
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultifunctional lignin-based nanocomposites and nanohybrids
Erlantz
Lizundia
 *ab,
Mika H.
Sipponen
c,
Luiz G.
Greca
d,
Mikhail
Balakshin
d,
Blaise L.
Tardy
d,
Orlando J.
Rojas
de and
Debora
Puglia
*f
*ab,
Mika H.
Sipponen
c,
Luiz G.
Greca
d,
Mikhail
Balakshin
d,
Blaise L.
Tardy
d,
Orlando J.
Rojas
de and
Debora
Puglia
*f
aLife Cycle Thinking group, Department of Graphic Design and Engineering Projects, Faculty of Engineering in Bilbao, University of the Basque Country (UPV/EHU), Bilbao 48013, Spain. E-mail: erlantz.liizundia@ehu.eus
bBCMaterials, Basque Center Centre for Materials, Applications and Nanostructures, UPV/EHU Science Park, 48940 Leioa, Spain
cDepartment of Materials and Environmental Chemistry, Stockholm University, Svante Arrhenius väg 16C, SE-106 91 Stockholm, Sweden. E-mail: mika.sipponen@mmk.su.se
dDepartment of Bioproducts and Biosystems, School of Chemical Engineering, Aalto University, P.O. Box 16300, FI-00076 Aalto, Finland. E-mail: luiz.greca@aalto.fi; mikhail.balakshin@aalto.fi; blaise.tardy@aalto.fi; orlando.rojas@aalto.fi
eBioproducts Institute, Department of Chemical and Biological Engineering, Department of Chemistry, and Department of Wood Science, University of British Columbia, 2360 East Mall, Vancouver BC V6T 1Z4, Canada. E-mail: orlando.rojas@ubc.ca
fCivil and Environmental Engineering Department, University of Perugia, Strada di Pentima 4, 05100, Terni, Italy. E-mail: debora.puglia@unipg.it
First published on 20th August 2021
Abstract
Significant progress in lignins valorization and development of high-performance sustainable materials have been achieved in recent years. Reports related to lignin utilization indicate excellent prospects considering green chemistry, chemical engineering, energy, materials and polymer science, physical chemistry, biochemistry, among others. To fully realize such potential, one of the most promising routes involves lignin uses in nanocomposites and nanohybrid assemblies, where synergistic interactions are highly beneficial. This review first discusses the interfacial assembly of lignins with polysaccharides, proteins and other biopolymers, for instance, in the synthesis of nanocomposites. To give a wide perspective, we consider the subject of hybridization with metal and metal oxide nanoparticles, as well as uses as precursor of carbon materials and the assembly with other biobased nanoparticles, for instance to form nanohybrids. We provide cues to understand the fundamental aspects related to lignins, their self-assembly and supramolecular organization, all of which are critical in nanocomposites and nanohybrids. We highlight the possibilities of lignin in the fields of flame retardancy, food packaging, plant protection, electroactive materials, energy storage and health sciences. The most recent outcomes are evaluated given the importance of lignin extraction, within established and emerging biorefineries. We consider the benefit of lignin compared to synthetic counterparts. Bridging the gap between fundamental and application-driven research, this account offers critical insights as far as the potential of lignin as one of the frontrunners in the uptake of bioeconomy concepts and its application in value-added products.
 Erlantz Lizundia | Erlantz Lizundia is Associate Professor at the Faculty of Engineering in Bilbao, University of the Basque Country (UPV/EHU). He received his PhD in Advanced Materials Engineering in 2011 at the UPV/EHU. He joined the University of the British Columbia (Canada) and ETH Zurich (Switzerland) in 2016 and 2018 as a visiting scientist. His work aims to develop renewable-material based multifunctional nanohybrids for energy storage, catalytic, optical and environmental remediation applications using biomimetic, bioeconomy and circular economy approaches. Erlantz also works on the ecodesign of novel materials and technologies such as recycling and energy storage through life cycle assessment (LCA). |
 Mika H. Sipponen | Mika H. Sipponen is a tenure track Assistant Professor at the Department of Materials and Environmental Chemistry in Stockholm University. He leads the Sustainable Materials Chemistry (SUSMATCHEM) research group focusing on development of lignin-based functional materials. Sipponen obtained his M.Sc. (Tech.) and D.Sc. (Tech.) degrees from Aalto University (Finland) in 2010 and 2015 under supervision of Prof. Simo Laakso. In 2016, he worked as Research scientist at VTT (Finland) prior to a three-year post as Academy of Finland postdoctoral researcher. His current research programme builds on the experience gained from industrially aligned research on applied biochemistry and biomass processing. |
 Luiz G. Greca | Luiz G. Greca is a PhD candidate under the supervision of Prof. Orlando Rojas at the Department of Bioproducts and Biosystems at Aalto University, Finland. He received his Mechanical Engineering degree from the Pontifical Catholic University of Paraná (Brazil) in 2016. His research interests include mechanical characterization of biobased materials, and the use of wetting and interfacial interactions for controlling the assembly of biobased colloids. |
 Mikhail Balakshin | Mikhail Balakshin is a Professor of Practice in Lignin Chemistry at Aalto University. He graduated from Leningrad Forest Technical Academy (Russia) with an Engineering Degree in Pulp and Paper Industry and a PhD in Wood Chemistry. His major academic achievements are in development of state-of-the-art analytical methods in lignin characterization and their implementation in fundamentals of various lignocellulosic conversion processes as well as elucidation of the correlation between the structure of lignin with processing parameters and structure-performance relationships in lignin applications. He also held senior R&D positions at biorefinery companies in Russia, Canada and USA focusing on process development and industrial applications of lignins and other biomass components. |
 Blaise L. Tardy | Blaise L. Tardy is currently a research fellow at Aalto University. He obtained his M.Sc. in bioengineering from the Swiss Federal Institute of Science and Technology (EPFL, Switzerland (2009)), and his Ph.D. degree in chemical and biomolecular engineering from The University of Melbourne, Australia (2015). His current research interests include the formation of structured materials from bio-based nano and microparticles as well as the association between supramolecular interactions and the properties of bioproducts. |
 Orlando J. Rojas | Orlando J. Rojas is a professor and Canada Excellence Research Chair at the University of British Columbia, with joint appointments with the Departments of Chemical and Biological Engineering, Wood Science and Chemistry. He is adjunct professor in the Departments of Bioproducts and Biosystems of Aalto University and Chemical and Biomolecular Engineering of North Carolina State University. Dr Rojas is the recipient of the 2018 Anselme Payen Award and is elected Fellow of the American Chemical Society and the Finnish Academy of Science and Letters. Dr Rojas is Associated Editor of ACS journal Biomacromolecules and he is the scientific director of UBC's Bioproducts Institute and research PI of the Finnish Materials Bioeconomy Flagship (FinnCERES), making part of the Boreal Alliance initiative. |
 Debora Puglia | Debora Puglia is Associate Professor at the Civil and Environmental Engineering Department, University of Perugia, where she received her PhD in Industrial Engineering in 2003. Her main current research activities are related to the extraction of lignocellulosic nanostructures from waste (agricultural and food sources) and their use in composite and nanocomposite biobased materials for food packaging, biomedical and plant protection applications. She also works on the combined use of thermosets and carbon based nanostructures. |
1. Introduction
In the last ten years the subject of “lignin” has re-surfaced in the fora of renewable polymers and associated materials. A reason for such interest relates to the impending availability of lignins from the next generation bioproduct mills or biorefineries, whose economic viability will hinge on finding new uses for this abundant biomacromolecule.1,2 Lignins are in fact one of the most important streams in the forest industry.3,4 Not surprisingly, the most recent publications related to lignin utilization include widespread opportunities going from chemical engineering, energy and fuels, green sustainable chemistry and technology, polymer science, physical chemistry, biochemistry and materials science.5 In such diverse fields, consideration has been given to lignin depolymerization,6e.g., to produce platform chemicals in large volumes.7,8 The opposite extreme, equally relevant, proposes the uses of lignin as a colloidal material or in the form of a polymeric matrix that can lead to lignin-based functional materials.9 In both cases, the so-called lignin-centered biomass fractionation and transformation or modification, using green chemistry routes or otherwise, are central aspects under scrutiny. Recent investments to pulp mills signal lignin extraction in addition to its utilization as a renewable option to energy generation. Such efforts are aimed at reducing the dependency on fossil carbon feedstocks and to fully utilize the plant-based resources, for example, in products relevant to the resin and plastic industries. Many applications such as biomedical and nutritional materials under development strive to take advantage of lignins’ antioxidant activity,10–13 or its high carbon content attractive to carbonaceous materials in the form of fibers and electroactive components.14Owing to its amphiphilic structure, lignins have been shown to be surface-active and are able to improve the strength at interfaces, for example, in adhesives.4 In these and other applications, however, there is the challenge of substituting molecules that are in current use, in stablished processing units, which should be adapted or retooled. In addition, a major roadblock is the chemical and structural variability of technical lignins, depending on the plant source, the nature of the process used in their extraction and other variables.15–17 Lignin complexity, variability and its elusive chemical structure, which remains a subject of discussion, compounds such challenges. On the other hand, the expected availability and low cost of lignin represent an incentive, together with the fact that lignin's property profiles can be tailored by controlling the variables listed before, paradoxically the same that have delayed lignin's wider adoption.
The complex phenolic makeup of lignin is quite challenging, if not impossible to achieve synthetically and one has to wonder why such complexity has not been frequently exploited in polymer architecture and functional groups. Along with its high density in hydroxyl and carboxyl substituents, there are plenty of reasons to consider when taking lignin as a precursor and as a reactive scaffold that can unleash development in the next generation materials. This consideration is particularly timely under the emerging concept of the circular bioeconomy. Lignin's own features are further expanded if one considers the bonding patterns and linkages with carbohydrates, for instance, in lignin-carbohydrate complexes,18,19 which are finding traction in exploiting the advantage that each component bring in given applications.
Lignin has attracted and increasing attention from both fundamental and applied fields as it is available in large quantities from renewable sources and can be potentially applied in many fields thanks to its special physico-chemical properties. In this review, as schematically summarized in Fig. 1, we focus on the fundamentals and prospective application of lignin as an added value material. As requirements and mechanistic of lignin-based materials significantly vary, the discussion is structured by lignin fundamentals, lignin-nanocomposites and lignin-nanohybrids, to finalize with the most common applications of those materials. Besides its presentation in the so-called black liquors or in solution, lignin solids are produced upon drying, as amorphous powder. Moreover, recent efforts have been applied in the production of lignin in other forms, such as spherical particles, responding to the possibility of capitalizing the interrelation that is known to exist between shape and function. Together with the exquisite opportunities expected from lignin, as a polyphenolic molecule, one should add its self-assembly and supramolecular organization, alone or in combination with polysaccharides, for example in composites. These aspects are discussed in this review in the section “Lignin fundamentals”. In the next section, “Lignin-nanocomposite approach”, we consider lignin synergies with polysaccharides, proteins, other biopolymers and synthetic polymers in the form of the most traditional types of lignin-based materials, where lignin is applied as a reinforcing phase to develop (nano)composites. We also consider the recent works highlighting the many new opportunities arising from the formation of hybrid-materials involving lignin interactions at the nano- to micro-scale with a secondary organic or inorganic component at the nanometer or molecular level. Accordingly, in the section “Lignin-nanohybrid approach” we show how lignin is used as a renewable matrix to host either inorganic or organic nanoparticles, are addressed. Due to the sheer volume of work in this area, lignin-nanohybrids are here grouped into those bearing metal oxides, zero-valent metals, carbonaceous nanoparticles and biobased nanoparticles. Hereafter, the more imminent realm of applications is highlighted, covering the reduced flammability of lignin-based materials, food packaging applications, plant and crop protection ability, energy storage, and health. Finally, the full utilization of lignin, in these and other applications, is justified considering the environmental benefits that can be gained, as shown by gains in net carbon storage and in the reduction of greenhouse gases. This specially applies when lignin fractionation and associated bioproduct developments are integrated under the biorefinery concept, following the purpose of fully utilizing the component of plants that are sustainably harvested to fulfill most of our current and future material needs.
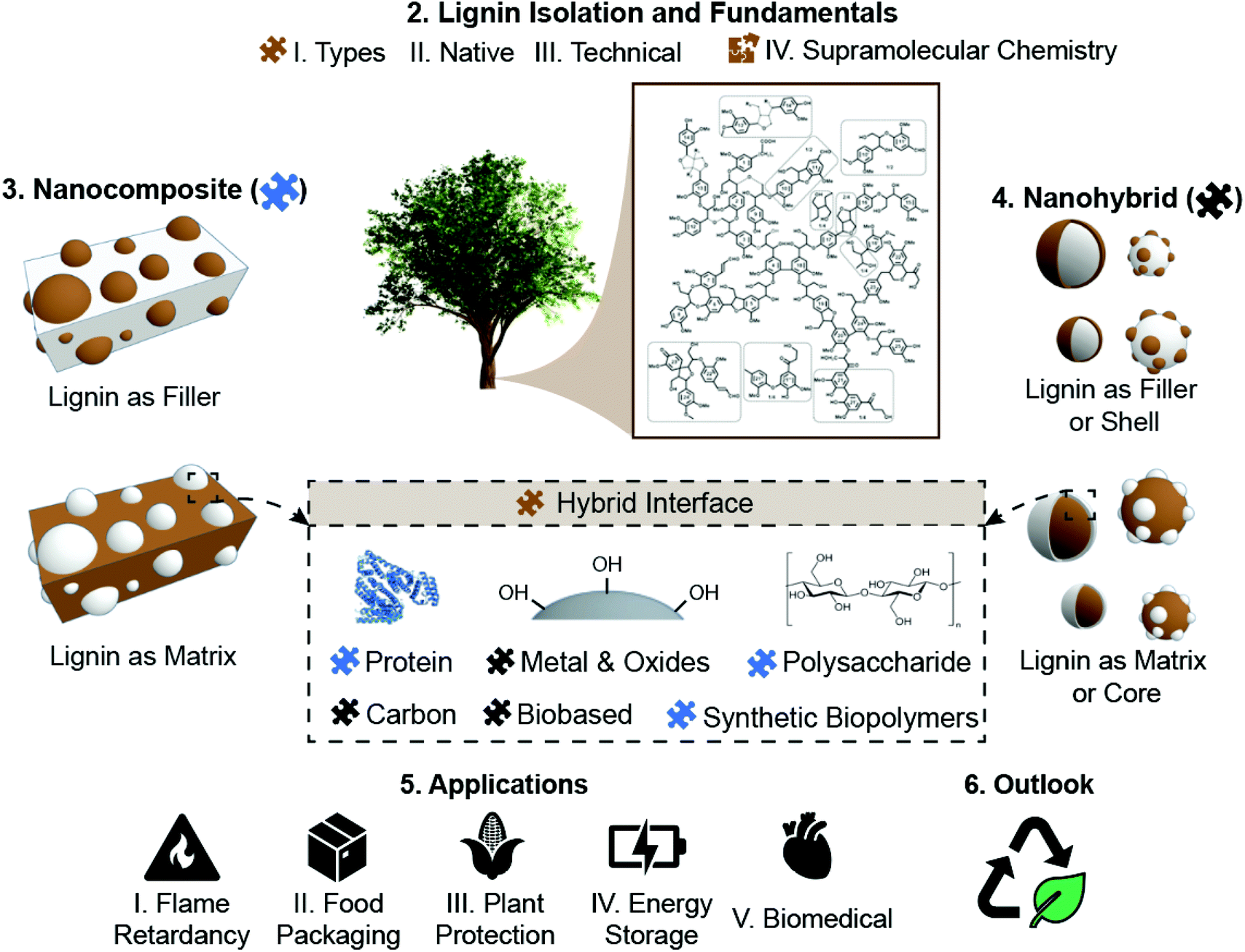 | ||
| Fig. 1 Schematic representation of the valorization of lignin in the form of nanocomposites and nanohybrids, as well as its associated production and fundamental considerations and applications. The main sections of the Review are summarized, with specific subsections for the main chapters. | ||
During the recent years, there have been many reviews on lignin nanoparticles and their applications,9,20–28,29 chemical and microbial transformation of lignin to chemicals and polymer precursors,30,31 and chemical functionalization and processing of lignin into various materials (not in the nano-scale).2,32–40 Though many of the prior reviews discuss formulation of lignin-based polymers and composite systems,41–49 the literature is lacking a review on nanocomposites and hybrid materials based on lignin. These two classes of materials are quickly gaining momentum. The present work is a comprehensive review of lignin building on the recent progress that links (1) progresses in fundamental understanding of the structure and properties of technical lignins as a function of their isolation and modifications and (2) their assembly to nano- and miscroscaled building blocks with (3) the formation of lignin-based composites, inclusive of hybrid nano- and microparticles. We thereafter thoroughly review recent progresses in lignin-based nanocomposites and nanohybrids when lignin is compounded with the main classes of inorganic and organic materials.
2. Lignin fundamentals
2.1. Lignin types
Understanding the molecular structures of lignins is of primary importance for fundamental reasons, and to access lignin reactivity and valorization. Specifically to lignin particles, it has been shown that lignin structure and other characteristics affect LMNP properties.50 Thus, it is critical to discuss variations in different lignin types, from native ones to those obtained by commercial-scale processes as well as new emerging technologies (lab or pilot scale).Lignin is an irregular aromatic polymer consisting of p-hydroxyphenyl (H), guaiacyl (G) and syringyl (S) type phenylpropane (or C9) units, correspondingly (Fig. 2a).15 Softwood (SW) lignins are mainly G-type with small amounts of H-units, while hardwood (HW) lignins have different proportions of S- and G-units (S/G ratio) (Table 1). Non-wood lignins are often called of HGS-type. These monomeric units are linked in lignin macromolecule by various ether and C–C bonds (Fig. 2b, 3 and Table 1).15,16
 | ||
| Fig. 2 (a) Main monomeric lignin blocks of p-hydroxyphenyl (H), guaiacyl (G) and syringyl (S) type.; (b) main structures in native and technical lignins (by a semiquantitative HSQC NMR method). Numbers indicate the amounts of specific units per 100 Ar (i.e., mol%).2 | ||
| Moieties/range | Alcell | Indulin | AKL | SEPL | Sucrolin | SBL | AMWL | PMWL |
|---|---|---|---|---|---|---|---|---|
| a Corrected for sugar content. b The number of C9-units involved in resinol structures; as the structure is symmetric, the number of resinol structures is ½ of the C9-units involved. Abbreviations: AKL – aspen Kraft lignin, SEPL – steam explosion poplar lignin, SBL – soda bagasse lignin, AMWL – aspen MWL, PMWL – pine MWL59 | ||||||||
| Total CO | 29 | 15 | 21 | 23 | 30 | 19 | 16 | 20 |
| Non-conj. CO | 15 | 7 | 11 | 11 | 17 | 7 | 3 | 3 |
| Conj. CO | 14 | 8 | 10 | 12 | 13 | 11 | 13 | 17 |
| Total COOR | 21 | 17 | 28 | 22 | 38 | 37 | 13 | 6 |
| Non-conj. COOR | 17 | 15 | 25 | 18 | 27 | 27 | 8 | 4 |
| Conj. COOR | 4 | 2 | 3 | 4 | 11 | 10 | 5 | 2 |
| Total OH | 103 | 115 | 107 | 124 | 92 | 96 | 156 | 140 |
| Aliph. OH | 33 | 49 | 31 | 61 | 43 | 37 | 134 | 107 |
| OHpr | 19 | 31 | 17 | 33 | 19 | 17 | 72 | 67 |
| OHsec | 14 | 18 | 14 | 28 | 24 | 20 | 62 | 40 |
| Phenolic OH | 70 | 66 | 76 | 63 | 49 | 59 | 22 | 33 |
S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :G-ratio :G-ratio |
1.18 | NA | 1.31 | 1.63 | 0.53 | 0.51 | 2.11 | NA |
| OMe | 103 | 81 | 120 | 126 | 81 | 92 | 164 | 97 |
| ArH | 202 | 234 | 199 | 201 | 207 | 213 | 221 | 253 |
| DC, % | 44 | 66 | 44 | 37 | 58 | 53 | 11 | 43 |
| β-5 | 3 | 4 | 2 | 2 | 1 | 1 | 2 | 10 |
| β-βb | 3 | 4 | 5 | 4 | 2 | 1 | 8 | 4 |
| β-O-4 | 7 | 7 | 1 | 17 | 4 | 2 | 52 | 42 |
| Oxygen. Aliph. | 82 | 93 | 93 | 128 | 64 | 52 | 237 | 214 |
| Saturated Aliph. | 149 | 109 | 145 | 116 | 161 | 140 | 56 | 32 |
| Side chain length | 281 | 233 | 269a | 289 | 293 | 248 | 322 | 272 |
| Alkyl ethers | 50 | 44 | 54a | 68 | 21 | 15 | 103 | 107 |
| Sugars | <1 | ∼1 | 4 | <1 | <1 | ∼1 | <1 | ∼1 |
| MAr | 178 | 173 | 201 | 194 | 203 | 195 | 218 | 180 |
2.2. Native lignins
The main linkages in native lignins are β-O-4 linkages, followed by 5-5, β-5 (SW), β-β (HW) etc. (Fig. 2b, 3 and Table 1).51–53 Milled Wood Lignin (MWL) and cellulolytic enzyme lignin (CEL) preparations are most commonly used to access the structure of native lignins (Table 1) when the analysis requires a soluble lignin preparation (e.g., for various high-resolution NMR methods).54 However, one should consider specific differences between in situ native lignin (and isolated lignin preparations.55,56Despite the lack of direct evidence, lignin is usually considered as a three-dimensional, cross-linked macromolecule.57 However, this classical vision has been challenged; spruce MWL was proposed to be linear.58 Related propositions of rather linear lignins have been forwarded in a recent review by Ralph et al., who assumed models of native lignins, based on the knowledge available on lignification theory.52 However, the data on the absolute molecular mass of spruce milled wood lignin (MWL) along with the quantification of terminal groups, clearly indicate that MWL is heavily branched and cross-linked (with ∼36% of lignin units partaking in these linkages) (Fig. 3).53 The very branched/crosslinked lignin character will likely remain and be even intensified after biomass processing, which result in so-called technical lignins.
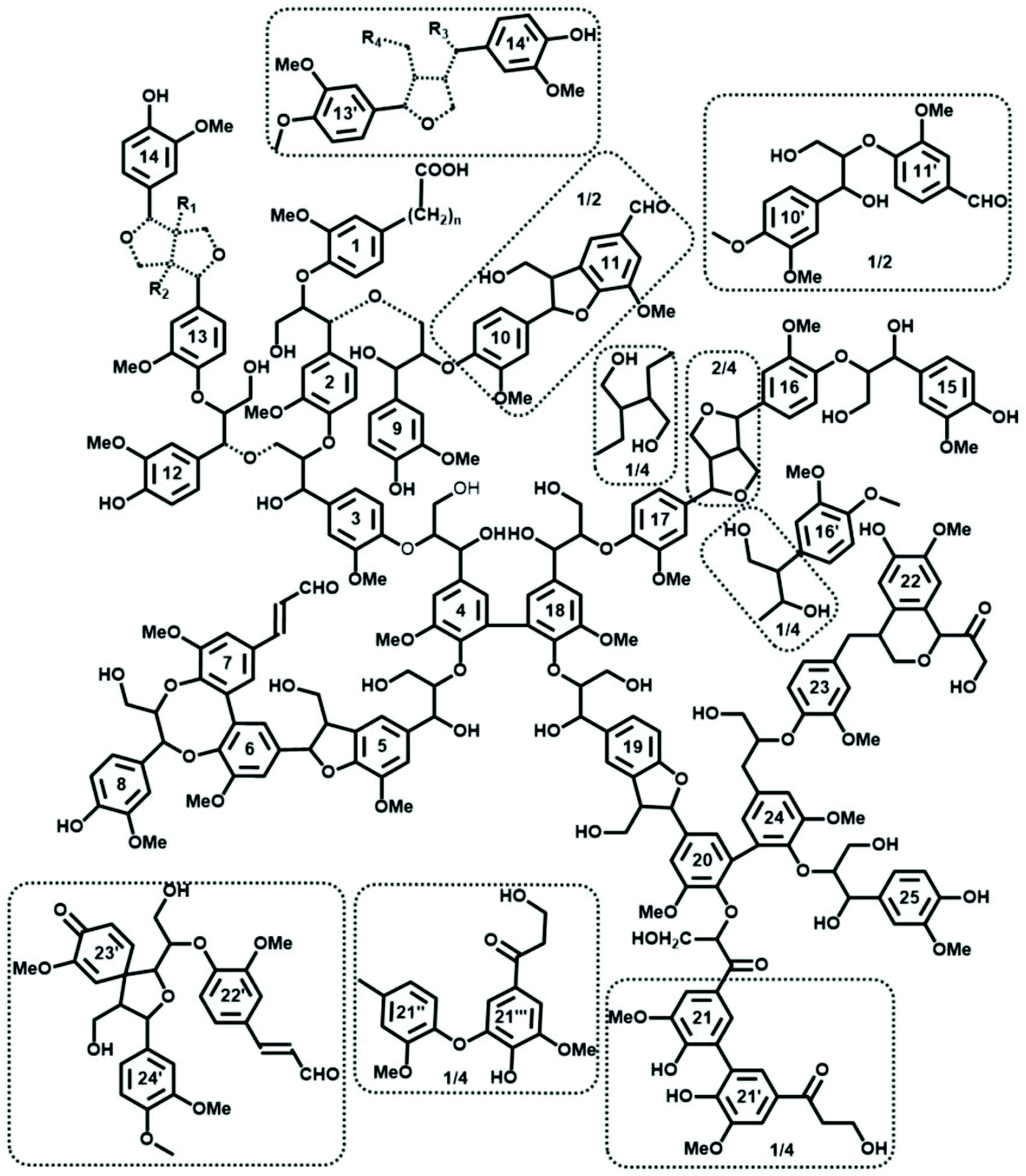 | ||
| Fig. 3 Tentative quantitative structural model of spruce MWL, in agreement with currently available structural data on lignin moieties and their occurrence frequencies. Reproduced with permission from ref. 53. Dashed bond: different structure agreeing with available information; the exact structure of the Alk–O–Alk moieties (units 2–9, 3–12, 13–14) is not well defined, therefore the model indicates a general type of units rather than insisting on a precise structure. Minor structures (below 4/100 Ar) are given in the dotted boxes. | ||
2.3. Technical lignins
Most of the lignins available for practical and commercially relevant applications are technical lignins, obtained after chemical processing of biomass. Technical lignins differ dramatically from the corresponding native ones as a result of a combination of multiple reactions, including catalyzed biomass hydrolysis, condensation of lignin fragments, decrease in native lignin functional groups, formation of new functional groups, and others. The technical lignins are appreciably more heterogeneous in terms of chemical structure and molecular mass than the native lignins, possessing a high variety of structural moieties present in rather small amounts (Fig. 2b and Table 1).60,61Technical lignins can be classified from different points of view (Table 2). From a practical point of view, there are technical lignins that originated from industrially available pulp and paper processes. These are kraft and soda lignins (from kraft and soda pulping, respectively) and lignosulfonates (sulfite pulping). On the other hand, there is a big group of technical lignins from various emerging biorefining processes, such as those obtained from the different variations of acid hydrolysis (AH), steam explosion (SE), and organosolv (OS) pretreatment.
| Lignin type | Scale | Chemistry | S-content | Purity |
|---|---|---|---|---|
| Kraft | Industrial | Alkaline | Moderate | Moderate |
| Soda | Industrial | Alkaline | Free | Moderate-low |
| Lignosulphonate | Industrial | Acid | High | Low |
| Organosolv | Pilot/demo | Acid | Free | High |
| Hydrolysis | Industrial/pilot | Acid | Low-free | Moderate-low |
| SE | Pilot | Acid | Low-free | Moderate-low |
| AFEX | Pilot | Alkaline | Free | Moderate-low |
In terms of process chemistry and, correspondingly, the lignin chemical structure, lignins can be derived from acidic or alkaline-based processes. In addition to the process nature, the origin of the feedstock has naturally an important impact on the structure of technical lignins. Another factor to classify technical lignins, especially in view of their application, is the presence or absence of sulfur in their structure (see Table 2).
All pulping and biorefinery processes result in significant lignin degradation. These reactions are always accompanied with a decrease in oxygenated aliphatic moieties in general, specifically β-O-4 units and aliphatic OH (primary and especially secondary ones), and an increase in phenolic OH, COOH, saturated aliphatic moieties, degree of condensation (DC) when compared to native lignins (Table 1).59 However, there are significant structural differences between various technical lignins. The degree of lignin degradation is dependent on the process utilized and its severity. Among ten different pulping and biorefinery lignins investigated, SE lignins are the least degraded while aspen kraft lignin (AKL) underwent the most severe degradation. A specific characteristic of the acid-derived lignins (organosolv ones and Sucrolin isolated from acid hydrolysis of bagasse) was high amounts of CO groups. Also, a typical feature of the ethanol-based organosolv lignins is the presence of EtO-groups.62
It is worthy to address rather popular recent claims on production of “native-like” lignins in different biorefinery processes. This conclusion came from the methodology based on relative proportion of β-O-4 units vs. other native lignin units remaining in technical lignins, detected in 2D spectra when a few major native lignin units (see Fig. 2b, first row) were assumed as 100%. Very minor lignin transformation during a process is concluded from still predominant relative amount of β-O-4 structures in this normalized approach. However, absolute quantification (per monomeric lignin unit or/and in mmol g−1 lignin) often provides a completely different picture; although β-O-4 are still major lignin units, their amounts decrease dramatically, in one order of magnitude (Fig. 2b and Table 1). This was recently confirmed with an appropriate 2D NMR methodology. Although the authors claimed a “new concept” for biorefinery lignin structure,63 extensive degradation of lignin during different chemical processes is a well-known fact consistently reported for many decades using various analytical methods.17
Certain lignin modifications, for instance alkylation and acylation, are able to upgrade lignin towards important applications, such as thermoplastic blends, carbon fibers, surfactants, and sorbents.3,4,67–70 Usually, a degree of substitution (DS) of 100% is targeted, meaning that all available hydroxyl groups are functionalized. However, such complete lignin modifications typically require expensive or/and toxic chemicals and therefore they may not be easily viable on an industrial scale. In contrast, a simpler process for partial lignin derivatization (alkylation or acylation) has been proposed recently.71,72 Although only partial derivatization occurs, it can be sufficient for achieving the targeted lignin performance. Other characteristics of lignin, such as molecular weight, glass transition temperature (Tg), and molecular structure, can be tuned by the process variables. The combination of lignin modification and extraction in a very simple approach (reactive extraction) that uses inexpensive chemicals allows for a process of significantly lower costs compared to other known processes for functionalized lignins.
