
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceContinuous flow heterogeneous catalytic reductive aminations under aqueous micellar conditions enabled by an oscillatory plug flow reactor†
Michaela
Wernik
ab,
Gellért
Sipos
cd,
Balázs
Buchholcz
e,
Ferenc
Darvas
cd,
Zoltán
Novák
 f,
Sándor B.
Ötvös
*ab and
C. Oliver
Kappe
*ab
f,
Sándor B.
Ötvös
*ab and
C. Oliver
Kappe
*ab
aInstitute of Chemistry, University of Graz, NAWI Graz, Heinrichstrasse 28, A-8010 Graz, Austria. E-mail: sandor.oetvoes@uni-graz.at; oliver.kappe@uni-graz.at
bCenter for Continuous Flow Synthesis and Processing (CCFLOW), Research Center Pharmaceutical Engineering GmbH (RCPE), Inffeldgasse 13, A-8010 Graz, Austria
cThalesNano Inc., Záhony u. 7, 1031 Budapest, Hungary
dComInnex Inc., Záhony u. 7, 1031 Budapest, Hungary
eInnostudio Inc., Záhony u. 7, 1031 Budapest, Hungary
fELTE “Lendület” Catalysis and Organic Synthesis Research Group, Faculty of Science, Institute of Chemistry, Eötvös Loránd University, Pázmány Péter stny. 1/a, 1117 Budapest, Hungary
First published on 8th July 2021
Abstract
Despite the fact that continuous flow processing exhibits well-established technical advances, aqueous micellar chemistry, a field that has proven extremely useful in shifting organic synthesis to sustainable water-based media, has mostly been explored under conventional batch-based conditions. This is particularly because of the fact that the reliable handling of slurries and suspensions in flow has been considered as a significant technical challenge. Herein, we demonstrate that the strategic application of an oscillatory plug flow reactor enables heterogeneous catalytic reductive aminations in aqueous micellar media enhancing mass transport and facilitating process simplicity, stability and scalability. The micellar flow process enabled a broad range of substrates, including amino acid derivatives, to be successfully transformed under reasonably mild conditions utilizing only very low amounts of Pd/C as a readily available heterogeneous catalyst. The preparative capabilities of the process along with the recyclability of the heterogenous catalyst and the aqueous reaction media were also demonstrated.
Introduction
Whilst solvent-free reactions are typically limited to a few special cases,1 organic solvents constitute the largest part of waste generated in synthetic chemistry.2 Therefore, the use of water as solvent has attracted a great interest towards more sustainable manufacturing practices.3 The application of water as a naturally abundant reaction medium exhibits obvious advantages concerning environmental, safety and economic aspects in comparison with traditional organic solvents.4 However, water is often overlooked as a solvent due to the insolubility of most organic substrates and the sensitivity of numerous reactions towards moisture. To circumvent these limitations, aqueous micellar conditions have extensively been investigated with various surfactants being employed to form micellar arrays as amphiphile nanoreactors.5 The lipophilic part of such micelles functions as the ‘organic solvent’ which serves as a non-aqueous environment. In this manner, even water sensitive reactions can be performed in aqueous media.6 It was verified that the particle size and the shape of micelles is crucial in such transformations,7 therefore novel surfactants have been designed that facilitate fine-tuning of micelle properties and enabled an array of chemistries, including various cross-couplings, C–H activations, amide bond formations, oxidations and reductions, to be realized under aqueous micellar conditions.8In micellar chemistry, an aggregate of surfactant molecules forms a colloidal suspension generating a biphasic mixture where mass transfer of reactants is a limiting phenomenon.3a This is especially pronounced in cases where micellar chemistry is performed in the presence of heterogeneous catalysts which must maintain close contact with the substrates encompassed in the hydrophobic pocket of the nanomicelles.9 Aqueous micellar catalysis is typically performed under conventional batch conditions where mixing and mass transfer is dependent on the batch size.5,8 Consequently, the scale-up is often challenging due to these physical limitations.10 In contrast, continuous flow reactors offer unique advantages to improve transport phenomena between phases, and due to their extraordinary surface-area-to-volume-ratio, heat transfer is also increased considerably.11 As a result, process intensification and simplification become feasible and scalability is also straightforward, whilst improving product quality and uniformity by better control over the reaction parameters. However, the handling of solids, such as slurries and suspensions, in flow reactors is perceived as one of the main challenges in continuous processing due to well-known clogging and reactor fouling issues.12 Despite the fact that numerous reactor concepts have been developed to handle solid–liquid biphasic mixtures,13 there is only one recent example for performing heterogeneous micellar catalysis under flow conditions, in which Suzuki–Miyaura couplings were achieved utilizing a series of continuous stirred-tank reactors (CSTRs) in the presence of Fe/Pd nanoparticles as heterogeneous catalyst.14 Although CSTRs offer a practical solution for handling solids under flow conditions,15 enlarging the tank size of each unit may result in inferior mixing behavior. Therefore, our aim was to take a different approach and to develop an inherently scalable continuous process for performing heterogeneous micellar catalysis in water using an oscillatory plug flow reactor (OFR). On the basis of our earlier experiences on continuous flow photocatalytic processes involving solids in OFRs,16 we speculated that the intense mixing by the use of pulsation superimposed to the net flow of the reaction stream would enhance mass transport, whilst ensuring a stable suspension of the aqueous solid–liquid mixture alongside the reactor. Additionally, by exclusion of moving parts from the reaction zone together with the optimization of the pulsation parameters, we anticipated a robust setup for handling biphasic micellar media facilitating minimal axial dispersion and a reasonable residence time distribution.
As a model for our proof of the concept study, we selected reductive amination,17 which is one of the key approaches for C–N bond construction and thus plays a paramount role in pharmaceutical and medicinal chemistry.18 Despite the fact that during initial imine formation stoichiometric water is released and thus reductive aminations performed in water is at first instance not beneficial,19 Lipshutz and co-workers recently reported a batch process for reductive aminations under aqueous micellar conditions.20 In this study, Pd/C was employed as a readily available heterogeneous catalyst and Et3SiH as reductant along with the rationally designed surfactant TPGS-750-M, which comprises a lipophilic vitamin E portion combined with a hydrophilic polyethylene glycol (PEG) side-chain (Fig. 1A).21 These literature findings served as starting point for our investigations to develop a novel continuous flow protocol for heterogeneously catalyzed reductive aminations in water enabled by micellar chemistry (Fig. 1B). Our results are presented herein.
