362 Search PubMed.
- X. Ding, I. Gueorguieva, J. A. Wesley, L. J. Burns and C. A. Coutant, AAPS J., 2015, 17, 1395–1406 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, in Electrochemical Methods Fundamentals and Applications, Wiley, New York, 1980, 2nd edn, 2001, ch. 11, pp. 430–432 Search PubMed.
- A. Angulo, P. van der Linde, H. Gardeniers, M. Modestino and D. Fernández Rivas, Joule, 2020, 4, 555–579 CrossRef CAS.
- A. Taqieddin, R. Nazari, L. Rajic and A. Alshawabkeh, J. Electrochem. Soc., 2017, 164, E448–E459 CrossRef CAS PubMed.
- D. Zhang and K. Zeng, Ind. Eng. Chem. Res., 2012, 51, 13825–13832 CrossRef CAS.
- M. M. Baizer, J. Electrochem. Soc., 1964, 111, 215–222 CrossRef CAS.
- D. S. P. Cardoso, B. Šljukić, D. M. F. Santos and C. A. C. Sequeira, Org. Process Res. Dev., 2017, 21, 1213–1226 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, in Electrochemical Methods Fundamentals and Applications, Wiley, New York, 2nd edn, 2001, ch. 9, pp. 341–342 Search PubMed.
- C. Edinger, V. Grimaudo, P. Broekmann and S. R. Waldvogel, ChemElectroChem, 2014, 1, 1018–1022 CrossRef CAS.
- C. Gütz, V. Grimaudo, M. Holtkamp, M. Hartmer, J. Werra, L. Frensemeier, A. Kehl, U. Karst, P. Broekmann and S. R. Waldvogel, ChemElectroChem, 2018, 5, 247–252 CrossRef.
- C. Gütz, M. Selt, M. Bänziger, C. Bucher, C. Römelt, N. Hecken, F. Gallou, T. R. Galvão and S. R. Waldvogel, Chem. – Eur. J., 2015, 21, 13878–13882 CrossRef PubMed.
- T. J. P. Hersbach and M. T. M. Koper, Curr. Opin. Electrochem., 2021, 26, 100653 CrossRef CAS.
- US EPA, Lead and Copper Rule, https://www.epa.gov/dwreginfo/lead-and-copper-rule, Accessed 1/5/2021.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Norskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- P. Quaino, F. Juarez, E. Santos and W. Schmickler, Beilstein J. Nanotechnol., 2014, 5, 846–854 CrossRef PubMed.
- M. Sigma, TraceCERT® CRMs for quantitative NMR (qNMR), https://www.sigmaaldrich.com/analytical-chromatography/analytical-standards/application-area-technique/organiccrm.html, Accessed 2/8/2020.
- P. K. Glasoe and F. A. Long, J. Phys. Chem., 1960, 64, 188–190 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional thermocatalytic and electrochemical hydrogenation results, NMR spectra of reactants and products, complete process modelling in ASPEN and technoeconomic analysis. See DOI: 10.1039/d1gc02225c |
| This journal is © The Royal Society of Chemistry 2021 |