An approach to improve lignin reactivity in various applications via its depolymerization is very popular. In spite of great technical progress in the area, significant challenges are still remained. Even more importantly, this direction looks very problematic from the economic point of view due to low yields of the target products resulting in low revenue for the lignin feedstock used.2 In contrast, the use of spherical lignin particles (LPs) in different high-value applications can overcome low lignin reactivity without complex and expensive lignin depolymerization and thus looks as a more reasonable alternative.9
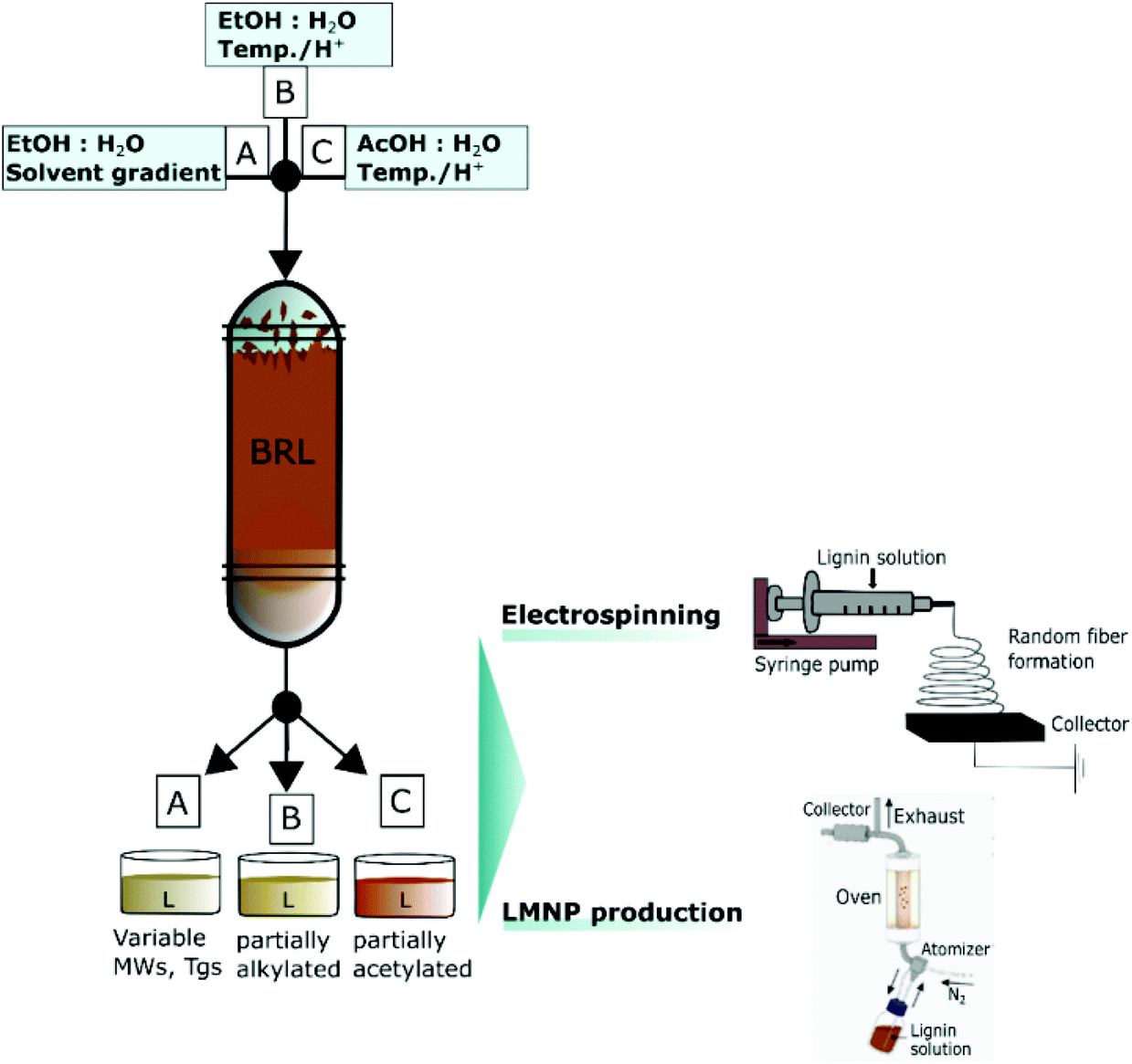 | ||
| Fig. 4 One-pot purification, fractionation and functionalization of crude biorefinery lignins (BRL) and direct use of these solutions for LMNP production or nanofiber production by electrospinning. Reproduced with permission from ref. 2. | ||
2.4. Self-assembly/supramolecular-assembly
Contrary to conventional synthetic polymers, lignins are extremely diverse in terms of functional groups, chemical bonds, and overall architecture as associated with their yet undefined branched structure. While this limits their fine-tuned engineerability and bulk processability, due to difficulties in predicting their behavior or the properties of their constructs, a vast variety of non-covalent interactions can be achieved with lignins. These will significantly affect the properties of materials they form. Thereafter, supramolecular interactions will occur between lignin macromolecules, with other polymers, particles, and at interfaces with varying surface chemistry (i.e. type of functional groups and their surface topology). Herein, we introduce the various functional groups of lignins relevant to their interactions in material formation. We also discuss findings on how the preparation of nanoparticles can affect the functional groups displayed and their size and compaction. The latter are key parameters in the formation of composites, and nanobiohybrids, and the vast majority of the studies put forward in sections 3 to 5 are exploiting these non-covalent interactions to form materials as detailed across the review.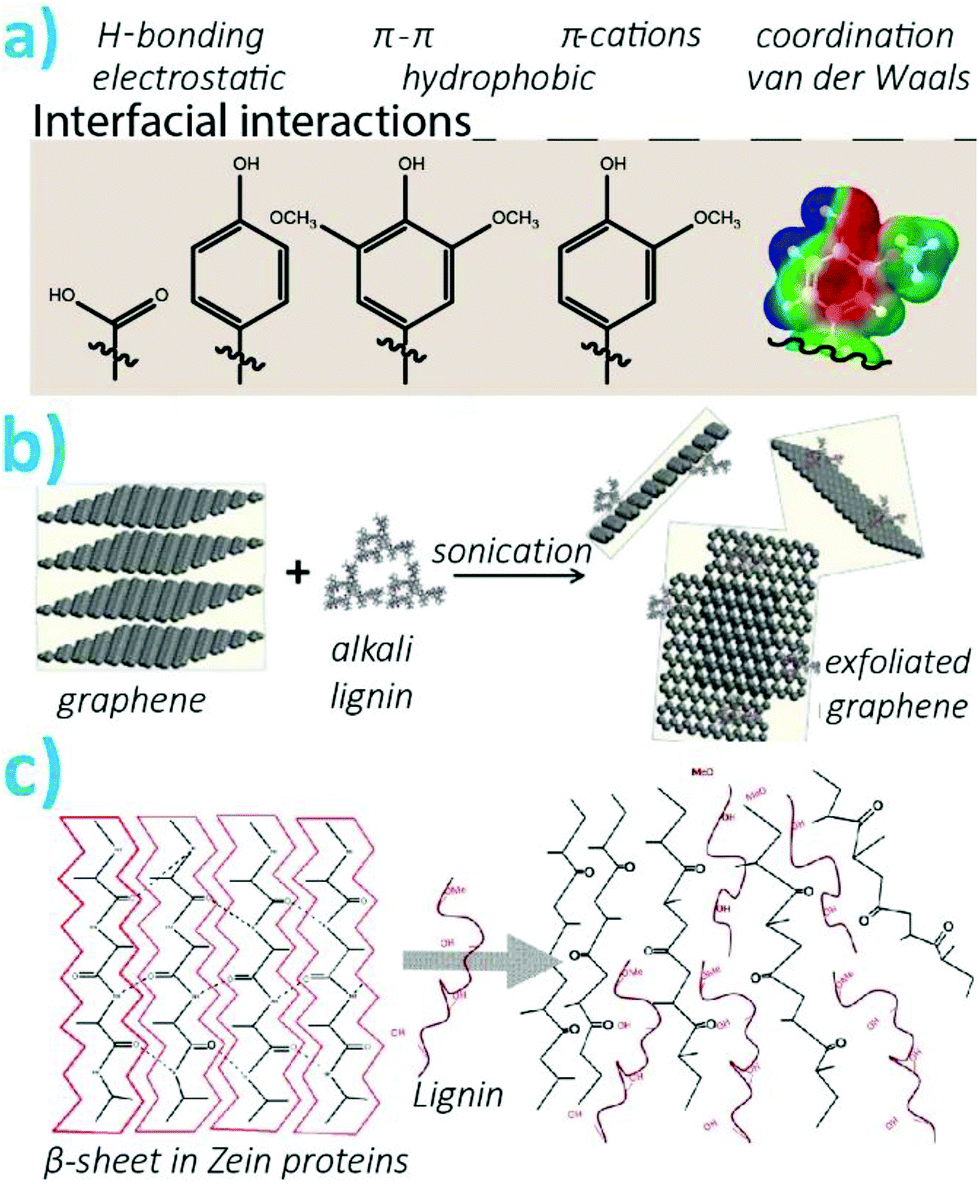 | ||
| Fig. 5 (a) Interfacial supramolecular interactions as driven by the functional groups present on lignins. On the right-hand side, the electrostatic potential of a guaiacyl group is shown, illustrating the complex potential interactions offered by lignins (blue = positive, red = negative). Reproduced with permission from ref. 21 (b) Schematic illustration of graphene exfoliation as promoted by alkali lignin. Reproduced with permission from ref. 89 (c) Schematic illustration of the disruption of the quaternary structure of proteins by lignin. Reproduced with permission from ref. 91. | ||
The supramolecular interactions between lignins and other polymers or interfaces can be optimized by judicious choice of the type of lignin, their modification, or the approach chosen to synthesize the particles. Such supramolecular complexation will play an important role on the cohesion of the composites they form. For instance, when interfacing with polylactic acid (PLA), H-bonding and hydrophobic interactions with lignins will determine the uniform response of the composite and hybrid materials under stress.87 Furthermore, toughening mechanisms from the dynamics of supramolecular interactions can result in extreme enhancement of mechanical properties. This was demonstrated with the combination of lignosulfonates and polyvinyl alcohol (PVA) reaching extreme toughness to strain ratios.88 Substituted aromatics of lignins are auspicious to π–π interactions between carbonaceous materials and lignins. This was demonstrated for the exfoliation and stabilization of graphenes nanosheets (Fig. 5b).89 In another effort, introduction of lignins to compatibilized carbon additives within a polypropylene matrix improved the interfacial shear by up to two-fold.90 As most polyphenols, lignins can interact strongly with proteins through non-specific interactions. Lignins were shown to denaturate quaternary motifs in proteins, specifically, the lignins intercalated within beta-sheets and alpha-helices in zein proteins (Fig. 5c).91
When considering oxide surfaces, however, lignin has a rather high specificity towards metal oxides. For instance, the adhesion strength of alkali lignins (purified from pulping black liquor) with metallic oxides, such as alumina or manganese oxide, was up to three folds higher than that with non-metallic oxides such as silica.92 This was attributed to the π–cation interaction overcoming electrostatic repulsion. Other hydroxyl-surfaces such as cellulose or chitosan were also shown to have a strong interaction with lignins, although these are considered to be the result of a specific interaction configuration involving multiple binding sites oriented in three dimensions. Such supramolecular interaction was exploited to form cellulose–lignin hydrogels used for fractionation of lignins based on specific interactions.93 Furthermore, there are many examples of lignin-silica hybrid materials and lignin-capped metal oxide nanoparticles such as magnetite, as recently reviewed.79 One recent example is the use of lignin to coat silica particles that greatly increased their adsorption capacity towards cobalt(II) ions from water.94 The adsorption capacity of 18 mg Co(II) per gram of dry adsorbent at room temperature as three times as high as that of the lignin or silica starting materials together.
In the supramolecular interaction of lignins with other interfaces, the functional group exposed and their configuration will be of key importance. This will completely determine the development of the interphase in nanocomposites but also the mechanical properties of the composite as associated with the adhesive properties at the hybrid interfaces. For lignin films formed with a root mean-square roughness below 1.5 nm contact angles of 46, 52.5, and 55.5° were measured for softwood Kraft, and hardwood milled wood lignins, respectively. These values suggest an intermediate wettability, however, they are only indicative of the groups self-assembled at the interface, which can be influenced by residual contaminants.103 Thereafter, the surface decoration of lignins with functional groups is related to the assembly strategy, as associated with the amphiphilic nature of lignins. For instance, with the hydrophilic group of lignins being oriented towards the outside of the surface for precipitation-driven strategies in polar solvents,104 and with the hydrophilic groups of lignins being oriented towards the inside of the material for aerosol-formed particles, as a result of the air–water interface.9 However, even in the case of the aerosol-formed particles an anionic zeta potential in the range of −30 mV is reported, which suggest charge migration upon exposure to solvent.95 The latter is likely to occur as a result of the small molecular weight of lignins, for instance compared with that of polystyrene in latex particles. This phenomenon is likely to be true for most lignin particles preparation as a zeta potential between −40 mV and −60 mV were reported. In fact, low molecular weight lignin molecules enriched in carboxylic groups form the outer surface of lignin nanoparticles.13 This observation is true even for particles obtained from separated fractions of the same lignins.105 For most particles, the zeta potential will have a principal sigmoidal inclination around pH 4 indicating that the carboxylate groups are responsible for their colloidal stabilization. Interestingly, a secondary inflection is observed above pH 11, corresponding to desorption of the more hydrophilic chains from the particles, resulting to an overall decrease in surface charges.96 The latter is combined with an increase in solubility as the pH increase beyond the pKa of the phenolic acids. When hydrolysates obtained by solvo-thermal processing of corncob, corncob residue, vinasse, pubescens, pine, and birth were used to form lignin particles by the solvent-shifting method. Only corncob residue and pine could form stable spherical nano/microparticles.106 This was attributed to the predominant presence of unoxidized Cα, the abundance of guaiacyl units, and the amphiphilic, polydisperse, nature of the macromolecules. These were shown to have a significantly higher impact than the average molecular weight of the lignins.
Self-assembly kinetics of lignins into nano- and microparticles significantly impact the surface chemistry of the constructs.107–110 The assembly conditions have been found to significantly affect the homogeneity of the particles distribution, their charges, and their sizes. The two approaches which have been evaluated the most for particle preparation are acid neutralization, or pH-shifting, and solvent-shifting. In the first case, smaller particle sizes were obtained, as small as 21 nm and typically below 150 nm in diameter. The size was also found to decrease when fractionating Kraft lignins, with fractions with a higher MW leading to smaller particles, as obtained from pH-shifting (Fig. 6a).111 A similar trend, albeit with larger sizes was observed with a similar fractionation process but with organosolv lignins.105 However, the zeta potential (ca. −50 mV) and the polydispersity index (PDI) below 0.1 was measured to be nearly identical for particles obtained from each fractions. Increasing the rate of acidification of lignins led to larger particles while increasing the concentration above the overlap concentration led to a reduction in particle size.84 Noteworthy, with stronger acids, the larger non-spherical particles are formed from smaller aggregates.112 The same trend was noted across a range of lignins including kraft lignins, alkali lignins, enzymatic hydrolysis lignins and pretreated (hot acid extraction) poplar lignins, when comparing the final pH of precipitation. Lower pH values lead to larger particles. Larger sizes of particles obtained at a pH <4 were observed for Kraft lignins compared with the three others lignins evaluated, while a smaller size was obtained for the particles formed at a final pH of 9.2 for Kraft lignins (Fig. 6b). This suggests that a smaller subset of lignins may be included in the formation of particles at a higher pHs.113 Interestingly, pH-shifting (decrease) in conjugation with solvent-shifting led to a decrease in size with increased acidity.114 The latter seminal work induced a wide range of studies on the effect of pH-shifting on particles formation.23 Importantly, these particles present the optimal properties as filler for nanocomposites as they are expected to present the most compact morphology and the smallest sizes, hence maximizing the available interphase to modulate the mechanical properties of the mixed systems.
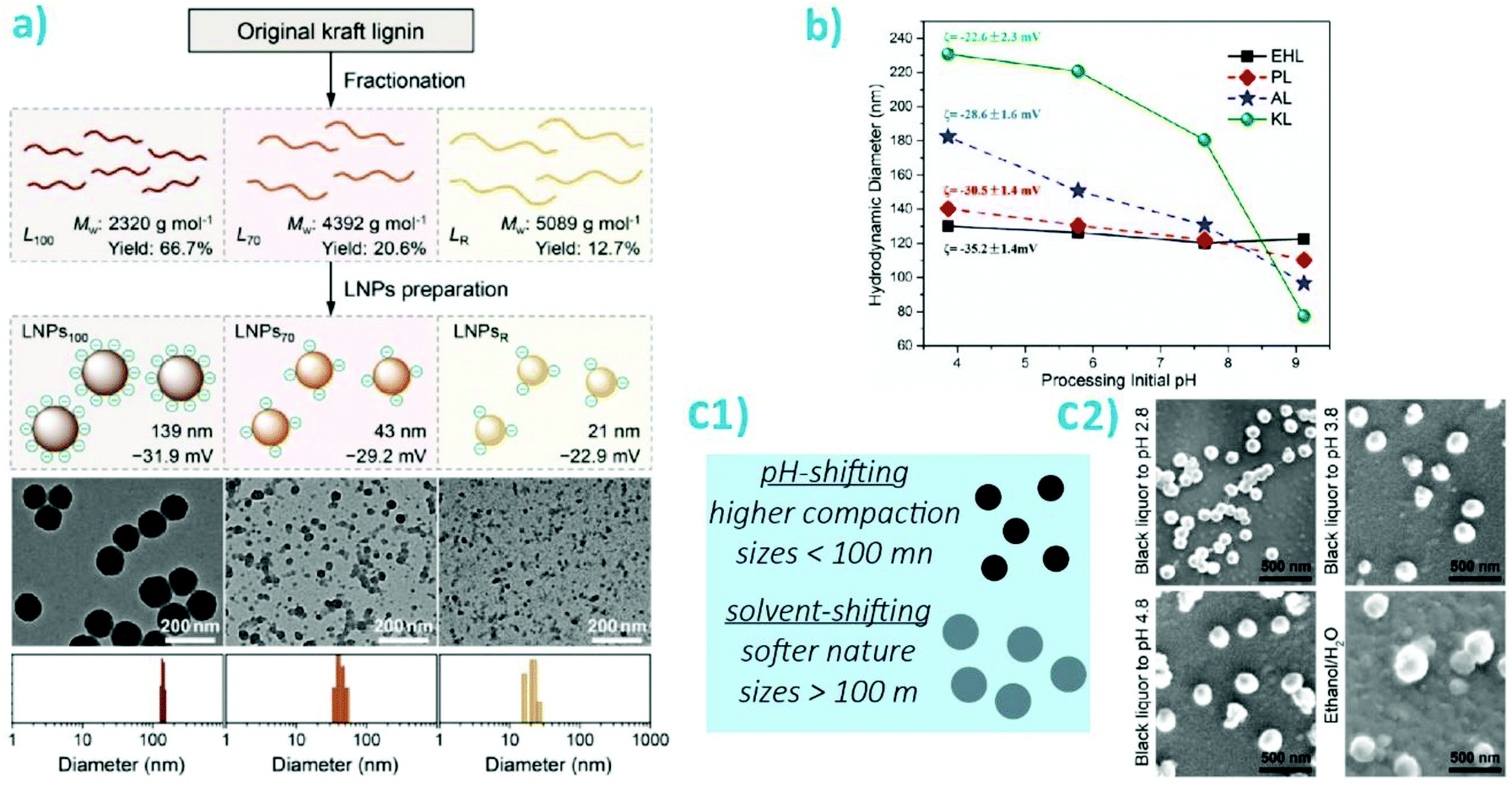 | ||
| Fig. 6 Illustration of the physico-chemical properties of lignin nanoparticles as a function of preparation parameter. (a) Fractionation followed by pH-shifting (acid precipitation) and resulting nanoparticles as represented schematically and via transmission electron microscopy images. Reproduced with permission from ref. 111. LNP correspond to lignin nanoparticles; (b) illustration of diameter and zeta potential of particles obtained by acid precipitation of enzymatic hydrolysis lignins (EHL), spent liquor lignins (PL), Kraft lignins alkalized (sodium, AL), or Kraft lignins (KL) as a function of pH in the precipitation bath. Reproduced with permission from ref. 113; (c1) depiction of main differences between pH-shifting and solvent-shifting preparations; (c2) Representative scanning electron microscopy images of particles obtained from acidification of black liquor or solvent-shifting. Reproduced with permission from ref. 116. | ||
In contrast with pH-shifting, the particles obtained by using solvent-shifting, where dissolved lignins are colloidally precipitated by reducing the volume fraction of the “good” solvent, are typically significantly larger, often 100–300 nm in radius.13,115 These results were independent on the lignin source. These particles generally present a less compact structure, as indicated by the outward diffusion of molecules and macromolecules from their core.13,115 In a different effort, fractionation of Kraft lignins led to the same trend in terms of particle size, although the particles were obtained by solvent-shifting.111 Solvent shifting led to a lower zeta potential for the smaller particles (from larger Mw fractions), suggesting that fractionation can effectively increase the hydrophobicity of the particles given the adequate particle formation mechanism (Fig. 6c). This trend was further confirmed in a study that compared pH-shifting with solvent shifting, where particles precipitated from black liquor in water ethanol mixtures had sizes of ca. 260 nm while those obtained by acidification reached sizes of ca. 150 nm (Fig. 6a).116 In this effort, the presence of remaining hemicelluloses was found to positively impact colloidal stability. It is expected that particles formed by acidification or solvent polarity shifting of dissolved lignins will have sharply different properties. Although, importantly, while the plant-source for the lignins mattered little on the properties of the formed particles, particularly their surface chemistry, the final properties of the self-assembled particles such as size morphology and surface chemistry, were more significantly affected by the functional groups, pH-response, and solvent-dependent solubility as imparted by the extraction process.27
For integrated processes (i.e. one main step), for instance, where steam explosion is combined with organosolv extraction, the particle size decreased from 3–5 μm with a PDI above 0.48 to particle sizes as low as 200 nm and a PDI of 0.34.117 Additionally, the largest well-defined spherical particles were obtained by a one-step aerosol flow reactor (>20 μm), which enable a high versatility in composition of the particles albeit a reduced control over size and surface chemistry, as associated with the receding air–water interface during rapid solidification.95,118
The assembly regime of lignins also provides important insights on the extent of compaction of the particles, and therefore their softness. For instance, rapid diffusion limited aggregation will lead to the formation of looser particles than the slower reaction limited aggregation, for instance when lignin solution undergoes slow solvent exchange.119 The type of lignin will substantially affect compaction too, with the higher repulsions between highly charged lignins or the presence of larger branched chains leading to less compact assemblies. For instance, softwood Kraft lignins were found to form less compact aggregates than hardwood organosolv lignins when evaluating concentrated bulk solutions.120 Also, model coniferyl alcohol and coniferyl aldehyde polymerized into dehydrogenation polymers (using peroxidation) formed more compact aggregates than softwood kraft lignin.121 Furthermore, reducing charge repulsion, for instance by addition of acids, results in a compaction proportionally improved by increasing the acidification rate.102 Overall, decreasing self-repulsion and aggregation rate while increasing self-interaction will result in the most compact assemblies, with the potential to tune the mechanical properties of the resulting nanoparticles and the nanocomposites as well as nano-biohybrids they form. In accounting for the kinetics of self-assembly, sequential nucleation-growth mechanisms may have detrimental effects on the mechanical properties of the particles due to the formation of discontinuous network. However, this mechanism favors encapsulation of secondary cargo for controlled release applications.13 For the commercially obtained powder of alkali lignin, elastic moduli of up to 14 GPa were measured in dry conditions, which were reduced to ca. 2 GPa upon adsorption of 18% moisture, suggesting significant swelling and softening.122 However, this upper bound of mechanical properties suggest their potential to fine tune the mechanical rigidity of the constructs formed from nanoparticles across a broad range, when compaction of the lignins is maximized and hygroscopic behavior is minimized.
3. Lignin-based nanocomposites
Composites cover a wide range of component dimensions, while hybrids entail constituents at the nanometer and molecular level. Dealing with lignin, functionality is key to access both composite and hybrid materials. Blending of lignin with separate fractions in composites will be introduced in this section, while the preparation of nanosized lignin hybrids will be described in section 4. Composite materials offer feasible routes to large scale applications, in several product categories, including 3D printing.123 In this context, scalability and environmental sustainability is of central importance. A recent review on life cycle assessment (LCA) of lignin and derived products concluded that lignin offers advantages over fossil-based products when it comes to climate change impact.124 To date, only a few techno-economic (TEA),125 and LCA126 studies have focused on LNPs. The key hotspots appear to be solvent recycling and concentration or drying of LNPs. In addition, wastewater treatment deserves more attention even if the ultimate target is to develop closed-loop processes. In the short and long term, the application of lignin nanoparticles will be further advanced and LCA of final products containing LNP will be essential for process development. The data and insights obtained via LCA and related models will be essential to support critical decisions as far as technology and process developments. In this chapter, we discuss selected examples of lignin and nanolignin in different types of nanocomposites.3.1. Polysaccharide biobased blends and complexes (lignin/cellulose, lignin/starch, lignin/chitosan) – composites and nanocomposites
| Lignin | Lignin content (wt%) | Cellulose | Composite type | Functionality | Ref. |
|---|---|---|---|---|---|
| a Isolated by extraction from steam-pretreated poplar wood. SKL, softwood kraft lignin. HKL, hardwood kraft lignin. KL, kraft lignin (unspecified origin). OSL, organosolv lignin. LS, lignosulfonate. LPs, lignin particles (nano/micro), CML, carboxymethylated lignin. LNPs, lignin nanoparticles. HT, hydrothermal. MCC, microcrystalline cellulose. HPMC, hydroxypropylmethylcellulose. CNF, cellulose nanofibrils. TEMPO, (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl. CMC: carboxymethyl cellulose. | |||||
| Na-LS | 33 | MCC | Membrane | Antifouling, adsorption | 128 |
| LPs | 50–100 | CNF | Membrane | UV barrier, antioxidant activity | 12 |
| Na-CML, Al-CML | n.a. | Cellulose acetate | Membrane | Mechanical reinforcement | 132 |
| SKL, OSL, HL (propionated) | 1.0 | Cellulose acetate | Membrane | Mechanical reinforcement, water resistance, improved metal ion removal (lignin-dependent) | 133 |
| SKL, OSL, HL (as such and propionated) | 1.0 | Cellulose acetate | Membrane | Mechanical reinforcement | 134 |
| KL, AL, OSLa | 4.8 | MCC | Film | UV barrier | 135 |
| Propargylated lignin | 0.5–2.0 | MCC (azide-modified) | Film | UV barrier | 136 |
| SKL LNPs | 1.0–50 | CNF | Film | UV barrier, mechanical reinforcement, water resistance, antioxidant activity | 127 |
| Carbonized SKL LPs | 15 | CNF | Film | Electrical resistance | 137 |
| KL, Na-LS | 1.6–25 | TEMPO-CNF | Film | Mechanical reinforcement | 138 |
| SKL (raw and acetylated) | 1.0–9.1 | CNC | Film | UV barrier | 139 |
| OSL | 9.1–91 | CNC | Film | UV barrier | 140 |
| SKL, HKL | 2.4 | CNF + starch | Film | Thermal stability | 141 |
| OSL esters | 0–100 | Cellulose acetate, oleate | Film | Water resistance | 142 |
| OSL (corncob, HT + EtOH cook) | 0–33 | CMC | Film | Water resistance, reduction of water vapor permeability | 143 |
| KL, OSL (various grades) | n.a. | HPMC | Film | Antioxidant, antimicrobial | 144 |
| OSL (aspen) | 0–100 | hydroxypropyl cellulose | Film | Filler | 145 |
The functionality provided by lignin to cellulosic composite films typically includes UV-barrier,127,135,136 mechanical reinforcement,132–134 water resistance,140,143 antioxidant activity,12 adsorption and antifouling capacity,11,128 and antimicrobial activity.144 Many of these properties can be associated with the complex aromatic structural units of the lignin oligomer or polymer chains rich in substituted phenolic hydroxyl groups. Colburn et al. prepared composite membranes containing sodium lignosulfonate in regenerated cellulose matrix.128 The composite membranes showed a higher volumetric flux for water and aqueous humic acid solution (0.1 g L−1) compared to that of the membrane consisting of cellulose only. Manjarrez Nevárez et al. fabricated composite membranes by solvent casting 1 wt% propionated lignin (either softwood kraft lignin (SKL), organosolv lignin (OSL), or HL) in cellulose acetate matrix.133 Incorporation of propionated SKL doubled the Young's modulus (1.9 GPa) compared to the value (0.94 GPa) of the pure cellulose acetate membrane, while propionated HL had only a minor impact on the Young's modulus. On the other hand, without propionation, hydrolysis lignin resulted in highest Young's modulus (2.98 GPa) and stress at break (128 MPa) among the films containing crude lignins. Although it was not elucidated if these differences are related to compositional variations between the lignins, propionation seem to reduce the strength of lignins for supramolecular interactions. The results also show that even a low concentration of lignin (1 wt%) can alter the mechanical properties of cellulosic membranes.
One of the classical challenges of cellulosic composites has been to achieve high lignin content and uniform distribution of lignin in the fibrous matrix. The advent of lignin nano/microparticles has greatly facilitated overcoming these challenges by enabling preparation of well-dispersed composites without severe aggregation of the lignin particles (Fig. 7). Farooq et al. prepared CNF-colloidal lignin particle (CLP) composite films with theoretical lignin content from 1 wt% to 50 wt%.127 They showed that 10 wt% of CLPs maximized ductility of the films prepared from mechanically fibrillated CNFs, regardless of the surface charge of the particles (anionic or cationic) (Fig. 7a and b). CLPs were superior to the crude lignin as a reinforcing component in films with respect to their capability to improve toughness and selective UV-barrier properties (Fig. 7c). The composite films exhibited antioxidant activity that increased non-linearly with increasing (theoretical) lignin content (Fig. 7d). The authors concluded, based on the multiscale visualization and analysis of the films (Fig. 7e–g), that CLPs interacted with CNF mainly via weak hydrogen bonding interactions. In the presence of moisture, a ball bearing lubrication effect was responsible for the increased ductility of the composite films. Later, Cusola et al. also prepared composite films consisting of CNF and spherical lignin particles (Fig. 7h–k).12 In their case, the LPs originated from an aerosol process were larger and less monodisperse than the CLPs used by Farooq et al. Another difference is the lignin content of the composite films. Cusola et al. used 50–100 wt% of LPs while the purpose of CNF was to reinforce the particulate network by wrapping adjacent LPs within the nanofibrils. The same effect was observed by Mattos et al.146 where the authors also showed that the polydispersity in size of particles was significantly impairing the cohesion of the composites, where particle sizes <50 nm were reducing the constructs’ toughness. The authors showed that a combination of LPs, CNF, and the wet strength agent polyamidoamine-epichlorohydrin resin at respective weight percentages of 90%, 7%, 3% formed cohesive films that did not visibly disintegrate when submerged in water. In addition, their use as filtration membranes for the rejection of radical cation dye solution was demonstrated (Fig. 7m and n).
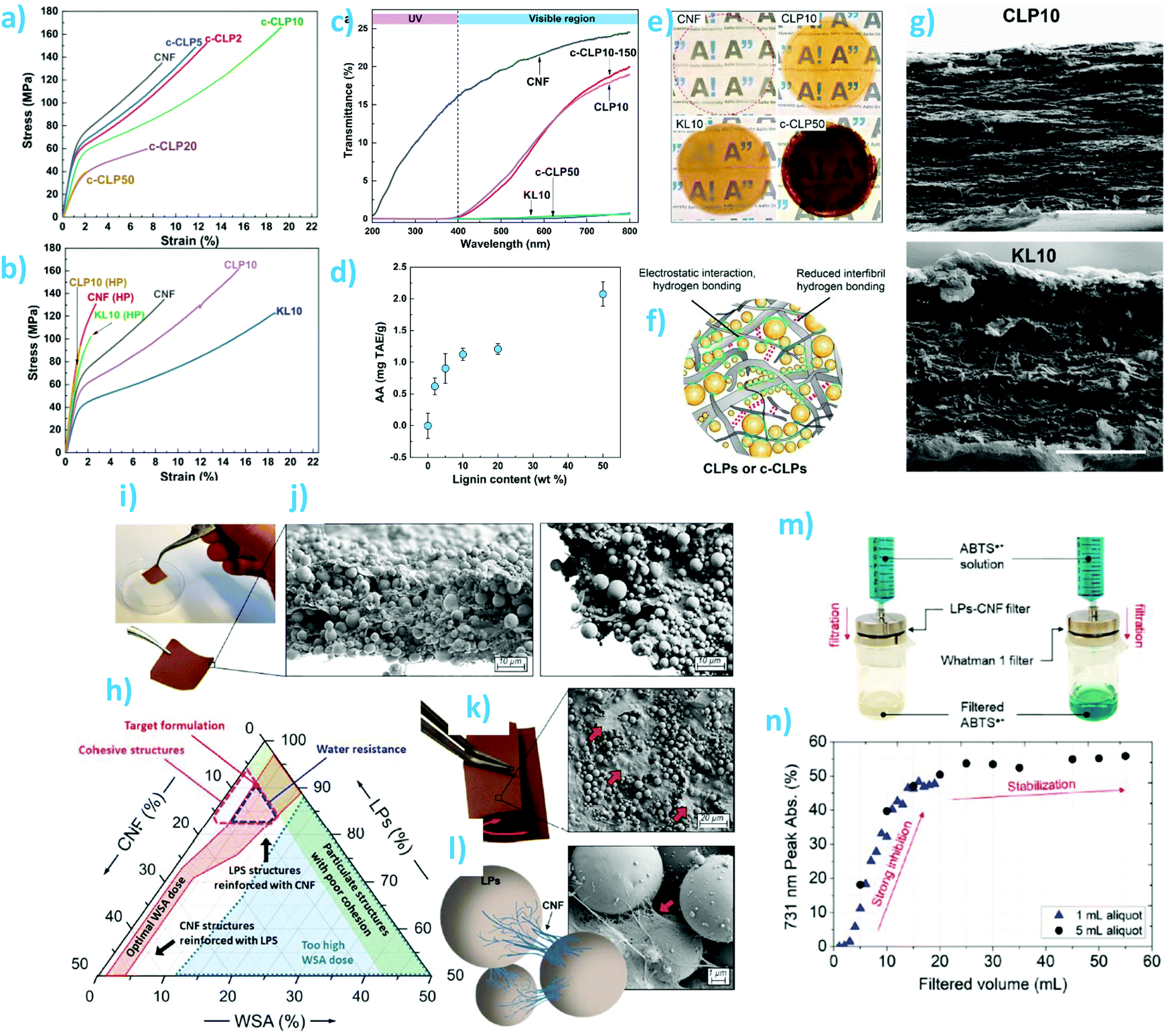 | ||
| Fig. 7 Composite films and membranes from nanocellulose and spherical lignin particles. CNF-CLP nanocomposite films: (a and b) tensile stress–strain curves of various film compositions. (c) Optical transmittance of CNF film and nanocomposite films containing different lignin contents and morphologies. (d) Antioxidant activity of the films as a function of their theoretical lignin content. (e) Photographs of the CNF film and CNF-lignin composite films. (f) Schematic overview of possible interactions of cationic CLPs and CNF. (g) SEM images of composite films containing 10 wt% of CLPs or crude softwood kraft lignin. Reproduced with permission from ref. 127 Lignin nano/microparticle-CNF films: (h) Ternary phase diagram of CNF, LPs, and wet strength additive. (i) Photograph of the self-standing membrane at 8 wt% CNF content. (j and k) SEM images of the cross sections and top surfaces of the membrane. (l) Schematic diagram and SEM images of bridging of LPs by CNF. (m) Use of the membranes as antioxidant filtration membranes for ABTS˙+ radical cation solution. (n) Evolution of absorbance reduction as a function filtration volume. Reproduced with permission from ref. 12. | ||
Chemical cross-linking is a common approach to prepare hydrogels,147 which represent another type of cellulose–lignin composite,148–150 in contrast to the composite films and membranes presented above (fabricated using non-covalent methods). Hydrogels are often cross-linked and dewatered to give rise to aerogels that preserve the porous morphology of the original hydrogel. A summary of these cellulose–lignin composites is presented in Table 4. In the case of hydrogels, covalent cross-linking is often used to increase the stability of lignin in the hydrated cellulose fiber matrix. As mentioned above, the cross-linked hydrogels are also viable precursors for aerogels or xerogels. Regardless of the fabrication method or material type, mechanical, thermal, and chemical stability of the composite are often important for practical applications such as adsorption,151–153 controlled release of loaded cargo,154 fractionation of lignin,93 and enzyme immobilization.155
| Lignin | Matrix | Material | Functionality | Ref. |
|---|---|---|---|---|
| AL, alkali lignin. LCC, lignin-carbohydrate complex. EHL, enzymatic hydrolysis lignin. PEG, polyethylene glycol., SEL, Swelled Enzyme Lignin. TOFA, tall oil fatty acid. CCL, choline lactate-lactic acid. LS, Lignosulfonate. SKL, softwood kraft lignin. | ||||
| AL | Cellulose pulp fibers, CNF | Hydrogel | Adsorption (Cu2+) | 151 |
| SEL | MCC | Hydrogel | Controlled release (tannins) | 154 |
| AL (HT-pretreated corncobs) | MCC | Hydrogel | Fractionation of lignin | 93 |
| AL | MCC | Hydrogel | Enzyme immobilization | 155 |
| AL-PEG-NH2 | TEMPO-CNF + PNIPAM | Hydrogel | pH and T responsiveness | 147 |
| AL (soda pulping of pine and eucalyptus), CCL, EHL | HEC + PVA | Hydrogel | Adsorption of dye | 152 |
| SL | WS soda pulp fibers | Aerogel | Adsorption of dye | 153 |
| LCC | CNC | Aerogel | Cell culture medium | 156 |
| SL | Wood fiberboard | Fiberboard | Binder | 157 |
| Willow lignin (extracted with IL) | Cellulose pulp fibers | Microfibers | Filler, increased thermal stability and carbon yield | 158 |
| TOFA-esterified SKL LNPs | Cellulose pulp fibers | Suspension | Antimicrobial | 148 |
| LS + chitosan | Cellulose pulp fibers | LbL-coated fibers | Antibacterial | 159 |
| Hydrotropic lignin | Dissolving pulp fibers | Beads | Antimicrobial | 131 |
| Eucalyptus lignin and nanolignin | CNC | Microparticles | Water resistance (hydrophobized particles) | 149 |
| Pine acetic acid lignin (acetylated) | Cellulose acetate | Powder | Filler | 150 |
Dai et al. prepared composite hydrogels by using epichlorohydrin (ECH) to cross-link alkali lignin and microcrystalline cellulose (MCC) (Fig. 8a). MCC was first dissolved in aqueous sodium hydroxide-urea (6 wt% NaOH, 4 wt% urea) solution using a freeze–thaw method and subsequently cross-linked with ECH in the presence of lignin.93 The weight percentages of lignin in three different formulations were 1.3%, 2.6%, and 5.2% relative to the total dry weight of hydrogel. The purified hydrogels were characterized for rheological properties and thermal stability, and subsequently used to fractionate the same alkali lignin that was used in its synthesis (Fig. 8b). The rheological tests of the lignin-containing hydrogels revealed elastic network structure in the low strain range, while a collapse of the material was observed beyond a strain of 3%, which was ascribed to the gel–sol transition. The appearance and microscaled morphology of the hydrogels changed depending on the lignin content. Higher lignin content rendered the hydrogels darker and decreased the pore cell diameter (Fig. 8c). In turn, when these hydrogels were used to adsorb and filter aqueous ethanol solution of alkali lignin, separation of low and high molecular weight lignin fractions increased as the lignin content of the hydrogel increased (Fig. 8d). However, despite comprehensive characterization by NMR spectroscopy, the authors could not explain the association of the lower molecular weight fraction in the hydrogels since the two separated fractions was almost identical with respect to their content and type of functional groups.