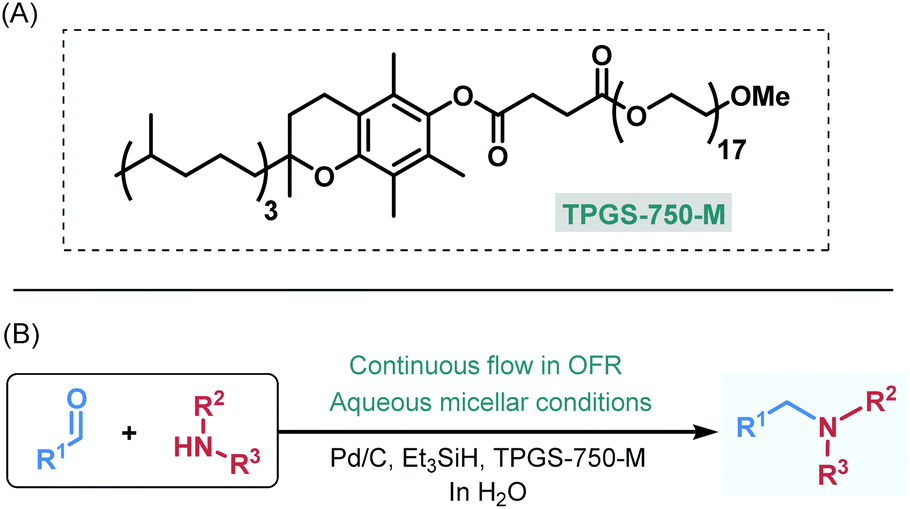 | ||
| Fig. 1 Surfactant TPGS-750-M (A), and general scheme of the present work (B). | ||
Results and discussion
Initially, batch screening of the most important reaction conditions was performed utilizing the reductive amination of benzaldehyde with aniline as a simple model in the presence of 1.2 equiv. of Et3SiH and various amounts of 5 wt% Pd/C (Table 1). Although, the direct use of hydrogen gas may have been more atom economic, we selected Et3SiH as reductant particularly because of safety as well as scalability aspects, and also due to the fact that the direct involvement of a gaseous reagent would have made mixing and mass transfer even more challenging under heterogeneous micellar conditions.|
|
|||||
|---|---|---|---|---|---|
| No.a | Surfactant | Pd/C [ppm] | T [°C] | Conv.b [%] | Select.c [%] |
| a 1 mL scale, stirring at 500 rpm. b Consumption of starting material, based on GC-FID area. c Selectivity, determined by GC-FID area. (The corresponding imine was detected as the only side product.) d 1 wt% TPGS-750-M. e 30 min reaction time. | |||||
| 1 | — | 2000 | 50 | 96 | 27 |
| 2 | SDS | 2000 | 50 | 90 | 32 |
| 3 | Brij S 100 | 2000 | 50 | 96 | 17 |
| 4 | Kolliphore EL | 2000 | 50 | 97 | 19 |
| 5 | TPGS-750-M | 2000 | 50 | >99 | 95 |
| 6 | TPGS-750-M | 1500 | 50 | >99 | 99 |
| 7 | TPGS-750-M | 1000 | 50 | >99 | 99 |
| 8 | TPGS-750-M | 500 | 50 | 98 | 96 |
| 9d | TPGS-750-M | 500 | 50 | 99 | 73 |
| 10 | TPGS-750-M | 500 | 25 | >99 | 25 |
| 11e | TPGS-750-M | 500 | 60 | >99 | 86 |
Upon investigating the effects of different amphiphiles, the corresponding imine intermediate was readily formed in all reactions, but its subsequent reduction to 1a proved highly dependent on the surfactant applied (entries 1–5). It was verified that TPGS-750-M is superior in this transformation compared with other surfactants as well as with the on-water (surfactant-free) reaction after 60 min at 50 °C. In fact, SDS, Brij S 100 and Kolliphore EL did not result any notable micellar accelerating effect and gave results comparable to the on-water reaction (90–97% conversion and 17–32% selectivity), which may be explained by the unfavorable hydrophilic–lipophilic balance of the surfactants and/or by inadequate size of the micelles formed. Gratifyingly, a catalyst loading of 500 ppm proved sufficient (entries 5–8), which is a significant decrease compared with earlier metal-catalyzed reductive aminations and is even lower than the amount reported earlier for reductive aminations under micellar conditions.17,20 The extraordinarily low catalyst loading can be explained by the presence of the PEG-containing nanomicelles formed by TPGS-750-M, which have a tendency to aggregate around palladium nanoparticles thus facilitating close contact between the catalyst and the reaction partners housed within the micelles.9 The reaction worked the best with 2 wt% of TPGS-750-M (entry 8); the use of only 1 wt% surfactant resulted in a significant decrease in amine formation (entry 9). Successful reduction of the imine intermediate required gentle heating, 50 °C or 60 °C was therefore considered as an optimum value (entries 8, 10 and 11). Besides activated charcoal, various further catalyst supports were tested under batch conditions. SiO2, BaCO3 and Al2O3 provided comparable results to activated charcoal, whereas BaSO4 resulted incomplete imine reduction (Table S1†).
