
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the selectivity of biomass oxidation over oxygen evolution on NiO–OH electrodes†
Laxman
Gouda
ab,
Laurent
Sévery
a,
Thomas
Moehl
a,
Elena
Mas-Marzá
b,
Pardis
Adams
 a,
Francisco
Fabregat-Santiago
*b and
S. David
Tilley
*a
a,
Francisco
Fabregat-Santiago
*b and
S. David
Tilley
*a
aDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland. E-mail: david.tilley@chem.uzh.ch
bInstitute of Advanced Materials (INAM), Universitat Jaume I, Avda. V. Sos Baynat s/n, 12006 Castelló de la Plana, Spain. E-mail: fabresan@uji.es
First published on 29th September 2021
Abstract
Electrochemical reactions powered by renewable electricity are an important means of reducing the carbon footprint of large-scale chemical processes. Here, we investigate the efficient conversion of biomass-derived 5-hydroxymethylfurfural (HMF) to 2,5-furandicarboxylic acid (FDCA), an important building block in the polymer and pharmaceutical industries, using a cheap and abundant nickel-based electrocatalyst. We elucidate the key factors for tuning the chemical selectivity for HMF oxidation over the competing oxygen evolution reaction (OER) at the catalyst surface. We show that the selectivity for HMF oxidation is enhanced by removing trace impurities of iron species as well as adjusting the composition of the alkali hydroxide electrolyte solution. LiOH solution without iron impurities is more favorable for HMF oxidation, whereas CsOH solution with iron species present is more active for the OER and unfavorable for HMF oxidation. Under optimized conditions, HMF oxidation in 1 M LiOH electrolyte solution without iron (pH 14) achieved 98% faradaic efficiency for the production of FDCA. The principles used in this work can be applied to other electrosynthetic reactions, in particular where the OER is the main competing side reaction.
1. Introduction
Electrifying organic synthesis has undergone a revival in the last few decades, emerging as a green method for industrial scale chemical synthesis with low pollution impact.1 The attractive feature of electrochemical synthesis is that it can be powered by cheap renewable energy sources (photovoltaics, photoelectrochemical cells, windmills, hydropower), which motivates the use and scale-up of electrochemical technologies for industrial applications.2–4 Generally, electrosynthesis proceeds with coupled oxidation and reduction reactions on two electrodes, which is similar to the electrolysis of water with the oxygen evolution reaction (OER) at the anode and the hydrogen evolution reaction (HER) at the cathode.2 The synthesis of hydrogen from renewable sources is of high interest not only as a potential source of fuel for transportation and heating, but also for chemical processes such as the Haber–Bosch ammonia synthesis, crude oil refining, metal production, methanol production, food processing, and electronics.5,6 The vast majority of hydrogen today is produced from natural gas (96%), with the remainder produced by electrolysis.7 Therefore, a green synthesis of hydrogen is needed to reduce the carbon footprint of many important chemical process.8Regarding oxygen production, natural photosynthesis is sufficient to supply the global requirements of the chemical industry. Therefore, in electrolysis, instead of using the oxidation equivalents to drive the notoriously demanding OER (and yielding a low value product), the OER could be replaced by a useful alternative reaction leading to added-value products.3,9 One option for this alternative reaction is the oxidation of lignocellulosic biomass, an abundant and renewable chemical feedstock for many commodity chemicals.3,10 Among biomass-based commodity chemicals, 2,5-furandicarboxylic acid (FDCA) belongs to the most economically valuable ones.11 The biomass derived product FDCA is used to fabricate the polymer polyethylene furanoate (PEF). The properties of PEF are superior in many ways to those of the petrochemical-based polyethylene terephthalate (PET), such as lower melting point, higher glass transition temperature, better barrier properties to O2 and CO2, and 100% recyclability with reduced carbon footprint, which would enable the fabrication of lighter and better plastic containers and packagings.12–14 It is estimated that 30.3 million tons of PET are produced per year globally.12 Therefore, PEF has the potential to replace PET as a renewable alternative.15 The building block FDCA is synthesized from 5-hydroxymethylfurfural (HMF), which is derived from lignocellulosic biomass.16
