
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sustainable catalytic epoxidation of biorenewable terpene feedstocks using H2O2 as an oxidant in flow microreactors†
Joshua D.
Tibbetts
 ab,
William B.
Cunningham
ab,
Massimiliano
Vezzoli
c,
Pawel
Plucinski
c and
Steven D.
Bull
*a
ab,
William B.
Cunningham
ab,
Massimiliano
Vezzoli
c,
Pawel
Plucinski
c and
Steven D.
Bull
*a
aDepartment of Chemistry, University of Bath, Bath, UK. E-mail: s.d.bull@bath.ac.uk
bCentre for Sustainable Chemical Technologies, University of Bath, UK
cDepartment of Chemical Engineering, University of Bath, Bath, UK
First published on 6th July 2021
Abstract
Solvent-free continuous flow epoxidation of the alkene bonds of a range of biorenewable terpene substrates have been carried out using a recyclable tungsten-based polyoxometalate phase transfer catalyst and aqueous H2O2 as a benign oxidant. These sustainable flow epoxidation reactions are carried out in commercial microreactors containing static mixing channels that enable common monoterpenes (e.g. untreated crude sulfate turpentine, limonene, etc.) to be safely epoxidized in short reaction times and in good yields. These flow procedures are applicable for the flow epoxidation of trisubstituted and disubstituted alkenes for the safe production of multigram quantities of a wide range of epoxides.
Introduction
Terpenes and terpenoids are produced in large amounts as secondary metabolites from plants and microbes, which are widely used as biorenewable feedstocks for production of chemicals, flavours, fragrances, drugs and polymers.1–3 Several unsaturated monoterpenes are currently available in large volumes as waste by-products from the forestry (e.g. α-/β-pinene from paper milling) and agricultural (e.g. limonene from citrus juice production) industries.4 The largest biorenewable monoterpene source is turpentine, with an estimated 230![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes of Crude Sulfate Turpentine (CST) produced annually as a co-product from the Kraft paper pulping process. Another 100000 tonnes of gum turpentine are produced through distillation of resin harvested from tapping trees in forestry plantations. CST (major components: α-/β-pinene and 3-carene; exact composition dependent on geographic origin) is currently available at low-cost (∼$220 per tonne), which is used for production of a range of value-added biorenewable chemical products (e.g. flavours, fragrances and vitamins).5 Consequently, the development of new efficient catalytic protocols to transform abundant monoterpene feedstocks into synthetically versatile intermediates is important, since they would broaden the range of biorenewable products available from terpene biorefineries, thus helping improve their economic viability.6,7
000 tonnes of Crude Sulfate Turpentine (CST) produced annually as a co-product from the Kraft paper pulping process. Another 100000 tonnes of gum turpentine are produced through distillation of resin harvested from tapping trees in forestry plantations. CST (major components: α-/β-pinene and 3-carene; exact composition dependent on geographic origin) is currently available at low-cost (∼$220 per tonne), which is used for production of a range of value-added biorenewable chemical products (e.g. flavours, fragrances and vitamins).5 Consequently, the development of new efficient catalytic protocols to transform abundant monoterpene feedstocks into synthetically versatile intermediates is important, since they would broaden the range of biorenewable products available from terpene biorefineries, thus helping improve their economic viability.6,7
Epoxides are one of the most synthetically useful functional groups,8,9 so the availability of sustainable catalytic protocols that can be used to epoxidize the alkene bonds of monoterpene feedstocks to produce monoterpenoid epoxides is highly desirable.10 Current examples of potentially useful terpene epoxides include α-pinene oxide which is used as an intermediate to produce commercially valuable fragrance compounds,11 and limonene 1,2-oxide which can be used as a monomer for the sustainable synthesis of biorenewable polymers.12 We have recently described optimal biphasic solvent-free protocols that use a preformed tungsten based Venturello Phase Transfer Catalyst (VPTC) and 30 mol% H2O2 as a benign terminal oxidant to carry out batch catalytic epoxidation of the alkene bonds of biorenewable terpene feedstocks (including untreated CST, see Fig. 1).13 Variations of this batch catalytic epoxidation process have been used to epoxidize the tri- and/or di-substituted alkene bonds of a wide range of terpenes, exhibiting good tolerance of common terpenoid functional groups (e.g. alcohols and esters). Catalyst recycling studies revealed that the VPTC could be recycled up to three times for the sequential production of monoterpene oxides with only minimal losses in epoxide yield or catalyst activity.13 Treatment of crude epoxides (no work-up) with a heterogeneous acid catalyst (Amberlyst-15) was also used to prepare their corresponding terpene-anti-diols in good yields.13
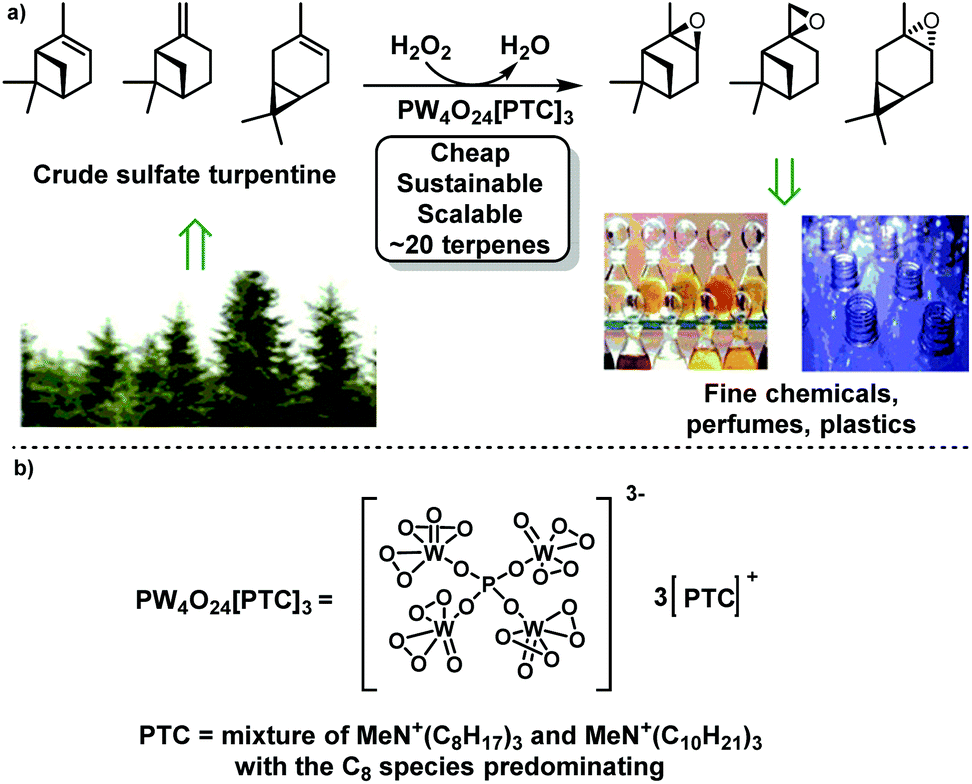 | ||
| Fig. 1 (a) A recyclable tungsten-based polyoxometalate catalyst using H2O2 as a stoichiometric oxidant for the solvent free batch epoxidation of the alkene bonds of monoterpenes present in untreated CST. (b) Structure of Venturello Phase Transfer Catalyst (VPTC) used in H2O2 based epoxidation reactions. | ||
The scope and versatility of the VPTC/H2O2 catalytic system means that it has been widely used for batch epoxidation of the alkene bonds of a wide range of substrates.14–16 However, we (and others17,18) have described that scale up of batch biphasic VPTC/H2O2 epoxidation reactions is challenging, requiring slow dropwise addition of the H2O2 oxidant and careful control of reaction temperature to prevent dangerous delayed onset thermal runaways from occurring.19 The potential benefits of carrying out catalytic epoxidation reactions using H2O2 as an oxidant in flow reactors are well established (for literature examples, see Table 1, entries 1–7), including: excellent mass and heat transfer; reduced reaction times; more efficient temperature control; improved safety profiles; and the ability to telescope reactions to generate epoxide derivatives.20–28 Therefore, the ability to carry out continuous VPTC/H2O2 mediated epoxidations of monoterpenes (and other types of alkenes) in flow reactor systems is desirable, since it would enable this catalytic system to be used safely for large scale epoxide manufacturing. Consequently, we now describe that preformed VPTC/H2O2 systems can be used for the flow epoxidation of biorenewable terpene feedstocks in inexpensive continuous flow microreactors containing static mixing units, with these simple flow protocols also applicable for epoxidation of other types of alkene substrate.29
| Entry | Substrate | Reactor type | Conditions | Yield | Ref. |
|---|---|---|---|---|---|
| 1 | Styrene | Corning advanced flow G1 reactor | 15% 2,2,2-trifluoroactephenone, 3 equiv. H2O2, MeCN/t-BuOH, 80 °C, 3 min | 92% | 22 |
| 2 | Cocoa butter (cis-alkenes of palmitic, stearic and oleic acid side chains) | Vapourtec PTFE tubular reactor (1 mm i.d. × 12.7 m length) | 2.5% tungsten powder, 1.5% surfactant, 4% H3PO4, 8 equiv. H2O2, 80 °C, 80 min | 77% | 23 |
| 3 | Soybean Oil (cis-alkenes of α-linoleic, linoleic and oleic acid side chains) | Microflow slit plate mixer | 3% H2SO4, 3% EDTA-2Na, 8 equiv. H2O2, 8 equiv. HCOOH, 75 °C, 7 min | 7.3 (epoxy number) | 24 |
| 4 | Soapstock fatty acids (cis-alkenes of linoleic, oleic and linolenic acid side chains) | Microchannel bioreactor (0.8 mm i.d. × 1000 mm) | 0.3 wt% Candida rugosa lipase, 1.6 equiv. H2O2, 36 °C, 51 min | 85% | 25 |
| 5 | Cyclohexene | Glass capillary microreactor (3 mm i.d. × 36 mm length) packed with enzyme catalyst | 100 mg Novozym® 435, 2 equiv. H2O2, 2.6 min | 98% | 26 |
| 6 | Cyclohexene | Micro-flow capillary tube | 3% DCC, 8 equiv. Urea H2O2, 80 °C, 12 min | 96% | 27 |
| 7 | Cyclohexene | Vapour phase flow reactor (fluorinated ethylene propylene tubing) | Ti/Nb-SiO2, 3 kPa H2O2 and 3 kPa cyclohexene, 120 °C, 200 min | 50% selectivity (+∼50% diol) | 28 |
| 8 | Cyclohexene | LTF microreactor (7.5 mL) | 2% PW 4 O 24 [PTC] 3 , 1.6 equiv. H 2 O 2 , 50 °C, 17 min | 91% | This work |
Results and discussion