Having acquired a clear picture on the effects of the reaction conditions of the reductive amination between benzaldehyde and aniline under aqueous micellar conditions in batch, the reaction was next repeated on different scales ranging from 1 mL to 100 mL, with the aim to investigate mixing effects and also to explore possible limitations in scalability. As shown in Fig. 2, the yield drastically decreased with the batch size despite adjustment of the stirring rate upon scale-up. For example, at 800 rpm stirring rate, the reaction furnished 92% 1a formation on 1 mL scale; however, yield decreased steeply to 62%, 18% and 14% upon increasing the batch size to 10 mL, 50 mL and 100 mL, respectively. It was also observed that in case of reactions performed on the same scale, the yield was clearly increasing with the stirring speed. These findings demonstrate that the biphasic micellar reaction is strongly dependent on mixing, which implies an obvious limitation of direct scalability under batch conditions. In fact, in typical applications of micellar reaction technology on scale, organic co-solvents are employed with the aim to improve physical properties of the bulk reaction medium and thus to facilitate mass transfer during the process.10,22
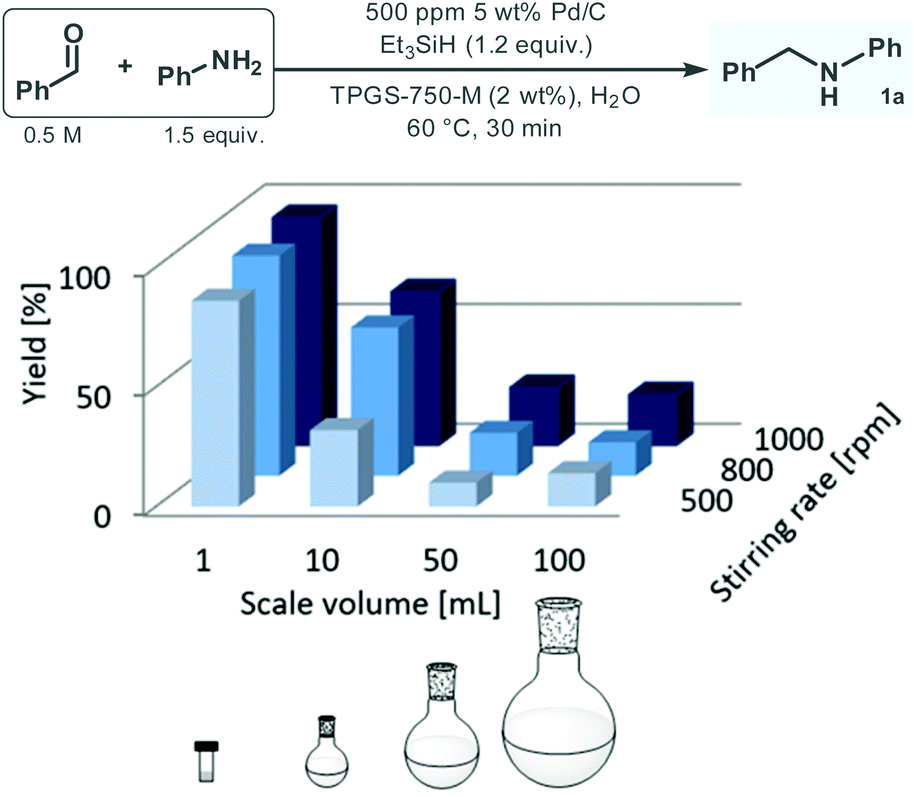 | ||
| Fig. 2 Investigation of the reductive amination of benzaldehyde with aniline under aqueous micellar conditions at different batch scales (yield was determined by GC-FID area%). | ||
With sufficient data on the batch reaction in our hands, we next turned our attention to continuous flow method development. For this purpose, a commercially available OFR (HANU™ HX 15 Flow Reactor, Creaflow) was chosen which comprises a linear plate-like process channel equipped with a series of cubic static mixing elements. Besides a metering pump delivering the net flow of the reaction components, the system contained an oscillatory diaphragm pump providing a tunable symmetrical pulse. In cooperation with the well-defined static mixing elements, this ensured split and recombine mixing of the process stream. Additionally, strategically placed vibrator motors facilitated suspension of particles in the peripheral tubings of the system, whilst the narrow reaction channels increased pulsation velocity thereby mitigating the risk of settling even at low flow rates. In a typical experiment, the slurry of the starting materials was continuously streamed through the reactor using a peristaltic pump. The system also comprised a 3-bar backpressure regulator (BPR) to prevent cavitation or suction of air from the reactor outlet during backward pulsation. The HANU reactor was originally designed for multiphasic flow photochemistry.16,23 However, we anticipated that because of the beneficial combination of active and passive mixing, it would enable a significantly improved mass transfer compared with batch processes and CSTRs, and would ensure a stable suspension of the biphasic medium alongside the reactor, thereby furnishing a robust and scalable platform for performing aqueous micellar reactions under continuous flow conditions. The schematic representation of the flow setup is depicted in Fig. 3.
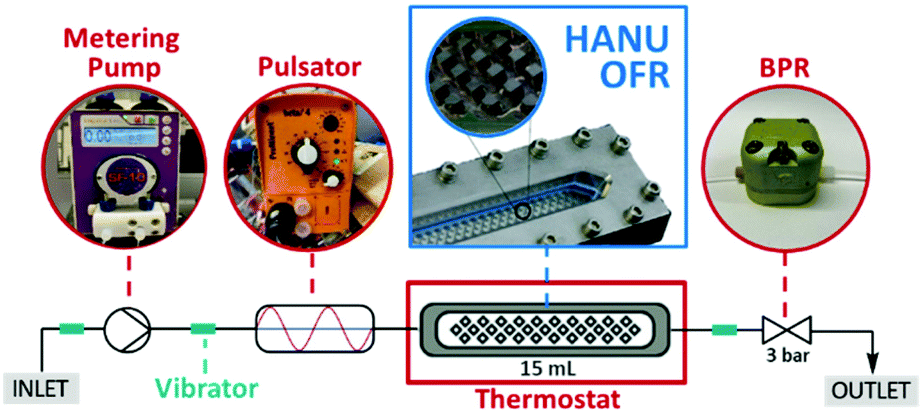 | ||
| Fig. 3 Schematic representation of the continuous flow setup used for reductive aminations under aqueous micellar conditions. | ||
After setting up the OFR, a flow optimization study was performed, which initially focused on pulsation amplitude (mL displaced per pump stroke) and frequency (number of strokes per second, Hz), the novel process parameters in this reactor setup. Fine-tuning of these parameters allowed us to establish a stable suspension of the biphasic reaction mixture, whilst minimizing axial dispersion in the reaction zone. As far as residence time distribution in the OFR is concerned, our control experiments in accordance with literature findings suggested that lower amplitudes typically imply narrow distribution curves, while the applied pulsation frequencies exert a similar trend but to a lower extent (Fig. S3†).16b,23a Upon investigating the effects of the pulsation parameters on the Et3SiH-mediated, Pd/C-catalyzed reductive amination of benzaldehyde with aniline in the presence of TPGS-750-M as surfactant, inconsistent conversions and an intense foam formation was found at lower amplitudes (Table 2, entries 1 and 2). Gratifyingly, by increasing the amplitude to 0.24 mL (approx. 50% of displaced volume), quantitative and selective reaction was achieved, whilst maintaining a stable suspension of the reaction mixture (entry 3). To further decrease the potential of foam formation, the frequency was reduced to 0.6 Hz (entry 4).