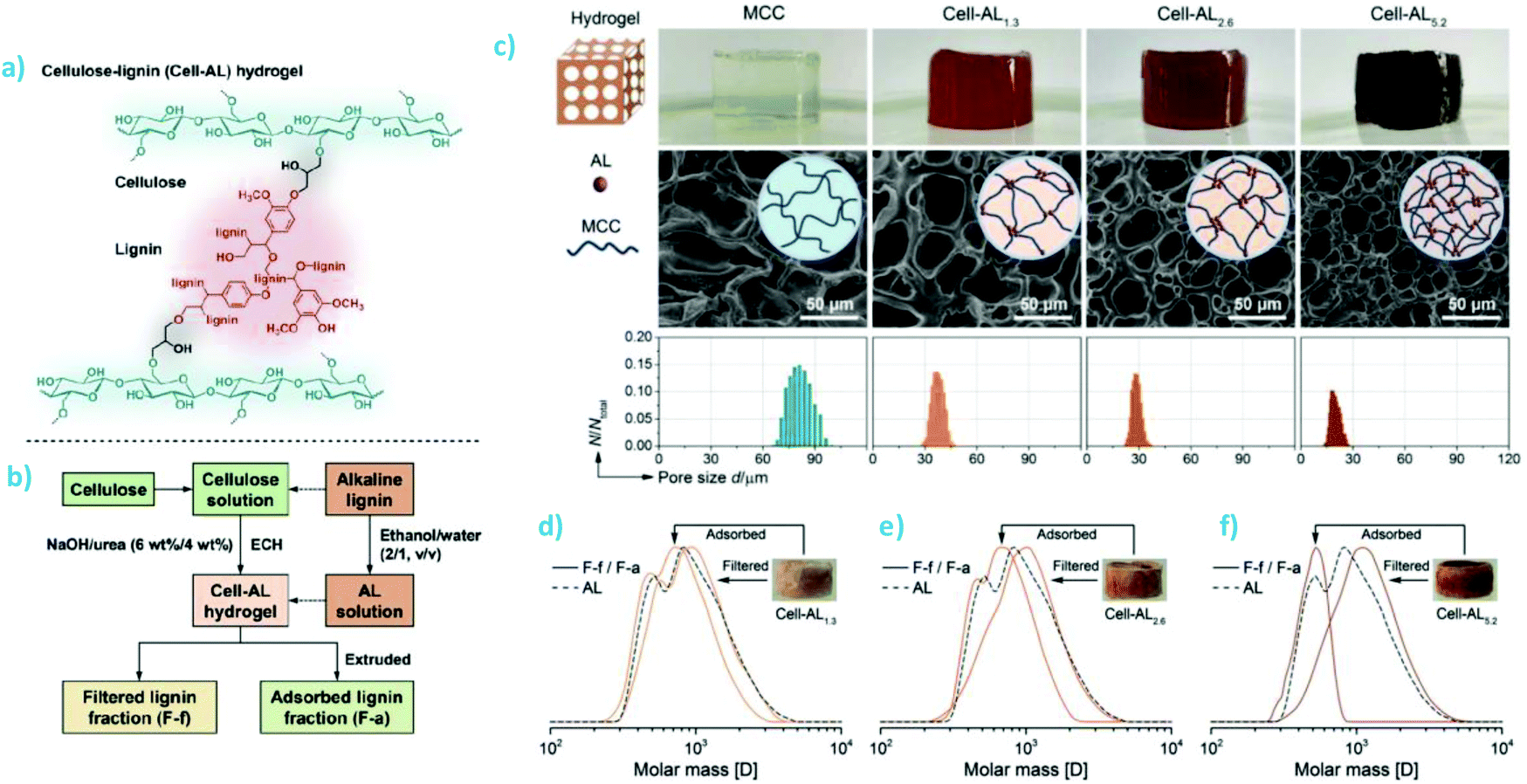 | ||
| Fig. 8 Synthesis of cellulose–lignin hydrogels for fractionation of lignin. (a) Cross-linking of cellulose and lignin using ECH. (b) Flow chart of synthesis and application of the composite hydrogels. (c) Appearance of MCC and the MCC-AL hydrogels with different lignin contents. (d) Size-exclusion chromatography traces showing fractionation of the original AL solution using the hydrogels with (d) 1.3%, (e) 2.6%, and (f) 5.2% of lignin. Reproduced with permission from ref. 93. | ||
Baumberger and co-workers have made a significant contribution to the fundamental understanding, processing, and applications of lignin-starch composites and blends.163 One of the key questions has been the compatibility of lignin in a starch matrix. Due to the heterogeneity and broad molecular weight distribution of lignin, incomplete compatibility has been reported in many cases. Changes observed in mechanical properties suggest a certain level of molecular interactions between various starches and lignin. It has been found that the interactions of lignin and linear amylose differ from those of lignin and branched amylopectin in that in the latter case the association is stronger.163 However, the structural heterogeneity of lignin and the variation of molecular weight of lignin from different plant sources and processes make it difficult to predict these interactions. For instance, despite their usual incompatibility, a low molecular weight kraft lignin fraction isolated at early stages of delignification was found to act as a starch plasticizer, increasing elongation at break while decreasing stress at break.164
There are a number of more recent studies that have pushed the frontier by introducing new and modified lignins to starch-based composites. Kaewtatip and Thongmee compared kraft lignin and acetylated kraft lignin as fillers in thermoplastic starch (TPS) composites at 5 wt% lignin content.165 They found that tensile strength of the composite containing acetylated lignin was 32% higher than that of the pristine TPS. This increase was almost two times as high as that obtained with the non-acetylated lignin, and indicated that H-bond acceptors (as in acetates) favored plasticizing over H-bond donors (as in hydroxyl groups). The respective water absorption capacities of TPS, TPS–lignin, and TPS-acetylated lignin were 23.8%, 18.6%, and 14.3%. The highest water resistance was due to the acetylation of the aliphatic and phenolic hydroxyl groups that were no longer available for hydrogen bonding with water. Stevens et al. prepared starch–lignin foams by dissolving softwood kraft lignin in ammonium hydroxide in the presence of corn starch, and heating the mixture first at 70 °C to gelatinize the starch and then at 170 °C to evaporate water.161 The cellular structure of the resulting foams did not reveal lignin aggregates on the walls, indicating an absence of any deleterious effects on morphology when 20% of the starch was replaced with lignin.
| Lignin | Chitosan | Material geometry | Functionality | Ref. |
|---|---|---|---|---|
| AL | 95% deacetylation. | Extruded pellets | Adsorption of dyes | 171 |
| AL | 95% deacet. | Powder | Adsorption of dyes | 172 |
| KL | 85% deacet. | Film | Antioxidant, antimicrobial | 144 |
| Acetic acid-soluble lignin | M w 340 kDa, 90% deacet. | Film | Thermal stability, ductility | 173 |
| SL | M w 270 kDa, 90% deacet. | Film | Antioxidant | 174 |
| SL | M w 270 kDa, 90% deacet. | Film | Antioxidant | 175 |
| HKL, OSL | M w 500 kDa, 98% deacet., microcrystalline | Film | UV barrier, mechanical reinforcement | 166 |
| Ca-LS | M w 300 kDa, 95% deacet., microcrystalline | Film | Controlled WCA | 176 |
| SL | M w 190–310 kDa, 79% deacet. | Film | Antimicrobial food packaging | 177 |
| Na-LS | ≥90% deacet. | Membrane | Proton exchange | 178 |
| AL | M w 890 kDa, 90% deacet. | Hydrogel, film | Wound healing | 167 |
| LS | Chitosan + PVA | Hydrogel | Wound healing | 179 |
| AL (HT-pretreated corncobs) | Carboxymethyl chitosan (DS ≥80%) | Hydrogel | Encapsulation (trans-resveratrol) | 180 |
| Na-LS | M w 190–310 kDa, 75–85% deacet. | Hydrogel | Adsorption (Cu2+, Co2+) | 181 |
| LS | 97% deacet. | Aerogel | Adsorption of dyes | 182 |
| AL | M w 190 kDa, 100% deacet. | Microfibers | N/A | 183 |
| SL | M w 190–310 kDa, 75–85% deacet. | Electrospun fibers | N/A | 184 |
| LS | Medium Mw | Nanoparticles | N/A | 185 |
| Ca-LS | >75% deacet. | Nano/micro particles | Carrier for RNase A | 186 |
| OSL | Low Mw, 80% deacet. | 170 | ||
| Na-LS | M w 50–190 kDa, 75–85% deacet. | Nanospheres | Antimicrobial | 187 |
| SKL | M w 100–300 kDa, ≥90% deacet. | Pickering emulsion | Encapsulation (ciprofloxacin) | 168 |
| Na-LS | M w 30 kDa, 90–94% deacet. | Nanoemulsion | Encapsulation (deltamethrin) | 169 |
| NH4-LS | 95% deacet. | Viscous solution | Adhesive | 188 |
An interesting development is the use of chitin nanofibers to nucleate and grow lignin nanoparticles.189 In this vein, other interesting applications is that of chitosan–lignin composites for emulsion stabilization and encapsulation of active payload. Nguyen et al. used high pressure homogenization to prepare corn oil nanoemulsions encapsulating deltamethrin.169 The nanoemulsion was stabilized by coating chitosan at the oil–water interface and thereafter adsorbing lignosulfonate as an outermost polyelectrolyte layer that provided efficient photoprotection of deltamethrin. The amount of non-degraded deltamethrin after 60 min UV irradiation was approximately 95% in chitosan–lignin coated nanoemulsion droplets compared to only 59% in the case of the free-form deltamethrin.
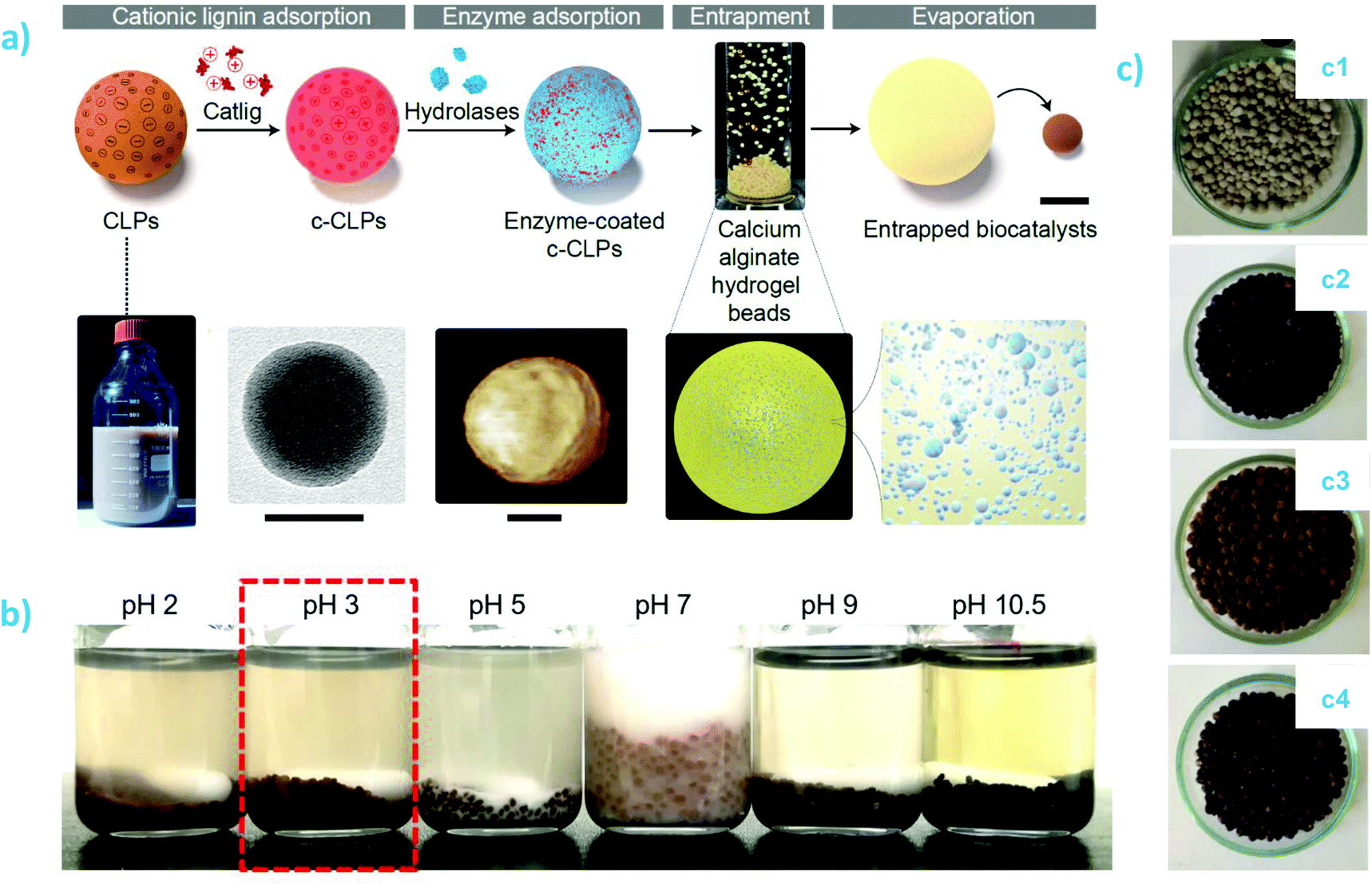 | ||
| Fig. 9 Lignin-polysaccharide composites: (a) Preparation of biocatalytic composite beads containing enzyme-coated cationic lignin nanoparticles embedded in calcium alginate matrix. The scale bars are 1 mm (composite bead, top row) and 50 nm (TEM and AFM images); and (b) pH stability of lipase@cationic LNP-alginate beads (0.2 M aqueous buffer, 72 h, RT). Reproduced with permission from ref. 192 (c) Appearance of composite coatings on triple superphosphate fertilized granules (TSP, c1) and TSP-coated with lignin-alginate (c2), lignin-carboxymethyl cellulose (c3), and lignin-carrageenan (c4). Reproduced with permission from ref. 193. | ||
The second prominent approach to form lignin-polysaccharide hydrogels involves chemical cross-linking of the two components with epichlorohydrin. Many different types of lignins have been cross-linked this way with polysaccharides such as hyaluronan,194 xanthan gum,195 and agarose.196 Another approach is to entrap lignin and polysaccharides, such as hemicellulose maleinate, within a covalent polymer network synthesized from acrylic acid monomers.197 There are also a few examples of composite manufacture without any ionic or covalent cross-linkers when using alginate, carrageenan, and agar.193,198
The functionality provided by lignin to these composites includes increased adsorption capacity to dyes,197 and other organic pollutants,199 carrier systems for active compounds,193 thermal stability and thermal insulation,191,198,200 as well as mechanical reinforcement.196,201 Although not emphasized in most of these studies, the incorporation of lignin in the composites additionally imbues the composites with antioxidant activity and UV-barrier properties. Lignin content is the most prominent factor that defines many functional properties of the composites. The weight fraction of lignin varies broadly from 1 to 90% relative to the total dry weight of the composite. Another important determinant is the type of lignin, and more studies should be made to compare, side by side, SKL, AL, lignosulfonates, and non-industrial lignins isolated from different biomass sources to draw conclusions on the most suitable lignin sources as well as structure–function relationships.
Lignin and lignin-derived biopolymers may offer opportunities for commercial exploitation. They can be used in widely different areas, pulp and paper, plastic, agriculture and others, such as water remediation, biopolymer-based blends (with chitosan, starch and cellulose–lignin composites), all of which have shown promising application potential.
3.2. Protein biobased blends (lignin/plant and animal proteins) – composites and nanocomposites
Protein–lignin blends have also been utilized for the fabrication of composites and nanocomposites. Interest in blending lignins with proteins comes not only from the sustainability aspect, but also from the unique structural characteristics of proteins. Proteins consist of peptide assemblies, which are formed from the vast combination of amino acids.202 Such structural diversity confers proteins and their assemblies a nearly limitless range of possible structures, functions and properties that are also key to sustaining vital biological roles. This well-known fact makes proteins abundant across the natural world, being found in essentially all living organisms. Wider utilization of such materials, however, is mostly hindered by challenges related to their extraction,203 synthesis and purification, or assembly into functional materials.204,205Three main strategies, or a combination thereof, are often used for assembling protein-based materials, i.e. solvent casting, thermoforming and extrusion.206 Certain solvents, pH conditions or high temperatures are often used for denaturating proteins, thus promoting conformational changes and new interactions previously inexistent to the native protein. Temperature and shear stresses are also known to result in higher plasticity, with the former also contributing to increasing the degree of cross-linking.205 Regardless of the processing strategy, additives are essential for improving processability and materials properties, as brittleness and poor water resistance are recurring challenges. In such a context, lignins have been utilized as a sustainable additive for protein-based biodegradable and functional materials. Overall, the most common types of animal- and plant-based proteins investigated for bionanocomposites, especially for packaging and bioplastics applications, are gelatin and soy proteins.205
Ojagh et al. investigated fish gelatin-soda lignin (Protobind 1000) films for the preservation of salmon muscle during high hydrostatic pressure treatment.210 This treatment is used for extending the shelf-life for additive-free foods and utilizes pressures above 200 MPa. The films were prepared using sorbitol and glycerol as plasticizers and at pH 11 for solubilizing lignin. It was shown that such films were effective in reducing lipid oxidation during 23 d of refrigerated storage and could be easily separated from the fish meat. Furthermore, Núñez-Flores et al. showed that lignin-containing gelatin films presented antioxidant capacity at non-cytotoxic concentrations for fibroblast 3T3 cells.211 In fact, the concentration at which lignosulfonate was shown to be cytotoxic was 15- to 20-fold higher than that required for producing the antioxidant effect. Better water resistance was also achieved, as the gelatin-lignosulfonate blends containing 20 wt% lignin decreased water solubility in ca. 50% when compared to lignin-free gelatin films. Moreover, in the study by Belgodere et al., lignosulfonate and alkali lignin also did not show cytotoxicity against adipose-derived stromal cells, while resulted in gels with higher stiffness and attenuated shear-thinning behavior.212
From the mechanical properties perspective, gelatin–lignin films can have robust performance, mostly because of H-bonding and hydrophobic interactions.213 In the work of Mehta & Kumar, gelatin-alkali lignin blends were mixed with a biobased ionic liquid (choline citrate) additive, resulting in high tensile strength (ca. 70 MPa) and large elongation at break (ca. 200%).214 The ionic liquid was said to increase the amount of hydrogen-bonds in the system and to induce antimicrobial activity. Although such antimicrobial activity might have been influenced by leaching of the ionic liquid, choline-based ionic liquids are considered of low to negligible toxicity and good biodegradability.215 The same study also observed the UV-blocking properties of the composite film, which is another important performance aspect needed for packaging materials. 98% blocking efficiency for UV-B light was achieved by the addition of 1.5 wt% lignin, resulting in a sun protection factor of about 45.214
Interestingly, gelatin–lignin blends have also been used for their binding capacity at high temperatures. Such blends were used for binding anthracite fines into robust bricks used for substituting foundry coke.216,217 As shown by the compressive strength of the bricks after pyrolysis, hardwood lignin performed better than softwood lignin (ca. 6.5 MPa against ca. 3.2 MPa). The good mechanical stability and char formation capacity brought by lignin was key for making anthracite bricks of high energy density that displayed comparable or higher combustion efficiency than coke in full scale tests involving ca. 8 tons of anthracite.217 Zhao et al. recently studied the synergistic interactions occurring between lignin and collagen during pyrolysis.218 In short, they concluded that collagen effectively enhance the fusing propensity of lignin, resulting in more aromatic rings and char yield than pyrolyzed lignin or collagen alone. Such efforts have also been used for the development of other lignin-gelatin or lignin-collagen precursors for carbon-based materials, such as in the case of specialty graphites and graphite electrodes.219
Processing of lignin-soy protein composite films and coatings can be carried by aqueous solution casting using nearly ambient temperature,222 or by compression molding at temperatures exceeding 100 °C.223 For adhesives, hot pressing at high temperatures is also often utilized.224–226 For such application, Pradyawong et al. tested kraft lignin particles ball milled to different sizes (i.e. 35, 19 and 10 μm) as reinforcement fillers.224 They observed that the smaller particles at 20 wt% resulted in the best performance, with a 53% higher wet strength in relation to pure soy protein adhesives without reinforcement.
Interestingly, nanofibers could also be produced via solution electrospinning (Fig. 10a).227,228 Salas et al. used an aqueous solvent consisting of 10 vol% acetonitrile in 0.1 M NaOH to dissolve polyethylene oxide (PEO), soy protein (or glycinin, one of the main constituents of soy proteins) and kraft lignin.227 PEO (400 kDa) was used at a concentration of ca. 10 wt% of the total mass of the lignin-protein mixture for improving miscibility and hydrogen bonding in the system. At increasing lignin amounts, from ca. 20 to 80 wt%, the diameter of the nanofibers increased from about 124 nm to 400 nm. Furthermore, wetting properties of the fiber mats were similar to those from adhesives reported by Pradyawong et al.224 with lower lignin amounts having higher contact angles (Fig. 10b). Such results were attributed to the rearrangement of the hydrophobic parts of the denatured proteins towards the surface of the composite fibers.227 The small differences between glycinin and the soy protein isolate might be explained by structural and conformational differences of the two main protein components of soy (glycinin and β-conglycinin) upon adsorption.229
 | ||
| Fig. 10 (a) Electrospun soy protein isolate-kraft lignin fibers containing 10 wt% PEO. I1, I2, I3 and I4 correspond to increasing lignin concentrations of 22, 50, 67 and 78 wt% (lignin + protein), respectively. (b) Water contact angles of the electrospun materials produced from soy protein isolate or from glycinin at different protein–lignin ratios. Reproduced with permission from ref. 227. | ||
Recent advances in the field of biopolymer processing are highly promising for the design of lignin based, sustainable, low cost composite materials. Rational next steps in sought developments for biobased polymers/lignin composites need to be focused in facile methods that offer effective lignin fractionation and facile chemical modifications. All of the above need be coupled with detailed evaluations of the polymer and other physicochemical characteristics of resulting materials. Research on lignin-based composite should be conducted to break through the biggest limitation of polymer material blending “trade-off” effect, and to allow for its large-scale application, to protect the environment, save resources, and for green and sustainable development. If this is properly done, it is anticipated that lignin-based materials will continue to see a rapid expansion towards novel advanced and demanding applications. We strongly believe this could lead to the creation of a wave of lignin-based bioproducts.
3.3. Lignin and synthetic biopolymers in composites and nanocomposites
Herein the use of synthetic biopolymers is beneficial since besides carbon cost considerations, the environmental friendliness of the formed materials might be positively impacted. Thereafter, three aspects of the technical issues associated with lignin based composites and nanocomposites and their prospective applications should be considered, namely, the use of lignin (modified or not) to fabricate lignin-based thermoplastics, the theoretical basis for blends with high lignin contents, and the methods for realizing efficiently biodegradable lignin-based plastic materials.234 Actually, there is a considerable confusion in the literature about the definition of the polymer/lignin combination. Authors identify it either as a composite or as a blend. The differences should be considered depending on the interactions, which determine the mutual solubility of the phases and the strength of interfacial adhesion in heterogeneous systems.235Looking at blend preparation, we will also consider here the development of lignin-based thermoplastics with prevalent lignin component or, at least, high lignin content (more than 30 wt%), to identify lignin as a matrix and not as a filler. To achieve large-scale application of thermoplastic materials with high lignin content, the inherent thinking must be fundamentally novel. As stated earlier by Falkehag et al., “in the attempted uses of lignin to meet polymer or materials needs, one should not just try to ‘replace’ a synthetic component, but to take new innovative approaches where the uniqueness of lignin as a macromolecule should be exploited”.236
Lignin contains a variety of potential polymerization sites (abundant phenolic and aliphatic hydroxyl groups), with different polarities, that should be considered when improvement of miscibility needs to be reached.237 It is known that lignin molecule is relatively polar, due to its numerous hydroxyl groups, and generally presents good compatibility with polar polymer matrices. Therefore, lignin is immiscible with most polymers: this is often related to strong intermolecular interactions of lignin which result in poorer interactions between the polymer and lignin. Moreover, lignin tends to aggregate in biopolymers, such as biomass based or petroleum-based biomaterials, owing to the π–π stacking of hydrogen bonding, its aromatic rings and van der Waals forces between the polymer chains, which can impair the properties of the resulting composites. At present, there are three major approaches to enhance the miscibility of two polymeric matrices that remain useful even when one of the two components, actually the prevalent one, is lignin-based.
Even being at the limit of considering lignin as a filler instead of a matrix, excluding the chemical modification of the lignin to reduce its polarity, two useful approaches can be considered to enhance the compatibilization with biobased polymers: graft copolymerization of lignin with other biopolymers, and addition of compatibilizers to improve interfacial compatibility with biobased matrices.238,239 Reducing the analysis to the last two methodologies, graft copolymerization of lignin is certainly a common modification method to form, in general, stable covalent bonds between lignin and polymers. Graft copolymerization provides possibilities to combine advantages of physical and chemical properties of both natural lignin and synthetic polymers:240 generally, well-defined graft copolymers can be prepared via a “grafting through” process, a “grafting from” controlled polymerization process or a “grafting to” process such as “click chemistry” (Fig. 11a). After activation, the hydroxyl functional groups in lignin form seeding sites for the subsequent graft copolymerization using lignin macromolecules as initiators. The graft copolymer is usually composed of lignin as the backbone chain and one or more types of polymers as the branches linking to the backbone by covalent bonding. The properties of the graft copolymer depend on the graft groups, the graft length, and the graft density.240
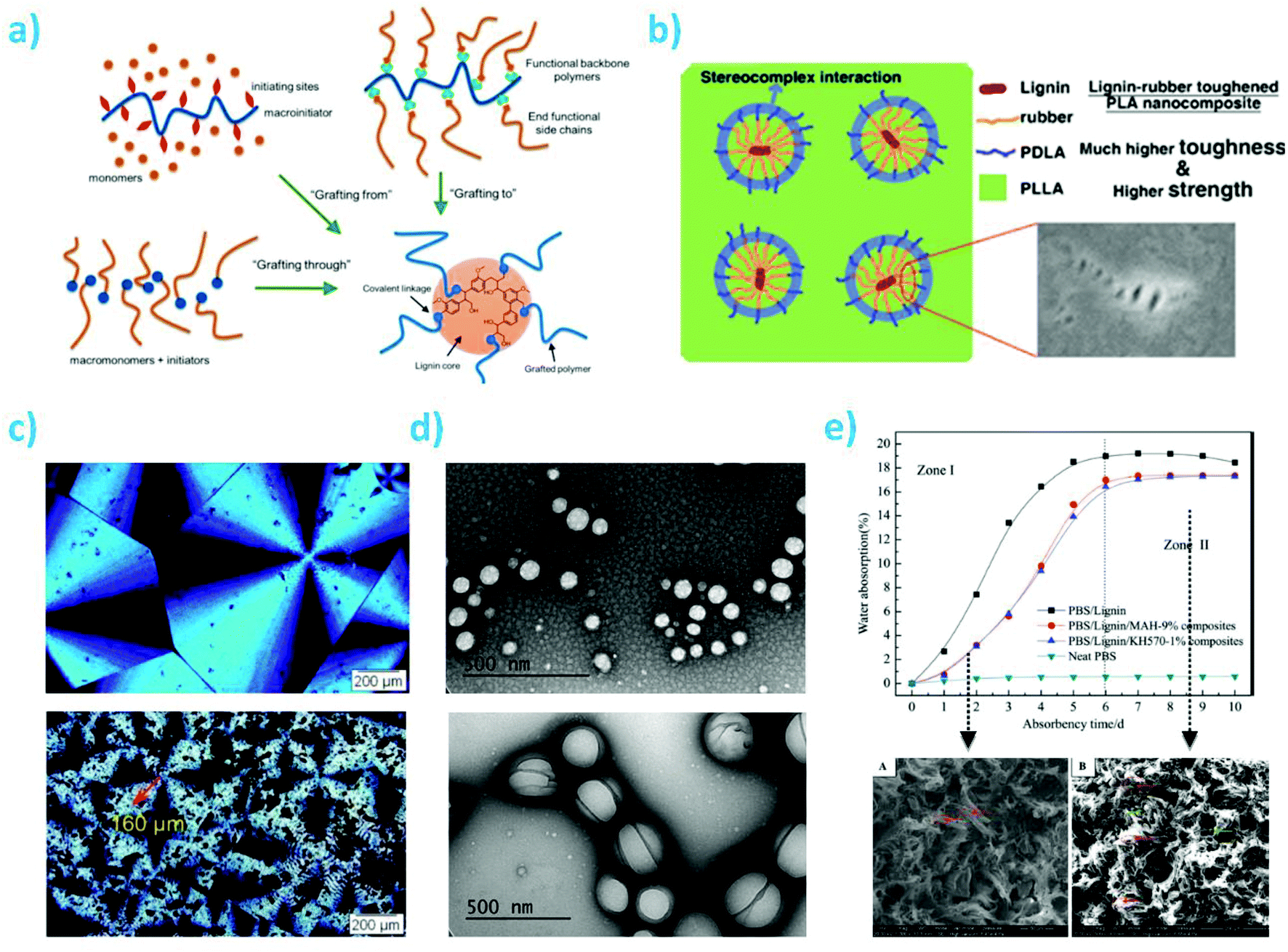 | ||
| Fig. 11 (a) Strategies for lignin based graft copolymers synthesis; reproduced with permission from ref. 252; (b) PDLA segments contribution to interfacial interactions between lignin-rubber-PDLA and PLLA matrix by stereocomplexation: poly(ε-caprolactone-co-lactide) (PCLLA) rubbery layer formed via lignin-initiated ring opening copolymerization of an ε-caprolactone/L-lactide mixture, followed by the formation of poly(D-lactide) (PDLA) outer segments via the polymerization of D-lactide; reproduced with permission from reproduced with permission from ref. 242; (c) POM micrographs of PHBV and PHBV-g-lignin crystallized at 90 °C, reproduced with permission from ref. 243; (d) TEM pictures of biopolymeric nanoparticles from LGN/PLGA 1:2 w/w and LGN/PLGA 1:6 w/w ratios; reproduced with permission from ref. 246; (e) water absorption profiles of polybutylene succinate (PBS):lignin at 50:50 wt/wt in presence of silane and maleic anhydride (MAH) compatibilizers and their related morphologies PBS/lignin and PBS/lignin + MAH (9%). Reproduced with permission from ref. 253. | ||
Grafting from ring opening polymerization (ROP) of cyclic ester on lignin was performed by Ren et al.241 and Sun et al.242 Poly(ε-caprolactone-co-L-lactide) (PCLLA) was grafted first on a lignin in the presence of tin(II)2-ethylhexanoate (Sn(Oct)2 as catalyst. Then, D-lactide (DLA) was further polymerized on lignin graft-PCLLA outer segments. The prepared lignin-graft-PCLLA showed rubber-like behavior that is mainly induced from characteristic soft properties of PCLLA without lignin. Subsequently polymerized DLA possesses a high degree of crystallites which yields overall semicrystalline properties of finally produced lignin-graft-(PCLLA-co-PDLA). The stereocomplex crystallites region of the lignin graft polymer accounts for the superior mechanical properties and thermal resistance compared with homo PLA. Therefore, complexation between the PDLA and PLLA matrix contributes to strong interactions at the interface of the lignin-rubber copolymer and PLLA (Fig. 11b).
In situ free radical reaction has been also considered as a suitable methodology to graft biopolymers to lignin. In this sense, Luo et al.243 reacted softwood kraft lignin to another radical containing poly(3-hydroxybutyrateco-3-hydroxyvalerate) (PHBV). For both PHBV and lignin, dicumyl peroxide (DCP) was mixed to the polymers at 185 °C to generate radicals. The incorporation of lignin improved the Young's modulus as compared with PHBV homopolymer, while both tensile strength and strain at break decreased. A key reason for the improved properties was the reduction in the crystallinity degree through grafting, as revealed by polarized optical microscope (POM) observation, where it was found that spherulites in grafted biopolymers were significantly greater in number and smaller in size than those found in neat PHBV and simple blend (Fig. 11c). Panesar et al. reported their attempts at the synthesis of poly(vinyl acetate) grafted kraft lignin copolymers.244 The thermal stability of the grafted copolymer was slightly inferior to that of the pure polyvinyl acetate. However, the glass transition temperature (Tg) increased substantially after the introduction of aromatic rich rigid lignin structures. Later, Chung and coworkers also reported an increase in the Tg of polylactic acid when grafted on lignin. They described a 40 °C increase in Tg when the lignin content was increased from 1 wt% to 50 wt%.245 They developed a catalytic and solvent-free method for the synthesis of a lignin-g-poly(lactic acid) copolymer to improve the miscibility of lignin with other biopolymers. In their method, graft polymerization of lactide onto lignin was catalyzed by triazabicyclodecene (TBD). Pre-acetylation treatment or varying the lignin/lactide ratio demonstrated to be useful to control the PLA chain length. It was evident that a high grafting efficiency and preferential grafting on lignin was on aliphatic hydroxyls over phenolic hydroxyls. In all cases enhanced elasticity, superplasticizer capability, UV absorbent capabilities and surfactant capabilities have been found for lignin-based graft copolymers.240
Lignin has been also covalently linked to poly(lactic-co-glycolic acid) (PLGA) for the synthesis of polymeric nanoparticles without the need for surfactants for stabilization purposes.246 Lignin-graft-PLGA NPs were made in a wide range of sizes (Fig. 11d) and surface charges by changing the lignin/PLGA ratio, addition of more PLGA resulted in an increase in the nanoparticle diameter. Nanofibers were also produced starting from PHBV grafted lignin: in Kai et al.,247 β-butyrolactone was grafted onto the lignin core by using solvent-free ROP, and then different amounts of lignin-PHB copolymers were blended into PHB and then engineered into nanofibers via electrospinning. A further evolution of grating procedures relies in the possibility of using hybrid lignin (in combination with silica) as a polyol macroinitiator for ROP of ε-caprolactone monomer.248 The authors observed that decreasing the monomer/OH ratio from 22 to 5.5, melting temperatures (Tm) and degree of crystallinity (Xc) drop from 57 °C and 70% to 55 °C and 57%, respectively.