There are several ways to convert HMF into FDCA, such as aerobic oxidation, thermal treatment, and high temperature catalytic oxidation. These methods are sufficient to reach a large industrial scale, although the long processing time, high operating temperatures, and by-product formation make these processes cost-inefficient.16,17 An alternative approach is electrochemical oxidation, where HMF oxidation is driven by electrical potential, eliminating the use of chemical oxidants for the process.18 Considering its potential for low cost, high yield and the scalability of this method, the electrocatalytic approach to biomass oxidation has attracted notable attention.19 Electrosynthesis coupled with HER presents an elegant solution to provide renewable hydrogen for the chemical industry along with the synthesis of valuable biomass-derived chemical products.10,20
As early as 1991, electrocatalytic oxidation of HMF to FDCA was first reported by Grabowski and co-workers.21 Typically, electrochemical HMF oxidation is performed in basic media and can take place over two distinct reaction pathways, as shown in Scheme 1.16 It has been reported that electrochemical HMF oxidation in aqueous solution competes with water oxidation under the required operating potentials, resulting in relatively low FDCA yields.22 To circumvent competition with the OER, redox mediators such as TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl) have been used for indirect HMF oxidation, with excellent conversion efficiencies.20 However, the use of organic redox mediators is considered to be cost-prohibitive on industrial scale. As an alternative, nickel (and its corresponding oxides and hydroxides, specifically nickel oxide hydroxide NiO–OH) is a cheap, abundant and widely explored redox catalyst for alcohol oxidation reactions.23 The first reported electro oxidation of HMF with NiO–OH gave only 71% FDCA yield at pH 14, however subsequent advancement in the NiO–OH chemistry, gave more than 90% FDCA yield.21,24 NiO–OH is also a good water oxidation catalyst, and therefore a strategy that favors the organic oxidation reaction (alcohol and aldehyde functional groups oxidation) over the OER is required.25 There are two main strategies reported in the literature to tune the selectivity: tailoring the electrolyte composition and electrolysis conditions, or altering the nickel surface structure with different starting materials (i.e. NiP, NiS and NiFe).10,26–28 In any case, the main active species in Ni based catalysts is amorphous β-NiO–OH.24,25,29
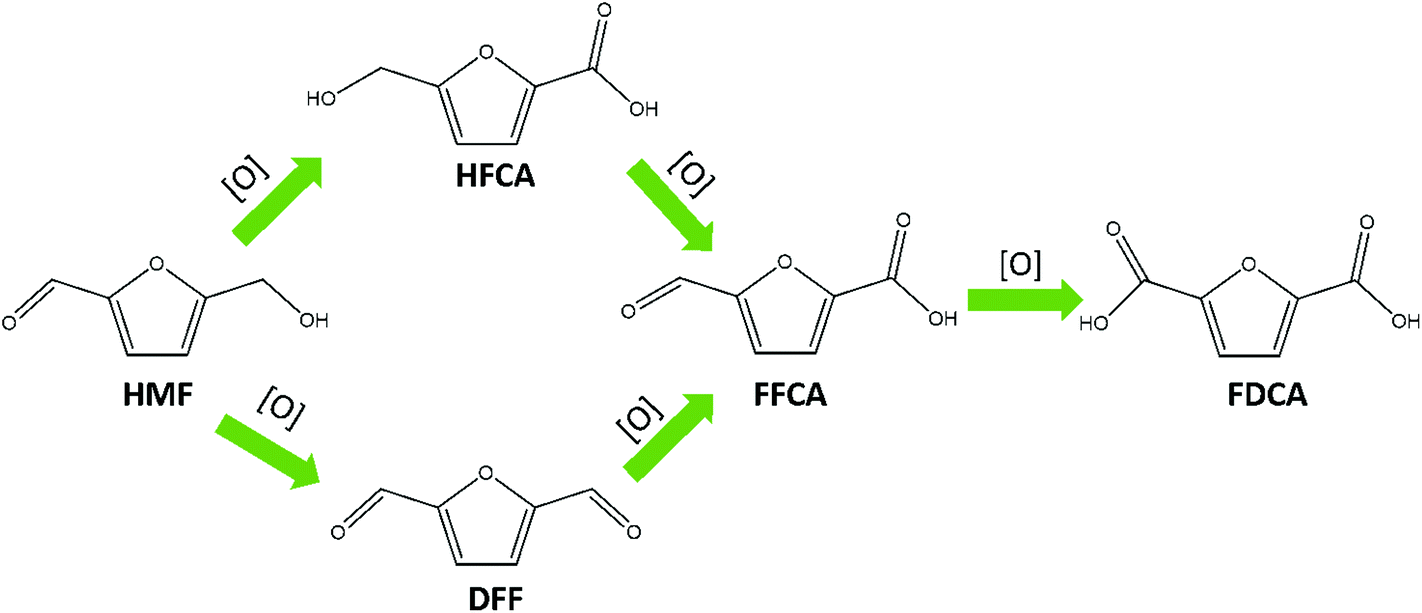 | ||
| Scheme 1 Electrochemical HMF oxidation reaction pathways. HMF is converted to DFF (diformyl furan) or HFCA (hydroxymethyl furan carboxylic acid), which are then further oxidized to FFCA (formyl furan carboxylic acid) and finally to FDCA (2,5-furandicarboxylic acid). | ||
Most of the organic oxidation reactions using NiO–OH are reported at pH > 11, where this catalyst also has optimal activity for the OER reaction.30 Although the overpotential for the HMF oxidation reaction can be much higher than for the OER, it is thermodynamically easier, and therefore selective HMF oxidation over the OER can be achieved.31,32 The OER onset of NiO–OH can be tuned by varying the electrolyte solution composition with different cations (Cs+, K+, Na+ and Li+) as well as by incorporating iron into the catalyst, transforming the NiO–OH to Ni1−xFexOOH.33–37 Thus, the competition between the two electrochemical oxidation reactions at the NiO–OH surface can be tuned by controlling the traces of iron and the alkali metal cation in the electrolyte solution.