Previous studies using the VPTC/H2O2 system for batch epoxidation reactions had established that the overall rate of epoxidation was highly dependent on the rate of stirring of these biphasic reactions.13 Consequently, it was decided to use a commercial flow microreactor containing static mixing channels to carry out flow epoxidation reactions, since this would ensure efficient mass and heat transfer between organic and aqueous streams, as well as allowing for efficient control of reaction temperature. The microreactor system used contains separate channels (2.2 × 2.2 mm) that allow discrete preheating of organic (terpene and catalyst, solvent free) and aqueous (aq. 30 wt% H2O2) input streams. Both these organic and aqueous streams are then flowed into a single static mixer channel that results in efficient mass transfer between the two phases (under a positive pressure of up to 7 bar) (see Fig. SI1 and SI2† for details of flow reactor design). Use of this flow reactor design results in fast solvent-free catalytic epoxidation reactions under good thermal control to produce an epoxide and water as the only by-product. Sampling ports were used for reaction monitoring, with reaction data (e.g. alkene conversion rates, product ratios, etc.) acquired at different time points used to modify reaction conditions (e.g. catalyst loadings, flow rates, etc.) to enable flow epoxidation conditions of individual alkene substrates to be optimised.Previous batch epoxidation conditions developed for mono-epoxidation of the trisubstituted alkene of limonene that produce good yields of limonene 1,2-oxides in ∼1 h were chosen as a starting point to commence the flow epoxidation studies.13 Therefore, an organic flow stream containing 1 mol% PW4O24[PTC]3 catalyst dissolved in limonene and an aqueous stream containing 30 wt% H2O2 (pH 7.0) was trialled. The organic and aqueous streams were preheated to 50 °C, with both streams then flowed into the static mixing channel (7.5 mL total volume, 50 °C) of the reactor where epoxidation occurs. Limonene consumption levels at a relatively low flow rate of 9 mL h−1 proved to be sluggish, requiring >30 min to reach completion. Conversely, a higher flow rate of 54 mL h−1 resulted in a faster epoxidation rate, however the total volume of the static reactor gave insufficient residence time to allow for full limonene consumption. Therefore, a flow rate of 27 mL h−1 was chosen, which resulted in total consumption of limonene in ∼17 min, with this median flow rate subsequently used to carry out all further flow optimisation studies (see Fig. SI3† for flow rate studies). Incremental increases in the temperature of the flow epoxidation reaction from 25 to 50 °C resulted in a corresponding increase in the rate of limonene epoxidation, with temperatures up to 50 °C resulting in >90% selectivity for formation of limonene 1,2-oxides (55:45 mixture of diastereomeric epoxides 1a and 1b). Raising the temperature above 50 °C led to a decrease in selectivity for production of limonene 1,2-oxides 1a/1b (<75% at 60 °C), with losses in epoxide yield caused by competing epoxidation/hydrolysis reactions producing anti-diols 2 and diastereomeric bis-epoxides 3 as unwanted side-products. Increasing the temperature of the static mixer further to 70 °C resulted in thermal decomposition of hydrogen peroxide (oxygen bubbles observed in reactor channels) that resulted in lower limonene consumption rates (see Fig. SI4† for flow temperature studies). Use of 1 mol% VPT catalyst proved optimal for carrying out the flow epoxidation of limonene, with higher catalyst loadings leading to faster limonene consumption, however lower yields of limonene 1,2-oxides 1a/1b were obtained due to greater amounts of unwanted diols 2 and bis-epoxides 3 being formed as by-products (see Fig. SI5 and SI6† for catalyst loading studies).30 Increasing the amount of commercial 30 wt% H2O2 (pH 7.0) oxidant from 1.0 to 1.6 equiv. had a positive effect on limonene conversion rates, with only a 2% decrease in selectivity for 1a/1b observed at higher H2O2 levels (see Fig. SI7† for H2O2 concentration studies). These screening studies led to optimal conditions for flow epoxidation of limonene being established as 1 mol% VPTC and 1.6 equiv. H2O2 (pH 7.0) at 50 °C using a 27 mL h−1 total flow rate that produced a residence time of 16.7 min in a 7.5 mL volume microreactor. These conditions gave 89% limonene conversion, producing limonene 1,2-oxides 1a/1b with >90% selectivity in <20 min (Fig. 2).