If the grafting procedures are at the boundaries of considering lignin as a matrix in the blend or composite, lignin has been indeed successfully blended with many different biodegradable polyesters, such as poly(lactic acid), poly(caprolactone), poly(hydroxybutyrate), poly(butylene succinate) and poly(3-hydroxybutyrate-co-hydroxyvalerate), in presence of different compatibilizers,249 to produce environmentally friendly materials. Actually, due to its inherent fragility, most of the cited papers considered the widespread use of the synthetic biopolymers, instead of prevailing lignin. Few examples can be found where lignin was effectively the predominant matrix. Examples of thermoplastic polymers made with 85 wt% kraft lignin content and even 100 wt% alkylated lignin can be found.250 The 85 wt% kraft lignin polymer was a blend of underivatized industrial kraft lignin with poly(vinyl acetate) (90000 gmol−1) and diethyleneglycol dibenzoate in a 16:2:1 weight ratio; in those works, the authors demonstrated that the properties of these high lignin content thermoplastic polymers can be assimilated to the performance of currently existing petroleum-based polymers.
As stated before, one strategy to be used for improving blend compatibility between lignin and polymers is the use of compatibilizers based on esterified version of the same synthetic fraction. For instance, maleated polycaprolactone was shown to result in blends with better properties than when no compatibilizer was used.251 It was shown that thermal-molded blend sheets of organosolv lignin esters of n = 3–5 with PCL attained ≥500% of elongation at rupture at 20 °C even when the blends contained 50 wt% of the respective esterified lignin components, reflecting the good miscibility of these organosolv lignin esters with PCL.
Nitz et al. melt-blended biodegradable aromatic polyester with hardwood organosolv lignin and soda lignins from sisal and abaca to obtain materials with up to 50 wt% lignin contents.254 The blends of the polyester and any of the lignins at 30% or lower exhibited elongation at break above 500% and very high impact strength. At up to about 40% lignin content, the materials had about the same or slightly higher Young's modulus and yield stress as the neat polyester. At 50% lignin content modulus and yield stress were significantly higher, particularly in the case of lignin from sisal.
Polycaprolactone and hardwood organosolv lignin showed an increase in Young's modulus (particularly when lignin content was 60–70 wt%), a moderate decrease in yield stress, a significant decrease in impact stress, while maintaining a high strain.255 The effect of a relatively high amount of grafted lignin (20%) was also considered by Kumar et al.:256 the combination of E-beam irradiation in the presence of 3 phr triallyl isocyanurate (TAIC) gave, as a result, the formation of PLA-TAIC-Lignin crosslinked structures which act as an interface between the dispersed lignin phase and PLA matrix and hence improved their compatibility in the resulting blend.
Polybutylene succinate (PBS) was also considered in combination with 50 wt% of lignin,253 where the authors demonstrated that maleic anhydride (MAH) grafted polymers and silanes induced significant improvements in flexural and tensile behavior. Furthermore, both lignin treatment and MAH addition can lead to a reduction in composites hydrophilicity (Fig. 11e). A recent work was published dealing with the incorporation of high fractions of lignin (50–70 wt%) in different bio-polyamides,257 demonstrating that an organosolv hardwood lignin can be successfully blended, with the maximum processable lignin/PA blend ratio being 70/30 wt%, justifying, on the basis of reduced crystallinity and viscosity, the use of these blends as precursors for carbon fiber production. Blends containing poly(hydroxybutyrate) (PHB) and L-ER (variable content between 10–35 wt%) were also prepared and the interaction of epoxy groups of L-ER either with the end groups of the PHB or with diaminodiphenylmethane (a cross-linking agent), achieved a satisfactory homogeneity of the blends, totally degradable after 45 days.258
Rare examples of biopolymeric nanocomposites containing grafted or high lignin content can be found, essentially related to the mixing of clay, such as in the case of organo-modified clay.259 The prepared lignin/organoclay mixtures were mechanically mixed and subjected to subsequent melt intercalation. In general, thermal extrusion of the lignin/organoclay mixtures led to a slight increase in Tg, while the result of lignin condensation reactions increased the interaction with the organoclay and caused a significant intercalation of lignin into the galleries of the silicate layers of the organoclays.
In conclusion, although the physiochemical properties of lignin do not render it the best candidate for simple blending with other polymers, the simplicity of this concept has led to attempts of its blending for various uses. Romhányi and colleagues shed some light on the resulting properties of these systems,260 by concluding that miscibility, structure, and properties can be predicted with simple theories that preliminary look at the interaction strength parameters, such as in the case of evaluation of Flory–Huggins interaction parameter. Several efforts indicate the use of lignin in composites and blends, either as a matrix material, additive, filler, or strengthening agent. Mixtures involving lignins have been shown with bio/synthetic polymers. In most lignin/polymer blends, poor mechanical features are observed, even when compatibilizer are used. Efforts have been made to chemically modify lignin, e.g., with given functional groups targeting enhanced miscibility, compatibility, and superior performance in lignin-polymer systems.
3.4. Lignin as filler in natural matrices – composites and nanocomposites
Lignin has been applied to natural matrices originated from multiple sources, from animal to plant origin. However, cases where natural materials are isolated in large scale and utilized in material production that outperform synthetic ones are not very common. One recent example relates to the synthesis of elastomers based on castor oul and lignin.261 Natural rubber (NR) is also a prime example of a material with such characteristic and is the focus of this session where lignin is used as an alternative filler in natural rubber composites. NR has been traditionally produced from Hevea Brasiliensis in regions of South America, South East Asia and small areas in Africa. It is mostly composed of poly(cis-1,4-isoprene) particles of ca. 1 μm and 0.2 μm stabilized by rubber elongation factor and small rubber particle protein (respectively, HbREF and HbSRPP) assembled into a monolayer at the particle surface. Such proteins are also known to promote rubber biosynthesis.262 Despite of the vast number (>2.500) of plants species from where NR can be extracted, many of them do not generate high enough yields or materials with the necessary quality. The molecular weight of poly(cis-1,4-isoprene) produced by H. Brasiliensis is about 1.3 MDa, while that produced by other plants may be much lower (e.g. <10 kDa), which may impair mechanical properties.263 Alternative sources of NR, such as Parthenium argentatum, exist and are under frequent evaluation.Considering production volume and function, tires are among the most important products containing NR. Tires are mostly composites having a range of additives, such as sulfur, metal wires, carbon-black, inorganic particles and others intended to aid processing or to improve durability and performance. Importantly, not only for tire applications, vulcanization is widely utilized for enhancing mechanical properties. This process is made by adding elemental sulfur and processing at temperatures around 140–200 °C, resulting in the NR crosslinking. Enhancement of mechanical performance is also achieved by adding ca. 20 parts per hundred rubber (phr – conventional measure used for NR composites) of carbon black (CB) as a reinforcing filler. CB is composed of roughly spherical particles of a few tens to a few hundreds of nanometers organized in the form of particle aggregates surrounded by a thin layer (ca. <3–10 nm) of tightly bound NR.264 Interestingly, such layer has been reported to have a much larger elastic modulus (ca. 1 GPa) compared to the neighboring NR layer (ca. 60 MPa) or to the main NR matrix (8 MPa).265 When CB accounts for less than ca. 40 phr, the composite mechanical properties are dominated by hydrodynamic interactions between the CB aggregate and the matrix. On the other hand, when the CB content of the composition is higher than that, such aggregates form a network that further reinforce the composite.264
CB has been traditionally produced from fossil fuel-based sources and is also known to release considerable amounts of CO2 during production. Therefore, since the 1940s, lignins have been evaluated as a possible replacement for CB.266 Different mixing methods have been tested for achieving proper dispersion of lignin in the NR matrix. Optimal mixing is usually achieved by a coprecipitation method, where lignin is dissolved in an alkaline aqueous solution and then mixed with the NR latex. Such mixture is then precipitated by slowly adding an acid (e.g. H2SO4).267,268 The precipitate is then used for further drying and vulcanization. This method results in composites with smaller and more homogeneous particles aggregates than via dry mixing, as shown by Barana et al. (Fig. 12a1,2).267 Also, Ikeda et al. demonstrated that such better dispersion directly affected the tensile strength of composites having 5, 10, 20 or 40 phr of Kraft lignin (Fig. 12b1,2).268 In fact, it was observed that the lignin particles dispersed through coprecipitation had a similar network-forming behavior and reinforcing mechanism to that of CB. The importance of surface functional groups and interfacial affinity was demonstrated in the work of Jiang et al. They made hybrid cationic colloids by slowly adding an alkaline solution (pH 12) of sulfate lignin to an equally alkaline solution of poly(diallyldimethylammonium chloride) under vigorous stirring. The resulting cationic particles suspension was added to the latex and further processed into vulcanized NR composites containing 1, 3, 5 and 7 phr of filler. When compared to unmodified sulfate lignin processed through the same coprecipitation method, the cationic particles resulted in better dispersion, mechanical properties, thermal stability and degree of crosslinking.
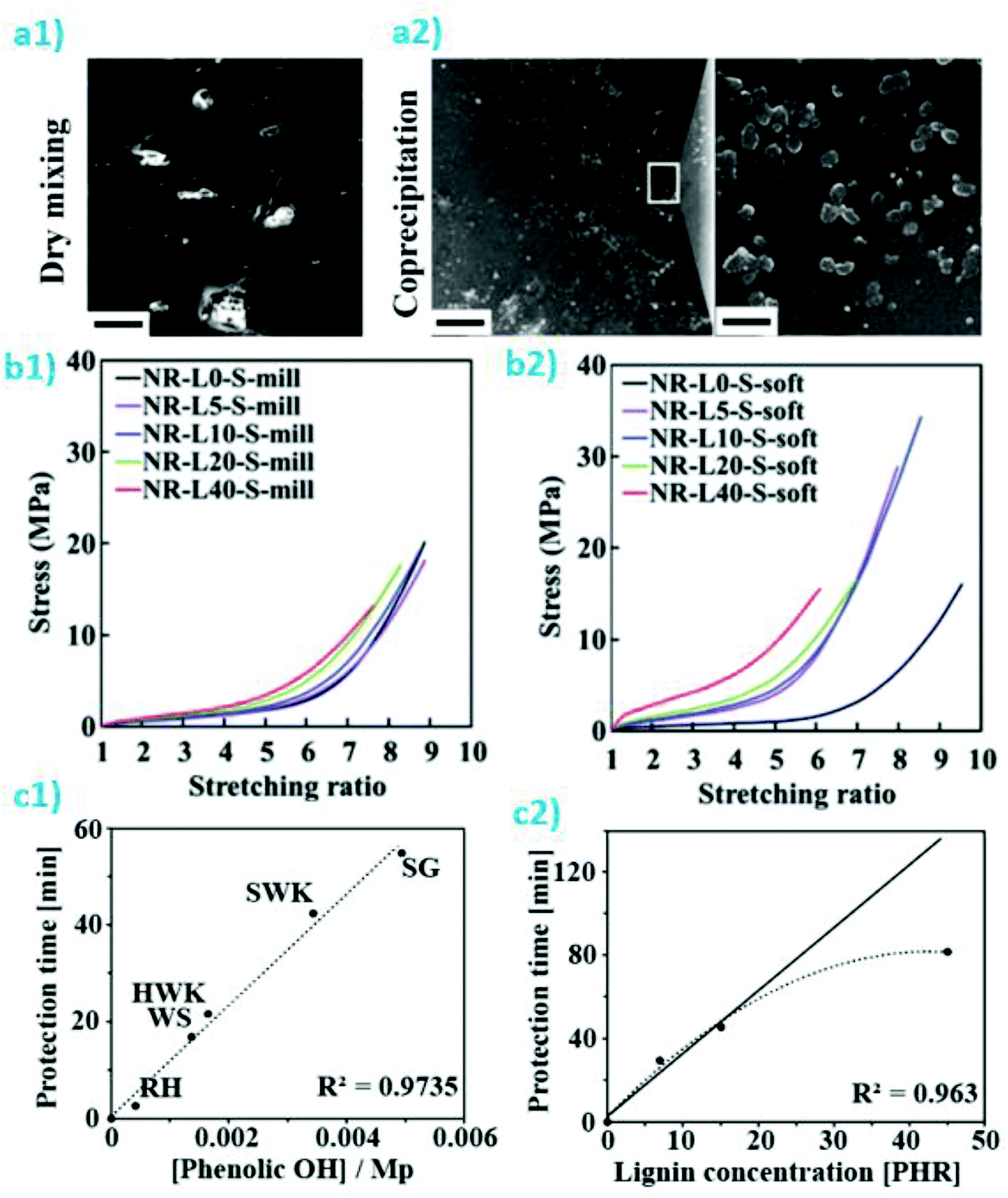 | ||
| Fig. 12 SEM images of a NR composite reinforced by 15 phr soda grass lignin particles mixed via (a1) dry mixing or (a2) coprecipitation. Reproduced with permission from ref. 267. The scale bars are 40 μm (left and center) and 4 μm (right), respectively. Tensile tests from NR composites having 0, 5, 10, 20 and 40 phr sodium lignosulfonate and processed via (b1) dry mixing or (b2) coprecipitation. Reproduced with permission from ref. 268. Oxygen induction time results depicting the onset of exothermal peak of samples heated to 170 °C in nitrogen and further isothermally exposed to oxygen atmosphere: (c1) results as a function of phenolic hydroxyl groups and molecular weight from lignins of different origins (i.e. RH = rice husk, WS = wheat straw, HWK = hardwood from Kraft process, SWK = softwood from Kraft process, and SG = soda grass). (c2) results as a function of SWK lignin concentration. Reproduced with permission from ref. 267. | ||
Antioxidant properties have been shown to correlate with lignin type and network formation. Barana et al. compared the performance of five different types of lignins (i.e. soda grass, softwood kraft, hardwood kraft, steam exploded wheat straw, and rice husk from a mild sodium hydroxide extraction) at 15 phr in composites produced through the coprecipitation method.267 They noticed that, despite of the different botanical origin and extraction process, smaller molecular weight and higher content of phenolic hydroxyl groups resulted in higher oxygen induction time (Fig. 12c1). These results were mostly attributed to the well-known antioxidant activity of phenolic hydroxyl groups as well as to the higher mobility and solubility of low molecular weight lignins in the rubber matrix. Upon heat exposure during the processing, such antioxidant effect protected the composite and ultimately resulted in better mechanical properties. Importantly, improvement in antioxidant activity did not exactly correlate with the loading of lignin particles at concentrations higher than 15 phr (Fig. 12c2). At such high concentrations, upon particle addition, the size of aggregates increases and network formation results, similarly to CB. When this occurs, particles addition does not effectively translate to increased interfacial area between filler and matrix, which is of fundamental importance for the antioxidant effect.
In conclusion, lignin can be effectively used for improving mechanical and thermal properties of NR composites. Surface functionalization can be effective for aiding in the vulcanization process. The coprecipitation method showed some of the best results for achieving proper dispersion, similar to that of CB fillers. This method, however, may be impractical for industrial application.269 Therefore, further research is needed aiming at refining the processing conditions not only for optimal performance, but also for industrial implementation. For further insights on the state of the art on such composites, the reviews by Roy et al. and Aini et al. are suggested.269,270
3.5. Lignin as filler in biobased synthetic matrices – composites and nanocomposites
As summarized in a recent review on lignin valorization,45 lignin can enhance the mechanical and thermal stability of polymer blends or composites, acting as an adsorbing agent, reactive component in various resins, UV blocker, antimicrobial agent or flame retardant. However, it has been observed that the use of irregular lignin powders hinders high loads in composites, mostly because of the natural poor compatibility, so it has been concluded that controlled surface shape and chemistry could overcome these issues and win on miscibility crucial effect with the matrix.9 Many literature works reported the combination of lignin fillers, both at the micro and nanoscale, all of them claiming that blending with lignin is a reasonable way to improve both the compatibility and the mechanical properties of reference biopolymers and reduce, at the same time, the costs. Results on PLA,271 polybutylene adipate terephthalate (PBAT),272 PHB,273 are examples of how lignin can be largely effective in enhancement of thermal and mechanical behavior of biopolymers.To deepen the analysis and limit the discussion to innovative results on biopolymeric matrices, we here considered the combinations of lignin with synthetic biopolymers in a hybrid configuration, where lignin filler was even added in the presence of organic or inorganic counterparts. Organic–inorganic hybrid materials have received wide-ranging attention in recent years, so the preparation of lignin/inorganic nanocomposites provides a new approach for high-value application of industrial lignin, potentially combining the advantages of all components to access complementary properties and synergetic effects.10,79 Emphasis is put largely on the chance of using lignin as a precursor for metal or metal oxide synthesis to be further incorporated in polymeric matrices,274 although a few works are available dealing with the incorporation of lignin,275 in the presence of other inorganic nanoparticles, with the main aim of synergistically combine fillers to enhance structural and functional properties of biodegradable polymer matrix.
When SiO2 was mixed with macro-scaled lignin particles and combined in PLA,276 the results evidenced how the presence of hybrid filler in PLA matrix influences the formation of transcrystalline layer and thereafter the supramolecular structure of composites. The same authors investigated the biodegradation behavior of SiO2–lignin/PLA composites,277 showing how composite with 2:1 hybrid filler was the most thermally stable (comparable to unfilled PLA) and underwent biodegradation process most effectively. This combination is also effective from the point of view of fire retardancy.278
At the nanoscale, Fal et al. prepared nanocomposites based on plasticized PLA and various mass fractions of silica–lignin (SiO2L) by melt blending and injection molding:279 experimental results clearly showed that, depending on the mass fraction, the electrical conductivity of the systems can increase by several orders of magnitude. The most favorable effect of SiO2L nanoparticles was observed at the two highest mass fractions tested (10 and 15 wt%). Also TiO2 was considered in an hybrid configuration: as separated filler in polyurethane,280 or a decoration for lignin particles (TiO2@lignin), that were synthesized successfully by hydrothermal method in aqueous solution to improve the UV shielding performance of lignin particles. Poly(propylene carbonate) (PPC) composite films with different contents of TiO2@lignin were prepared via a blade-casting method.281 The results confirmed that the presence of TiO2@lignin could significantly improve the thermal stability of the PPC based composite films.
Results of how hybrid polymeric composites containing metal oxide and lignin can perform are available in the works of Klapiszewski et al.,282–284 where the authors showed that the use of lignin did not deteriorate the mechanical properties of the composites and contributed to enhanced antimicrobial activity of the films. From the same group we have information on hybrid MgO/lignin,285 and organically modified silica with gold nanoparticles in the presence of lignosulfonate,286 where it was shown that lignosulfonate can be used as an effective reducing agent for gold ions and simultaneously as a stabilizing agent for gold nanoparticles (AuNPs).
With the main aim of designing and preparing lignin and other biomass-based polymer nanocomposites, graphene oxide (GO)–lignin hybrids were considered as reinforcement in PVA matrix via solvent casting method. Due to the strong interfacial interaction and the synergistic effect generated from the combination of the GO and AL, thermal stability, Young's modulus and tensile strength of all the resulting PVA/GO–AL nanocomposites were largely enhanced.287 3D porous graphene/lignin/sodium alginate nanocomposites were also successfully synthesized via a green hydrothermal self-assembly of a mixture of GO solution in the presence of lignin and alginate, followed by ionic (Ca2+) cross-linking.288 The as-prepared material was a three-dimensional porous graphene that could be used as adsorbent for removing heavy metal ions from aqueous solutions, providing an extensive and facile route to prepare high-performance adsorption material.
Organosolv lignin was also used as a reducing agent for the preparation of silver NPs to be introduced in PLA. In that work, Shankar et al.275 confirmed that mechanical and water vapor barrier properties of the composite films increased after the incorporation of lignin and AgNPs. The films containing AgNPs exhibited also potent antibacterial activity against Escherichia coli and Listeria monocytogenes. A novel and facile method to synthesize antibacterial microencapsulated phase-change materials (microPCMs) decorated with silver particles, where lignin was acted as Pickering emulsion stabilizer and as a reducing agent for silver, was considered by Li et al.289 Their results showed that the lignin particles, embedded in the microPCMs shell and utilized to reduce silver ions, resulted in silver particles decorated microPCMs (Ag/lignin microPCMs) (Fig. 13a). These microparticles exhibited a well-defined core–shell spherical morphology and presented good antibacterial activity, showing great potential in industrial applications such as biomedical, textile and construction areas. Furthermore, lignin, when mixed with PVA to prepare electrospun nanofilaments, was combined with AgNPs, revealing good miscibility of the lignin and silver NPs with PVA.290 The antimicrobial activity of the PVA–lignin nanofibers containing silver nanoparticles revealed growth inhibition against Bacillus circulans and Escherichia coli. These results suggest that PVA–lignin nanofiber containing silver nanoparticles may have potential applications as membrane filtration, antimicrobial fabrics, and wound dressing material in other biomedical applications, which will be explored in the near future.
 | ||
| Fig. 13 (a) Optical images of (I) lignin microPCMs and (V) Ag/lignin microPCMs; SEM images of lignin microPCMs and (II and III) before and (IV) after washing with NaOH; SEM images of Ag/lignin microPCMs before (VI and VII) and (VIII) after washing with NaOH, reproduced with permission from ref. 289. (b) Advancing and receding contact angles, and snapshots taken after the first second of contact between a water droplet and the surface of the mats, Reproduced with permission from ref. 291. (c) Schematic representation of surface composition of composite electrospun lignin/PVA fibers as a function of bulk lignin content, FESEM micrograph of 20:80/15% lignin/PVA/CNC fibers. Reprinted with permission from. Reproduced with permission from ref. 292. | ||
Many contributions are definitely available on the combination of cellulose and lignin, both at micro and the nano-scale. The works of De Oliveira Santos et al.291,293 investigated the final surface properties of mats based on polyethylene terephthalate (PET) that can be tuned by adding cellulose and/or lignin to the electrospun solutions as well as by varying the dissolution time (Fig. 13b), conditions that could be exploited to develop bio-based mats with different properties. Defect-free electrospun fibers from aqueous dispersions of lignin, PVA, and CNCs were produced:294 the results revealed that the thermomechanical performance of lignin: PVA electrospun mats was improved upon the addition of lignin, because of the strong molecular interaction with PVA. The addition of CNCs further improved such properties. It was observed that CNCs also lead to the stabilization of the matrix against water absorption. The same group investigated the surface polymer distribution, with conclusions on the effect of lignin and CNCs concentration on fiber radii and identification of a threshold around 50 wt% bulk composition for partitioning of PVA and lignin components on the surface below and above this value (Fig. 13c).292
Recent years have seen several developments toward the production of lignin-based hybrid materials with controlled architecture, down to the nanoscale. This field, which is still largely underexploited for lignin-based materials, is of particular interest to bring lignin to high added-value applications.
4. Lignin-based nanohybrids
4.1. Metal oxide–lignin hybrids
Thanks to its many oxygen functionalities provided by the phenol, carbonyl, hydroxyl, aldehyde and methoxyl groups, lignin shows a high adsorption capacity towards metal ions and organic dyes. Taking advantage of such adsorption ability, most of the developed metal oxides–lignin nanohybrids have been aimed to wastewater treatment. In this context, Li et al. prepared magnetic lignin spheres (MLS) by attaching 11 nm diameter Fe3O4 nanoparticles onto lignin hollow microspheres of 500–1700 nm diameter via electrostatic interactions with the electron-rich oxygen and hydroxyl groups of lignin (Fig. 14a).295 The rough surface and tiny holes onto lignin spheres provide further immobilization points for Fe3O4 NPs, yielding lignin hybrids with magnetization saturation (Ms) values of 22.7 emu g−1. Thanks to the high adsorption capacity of lignin towards organic dyes and the associated Ms, low cost and reusable highly adsorptive magnetic materials for organic dye removal (methylene blue and Rhodamine B) were obtained, solving the often difficult recovery process of adsorbents from the polluting medium. Similarly, Geng et al. used a one-step fabrication approach to obtain magnetic lignosulfonate with a Ms value as high as 44 emu g−1 for pollutant treatment. To that end, FeSO4·7H2O and FeCl3·6H2O were added into sodium lignosulfonate dissolved in water and after NaOH addition and subsequent ultrasonication, magnetic lignin with a curious sunflower morphology comprising homogeneously distributed Fe3O4 NPs was obtained (Fig. 14b).296 Interestingly, this process avoids the need of an oxygen-free environment to protect Fe(II) from oxidation, while no acid was added to preserve Fe(II) and Fe(III) from hydroxide formation. Thanks to the negatively charged surface provided by the sulfonate, magnetic lignin showed a great potential to adsorb positively charged substances such as the highly toxic and carcinogenic Cr(VI) metal ion. | ||
| Fig. 14 Morphology of diverse metal oxide@lignin NPs: (a) SEM image of magnetic lignin spheres having Fe3O4 NPs attached on the smooth surface (yellow circle) and into surface holes (green circle). Reproduced with permission from ref. 295; (b) TEM image depicting the sunflower morphology of magnetic lignosulfonate NPs comprising Fe3O4. Reproduced with permission from ref. 296; (c) TEM image of Fe2O3@lignosulfonate@polystyrene composite latex particles prepared by Pickering miniemulsion polymerization. Reproduced with permission of Elsevier from ref. 301; (d) HRTEM image on an individual Fe3O4@lignin NP. Reproduced with permission from ref. 303 and (e) HRTEM image showing the lignin coating onto a Cu2O NP. The lattice spacing is about 0.24 nm, in agreement with the crystal lattice theoretical for the (111) plane of Cu2O. Reproduced with permission from ref. 305. | ||
Pollutant removal can be also performed through catalysis, where the intended pollutant is decomposed into harmless moieties. To that end, palladium was coated onto Fe3O4–lignin NPs to obtain magnetically reusable materials for the catalytic reduction of Cr(VI).297 Lignin was activated to yield calcium lignosulfonate to facilitate its bonding to Fe3O4, while PdCl2 was used as precursor in the presence of Fe3O4–lignin to obtain Pd@Fe3O4–lignin without the need of additional reducing agents or surfactants. NPs of 20–25 nm having Ms values of ∼30 emu g−1 were obtained. Provided by the three-dimensional amorphous structure of lignin, obtained nanohybrids are stable, present a high surface area and can be easily recovered using external magnets. Such nanohybrids were proven efficient for Suzuki–Miyaura reactions, showing superior reaction yields compared with other catalysts.
Lignin can be used as stabilizer in oil-in-water emulsions.298–300 As shown in Fig. 14c, Fe2O3@lignosulfonate@polystyrene composite latex particles were prepared through Pickering miniemulsion polymerization.301 Negatively charged lignosulfonate (−34 mV) and positively charged 23 nm (diameter) γ-Fe2O3 NPs (+45 mV) were combined through electrostatic interactions to modify the amphiphilicity of γ-Fe2O3 and allow its use as Pickering miniemulsion polymerization stabilizers with no need of auxiliary surfactants. After polymerization, latex particles with interesting strawberry-like morphologies and superparamagnetic properties (Ms of 30.9 emu g−1) were obtained. The size of the latex particles could be controlled in the 121 to 245 nm range with increasing γ-Fe2O3 concentration.
The numerous and diverse functional groups comprising carbonyl, methoxy, carboxyl and hydroxyl groups of lignin provide a unique platform for the design of advanced sensing materials. In this sense, nanohybrids based on 8–12 nm Fe3O4 spherical NPs coated with lignin and polydopamine having a zeta potential of −27.5 mV were used as multifunctional substrates to immobilize glucose oxidase (GOx). Thanks to the notable immobilization effectiveness of 29.44 ± 2.39 mg enzyme per g of support, a biosensor with a Limit Of Detection (LOD) of 10 mM through a photometric assay was constructed by mixing synthesized nanohybrids with a carbon paste electrode and ferrocene.302 Fe3O4 and lignin were also combined via self-assembly to obtain materials for colorimetric H2O2 detection with a LOD of 2 μM.303 The size of the spherical lignin coated with tiny Fe3O4 NPs changed from 153 to 764 nm as a function of lignin concentration (see Fig. 14d). Lignin provides high dispersion stability of magnetic NPs in water as a result of the electrical double layer repulsion arising from the deprotonation of phenolic hydroxyl and carboxyl groups of lignin. Thanks to such dispersion stability, resulting nanohybrids showed a good peroxidase mimic activity, improving the LOD of bare Fe3O4 NPs. Magnetic microspheres having a silica-coated poly(styrene-co-acrylic acid) core and Fe3O4/Au shell were modified with aminated lignin using a two-step carbodiimide coupling process for surface enhanced Raman scattering (SERS) sensing of 2,4,6-trinitrotoluene (TNT).304 Au NPs can act as efficient SERS active nanostructures as they remain closely packed on the surface of the microspheres. Because of the high affinity of lignin towards TNT through hydrogen bonding, its presence provided a high selectivity and a significant enhancement of the SERS signal (8.6 × 108, two orders of magnitude above the NPs without lignin), reaching a LOD as low as 1 pM.
Several pH-responsive nanohybrids based on lignin and metal oxide NPs have been developed so far. In this context, magnetic lignin NPs useful for Cellic CTec2 cellulase immobilization and recovery were obtained by coating quaternized lignosulfonate onto Fe3O4 NPs using a simple co-precipitation approach.306 Due to the presence of both negatively and positively charged groups of quaternized lignosulfonate, the obtained magnetic nanohybrid showed different binding capacity towards cellulase at different pH values. Moreover, the quaternary ammonium group provided a strong attraction towards cellulose, allowing easily recoverable materials through external magnetic fields with immobilization values as high as 55.5%. In another work, Fe3O4 NPs were coated with lignin and lignin amine via a one-step co-precipitation and complexation to obtain magnetic materials with pH-dependent adsorption capacity.307 By coordination, the phenolic groups of lignin and lignin amine can adsorb Fe ions, avoiding NP aggregation during nucleation and subsequent growth. The thickness of the lignin grafting layer could be controlled by modifying the amount of incorporated lignin during the synthesis. Because of the interaction of dyes with lignin via π–π stacking and electrostatic attraction interactions, and the large active surface area of the obtained nanohybrids, adsorption capacities as high as 211 mg g−1 for methylene blue were achieved. Interestingly, 90% of adsorbates can be released by simply changing the solution pH due to the fact that a pH change modifies the ionization degree of lignin and lignin amine coating.
Other lignin@metal oxide nanohybrids have also been prepared. For instance, Liu et al. functionalized cerium oxide (Ce2O) NPs with aminated lignin through an in situ precipitation approach to obtain materials for wastewater treatment.308 The prepared nanohybrids showed a diameter of 15 nm and a BET surface area of 90 m2 g−1, providing a 14-fold increase in phosphate adsorption capacity in comparison with bare lignin. Moreover, the inherent adsorbent capacity of Ce2O towards phosphate was upgraded using lignin as a support material, which avoids NP agglomeration and provides functional groups for the complexation between the Ce–OH of the nanohybrids and phosphate via Ce–O–P coordination bonds. In another work, the abundant oxygenous hydroxyl groups of lignin were used as a reducing agent for Cu(II) ions into cuprous oxide (Cu2O) NPs, bypassing the use of environmentally toxic reducing agents required to synthesize Cu2O NPs.309 At the same time, the oxygen moieties in lignin play a stabilizing role, yielding broccoli-like NPs of 100–200 nm wrapped by a lignin layer. Obtained nanohybrids showed a good bactericidal activity against E. coli and S. aureus and a low cytotoxicity. Zhou et al. used 605 ± 5 nm lignin nanospheres as a supporting material to prevent catalyst deactivation resulting from Cu2O agglomeration.305 The polar phenolic and alcoholic hydroxyl groups in lignin reduce cupric sulfate in water media at room temperature to yield 20 nm Cu2O NPs, while the 3D molecular structure of lignin provides a stable support for catalysis, resulting in raspberry-like catalysts covered by a thin lignin layer (Fig. 14e). When cuprous oxide was used as a catalyst for “click” reactions (involving the formation of irreversible carbon–heteroatom and carbon–carbon bonds), yields up to 99% were obtained under solvent-free conditions. Moreover, catalytic efficiency remained unchanged after several cycles, highlighting the stable structure provided by the lignin nanospheres.
Lignin could be used as a template to synthesize mesoporous titania (TiO2) NPs with increased photo-catalytic activity.310 To that end, a hydrolysis precipitation method using TiCl4 as a reactant and lignin as a template followed by calcination at 500 °C was applied. The presence of lignin creates mesoporous catalysts having a loose structure with abundant uniform nano-pores, reaching specific surface areas (SSA) of 166 m2 g−1. This structure proved beneficial not only to enhance the surface permeability for pollutants but also to allow a quick transfer of light-excited carriers to the TiO2 surface, limiting carrier recombination rate and boosting photocatalytic reactions. Additionally, the presence of trapped lignin and its pyrolyzed carbon was found to delay the anatase-to-rutile transition during calcination, providing a larger fraction of the more active anatase within the mesoporous titania. As a result, when irradiated with UV light, 98% of phenols can be degraded in 120 minutes in comparison with the extent of 76% degradation when no lignin was used. Conversely, lignin can also provide a protecting layer to quench the photocatalytic activity of metal oxides, which is of prime interest in sunscreen formulations, which incorporate TiO2 to reflect and scatter UV radiation but should avoid undesired photocatalytic reactions which produce reactive oxygen species (ROS) which can damage living tissues. Accordingly, Morsella et al. coated anatase (20–40 nm) and rutile (80–100 nm) TiO2 NPs by co-precipitation of lignin under appropriate pH values in the presence of titania NPs.311 A complete inhibition of the production of singlet oxygen by TiO2 was observed, while its photo-protection character was kept unchanged. Such results were ascribed to the dissipation of photogenerated electrons upon UV irradiation by the lignin phase, where lignin acted as a free radical scavenger layer.
Similarly to TiO2, mesoporous ZnO NPs with high sunlight photocatalytic activity were fabricated using lignin-amine as a template.312 Following a solid-state reaction between zinc nitrate [Zn(NO3)2], sodium oxalate (Na2C2O4) and lignin-amine, and a subsequent calcination process, ZnO NPs with a SSA up to 35.5 m2 g−1 (in comparison with 14 m2 g−1 with no lignin-amine) and a red-shifted absorption wavelength were obtained, enhancing the absorption of visible-light. These improvements were ascribed to a combination of the electrostatic attraction for zinc ions and oxalate ions of lignin-amine with the steric hindrance effects of lignin, which ensures small and uniform ZnO NPs. As a result, 96.5% of methyl orange was degraded after 6 h of solar illumination. ZnO is also widely used as a sunscreen additive to prevent skin damage as it shows a good UVA radiation blocking ability. The skin photoprotection of ZnO was upgraded by combining flake shaped ZnO NPs with spherical lignin NPs.313 Lignin NPs provided a marked absorption in the UV-B and UV-C regions (reaching sun protection factors up to 8.5), resulting in hybrid materials which can absorb throughout the entire UV region.
