
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selectivity switch by phase switch – the key to a high-yield furfural process†
Luca
Ricciardi
a,
Willem
Verboom
 *a,
Jean-Paul
Lange
*bc and
Jurriaan
Huskens
*a
*a,
Jean-Paul
Lange
*bc and
Jurriaan
Huskens
*a
aMolecular NanoFabrication group, Department of Molecules & Materials, MESA+ Institute for Nanotechnology, University of Twente, P.O. Box 217, 7500 AE, Enschede, The Netherlands. E-mail: w.verboom@utwente.nl; j.huskens@utwente.nl; Tel: +31-53-489-2977 Tel: +31-53-489-2995
bSustainable Process Technology group, University of Twente, P.O. Box 217, 7500 AE, Enschede, The Netherlands. E-mail: j.p.lange@utwente.nl; Tel: +31-20-630-3428
cShell Global Solutions International B.V. Grasweg 31, 1031 HW Amsterdam, The Netherlands
First published on 6th September 2021
Abstract
We show the effective conversion of xylose into furfural, with a selectivity >90 mol%, in an aqueous–organic three-solvent system, which is composed of an apolar aromatic solvent, a polar organic solvent and acidic water and helps in catalyzing the formation of furfural. We couple this with a pre-extraction step of xylose as a boronic diester into a promising integrated process for valorising a diluted xylose hydrolysate. This process promises facile recovery of furfural and good recycling of all solvent components. The use of the boronic diester was found to be irrelevant for obtaining high selectivity, as its hydrolysis under the reaction conditions is fast. Surprisingly, the >90 mol% selectivity requires the three-solvent system to transition from biphasic to monophasic under the reaction conditions. Phenylboronic acid (PBA) used to extract xylose was found to be instrumental to this transition; PBA-lean media (<20 mM PBA) remain biphasic under the reaction conditions and deliver only 70 mol% selectivity. Water partial pressure measurements across the phase transition temperature confirm the occurrence of the phase transition. The increase of the apolar nature of the reaction medium when transitioning from biphasic to monophasic operation, reached upon mixing of the aromatic solvent with the water–polar organic phase, is likely responsible for the improved selectivity. The presence of an aromatic solvent in the mixture is important, probably due to its interaction with PBA that is instrumental to achieving the phase transition. A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 toluene–sulfolane–water (pH = 1) mixture and [PBA] > 20 mM resulted in the highest observed xylose-to-furfural selectivity (95 mol%).
:1:1 toluene–sulfolane–water (pH = 1) mixture and [PBA] > 20 mM resulted in the highest observed xylose-to-furfural selectivity (95 mol%).
Introduction
Furfural is among the top value-added chemicals that could serve as renewable carbon sources for energy and chemical production.1,2 Furfural is currently produced by acid-catalysed dehydration of xylose present in lignocellulosic biomass.1–4 The xylose-to-furfural selectivity is, however, severely limited by side-reactions, in particular the formation of solid humic by-products, observed consistently under several reaction conditions.1–6To address these problems and to enhance the production of furfural, various studies have introduced the use of an organic solvent in the process.6–14 A common strategy to improve the efficiency of xylose dehydration is to perform the reaction in a biphasic system,6–12 with the aim to extract furfural as soon as it is formed and thereby to protect it from degradation.11,12 Accordingly, the xylose-to-furfural selectivity is increased to 65–70 mol%, which is a significant improvement over the 50 mol% observed when xylose is dehydrated in water.12
Alternatively, the reaction can be performed under monophasic conditions, using polar aprotic solvents (e.g., DMSO and sulfolane), optionally with limited amounts of water, delivering furfural with a selectivity of up to 80–85 mol%.16–21 An explanation for this improved selectivity is that aprotic organic solvents suppress the formation of acyclic sugar, which is more prone to degradation, while the limited amount of water directly interacts with the sugar influencing the mechanism of dehydration.16,17 However, this approach requires xylose to be recovered from the dilute aqueous feed, which severely increases the cost of the sugar.1,2 Additionally, these solvents cannot be considered green, due to their detrimental effect on human health and hazard to the environment, and their separation from water streams requires a high energy input.22
In the context of fructose dehydration to hydroxymethylfurfural (HMF) under biphasic conditions, Román-Leshkov and Dumesic introduced the use of a polar organic solvent (e.g. 1-butanol) that gave a monophasic system under the reaction conditions.15 However, the mutual solubility of water and the organic solvent at higher temperatures resulted in the formation of more degradation products, possibly due to rehydration.15
Here, we study the use of a three-solvent mixture, using water (with an acid to induce the reaction), an apolar, aromatic organic solvent, and a polar aprotic solvent, in the conversion of xylose to furfural, with the aim to integrate this step into a process with a pre-extraction step of xylose as a boronic diester (Fig. 1). In a previous study,23 we have reported the extraction of xylose from an acidic aqueous feed into an organic aromatic solvent as a boronic diester (PBA2X) and its conversion under biphasic conditions. In the present study, upon adding a third, polar aprotic, solvent, we observed a temperature-dependent switching from biphasic to monophasic in the presence of phenylboronic acid (PBA), and we found a simultaneous strong effect on the xylose-to-furfural selectivity. We explore the conditions under which this phase transition occurs and try to rationalize its effect on the selectivity. Based on these findings, we provide a concept of an integrated process that benefits from both the diester extraction and the highly selective monophasic conversion and allows the separation of the solvents from the aqueous waste stream and the recycling of both solvents and additives.