Our simple approach uses an electrodeposited Ni(OH)2 film as a precursor to study the NiO–OH electrode systematically for HMF oxidation, under strongly basic conditions. We performed in situ studies to explore HMF oxidation vs. self-degradation as well as the selectivity between HMF oxidation and the competing OER. The key finding is that removal of traces of iron from the electrolyte solution enables high chemical selectivity of HMF oxidation over water oxidation, which enables the practical use of earth abundant Ni-based anodes in aqueous electrochemical cells targeting oxidation of organic molecules. High chemical selectivity of NiO–OH for HMF oxidation over water oxidation can be then achieved by smaller alkali cations in combination with an electrolyte solution free of traces of iron.
2. Results and discussion
Electrochemical HMF oxidation and OER on NiO–OH thin film electrodes
Fig. 1 shows a general scheme of the chemical transformations in a single compartment cell with a NiO–OH anode and a Pt cathode, and the individual redox reactions of the HER, OER, and HMF oxidation reaction are given in Fig. S1.† The topography of the electrodeposited films after chemical activation (see ESI†) was determined by atomic force microscopy, which confirms that the resulting nickel oxide layer is uniform with small grains (Fig. S2a†). The roughness of the electrode is mainly determined by the underlying FTO substrate. Energy dispersive X-ray analysis also confirms the Ni on FTO electrode after chemical activation (Fig. S2b†). X-ray photoelectron spectroscopy (XPS) analysis indicates that before use the film is entirely in the NiII oxidation state, while after use a mixture of NiII and NiIII is observed (Fig. S3†).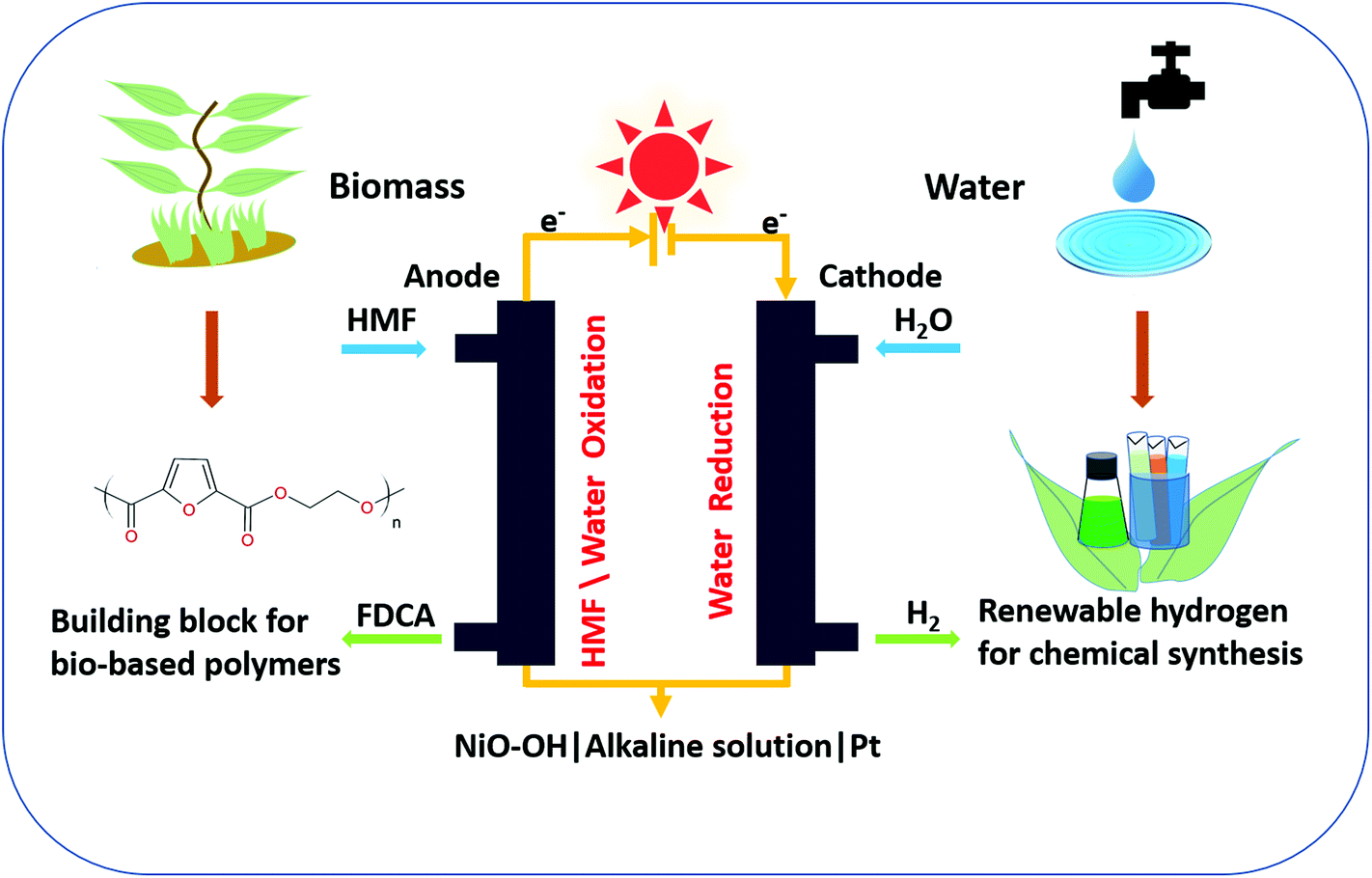 | ||
| Fig. 1 General scheme of the complete process of FDCA + H2 production through a single chamber electrolytic cell for HMF/water oxidation on a NiO–OH electrode and water reduction at a Pt electrode under alkaline conditions. | ||
A typical cyclic voltammetry (CV) measurement under stirring of a NiO–OH electrode after activation in the standard iron-free electrolyte solution (1 M LiOH) is shown in Fig. 2a (black line). The main features in this CV are the Ni2+/Ni3+ redox peak between 1.2 to 1.4 V and the OER onset at 1.6 V vs. RHE (with ∼350 mV overpotential at a current of 0.1 mA cm−2, see also Fig. S4†).29,35 A broad oxidation peak is also visible at around 1.8 V, which corresponds to the higher oxidation state Ni3+/Ni4+.35 Upon addition of HMF, an oxidative wave appears with a peak at 1.47 V vs. RHE, merging with the OER at more positive potentials (Fig. 2a; red trace). After 300 CV scans, the HMF oxidation peak is no longer visible (Fig. 2a; green trace), indicating nearly complete consumption or degradation of the substrate (vide infra for details in the quantification of products). The consecutive CVs in Fig. 2b show that after the reaction, the Ni2+/Ni3+ redox peak has shifted to slightly more positive potentials (by ∼50 mV) while the OER shows an earlier onset potential (∼50 mV).
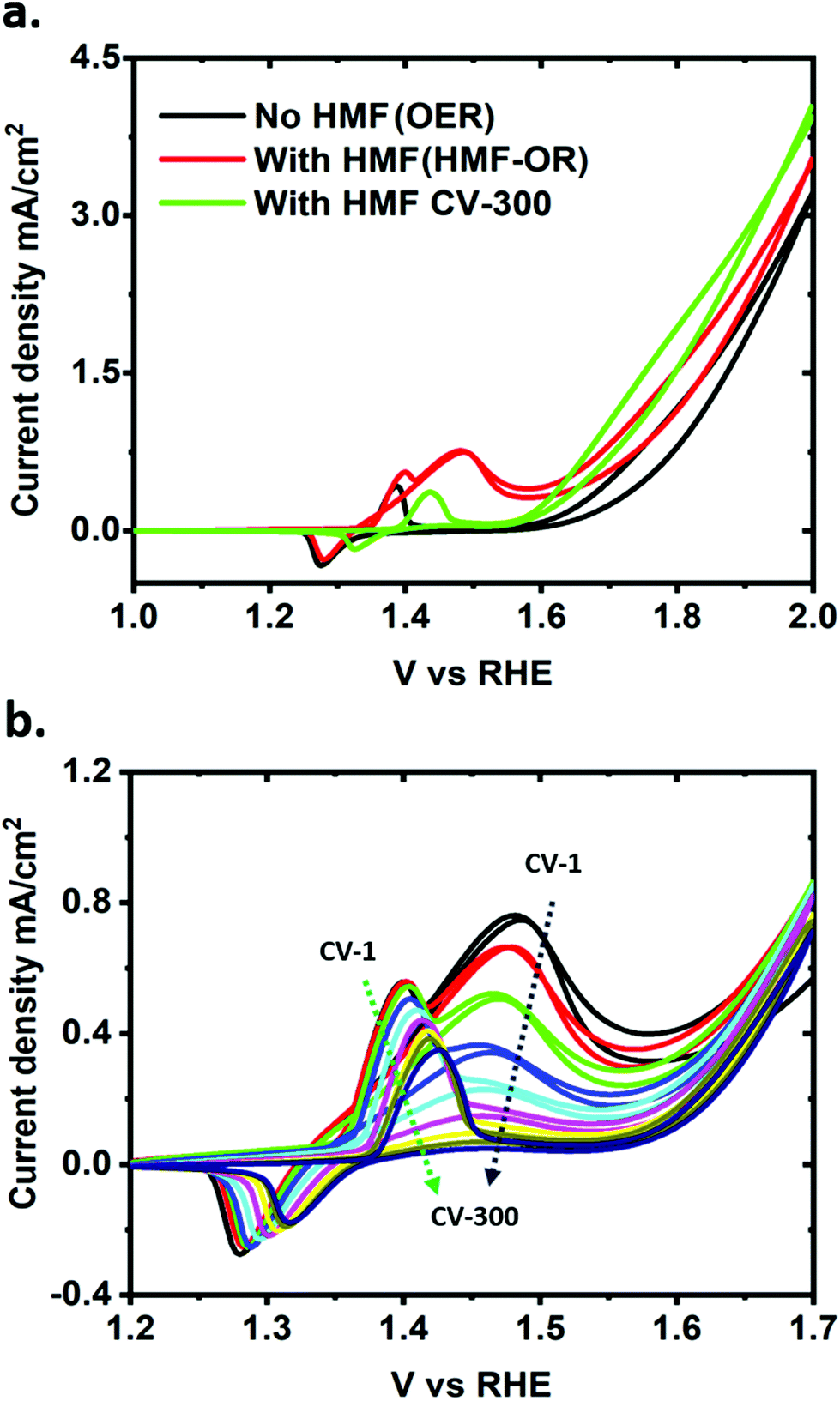 | ||
| Fig. 2 (a) CV of OER in iron-free 1 M LiOH solution, with 5 mM HMF added, and after 300 CV scans. (b) CV scans of HMF oxidation with an interval of 50 cycles. CVs were carried out at a scan rate of 5 mV s−1 while stirring the solution. | ||
Normally, trace impurities such as iron in the electrolyte solution can be adsorbed at the catalyst surface or, as reported, incorporated into the NiO–OH crystal lattice, which can lead to shifts in the OER onset and Ni redox peak.34,38,39 Since we used iron purified electrolyte for our experiments, the only source of iron impurities could be from the HMF. We therefore performed inductively coupled plasma mass spectroscopy (ICP-MS) of the HMF starting material. Indeed, we found that for a 5 mM solution of HMF, traces of iron were present at a concentration of 2.2 nM, which likely gives rise to the shift in the nickel redox peak.