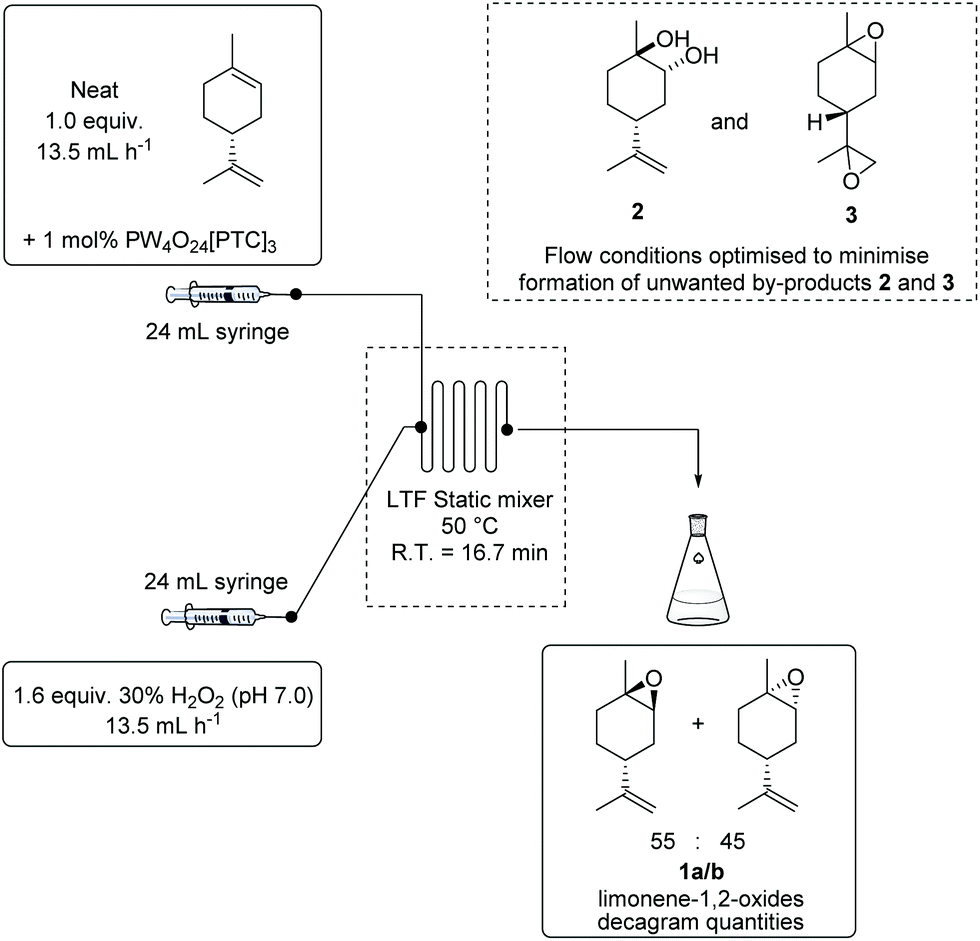 | ||
| Fig. 2 Optimal conditions used for the continuous flow epoxidation of limonene using 1 mol% PW4O24[PTC]3 catalyst and 30 wt% H2O2 (pH 7.0) as oxidant. | ||
These conditions were then used as a starting point to optimise flow epoxidation of six other monoterpenes in 92–100% conversions (Fig. 3). 2 mol% VPTC was used to epoxidize the alkene bond of 3-carene to give 3-carene oxide 4 in 97% conversion and 98% selectivity in <20 min. Previous batch epoxidation studies had shown that α-pinene oxide 5 produced under VPTC/H2O2 conditions underwent rapid hydrolysis to its corresponding anti-diol. However, this unwanted epoxide ring-opening rection could be suppressed by inclusion of 30 mol% Na2SO4 as an additive, which increases the ionic strength of the aqueous H2O2 phase to minimise hydrolysis of the epoxide.13,31 Attempts to carry out additive-free flow epoxidation of α-pinene also produced α-pinene anti-diols as major products (<20% α-pinene oxide 5). However, incorporation of 30 mol% Na2SO4 in the aqueous H2O2 (pH 7.0) flow stream and use of a higher 3 mol% VPTC loading resulted in excellent 95% conversion and 95% selectivity for formation of α-pinene oxide 5 (see Fig. SI8 and SI9†). Selective flow mono-epoxidation of the trisubstituted alkene bond of myrcene could also be achieved, producing myrcene mono-epoxide 6 in 92% conversion and 97% selectivity, with its diene fragment left intact for further functionalisation.