4.2. Metal–lignin hybrids
The general preparation of metal NPs involves the reduction of metal ions into the zero-valent state followed by a stabilization step. Typically, these syntheses are performed in the presence of toxic agents (such as hydranize or dimethyl formamide) and/or capping agents, making their fabrication tedious, expensive and non-sustainable. In this context, lignin can be used to minimize the environmental impact of NP synthesis as it can act as a reducing, capping and stabilizing agent. Additionally, lignin provides a substrate where NPs could be anchored, avoiding their aggregation. Therefore, many efforts have been carried out to develop zero-valent metal NPs with the help of lignin.Owing to their strong antibacterial property and wide use in the biomedical and packaging field, silver (Ag) NPs are the most common noble metals synthesized in the presence of lignin. The antibacterial activity of Ag NPs is ascribed to the biologically active Ag+ ions, which can be released during the surface oxidation of silver atoms.314 Ag NP synthesis usually comprises a bottom-up reduction of silver salts in the presence of capping agents to control particle size and shape while favoring a good dispersion of the particles. Hydroxyl-containing groups present a great potential to reduce silver cations into silver nanospheres,315 opening the path to the use of lignin as a binding, complexing, reducing or capping agent for the synthesis of silver NPs. For instance, Hu et al. used AgNO3 as a precursor and alkali lignin as dual reducing and capping agent to obtain polydispersed Ag NPs at pH < 7.5 (slow pseudo-first order kinetic reaction), while pH values >8.7 allowed the fabrication of monodisperse and large Ag NPs (quick self-catalyzed reduction of Ag+ onto Ag2O surfaces).316 Lignin has been used as environment-friendly reducing and stabilizing agent to synthesize spherical Ag NPs with diameters of 24 nm under microwave conditions (10 min at 60 °C using 50 mg of lignin and 0.3 mM of Ag+).317 During microwave irradiation, the phenolic lignin of α-aryl ether and alkyl ether changed promoted the fracture of the ether bond, yielding a quinone methide and eventually formaldehyde, which reduces Ag+ ions to Ag0, while its spatial three-dimensional structure provides lignin a great stabilizing function, preventing Ag NPs from aggregation even after centrifugation. As a result of the complexation between the carboxyl/hydroxyl groups of lignin and Hg2+, lignin-capped Ag NPs presented a LOD of 23 nM for Hg2+. Interestingly, the shift in the surface plasmon resonance (SPR) band of silver was highly selective to Hg2+, as no color changes were observed in the presence of 19 other metal ions.
In spite of the broad use of Ag NPs in the biomedical field, their use raises serious concerns as they could persist in the environment for long times, resulting in hazards to living organisms and diverse ecosystems.318 In this context, Richter et al. infused silver ions into degradable lignin (Indulin AT) NP cores using an aqueous solution of AgNO3. These NPs were then coated with a cationic polyelectrolyte to limit the inherent toxicity of Ag while ensuring a good adhesion of the nanohybrids to bacterial cell membranes, killing a broad spectrum of bacteria.319 In comparison with Ag NPs, which release Ag+ ions during post-utilization (Fig. 15a), prepared lignin-Ag nanohybrids coated with a cationic polyelectrolyte layer (84 ± 5 nm in diameter) attached well onto negatively charged bacterial cell walls through electrostatic attraction (Fig. 15b). The prepared materials lost their antimicrobial activity after use and can be degraded once disposed. Interestingly, NPs exhibited broad-spectrum biocidal action using 10 times less silver than when using conventional silver NPs. Following a similar approach, lignin NPs were coated with a cationic polymer layer and infused with silver.320 Spherical silver carboxylate colloidal lignin NPs with sizes ranging from 60 to 200 nm were prepared after a deprotonation reaction of an organic solution of anhydrous lignin and subsequent ion exchange with silver nitrate and solvent exchange. During reaction, carboxylic and phenolic groups of lignin are deprotonated with sodium methoxide to yield sodium carboxylates and phenolates where Ag is ionically bound via ion exchange with AgNO3. Prepared NPs can be stored as aqueous dispersions for long periods of time with no loss of their antibacterial activity as they only release Ag+ ions in the presence of counter-cations, i.e., under physiological conditions. As a result of the strong binding of Ag+ with the carboxylated groups present onto lignin NPs, longer and sustained activity times are obtained against Gram-positive and Gram-negative strains.
 | ||
| Fig. 15 Lignin for the synthesis of noble metal NPs: (a) Scheme summarizing the bactericidal action of Ag (left column) and lignin-Ag NPs (right column). Reproduced with permission of Nature Publishing Group from ref. 319. (b) Schematic diagram for the reaction mechanism of alkali lignin and Ag2O in DMSO solvent to yield Ag NPs. Reproduced with permission from ref. 321. (c) HRTEM images of kraft lignin–AuAg core–shell NPs. Reproduced with permission from ref. 322. (d) SEM image of Pd@LPR nanocomposite. Reproduced with permission from ref. 323. | ||
Combining the good adsorption capability of silica and lignin, hybrid Ag NPs were grafted onto silica/lignin nanohybrids to prevent undesired desorption of silver ions and obtain stable antibacterial materials.324 Silica was modified with aminosilane and then lignin was oxidized using a sodium periodate solution to afford a better Ag–lignin interaction. The cost-efficiency of the process was improved by the presence of lignin, which favors silver NP dispersion due to the steric repulsion provided by its negatively charged surfaces, affording the use of less quantity of Ag NPs for a given antimicrobial activity. As the nanosilver remains trapped inside the lignin, its excessive exposure with microbes is reduced, providing materials with extended longevity of antibacterial activity. In another work, the tendency to form large-size aggregates of carrier-free nanocrystalline metal NPs was solved using a polyacrylamide-modified lignin hydrogel as a catalyst carrier.325 The abundant amino groups can disperse Ag+ ions homogeneously to limit the growth of silver NPs and avoid their large-scale agglomeration during complexation and reduction of AgNO3 with NaBH4.326 As a result, spherical Ag NPs of 3–6 nm in diameter were synthesized, which presented good catalytic activity towards the dynamic hydrogenation reduction of p-nitrophenol (97% conversion after 10 cycles). Although typically salts such as AgNO3 are used as precursors for Ag NP formation, Ag2O could be also used. In this context, spherical 20 to 24 nm silver NP were synthesized for Hg2+ colorimetric sensing using alkali lignin (Mw: 4.300 g mol−1; –COOH content of 2.01 mmol g−1) as reducing and stabilizing agent and Ag2O as silver precursor and catalyst for the reduction process of Ag+ due to the metal–metal oxide interactions.321 After 12 h reaction at 25 °C in dimethyl sulfoxide (DMSO), a reduction capacity of alkali lignin to Ag+ up to 8 mM g−1 was reached, which is one of the highest values reported so far. This fact arises from the Lewis-based character of DMSO, which reactivates hydroxyl derivatives and enhances the reducing capacity of alkali lignin (Fig. 15b). Importantly, this process avoids the use of inorganic ions and replaces the traditional NaOH during Ag NP synthesis by lignin.
Inspired by dynamic plant catechol chemistry, lignin combined with Ag NPs was applied as a new design strategy to create long-lasting reductive-oxidative microenvironments. 12–145 nm core–shell Ag–lignin NPs generated free radicals and triggered self-gelation of a polymeric hydrogel under ambient environment through a redox catechol chemistry.327 In this approach, the phenol or methoxy groups in lignin reduce Ag+ to metallic silver NPs, while the functional groups of the lignin were oxidized to the corresponding redox-active quinone/hydroquinone. Ag NPs form photogenerated electrons because of their SPR, which convert the quinone/hydroquinone groups in lignin into catechol groups. As a result, mechanically tough hydrogels with good adhesion to many material surfaces and tissues arising from the catechol functional groups were obtained. Interestingly, catechol groups were continuously generated, endowing long-term and repeatable adhesiveness to the hydrogel, which also shows bactericidal ability from the Ag+.
Although most of the works on noble metal NP synthesis have been focused on silver, other elements have also been investigated. Given its non-toxic character, therapeutic activity, antioxidant/antimicrobial activity and special optical properties, gold is one good example.328 Unfortunately, gold NPs tend to aggregate into bulkier materials, limiting their SSA and thus effectiveness. Therefore, gold NPs were anchored to lignocelluloses fibers removing the need for external immobilizing agent for active packaging applications.329 When chloroauric acid (HAuCl4) is added into aqueous unbleached softwood kraft pulp slurry, the aromatic methoxy and phenol groups of lignin reduce Au3+ to Au0 at the same time that they bind formed NPs onto the surface of the fibers. NP formation can be easily followed by naked eye as the color gradually changes from light brown to dark purple due to the SPR of the Au NPs at 537 nm. In another work, a one-pot thermal and photochemical synthetic approach was followed to obtain colloidal lignin-stabilized gold NPs, where lignin acted as a reducing and capping agent.330 HAuCl4 was reduced to Au in few minutes to yield NPs having 12 to 25 nm in diameter and Zeta-potential values from −54 to −27 mV. Thanks to the stabilizing effect provided by lignin, the obtained nanohybrids remained well-dispersed in different biological media such as phosphate buffered saline, Luria Bertani (LB) broth and Dulbecco's Modified Eagle's Medium (DMEM) for more than a week, allowing their use in diverse clinical settings. Importantly, obtained nanohybrids showed photoinduced antimicrobial activity and the outer lignin layer enhanced the interaction with the bacterial cell wall, providing a good selectivity towards different type of bacteria.
To enhance the catalytic performance, selectivity and stability of gold NPs, lignin has also been used to prepare gold-based bimetallic nanohybrids. These hybrids present combinational interactions between two or more metallic electronic states which are different from the individual metals, providing unique physicochemical and surface characteristics to the resulting hybrids. For instance, Au–Pd nanoalloys with a core–shell structure were obtained after HAuCl4 reduction in the presence of an aqueous basic lignin solution and subsequent Pd(NH3)2Cl2 reduction under microwave.331 This method avoids the use of toxic reducing agents or additional stabilizers, resulting in nanoalloys ranging from 20 to 50 nm. Moreover, the obtained NPs outperformed the single-component catalysts in reduction of 4-NP to 4-AP, and were stable in aqueous dispersions thanks to the stabilizing effect of lignin. Lignin-stabilized Au–Ag bimetallic nanocomplexes were synthesized using lignin as the sole source for reducing, capping and stabilizing effect towards the nanoagents.322 Following a seed mediated growth, pre-synthesized lignin–Au was used as seeds for the reductive deposition of Ag, and yielding 36 nm core–shell Au–Ag nanohybrids (see Fig. 15c for the corresponding HRTEM image). The phenolic and flavonoid groups provided by the outer lignin layer, together with the bimetallic character of the nanohybrids resulted in materials displaying half maximal inhibitory concentration (IC50) values against tested microbial strains 28 times lower than those compared with monometallic silver NPs, highlighting the remarkable antimicrobial activity obtained.
Palladium (Pt) and platinum (Pd) NPs were synthesized using lignin and fulvic acid as both reducing and stabilizing agents.332 As water-soluble lignosulfonate derivatives contain easily oxidizable functional groups bearing –OH moieties, which can reduce high-valent metal ions such as PtIV and PdII into their zero-state, H2PtCl6 and PdCl2 were used as precursors to obtain 16–20 nm Pd and Pt NPs (80 °C in water under aerobic conditions). The obtained materials were effective towards the catalytic reduction of 4-nitrophenol and aerobic oxidation of alcohols. Spherical Pd NPs can be prepared using water-soluble lignin as a reducing and stabilizing agent under green conditions (short time, moderate temperature, water as a solvent and aerobic conditions).333 The reduction of PdII to zero-valent Pd proceed in 3 h at 80 °C, yielding NPs of 8–14 and 16–20 nm when Kraft lignin and lignosulfonate are used as reducing and stabilized agents, respectively. The obtained materials were useful for carbon coupling reactions under mild conditions, reaching conversions higher than 90% at yields of 72%. Pd NPs have also been supported onto lignin-based phenolic resin (LPR) nanospheres after an in situ reduction of PdCl2.323 The amphiphilic character of lignin decreases the surface tension of phenolic resin spheres and dissociates during curing, yielding small (200–500 nm) and rough LPR nanospheres where Pd2+ can be then reduced (via binding to the negatively charged nanosphere groups via electrostatic interactions) (Fig. 15d). The SSA of 23 m2 g−1 provided by lignin nanospheres and the good reducing activity of lignin resulted in a high-density coating of 30 nm spherical Pd NPs, which showed a high efficiency for the elimination of heavy metal and organic pollutants (reduction of highly toxic Cr(VI) into inert Cr(III) and two model dyes).
Regarding lignin-assisted multicomponent hybrids, spherical 5 nm bimetallic lignocelluloses-stabilized Pd–Fe NPs with different Fe contents (1–75 at%) were grown onto lignocelluloses fibers through wet chemical synthesis.334 The obtained NPs were composed by a zero-valent Pd–Fe alloy covered by a thin Fe3O4 layer. Importantly, lignin lowers the tendency of nano zero-valent iron (nZVI) to oxidation, thus lowering its catalytic and magnetic performance. Thanks to the tiny size of the synthesized materials and the protecting role of lignin against oxidation, the obtained nanohybrids present remarkable superparamagnetic properties, where the magnetic moment of the material fluctuates in response to thermal energy while keeping the internal ordered structure.335 Lintinen et al. prepared iron–lignin nanohybrids upon the reaction of iron isopropoxide and lignin in a THF solution.336 Synthetized Fe:lignin nanohybrids were stable in aqueous dispersions, and depending on the condensation and hydrolysis reaction parameters (molecular ratio, concentration and temperature), nanoparticles presenting a fishnet-type open network structure, small solid particles with a diameter of 10–30 nm, spheres of 50–400 or hollow structures were achieved.
Other zero-valent metal NPs (25 nm) such as Cu, Ni, Co, Ag and Zn have been synthesized using lignocellulosic biomass as support and CuSO4, NiSO4, CoNO3, AgNO3 and ZnNO3 as precursors.337 The combination of salts (CuSO4 + NiSO4, CuSO4 + CoNO3, CuSO4 + AgNO3 and CuSO4 + ZnNO3) was also investigated to afford Cu + Co, Cu + Ag, Cu + Ni and Cu + Zn NPs, respectively. Overall, lignin serves not only as a reducing agent but also as a supporting material to prevent the zero-valent NPs from oxidation and agglomeration, keeping their catalytic performance intact. In another work, a low energy and solvent-free mechanochemical grinding (29.5 Hz, 90 min) has also been pursued to obtain highly monodispersed Au, Pd, Ag, ruthenium (Ru) and rhenium (Re) NPs using lignin as both reducing and stabilizing agent.338,339 It was shown that mechanical breakdown of lignin yields phenoxy radicals which can be oxidized and behave as reducing agents. Au NPs of 15 nm (HAuCl4 precursor), Pd of 3 nm (PdCl2 precursor), Ag of 1–30 nm (AgNO3 precursor), Ru of 7 nm (RuCl3 precursor) and Re of 6 nm (Re(CO)5Br precursor) were obtained. Upon milling inorganic precursors in the presence of lignin, organometallic precursors yielded NPs encapsulated by lignin, while ionic precursors resulted in matrix-free NPs. As mechanochemical synthesis is atom economical, requires no solvent and needs a lower energy input, this procedure presents a bright future to develop lignin-metallic zero-valent NPs, albeit energy consumption required for milling should be considered.
The unique benefits/advantages of low cost, renewable lignin as support of metal/metal oxide nanomaterials and nanocomposites make them tremendously attractive for a diversity of catalytic applications. Associated systems have been shown for their adsorption capacities/efficiencies that are strongly dependent on the surface properties of the nanoparticles. Lignin and lignin-derived (nano)materials can efficiently uptake, adsorb and/or remove toxic heavy metal ions and organic/azo dyes via chelation, ion exchange, π–π stacking, hydrogen bonding, and/or electrostatic interaction. As the low cost of fabrication is crucial for a wider utilization, future efforts need to be in place to refine the economic viability of these renewable systems.
4.3. Carbon–lignin hybrids
Similar to noble metals and metal oxides, lignin has been hybridized with diverse carbonaceous nanofillers to improve their multifunctional properties. On itself, ligin can be used as a precursor to carbon nanotubes (CNTs),340 that is, quasi-1D allotropes of carbon that have attracted a great deal of attention since their discovery by Iijima in 1991,341 as well as graphene, a 2D allotrope of carbon with remarkable electrical conductivity, high mechanical strength and high SSA.342 Unfortunately, the application of these carbon allotropes has been limited due to their tendency to form bundled structures. Many different de-bundling strategies have been pursued to obtain dispersible nanocarbons, which mainly rely on the application of complex chemical routes for surface functionalization or use of non-renewable surfactants as dispersing agents.343 In this context, lignin has been used as an additive to improve the dispersion of CNTs in water.344 When compared with the commonly used sodium dodecylbenzenesulfonate (SDBS) surfactant, the use of lignin allows conducting fluids with lower viscosities, facilitating its processing. This improvement arises from the efficient dispersing and stabilizing effect of lignin, which avoids the formation of aggregated networks at high volume fractions. In another work, CNTs were functionalized with kraft lignin to improve their dispersion in organic (DMSO) and aqueous (0.1 M NH3) solutions.345 A dense and uniform adsorption of lignin on CNT surface was observed, where the aromatic moieties of lignin interact via π-stacking with the hydrophobic CNT surface while the hydrophilic backbones and phenolic –OH groups confer water solubility at high pH values. Because of the combination of the lignin-derived quinine moieties and CNTs, the obtained nanohybrid presented persistent reversible redox behavior, providing a 100% increase in the capacitance in comparison with non-functionalized CNTs. CNTs have also been modified with lignin to improve their inherently poor compatibility with polymeric matrices.346 To that end, sulfonated lignin was used as a natural polyelectrolyte to modify CNT surfaces via non-covalent functionalization, where the lignin's aromatic structures are highly compatible with CNTs through strong van der Waals and π–π stacking forces (Fig. 16a shows the lignin coating layer). As a consequence, when incorporated into PVA, modified CNTs composites displayed enhanced mechanical properties. Lignin has been mixed with CNTs not only to enhance their dispersability but also to provide additional functionalities. For instance, CNTs were modified with lignin in the presence of acid chloride under basic conditions to obtain materials which selectively absorbed oils and up to 10 different organic solvents but not water.347 Lignin was covalently linked to CNTs, which induced superhydrophobicity to lignin, thereby improving its oil/water selectivity, reaching sorption capacities up to 23 g g−1. Fe3O4 NPs were then incorporated into the CNT-g-lignin to enable an easy recovery of the absorbent via external magnetic fields. | ||
| Fig. 16 Carbon–lignin nanohybrids: (a1) HRTEM images of pristine and (a2) lignin functionalized CNTs. Reproduced with permission from ref. 346. (b) Photograph of the carnivorous Venus flytrap plant and SEM image of the graphene used to confine lignin. Reproduced with permission from ref. 348. (c) Scheme showing preparation process together with the interaction interactions between lignin nanocrystals and RGO. Reproduced with permission from ref. 350. (d) Schematic mechanisms for the synchronous preparation of LNP and CQDs from lignocelluloses via a one-pot microwave synthesis. Reproduced with permission from ref. 352. | ||
Owing to the reversible redox reactions in lignin, graphene in combination with lignin has been used to develop materials for energy storage. To simultaneously solve the low electric conductivity of lignin and its tendency to dissolve in the aqueous media, a hierarchical graphene cage was synthesized to capture lignin similarly to the carnivorous Venus flytrap plant (Fig. 16b).348 Lignin was trapped and wrapped into the graphene structure, preventing its dissolution by the electrolyte and providing efficient electron transport pathways for electrochemical reactions. Strong non-covalent π–π interactions were achieved because of the large number of aromatic rings in lignin. Interestingly, the estimated fabrication cost was as low as 4 Euro cents per g as only graphite, 30% H2O2 and FeCl3 are required. Another simple, quick and environmentally-friendly approach for the fabrication of stable graphene dispersion was patented in 2015.349 A redox reaction of lignin liquor dissolved in an alkaline solution with an aqueous graphene oxide solution at 110 °C was applied, where lignin behaved as a reduction and stabilizing agent. Other forms of graphene have been hybridized with lignin to novel multifunctional materials. For instance, reduced graphene oxide (RGO) has been combined with electroactive lignin nanocrystals to fabricate pseudocapacitor materials exploiting the fast and reversible redox charge transfer of surface-confined quinone and the interaction with highly-conducting RGO (Fig. 16c).350 The irreversible aggregation of RGO after the conversion of exfoliated graphene oxide (GO) was prevented by lignin nanocrystals, which remained strongly confined on the surface of RGO through combined π–π and hydrophobic interactions. Therefore, charges were transferred from the electron-withdrawing groups of lignin to RGOs, leading to fast/reversible pseudocapacitance and long-term stability. A protective layer of graphene nanoplatelets (GnPs) and CNTs with pre-adsorbed alkali lignin was coated onto flexible papers to provide self-extinguishing properties to the substrate.351 Lignin functionalization had a dual role of improving dispersion of GnP/CNT onto hydrophilic cellulose substrate (providing a physical barrier to limit the permeation of combustion gases) and providing a material with good char-forming capacity (limiting the transfer of oxygen and heat), resulting in fire-retardant properties (burned area was decreased from 100% to 21% after 15 s of ignition).
It should be considered that most of the reported works rely on the modification of pre-synthesized carbon nanomaterials with lignin, while the synchronous preparation of lignin and carbonaceous materials has received little attention so far. In this context, Si et al. used natural lignocelluloses as a source to obtain monodisperse LNPs and nitrogen-doped carbon quantum dots (CQDs) via a one-pot microwave synthesis.352 During the reaction, an acid catalytic ethanol-water co-solvent system dissolves the lignin fraction to form LNPs of 100 nm in diameter (formed via layer-by-layer self-assembly from inside to outside based on π–π interactions) (Fig. 16d). At the same time, an acidolysis process releases monomolecular compounds which acted as precursors for 2–3 nm CQD formation after dehydration, polymerization, condensation, aromatization and carbonization. Interestingly, LNPs were highly stable due to an electrical double-layer repulsion provided by the surface phenolic hydroxyl and carboxyl groups, while CQDs had upconversion photoluminescence.
The calcination of biopolymers such as cellulose and chitin has been widely explored to obtain nanoscale carbonized replicas for diverse applications.353,354 Thanks to its high carbon content (nearly one third of the organic carbon in earth), a high concentration of hydroxyl groups, low-cost and bio-renewable character, lignin has been used as a precursor to obtain carbonaceous structures. Using Pluronic P123 and silica NPs as templates, hierarchical porous carbon monoliths with tunable mesopore sizes and large SSA were obtained by carbonization of kraft lignin under an argon environment at 900 °C followed by template removal with 2 M NaOH.355 During self-assembly, Pluronic P123 and silica NPs acted as a scaffold to support the framework during pyrolysis, while the micelle domains (in the case of lignin/P123) and silica NPs were responsible for producing the mesopores in the resulting carbon. As shown in Fig. 17a, pore-morphology can be controlled upon the incorporation of silica NPs with different sizes. The obtained monolith was mechanically stiff, presented SSAs up to 635 m2 g−1 and showed an electrical conductivity of 1300 S m−1. As a result, such porous structures facilitate ion diffusion and enhance electrolyte/electrode contact area, making these materials suitable for energy storage.
 | ||
| Fig. 17 Carbon structures obtained from lignin: (a) Schematic representation of the porous carbon monolith formation using silica as pore-forming agent. SEM images corresponding to following samples are shown from left to right: no silica, 200 nm silica, 100 nm silica, 7 nm silica. Reproduced with permission from ref. 355. (b) HRTEM image of Co@C catalyst. Reproduced with permission from ref. 356. (c) Schematic representation summarizing lignin-intercalated GO film fabrication. Reproduced with permission of John Wiley & Sons Ltd from ref. 362. (d) Scheme showing the fabrication for the preparation of NiO/hierarchical mesoporous carbon nanospheres derived from lignosulfonate. Reproduced with permission from ref. 363. | ||
Not only monolithic carbon but also several carbon-metal and carbon-metal oxides hybrids have been synthesized from lignin. Co@C core–shell NPs for catalysis were prepared through a low temperature carbonization using cobalt nitrate as Co precursor and lignin as the carbon source.356 Co NPs were wrapped by several defective graphitic layers (16 nm thick shell) to yield nanohybrids of 20–150 nm having SSAs up to 236 m2 g−1 (see Fig. 17b for a representative HRTEM image). Such core–shell structure is especially useful for catalysis as the graphitic shell enhances the electronic conductivity between the Co catalyst and the surrounding environment, avoids NP aggregation during reaction and provides an increased adsorption space. Similarly, Fe NPs have been encapsulated by a graphitic carbon shell derived from lignin to obtain catalytic materials.357 To that end, lignin and ferric nitrate were mixed and submitted to a flash pyrolysis process, yielding 9 nm diameter metal cores wrapped by 1 to 5 graphitic layers (SSAs of 158 m2 g−1). Core–shell nanohybrids comprising a lignin-derived carbon external layer have been also developed for other applications. Microporous magnetic carbon materials derived from lignin were fabricated by coating lignin onto iron oxide NPs via precipitation and subsequent calcination of the complex under a nitrogen atmosphere at 500 °C for 3 h.358 Such carbonization conditions do not remove the O–H groups and C–C bonds of the native lignin from the surface and allow the formation of amorphous carbon with abundant pores of 77 nm in average, providing suitable supports for the removal of pollutants (e.g., adsorption of methyl orange of 113 mg g−1).
Because of their inherent catalytic activity, metal oxides such as TiO2 and ZnO have a good potential for environmental remediation applications. However, their performance is hampered by a fast recombination of photogenerated carriers, limited absorption within the solar spectrum and marked aggregation. Several works have faced these issues by supporting such NPs onto a porous carbon framework derived from lignin. TiO2/carbon nanohybrid fibers were prepared through electrospinning followed by carbonization under nitrogen atmosphere.359 The highly graphitized fibers had 207 nm in diameter and presented a homogeneous TiO2 NP deposition of 13.65 wt%. Good photocatalytic performance was obtained thanks to the combination of highly conducting and adsorbent carbonaceous support and catalytically active TiO2. Using a solvothermal approach, flower-like carbon consisting of 2D corrugated nanosheets were applied to support 10 nm ZnO NPs with Mg(OH)2 as structure inducer, providing materials with a SSA of 852 m2 g−1 but avoiding the characteristic stacking of many 2D materials.360 The oxygen-rich functional groups of the produced carbon provided an intimate contact with the pollutant, shifted the optical absorption into the visible region, and enhanced the separation of photogenerated electron–hole pairs as a result of the ZnO-carbon interface, increasing the catalytic activity 2.1 times in comparison with pure ZnO. Wang et al. synthesized nanohybrids containing highly dispersed ZnO NPs embedded onto lignin-derived carbon nanosheets for catalytic decomposition of organic pollutants.361 To enhance the interaction between lignin and the negatively charged ZnO, lignin was quaternized. Using zinc oxalate (ZnC2O4) as the precursor, lignin carbonization and ZnO growth occurred simultaneously at a relatively mild temperature of 550 °C (in comparison with 700–900 °C shown in other works). Interestingly, the CO2 and H2O gas originating from the ZnC2O4 decomposition resulted in a loose structure with a SSA of 140 m2 g−1, while the UV light absorption was markedly increased thanks to the benzene ring structure and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CO bonds originating from lignin, reaching a 5-fold enhancement of photocatalytic activity when compared with pure ZnO.
C and CO bonds originating from lignin, reaching a 5-fold enhancement of photocatalytic activity when compared with pure ZnO.
Other lignin-derived carbon nanohybrids have also been reported. For instance, alkali lignin was transformed into 1–4 nm luminescent graphene quantum dots (GQDs) via a two-step bottom-up approach involving hydrothermal re-fusion.364 Alkali lignin was fractionated with simultaneous S, N co-doping by esterification to generate lignin NPs which were catalytically cleaved via hydrolysis. Further hydrothermal treatment led to additional π–π stacking that boosted C–N, C–S and –C–SOx– bond formation, resulting in the development of S, N co-doped GQDs. Obtained GQDs exhibited a bright fluorescence with a quantum yield of 21%, water-dispersability and long photostability. Because of the sulfinyl group at the defect sites in GQDs could bind H2O2, this peroxide could be detected at concentrations as low as 0.13 nM through a luminescent quenching process. Pd NPs were supported onto a porous carbon framework using Pd(NH3)Cl2 as a precursor and lignin as a reducing/stabilizing agent and a carbon source.365 Using silica NPs as a hard-template, 3D interconnected mesoporous–macroporous structures with SSAs up to 1.467 m2 g−1 were achieved. An 80–100 nm thick highly graphitized layer composed of graphene-like lignin carbon sheets was deposited onto a copper foil by annealing lignin at 1000 °C in an argon atmosphere to obtain composites with a thermal conductivity of 478 W m−1 K−1, which is 43% higher than the neat copper foil.366 The graphitization process of lignin resulted in deoxygenation and dehydrogenation, while copper acted as support to enhance lignin graphitization.
Several reports discuss lignin carbonization to develop materials for energy storage applications.367 Although related efforts are addressed in detail in section 5.4, their synthetic approach is described here. A composite film of lignin and GO was simultaneously carbonized and activated at 750 °C under nitrogen atmosphere for 1 h to reduce GO and pyrolyze lignin to carbon (Fig. 17c).362 KOH serves to dissolve lignin and activate the obtained carbon generating 30–80 μm thick films with a remarkable SSA of 1.744 m2 g−1. In such binder-free hybrids, lignin-derived amorphous carbon acts as an intercalating agent to avoid restacking and agglomeration of the graphene sheets. Thanks to the large surface areas and absence of graphene stacks, the obtained hybrids were attractive for supercapacitor applications. Zhou et al. prepared hierarchical mesoporous carbon nanospheres hybridized with NiO NPs with SSAs of 852 m2 g−1 (Fig. 17d).363 Lignosulfonate nanospheres were treated at 800 °C in a nitrogen atmosphere and after in situ loading of Ni(OH)2 onto carbon spheres, and additional carbonization at 450 °C in a nitrogen atmosphere was applied to obtain highly graphitized porous carbon with some defects and sp2 hybrid structure, providing active sites for further reaction. The synergetic effect from the NiO transition metal, which presents a high theoretical capacity with the large conductivity and surface area provided by the carbon nanospheres enhanced the electrochemical performance of the resulting material.
Lignin-based carbon materials is a cross-cutting subject involving chemistry, chemical engineering, physics, engineering and other disciplines. How to control the preparation of lignin-carbon hybrids with excellent performance and good stability is still a challenging subject.
4.4. Biobased nanoparticles–lignin hybrids
Sipponen et al. reported an “all-lignin” approach for the preparation of cationic colloidal lignin particles.368 Softwood kraft lignin was cationized by an epoxy ring-opening reaction with glycidyltrimethyl ammonium chloride in aqueous sodium hydroxide solution. The water-soluble fraction of cationic lignin was thereafter was adsorbed on spherical lignin nanoparticles to reverse their surface charge from anionic to cationic. The minimum amount of cationic lignin needed to obtain stable dispersions was 40 mg g−1 relative to the LNPs. These cationic particles were later demonstrated as efficient (91%) adsorption of Lipase M for an enzymatic synthesis of butyl butyrate.192 Cationic LNPs have also been prepared by using synthetic cationic polymers such as poly(diallyldimethylammonium chloride),97,170 as well as natural polymers like chitosan,170 Concanavalin A (lectin),369 and a variety of proteins.370 LNPs with a cationic surface have been found to exhibit benefits in stabilization of Pickering emulsions,168,368 enzyme immobilization by adsorption,192,371 and more recently as agglomeration agents that facilitate virus removal from contaminated water.372 A new frontier in the context of LNPs is the synthesis of particles that resist dissolution in organic solvents and under extreme pH conditions. Zou et al. co-precipitated bisphenol A diglycidyl ether with softwood kraft lignin to produce hybrid particles that could be thermally cross-linked for improved stability.373 Recently, Moreno et al. showed that particles analogous to LNPs, but prepared using lignin oleate, contain a hydration barrier layer resulting from the hydrophobic acyl chains that hinder protonation and deprotonation of carboxylic acid and phenolic hydroxyl groups under acidic and alkaline conditions.374 The OLNPs (oleic–lignin nanoparticles) were surface-modified without any cross-linking directly in aqueous dispersions using epoxy ring-opening reactions at pH 2 and pH 12. The resulting methacrylated and cationized particles showed competitive performance as curable anticorrosion coatings and pH-controlled switchable cationic/anionic adsorbents for rapid water purification.
One of the most interesting properties of LNPs is their ability to act as interfacial stabilizers. Zou et al. developed Pickering emulsion capsules that retain their integrity upon drying and rewetting.168 The amount of chitosan required to coat LNPs and to obtain stable emulsions was 50–100 mg g−1 relative to the dry weight of LNPs (Fig. 18a). The coating of LNPs with chitosan increased the particle diameter from 97 nm to 120–130 nm, while the net charge was reversed from anionic to cationic with 10–20 mg g−1 chitosan to LNP mass ratio (Fig. 18b). Only 0.2 wt% of the particles was required relative to the weight of olive oil to stabilize Pickering emulsions. This concentration was notably lower compared to 1.7 wt% of cationic lignin–coated LPNs that gave only partial stabilization of olive oil/water Pickering emulsion.368 The Pickering emulsions stabilized by chitosan-coated LNPs were characterized for their macroscopic or microscopic appearance (Fig. 18c–e), and could be stored at room temperature for prolonged time without significant changes in size distribution of the emulsion droplets after 60 d (Fig. 18f). Moreover, the Pickering emulsion droplets could be electrostatically stabilized by using sodium triphosphate, which allowed for drying and rewetting of the emulsions while retaining their spherical microdroplete shapes (Fig. 18g). Moreno and Sipponen recently reported the use of chitosan-coated LNPs for the adsorption of glucose oxidase, and further use of the GOx-chi-LNPs for the stabilization of Pickering emulsion polymerization of a series of vinyl monomers.375 These multifunctional hybrid particles enabled in situ degassing (oxygen removal) by the catalytic activity of GOx and synthesis of well-defined polymers using single electron transfer-living radical polymerization (SET-LRP). Later, the same group extended this biocatalytic hybrid LNP approach to the synthesis of polymer nanocomposites in polystyrene and polybutylmethacrylate matrices.376 The thermally reformable composites contained up to 35 wt% of lignin and surpassed the pristine polymers in mechanical properties (tensile strength and toughness).