|
|
||||||
|---|---|---|---|---|---|---|
| No.a | Catalyst | A [mL] | ν [Hz] | t res [min] | Conv.d [%] | Select.e [%] |
| a 25 mL scale. b Pulsation amplitude (mL displaced per pump stroke). c Frequency (number of strokes per second). d Consumption of starting material, based on GC-FID area%. e Selectivity, determined by GC-FID area% (the corresponding imine was detected as the only side product). f 250 ppm catalyst. g Stability test, 60 mL scale. h Corresponding to 500 μL min−1 flow rate. i Corresponding to 750 μL min−1 flow rate. | ||||||
| 1 | 5 wt% Pd/C | 0.12 | 1.5 | 30h | >99 | 57–99 |
| 2 | 5 wt% Pd/C | 0.20 | 1.5 | 30h | >99 | 90–99 |
| 3 | 5 wt% Pd/C | 0.24 | 1.5 | 30h | >99 | >99 |
| 4 | 5 wt% Pd/C | 0.24 | 0.6 | 30h | >99 | >99 |
| 5 | 5 wt% Pd/Al2O3 | 0.24 | 0.6 | 30h | >99 | 62 |
| 6 | 5 wt% Pd/BaCO3 | 0.24 | 0.6 | 30h | >99 | 95 |
| 7g | 5 wt% Pd/C | 0.24 | 0.6 | 30h | >99 | >99 |
| 8 | 1 wt% Pd/C | 0.24 | 0.6 | 30h | >99 | >99 |
| 9 | 5 wt% Pd/C | 0.24 | 0.6 | 20i | 99 | 90 |
| 10f | 5 wt% Pd/C | 0.24 | 0.6 | 30h | 99 | 72 |
One of the main challenges of continuous flow process development under heterogeneous catalytic micellar conditions was occasional settling and deposition of particles along the reactor. We anticipated that in addition to fine-tuning of the pulsation parameters, the stability of the reaction suspension can also be facilitated by varying the nature of solids involved in the reaction. Therefore, the effects of catalyst supports which provided good results in batch were re-evaluated under continuous flow conditions. Although with 5 wt% Pd/Al2O3 and 5 wt% Pd/BaCO3 (500 ppm both) amine 1a was furnished with quantitative conversion and with reasonable selectivities (62% and 95%, respectively) under flow conditions too (entries 5 and 6), due to the high density of these support materials (Al2O3: 4.00 g mL−1; BaCO3: 4.49 g mL−1), settling of particles and clogging occurred in both cases preventing any practical applications (Fig. S4†). In contrast, with 5 wt% Pd/C (density of activated charcoal: 2.01 g mL−1), stable and clogging-free operation was achieved along with quantitative and selective formation of 1a (entry 7). Upon comparing 1 wt% and 5 wt% Pd/C (500 ppm Pd in both cases), no difference in stability of the suspension was found, further experiments were therefore carried out with 5 wt% Pd/C (entry 8). In further attempts to optimize the reaction conditions, the application of 250 ppm 5 wt% Pd/C and the decrease of residence time to 20 min resulted in somewhat reduced yields (entries 9 and 10), therefore 500 ppm catalyst loading and 30 min residence time (corresponding to 500 μL min−1 flow rate) were selected as optimum values. Regarding the effects of other surfactants than TPGS-750-M in flow mode, a similar negative trend could be observed than in batch together with occasional clogging (Table S2†).
In order to demonstrate the applicability of the process, a range of aldehydes and amines were next submitted to reductive amination under optimized micellar flow conditions.24 Besides benzaldehyde, its 4-fluoro- as well as 4-methoxy-substituted derivative were quantitatively and selectively reacted in reductive amination with aniline furnishing the corresponding secondary amines (1b and 1c) in high yields (84% and 81%, respectively; Fig. 4A). 1-Naphtaldehyde exhibited somewhat lower reactivity in reaction with aniline; however, upon increasing the catalyst loading to 1000 ppm, amine 1d could successfully be isolated in 58% yield. Furfural and 2-phenylpropanal showed high conversions and good selectivities, and resulted in amines 1e and 1f in 73% and 61% yield, respectively. Reductive amination of various aliphatic aldehydes with aniline also furnished good results. 2-Ethylhexanal reacted smoothly to give amine 1g in 75% yield. In reactions of cyclopentane- and cyclopropanecarbaldehyde, the corresponding amines (1h and 1i) were isolated in yields >70%; however, in these cases the catalyst loading was increased to 2000 ppm.
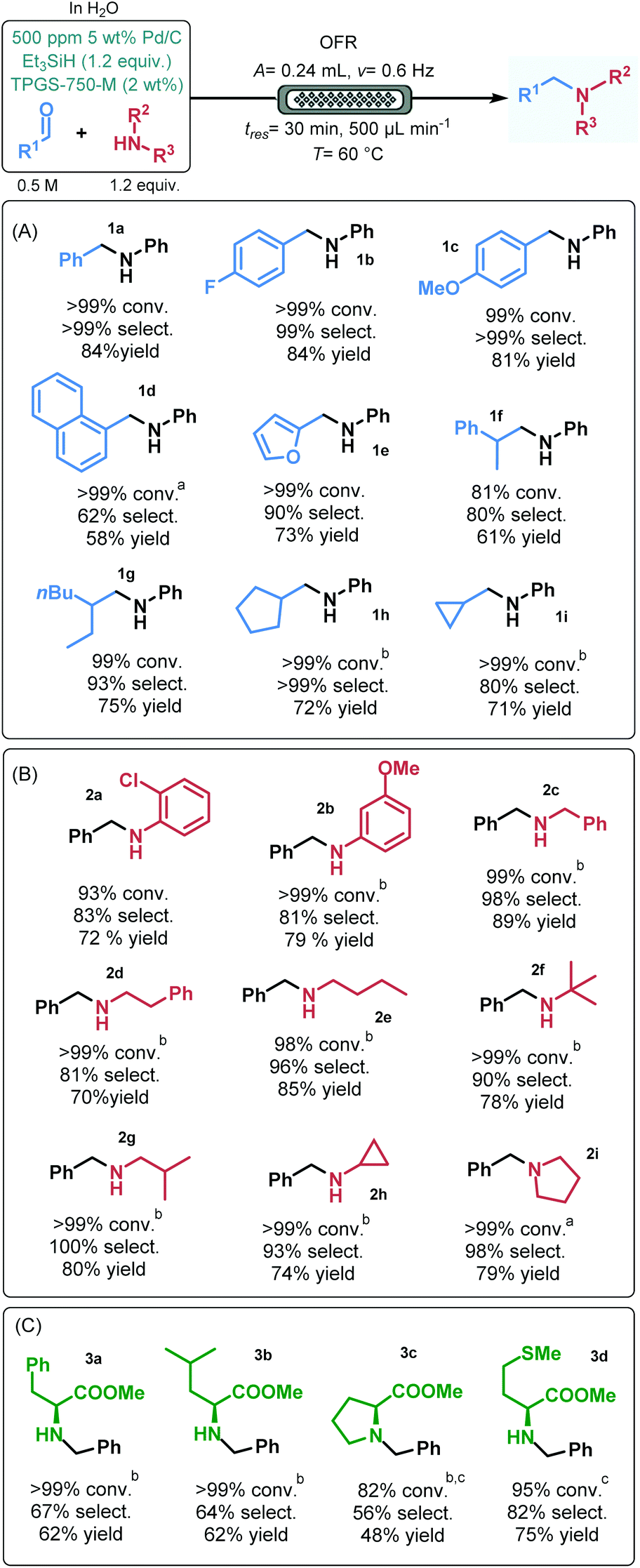 | ||
| Fig. 4 Substrate scope of the continuous flow Pd/C-catalyzed reductive amination under aqueous micellar conditions (conversion and selectivity were determined by GC-FID area%; yields shown are isolated yields). a1000 ppm 5 wt% Pd/C. b2000 ppm 5 wt% Pd/C. cAt 65 °C. | ||
The scope of amines was investigated using benzaldehyde as reaction partner (Fig. 4B). Various substituted aniline derivatives as well as benzylamine and phenylethylamine gave excellent conversions and high selectivities and resulted in the appropriate amines (2a–d) in 70–89% yields. Reductive amination of a diverse set of aliphatic amines, including open chain, α- and β-branched as well as alicyclic primary amines went smoothly under flow conditions. In these reactions, selectivities were ≥90% and the corresponding secondary amine products (2e–h) were isolated in yields up to 85%. Additionally, the secondary amine pyrrolidine was quantitatively and selectively converted to tertiary amine 2i and furnished 79% yield. Notably, for most of the amine substrates investigated, the catalyst loading was increased to 1000 or 2000 ppm.