 | ||
| Fig. 3 Catalytic flow epoxidation reactions used for the synthesis of a range of trisubstituted and disubstituted epoxides 1a/b and 4–13. Standard flow epoxidation conditions (30 wt% H2O2 (pH 7.0), 50 °C. Residence Times (R.T.) were 16.7 min unless otherwise stated. FR1 and FR2 = Flow Rates 1 and 2. See ESI† for individual FR and R.T. values. Conversion values refer to levels of alkene consumption. Selectivity values refer to amount of epoxide product relative to substrate consumption levels. | ||
Bis-epoxidation of both trisubstituted alkenes bonds of γ-terpinene was achieved by increasing the amount of 30 wt% H2O2 used as oxidant from 1.6 to 3.2 equiv., with γ-terpinene-bis-epoxide 7 produced in >99% conversion and 60% selectivity. The other products formed were γ-terpinene-monoepoxides that could be easily separated by distillation/chromatography and resubjected to the flow epoxidation conditions to produce more bis-epoxide 7 as required. The diastereoselectivity of this bis-epoxidation reaction arises from coordination of the tungsten catalyst to the oxygen atoms of either mono-epoxide, which directs alkene epoxidation to the same face to exclusively afford the syn-bis-epoxide 7. These flow epoxidation reaction conditions were also used to epoxidize both trisubstituted alkene bonds of acyclic farnesene (leaving the less reactive diene fragment intact), with farnesene bis-epoxide 8 generated in 99% conversion and 72% selectivity (along with ∼25% mono-epoxides).
The less electron rich gem-disubstituted alkene bond of (−)-isopulegol was flow epoxidized using 1.6 equiv. H2O2 to afford isopulegol epoxides 9a/b as a 60:40 mixture of diastereomers in 92% conversion and 97% selectivity. Previous batch epoxidation reactions of the exocyclic alkene bond of β-pinene using the VPTC/H2O2 epoxidation protocol had proven challenging, primarily due to the reactive β-pinene oxide undergoing hydrolysis to its corresponding β-pinene diol. Similar problems were encountered using our standard flow epoxidation conditions which also produce large quantities of β-pinene diol. However, use of higher 5 mol% VPTC loadings, 1.6 equiv. of more concentrated aq. 50 wt% H2O2 (18 M, (pH 7.0)), a higher temperature of 75 °C and shorter static mixer residency times of 1.65 min enabled 55% conversion of β-pinene into β-pinene oxide 10 with excellent 99% selectivity levels (Fig. 3). These flow conditions proved optimal, with attempts to increase β-pinene conversion levels by reducing flow rates, increasing catalyst loadings/temperature, and changing static mixer residency times leading to lower yields of β-pinene oxide 10.