To explore more about the observed peak shifts we additionally performed prolonged OER at 1.8 V vs. RHE with freshly prepared electrodes both with and without iron in the electrolyte solution. The electrolyte solutions “with iron” were prepared by adding intentionally a known amount of iron to iron-free electrolyte solutions (with a ratio of nickel to iron atoms of either ∼200 or ∼100 to 1 in contrast to the ratio of nickel to iron in the iron purified electrolyte solution of at least 4700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (see Experimental section in ESI and Table S1†). The NiO–OH redox peak in iron-free electrolyte solution did not change its redox potential in this prolonged OER experiment while the OER showed only a minor shift to lower onset potential (Fig. S5a†). For the case with the intentionally added iron into the electrolyte solution (Fig. S5b†), we likewise did not observe a clear shift of the redox peak of Ni(OH)2/NiO–OH (2+/3+), although the onset of the OER in this case clearly shifted to a lower onset potential. Generally, the Ni2+/Ni3+ redox peak shift has been found to be very sensitive to iron concentration, microenvironments and deposition methods.40 The shift of the OER onset potential to more negative potentials is a clear indication of iron incorporation into the NiO–OH crystal lattice.40
:1 (see Experimental section in ESI and Table S1†). The NiO–OH redox peak in iron-free electrolyte solution did not change its redox potential in this prolonged OER experiment while the OER showed only a minor shift to lower onset potential (Fig. S5a†). For the case with the intentionally added iron into the electrolyte solution (Fig. S5b†), we likewise did not observe a clear shift of the redox peak of Ni(OH)2/NiO–OH (2+/3+), although the onset of the OER in this case clearly shifted to a lower onset potential. Generally, the Ni2+/Ni3+ redox peak shift has been found to be very sensitive to iron concentration, microenvironments and deposition methods.40 The shift of the OER onset potential to more negative potentials is a clear indication of iron incorporation into the NiO–OH crystal lattice.40
Investigating the CVs further, another interesting observation is found. The oxidative wave for HMF at the NiO–OH surface occurs not only when scanning to more positive potentials but also on the negative sweep, which is unusual. A similar behavior has been observed with glucose oxidation on NiO electrodes.41,42 Though in the literature this is often only explained in terms of binding of an intermediate to the catalyst surface, a more sophisticated treatment of such observations can be found in oscillating electrochemical reactions.43–46 Generally, oscillating (electro)chemical reactions, like the Belousov–Zhabotinsky reaction, are based on an auto-catalytic step in the reaction mechanism itself. Such behavior can be described in more simplified reaction schemes/kinetics like the Bruesselator or the Oregonator.47,48 On the other hand oscillations in electrochemical reactions can also appear if the electrode or the catalyst surface and therefore the active sites for catalysis are blocked by reaction intermediates. Known examples include formic acid, formaldehyde, methanol, ethylene glycol, ethanol, glycerol or glucose and their oxidation on Pt.46 To verify the oscillating behavior of our electrochemical reaction, we performed slow galvanodynamic scans (50 nA s−1) and observed the evolution of the potential (Fig. S6†). Clearly potential oscillations were observed during the current scan, supporting our assumption.
This oscillatory behavior suggests a potential-dependent binding of HMF reaction intermediates that partially block the active sites on the catalytic surface and is consistent with our observation of the less-than-expected amount of substrate and product in the reaction solution under electrical polarization, shown in Fig. 3b (see discussion below). Furthermore, no cathodic reduction peak for the back reaction of FDCA or the intermediates to HMF was observed in the CV, indicating that the oxidation of HMF at the NiO–OH electrodes is an irreversible process.