Natural amino acids are frequently employed as chiral building blocks in the synthesis of various biologically active compounds and active pharmaceutical ingredients.25 To verify the scope and applicability further, a small library of N-benzylated amino acid derivatives was prepared by reductive amination of benzaldehyde with L-phenylalanine, L-leucine, L-proline and L-methionine methyl esters under micellar flow conditions (Fig. 4C). To our delight, the desired adducts (3a–d) could successfully be prepared in yields up to 75%; however, in these cases, imine formation was more difficult than in reactions of simpler model substrates (Fig. 4A and B), as indicated by the appearance of benzyl alcohol as a side product arising from competing aldehyde reduction. In these reactions, catalyst loading was typically set to 2000 ppm, and in some instances, temperature was increased to 65 °C.
To verify the stability and preparative capabilities of the developed flow process, the reductive amination of 4-fluorobenzaldehyde with aniline was performed as a long-run. Considering that the viscosity and density of the starting material solution may change over time due to background imine formation, a modified three-feed system was used to separately introduce a suspension containing the aqueous surfactant solution and the Pd/C catalyst as feed 1, a neat mixture of the aldehyde and the reductant as feed 2 and neat aniline as feed 3 (Fig. 5A). All other reaction conditions were set to the previously optimized values. A 7 h reaction window was monitored under steady state conditions with conversion and selectivity being checked in every hour.26 To our delight, the system proved very robust during the experiment, no clogging or other issues occurred. Since quantitative conversion and a selectivity of ≥98% were achieved in all fractions, after extractive work-up, a simple silica filtration was sufficient to remove any excess components and to achieve analytically pure product. The isolated yield was thus readily determined in all portions collected providing in total 15.0 g of amine 1b (85% overall yield). This correlates to a space–time–yield (STY) of 143 g L−1 h−1 and a productivity of 2.14 g h−1. Practically no leaching occurred during the experiment as verified by a palladium content of 1.4 ppm measured in the crude product, which could readily be decreased to <0.05 ppm by simple silica gel filtration. These amounts meet the current FDA guidelines on maximum acceptable limits of residual palladium in drug substances and excipients.27 Importantly, the process generated a low amount of waste as indicated by an E-factor of 1.2, which is significantly lower than typically found in pharmaceutical manufacturing.28
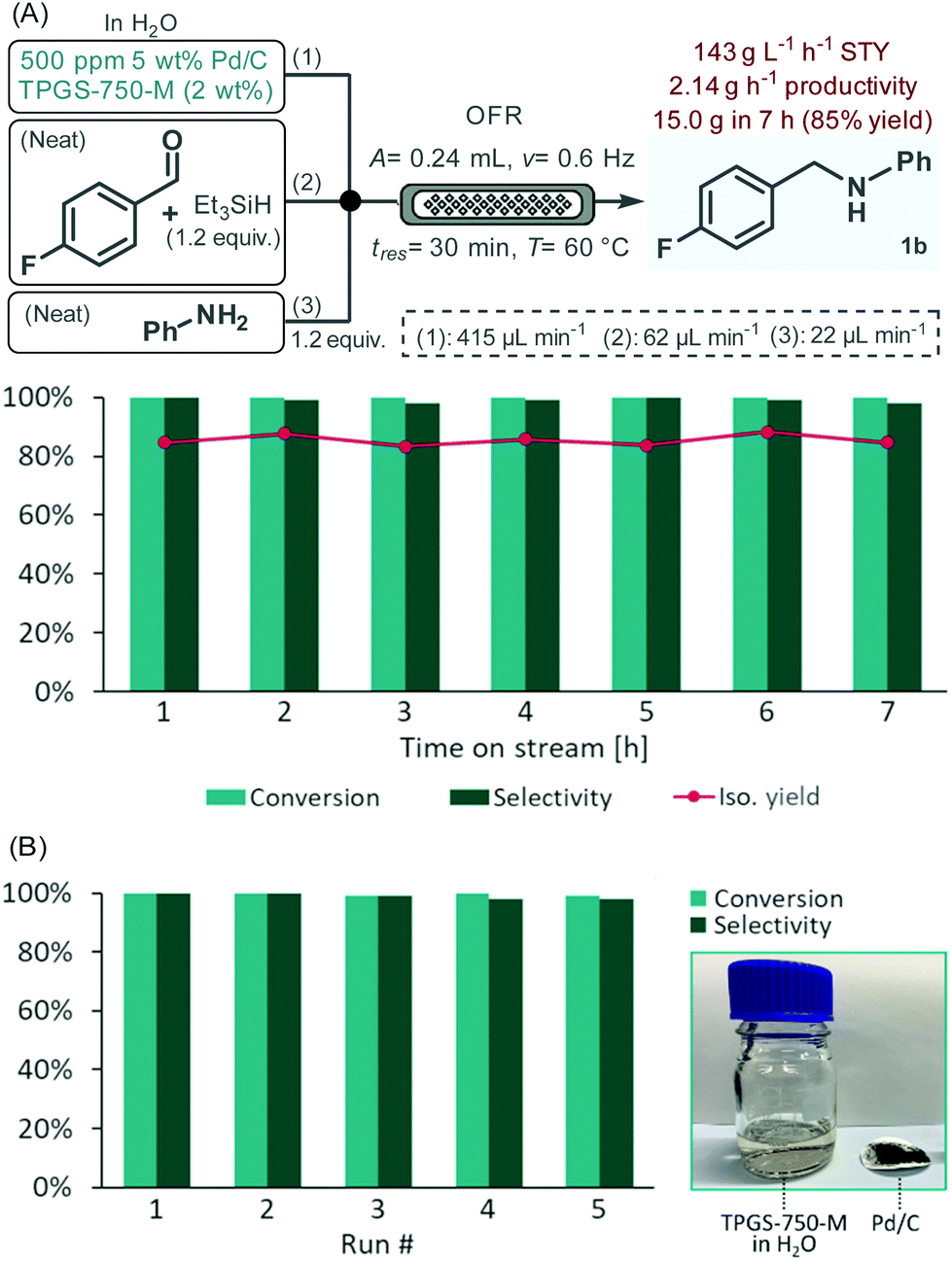 | ||
| Fig. 5 Long run experiment under optimized flow conditions (A), and recycling of the catalyst as well as the surfactant solution in the same reaction (B). Conversion and selectivity were determined by GC-FID area%. | ||
With the aim to further enhance the sustainable aspects of the current process, the recycling of the heterogeneous catalyst as well as of the surfactant solution was attempted utilizing the same reaction and the same conditions than in the long-run. Between each cycle, the amine product was removed from the reaction mixture by extraction with EtOAc; the catalyst was filtered off and washed with minimal amounts of EtOAc and water, dried at room temperature and used again in the next run. The residual organic solvent was removed from the surfactant solution under reduced pressure before reusing. In this manner, 91–95% of Pd/C along with >99% of the surfactant solution could be recovered in each run (see Table S3†). Gratifyingly, no loss of the initially quantitative conversion occurred during five consecutive runs, and selectivity remained steady at >98% (Fig. 5B).
Conclusions
A continuous flow process has been demonstrated for reductive aminations under heterogeneous catalytic micellar conditions in water using an OFR. The process relied on Et3SiH as reducing agent along with ppm amounts of Pd/C as heterogeneous catalyst and TPGS-750-M as a commercially available surfactant. Due to the beneficial combination of an oscillatory flow regime and multiple static mixing elements, the flow system proved as a robust and scalable platform for performing reactions under aqueous micellar conditions, whilst ensuring enhanced mixing and mass transfer between the reacting phases. Following thorough optimization of the most important reaction conditions, such as pulsation parameters, temperature, residence time and catalyst carrier, high yielding reductive aminations