The synthetic utility of the VPTC/H2O2 catalytic system used in this flow epoxidation study is well established, and it has been used to carry out batch epoxidation reactions of a wide range of alkene substrates.14–18 Consequently, we decided to carry out a brief exploration of the scope and limitation of the protocol to flow epoxidize four non-biorenewable substrates containing challenging disubstituted and monosubstituted alkene substitution patterns. Flow epoxidation of the cis-disubstituted alkene bond of cyclohexene (benchmark substrate used in previous flow epoxidation studies, see Table 1) gave cyclohexene oxide 11 with excellent 93% conversion and 98% selectivity. The (E)-alkene bond of trans-ethyl-3-hexenoate proved less reactive, with flow-epoxidation affording the desired trans-epoxide 12 in only 26% conversion, but with 100% selectivity levels. Exclusive formation of trans-epoxide 12 means that better yields of this epoxide should be available using longer flow reaction times in a microreactor with a larger volume. Flow epoxidation of the gem-disubstituted alkene functionality of α-methylstyrene required incorporation of 30 mol% Na2SO4 as an additive to prevent unwanted epoxide hydrolysis, which produced epoxide 13 in 55% conversion with 89% selectivity. Unfortunately, the alkene bond of 1-octene proved unreactive towards the flow epoxidation conditions, with electron poor monosubstituted alkenes also found to be unreactive in batch VPTC/H2O2 epoxidation systems.13
The flow epoxidation protocol was then applied to untreated industrial CST obtained from a Swedish paper mill, which is the largest biorenewable monoterpene feedstock available for use in a terpene biorefinery. Pleasingly, use of 5 mol% catalyst, 2.2 equiv. of 30 wt% H2O2 (pH 7.0), 30 mol% Na2SO4, 60 °C, 15 min residence time resulted in 95% consumption of its three major components [α-pinene (40%), 3-carene (39%), β-pinene (10%), other monoterpenes (11%)]. This resulted in an excellent selectivity profile for production of 3-carene oxide 4 (99%) and α-pinene oxide 5 (63%), whilst its β-pinene fraction was converted into β-pinene diol (Fig. 4 and Fig. SI10†). The major epoxide products could be separated by fractional distillation of the crude product under reduced pressure to afford 3-carene oxide 4 (bp 87 °C at 50 mm Hg) and α-pinene oxide 5 (bp 102 °C at 50 mm Hg), with small amounts of pinane diols recoverable from the distillation residue. Pleasingly these flow oxidative conditions also eliminated the noxious sulfurous odour caused by the sulfur impurities (e.g. Me2S, Me2S2, etc.) present in the CST feedstock (e.g. through oxidation to DMSO).
 | ||
| Fig. 4 Flow epoxidation of the α-pinene, β-pinene and 3-carene components of untreated CST. | ||
The yields of the eight terpene epoxides 1 and 4–10 produced in these flow epoxidation protocols are similar (cf. 92% (batch) vs. 95% (flow) for 3-carene oxide 4), or better (cf. 63% (batch) vs. 90% (flow) for isopulegol epoxides 9a/b) than those obtained in their corresponding batch epoxidation reactions.13 Flow epoxidation rates for the terpene substrates were generally an order of magnitude faster than for their corresponding batch epoxidation reactions, with flow epoxidation reactions generally complete within 20 min, whilst the batch epoxidation reactions required between 1–18 h to proceed. The efficiency of these VPTC/H2O2 catalytic flow-epoxidation reactions of cyclohexene (see Table 1, entry 8) also compare favourably (e.g. lower temperature, low recyclable catalyst loadings, fast conversion rates, lower equiv. of H2O2, etc.) with previous reports where H2O2 has been used as an oxidant in catalytic flow epoxidation reactions of alkene substrates that are liquid at ambient temperatures (see Table 1, entries 1–8 for comparison).22–28
The smaller reaction volumes, static mixing channels and heat exchangers present in the microreactors result in highly efficient mass transfer and excellent temperature control in these flow epoxidation reactions. This means that it should be much safer to scale-up VPTC/H2O2 catalysed flow epoxidation reactions, since it should avoid dangerous thermal runaways that can be difficult to control in large-scale batch epoxidation reactions.13 The scale-up potential of this flow epoxidation protocol was demonstrated for continuous epoxidation of 13.5 mL of limonene over a 1 h period, which safely produced decagram quantities of limonene 1,2-oxides 1a/b. Vacuum distillation of the resultant crude epoxidation product enabled 9.0 g of pure limonene 1,2-oxides 1a/b to be isolated in 71% yield. The VPT catalyst recovered from this limonene flow epoxidation reaction could be reused to produce a second batch of limonene 1,2-oxide in 83% conversion. 1H NMR spectroscopic analysis of the recovered catalyst revealed some degradation had occurred, however the VPTC was still present as the major component (see Fig. SI11 and SI12† for details).32 Flow reactions in microreactors can be scaled up using a numbering up principle, with multiple flow reactions run in parallel to generate large amounts of product. Based on results obtained for the flow epoxidation of limonene, it can be calculated that simultaneous use of 111 microreactors could be used to produce 1 kg of limonene 1,2-oxides 1a/b per hour, which if operated continuously would generate around 8 tonnes of these epoxides annually.