 | ||
| Fig. 1 Conceptual representation of the solvent composition of the system and its phase behaviour, during the preparation of a boronic ester feed (of xylose and PBA) in an aromatic solvent, PBA-induced transition to a monophase at the reaction temperature, and separation of the produced furfural during work-up. | ||
Results and discussion
Selectivity switch in the dehydration of xylose to furfural in a three-solvent system
When aiming for a process to convert xylose from biomass into furfural, the extraction of xylose from an acidic aqueous hydrolysate will deliver a toluene phase containing PBA2X.22 This extraction allows one to naturally consider a biphasic system to convert xylose upon back-extraction into acidic water, but the xylose-to-furfural selectivity under biphasic conditions is typically only about 70%.11 We therefore considered adapting the solvent to convert the back-extracted xylose. We started with a solution of PBA2X in toluene in the present study. However, it has to be noted that choosing the diester as the starting material is not an essential element to provide the selectivity observed in this process. As we will show, (i) the hydrolysis of the diester in a solvent mixture that contains water is fast compared to the conversion of released xylose into furfural and (ii) it is free PBA (not its ester) that provides control over the temperature-induced biphasic to monophasic transition that in turn provides the high selectivity. Many experiments were therefore conducted by simply adding xylose to the water phase and PBA to toluene before mixing the solvents, to allow, for example, variation of the xylose–PBA ratio.Performing the hydrolysis and conversion of PBA2X in a 1:1:1 toluene–sulfolane–water (water at pH = 1, 97 mM of PBA2X in the reaction mixture) system led to an unexpectedly high furfural selectivity of approx. 95 mol% at full xylose conversion, after approx. 3.5 h (Fig. 2). At room temperature, the mixture was biphasic, with toluene forming the organic phase using the sulfolane–water mixture as the polar aqueous phase. However, at the reaction temperature (180 °C), formation of a single phase was observed. In contrast, operation in 1:1 toluene–water (pH 1), which is biphasic at the reaction temperature, resulted in only 70 mol% selectivity.11
 | ||
| Fig. 2 Relative concentrations (in mol% of the starting total xylose concentration of 97 mM) of furfural, xylose and unconverted PBA2X, and the selectivity toward furfural vs. time. Reaction performed in a 1:1:1 toluene–sulfolane–water (pH = 1) system at 180 °C. Values calculated from NMR analysis of aqueous and toluene phases at r.t. (lines are to guide the eye). | ||
To understand the origin of the high furfural selectivity in the former case, the progress of the reaction was followed in time (Fig. 2), showing a fast (<30 min) release of the sugar from its boronate ester and partitioning into the aqueous phase (upon analysis at room temperature at which the mixtures are always biphasic), followed by a slower conversion of the released xylose into furfural (Fig. S1†).
Remarkably, the xylose-to-furfural selectivity remained >90 mol% over the entire course of the reaction until full conversion. The fast hydrolysis of the boronate ester form of the xylose, PBA2X, into xylose in water, which is practically complete within the first 30 min, indicates that the use of PBA2X as the starting material is not essential to achieve the observed selectivity. Indeed, performing the reaction in the same solvent mixture, but starting from free xylose in water (pH = 1) and PBA in toluene (with a xylose concentration in the system of 97 mM and a PBA concentration of 194 mM), gave the exact same furfural selectivity.
In contrast, performing the reaction starting from free xylose in water (pH = 1), using the same solvent system and experimental conditions, but in the absence of PBA, gave only a 70 mol% yield of furfural, comparable to the selectivity obtained under commonly applied water–toluene biphasic conditions.11 This suggests that PBA, originating from the hydrolysis of PBA2X, is an essential component to obtain the high selectivity, even though ester formation is not.
To investigate the effect of the reaction medium and the role of PBA in it, the xylose dehydration reaction was performed at a constant xylose concentration and using the same experimental conditions and solvent system. PBA was added to the toluene phase reaching concentrations in the reaction mixture that varied from 0 to 233 mM (Fig. 3). While the final furfural selectivity was always close to 70 mol% at [PBA] < 20 mM in the reaction mixture, it increased markedly and in a stepwise manner to >90 mol% at PBA concentrations in the reaction mixture above 20 mM.