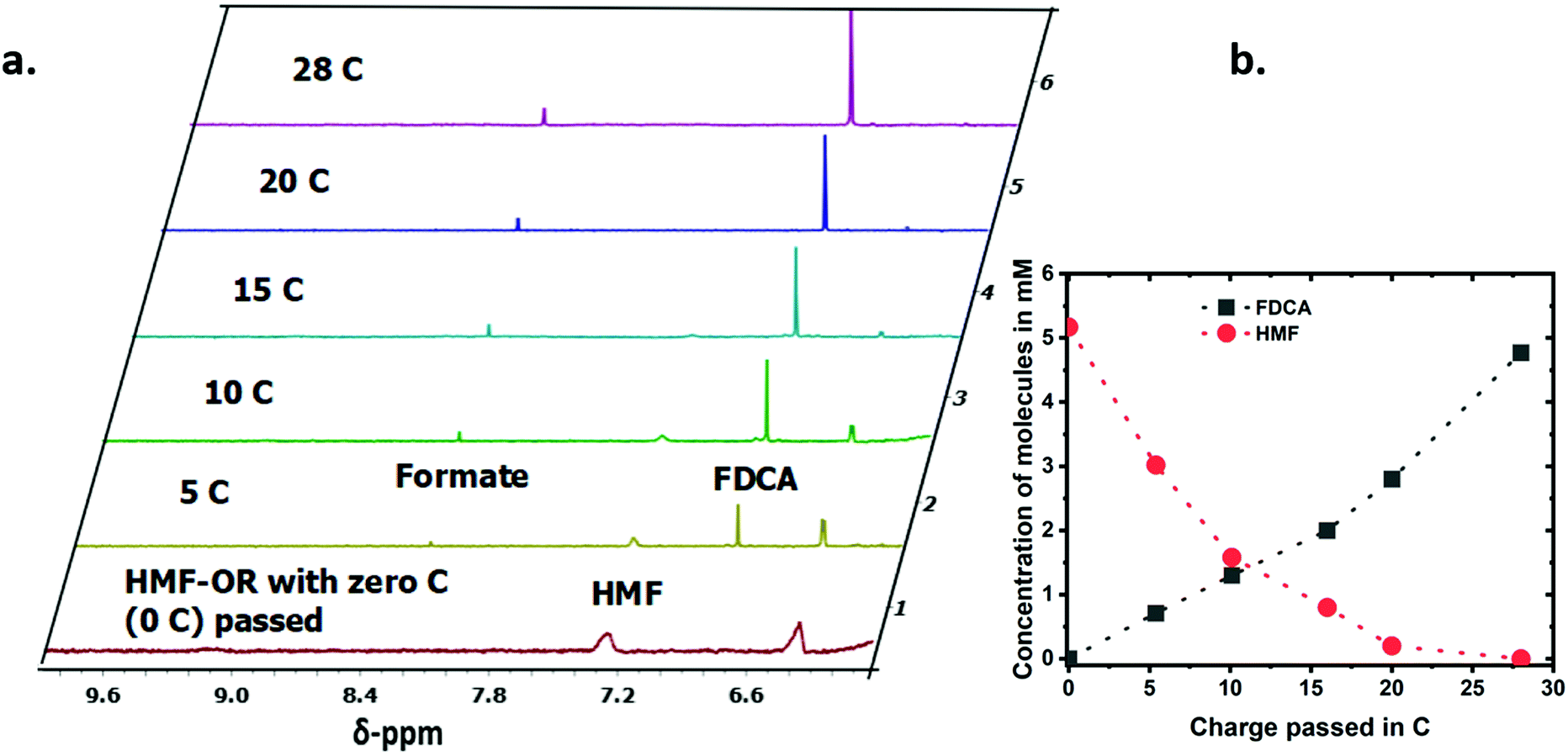 | ||
| Fig. 3 (a) 1H-NMR spectra for the HMF–OR with respect to charge passed (C) during the electrochemical reactions in iron-free 1 M LiOH electrolyte with 5 mM HMF. (b) Concentrations of HMF and FDCA during the electrochemical HMF oxidation reaction. | ||
Product quantification and degradation studies by 1H-NMR and UPLC
To analyze the progress of the reaction for the CV measurements presented in Fig. 2, we used ultrahigh-pressure liquid chromatography (UPLC) after 160 and 300 CV cycles. With an increasing number of potential cycles, the UV/Vis peak in the UPLC analysis related to the absorption of HMF showed the expected decrease while the absorption for the FDCA increased (Fig. S7†). The quantification was made by using a standard calibration curve for each molecule. For a better understanding and quantification of the conversion reaction we have performed chronoamperometric measurements at 1.5 V vs. RHE and analyzed the reaction solution every ∼5 C passed by UPLC and 1H-NMR. A total of 28 C were passed during the experiment, which typically lasted about 20 hours (∼0.8 C per hour). Fig. 3a shows the 1H-NMR of the reaction mixture. The 1H-NMR peaks were assigned based on measurements of the pure molecules in the same electrolyte solutions (Fig. S8†). The two peaks associated with the furan protons of HMF at 6.4 ppm and 7.3 ppm showed a steady decrease with the amount of charged passed in the chronoamperometric measurements. On the other hand, the peak of the furan protons of the FDCA at 6.8 ppm increased with longer reaction time. During electrooxidation of HMF under these experimental conditions, two competing reactions can occur simultaneously for the HMF, the electrooxidation and its (undesired) degradation.16 The degradation reaction of the organic substrate could be detected even without any applied potential, which is exemplified in Fig. S9† with an in situ degradation experiment of HMF in an NMR tube with 1 M LiOD in D2O. Clearly, formate is formed as a degradation product. Though at the moment more experiments have to be performed to fully understand the formation of formate under these conditions, the amount of formate found in our chronoamperometric experiments suggests that out of one HMF molecule six formate molecules can be formed. At the end of the reaction, the concentration of the FDCA produced during the electrochemical reaction was roughly equivalent to the concentration of HMF consumed, as presented in Fig. 3b and Fig. S10,† which implies that the rate for electrooxidation of HMF is high enough so that the degradation or other side reactions play only a minor role.The products formed during chronoamperometric studies were also quantified by UPLC and showed a similar trend as in the 1H-NMR spectra (Fig. S11†). Upon close inspection, one can observe both for the 1H-NMR and the UPLC measurements that initially the formation of FDCA is not 1:1 with respect to the amount of HMF converted. At the intermediate data points, obtained by sampling the solution during the electrolysis, the total concentration of HMF and of the products detected does not match the initial starting concentration. The last data point – obtained after stopping the electrolysis, releasing the potential, and equilibrating the electrolyte solution under continuous stirring – shows that all HMF has been transformed to either FDCA or formate. The amount of FDCA determined in this last datapoint of the UPLC suggests that partially oxidized HMF species and/or FDCA are trapped and accumulate at the electrode surface under applied potential as no peaks for intermediate species could be observed in the aliquots measured by 1H-NMR. This observation is also consistent with the unusual CV behavior shown in Fig. 2 and the oscillatory behavior shown in Fig. S6.†
Tuning the oxidation reaction from OER towards HMF oxidation