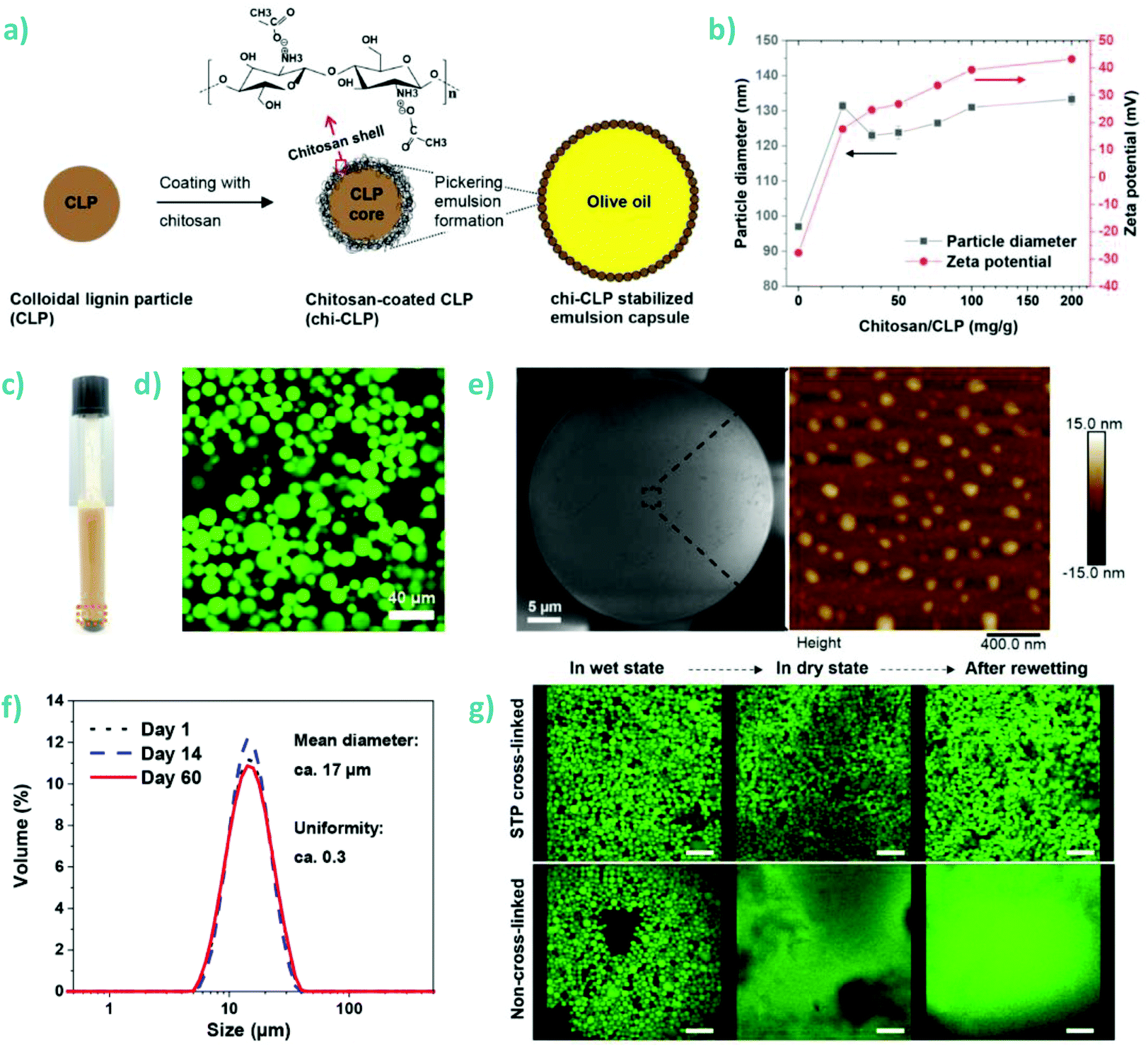 | ||
| Fig. 18 Chitosan-coated LNPs and their use for stabilization of olive oil/water Pickering emulsions: (a) Schematic of the preparation of chitosan-LNPs in capsules obtained from Pickering emulsions. (b) Change of nanoparticle diameter and zeta-potential as a function of chitosan to colloidal lignin particle mass ratio. (c) Appearance of the Pickering emulsion directly after its preparation by ultrasonication. (d) Confocal laser scanning microscopy (CLSM) images of the Pickering emulsion. (e) Cryo-SEM image of a single emulsion droplet and Atomic force microscopy image of the surface of a single emulsion droplet revealing the presence of chi-LNPs embedded at the oil–water interface. (f) Storage stability of the Pickering emulsion as followed measuring droplet size distribution at within 60 d, (g) CLSM images of the emulsion droplets stabilized by sodium triphosphate (STP) that withstand drying and rewetting in contrast to non-cross-linked emulsion droplets. All the images are reproduced with permission from ref. 168. | ||
Besides enzymes, non-catalytic proteins are interesting biological macromolecules to modify the surface of LNPs. Proteins form a corona over particles in biological tissues, and therefore it is of fundamental importance to understand protein–LNP interactions. Leskinen et al. studied adsorption of various proteins on colloidal lignin particles.370 Using QCM-D the authors found that the adsorbed mass of random coil proteins gelatin and casein on softwood kraft lignin was higher compared to the globular folded proteins conalbumin and albumin. Bovine serum, a protein mixture, was in between the two groups with respect to the adsorption capacity of the lignin particles. However, the adsorption studies using QCM-D were conducted on spin-coated model films prepared from lignin, similar to other efforts aimed to elucidate the effect of composition and electrostatic screening on enzyme binding.377 As LNPs are known to have more hydrophilic surfaces compared to bulk lignin, further studies on protein adsorption would benefit form the recently described model films from colloidal lignin particles.104 There are many opportunities for fundamental studies to shed more light on lignin-protein interactions, which is becoming increasingly important as the biomedical applications of LNPs are rapidly developing.48,378
Nanocellulose–lignin hybrids are produced via deconstruction of unbleached or partially bleached pulp, making the process attractive from an environmental and economic standpoint. Such advantages derive mostly from the high yield, reduced chemical processing and lower energy input required for defibrillation, if compared to the production of purified nanocelluloses.379 Among other reasons, such lower energy input is often attributed to the presence of lignin and hemicelluloses in unbleached or incompletely bleached pulps, which helps decreasing cellulose-cellulose interactions that counteract defibrillation efforts. There is no consensus, however, on the effect of lignin on ease of defibrillation. A detailed review discussing such aspects related to the effect of lignin and hemicelluloses on fibrillation has been recently published by Albornoz-Palma et al.380
Importantly, composition-processing correlations affect not only the economics of pulp deconstruction, but also the lignin-nanocellulose hybrid morphology. By controlling the bleaching and pretreatment time or severity, biomass of varying lignin and hemicellulose content can be used for further deconstruction. Interestingly, lignin-rich spherical nano and microparticles can be observed on the surface of hydrothermally pretreated biomass.381,382 Selig et al. and Donohoe et al. attributed the formation of such particles to the heating of lignin above the glass transition temperature, leading to the coalescence of lignin into droplets that migrate outside of the cell wall (Fig. 19a),381,382 where they eventually get deposited. Such hypothesis was also later confirmed by Pingali et al.383
 | ||
| Fig. 19 Confocal microscopy of E. Globulus treated by autohydrolysis at two different severities (S), (a1) S = 3.89 and (a2) S = 4.78, showing the formation of lignin aggregates at the fiber surface for S = 4.78. Reproduced with permission from ref. 398. The S factor is a function of treatment temperature and time. Scale bars are 40 μm. (b) Usual appearance of LCNF suspensions. Reproduced with permission from ref. 392. (c) AFM image and (d) height distribution of LCNF having 17 wt% lignin produced from unbleached kraft pulp, treated with maleic acid (120 °C during 120 min) and via microfluidization. Reproduced with permission from ref. 392. Spherical particles were attributed to lignin coalescence. AFM images from LCNF samples containing (e) 13.6 wt% lignin, (f) 9.5 wt% lignin, (g) 7.3 wt% lignin, produced from reed flour via ball-milling at room temperature in 1, 4 and 7 wt% NaOH (samples were sonicated to further improve dispersion). Reproduced with permission from ref. 393. Lignin particles are not clearly visible as in the pretreated LCNF. | ||
Although the spherical particles are mostly composed of lignin, they may contain trace amounts of carbohydrates and humins and can be generated in different preprocessing conditions. Organosolv treatment, for instance, have also been observed to form such spherical particles, whereas dissolved lignin may precipitate during the washing or cooling steps.384 Furthermore, lignin-carbohydrate complexes, known to originate from covalent linkages between lignin and hemicelluloses,385 may form core–corona structures, where lignin particles are coated by a polysaccharide corona.386 Spherical particles can also be generated though the degradation of carbohydrates (mostly hemicelluloses) during biomass pretreatment, leading to formation of lignin-like aromatic structures.387,388 The terms pseudo-lignin or humins are generally used to describe such degradation products that resemble lignin not only structurally, but also in terms of solubility properties. Pseudo-lignin is acid-insoluble and not easily distinguishable from Klason lignin.388 Importantly, similarly to lignin, it readily binds enzymes and negatively affects enzymatic hydrolysis of biomass.388–391 The precise identification of these distinct types of spherical particles, having similar morphology and originating from similar processing conditions, may not be promptly assumed solely based on microscopy evidence, as in some cases they may coexist, depending on the pretreatment applied.
Lignocellulose hybrid particles are most often used in the form of lignocellulosic nanocrystals (LCNC) or lignocellulosic nanofibers (LCNF). LCNF and LCNC are fabricated using similar methods to those traditionally employed for nanocellulose production, but result in suspensions having the brownish coloration usually observed in lignin (Fig. 19b). LCNF are produced through fibrillation, which is commonly achieved through mechanical methods, e.g. microfluidization,392 or milling.393 For LCNC, acid hydrolysis is used.392 Although lignin and residual hemicelluloses may negatively affect the yield of conventional acid hydrolysis used for bleached pulp, yields of up to 66% are observed through the use of deep eutectic solvents.394
Interestingly, spherical colloidal particles surrounding the nanocrystals and nanofibers are often observed in both LCNC and LCNF (Fig. 19c and d). Considering the wide range of raw materials and processing conditions utilized across literature, it is difficult to precisely evaluate the origin and composition of such spherical colloids merely based on morphological observations. Nevertheless, given their similarity to those nano and microspheres obtained in the pretreatment of biomass, it is reasonable to assume that they are rich in lignin, pseudo-lignin or a mixture of both and that their composition should be carefully evaluated on a case-by-case basis depending on the processing conditions. Kumagai et al.395 for instance, observed an increased Klason lignin content (indicating the formation of pseudo-lignin) in relation to the untreated biomass and also an increased amount of spherical particles when severe pretreatment temperatures were used.396 Such spherical particles, however, are not always observed in LCNF suspensions, particularly when high temperature treatments are not employed. Liu et al.393 reported the preparation of LCNF through a method involving ball-milling of reed straw under mild-alkaline conditions, resulting in hybrid nanofibers containing high amounts of lignin (i.e. from 7 to 16 wt% depending on the concentration of NaOH) and no clear occurrence of spherical particles (Fig. 19e, f and g). The lignin derived from such process retained a native-like structure with minimal degradation or condensation. In the study by Diop et al.397 processing at ambient conditions had similar results, with no clear occurrence of spherical particles. Such results indicate that hybrid lignocellulosic nanoparticles might also exist with a morphology and structure resembling that occurring in nature, with lignin more uniformly coating nanocellulose if compared to the case where large segregated spherical particles occur.
Regardless of morphological aspects, lignocellulosic hybrid nanoparticles have shown good performance for a wide range of applications. Although lignin nanoparticles alone can be used as reinforcing fillers,399 the outstanding mechanical properties of nanocelluloses – outperforming most natural and synthetic polymers – make them more suitable for sustainable nanocomposites.400 The high polar character of cellulose, however, hinder good compatibility with hydrophobic polymeric matrixes, making necessary the use of compatibilizers (coupling agents).401 In such a context, the amphiphilic nature of lignins provides a better interface between cellulose nanoparticles and the more hydrophobic polymers. Therefore, LCNCs have been tested as reinforcing fillers for a wide range of natural and synthetic polymeric nanocomposites, including polypropylene (PP),402 PS,403 epoxy,404 PLA,405 PVA,406 polycaprolactone,407 or starch,408 and others. For such applications, proper dispersion of the filler in the matrix is essential for improving mechanical and thermal properties, e.g. as shown in the work by Iwamoto et al.409 Good dispersion was also achieved by Ballner et al. through the use of Pickering emulsions.403 In such a case, LCNF was used to stabilize styrene in water emulsions that were further polymerized into PS microspheres coated with the LCNF. The microspheres were hot-pressed to produce reinforced polymer sheets. The bending strength and impact toughness of the composites were superior to that of neat polystyrene sheets. Such approach elegantly highlights the use of the amphiphilic nature of lignocellulosic hybrid particles in two possible applications, i.e., emulsions and composites. Other important applications include adhesives,410 superhydrophobic coatings,411 oil barrier coating,412 nanopapers,413 and others.
In summary, understanding the effects of pretreatment and deconstruction methods in the preparation of lignocellulosic hybrid particles is crucial for optimizing materials performance and applications. For instance, if lignin spheres coat nanocellulose particles, the overall surface properties (e.g. surface charge, wettability and adhesion properties) may largely differ from the case where lignin more uniformly surround nanocellulose. Both of such morphologies may occur depending on the pretreatment and process parameters used for particle formation. Therefore, this consideration is of fundamental importance as lignocellulosic particles applications largely rely on the surface properties brought by the reduced polarity and amphiphilicity of lignin, as investigated by AFM force measurements of lignocellulosic fibers.414 Furthermore, much can be learned from the rich literature on parallel efforts related to the morphological evolution in biomass pretreatments aiming at lignin and hemicellulose redistribution or removal.396,398 Importantly, expanding our understanding on how pretreatment conditions and raw material composition affect the morphology and properties of lignocellulosic nanoparticles is needed. This knowledge could thus be translated into materials with optimized properties.
5. Applications of lignin composites and hybrids
Lignins have found use in a range of applications such as fuels, superplasticizers, precursor of aromatic flavoring agents, etc. Much of these applications rely on the macromolecular properties of lignins and their functional groups. Thereafter, controlling the aggregation of lignins into nano- and microparticles, including hybrids, enables a wider range of applications. In this section we put forward a subset of these applications and highlight the main developments towards these types of materials, where lignins enable to increase their sustainability, overall performance, or both.5.1. Flame retardancy
Owing to it oxygenated aromatic nature, lignin is a natural char-forming compound and as such of interest for flame retardant materials. Progress in lignin-based flame retardants was reviewed by Yang et al.415 There is a renewed interest in this research area as indicated by the studies published since their recent review.416,417 To contribute to the development of scalable materials, synthesis of lignin-based flame retardants should increasingly rely on green and sustainable chemistry. In particular, the number of synthetic steps should be minimized along with the solvents and reagents used, and the proportion of lignin in the multicomponent flame retardants should be as high as possible.The most straightforward approach is to use lignin as such in fire retardant composites and coatings. The combustion of solid material involves the generation of flammable gases that ignite and burn and cause simultaneous thermochemical changes in the condensed phase. A fire-retardant material can inhibit the generation of flammable gases or promote the formation of a protective char layer over the bulk material. Due to their rigid aromatic structures, many different types of lignins are known to form a high proportion of char upon thermal decomposition. It is thus not surprising that there are many studies describing lignin as a char forming agent in fire retardant systems.418,419
Lignosulfonates seem to be more thermally stable compared to all other technical lignin types when judged based on the percentage of char residues by thermogravimetric analysis. For example, the char yield at 500 °C of a sodium lignosulfonate was 58% compared to 41% for SKL. These differences in char formation from different lignin types might also be related to the content of inorganic impurities arising from the lignin extraction processes. It is evident, however, that the thermal stability of lignin is due to the formation of polycyclic aromatic hydrocarbons under oxygen-limited conditions,420 and is reflected to the fire retardancy of various polymer composites containing lignin. For instance, when 20 wt% of lignin was mixed within a polyamide matrix, the highest char yield at 600 °C was obtained with sodium lignosulfonate (13.2%) and the lowest with a purified SKL (9.8%).421 Soaking of a cotton fabric in sodium lignosulfonate at 30 wt% loading concentration decreased the peak heat release rate (PHRR) and total heat release (THR) in a cone calorimeter test by 26% and 58%, respectively.419 When melt extruded with PLA, the presence of 5 wt% of SKL resulted in a char residue fraction of 4.2% at 500 °C, whereas PLA alone degraded completely under the same conditions.422
Lignin has also been chemically modified to improve its fire-retardant performance. These modified lignin polymers typically carry nitrogen (N), phosphorous (P), and boron (B) elements. These compounds release non-combustible gases that increase the limited oxygen index (LOI), i.e., the volume fraction of oxygen required to ignite the material. The majority of these lignin-based fire-retardant polymers are P-lignin,423 N-lignin,424 and PN-lignins.417,425 There are also works incorporating siloxane,426 and boron427 in lignin-based fire retardant materials. Though lignin itself has been found to exhibit intumescent behaviour,428 mainly chemically modified lignins have been evaluated in intumescent coating systems that swell upon thermal exposure and form a protective carbonaceous char layer.424,429 Overall, there is room for more studies towards improving the fire retardant properties of lignin, for instance, to enable development of coatings that are stable in outdoor conditions.
5.2. Food packaging
The continuous demand for antimicrobial packaging and the use of semi-prepared fresh foodstuffs with high nutritional value have promoted the present tendency in food packaging application to design new systems with modulated characteristics, in order to improve the shelf-life of packaged food products.430 The increasing responsiveness regarding synthetic antioxidant application not only for food packaging, but also for stabilization of polymers, stimulated the interest towards natural antioxidants, in particular natural polyphenols. This demand for natural polyphenols is spotlighted by their biodegradability and low toxicity and lignin, as natural product with antiparasitic, antifungal, and antioxidant properties, take the active role in this specific sector.144Lignin has been considered as active ingredient in polymeric formulations, both at the micro and nanoscale, that has been properly reviewed in several contributions.431 On the other hand, less attention has been given to the possibility of combining lignin in a hybrid configuration, so this section will only report on the available literature related to the synergic use of lignin in the presence of other organic/inorganic reinforcements for packaging purposes. Thinking about natural based reinforcements, the use of cellulose was unsurprisingly aligned with lignin. In this specific case, cellulose/lignin formulations in the absence of a polymeric substrates, such as in the case of the work of Tayeb et al. are out of the scope of this specific section, so the analysis will be mainly devoted to the use of these lignocellulosics fillers in polymeric substrates.127,412 Example of this approach can be found in the work of Yang and co-authors, that investigated the effect of adding lignin nanoparticles and cellulose nanocrystals (1 and 3 wt%) in PLA based films prepared by extrusion.432 This work evaluated the antioxidant, antimicrobial, migration and disintegrability activities of ternary nanocomposite films. Data from antimicrobial tests revealed a capacity to inhibit the Gram-negative bacterial growth of Xanthomonas axonopodis pv. vesicatoria and Xanthomonas arboricola pv. pruni over time, offering innovative control against dangerous bacterial plant pathogens. The same authors studied also the effect of adding lignin nanoparticles in binary and ternary nanocomposites composed by PVA and chitosan polymeric blend:433 antimicrobial test revealed a capacity of these composites to neutralize the bacterial growth of Gram-negative bacterial plant/fruit pathogens.
Lignin has also been combined in a core–shell configuration with melanin (LMNP), to synthesize nanoparticles through spontaneous deposition of polydopamine (PDA) on the surface of lignin nanoparticles via a nanoprecipitation method, to be integrated in a series of PBAT-based nanocomposites.434 PBAT–LMNP films exhibited a clear superiority over the reference system, both in maintaining the UV shielding efficiency and the desirable mechanical performance, providing a promising solution to obtain novel cost-effective nanofillers with attractive functions and biodegradability.
Flower-like lignin/ZnO hybrids were also used as active fillers in polyurethane,435 where a simple, sustainable, and environmentally friendly in situ precipitation method was developed to prepare lignin/ZnO polyurethane composite (Fig. 20a and b). The results showed the excellent interfacial contact between lignin and ZnO, which resulted in excellent synergistic UV-absorption properties. Furthermore, the mechanical properties of the polyurethane (PU) were also greatly improved when blended with only 0.6 wt% quaternized alkali lignin (QAL)/ZnO composite.
 | ||
| Fig. 20 (a) (a and c) SEM images of (a) quaternized alkali lignin (QAL)/ZnO composite and (c) ZnO. (b and d) TEM images of (b) QAL/ZnO composite and (d) ZnO, Reproduced with permission from ref. 435. (b) UV light transmittance curves, digital photos, and SEM images of PU, PU + ZnO, and PU + QAL/ZnO films, Reproduced with permission from ref. 435. (c) Process for incorporation of deep eutectic solvent and ethanol-organosolv extracted technical lignins into PVA, reproduced with permission from ref. 399. | ||
Other examples of hybrids lignin based formulations for specific applications can be found in,399 where the authors, inspired by the example of nanocellulose biopolymer, considered lignin nanoparticles, prepared through self-assembling of deep eutectic solvent and ethanol-organosolv extracted technical lignins, and incorporated them into poly(vinyl alcohol) (Fig. 20c). The films showed additional UV-shielding efficacy (∼80% at 400 nm with 4 wt% of LNPs) and antioxidant functionalities. An approach for obtaining antioxidant active packaging to be used in food preservation was the grafting of metallic particles on lignocellulosic pulp. In details, lignocellulose fibers were grafted with AuNPs, directly synthesized and anchored without an external immobilizing agent on unbleached softwood pulp.329 As a consequence, AuNP–pulp nanocomposite paper showed exceptional antioxidant performance with a radical scavenging rate of over 98%, which is attributed to the synergistic effects of adsorption by the fiber–fiber network and subsequent catalytic activity of the AuNPs. Enhancements in terms of the film flexibility were obtained by combining lignin nanoparticles and boron nitride (BN)-OH/PVA composite film:436 these films, fabricated through vacuum filtration in conjunction with chemical cross-linking, showed multiple improved properties, confirming their promising role as packaging material.
These works confirm how the addition of lignin, especially nano-scaled, is able to address and solve the concerns associated with poor thermal stability, low mechanical strength, and brittleness of conventional polymeric composite films. Moreover, the synergy with other inorganic nanoparticles opens new outstanding use to a broad spectrum of applications that range from the traditional food sector to electronic and thermal resistant packaging that may count on inherent thermal stability of the lignin component.
5.3. Plant protection
The increasing demand for plant protection products, and the emerging problems related to the environmental impacts, have stimulated academic researchers to develop bio-based systems for plant protection.437 In this context, natural phenolic compounds and lignin-based materials have gained much attention due to their interesting properties: lignin-based controlled release of pesticides takes the advantage of lignin fragmentation and re-polymerization, that allows a precise control with flexibilities, helping to achieve desired release rates.26 As a phenolic compound, lignin presents a chemical structure able to act as a plant protector against external environmental aggressions, such as insects, weather, bacteria and fungi.438 Yang and co-authors were among the first researchers to study the antimicrobial activity of lignin nanoparticles against plant pathogenic bacteria.112 In this work, the authors demonstrated that the lignin nanoparticles positively acted as an antimicrobial agent towards Pseudomonas syringae and Xanthomonas axonopodis pv. vesicatoria strains when the aqueous concentration of lignin nanoparticles increased from 5% to 8 wt%, and Pseudomonas syringae was found to be the most vulnerable strain at both ratios. Lignin-based nanocapsules (LNCs) have been also applied as biocompatible vectors to deliver bioactive compounds, such as gibberellic acid (GA), to modulate the plant-growing process, as proposed by Falsini and co-authors.439 Cargo release and cytotoxicity assays were carried out in two model plants, Eruca vesicaria and Solanum lycopersicum, using lignin concentrations up to 1% w/v. The different seeds were treated with Ga-loaded LNCs, and the seedlings showed tolerance without symptoms of toxicity. Additionally, fluorescently-labeled LNCs showed the capacity to enter and accumulate in seeds and seedlings. The hydrophilic groups in the lignin chemical structure, which are localized on the external surface of LNCs in aqueous system, could positively influence the processes associated to plant germination and growth by increasing water availability.An innovative valorization of a lignin nanocarrier was proposed as curative treatment of the grapevine trunk affected by Esca infection by Fischer et al.440 that fabricated an enzyme-responsive lignin nanocarrier loaded with a minimal quantity of fungicide (pyraclostrobin) via miniemulsion polymerization, providing an aqueous solution with a high encapsulation efficiency (Fig. 21a and b). These nanosystems were first studied in vitro, and then injected in planta (in vivo) into Vitis vinifera L. cv. ‘Portugieser’ plants. The study reported that a single trunk injection resulted in a significant reduction of Esca-associated pathogens, including the tracheomycotic Phaeoacremonium minimum and Phaeomoniella chlamydospora, and the evaluation was maintained over a period of five years. This curative treatment could also be used as a preventive treatment, since no drug is released without the secretion of lignin-degrading enzymes during the Esca infection. This general strategy can be extended for other plant diseases, and decrease environmental pollution induced by the overuse of agrochemicals.
 | ||
| Fig. 21 (a) Concept of the nanocarrier-mediated drug delivery: green pathway: degradation of a substrate leads to growth of the pathogen (infection, disease); orange pathway: a negative feedback loop is initiated by masking a drug that Fights the pathogen in nanocarriers based on the food source of the pathogen, reproduced with permission from ref. 440. (b) Concept shown in (a) used to Fight the fungal disease “Esca”: fungicide-loaded lignin NCs release the drug load only when the Esca fungi release lignin-degrading enzymes, Reprinted with permission from ref. 440. (c) Lignosulfonate granule production using Myceliophthora thermophila laccase as a biocatalyst. loaded with 2 g·kg−1 3,6-dichloro-2-methoxybenzoic acid (Dicamba), a broad-spectrum herbicide, images of the granules before and after release experiments in different conditions, and after drying, Reproduced with permission from ref. 444. (d) Potential nutrient release mechanism of coated-lignin slow-release fertilizer. Reproduced with permission from ref. 449. | ||
Machado et al.441 prepared chemically cross-linked lignin nanocarriers (NCs) by azo-Michael addition in miniemulsion, followed by solvent evaporation. The cross-linking of lignin was achieved with the bio-based amines (spermine and spermidine). Several fungicides were encapsulated in situ during the miniemulsion polymerization, demonstrating the versatility of the method. Lignin NCs with diameters of 200–300 nm (determined by dynamic light scattering) were obtained, with high encapsulation efficiencies (70–99%, depending on the drug solubility). Lignin NCs successfully inhibited the growth of Phaeomoniella chlamydospora and Phaeoacremonium minimum, which are ligninase-producing fungi associated with the worldwide occurring fungal grapevine trunk disease Esca. In planta studies proved their efficiency for at least four years after a single injection into Vitis vinifera (“Portugieser”) plants on a test vineyard in Germany. The lignin NCs are of high interest as biodegradable delivery vehicles to be applied by trunk injection against the devastating fungal disease Esca but might also be promising against other fungal plant diseases. Peil et al. encapsulated the Trichoderma spores in lignin-based polyelectrolyte shells via the LbL technique creating a surfactant-free, self-stabilizing spore dispersion.442 The lignin shell transfers the spores in a resting state without inflicting damage. The resulting spore dispersion was colloidally stable over several months and applicable via trunk injection. Once injected, encapsulated spores fulfill the “Trojan horse” role. If the plant is infected by Esca pathogens, lignin-degrading enzymes, produced by the pathogens themselves, degrade the lignin shell and trigger the germination process. An extensive review on methods for active loading of lignin-based materials can be found in Sipponen et al.443
Besides the use of lignin as a carrier to deliver plant growth regulators, herbicides and pesticides, lignin-based materials can also be used to control the release of different fertilizers444 (Fig. 21c). In this context, Legras-Lecarpentier et al. proposed the synthesis of 100% lignosulfonate-based granules as fertilizer storage and controlled and sustained release systems, using an enzymatic polymerization approach.445 Release studies indicated that the lignosulfonate-based granules released more than 94% of the potassium nitrate and potassium phosphate during the first 10 d. Adding 1% w/w alginate into the polymerized lignosulfonates allowed obtaining bigger and perfectly stable granules. Moreover, the presence of increasing alginate concentrations allowed for the prolonged release rate of both fertilizers over 25 d, due to the different swelling behaviors of the coatings. This study showed for the first time the possibility of synthesizing fully biodegradable fertilizer delivery systems using completely green enzymatic technology. Luo et al. encapsulated pyraclostrobin in a lignin-modified polymer nanocapsule (NCS) to produce a nanoscale-delivery system with excellent soil mobility.446 These nanocapsules exhibited a stable core–shell structure and rapid release performance. In addition, they also increased the distribution of particles on the surface of target organisms and enhanced the soil mobility of pyraclostrobin, mainly benefitting from the nanocapsule's negatively charged polymer shell and nanoscale size. Pot experiments showed that, compared to treatment with nanosized emulsion in water and micron-grade microcapsule suspension (CS) of pyraclostrobin, the NCS provided improved control efficacy on tomato crown and root rot. They also showed lower pesticide residue in the soil than CS treatment.
In the case of hybrid lignin composites, examples of use can be found in Sun et al.447 where a novel composite porous hydrogel (LBPAA) based on lignin and polyacrylic acid was developed for use as the support frame of a pore structure for water delivery. The LBPAA was analyzed to determine its water-swelling and slow release properties. The controlled release properties of LBPAA were evaluated through experiments in relation to the cumulative release of pesticides, with particular emphasis on environmental effects and release models. The porous LBPAA hydrogel showed improved properties compared to the sole polyacrylic acid, and could therefore be considered an efficient material for application in controlled release systems in agriculture. Flores-Céspedes et al. successfully entrapped an insecticide, imidacloprid, in lignin−PEG and ethylcellulose to obtain imidacloprid monolithic systems and coated granules, respectively, demonstrating the existence of a uniform film able to supply the appropriate amount of imidacloprid in each specific agronomic practice to increase the efficiency of this bioactive material and minimize the risk of environmental pollution.448
Li et al. prepared a lignin/clay composite material with a porous structure that acted as an efficient slow-release agent for urea.450 Leaching experiments showed that when lignin/clay/urea was 2:1:4, the prepared lignin/clay composite materials exhibited more efficient slow-release of the urea molecules. Furthermore, the lignin/clay composite material could adsorb water from the air, which may aid water retention by dry soil. Soil urease activity inhibition experiments showed that the composite materials could reduce the urease activity, leading to the transformation of urea into volatile ammonia. Jiang et al. prepared an alkaline lignin-poly(vinyl alcohol)-urea (AL-PVA-U) film by crosslinking with formaldehyde.451 The results showed that the addition of alkali lignin limited the release of nitrogen in the AL-PVA-U film and resulted in an increase of the span for controlled release. Jiang et al. also measured the release of K and Mg from an alkaline lignin/PVA cross-linked slow-release film by using the leaching experiment.452 It was found that the addition of alkali lignin was unfavorable for K release, whereas an appropriate amount of alkali lignin favored Mg release, compared with the cross-linked PVA film alone. The mechanism of the slow-release of K from the film mainly involved the physical barrier effect of the molecular structure, whereas for Mg release, in addition to the physical barrier effect, the chelation of lignin also played a part. Ma et al. prepared polyacrylic acid-grafted alkali lignin-based (ALS-G-P(AA)) iron fertilizer by grafting an acrylic monomer onto amorphous alkali lignin.453 Under the conditions of 60% neutralization degree of acrylic acid, 0.06 g of initiator, 0.03 g of cross-linking agent, temperature of 60 °C, and alkaline lignin grafting ratio of 25%, the water absorption of the resulting ALS-G-P(AA) iron fertilizer was 1017 g g−1 and the iron ion content was 7.74 mg g−1. The effective period of the water-saturated ALS-G-P (AA) iron fertilizer was twenty-two d.
Novel hybrid organic–inorganic materials obtained by combining copper(II) salts with two types of technical lignins were tested for their antifungal and antibacterial profile against a vast panel of pathogens of agronomical interest.437 Furthermore, preliminary tests on crops in a greenhouse were performed, showing that lignin@Cu had better performances than a commercial pesticide based on copper(II) hydroxide on tomato plants against Rhizoctonia solani, indicating a great potential of these materials as plant protection products.
In a recent review from Chen et al.449 the preparation principles of lignin-based slow/controlled release fertilizer were summarized (Fig. 21d). Although the evaluation standards and methods of lignin-based slow/controlled release fertilizers need to be further improved, it is believed that more and more scholars will pay more attention to them to accelerate the development and application of lignin-based slow/controlled release fertilizers, so as to improve their relevant standards, since lignin-based slow/controlled release fertilizer has a very broad application prospect in agro-ecological environmental protection and sustainable agricultural development.
5.4. Energy storage applications
Energy storage is a highly relevant area of research as efficient energy storage materials and devices will boost the transition towards a decarbonized society; i.e. powering electric vehicles (EVs) or storing the electricity generated from renewable resources. In this field, electrochemical energy storage (EES) systems are especially relevant. Depending on their energy/power densities and energy storage mechanism, they can be categorized into three main systems comprising capacitors, supercapacitors, and batteries. Because of their high energy density, wide operational voltage range, low self-discharge rate and long cycle life, lithium-ion batteries (LIBs) dominate the battery market, although other emerging alternatives such as sodium-ion batteries (NIBs) or zinc-ion batteries (ZIBs) could be found. Typically, such batteries are composed by two electrodes (the active anode and cathode) separated by a porous membrane (usually glass fiber or polyolefin-based) soaked into a liquid electrolyte, which physically isolates both electrodes but permits ion transfer between them.454Regarding separators, a high-porosity nonwoven lignin/polyacrylonitrile composite was fabricated by electrospinning to obtain a porosity of 74% with 0% dimensional shrinkage at 150 °C (42 and 32% for the commercial PP separator, respectively).455 Lignin markedly enhances the electrolyte affinity of the separator, reaching electrolyte uptake as high as 790%. As a result, the ionic conductivity is enhanced by two orders of magnitude, yielding a capacity of 110 mA h g−1 at 2C (∼90 mA h g−1 for the PP separator) when mounted into a Li/LiFePO4 battery. Similarly, a nonwoven lignin-polyvinyl alcohol nanofiber separator showing a 1 M LiBF4 EC/DMC electrolyte uptake of 533% (108% for commercial Celgard) and small thermal shrinkage has been prepared for LIBs using a water-based process.456 Li/Li(Ni0.33Mn0.33Co0.33)O2 half cells with the lignin-based separator delivered a capacity of 91 mA h g−1 at 2C (48 mA h g−1 for Celgard). Interestingly, lignin protects the separator against electrochemical side reactions that may increase interfacial resistance. Besides increasing electrolyte uptake and providing an increased thermal stability to the separator, lignin can be used as a functional coating to suppress dendrite formation. Accordingly, lignin-Nafion porous membranes have been used to enhance the life cycle of ZIBs.457 Lignin increases ionic conductivity from 4.6 × 10−6 to 1.4 × 10−3 S cm−1 while promoting the formation of a zinc layer hydroxide as a result its interaction with Zn2+. In turn, the latter provides a stable solid-electrolyte interface, which extends the cycle life over 400 h in comparison with <200 h for commercial separators (see Fig. 22a). Other use of lignin as a functional coating was shown by Zhang et al., who deposited lignin NPs onto a Celgard separator by simple filtration to prevent the worrisome shuttle effect caused by soluble polysulfides in lithium–sulfur (Li–S) batteries.458 Such an effect was ascribed to the abundant electron-donating groups of lignin that induce chemical binding of polysulfides and hinder the shuttle effect, delivering ∼487 mA h g−1 at 2C in comparison with 424 mA h g−1 for the non-coated Celgard.
 | ||
| Fig. 22 Lignin for energy storage applications: (a) Stripping/plating for the separator/membranes to assess their life cycles. Reproduced with permission from ref. 457. (b) Rate capacity of lignin-based electrolyte in Li/LiFePO4 cell compared with commercial separator. Reproduced with permission from ref. 459. (c) Comparative cycling performance at a current density of 0.1 A g−1 of pure NiO, hierarchical mesoporous carbon (HMPC) and NiO/HMPC. Reproduced with permission from ref. 363. (d) Cyclic voltammetry curves of GO/lignin supercapacitors measured at 20 mV s−1. Numbers represent initial lignin % concentration. Reproduced with permission from ref. 362. | ||
A step forward in the development of efficient and safer separators may come from the development of gel polymer electrolytes (GPEs), which are considered an effective solution to build safer batteries. In this context, a free-standing lignin-based GPE was obtained mixing lignin and poly(N-vinylimidazole)-co-poly(poly(ethylene glycol) methyl ether methacrylate) in water and subsequent casting and drying.459 After activation by organic electrolyte, a remarkable lithium ion transference number of 0.63 (0.27 for the commercial separator), ionic conductivity of 6.3 × 10−4 S cm−1 at 30 °C, and electrochemical window up to 4.7 V vs. Li/Li+ were obtained. Lignin-electrolyte showed a discharge capacity of 150 mA h g−1 at 1C into a Li/LiFePO4 cell for more than 450 cycles (see Fig. 22b for the rate capacity). Thanks to the large number of hydroxyl groups in lignin that promote anion dissociation from lithium salt and its GPE character, a remarkable rate capacity of 110 mA h g−1 at 10C and Li dendrite growth inhibition were observed.