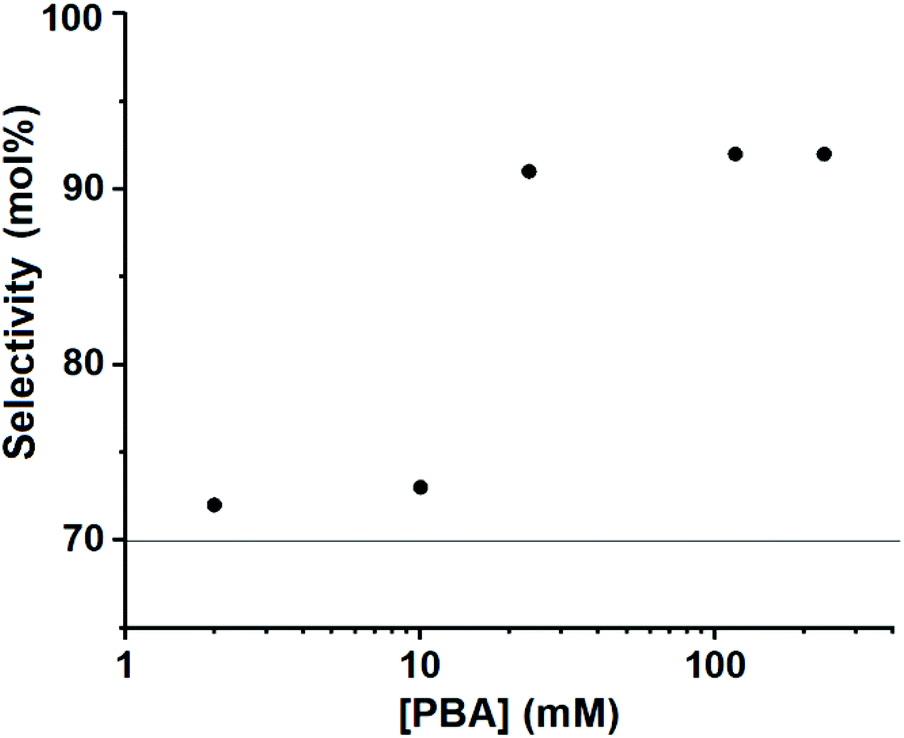 | ||
| Fig. 3 Xylose-to-furfural selectivity (mol%) vs. PBA concentration (mM) in the reaction mixture; reaction performed at 180 °C for 3–4 h (full conversion achieved in all cases) in a 1:1:1 toluene–sulfolane–water (pH = 1) solvent system with free xylose (97 mM in the reaction mixture) and free PBA at varying concentrations. The horizontal line at approx. 70 mol% represents the selectivity obtained at a PBA concentration of 0 mM. | ||
Phase change of the three-solvent mixture as a function of PBA concentration and temperature
The sharp selectivity transition observed above triggered us to investigate the phase behaviour of the reaction mixture as a function of PBA concentration and reaction temperature. Visual observation of the number of phases of the reaction mixtures was performed, and the xylose-to-furfural selectivity was assessed (Fig. 4). Datapoints were found distributed over those with biphasic and monophasic behaviour, and a dividing line became apparent. Complementary experiments in glass capillaries containing the solvent–PBA mixtures (without xylose) indeed showed that the 1:1:1 toluene–sulfolane–water (pH = 1) system transitions from biphasic to monophasic, depending on the temperature and the PBA concentration (Fig. S2, Videos S1 and S2†). Furthermore, these experiments showed no partial mixing prior to the phase transition from biphasic to monophasic. The transition itself is very fast and occurs in a 2–3 s time interval. After the transition, the single phase is clear and does not cause diffraction of light, which is a good indication that the single phase is not an emulsion. Upon cooling, the monophasic system turns back to biphasic at the same transition temperature.
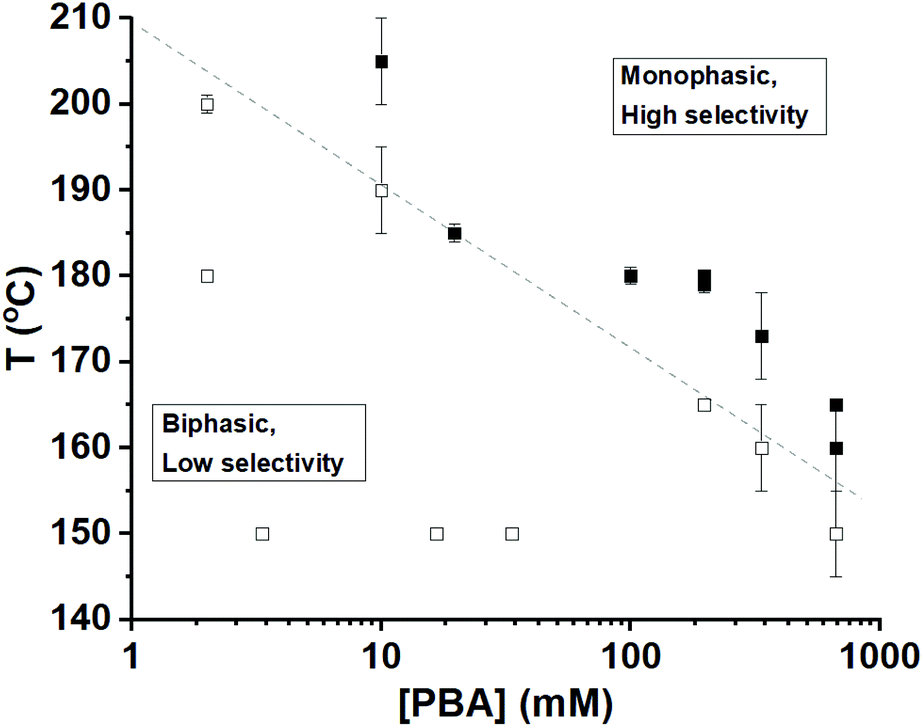 | ||
| Fig. 4 Determination (by visual inspection) of the phase behaviour of the 1:1:1 toluene–sulfolane–water (pH = 1) three-solvent system as a function of the PBA concentration in the reaction mixture and the reaction temperature, in the absence of xylose. The markers represent experimental points at which the number of phases was determined, with the solid and open symbols indicating monophasic and biphasic conditions at the observation temperature, respectively. For the data points with a small error bar for the temperature, the determination of the transition was achieved applying a gradual temperature variation to the mixture in glass capillaries (as described in the ESI†). In all other cases, the transition temperature was determined at a larger scale using pressure-resistant vials, with no gradual temperature variation, hence the larger error bars. The dashed line serves as a guide to the eye, indicating the transition in phase behaviour. | ||
Apparently, the higher the PBA concentration, the lower the temperature needed to transition from biphasic to monophasic. All points in Fig. 4 that correspond to monophasic operation (above the dashed line) gave xylose-to-furfural selectivities ranging from 89% to 96%, while the reactions under biphasic operation (below the line) gave selectivities between 65% and 73% (Table S1†). In neither regime, we observed a dependence of the selectivity on the PBA concentration. Thus, we conclude that the step in the selectivity graph observed in Fig. 3 coincides with the conversion from a two-phase to a one-phase reaction mixture, which can be viewed in Fig. 4 as going horizontally from left to right at a reaction temperature of 180 °C.