The deprotonated negatively charged NiO–O− surface species (superoxo species) is reported to be the catalytic active site for the OER and HMF oxidation reactions.24,30 Koper et al. showed that the correlation between alkali metal cation dependent stability of the NiO–O−–M+ species at the catalyst surface and the catalytic activity for OER followed the trend Cs+ > Na+ > K+ > Li+.33 The authors argue, based on Raman measurements, that the origin of the improvement is due to a better stabilization of the NiO–O–M intermediate by larger cations.32 Another literature report provided evidence that the same intermediate is involved in organic alcohol oxidation (methanol oxidation versus OER on NiO).49 Nevertheless, differential effects of the alkali ion on HMF oxidation versus OER could be used to further separate the onset potential of the oxidation reactions and thereby improve the selectivity for the HMF oxidation reaction over OER while maintaining a high reaction rate.As discussed earlier, traces of iron in electrolyte can interact with the NiO–OH catalyst and be incorporated into the catalyst structure, which promotes a synergetic effect on the OER.38 Based on the literature, the role of iron is still under debate. However, it is very well understood that iron incorporation reduces the covalent character of the Ni–O bonds, which energetically favors the adsorption of oxygen intermediates, and thus enhances molecular oxygen evolution.35,40 Therefore, iron traces at the NiO–OH matrix synergistically lowers the OER overpotential. In this scenario, the competing OER process reduces the HMF conversion on the NiO–OH surface. Based on these observations we can infer that iron-free LiOH should be the best choice for electrochemical HMF oxidation with a minimal contribution from the competing OER. Thus, we systematically investigated the chemical selectivity between electrooxidation of HMF and OER activity with varying alkali cations with and without traces of iron in the electrolyte solution. For clarity, we have plotted the electrochemical data using only the smallest and largest cation size with and without traces of iron in the electrolyte solution (Fig. 4). The comparison with all four alkali cations can be found in the ESI (Fig. S12 and S13†). We observe a shift in the OER onset with different alkali cation similar to a previous report (Fig. 4a and b).32 Thus, the presence of Li+ in the electrolyte solution leads to a higher OER overpotential as compared to Cs+. The shift in the OER onset follows the size of the cations used for the electrolyte solutions as can be observed in Fig. 4 and S12.† This is exemplified in the negative shift of the OER onset potential in the presence of iron and Cs+, appearing very close to the Ni2+/Ni3+ redox peak as shown in Fig. 4b and d. Fig. 4d shows that OER with Cs+ and HMF in solution is the dominant reaction under such conditions as compared to HMF oxidation. The trend of the alkali cation size favoring OER is clearly more pronounced with trace iron present and without HMF in solution (Fig. 4b and c and S13b and d†).
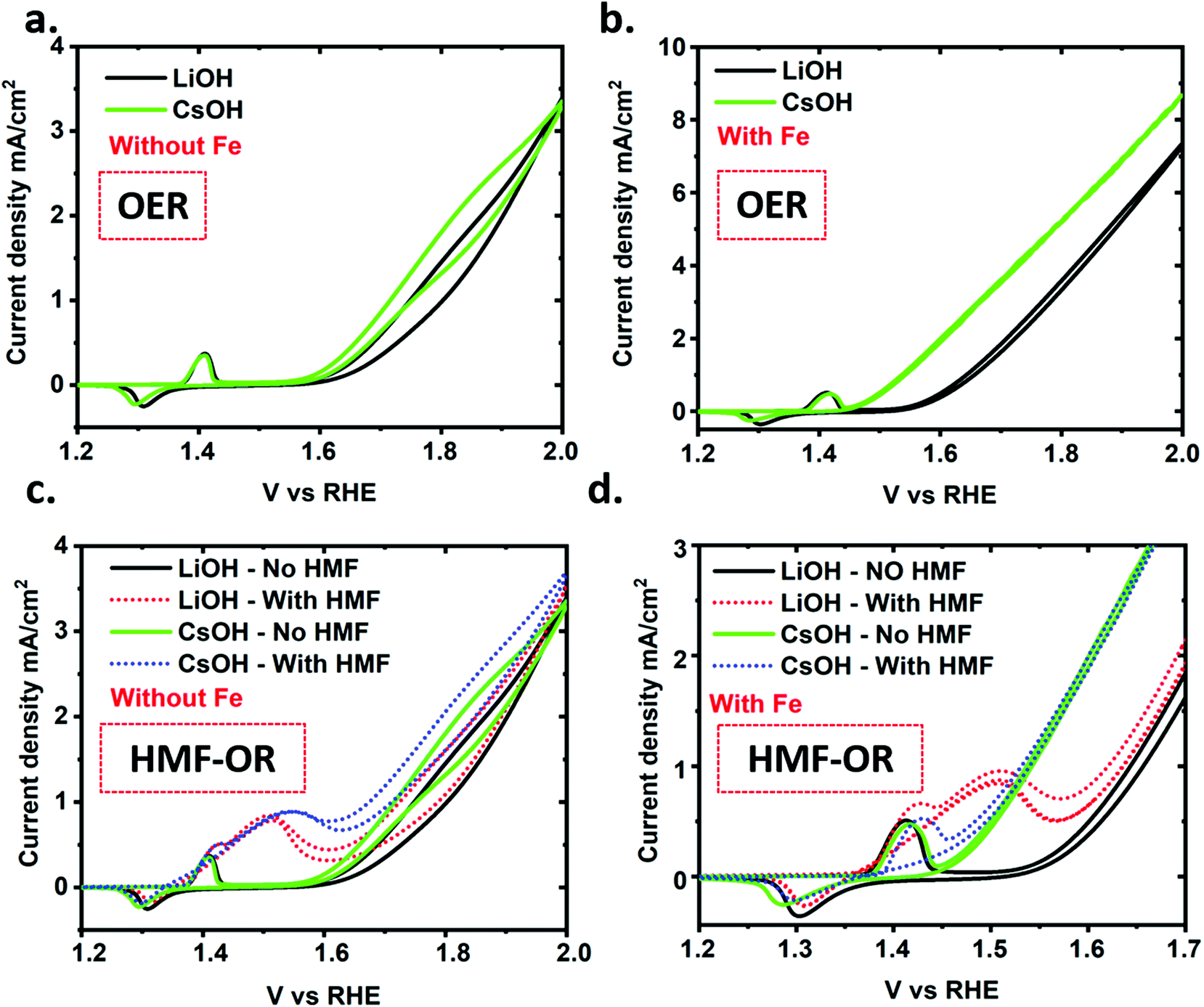 | ||
| Fig. 4 (a) and (b) CVs (under stirring) of OER reaction on NiO–OH electrodes in 1 M LiOH and CsOH electrolyte solution (without and with iron in solution, respectively). (c) and (d) are measured under similar conditions but with 5 mM HMF also in solution. | ||
Interestingly, the hysteresis of the CV curve after OER onset (in the region from 1.6 V to 2 V) is clearly visible in the samples without iron. In the samples with iron, as the faradaic current increases, this effect is reduced until it is almost negligible. This fact could be related to some structural transformation of the catalyst, or the activation of the Ni3+/Ni4+ redox state.38 In any case, the potential interest of this process in the OER is beyond the scope of this study.