were achieved applying comparatively mild conditions, whilst maintaining a stable suspension of the biphasic micellar media alongside the reactor. Reductive aminations of a diverse set of aldehydes with various amines were achieved, including a number of amino acid derivatives as pharmaceutically relevant building blocks. The preparative utility of the flow process was verified by a 7 h long-run furnishing straightforward multigram-scale production with low waste formation. The sustainable nature of the process was further elaborated by successfully recycling the heterogenous catalyst as well as the aqueous surfactant solution over 5 consecutive runs.To the best of our knowledge, this is the first demonstration of aqueous micellar catalysis in an OFR system. Notably, the process developed herein offers a straightforward scaling-up strategy by using a commercially available pilot-scale reactor, which maintains all the major process characteristics, including channel dimensions, residence time distribution and heat- and mass transfer.23a,29 We are currently investigating these scale-up possibilities.
Experimental section
General information on the flow setup
Flow experiments were carried out in a commercially available OFR, consisting of a HANU HX 15 Flow Reactor (Hastelloy, 15 mL internal volume) containing 2 × 2 × 2 mm cubic static mixers and a pulsator (customized ProMinent Beta/4 pump, PTFE/carbon pump head) from Creaflow. The feed mixtures were introduced using a Vapourtec SF-10 peristaltic pump and a Syrris Asia syringe pump module. Pressure was applied by an adjustable Zaiput BPR. Thermal regulation was managed by using a Huber CC304 thermostat. Parts of the setup were connected using 1/8′′ OD (1.6 mm ID) or 1/16′′ OD (0.8 mm ID) PTFE or PFA tubes along with PEEK or stainless steel fittings.Representative procedure for the batch experiments
A vial containing 500 ppm 5 wt% Pd/C (0.05 mol% Pd) was charged with 0.5 mmol of benzaldehyde (1 equiv.), 0.75 mmol of aniline (1.5 equiv.) and 1 mL of a 2 wt% TPGS-750-M solution in H2O. After the addition of 0.6 mmol Et3SiH (1.2 equiv.) the vial was sealed and the mixture was heated in an aluminum block at 60 °C for 30 min. The reaction mixture was next extracted with 1.5 mL EtOAc and the organic phase was filtered using a syringe filter (0.45 μm), dried over Na2SO4, diluted with acetonitrile (10 μL in 1 mL), and submitted to GC-FID analysis.Representative procedures for the flow experiments
For investigating the substrate scope under flow conditions, a single-feed setup was used as shown in Fig. 4 and also in Fig. S1.† A Duran bottle was charged with 31.5 mg 5 wt% Pd/C (500 ppm or 0.05 mol% Pd), 30 mmol of the corresponding aldehyde (1 equiv.), 36 mmol of the corresponding amine (1.2 equiv.), 60 mL 2 wt% aqueous TPGS-750-M solution and 15 mmol Et3SiH (1.2 equiv.). While being stirred using a magnetic stirrer, the mixture was continuously pumped through the OFR at 500 μL min−1 flow rate, while maintaining 3 bar pressure and 60 °C temperature. During the reaction, the pulsator was set to 0.24 mL (approx. 50%) pulsation amplitude and 0.6 Hz (20%) pulsation frequency. The product mixture was collected for 20 min under steady state conditions and was next extracted with a minimal amount of EtOAc (2 × 5 mL). The organic layers were combined and the solvent was removed under reduced pressure. The obtained material was purified by flash chromatography on silica gel using mixtures of cyclohexane and EtOAc as eluent to afford the corresponding analytically pure product.For the long-run experiment, a modified three-feed system was used as shown in Fig. 5 and also in Fig. S5.† Feed 1 consisted of the aqueous surfactant solution (2 wt% TPGS-750-M; 240 mL) and Pd/C (126 mg 5 wt% Pd/C – 500 ppm or 0.05 mol% Pd). Feed 2 contained a neat mixture of 4-fluorobenzaldehyde and 1.2 equiv. of Et3SiH, while feed 3 comprised neat aniline. Feed 1 was pumped at a flow rate of 415 μL min−1, feed 2 at 62 μL min−1 and feed 3 at 22 μL min−1. Similarly as in the single feed experiments, the flow reactor was pressurized at 3 bar and heated at 60 °C, while the pulsator was set to 0.24 mL (approx. 50%) pulsation amplitude and 0.6 Hz (20%) pulsation frequency. The product mixture was collected for 7 h under steady state conditions into separate fractions in every hour. Each fraction was extracted with a minimal amount of EtOAc (2 × 10 mL). The organic layers were combined and the solvent was removed under reduced pressure. The obtained material was purified by filtration through a silica plug in order to afford the corresponding analytically pure product (15.0 g, 85% yield over 7 h collection time).