We found indications that an increase in the temperature promotes the uptake of PBA in the aqueous–organic phase just before reaching the phase switching temperature. The relative concentrations of boronic acid in the two phases, prior to the phase transition, appear to be strongly temperature-dependent. The partitioning as a function of temperature was investigated using a coloured analogue of PBA, 10-bromoanthracene-9-boronic acid (BBA), which allowed us to visualize the relative concentrations from the colour intensity (Fig. S3†). This analysis shows that there is a drastic increase in the boronic acid concentration in the aqueous phase at T > 150 °C. For PBA, the uptake in the water–organic solvent is probably already enhanced at a lower temperature because PBA is less hydrophobic than the coloured derivative, which is supported by the fact that some PBA can already be observed in the water phase at room temperature, whereas the coloured one cannot. PBA in itself can be seen as an amphiphile, having an aromatic moiety that interacts well with the aromatic organic solvent and a polar and ionizable headgroup that interacts well with water and polar solvents. The promoted dissolution of boronic acid in the aqueous phase at elevated temperatures is in part due to the increased importance of entropy in the partitioning but may also indicate an improved interaction with the water–organic mixture. This improved interaction in turn might promote the observed biphasic-to-monophasic transition. From the observed independence of the selectivity on the PBA concentration under monophasic operation mentioned before, we conclude that PBA itself is present in too low amounts to affect the overall solvent polarity, so that its role is primarily in effectuating the phase transition.
With the aim to get insight into the interactions between the three different solvents in the system and in particular about the solubilization of the water in it, we looked for eventual anomalies in the water partial pressure as the system transitions from a biphase to a monophase. These initial measurements cannot fully explain the impact of the phase switch on the conversion selectivity and their molecular origins. So more detailed studies are encouraged, and these should preferably be carried out under operating conditions. To this end, we analysed the water partial pressure at a 1:1:1 mixture of 1-methylnaphthalene, sulfolane and water (pH = 1) containing PBA at various concentrations (at 0, 23 and 117 mM in the mixture) and at temperatures ranging between 170 and 190 °C (Fig. S4 and Table S2†). 1-Methylnaphthalene was used instead of toluene because its lower vapour pressure will not significantly mask eventual changes in the water partial pressure. A statistically significant difference in the water partial pressures between the cases with and without PBA developed once the temperature exceeded 180 °C, and the systems that contained PBA showed a slightly higher water partial pressure (Fig. S4 and Table S2†). These data are in agreement with the occurrence of a transition to the monophase at 180 °C, and it indicates that the monophasic mixture accommodates water less well than the polar aqueous phase under biphasic conditions. Qualitatively, the poorer accommodation of water in the monophasic case is in agreement with the mixture that has to hold the water possessing a higher extent of organic solvent (comparing water in sulfolane to water in sulfolane plus 1-methylnaphthalene under biphasic and monophasic conditions, respectively).
Effects of water contents and solvent composition
To further characterize the effects of solvent composition on the system, experiments were performed with the aqueous phase (at pH = 1), but varying the polar and apolar organic solvents to compose the final three-solvent system, at a constant reaction temperature of 180 °C (Table 1; full xylose conversion was always achieved).:1:1 apolar–polar–aqueous three-solvent systems (180 °C, pH = 1). In all cases, the reaction was stopped at a time point shortly after full xylose conversion. The solvent polarity of the polar solvent is represented here by the water–octanol partition coefficient (−logP)
| Apolar solvent | Polar solvent | −logP |
Number of phases at the reaction T (°C) | Selectivity (mol%) | t (h) |
|---|---|---|---|---|---|
| Toluene | DMSO | 1.35 | 1 | 90 | 3 |
| Toluene | Sulfolane | 0.77 | 1 | 95 | 3.5 |
| Toluene | Dioxane | 0.42 | 2 | 60 | 3 |
| Toluene | GVL | 0.27 | 2 | 65 | 4 |
| Nitrobenzene | DMSO | 1.35 | 1 | 95 | 2 |
| 1-Methylnaphthalene | DMSO | 1.35 | 1 | 87 | 3 |
| 1-Methylnaphthalene | Sulfolane | 0.77 | 1 | 89 | 3.5 |
The behaviour of the different solvent mixtures containing toluene and a polar organic solvent shows a clear qualitative difference at room temperature and at the reaction temperature (Fig. S5 and Table S3†). All these systems are biphasic at room temperature; however, the relative volumes of the two phases differ from system to system. Dioxane partitions equally between water and toluene, γ-valerolactone (GVL) mixes exclusively with toluene, and DMSO and sulfolane mix exclusively with water. This behaviour relates well to the relative polarities of the organic solvents expressed e.g., in terms of water–octanol partition coefficient logP (Table 1).