The large volume expansion suffered by different host during charge–discharge and their poor intrinsic conductivity remains one of the most relevant challenges when designing high-performance anodes. This problem can be solved incorporating active materials, usually transition metal oxides, into a porous carbon host derived from lignin. In this context, ZnO NPs were supported onto porous carbon from lignin by solvothermal method to fabricate a LIB anode with high electrical conductivity and stability during lithiation/delithiation.460 The structure of the porous carbon provides shorter diffusion distance of Li+ and electrons and protects against volume changes, while it also acts as an active material of lithium storage, providing a discharge capacity of 898 mA h g−1 at 0.2 C after 100 cycles (84 mA h g−1 for pure ZnO). Similarly, lignin-derived hierarchical mesoporous carbon loaded with NiO NPs were applied as LIB anodes.363 As shown in Fig. 22c, obtained micropore-to-mesopore distribution and SSA of 852 m2 g−1 provides additional reactive interfaces and electron/ion channels for electrolyte ions, delivering a discharge capacity of 863 mA h g−1 at 0.1 A g−1 after 100 cycles, which is even above the NiO theoretical capacity of 718 mA h g−1 and surpasses the poor ∼300 mA h g−1 delivered by the NiO anode.
The redox quinine/hydroquinone moieties of lignin have been exploited to store an electric charge. Lignin was mixed with a conjugated polymer, poly(3,4-ethylenedioxythiophene) (PEDOT) via oxidative polymerization to fabricate cathodes for NIBs.461 Conductive PEDOT allows electron transfer and the presence of lignin provides an extra redox process increasing the specific capacity of the PEDOT electrodes, yielding a cathode material which keeps its redox activity and stability for over 150 cycles. Further, lignin-based anode materials for NIBs with a reversible capacity of 347 mA h g−1 were obtained after carbonization of lignin-nanospheres at 1.300 °C under nitrogen atmosphere.462 The moderate interlayer distances and rich defect sites obtained provide enhanced Na+ storage by intercalating into interlayers, while the nanoscale morphology reduced ion diffusion length.
Supercapacitors are being explored for energy storage as they show high-power density, rapid charging/discharging ability, safe operation and long cycle life. To date, the quinone redox function of lignin has been used for supercapacitor fabrication as quinone/hydroquinone couples provide a reversible single step two-electron two-proton redox reaction 2H+/2e, giving a charge density of 1787 C g−1. In their pioneering work, Milczarek and Inganäs prepared a composite cathode electrode comprising lignin and polypyrrole, where the quinone groups can reversibly accept and release electrons and protons during successive charge and discharge cycles.463 Similar performance has also been proven for other conducting polymers such as PEDOT, which reached specific capacitance values 170.4 F g−1.464 Regarding lignin nanohybrids comprising inorganic NPs, CNTs were functionalized with kraft lignin to exploit the reversible redox activity of lignin.345 An enhanced gravimetric capacitance from 75 for bare CNTs to 143 F g−1 for lignin/CNT nanohybrid was obtained due to the electrical double-layer capacitance (EDLC) contribution from CNTs and pseudocapacitance of quinone (CNTs provide a conducting network for electron transport to the potentially electroactive groups of lignin). To enhance the short life cycle, low cycling efficiency and high self-discharge rate of lignin originating from its dissolution by common electrolyte solvents, lignin has been confined into a graphene cage.348 Besides protecting lignin against dissolution, graphene provides efficient electron transfer pathways during electrochemical reaction while buffering the strain relaxation and volume changes, thus delivering a capacitance of 211 F g−1 at 1.0 A g−1 and allowing a pseudocapacitance retention of 88% after 15000 cycles at 2.0 A g−1. Transition metal oxides can be also used for supercapacitor applications. In this context, NiWO4 NPs were decorated with lignin and deposited onto an aluminum substrate to obtain a supercapacitor anode with a pseudocapacitance behavior, which resulted in an energy density of 2 W h cm−2 and a 97% capacitance retention after 1000 cycles.465 Lignin nanocrystals were confined on RGO to combine the fast and reversible redox charge transfer of surface-functionalized quinones and their interplay with electron-conducting RGOs.350 Interestingly, this work focused on the development of pseudocapacitors, which show improved capacitance over the supercapacitors that deliver charges through non-faradaic means (i.e., electrochemical double-layer capacitors). Through nanoscale confinement manipulation, a strong interaction of lignin with RGO is obtained via strong π–π and hydrophobic interactions between the aromatic backbone of lignin and RGO sheet, allowing a fast redox charge transfer which results in a pseudocapacitor with a maximum capacitance of 432 F g−1 at 1.0 A g−1 (six times larger than that or pure RGO).
Carbonaceous materials have been widely used as electrode materials for uses as (micro)electrodes,466 and supercapacitors, which tale advantage of processability, low cost and good chemical stability. Hierarchical porous carbon monoliths derived from lignin carbonization have been used directly as a supercapacitor electrode with no need of binders. Rectangular cyclic voltammetry curves indicating good EDLC behavior were observed. The obtained hierarchical 3D porous structure provides large surface area to enhance the ion storage space while affording ion buffering reservoirs to minimize the ion diffusion distance to the inner surfaces, resulting in a specific gravimetric capacitance of 200.2 F g−1 at 0.1 A g−1 (aqueous 6 M KOH electrolyte).355 A bacterial activation method has been applied to optimize the pore-structure of carbon from lignin and promote its graphitization, yielding carbon with a remarkable SSA of 1831 m2 g−1.467 The micropores formed during activation enable charge accumulation, the mesopores accelerate the ion diffusion and the macropores provide space for buffering/storing electrolyte ions. Moreover, the obtained carbon presented oxygen-containing functional groups, improving electrolyte wettability, decreasing interfacial charge transfer resistance and providing additional pseudocapacitance through the Faraday reaction that can combine with EDLC. Consequently, a capacitance of 428 F g−1 at 1.0 A g−1 was obtained in an aqueous 6 M KOH electrolyte (capacitance retention 96.7% after 10.000 cycles at 5.0 A g−1).
To further increase the capacitance, porous carbon from lignin has been used as a structural framework to host active NPs. In this context, ZnO NPs have been incorporated into lignin-derived porous carbon to obtain a maximum specific gravimetric capacitance of 193 F g−1 at 0.5 A g−1 (PVA/KOH gel electrolyte).468 Superior cycling was achieved owing to the carbon host that allowed quick transfer and diffusion for electrolyte ions, provided more active sites for charge accumulation, enhanced ZnO conductivity and relieved ZnO volume expansion during charge/discharge. Importantly, the obtained EDLC behavior with good reversibility resulted in a capacitance of 161 F g−1 at 2.0 A g−1 after 10000 cycles. Similarly, a 30–80 μm thick free-standing porous carbon/graphene hybrid prepared after KOH activation with a SSA of 1744 m2 g−1 has been applied as a binder-free supercapacitor electrode.362 Amorphous carbon avoids graphene aggregation, exhibiting a near-ideal capacity behavior with a capacitance of 162 F g−1 at an ultrahigh scan rate of 20.0 A g−1 (aqueous 1 M H2SO4 electrolyte, Fig. 22d). Ternary nanohybrids have also been investigated to enhance the capacitance (i.e. MnO2 NPs, reduced graphene oxide and porous carbon from lignin to yield 135 F g−1 at 2.0 A g−1),469 although the presence of further active materials does not per se guarantees an increased capacitance.
Although energy storage has been mainly focused on EES, harvesting and storing other energy types is of prime interest. Phase change materials (PCMs) are emerging as a suitable choice as they can store and deliver thermal energy during a solid–liquid transition.470 Lignin nanocapsules containing fatty acids prepared by co-precipitation have been employed as PCMs for thermal energy storage.471 Using water as a non-solvent, core–shell NPs having 122 to 190 nm in diameter and a controlled concentration of tall oil fatty acid, oleic acid or lauric acid stabilized by a 18 nm thick softwood Kraft lignin shell were fabricated. The nanoconfinement provided by lignin avoided phase separation during melting stage, resulting in enthalpy retention values of 99.7 and 98.1% for the solidification and melting after 290 cycles (in the dry form). Such stability was ascribed to the confinement effect of fatty acids provided by the lignin shell, resisting broadening and separation of the phase transition peaks. Interestingly, synthesized nanocapsules could also be used in aqueous systems, opening new opportunities in thermal energy management.
Solar energy harvesting is a prominent mean of sustainable power generation. Over the last years considerable efforts have been devoted to the development of efficient dye-sensitized solar cells (DSSCs), which show a photo-electricity conversion by photo-excitation of dyes.472 To avoid the use of scarce materials, lignin and its derivatives have been applied into solar cells. For instance, various types of lignophenol have been combined with nano-porous TiO2 to increase light adsorption (originating from the aromatic rings) and fabricate sensitized solar cells with a photo-electricity conversion efficiency of 3.61% under visible light irradiation.473 Carbon nanofiber mats having a SSA of 941 m2 g−1 and electric conductivity of 502 S m−1 obtained from lignin carbonization at 1500 °C were applied as DSSC counter electrodes to achieve a efficiency of 7.60%, matching the 7.67% showed by conventional Pt counter electrode (lignin-derived carbon efficiently collects electrons thanks to its low charge transfer resistance).474 In another work, modified alkali lignin was used as a p-type dopant for PEDOT to increase its conductivity.475 The electron transfer property resulting from lignin's aromatic structure can be exploited to obtain materials that act as a transfer medium for the conversion of photonic energy. Wu et al. showed that the rich phenolic hydroxyl groups in alkali lignin provide a hole mobility as high as 2.27 × 10−6 cm2 V−1 S−1, which is comparable to the mobility shown by conventional conjugated polymers. Therefore, perovskite solar cell anodes were prepared by combination of PEDOT and alkali lignin, reaching a power conversion efficiency of 14.94% in comparison with the 12.6% obtained for the common PEDOT:PSS based devices.
5.5. Biomedical
Lignin-based composites in various different morphologies have been identified as promising biomedical materials with antioxidant and antimicrobial activity, compatibility with mammalian cells, and physicochemical properties mimicking those of biological tissues. The literature includes antimicrobial composites in aerogel,476 nanofibers,477 and film,478 forms. Among these, Domínguez-Robles et al. recently reported softwood kraft lignin-poly(butylene succinate) composites that exhibited antioxidant activity and antibacterial properties.478 The extent that Staphylococcus aureus cells adhered on the injection molded composites was reduced approximately 90% regardless of their lignin content (2.5–15%). Instead, a linear increase in the melt flow index at 175 °C was found as a function of the increasing lignin content.Lignin-based composites have been proven compatible with mammalian cell lines including mouse fibroblasts,167 a rat neuronal cell line PC12,479 human dermal fibroblasts,479 human hepatocytes,156 human cardiac artery endothelial cells,480 human osteogenic sarcoma cells SaOS-2,481 a human hepatocellular carcinoma cell line HepG2,482 and human mesenchymal stem cells.479 Zhang et al. recently developed nanocomposites consisting of CNF, sodium alginate, and colloidal lignin particles and demonstrated their use as hydrogel inks for three-dimensional printing using a pneumatically driven extrusion process.482 The authors found that the incorporation of 1–17 wt% of CLPs (dry basis) increased viscosity of the hydrogels at a low shear rate and consequently provided a better shape fidelity and printing resolution to the scaffolds. The nanocomposite hydrogels showed no negative effect of CLPs on proliferation of HepG2 cells on the scaffolds that contained uniformly dispersed CLPs on their surfaces and within the matrix (Fig. 23a–c).
 | ||
| Fig. 23 Lignin-based composites for cell culture. CNF-alginate-CLP nanocomposites: (a) AFM height images of CNF-alginate (a1) and CNG-alginate-CLP hydrogel composites within lignin contents 1%, 4%, 7%, and 17% on dry basis (a2–5). (b) Photographs of the corresponding three-dimensionally printed hydrogel scaffolds. (c) Cell culture in the scaffolds as followed by optical density designating the extent of in vitro HepG2 cell proliferation. Reproduced with permission from ref. 482 SEM images of nanofibers produced from (d) polycaprolactone (PCL), (e) PCL blended with alkali lignin copolymer, (f) poly(L-lactic acid) (PLLA). (g) PLLA blended with alkali lignin copolymer. Fluorescence microscopy images of NIH/3T3 cells on the nanofibers: (h) PCL-alkali lignin, (i) PLLA-alkali lignin. Reproduced with permission from ref. 483. | ||
Spiridon and Tanase prepared PLA-lignin composites containing 7 or 15 wt% of birch wood organosolv lignin or softwood kraft lignin.481 The composites with 15 wt% lignin showed similar better mechanical properties compared to the ones with 7 wt% lignin. Tensile strength of the composites with 15 wt% of lignin was similar to that of the neat PLA but higher after 30 d immersion of the materials in a simulated body fluid. Despite these differences, all composites performed equally well in a cell culture test that measured metabolic activity of the SaOS-2 cell line. These results demonstrated that the incorporation of lignin did not cause any noticeable cytotoxic effects.
Kai and co-workers made a substantial contribution to the development of lignin-containing nanofibers.484 The lignin content in these materials was varied from 6% to 50%. For instance, lignin-based copolymers (lignin-poly(ε-caprolactone-co-lactide), were synthesized via solvent-free ring-opening polymerization and blended with polyesters such as poly(caprolactone) (PCL) and poly(L-lactic acid) (PLLA) by means of electrospinning. The resulting nanofibers formed fibrous scaffolds in which the fibril diameter was less than one micrometer (Fig. 23d–g). These composite nanofiber scaffolds were demonstrated as non-cytotoxic culture medium for attachment and proliferation of NIH/3T3 cells (Fig. 23h and i).483
6. Outlook
6.1 Current state
The main prospects of lignins are associated with their bio-sourced nature, high availability as by-products and co-products, and possible reintegration into the biospheres. While their biodegradability is the slowest among natural biopolymers,485,486 their end of life cycle highly benefit from their reduced hazards upon introduction into the ecosphere when compared with synthetic polyolefins. Commercially available lignins are separated in sulfur containing lignins, such as lignosulfonates (up to 8% sulfur) and kraft lignins (below 3% sulfur), and sulfur-free lignins such as soda lignin and other lignins obtained from modern biorefinery concepts.32 The largest volume of lignins extracted from biomass is Kraft lignins (55 Mton), followed by soda lignins (6 Mton), and lignosulfonates (1 Mton). Thereafter it is important to consider the fraction of lignin that is processed (i.e. dissolved and burned) or isolated as a biopolymer for further use. For instance, Kraft lignins are currently isolated at a yearly rate of ca. 0.2 Mton, whereas the rest is processed due to their high calorific value making them an excellent fuel for the classic Kraft mills. Lignosulfonates are almost entirely isolated, while Soda lignins are isolated at a considerably smaller rate, i.e. below 15 kton yearly. As a bioeconomical endeavor, the valorization of lignins has a high priority.487 Many lignins have been used in niche market and a wide range of applications is currently being explored. However, for their realization, performance-to-price considerations are always central. Thus far, lignosulfonates are the most exploited commercially, being heavily used as additives in a variety of applications. Kraft lignins are seeing a rise in interest for the development of materials, due to their relatively low cost, high potential availability, and versatile polymeric interactions.Economic strategies for valorization of different lignin types for different applications are illustrated in a plot showing estimated lignin production costs and potential production volume vs. the volume of potential applications and the potential revenue (Fig. 24). Biorefinery lignins (BRLs) have a big cost advantage over other lignin types. Crude BRLs can be easily upgraded (as described in section 2.4.1) and the low costs of the starting material result in different opportunities economically feasible.2
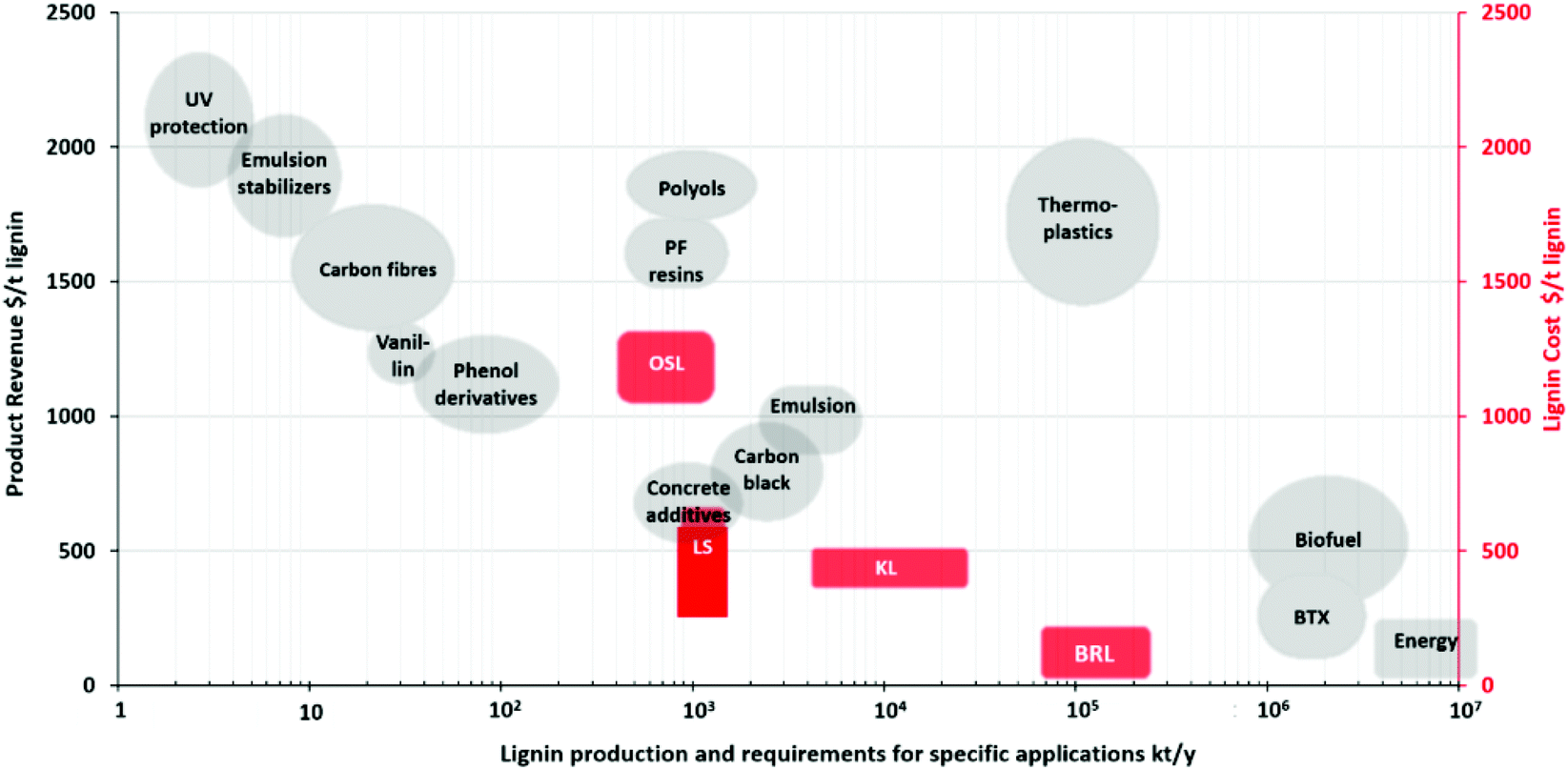 | ||
| Fig. 24 Evaluation of lignin market potentials based on publicly available information. Abbreviations: OSL – organosolv lignins, LS – lignosulfonates, KL – Kraft lignins, BRL – (crude) biorefinery lignins. Reproduced with permission from ref. 2. | ||
High-value/small market size applications (top left corner of the diagram in Fig. 24) would be interesting for specific customers and producers of lignin specialties, such as biomedical and active delivery applications. Lignin particles can be successfully used there.443 Although these applications are very attractive due to high value of the end-use products, the market size is rather limited as compared to potential lignin production market. The “sweet spot” for lignin producers, such as Kraft lignin and potential biorefinery lignin producers, would be in the areas of higher volume and still high value, such as replacement of phenol in PF resins (phenol replacement during resin formulation or direct PF resin replacement with lignin), polyols, emulsions or thermoplastic. Thermoplastics applications are of special interest due to their high market size and good value. Technically, lignin particles can be used very successfully in these applications. However, the LP production costs are close to the value of the final products. Therefore, further reduction in the production costs, e.g., via a few options described above, is critical for the economic feasibility.2,488
For the formation of nanocomposites and biohybrids, polymeric lignins are readily available as side streams and their cost is still “negligible” compared with their benefits in specific, potential applications. However, in the formation of materials with high performances, a significant challenge is imposed by the heterogeneous structure and low purity of some of the readily available lignins, which leads to the formation of materials with poor cohesive properties. As a result, in the formation of composites, a volume fraction substantially below 40% has to be maintained to optimize the benefits of the added lignins.489 However, beyond mechanical strength and toughness, lignins can improve thermal, flame, UV, and oxidative resistances.490,491 In the case of lignins used as a filler, fractionation and, potentially, subsequent particle formation are some of the most promising approaches for value added applications given that the properties can be more easily tethered through a more defined lignin chemistry and morphology. In turn, this may lead to materials with higher mechanical performances. In that endeavor, dry nano- and microparticles are readily usable in a variety of applications. These can be obtained from aerosol flow reactor or spray drying of dispersed particles. Both have a high potential for valorization of lignins in the context of composites and hybrids.125,488 Furthermore the environmental impact of the lifecycle of colloidal lignin particles was recently investigated taking the case of wheat straw lignins. Although limited data were available to build from, the biorefinery model, the main issues regarding environmental impacts were associated with the use and recovery of solvents (therein, water and ethanol) that impact wastewater generation and waterways contaminations beyond increasing general operating and purification costs. Regarding carbon costs, as with other studies including techno-economical assessment, the use of lignin as a substitute to synthetic polymers improved net carbon costs. Integration of the biorefinery in conjunction with other bioproducts further improved the carbon impact.126 Another life cycle assessment, this time considering Kraft lignin in blends concluded that replacement of synthetic phenols by lignin resulted in ca six fold decrease in aquatic acidification and tenfold decrease on impact on climate change.492
Despite the abundance of lignin as a renewable biopolymer, to date few applications have been successfully translated into commercialization, beyond those associated with lignin's high calorific value. However, lignin as a complex phenolic macromolecule presents a vast range of opportunities for the formation of advanced sustainable materials. These opportunities should be associated with extraction-dependent structure and fractionation approaches450 that are established under the biorefinery concepts, as a function of the sustainability of the process. As discussed herein, many of these opportunities are associated with the formation of hybrid-materials involving lignin interactions at the nano- to the micro-scales with secondary organic or inorganic components. We discussed how the strength and function of the materials formed can be connected by the component choice and the processes leading to material formation, i.e., when considering polysaccharides, proteins, carbonaceous interfaces, or metal oxides. Some of the impending applications are highlighted in this review, including those related to fire retardancy and flammability, food packaging, plant and crop protection, energy storage, and therapeutics. With the upcoming biorefinery concepts and the growing bioeconomy, a vast range of uses are expected to emerge provided that cost-performance and sustainability are fully balanced. The new developments in the area can benefit from by-design valorization of lignins, when compared with pulp and paper products that typically include lignin as a by-product. As these developments develop, studies on the sustainable sourcing of lignins and understanding the fundamental connection between its polymeric structure and its properties are required.
6.2 Challenges and future research directions
The potential of lignin as a sustainable material for different applications is highlighted through this Review. However, it is essential to accurately determine the environmental impacts of the processes involving lignin production and use, which demand specific and unambiguous metrics. In this context, life cycle assessment allows the quantification of the environmental burdens of a product or service throughout its life-cycle, from the raw material extraction to its EoL.493 To that end, the environmental impacts need to be divided between the co-products of the system, referred to as allocation. Accordingly, the multi-output character of lignin-centered biorefineries (many different products can be obtained), and the different possible allocations (functional unit or the unit to which impacts are normalized) make any comparison somehow difficult. Additionally, assessing the impacts is challenging due to data scarcity and the evolving character of the technology, particularly when the time from bench to market is measured in multiple years. Accordingly, above 85% of the LCA works provide the impacts in a cradle-to-gate perspective, leaving out of the scope the EoL.124 Although LCA allows determining additional impact categories such as eutrophication, land use, water use, or toxicity the attention has been mainly focused on the global warming potential (GWP) determination.494Among the different possible allocations, those associated with economic, energy, mass or allocation based on substituted impacts are of particular relevance. For instance, they allow a cross-comparison with other materials. Mass-allocation is commonly used because it is straightforward. A GWP of nearly 0.5 kg CO2 eq. per 1 kg of Kraft lignin has been achieved with a mass allocation approach.495 Other works have provided similar values for dry Kraft lignin, with impacts in the range of 0.1 to 2.7 kg CO2 eq. Cradle-to-gate Life Cycle Assessment of bio-adhesives for the wood panel industry.496 Organosolv lignin (from bark) presents a GWP of 1.4–2.1 kg CO2 eq. kg−1, where the type of solvent used significantly affects the results.497 To get a bigger picture, these results should be compared with the impacts originating from conventional polymers. For example, for 1 ton of polypropylene production, GWP values of 1.58–1.60 kg CO2 eq. kg−1 have been obtained using LCA,498 while polyethylene terephthalate encompasses ∼5.5 kg CO2 eq. emissions per 1 kg.499
Regarding lignin nanoparticles, Koch et al. recently reported GWP values ranging from 36.6 to 199 kg CO2 eq. for 1 kg of lignin nanoparticles suspended in water (obtained from wheat straw), depending on the amount of virgin ethanol and the thermal energy demand for the recovery.126 This impact is notably lower than the 800 kg CO2 eq. for 1 kg of nanofibrillated cellulose using TEMPO oxidation.500 Considering that most of the non-degradable plastic waste ends up incinerated/landfilled, or recycled (encompassing additional CO2 or CH4 emissions), renewable/biodegradable polymers, such as lignin, can contribute to decrease the carbon footprint of industries. Also noteworthy is the great potential of lignin to reduce CO2 emissions when replacing other materials. For instance, 23 kg CO2 eq. emissions per 1 kg of lignin could be saved when lignin replaces cotton.495 Generally, lignin-based products offer improved environmental performances in comparison with fossil-based and other bio-based products.
Short and mid-term commercial opportunities for lignins relate to uses such as those in the field of dispersants, wood adhesives, bitumen, polyols in polyurethanes, thermosets, animal feed, thermoplastics, BTX (benzene, toluene and xylenes) and lignin fibers, among others. Some of these applications represent relatively low-value products that may not be sufficient to justify the cost of lignin recovery and possible fractionation and modification. Hence, higher value products represent an opportunity. However, there is a tradeoff between cost of product and volume, often implying that process integration and multi-products are desirable. Here, we covered a segment of lignin utilization, namely, “Multifunctional lignin-based nanocomposites and nanohybrids”, which entails a high valorization potential but also challenged by the economy of scale. Regardless, it is clear that to make the upcoming biorefineries economically viable, all streams need to be optimized, demanding consideration of lignin beyond its energy value.2
A generic aspect that affects the possibility of adopting lignins in bioproducts relates to the economic feasibility and the barriers that exist to enter the marketplace. For instance, from the cost point of view, an obvious consideration beyond the access to lignin, is its physical state (dry versus wet), and the possible needs for packaging and transportation. Hence, any effort to upgrading the molecule at the source site is desirable. Meanwhile, the more technical challenges involve the requirement of producing lignin with uniform quality, for example, from black liquors and biorefinery streams. Indeed, owing to its biological origins and its processing history, lignin is a heterogenous, polydisperse polymer or a complex mixture of polymers with a wide range of molar masses. As reviewed here, there is a significant variation in structures (monomeric units and prevalence, interunit linkages), and potential diverse distributions of various functional groups (e.g., primary, secondary, and phenolic hydroxyl and carboxylate groups). Altogether, these features contribute to differences in lignin behavior, including its solubility and reactivity, both affecting its application in composites and hybrid materials. It is for this reason that lignin fractionation and modification with controlled substitution are necessary. Such processes are significant in adding value to the molecule. Producing more uniform molecular profiles and physical properties opens the possibility to fit target applications, most relevant to composites and hybrid products. Unfortunately, any added processing steps for lignin derivatization also increase the cost and environmental burden. For these reasons, it is important to factor the performance advantage that is achieved when lignin-based bioproducts displace those based on fossil carbon. So far, there is indication that from the industrial perspective, lignin-based biocomposites are in the upward trend, with several units integrating lignin with nanocelluloses and conducting successful trials where lignin has been used as adhesives, combined with bio-components in extrusion processes or to improve the performance of various thermoplastic materials. Likewise, uses in batteries are coming from extensive research, in the form of electrolyte-separator pair or in applications as those known for graphite, for example, in lithium-ion battery anodes. In fact, lignin can be considered as a precursor for carbon nanotubes and graphene. Finally, in composites or in hybrid forms, lignin is being applied in paints and coatings, to provide waterproofing and other properties.
Before ending this review, it is worth mentioning a quite timely aspect, i.e., the fact that plant-derived materials are at the center stage of innovation, in efforts to mitigate negative environmental impacts of traditional petroleum-based materials. In this vein, and even if not discussed herein, it is relevant to mention the possibility of harnessing nature's capacity to generate biopolymers for specific functions. This can be leveraged to isolate building blocks with given properties, which is best accomplished by following biocatalytic pathways that can yield materials with superior properties.501 While still in its infancy, this field is expected to expand and eventually offer interesting alternatives affecting the use of lignin.
We started the review by introducing lignin as a matrix component or as the dispersed phase (micro and nanoparticles) in composite and hybrids. In the latter cases, we discussed the synergies that can be exploited when combining lignin with proteins, metals and oxides, carbons, polysaccharides and synthesis polymers. Such combinations can lead to applications, for instance, relevant to the development of bioproducts that display flame retardancy or food packaging, plant protection, energy storage, and advanced materials, including biomedical. We hope the reader shares our appreciation that such developments require close consideration of lignin's properties (in solution or as particulate systems) and surface and interfacial interactions. They enable the design of materials, as discussed in the review, which offer great advantages considering circularity, end of life and the impact of substitution. As highlighted in this section, lignin processing and functionalization needs to be factored along with other metrics that include scalability, cost-performance balance, product-value and volume tradeoff (see Fig. 25). From the opportunity perspective, the current megatrends are noted to drive interest in lignin. As stated in a recent market report published at the time of the writing of this review, “2021 may well be a pivotal year for development of lignin-based products and markets”.502 However, any success in this direction will also depend on legislative mandates and incentives that support the development of bioproducts, applications, and markets. This includes composites and hybrid materials that might require either crude lignin or fractions subjected to processes that lead to the assembly of lignin into particles (bottom-up constructions enabled but supra-molecular and colloidal interactions) as well as other forms that benefit from derivatization, functionalization or depolymerization (top-down approaches).