Conclusions
Sustainable solvent free flow epoxidation of the di- and tri-substituted alkene bonds of various biorenewable terpenes to produce terpene oxides has been carried out using a recyclable tungsten-based polyoxometalate phase transfer catalyst and commercial H2O2 (pH 7.0) as an oxidant. Using these continuous flow epoxidation reactions in a flow reactor containing static mixing channels results in rapid mass and heat transfer between organic and aqueous streams that leads to fast epoxidation reactions and excellent temperature control. This enables epoxide products to be safely prepared in short reaction times, with the flow epoxidation protocol used to produce multigram quantities of limonene 1,2-oxide, with the recovered VPT catalyst then recycled for further epoxide production as required. The improved safety profile of these VPTC/H2O2 flow epoxidation conditions means they are better suited for large-scale manufacturing of epoxides than widely used biphasic batch epoxidation conditions that suffer from potentially dangerous thermal runaways caused by inefficient mixing and poor temperature control.Experimental
Representative flow epoxidation procedure of (R)-(+)-limonene
Two Little Things Factory (LTF) GmbH XXL-ST-04 microreactors were connected in series using PTFE 1/16-inch tubing to give a total reactor volume of 10 mL (see Fig. SI1 and SI2 ESI† for diagrams of microreactor setup). The reactor volume (and residence time) could be varied by allowing the reaction mixture to exit at any of the sampling ports. The PW4O24[PTC]3 (2.79 g, 2259 gmol−1, 1.24 mmol, 1 mol%) catalyst33 was dissolved in (R)-(+)-limonene (20 mL, 16.8 g, 124 mmol, 1 equiv.) and then loaded into a 24 mL syringe connected to the first input channel of the reactor using PTFE tubing. 30 wt% aqueous H2O2 (20 mL, 200 mmol, 1.6 equiv.) was basified to pH 7.0 using 3 M NaOH and then loaded into a second 24 mL syringe connected to the second input channel of the reactor using PTFE tubing. 30 mol% of Na2SO4 (relative to terpene substrate) was added to the aqueous H2O2 stream when epoxide products proved susceptible to hydrolysis. The syringes were pumped into the 50 °C heated static mixing reactor using syringe pumps at a rate of 13.5 mL h−1, with port sampling carried out after a combined 7.5 mL of reaction mixture had passed into the reactor (allowing for steady state equilibration). 1H NMR spectroscopic analysis of the organic layer allowed conversion levels and selectivities to be calculated. The organic layer of the preparative limonene epoxidation reaction was purified by distillation (or column chromatography) to give 9.0 g of (+)-limonene 1,2-oxide as a pale-yellow oil. The solid catalyst residue from this reaction was dissolved in a fresh batch of limonene and the flow epoxidation reaction repeated under identical conditions to give a second batch of limonene oxide in 83% conversion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank EPSRC for funding through “Terpene-based Manufacturing for Sustainable Chemical Feedstocks” EP/K014889 and the Centre for Doctoral Training in Sustainable Chemical Technologies (EP/L016354/1). Södra Forestry Cooperative are thanked for supplying an authentic industrial sample of CST.Notes and references
- N. Tsolakis, W. Barn, J. S. Srai and M. Kumar, J. Cleaner Prod., 2019, 222, 802–822 CrossRef CAS.
- W. Schwab, C. Fuchs and F. C. Huang, Eur. J. Lipid Sci. Technol., 2013, 115, 3–8 CrossRef CAS.
- M. Golets, S. Ajaikumar and J. P. Mikkola, Chem. Rev., 2015, 115, 3141–3169 CrossRef CAS PubMed.
- A. J. D. Silvestre and A. Gandini, in Terpenes: Major Sources, Properties and Applications in Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Amsterdam, 2008, ch. 2, pp. 17–38 Search PubMed.
- M. GScheidmeier and H. Fleig, Turpentines, in Ullman's Encyclopaedia of Industrial Chemistry, Wiley, 2012, pp. 537–550 Search PubMed.
- J. D. Tibbetts and S. D. Bull, Green Chem., 2021, 21, 597 RSC.
- J. D. Tibbetts and S. D. Bull, Adv. Sustainable Syst., 2021, 5, 2000292 CrossRef CAS.
- G. Sienel, R. Rieth and K. T. Rowbottom, Epoxides, in Ullmann's Encyclopedia of Industrial Chemistry, 2012, pp. 139–154 Search PubMed.
- R. Reeves and M. Lawrence, Epoxides, Synthesis, Reactions and Uses, Nova Science, 2017 Search PubMed.
- N. Ravasio, F. Zaccheria, M. Guidotti and R. Psaro, Top. Catal., 2004, 27, 157–168 CrossRef CAS.
- E. Breitmaier, Terpenes: Flavours, Fragrances, Pharmaca, Pheromones, John Wiley & Sons, 2006 Search PubMed.
- T. Stößer, C. Li, J. Unruangsri, P. K. Saini, R. J. Sablong, M. A. R. Meier, C. K. Williams and C. Koning, Polym. Chem., 2017, 8, 6099–6105 RSC.
- W. B. Cunningham, J. D. Tibbetts, M. Hutchby, K. A. Maltby, M. G. Davidson, U. Hintermair, P. Plucinski and S. D. Bull, Green Chem., 2020, 22, 513–524 RSC.