The choice of the polar solvent shows to be a crucial variable, with high selectivities obtained in the case of the most polar solvents, DMSO and sulfolane, and lower selectivities obtained in the case of the less polar ones, dioxane and GVL. When maintaining the polar organic solvent sufficiently polar, changing the aromatic counterpart did not seem to affect the final selectivity as much. These results further underline the results shown above that the crucial element influencing the xylose-to-furfural selectivity is the presence of a polar organic solvent in combination with an aromatic one so that a three-solvent mixture is obtained that transitions to a monophase at the reaction temperature, which is indeed observed in the case of DMSO and sulfolane, but not in the case of dioxane and GVL (Table S3†). The case of 1-methylnaphthalene–sulfolane–water (pH = 1) is specifically valuable as it allows easy distillation of furfural from the solvent mixture (specifically, 1-methylnaphthalene and sulfolane, obtained after phase separation achieved by cooling the reaction mixture), and it will be discussed in the process concept below.
These results relate well with the literature, in which most organic solvents and solvent mixtures show to favour high furfural selectivity.16–19 Comparing different solvent systems for xylose dehydration shows that fully organic (monophasic) and mostly organic transient monophasic conditions perform better than traditional biphasic conditions. Specifically, using a 1:1 mixture of water and 1-butanol, which is monophasic above 125 °C, for performing xylose dehydration to furfural results in an approx. 8 mol% selectivity improvement from traditional biphasic conditions (Table S4†). Closest to our finding, Román-Leshkov and Dumesic showed that, also in the case of fructose dehydration to HMF, an improvement in selectivity was observed when using a water/1-butanol system.15 Overall, the comparison shows that the three-solvent system proposed in this work gives a better furfural selectivity at milder temperatures than any of the systems reported earlier.16–19,24
The water content and the apolar nature of the phase in which the dehydration takes place appear to affect the xylose-to-furfural selectivity, as has also been reported for related systems. For instance, molecular dynamics simulations on the interaction of sugars (e.g., glucose) in progressively more organic solvent mixtures of water with DMSO, THF and DMF reveal that these solvents compete with water in forming the first solvation shell around the sugar, even when added in relatively low amounts (<40–50 v%).25 This promotes the dehydration of sugars by altering the relative stability of initial and transition states.16–19,26,27 In addition, high amounts of a polar organic co-solvent are reported to form water-rich local domains, influencing the energy barrier for the reaction.28 These findings agree qualitatively with our high selectivity observed when increasing the fraction of organic solvent after the transition to the monophase. On the other hand, it is difficult to rationalize solely by the water content why our monophasic operation, with one-third water in the mixture, performs also better than in a polar-organic solvent/mixture with less water. Yet, selectivity is also and foremost controlled by the good solubilization and protection of the produced furfural and by suppression of the formation of humins.11 Possibly we operate under more apolar conditions, by merging not only more water but also a more apolar organic solvent into the mixture, than is achieved in the polar-organic case. Such an enhanced apolar character of the phase in which the dehydration occurs would favour solubilization and stabilization of furfural, as well as a concomitantly better suppression of the formation of acyclic sugar, both leading to a better selectivity. Furthermore, control experiments show that performing the reaction in monophasic 1:1 DMSO–water or sulfolane–water systems, without the aromatic solvent but with the same initial PBA2X concentration, leads to lower xylose-to-furfural selectivities (Table S4†), confirming that the apolar nature of the mixture is important as well, not just the water content in it.
Conceptual integration of xylose extraction and monophasic conversion into a furfural manufacturing process
The three-solvent xylose-to-furfural conversion can be combined with a xylose-boronate ester extraction approach, to convert the diluted xylose hydrolysate to furfural at a high yield and with low energy demand.23 For the purpose of this study, a hydrolysate stream containing 4–5 wt% of xylose is chosen, as encountered in the industrial processing of biomass. Operating at a higher xylose concentration is not favourable because it requires reconcentration of the xylose syrup and results in a lower xylose-to-furfural selectivity (Fig. S6 and Table S5†).In the first step, xylose is extracted as the boronate diester by contacting the hydrolysate with a 1-methylnaphthalene (MN)/PBA solution. The resulting organic/boronate diester solution is mixed with an acidic sulfolane/water solution and heated to the reaction temperature to hydrolyse the ester and convert the released xylose into furfural. Sulfolane is chosen over DMSO due to its higher stability at high temperatures.29 The reactor effluent is then cooled to allow phase separation into an MN/PBA/furfural organic phase and a sulfolane/water/furfural phase. Furfural is separated from the organic phase by means of distillation, resulting in a furfural distillate, i.e., the desired product stream, and an MN/PBA bottom stream, which is to be recycled to the xylose extraction column. The sulfolane/water phase is sent to a distillation column to remove the unrecovered furfural before recycling it to the dehydration reactor. Indeed, much of the unextracted furfural can be distilled off as heterogeneous azeotrope with water that spontaneously splits into a water phase containing 8 wt% furfural and a furfural phase containing 6 wt% water.1,30 It is worth noting that this scheme does not require deep furfural recovery from the aqueous phase since some furfural can be recycled back to the dehydration step. The water product stream will need to be recycled to balance the water consumed to hydrolyze PBA2X to free xylose (Fig. 5).