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The CCFLOW Project (Austrian Research Promotion Agency FFG no. 862766) is funded through the Austrian COMET Program by the Austrian Federal Ministry of Climate Protection, Environment, Energy, Mobility, Innovation and Technology (BMK), the Austrian Federal Ministry for Digital and Economic Affairs (BMDW), and by the State of Styria (Styrian Funding Agency SFG). Z.N. acknowledges the support of National Research, Development and Innovation Office (NKFIH, K132077) and the ELTE Thematic Excellence Programme 2020 Supported by NKFIH - TKP2020-IKA-05. This research was partially financed by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund (2019-1.1.1-PIACI-KFI-2019-00070). The authors would like to thank Creaflow for the generous loan of the HANU Reactor used in this study. The assistance of Dr. Alejandro Mata-Gomez in the preliminary batch experiments is gratefully acknowledged. The authors also thank Prof. Walter Goessler for ICP-MS analysis.References
- (a) P. Cintas, S. Tabasso, V. V. Veselov and G. Cravotto, Curr. Opin. Green Sustainable Chem., 2020, 21, 44–49 CrossRef; (b) M. Tavakolian, S. Vahdati-Khajeh and S. Asgari, ChemCatChem, 2019, 11, 2943–2977 CrossRef CAS; (c) Z. Sainath and P. Pravinkumar, Curr. Org. Chem., 2019, 23, 2295–2318 Search PubMed; (d) M. B. Gawande, V. D. B. Bonifácio, R. Luque, P. S. Branco and R. S. Varma, ChemSusChem, 2014, 7, 24–44 CrossRef CAS PubMed.
- (a) H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929–1961 RSC; (b) M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberth, Green Chem., 2018, 20, 5082–5103 RSC; (c) D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC; (d) R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- (a) M. Cortes-Clerget, J. Yu, J. R. A. Kincaid, P. Walde, F. Gallou and B. H. Lipshutz, Chem. Sci., 2021, 12, 4237–4266 RSC; (b) T. Kitanosono, K. Masuda, P. Xu and S. Kobayashi, Chem. Rev., 2018, 118, 679–746 CrossRef CAS PubMed; (c) B. H. Lipshutz and S. Ghorai, Green Chem., 2014, 16, 3660–3679 RSC; (d) M. B. Gawande, V. D. B. Bonifácio, R. Luque, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 5522–5551 RSC; (e) A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725–748 CrossRef CAS PubMed; (f) D. Dallinger and C. O. Kappe, Chem. Rev., 2007, 107, 2563–2591 CrossRef CAS PubMed.
- R. Breslow, in Handbook of Green Chemistry, ed. P. T. Anastas, Wiley–VCH, 2010, pp. 1–29 Search PubMed.
- (a) B. H. Lipshutz, J. Org. Chem., 2017, 82, 2806–2816 CrossRef CAS PubMed; (b) B. H. Lipshutz, F. Gallou and S. Handa, ACS Sustainable Chem. Eng., 2016, 4, 5838–5849 CrossRef CAS; (c) G. La Sorella, G. Strukul and A. Scarso, Green Chem., 2015, 17, 644–683 RSC.
- (a) S. R. K. Minkler, N. A. Isley, D. J. Lippincott, N. Krause and B. H. Lipshutz, Org. Lett., 2014, 16, 724–726 CrossRef CAS PubMed; (b) A. Krasovskiy, C. Duplais and B. H. Lipshutz, Org. Lett., 2010, 12, 4742–4744 CrossRef CAS PubMed; (c) A. Krasovskiy, C. Duplais and B. H. Lipshutz, J. Am. Chem. Soc., 2009, 131, 15592–15593 CrossRef CAS PubMed; (d) S. Shirakawa and S. Kobayashi, Org. Lett., 2007, 9, 311–314 CrossRef CAS PubMed.
- (a) B. H. Lipshutz, N. A. Isley, J. C. Fennewald and E. D. Slack, Angew. Chem., Int. Ed., 2013, 52, 10952–10958 CrossRef CAS PubMed; (b) B. H. Lipshutz, S. Ghorai, W. W. Y. Leong, B. R. Taft and D. V. Krogstad, J. Org. Chem., 2011, 76, 5061–5073 CrossRef CAS PubMed.
- (a) T. Lorenzetto, G. Berton, F. Fabris and A. Scarso, Catal. Sci. Technol., 2020, 10, 4492–4502 RSC; (b) A. Steven, Synthesis, 2019, 2632–2647 CAS; (c) B. H. Lipshutz, S. Ghorai and M. Cortes-Clerget, Chem. – Eur. J., 2018, 24, 6672–6695 CrossRef CAS PubMed; (d) D. Paprocki, A. Madej, D. Koszelewski, A. Brodzka and R. Ostaszewski, Front. Chem., 2018, 6, 502 CrossRef CAS PubMed.
- B. H. Lipshutz, Johnson Matthey Technol. Rev., 2017, 61, 196–202 CrossRef CAS.
- (a) M. Parmentier, M. Wagner, R. Wickendick, M. Baenziger, A. Langlois and F. Gallou, Org. Process Res. Dev., 2020, 24, 1536–1542 CrossRef CAS; (b) D. J. Lippincott, E. Landstrom, M. Cortes-Clerget, B. H. Lipshutz, K. Buescher, R. Schreiber, C. Durano, M. Parmentier, N. Ye, B. Wu, M. Shi, H. Yang, M. Andersson and F. Gallou, Org. Process Res. Dev., 2020, 24, 841–849 CrossRef CAS.
- (a) M. Guidi, P. H. Seeberger and K. Gilmore, Chem. Soc. Rev., 2020, 49, 8910–8932 RSC; (b) L. Rogers and K. F. Jensen, Green Chem., 2019, 21, 3481–3498 RSC; (c) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed; (d) B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed; (e) V. Hessel, D. Kralisch, N. Kockmann, T. Noël and Q. Wang, ChemSusChem, 2013, 6, 746–789 CrossRef CAS PubMed.