- A. Mouret, L. Leclercq, A. Muhlbauer and V. Nardello-Rataj, Green Chem., 2014, 16, 269–278 RSC.
- C. Venturello, E. Alneri and M. Ricci, J. Org. Chem., 1983, 48, 3831–3833 CrossRef CAS.
- A. L. Villa, B. F. Sels, D. E. De Vos and P. A. Jacobs, J. Org. Chem., 1999, 64, 7267–7270 CrossRef.
- K. Sato, M. Aoki, M. Ogawa, T. Hashimoto, D. Panyella and R. Noyori, Bull. Chem. Soc. Jpn., 1997, 70, 905–915 CrossRef CAS.
- K. Sato, M. Aoki, M. Ogawa, T. Hashimoto and R. Noyori, J. Org. Chem., 1996, 61, 8310–8311 CrossRef CAS PubMed.
- Large scale batch epoxidation of limonene requires careful cooling to keep reaction temperatures below 30 °C and slow dropwise addition of H2O2 to prevent potentially explosive thermal runaway reactions from occurring, see: ref. 13 for details.
- J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8849–8869 RSC.
- L. de C. Alves, A. L. Desiderá, K. T. De Oliveira, S. Newton, S. V. Ley and T. J. Brocksom, Org. Biomol. Chem., 2015, 13, 7633–7642 RSC.
- W. Yuan, S. Zhou, Y. Jiang, H. Li and H. Zheng, J. Flow Chem., 2020, 10, 227–234 CrossRef CAS.
- D. D. Plaza, V. Strobel, P. K. K. Heer, A. B. Sellars, S. Hoong, A. J. Clark and A. A. Lapkin, J. Chem. Technol. Biotechnol., 2017, 92, 2254–2266 CrossRef CAS PubMed.
- W. He, Z. Fang, D. Ji, K. Chen, Z. Wan, X. Li, H. Gan, S. Tang, K. Zhang and K. Guo, Org. Process Res. Dev., 2013, 17, 1137–1141 CrossRef CAS.
- F. Mashhadi, A. Habibi and K. Varmira, Ind. Crops Prod., 2018, 113, 324–334 CrossRef CAS.
- C. Wiles, M. J. Hammond and P. Watts, Beilstein J. Org. Chem., 2009, 5, 27 Search PubMed.
- Y. Zhou, W. He, Z. Fang and K. Guo, ChemistrySelect, 2018, 3, 13530–13533 CrossRef CAS.
- S. Ahn, S. L. Nauert, K. E. Hicks, M. A. Ardagh, N. M. Schweitzer, O. K. Farha and J. M. Notestein, ACS Catal., 2020, 10, 2817–2825 CrossRef CAS.
- For previous flow epoxidation reactions of monoterpene substrates under different conditions, see: (a) Epoxidation of α-/β-pinene, camphene and limonene using an N-hydroxyphthalimide catalyst, molecular oxygen and 3 equiv. acetaldehyde in an oscillating disc reactor under Minisci radical conditions, ref: R. Spaccini, L. Liguori, C. Punta and H. R. Bjørsvik, ChemSusChem, 2012, 5, 261–265 CrossRef CAS PubMed; (b) Mono-epoxidation of limonene using a polybenzimidazole/molybdenum catalyst and 4 equiv. of tBuOOH in a flow reactor, ref: K. Ambroziak, R. Mbeleck, B. Saha and D. C. Sherrington, WO2011/012869A2, 2011.
- Different catalyst precursors can be used for VPTC preparation, however varying the tungsten source and the quaternary ammonium counterion did not affect the conversion/selectivity in these flow epoxidations reactions. See: D. C. Duncan, R. C. Chambers, E. Hecht and C. L. Hill, J. Am. Chem. Soc., 1995, 117, 681–691 CrossRef CAS.
- K. Sato, Y. Kon, H. Hachiya, Y. Ono, K. Takumi, N. Sasagawa and Y. Ezaki, Synthesis, 2012, 1672–1678 Search PubMed.
- Previous batch epoxidation catalyst recycling studies revealed that the VPTC could be recycled three times to produce consecutive batches of 3-carene oxide in 75–66% yields, with small losses in catalytic activity occurring between each consecutive epoxidation reaction. 1H and 31P NMR spectroscopic analysis of catalyst recovered after the third recycle indicated the presence of significant amounts of high boiling/oligomeric/polymeric terpene species and losses in POM counter-anion that are likely to be responsible for catalyst deactivation. See ref. 13 and 30.
- For a procedure describing the decagram scale synthesis of the VPTC used in this flow epoxidation study, see ref. 13.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc01734a |
| This journal is © The Royal Society of Chemistry 2021 |