 | ||
| Fig. 5 Conceptual process for the two-step furfural production based on the integration of xylose extraction as the boronate diester from an acidic hydrolysate followed by conversion of xylose into furfural in a highly efficient monophasic three-solvent mixture. | ||
The viability of this process concept will depend on the efficiency in closing the two recycle streams. Most critically, it will not allow significant losses of MN, PBA and sulfolane in the clean hydrolysate. To this end, it will not allow significant slip of sulfolane into the MN/PBA phase of the decanter, as eventual sulfolane contamination will likely end up in the clean hydrolysate stream. If the losses of sulfolane, MN and PBA are not negligible, we may need to consider a small finishing step, e.g., by adsorption, to further purify the clean hydrolysate and recover the lost sulfolane, MN and PBA. We note that the sequence of xylose extraction followed by dehydration allows leaving a number of hydrolysis by-products in the hydrolysate and, thereby, produces furfural with much higher purity. Boric acid has indeed been shown to extract xylose with high selectivity and leave behind most of the hexose impurities and acetic acid that are also formed during the hydrolysis step, limiting the efforts in the purification of both the reagent and product stream.23,31
:1 (v/v) mixture of water (pH = 1) and sulfolane. Accordingly, about 40% of the furfural was found in the MN-rich phase together with minor amounts of sulfolane (7% on intake) and water. The water/sulfolane phase contained then the remaining 60% of the furfural, together with modest amounts of PBA and negligible amounts of MN that will eventually build-up to steady-state in the recycle loop. These two phases need to be worked up to recover the furfural, recycle the sulfolane and MN/PBA, and eliminate excess water formed by the dehydration reaction.
:1:1 (v/v/v) MN/sulfolane/water (pH = 1) solvent system
| Component | [Conc.]wat+Sa (mM) |
[Conc.]MN (mM) | wt%wat-S | wt%MN |
K
d
[X] |
P
mol
|
|---|---|---|---|---|---|---|
| a Water/organic partition, as the volume of the polar phase is double the apolar one; this value implies a dilution. b This water/organic partition factor considers the amounts (in mmol) in the phases and not the concentrations, correcting for the fact that the polar phase is twice the volume of the apolar one. c A non-negligible amount of water is dispersed in the MN phase. | ||||||
| Furfural | 103 mM | 144 mM | 0.88 wt% | 1.3 wt% | 0.72 | 1.43 |
| Water | 27.6 M | 92 mMc | 44 wt% | 0.15 wt%c | 300 | 601 |
| Sulfolane | 5.1 M | 370 mM | 54 wt% | 4.0 wt% | 27.3 | 13.8 |
| PBA | 125 mM | 340 mM | 1.3 wt% | 3.7 wt% | 0.37 | 0.74 |
| MN | 3 mM | 7.0 M | 0.04 wt% | 91 wt% | 0.0004 | 0.0009 |
Recovering furfural from the aqueous/sulfolane phase is more challenging. However, much could be distilled off as an azeotrope with water, likely together with excess water which boils just a few degrees higher than the water/furfural azeotrope.1 Alternatively, furfural could be extracted by repurposing the clean MN/PBA. Control experiments showed that 5 extractions of the polar phase with a clean aromatic phase (MN) resulted in an approx. 90 mol% recovery of furfural (Fig. S7†). Furfural will be recovered to the extent that is economically attractive, and the remaining fraction will be recycled to the dehydration reactor.
The application of such a process concept promises the production of furfural with a selectivity approx. 20 mol% higher than the traditional industrial approaches, thus providing less waste.1,5 A preliminary economic analysis, which is detailed in the ESI† and follows the approach presented in the literature,33 suggests that the increase from 70 to 95 mol% selectivity in furfural provides sufficient economic room to pay for the increased complexity of the process proposed here. Based on premises detailed in the ESI,† particularly biomass and furfural market prices of $80 (±20) and $1400 (±200)/t, the selectivity increase more than doubles the room for conversion cost – i.e. investment, energy, and chemicals – from 161 (±140) $/t to 385 (±162) $/t, i.e., an increase by 224 (±22) $/t. Although simplistic, such analysis shows sufficient economic potential to warrant further research and develop a detailed economic assessment.
This process concept attempts to minimize the production of waste from feed and chemicals, which is a key aspect of green chemistry.34 Hence, it focuses on maximizing furfural selectivity and maximizing the recycling of all chemicals, solvents and PBA. The common solvents investigated here for proof-of-concept do not need to be the definitive ones. Now that the chemistry has been unraveled, future research should indeed refine the solvent selection by exploring greener solvents that are, for example, bio-based and, preferably, bio-degradable, while being inert under the present reaction conditions and providing comparable (or better) furfural selectivity and recycling.
Further development of the process concept described in Fig. 5 is the subject of future research. Such development needs to go beyond simple demonstration of the chemistry at a larger scale. It needs to prove the various separation steps, prove the closure of all recycles without detrimental build-up of impurities, unravel the scaling rules to design the major pieces of equipment, and much more. A small part of it, e.g., on the xylose extraction section, might be already available,23 but the rest is clearly outside the scope of the present proof-of-concept.
Conclusions
In this work, we have demonstrated the possibility to convert a xylose-diboronate ester, the product of xylose extraction, to furfural with >90 mol% yield by applying a three-solvent system that forms a single phase at the reaction temperature. Obtaining a single phase appeared to be essential to obtain high selectivity, and its occurrence is dependent on the PBA concentration and temperature. The interaction between PBA and the aromatic solvent is a likely trigger for the phase transition. Although not fully clear, we think that the increased apolar nature of the solvent mixture in which the dehydration occurs, achieved in monophasic operation, contributes to the observed improved selectivity. We show that high selectivities, at and above approx. 90 mol%, become feasible with a variety of solvent combinations and PBA concentrations. Specifically, the highest xylose-to-furfural selectivity (95 mol%) is obtained in a 1:1:1 sulfolane–toluene–water (pH = 1) system, using a PBA diester of xylose as the starting compound. The reaction conditions allow the use of solvents that ease the furfural recovery by splitting into two liquid phases upon cooling and allow easy distillation of furfural from the media.