When adding HMF to the electrolyte solutions, similar CV features are observed as discussed above. The electrochemical potential at which HMF oxidation current appears following oxidation of the Ni2+/Ni3+ redox peak and remains at nearly the same potential for all the cations used. Meanwhile, the onset potential of the OER has an extra slight negative shift for all cation sizes. In the case of the electrolyte with Fe and of Cs+, the peak of the HMF oxidation is hardly visible, suggesting that in this case OER dominates the reaction. While addressing the overall scheme, it is remarkable that, (except for Li cation) the larger the cation, the narrower potential difference between Ni redox peak/HMF oxidation potential and OER onset potential is observed. Thus, Ni redox peak and HMF oxidation potential shifts to higher potential while the OER onset shifts to lower potential. This fact suggests that on the Ni surface the HMF oxidation and OER occur by two independent mechanisms. Further investigation is needed to fully understand the origin of these mechanisms.
To analyze the conversion performance of these different electrolyte solutions, chronoamperometric oxidation experiments were carried out for 20 hours with the different electrolyte solution compositions at 1.5 V vs. RHE, where HMF has highest oxidation current and OER current is small. The conversion efficiencies are presented in Table 1 and Table S2.† As the competing OER decreases with the smaller cation size, the faradaic efficiency of HMF to FDCA conversion increases, and we observe the highest faradaic efficiency (and chemical yield) with LiOH and the lowest with CsOH (Table S3†).
| 1 M electrolyte solution with | Faradaic efficiency without iron (UPLC) % | Faradaic Efficiency with iron (UPLC) % |
|---|---|---|
| LiOH | 98 | 90 |
| NaOH | 95 | 88 |
| KOH | 93 | 85 |
| CsOH | 93 | <6 |
The data in Table 1 show that with traces of iron in the electrolyte solution, the OER can more easily compete with HMF oxidation and in the case of electrolyte solution containing cesium cation, the OER is in fact the dominant reaction at given applied potential (with only 6% FDCA formation). Thus, we observed a reduction in the faradaic efficiency for FDCA formation for all metal cations in the presence of iron impurities. Thus, while for LiOH the reaction still showed a high faradaic efficiency, for CsOH, the transformation to FDCA could not be completed and a variety of intermediate products such as HFCA, FFCA, and side products like formate and dimethylfuran were detected (Fig. S14 and Table S3†). Finally, our findings show that the faradaic efficiency (and chemical yield) change due to the variation of the alkali cation in the absence of trace iron in the electrolyte is small. However, the presence of trace iron activates the electrode for the competing OER the larger the alkali cation size is, lowering the yields and removing the possibility of selective HMF oxidation at any potential in the case of CsOH.
3. Conclusions
We investigated the chemical selectivity of NiO–OH towards HMF oxidation against the competing OER by systematically studying the influence of different alkali cations and traces of iron in the alkaline electrolyte solution. Small cation sizes favor HMF oxidation and thereby enable potentials to be used with minimal OER. The removal of traces of iron from the electrolyte solution further increased the conversion efficiency of HMF towards FDCA. We determined a faradaic efficiency of 98% for FDCA formation with electrolyte solutions of iron-free LiOH, whereas the lowest conversion efficiency of <6% was found for CsOH with traces of iron. Under the optimized conditions with iron-free LiOH, no intermediates of the HMF oxidation were observed. The presented investigation shows how the electrolyte solution and removal of impurities can lead to desired high conversion efficiencies at potentials with practical currents. Our results pave the way for future investigations in the field of organo-electrocatalysis in aqueous media in which the formation of value-added chemicals competes with the oxygen evolution reaction.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The University of Zurich and the University Research Priority Program (URPP) LightChEC are gratefully acknowledged for funding. The UZH-Center for Microscopy and Image Analysis and Jonas Zurflüh for HPLC analysis are acknowledged for their service. The Institute of Advanced Materials from Universitat Jaume I acknowledges Generalitat Valencia under project- GRISOLIAP/2018/A/070 and Ministerio de Economía y Competitividad (MINECO) under project ENE2017-85087-C3-1-R for financial support.References
- S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef PubMed.
- S. Ardo, D. Fernandez Rivas, M. A. Modestino, V. Schulze Greiving, F. F. Abdi, E. Alarcon Llado, V. Artero, K. Ayers, C. Battaglia, J. P. Becker, D. Bederak, A. Berger, F. Buda, E. Chinello, B. Dam, V. Di Palma, T. Edvinsson, K. Fujii, H. Gardeniers, H. Geerlings, S. M. Hashemi, S. Haussener, F. Houle, J. Huskens, B. D. James, K. Konrad, A. Kudo, P. P. Kunturu, D. Lohse, B. Mei, E. L. Miller, G. F. Moore, J. Muller, K. L. Orchard, T. E. Rosser, F. H. Saadi, J. W. Schüttauf, B. Seger, S. W. Sheehan, W. A. Smith, J. Spurgeon, M. H. Tang, R. Van De Krol, P. C. K. Vesborg and P. Westerik, Energy Environ. Sci., 2018, 11, 2768–2783 RSC.
- C. R. Lhermitte and K. Sivula, ACS Catal., 2019, 9, 2007–2017 CrossRef CAS.
- C. C. Elam, C. E. G. Padró, G. Sandrock, A. Luzzi, P. Lindblad and E. F. Hagen, Int. J. Hydrogen Energy, 2003, 28, 601–607 CrossRef CAS.
- C. Dolan, Road Map to a US Hydrogen Economy, Accesed 30/June/2020, http://www.fchea.org/us-hydrogen-study.
- N. Lazouski, M. Chung, K. Williams, M. L. Gala and K. Manthiram, Nat. Catal., 2020, 3, 463–469 CrossRef CAS.
- A. Nicita, G. Maggio, A. P. F. Andaloro and G. Squadrito, Int. J. Hydrogen Energy, 2020, 45, 11395–11408 CrossRef CAS.
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331–344 RSC.
- K. Sayama, ACS Energy Lett., 2018, 3, 1093–1101 CrossRef CAS.
- B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS PubMed.
- T. Werpy and G. Petersen, Off. Sci. Tech. Inf, 2004, p. 69 Search PubMed.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G. J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961–5983 RSC.