- (a) Y. Chen, J. C. Sabio and R. L. Hartman, J. Flow Chem., 2015, 5, 166–171 CrossRef; (b) K. J. Wu and S. Kuhn, Chim. Oggi, 2014, 32, 62–66 Search PubMed; (c) R. L. Hartman, Org. Process Res. Dev., 2012, 16, 870–887 CrossRef CAS.
- (a) P. Bianchi, J. D. Williams and C. O. Kappe, J. Flow Chem., 2020, 10, 475–490 CrossRef CAS; (b) M. Jiang and X.-W. Ni, Org. Process Res. Dev., 2019, 23, 882–890 CrossRef CAS; (c) P. Filipponi, A. Gioiello and I. R. Baxendale, Org. Process Res. Dev., 2016, 20, 371–375 CrossRef CAS; (d) S. Falß, G. Tomaiuolo, A. Perazzo, P. Hodgson, P. Yaseneva, J. Zakrzewski, S. Guido, A. Lapkin, R. Woodward and R. E. Meadows, Org. Process Res. Dev., 2016, 20, 558–567 CrossRef; (e) A.-K. Liedtke, F. Bornette, R. Philippe and C. de Bellefon, Chem. Eng. J., 2016, 287, 92–102 CrossRef CAS; (f) T. McGlone, N. E. B. Briggs, C. A. Clark, C. J. Brown, J. Sefcik and A. J. Florence, Org. Process Res. Dev., 2015, 19, 1186–1202 CrossRef CAS; (g) S. Mascia, P. L. Heider, H. Zhang, R. Lakerveld, B. Benyahia, P. I. Barton, R. D. Braatz, C. L. Cooney, J. M. B. Evans, T. F. Jamison, K. F. Jensen, A. S. Myerson and B. L. Trout, Angew. Chem., Int. Ed., 2013, 52, 12359–12363 CrossRef CAS PubMed; (h) D. L. Browne, B. J. Deadman, R. Ashe, I. R. Baxendale and S. V. Ley, Org. Process Res. Dev., 2011, 15, 693–697 CrossRef CAS; (i) S. Kuhn, T. Noël, L. Gu, P. L. Heider and K. F. Jensen, Lab Chip, 2011, 11, 2488–2492 RSC; (j) L. Liguori and H.-R. Bjørsvik, Org. Process Res. Dev., 2011, 15, 997–1009 CrossRef CAS.
- A. B. Wood, K. Y. Nandiwale, Y. Mo, B. Jin, A. Pomberger, V. L. Schultz, F. Gallou, K. F. Jensen and B. H. Lipshutz, Green Chem., 2020, 22, 3441–3444 RSC.
- (a) A. Pomberger, Y. Mo, K. Y. Nandiwale, V. L. Schultz, R. Duvadie, R. I. Robinson, E. I. Altinoglu and K. F. Jensen, Org. Process Res. Dev., 2019, 23, 2699–2706 CrossRef CAS; (b) Y. Mo, H. Lin and K. F. Jensen, Chem. Eng. J., 2018, 335, 936–944 CrossRef CAS; (c) M. R. Chapman, M. H. T. Kwan, G. King, K. E. Jolley, M. Hussain, S. Hussain, I. E. Salama, C. González Niño, L. A. Thompson, M. E. Bayana, A. D. Clayton, B. N. Nguyen, N. J. Turner, N. Kapur and A. J. Blacker, Org. Process Res. Dev., 2017, 21, 1294–1301 CrossRef CAS; (d) Y. Mo and K. F. Jensen, React. Chem. Eng., 2016, 1, 501–507 RSC.
- (a) P. Bianchi, J. D. Williams and C. O. Kappe, Green Chem., 2021, 23, 2685–2693 RSC; (b) C. Rosso, S. Gisbertz, J. D. Williams, H. P. L. Gemoets, W. Debrouwer, B. Pieber and C. O. Kappe, React. Chem. Eng., 2020, 5, 597–604 RSC.
- K. Murugesan, T. Senthamarai, V. G. Chandrashekhar, K. Natte, P. C. J. Kamer, M. Beller and R. V. Jagadeesh, Chem. Soc. Rev., 2020, 49, 6273–6328 RSC.
- O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS PubMed.
- (a) S. Sato, T. Sakamoto, E. Miyazawa and Y. Kikugawa, Tetrahedron, 2004, 60, 7899–7906 CrossRef CAS; (b) K. Manabe, S. Iimura, X.-M. Sun and S. Kobayashi, J. Am. Chem. Soc., 2002, 124, 11971–11978 CrossRef CAS PubMed.
- R. R. Thakore, B. S. Takale, G. Casotti, E. S. Gao, H. S. Jin and B. H. Lipshutz, Org. Lett., 2020, 22, 6324–6329 CrossRef CAS PubMed.
- B. H. Lipshutz, S. Ghorai, A. R. Abela, R. Moser, T. Nishikata, C. Duplais, A. Krasovskiy, R. D. Gaston and R. C. Gadwood, J. Org. Chem., 2011, 76, 4379–4391 CrossRef CAS PubMed.
- C. M. Gabriel, N. R. Lee, F. Bigorne, P. Klumphu, M. Parmentier, F. Gallou and B. H. Lipshutz, Org. Lett., 2017, 19, 194–197 CrossRef CAS PubMed.
- (a) W. Debrouwer, W. Kimpe, R. Dangreau, K. Huvaere, H. P. L. Gemoets, M. Mottaghi, S. Kuhn and K. Van Aken, Org. Process Res. Dev., 2020, 24, 2319–2325 CrossRef CAS; (b) Z. Wen, A. Maheshwari, C. Sambiagio, Y. Deng, G. Laudadio, K. Van Aken, Y. Sun, H. P. L. Gemoets and T. Noël, Org. Process Res. Dev., 2020, 24, 2356–2361 CrossRef CAS PubMed.
- In contrast to the optimization experiments, the amine excess was reduced to 1.2 equiv.
- R. Vardanyan and V. Hruby, Synthesis of Best-Seller Drugs, Academic Press, London, 2016 Search PubMed.
- The system reached steady state in 90 min.
- For more information, see the corresponding FDA guidance: https://www.fda.gov/media/135956/download.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- For further details see: https://www.creaflow.be/hanutm-hx-150-flow-reactors.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc02039k |
| This journal is © The Royal Society of Chemistry 2021 |