A conceptual process design for furfural production, based on these findings, a further analysis of the various losses and a preliminary economic analysis have been presented. This approach is potentially preferable to conventional biphasic systems (due to the high selectivity) and to the alternative monophasic organic operation (due to the minimal distillation duties required). The produced furfural can be recovered from the system with negligible losses of the solvents in the product and waste streams, and thus this process design minimizes waste and maximizes the recyclability. Additional research is needed to validate the process concept and deliver the information needed for designing the major pieces of equipment, with the possibility of implementing the use of bio-based and fully biodegradable solvent alternatives.22
Experimental
Chemicals
D-(+)-Xylose (>99%), D2O (99.9% atom D), toluene-d8 (99% atom D), dioxane (99.8%), 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt (TMSP, 98% atom D), dimethylsulfoxide (DMSO, 99%), sulfolane (99%), 10-bromoanthracene-9-boronic acid (BBA, 99%), γ-valerolactone (GVL, 99%), nitrobenzene (99%), 1-butanol (98%), and 1-methylnaphthalene (98%) were purchased from Sigma-Aldrich, while phenylboronic acid (PBA, 99%) and tetramethylsilane were obtained from Alfa Aesar. PBA2X was obtained as described previously.22Methods and equipment
All chemicals were used without further purification. 1H-NMR spectra were recorded using a 400 MHz Bruker spectrometer in a 1:1 H2O/D2O mixture with TMSP as the internal standard in the case of the aqueous phases or in a 1:1 mixture of toluene and toluene-d8 with dioxane and tetramethylsilane as internal standards in the case of the organic phases. These mixtures are composed of equal volumes (250 μL) of the sample and the deuterated solvent, containing the standard. In all cases, the reactions were performed using a heating mantle, stirring with a magnetic stirrer at 1000 rpm.
Xylose conversion to furfural
A 1:1:1 v/v/v solvent mixture (each solvent 2 mL) of an aromatic solvent (toluene, nitrobenzene, or 1-methylnaphthalene), a polar organic solvent (DMSO, sulfolane, GVL, or dioxane) and water (pH = 1 from added H2SO4), containing a total concentration of xylose of 97 mM (either in its free form or as the PBA diester), was heated between 150 °C and 205 °C, for the reaction times varying between 0.5 and 16 h. In the case of the experiments performed with free xylose as a starting material, PBA was also added to the mixture, at concentrations between 0 mM and 667 mM. To ensure maximum comparability between all the various experiments, the same total amount of solution (6 mL) was used at all times. Alternatively, 2 mL of an aqueous xylose solution (350 mM, pH = 1 from H2SO4) was mixed with 2 mL of a polar organic solvent (1-butanol, DMSO, sulfolane) and heated at temperatures between 180 °C and 200 °C between 3 and 5 h. Prior to the 1H-NMR spectroscopy analysis, all the reactions were stopped and cooled to r.t.
Determination of the phase behaviour of the three-solvent system
A pyrex capillary tube was filled with equal volumes of a PBA solution in toluene (from 6 mM to 750 mM), sulfolane and water (pH = 1 from H2SO4) for a total volume of approx. 1 mL. The capillary was sealed with a blowtorch (oxyacetylene flame), cooled to room temperature and then put into an oven, where the temperature was accurately controlled (from 25 °C to 220 °C). The observed transition from a biphase to a monophase was photographed and filmed (Fig. S2, Videos S1 and S2†). Alternatively, the same solvent mixture, for a total volume of 6 mL, was placed in a high pressure-proof hard glass vessel (ACE Glass Incorporated) and heated using a heating mantle (from 140 °C to 210 °C). In this case, the assessment of the phase behaviour is less precise and resulted in a wider range of temperatures for the transition to occur.Colorimetric analysis of the BBA concentration
5 mL of various solutions with BBA concentrations between 0.6 M and 2.6 M, both in MN and in a 1:1 sulfolane–water (pH = 1) mixture, were heated up to 180 °C. A 1:1:1 solvent mixture (each solvent, 2 mL) of 1-methylnaphthalene, sulfolane, and water (pH = 1) containing 0.5 M of BBA was heated between 25 and 180 °C. Photographs of these solutions were taken, at the mentioned temperature using the integrated camera of a smartphone and analysed using ImageJ, as described in the ESI.†
Evaluation of the partial vapour pressure of water
Three different 1:1:1 solvent mixtures (each solvent, 25 mL) of 1-methylnaphthalene, sulfolane, and water (pH = 1) containing 0 mM, 23 mM or 117 mM PBA were heated from 25 °C to 190 °C in a 200 mL autoclave reactor, equipped with a pressure sensor and two thermocouples (one to measure the temperature of the liquid phase and the other to measure the temperature of the gas phase). Temperature and pressure were monitored continuously in a time window between 3 and 5 h. All measurements were performed in triplicate. The obtained data were analysed using MatLab, as described in the ESI.†
Author contributions
J.L. conceived the project. J.H., W.V., J.L. and L.R. defined the approach and refined it as the project progressed. L.R. conducted all the practical experimental work and the analysis of the raw data. J.L. provided the process concept and the preliminary economics. Further data analysis, manuscript preparation and subsequent editing/improvement of the text were performed by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Shell Global Solutions International B.V. is gratefully acknowledged. L. R. is grateful for the fruitful discussions with Richard J. M. Egberink. L. R. acknowledges the help of S. van Wyk for the support in the experiments that resulted in Videos S1 and S2.† L. R. acknowledges the support of N. J. Overeem for the data analysis that resulted in Fig. S3.† L. R. acknowledges the help of T. te Molder for the support in the experiments and P. H. Hamming in the data analysis that resulted in Fig. S4.†Notes and references
- J.-P. Lange, E. van der Heide, J. van Buijtenen and R. Price, ChemSusChem, 2012, 5, 150–166 CrossRef CAS.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. López Granados, Energy Environ. Sci., 2016, 9, 1144–1189 RSC.