- S. K. Burgess, J. E. Leisen, B. E. Kraftschik, C. R. Mubarak, R. M. Kriegel and W. J. Koros, Macromolecules, 2014, 47, 1383–1391 CrossRef CAS.
- Z. Jia, J. Wang, L. Sun, F. Liu, J. Zhu and X. Liu, J. Appl. Polym. Sci., 2019, 136, 1–9 Search PubMed.
- United States Environ. Prot. Agency. Accessed 2020. https//www.epa.gov/facts-and-figures-about-materials-waste-and-recycling/plastics-material-specific-data.
- M. Sajid, X. Zhao and D. Liu, Green Chem., 2018, 20, 5427–5453 RSC.
- M. H. Jensen and A. Riisager, Advances in the synthesis and application of 2,5-furandicarboxylic acid, Elsevier B.V., 2020 Search PubMed.
- Y. Kwon, K. J. P. Schouten, J. C. Van Der Waal, E. De Jong and M. T. M. Koper, ACS Catal., 2016, 6, 6704–6717 CrossRef CAS.
- Z. Zhang and K. Deng, ACS Catal., 2015, 5, 6529–6544 CrossRef CAS.
- H. G. Cha and K. S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS PubMed.
- G. Grabowski, J. Lewkowski and R. Skowroński, Electrochim. Acta, 1991, 36, 1995 CrossRef CAS.
- S. Choi, M. Balamurugan, K.-G. Lee, K. H. Cho, S. Park, H. Seo and K. T. Nam, J. Phys. Chem. Lett., 2020, 2941–2948 CrossRef CAS PubMed.
- A. N. Vyas, G. D. Saratale and S. D. Sartale, Int. J. Hydrogen Energy, 2020, 45, 5928–5947 CrossRef CAS.
- B. J. Taitt, D. H. Nam and K. S. Choi, ACS Catal., 2019, 9, 660–670 CrossRef CAS.
- J. Huang, Y. Li, Y. Zhang, G. Rao, C. Wu, Y. Hu, X. Wang, R. Lu, Y. Li and J. Xiong, Angew. Chem., Int. Ed., 2019, 58, 17458–17464 CrossRef CAS PubMed.
- B. You, X. Liu, X. Liu and Y. Sun, ACS Catal., 2017, 7, 4564–4570 CrossRef CAS.
- B. You, N. Jiang, X. Liu and Y. Sun, Angew. Chem., Int. Ed., 2016, 55, 9913–9917 CrossRef CAS PubMed.
- W. J. Liu, L. Dang, Z. Xu, H. Q. Yu, S. Jin and G. W. Huber, ACS Catal., 2018, 8, 5533–5541 CrossRef CAS.
- B. Kim, A. Oh, M. K. Kabiraz, Y. Hong, J. Joo, H. Baik, S. Il Choi and K. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 10115–10122 CrossRef CAS PubMed.
- O. Diaz-Morales, D. Ferrus-Suspedra and M. T. M. Koper, Chem. Sci., 2016, 7, 2639–2645 RSC.
- S. D. Tilley, Adv. Energy Mater., 2019, 9, 1–13 Search PubMed.
- D. J. Chadderdon, L. Xin, J. Qi, Y. Qiu, P. Krishna, K. L. Moreb and W. Li, Green Chem., 2014, 16, 3778 RSC.
- A. C. Garcia, T. Touzalin, C. Nieuwland, N. Perini and M. T. M. Koper, Angew. Chem., Int. Ed., 2019, 58, 12999–13003 CrossRef CAS PubMed.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CrossRef CAS.
- J. Zaffran, M. B. Stevens, C. D. M. Trang, M. Nagli, M. Shehadeh, S. W. Boettcher and M. Caspary Toroker, Chem. Mater., 2017, 29, 4761–4767 CrossRef CAS.
- J. D. Michael, E. L. Demeter, S. M. Illes, Q. Fan, J. R. Boes and J. R. Kitchin, J. Phys. Chem. C, 2015, 119(21), 11475–11481 CrossRef CAS.
- M. Görlin, J. H. Stenlid, S. Koroidov, H. Y. Wang, M. Börner, M. Shipilin, A. Kalinko, V. Mrzin, O. V. Safonova, M. Nachtegaal, A. Uheida, J. Dutta, M. Bauer, A. Nilsson and O. D. Morales, Nat. Commun., 2020, 11, 6181 CrossRef PubMed.
- M. Görlin, P. Chernev, J. F. De Araújo, T. Reier, S. Dresp, B. Paul, R. Krähnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2016, 138, 5603–5614 CrossRef PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- S. Anantharaj, S. Kundu and S. Noda, Nano Energy, 2021, 80, 105514 CrossRef CAS.
- G. A. El-Nagar and C. Roth, Phys. Chem. Chem. Phys., 2017, 19, 2537–2548 RSC.
- A. M. Ghonim, B. E. El-Anadouli and M. M. Saleh, Electrochim. Acta, 2013, 114, 713–719 CrossRef CAS.
- M. Orlik, J. Solid State Electrochem., 2015, 19, 3203–3206 CrossRef CAS.
- R. Nagao, E. Sitta and H. Varela, J. Phys. Chem. C, 2010, 114, 22262–22268 CrossRef CAS.
- G. C. A. Ferreira, T. W. Napporn, K. B. Kokoh and H. Varela, J. Electrochem. Soc., 2017, 164, H603–H607 CrossRef CAS.
- G. B. Melle, T. Altair, R. L. Romano and H. Varela, Energy Fuels, 2021, 35(7), 6202–6209 CrossRef CAS.
- H. Degn, J. Chem. Educ., 1972, 49, 302–307 CrossRef CAS.
- M. A. Pellitero, C. Á. Lamsfus and J. Borge, J. Chem. Educ., 2013, 90, 82–89 CrossRef.
- H. B. Tao, Y. Xu, X. Huang, J. Chen, L. Pei, J. Zhang, J. G. Chen and B. Liu, Joule, 2019, 3, 1498–1509 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, HMF oxidation and OER reaction equations, AFM images of the electrodes, EDX measurements, CV of redox peaks of the nickel electrode, OER with and without iron, UPLC study of HMF oxidation, 1H-NMR spectra of HMF and its intermediates, 1H-NMR study of HMF degradation, 1H-NMR spectra of HMF oxidation for LiOH without iron and CsOH with iron, reaction mixture analysis by UPLC and 1H-NMR. See DOI: 10.1039/d1gc02031e |
| This journal is © The Royal Society of Chemistry 2021 |