- M. M. Antunes, S. Lima, A. Fernandes, M. Pillinger, M. F. Ribeiro and A. A. Valente, Appl. Catal., A, 2012, 417–418, 243–252 CrossRef CAS.
- P. Priecel, J. E. Perez Mejia, P. D. Carà and J. A. Lopez-Sanchez, Microwaves in the Catalytic Valorisation of Biomass Derivatives, Sustainable Catalysis for Biorefineries, ed. F. Frusteri, D. Aranda and G. Bonura, 2018, vol. 596, pp. 243–299, https://pubs.rsc.org/en/content/chapter/9781788013567-00243/978-1-78801-356-7 Search PubMed.
- L. Ricciardi, W. Verboom, J.-P. Lange and J. Huskens, ACS Sustainable Chem. Eng., 2019, 7, 14273–14279 CrossRef CAS.
- Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933–1937 CrossRef PubMed.
- F. Delbecq, Y. Takahashi, T. Kondo, C. C. Corbas, E. R. Ramos and C. Len, Catal. Commun., 2018, 110, 74–78 CrossRef CAS.
- R. Weingarten, J. Cho, W. C. Conner, Jr. and G. W. Huber, Green Chem., 2010, 12, 1423–1429 RSC.
- G. G. Millán, S. Hellsten, A. W. T. King, J.-P. Pokki, J. Llorca and H. Sixta, J. Ind. Eng. Chem., 2019, 72, 354–363 CrossRef.
- A. Parejas, V. Montes, J. Hidalgo-Carrillo, E. Sánchez-López, A. Marinas and F. J. Urbano, Molecules, 2017, 22, 2257–2275 CrossRef.
- L. Ricciardi, W. Verboom, J.-P. Lange and J. Huskens, ChemSusChem, 2020, 13, 3589–3593 CrossRef CAS PubMed.
- B. Li, S. Varanasi and P. Relue, Green Chem., 2013, 15, 2149–2157 RSC.
- Y. Nie, Q. Hou, W. Li, C. Bai, X. Bai and M. Ju, Molecules, 2019, 24, 594–612 CrossRef.
- C. Lansalot-Matras and C. Moreau, Catal. Commun., 2003, 4, 517–520 CrossRef CAS.
- Y. Román-Leshkov and J. A. Dumesic, Top. Catal., 2009, 52, 297–303 CrossRef.
- L. Shuai and J. Luterbacher, ChemSusChem, 2016, 9, 133–155 CrossRef CAS.
- B. R. Caes and R. T. Raines, ChemSusChem, 2011, 4, 353–356 CrossRef CAS.
- A. H. Motagamwala, K. Huang, C. T. Maravelias and J. A. Dumesic, Energy Environ. Sci., 2019, 12, 2212–2222 RSC.
- A. H. Motagamwala, W. Won, C. Sener, D. M. Alonso, C. T. Maravelias and J. A. Dumesic, Sci. Adv., 2018, 4, 9722–9730 CrossRef PubMed.
- A. S. Dias, M. Pillinger and A. A. Valente, J. Catal., 2005, 229, 414–423 CrossRef CAS.
- W. Wang, H. Li, J. Ren, R. Sun, J. Zheng, G. Sun and S. Liu, Chin. J. Catal., 2014, 35, 741–747 CrossRef CAS.
- F. Gao, R. Bai, F. Ferlin, L. Vaccaro, M. Li and Y. Gu, Green Chem., 2020, 22, 6240–6257 RSC.
- L. Ricciardi, W. Verboom, J.-P. Lange and J. Huskens, ACS Sustainable Chem. Eng., 2021, 9, 6632–6638 CrossRef CAS.
- C. Sener, A. H. Motagamwala, D. M. Alonso and J. A. Dumesic, ChemSusChem, 2018, 11, 2321–2331 CrossRef CAS PubMed.
- V. Vasudevana and S. H. Mushrif, RSC Adv., 2015, 5, 20756–20763 RSC.
- M. A. Mellmer, C. Sanpitakseree, B. Demir, P. Bai, K. Ma, M. Neurock and J. A. Dumesic, Nat. Catal., 2018, 1, 199–207 CrossRef CAS.
- J. J. Varghese and S. H. Mushrif, React. Chem. Eng., 2019, 4, 165–206 RSC.
- T. W. Walker, A. K. Chew, H. Li, B. Demir, Z. C. Zhang, G. W. Huber, R. C. Van Lehn and J. A. Dumesic, Energy Environ. Sci., 2018, 11, 617–628 RSC.
- D. L. Head and C. G. McCarty, Tetrahedron, 1973, 16, 1405–1408 CrossRef.
- I. Agirrezabal-Telleria, I. Gandarias and P. L. Arias, Bioresour. Technol., 2013, 143, 258–264 CrossRef CAS.
- R. Xing, W. Qi and G. W. Huber, Energy Environ. Sci., 2011, 4, 2193–2205 RSC.
- J.-P. Lange, ChemSusChem, 2017, 10, 245–252 CrossRef CAS PubMed.
- J.-P. Lange, Catal. Sci. Technol., 2016, 6, 4759–4767 RSC.
- P. Jessup, Green Chem., 2020, 22, 13–15 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc01752g |
| This journal is © The Royal Society of Chemistry 2021 |