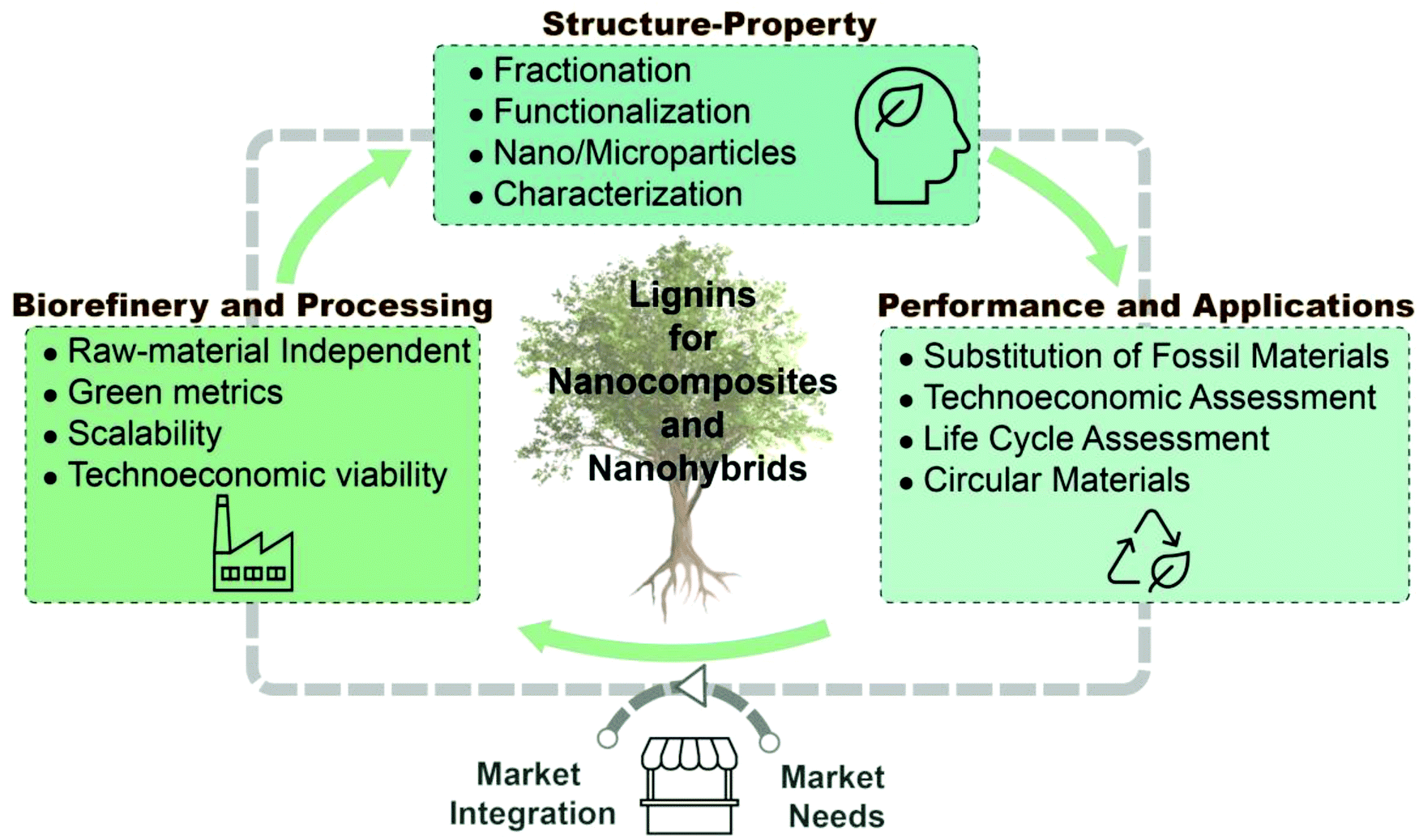 | ||
| Fig. 25 Schematic representation showing the main benefits arising from the use of ligning in nanocomposites and nanohybrids and the future challenges we envision toward practical implementation. | ||
Abbreviations
| ABTS | 2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) |
| AFEX | Ammonia fiber expansion lignin |
| AFM | Atomic force microscopy |
| AH | Acid hydrolysis |
| AKL | Aspen kraft lignin |
| AL | Alkali lignin |
| AMWL | Aspen milled wood lignin |
| BRL | Biorefinery lignins |
| CB | Carbon black |
| CCL | Choline lactate-lactic acid |
| SDBS | Sodium dodecylbenzenesulfonate |
| CLP | Colloidal lignin particle |
| CLSM | Confocal laser scanning microscopy |
| CMC | Carboxymethyl cellulose |
| CML | Carboxymethylated lignin |
| CNC | Cellulose nanocrystal |
| CNF | Cellulose nanofiber |
| CNTs | Carbon nanotubes |
| CQDs | Carbon quantum dots |
| DC | Degree of condensation |
| DCP | Dicumyl peroxide |
| DES | Deep eutectic solvent |
| DMEM | Dulbecco's modified Eagle's medium |
| DMSO | Dimethyl sulfoxide |
| DS | Degree of substitution |
| DSSCs | Dye-sensitized solar cells |
| ECH | Epichlorohydrin |
| EDLC | Electrical double-layer capacitance |
| EES | Electrochemical energy storage |
| EHL | Enzymatic hydrolysis lignin |
| FESEM | Field emission scanning electron microscope |
| GA | Gibberellic acid |
| GnPs | Graphene nanoplatelets |
| GO | Graphene oxide |
| GOx | Glucose oxidase |
| GPEs | Gel polymer electrolytes |
| GQDs | Graphene quantum dots |
| HKL | Hardwood kraft lignin |
| HMPC | Hierarchical mesoporous carbon |
| HPMC | Hydroxypropylmethylcellulose |
| HRTEM | High-resolution transmission electron microscopy |
| HW | Hardwood |
| HWK | Hardwood from Kraft |
| HT | Hydrothermal |
| MAH | Maleic anhydride |
| MCC | Microcrystalline cellulose |
| MLS | Magnetic lignin sphere |
| microPCMs | Microencapsulated phase-change materials |
| Mw | Weight average molecular weight |
| MWD | Molecular weight distribution |
| MWL | Milled wood lignin |
| NMR | Nuclear magnetic resonance |
| NCs | Nanocarriers |
| NP | Nanoparticles |
| NR | Natural rubber |
| KL | Kraft lignin |
| LCC | Lignin carbohydrate complex |
| LCNC | Lignocellulosic nanocrystals |
| LCNF | Lignocellulosic nanofibers |
| LIBs | Lithium-ion batteries |
| LMNPs | Lignin micro-nano particles |
| LNCs | Lignin-based nanocapsules |
| LNPs | Lignin nanoparticles |
| LOD | Limit of detection |
| LOI | Limited oxygen index |
| LPs | Lignin particles |
| LS | Lignosulfonate |
| NIBs | Sodium-ion batteries |
| OS | Organosolv |
| OSL | Organosolv lignin |
| PBAT | Polybutylene adipate terephthalate |
| PBS | Polybutylene succinate |
| PCL | Polycaprolactone |
| PCLLA | Poly(ε-caprolactone-co-L-lactide) |
| PCMs | Phase change materials |
| PDA | Polydopamine |
| PDI | Polydispersity index |
| PEDOT | Poly(3,4-ethylenedioxythiophene) |
| PEDOT:PSS | Poly(3,4-ethylenedioxythiophene) polystyrene sulfonate |
| PEG | Polyethylene glycol |
| PEO | Polyethylene oxide |
| PHB | Poly(hydroxybutyrate) |
| PHBV | Poly(3-hydroxybutyrateco-3-hydroxyvalerate) |
| PHRR | Peak heat release rate |
| PLA | Polylactic acid |
| PLGA | Poly(lactic-co-glycolic acid) |
| PLLA | Poly(L-lactic acid) |
| PU | Polyurethane |
| PVA | Polyvinyl alcohol |
| PMWL | Pine milled wood lignin |
| POM | Polarized optical microscope |
| PP | Polypropylene |
| PS | Polystyrene |
| QAL | Quaternized alkali lignin |
| ROP | Ring opening polymerization |
| QCM-D | Quartz crystal microbalance with dissipation monitoring |
| RGO | Reduced graphene oxide |
| RH | Rice husk |
| ROS | Reactive oxygen species |
| RT | Room temperature |
| SG | Soda grass |
| SBL | Soda bagasse lignin |
| SE | Steam explosion |
| SEM | Scanning electron microscopy |
| SERS | Surface enhanced Raman scattering |
| SEL | Swelled enzyme lignin |
| SET-LRP | Single electron transfer-living radical polymerization |
| SKL | Softwood kraft lignin |
| SPI | Soy protein isolate |
| SPR | Surface plasmon resonance |
| SSA | Specific surface area |
| STP | Sodium triphosphate |
| SW | Softwood |
| SWK | Softwood from Kraft |
| TAIC | Triallyl isocyanurate |
| TBD | Triazabicyclodecene |
| TEA | Techno-economic assessment |
| TGA | Thermogravimetric analysis |
| THF | Tetrahydrofuran |
| THR | Total heat release |
| T g | Glass transition temperature |
| T m | Melting temperature |
| TEM | Transmission electron microscopy |
| TNT | 2,4,6-Trinitrotoluene |
| TOFA | Tall oil fatty acid |
| TPS | Thermoplastic starch |
| UC | Unconfined compressive |
| UV | Ultra violet |
| WCA | Water contact angle |
| WS | Wheat straw |
| X c | Degree of crystallinity |
| ZIBs | Zinc-ion batteries |
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for funding support from Commission H2020 program ERC Advanced Grant (No. 788489, BioELCell), the Canada Excellence Research Chair initiative (CERC-2018-00006), and the Canada Foundation for Innovation (CFI). We are grateful for the support by the FinnCERES Materials Bioeconomy Ecosystem and NordForsk Project 82214 “High-Value Products from Lignin”. M. H. S. acknowledges Vetenskapsrådet (grant number 2020-03752) for financing this work.References
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- M. Y. Balakshin, E. A. Capanema, I. Sulaeva, P. Schlee, Z. Huang, M. Feng, M. Borghei, O. J. Rojas, A. Potthast and T. Rosenau, ChemSusChem, 2021, 14, 1016–1036 CrossRef CAS PubMed.
- J. H. Lora and W. G. Glasser, J. Polym. Environ., 2002, 10, 39–48 CrossRef CAS.
- W. G. Glasser, Front. Chem., 2019, 7, 1–17 CrossRef PubMed.
- D. Stewart, Ind. Crops Prod., 2008, 27, 202–207 CrossRef CAS.
- Y. Cao, S. S. Chen, D. C. W. Tsang, J. H. Clark, V. L. Budarin, C. Hu, K. C.-W. Wu and S. Zhang, Green Chem., 2020, 22, 725–736 RSC.
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2016, 9, 1544–1558 CrossRef CAS PubMed.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- M. Österberg, M. H. Sipponen, B. D. Mattos and O. J. Rojas, Green Chem., 2020, 22, 2712–2733 RSC.
- E. Lizundia, I. Armentano, F. Luzi, F. Bertoglio, E. Restivo, L. Visai, L. Torre and D. Puglia, ACS Appl. Bio Mater., 2020, 3, 5263–5274 CrossRef CAS.
- D. Ye, S. Li, X. Lu, X. Zhang and O. J. Rojas, ACS Sustainable Chem. Eng., 2016, 4, 5248–5257 CrossRef CAS.
- O. Cusola, O. J. Rojas and M. B. Roncero, ACS Appl. Mater. Interfaces, 2019, 11, 45226–45236 CrossRef CAS PubMed.
- M. H. Sipponen, H. Lange, M. Ago and C. Crestini, ACS Sustainable Chem. Eng., 2018, 6, 9342–9351 CrossRef CAS PubMed.
- I. Brodin, M. Ernstsson, G. Gellerstedt and E. Sjöholm, Holzforschung, 2012, 66, 141–147 CAS.
- E. Adler, Wood Sci. Technol., 1977, 11, 169–218 CrossRef CAS.
- J. Ralph, K. Lundquist, G. Brunow, F. Lu, H. Kim, P. F. Schatz, J. M. Marita, R. D. Hatfield, S. A. Ralph, J. H. Christensen and W. Boerjan, Phytochem. Rev., 2004, 3, 29–60 CrossRef CAS.
- A. Berlin and M. Balakshin, Industrial Lignins: Analysis, Properties, and Applications, in Bioenergy Research: Advances and Applications, ed. V. K. Gupta, M. G. Tuohy, C. P. Kubicek, J. Saddler and F. Xu, Elsevier, Amsterdam, 2014, pp. 315–336 Search PubMed.
- M. Lawoko, G. Henriksson and G. Gellerstedt, Biomacromolecules, 2005, 6, 3467–3473 CrossRef CAS PubMed.
- R. F. Helm, in Lignin: Historical, Biological, and Materials Perspectives, American Chemical Society, 1999, vol. 742, pp. 161–171 SE-5 Search PubMed.
- P. Duarah, D. Haldar and M. K. Purkait, Int. J. Biol. Macromol., 2020, 163, 1828–1843 CrossRef CAS PubMed.
- P. K. Mishra and A. Ekielski, Nanomaterials, 2019, 9, 243 CrossRef PubMed.
- W. Gao and P. Fatehi, Can. J. Chem. Eng., 2019, 97, 2827–2842 CrossRef CAS.
- S. Beisl, A. Miltner and A. Friedl, Int. J. Mol. Sci., 2017, 18, 1244 CrossRef CAS PubMed.
- L. E. Low, K. C. Teh, S. P. Siva, I. M. L. Chew, W. W. Mwangi, C. L. Chew, B.-H. Goh, E. S. Chan and B. T. Tey, Environ. Nanotechnol. Monit. Manag., 2021, 15, 100398 Search PubMed.
- Q. Tang, Y. Qian, D. Yang, X. Qiu, Y. Qin and M. Zhou, Polymers, 2020, 12, 2471 CrossRef CAS PubMed.
- J. Sameni, S. A. Jaffer, J. Tjong and M. Sain, Curr. Rep., 2020, 6, 159–171 Search PubMed.
- S. Iravani and R. S. Varma, Green Chem., 2020, 612–636 RSC.
- P. S. Chauhan, Bioresour. Technol. Rep., 2020, 9, 100374 CrossRef.
- W. D. H. Schneider, A. J. P. Dillon and M. Camassola, Biotechnol. Adv., 2021, 47, 107685 CrossRef CAS PubMed.
- S. S. Wong, R. Shu, J. Zhang, H. Liu and N. Yan, Chem. Soc. Rev., 2020, 49, 5510–5560 RSC.
- J. Rajesh Banu, S. Kavitha, R. Yukesh Kannah, T. Poornima Devi, M. Gunasekaran, S.-H. Kim and G. Kumar, Bioresour. Technol., 2019, 290, 121790 CrossRef CAS PubMed.
- D. S. Bajwa, G. Pourhashem, A. H. Ullah and S. G. Bajwa, Ind. Crops Prod., 2019, 139, 111526 CrossRef CAS.
- M. Sajjadi, F. Ahmadpoor, M. Nasrollahzadeh and H. Ghafuri, Int. J. Biol. Macromol., 2021, 178, 394–423 CrossRef CAS PubMed.
- A. Ekielski and P. K. Mishra, Int. J. Mol. Sci., 2021, 22, 63 CrossRef CAS PubMed.
- J. J. Liao, N. H. A. Latif, D. Trache, N. Brosse and M. H. Hussin, Int. J. Biol. Macromol., 2020, 162, 985–1024 CrossRef CAS PubMed.
- L. A. Zevallos Torres, A. Lorenci Woiciechowski, V. O. de Andrade Tanobe, S. G. Karp, L. C. Guimarães Lorenci, C. Faulds and C. R. Soccol, J. Cleaner Prod., 2020, 263, 121499 CrossRef CAS.
- D. Wang, S. H. Lee, J. Kim and C. B. Park, ChemSusChem, 2020, 13, 2807–2827 CrossRef CAS PubMed.
- S. Bertella and J. S. Luterbacher, Trends Chem., 2020, 2, 440–453 CrossRef CAS.
- J. Zhu, C. Yan, X. Zhang, C. Yang, M. Jiang and X. Zhang, Prog. Energy Combust. Sci., 2020, 76, 100788 CrossRef.
- N. Supanchaiyamat, K. Jetsrisuparb, J. T. N. Knijnenburg, D. C. W. Tsang and A. J. Hunt, Bioresour. Technol., 2019, 272, 570–581 CrossRef CAS PubMed.
- H. Sadeghifar and A. Ragauskas, Polymers, 2020, 12, 1134 CrossRef CAS PubMed.
- A. Moreno and M. H. Sipponen, Mater. Horiz., 2020, 7, 2237–2257 RSC.
- Z. Wang, M. S. Ganewatta and C. Tang, Prog. Polym. Sci., 2020, 101, 101197 CrossRef CAS.
- A. Grossman and W. Vermerris, Curr. Opin. Biotechnol., 2019, 56, 112–120 CrossRef CAS PubMed.
- M. N. Collins, M. Nechifor, F. Tanasă, M. Zănoagă, A. McLoughlin, M. A. Stróżyk, M. Culebras and C.-A. Teacă, Int. J. Biol. Macromol., 2019, 131, 828–849 CrossRef CAS PubMed.
- Y. Zhang and M. Naebe, ACS Sustainable Chem. Eng., 2021, 9, 1427–1442 CrossRef CAS.
- G. Parvathy, A. S. Sethulekshmi, J. S. Jayan, A. Raman and A. Saritha, Process Saf. Environ. Prot., 2021, 145, 395–410 CrossRef.
- R. Liu, L. Dai, C. Xu, K. Wang, C. Zheng and C. Si, ChemSusChem, 2020, 13, 4266–4283 CrossRef CAS PubMed.
- O. Yu and K. H. Kim, Appl. Sci., 2020, 10, 4626 CrossRef CAS.
- T. V. Lourençon, L. G. Greca, D. Tarasov, M. Borrega, T. Tamminen, O. J. Rojas and M. Y. Balakshin, ACS Sustainable Chem. Eng., 2020, 8, 1230–1239 CrossRef.
- M. Y. Balakshin, E. A. Capanema, R. B. Santos, H. Chang and H. Jameel, Holzforschung, 2016, 70, 95–108 CAS.
- J. Ralph, C. Lapierre and W. Boerjan, Curr. Opin. Biotechnol., 2019, 56, 240–249 CrossRef CAS PubMed.
- M. Balakshin, E. A. Capanema, X. Zhu, I. Sulaeva, A. Potthast, T. Rosenau and O. J. Rojas, Green Chem., 2020, 22, 3985–4001 RSC.
- E. Capanema, M. Balakshin, R. Katahira, H. Chang and H. Jameel, J. Wood Chem. Technol., 2015, 35, 17–26 CrossRef.
- H. Chang, E. B. Cowling and W. Brown, Holzforschung, 1975, 29, 153–159 CrossRef CAS.
- G. Zinovyev, I. Sumerskii, T. Rosenau, M. Balakshin and A. Potthast, Molecules, 2018, 23, 2223 CrossRef PubMed.
- H. Erdtman, J. Polym. Sci., Part B: Polym. Lett., 1972, 10, 228–230 CrossRef.
- C. Crestini, F. Melone, M. Sette and R. Saladino, Biomacromolecules, 2011, 12, 3928–3935 CrossRef CAS PubMed.
- M. Y. Balakshin and E. A. Capanema, RSC Adv., 2015, 5, 87187–87199 RSC.
- C. S. Lancefield, H. L. J. Wienk, R. Boelens, B. M. Weckhuysen and P. C. A. Bruijnincx, Chem. Sci., 2018, 9, 6348–6360 RSC.
- C. Crestini, H. Lange, M. Sette and D. S. Argyropoulos, Green Chem., 2017, 19, 4104–4121 RSC.
- A. Berlin, M. Balakshin, N. Gilkes, J. Kadla, V. Maximenko, S. Kubo and J. Saddler, J. Biotechnol., 2006, 125, 198–209 CrossRef CAS PubMed.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- J. Domínguez-Robles, T. Tamminen, T. Liitiä, M. S. Peresin, A. Rodríguez and A.-S. Jääskeläinen, Int. J. Biol. Macromol., 2018, 106, 979–987 CrossRef PubMed.
- E. A. Capanema and M. Yu, Balakshin. High purity lignin, lignin compositions, and higher structured lignin, PCT Int. Appl, WO2014144746A120140918, 2014 Search PubMed.
- H. Sadeghifar and A. Ragauskas, ACS Sustainable Chem. Eng., 2020, 8, 8086–8101 CrossRef CAS.
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS.
- D. A. Baker and T. G. Rials, J. Appl. Polym. Sci., 2013, 130, 713–728 CrossRef CAS.
- Y. Li and S. Sarkanen, Macromolecules, 2005, 38, 2296–2306 CrossRef CAS.
- M. Zhang and A. A. Ogale, Carbon, 2014, 69, 626–629 CrossRef CAS.
- M. Balakshin and E. A. Capanema, in 14th European Workshop on Lignocellulosics and Pulp, Grenoble, France, 2016.
- M. Y. Balakshin, E. A. Capanema, M. Colakyan and F. Lipiecki, Upgrading lignin from lignin-containing residues through reactive extraction, WO2015168571A1, 2015.
- M. Borrega, S. Päärnilä, L. G. Greca, A.-S. Jääskeläinen, T. Ohra-aho, O. J. Rojas and T. Tamminen, Langmuir, 2020, 36, 9675–9684 CrossRef CAS PubMed.
- J. D. Zwilling, X. Jiang, F. Zambrano, R. A. Venditti, H. Jameel, O. D. Velev, O. J. Rojas and R. Gonzalez, Green Chem., 2021, 23, 1001–1012 RSC.
- S. Beisl, J. Adamcyk and A. Friedl, Molecules, 2020, 25, 1388 CrossRef CAS PubMed.
- X.-J. Shen, T. Chen, H.-M. Wang, Q. Mei, F. Yue, S. Sun, J.-L. Wen, T.-Q. Yuan and R.-C. Sun, ACS Sustainable Chem. Eng., 2020, 8, 2130–2137 CrossRef CAS.
- M. A. Saiful Badri, M. M. Salleh, N. F. A. Md Noor, M. Y. A. Rahman and A. A. Umar, Mater. Chem. Phys., 2017, 193, 212–219 CrossRef CAS.
- A. Hasan and P. Fatehi, Appl. Clay Sci., 2018, 158, 72–82 CrossRef CAS.
- T. M. Budnyak, A. Slabon and M. H. Sipponen, ChemSusChem, 2020, 13, 4344–4355 CrossRef CAS PubMed.
- W. Brown, J. Appl. Polym. Sci., 1967, 11, 2381–2396 CrossRef CAS.
- F. Xiong, Y. Han, S. Wang, G. Li, T. Qin, Y. Chen and F. Chu, Ind. Crops Prod., 2017, 100, 146–152 CrossRef CAS.
- W. Zhao, L. P. Xiao, G. Song, R. C. Sun, L. He, S. Singh, B. A. Simmons and G. Cheng, Green Chem., 2017, 19, 3272–3281 RSC.
- Y. Deng, X. Feng, M. Zhou, Y. Qian, H. Yu and X. Qiu, Biomacromolecules, 2011, 12, 1116–1125 CrossRef CAS PubMed.
- C. Frangville, M. Rutkevičius, A. P. Richter, O. D. Velev, S. D. Stoyanov and V. N. Paunov, ChemPhysChem, 2012, 13, 4235–4243 CrossRef CAS PubMed.
- M. Balakshin, E. Capanema and A. Berlin, Stud. Nat. Prod. Chem., 2014, 42, 83–115 CAS.
- D. Tarasov, M. Leitch and P. Fatehi, Biotechnol. Biofuels, 2018, 11, 269 CrossRef PubMed.
- K. Lintinen, Y. Xiao, R. Bangalore Ashok, T. Leskinen, E. Sakarinen, M. Sipponen, F. Muhammad, P. Oinas, M. Österberg and M. Kostiainen, Green Chem., 2018, 20, 843–850 RSC.
- X. Zhang, W. Liu, D. Yang and X. Qiu, Adv. Funct. Mater., 2019, 29, 1806912 CrossRef.
- W. Liu, R. Zhou, D. Zhou, G. Ding, J. M. Soah, C. Y. Yue and X. Lu, Carbon, 2015, 83, 188–197 CrossRef CAS.
- H. Sakai, K. Kuroda, S. Muroyama, T. Tsukegi, R. Kakuchi, K. Takada, A. Hata, R. Kojima, T. Ogoshi, M. Omichi, K. Ninomiya and K. Takahashi, Polym. J., 2018, 50, 281–284 CrossRef CAS.
- M. Oliviero, L. Verdolotti, E. Di Maio, M. Aurilia and S. Iannace, J. Agric. Food Chem., 2011, 59, 10062–10070 CrossRef CAS PubMed.
- J. Wang, Y. Qian, Y. Deng, D. Liu, H. Li and X. Qiu, Appl. Surf. Sci., 2016, 390, 617–622 CrossRef CAS.
- L. Dai, W. Zhu, J. Lu, F. Kong, C. Si and Y. Ni, Green Chem., 2019, 21, 5222–5230 RSC.
- T. M. Budnyak, J. Piątek, I. V. Pylypchuk, M. Klimpel, O. Sevastyanova, M. E. Lindström, V. M. Gun'ko and A. Slabon, ACS Omega, 2020, 5, 10847–10856 CrossRef CAS PubMed.
- M. Ago, S. Huan, M. Borghei, J. Raula, E. I. Kauppinen and O. J. Rojas, ACS Appl. Mater. Interfaces, 2016, 8, 23302–23310 CrossRef CAS PubMed.
- M. Lievonen, J. J. Valle-Delgado, M. L. Mattinen, E. L. Hult, K. Lintinen, M. A. Kostiainen, A. Paananen, G. R. Szilvay, H. Setälä and M. Österberg, Green Chem., 2016, 18, 1416–1422 RSC.
- F. Chen, W. Liu, S. I. Seyed Shahabadi, J. Xu and X. Lu, ACS Sustainable Chem. Eng., 2016, 4, 4997–5004 CrossRef CAS.
- H. Li, Y. Deng, B. Liu, Y. Ren, J. Liang, Y. Qian, X. Qiu, C. Li and D. Zheng, ACS Sustainable Chem. Eng., 2016, 4, 1946–1953 CrossRef CAS.
- F. Xiong, Y. Han, S. Wang, G. Li, T. Qin, Y. Chen and F. Chu, ACS Sustainable Chem. Eng., 2017, 5, 2273–2281 CrossRef CAS.
- T. Kämäräinen, M. Ago, J. Seitsonen, J. Raula, E. I. Kauppinen, J. Ruokolainen and O. J. Rojas, Soft Matter, 2018, 14, 3387–3396 RSC.
- T. Kämäräinen, B. L. Tardy, S. J. Nikkhah and P. Batys, J. Colloid Interface Sci., 2020, 579, 794–804 CrossRef PubMed.
- M. Kannangara, M. Marinova, L. Fradette and J. Paris, Chem. Eng. Res. Des., 2016, 105, 94–106 CrossRef CAS.
- S. M. Notley and M. Norgren, Langmuir, 2010, 26, 5484–5490 CrossRef CAS PubMed.
- M. Farooq, Z. Tao, J. J. Valle-Delgado, M. H. Sipponen, M. Morits and M. Österberg, Langmuir, 2020, 36, 15592–15602 CrossRef CAS PubMed.
- Z. H. Liu, N. Hao, S. Shinde, Y. Pu, X. Kang, A. J. Ragauskas and J. S. Yuan, Green Chem., 2019, 21, 245–260 RSC.
- Z. Jiang, Y. Ma, X. Guo, J. Remón, D. C. W. Tsang, C. Hu and B. Shi, J. Hazard. Mater., 2021, 403, 123701 CrossRef CAS PubMed.
- T. E. Nypelö, C. A. Carrillo and O. J. Rojas, Soft Matter, 2015, 11, 2046–2054 RSC.
- C. Fritz, C. Salas, H. Jameel and O. J. Rojas, Nord. Pulp Pap. Res. J., 2017, 32, 572–585 CrossRef CAS.
- O. Cusola, S. Kivistö, S. Vierros, P. Batys, M. Ago, B. L. Tardy, L. G. Greca, M. B. Roncero, M. Sammalkorpi and O. J. Rojas, Langmuir, 2018, 34, 5759–5771 CrossRef CAS PubMed.
- M. Ago, B. L. Tardy, L. Wang, J. Guo, A. Khakalo and O. J. Rojas, MRS Bull., 2017, 42, 371–378 CrossRef CAS.
- M. Ma, L. Dai, J. Xu, Z. Liu and Y. Ni, Green Chem., 2020, 22, 2011–2017 RSC.
- W. Yang, E. Fortunati, D. Gao, G. M. Balestra, G. Giovanale, X. He, L. Torre, J. M. Kenny and D. Puglia, ACS Sustainable Chem. Eng., 2018, 6, 3502–3514 CrossRef CAS.
- L. Chen, X. Zhou, Y. Shi, B. Gao, J. Wu, T. B. Kirk, J. Xu and W. Xue, Chem. Eng. J., 2018, 346, 217–225 CrossRef CAS.
- T. Leskinen, M. Smyth, Y. Xiao, K. Lintinen, M. L. Mattinen, M. A. Kostiainen, P. Oinas and M. Österberg, Nord. Pulp Pap. Res. J., 2017, 32, 586–596 CrossRef CAS.
- Y. Deng, H. Zhao, Y. Qian, L. Lü, B. Wang and X. Qiu, Ind. Crops Prod., 2016, 87, 191–197 CrossRef CAS.
- M. Ma, L. Dai, C. Si, L. Hui, Z. Liu and Y. Ni, ChemSusChem, 2019, 12, 5239–5245 CrossRef CAS PubMed.
- L. Matsakas, A. Karnaouri, A. Cwirzen, U. Rova and P. Christakopoulos, Molecules, 2018, 23, 1822 CrossRef PubMed.
- B. L. Tardy, J. J. Richardson, J. Guo, J. Lehtonen, M. Ago and O. J. Rojas, Green Chem., 2018, 20, 1335–1344 RSC.
- M. Norgren and H. Edlund, Curr. Opin. Colloid Interface Sci., 2014, 19, 409–416 CrossRef CAS.
- D. R. Ratnaweera, D. Saha, S. V. Pingali, N. Labbé, A. K. Naskar and M. Dadmun, RSC Adv., 2015, 5, 67258–67266 RSC.
- S. M. Notley and M. Norgren, Holzforschung, 2012, 66, 15–622 Search PubMed.
- K. M. Hess, J. P. Killgore and W. V. Srubar, Cellulose, 2018, 25, 6345–6360 CrossRef CAS.
- R. Ajdary, N. Kretzschmar, H. Baniasadi, J. Trifol, J. V. Seppälä, J. Partanen and O. J. Rojas, ACS Sustainable Chem. Eng., 2021, 9, 2727–2735 CrossRef CAS.
- C. Moretti, B. Corona, R. Hoefnagels, I. Vural-Gürsel, R. Gosselink and M. Junginger, Sci. Total Environ., 2021, 770, 144656 CrossRef CAS PubMed.
- R. P. Bangalore Ashok, P. Oinas, K. Lintinen, G. Sarwar, M. A. Kostiainen and M. Österberg, Green Chem., 2018, 20, 4911–4919 RSC.
- D. Koch, M. Paul, S. Beisl, A. Friedl and B. Mihalyi, J. Cleaner Prod., 2020, 245, 118760 CrossRef CAS.
- M. Farooq, T. Zou, G. Riviere, M. H. Sipponen and M. Österberg, Biomacromolecules, 2019, 20, 693–704 CrossRef CAS PubMed.
- A. Colburn, R. J. Vogler, A. Patel, M. Bezold, J. Craven, C. Liu and D. Bhattacharyya, Nanomaterials, 2019, 9, 867 CrossRef PubMed.
- E. Lizundia, D. Puglia, T.-D. Nguyen and I. Armentano, Prog. Mater. Sci., 2020, 112, 100668 CrossRef CAS.
- T. Nesbakk and T. Helle, J. Pulp Pap. Sci., 2002, 28, 1–4 Search PubMed.
- K. Gabov, T. Oja, T. Deguchi, A. Fallarero and P. Fardim, Cellulose, 2017, 24, 641–658 CrossRef CAS.
- P. G. de Melo, M. Fornazier Borges, J. Afonso Ferreira, M. Vicente Barbosa Silva and R. Ruggiero, Int. J. Mol. Sci., 2018, 19, 1–13 Search PubMed.
- L. Manjarrez Nevárez, L. Ballinas-Casarrubias, O. S. Canto, A. Celzard, V. Fierro, R. Ibarra Gómez and G. González Sánchez, Carbohydr. Polym., 2011, 86, 732–741 CrossRef.
- L. A. M. Nevrez, L. B. Casarrubias, A. Celzard, V. Fierro, V. T. Mũoz, A. C. Davila, J. R. T. Lubian and G. G. Snchez, Sci. Technol. Adv. Mater., 2011, 12, 045006 CrossRef PubMed.
- Y. Guo, D. Tian, F. Shen, G. Yang, L. Long, J. He, C. Song, J. Zhang, Y. Zhu, C. Huang and S. Deng, Polymers, 2019, 11, 1–11 Search PubMed.
- H. Sadeghifar, R. Venditti, J. Jur, R. E. Gorga and J. J. Pawlak, ACS Sustainable Chem. Eng., 2017, 5, 625–631 CrossRef CAS.
- J. Köhnke, H. Rennhofer, C. Unterweger, N. Gierlinger, J. Keckes, C. Zollfrank, O. J. Rojas and W. Gindl-Altmutter, Nanomaterials, 2018, 8, 1055 CrossRef PubMed.
- Y. Liu, ACS Sustainable Chem. Eng., 2018, 6, 5524–5532 CrossRef CAS.
- M. Parit, P. Saha, V. A. Davis and Z. Jiang, ACS Omega, 2018, 3, 10679–10691 CrossRef CAS PubMed.
- A. Hambardzumyan, L. Foulon, N. B. Bercu, M. Pernes, J. E. Maigret, M. Molinari, B. Chabbert and V. Aguié-Béghin, Chem. Eng. J., 2015, 264, 780–788 CrossRef CAS.
- Y. Zhao, A. Tagami, G. Dobele, M. E. Lindström and O. Sevastyanova, Polymers, 2019, 11, 538 CrossRef PubMed.
- A. A. Younyia Mbiada, S. Musa, O. Richter, A. Kneer and S. Barbe, Polym. Renewable Resour., 2018, 9, 51–58 Search PubMed.
- M. Michelin, A. M. Marques, L. M. Pastrana, J. A. Teixeira and M. A. Cerqueira, J. Food Eng., 2020, 285, 110107 CrossRef CAS.
- A. Alzagameem, S. E. Klein, M. Bergs, X. T. Do, I. Korte, S. Dohlen, C. Hüwe, J. Kreyenschmidt, B. Kamm, M. Larkins and M. Schulze, Polymers, 2019, 11, 670 CrossRef CAS PubMed.
- T. G. Rials and W. G. Glasser, Polymer, 1990, 31, 1333–1338 CrossRef CAS.
- B. D. Mattos, B. L. Tardy, L. G. Greca, T. Kämäräinen, W. Xiang, O. Cusola, W. L. E. Magalhães and O. J. Rojas, Sci. Adv., 2020, 6, eaaz7328 CrossRef CAS PubMed.
- J. Lu, W. Zhu, L. Dai, C. Si and Y. Ni, Carbohydr. Polym., 2019, 215, 289–295 CrossRef CAS PubMed.
- H. Setälä, H.-L. Alakomi, A. Paananen, G. R. Szilvay, M. Kellock, M. Lievonen, V. Liljeström, E.-L. Hult, K. Lintinen, M. Österberg and M. Kostiainen, Cellulose, 2019, 27, 273–284 CrossRef.
- Z. Zhu, S. Fu and L. A. Lucia, ACS Sustainable Chem. Eng., 2019, 7, 5376–5384 CrossRef CAS.
- K. Peredo, D. Escobar, J. Vega-lara, A. Berg and M. Pereira, Int. J. Biol. Macromol., 2016, 83, 403–409 CrossRef CAS PubMed.
- L. Zhang, H. Lu, J. Yu, Y. Fan, Y. Yang, J. Ma and Z. Wang, Cellulose, 2018, 25, 7315–7328 CrossRef CAS.
- S. Huang, S. Shuyi, H. Gan, W. Linjun, C. Lin, X. Danyuan, H. Zhou, X. Lin and Y. Qin, Carbohydr. Polym., 2019, 223, 115080 CrossRef CAS PubMed.
- J. Xia, Z. Liu, Y. Chen, Y. Cao and Z. Wang, Cellulose, 2020, 27, 879–894 CrossRef CAS.
- D. Ciolacu, A. M. Oprea, N. Anghel, G. Cazacu and M. Cazacu, Mater. Sci. Eng., C, 2012, 32, 452–463 CrossRef CAS.
- S. Park, S. H. Kim, K. Won, J. W. Choi, Y. H. Kim, H. J. Kim, Y. H. Yang and S. H. Lee, Carbohydr. Polym., 2015, 115, 223–229 CrossRef CAS PubMed.
- H. Wu, Y. Xie, H. Zhao, X. Chen, C. Jiang, S. Bi and Y. Liu, BioResources, 2019, 14, 6465–6484 CAS.
- H. Zhu, Y. Li, B. Pettersson, L. Zhang and M. Lindström, J. Adhes. Sci. Technol., 2014, 28, 490–498 CrossRef CAS.
- S. Vincent, R. Prado, O. Kuzmina, K. Potter, J. Bhardwaj, N. D. Wanasekara, R. L. Harniman, A. Koutsomitopoulou, S. J. Eichhorn, T. Welton and S. S. Rahatekar, ACS Sustainable Chem. Eng., 2018, 6, 5903–5910 CrossRef CAS.
- H. Li and L. Peng, Carbohydr. Polym., 2015, 124, 35–42 CrossRef CAS PubMed.
- A. L. Santana and M. A. A. Meireles, Food Public Health, 2014, 4, 229–241 CrossRef.
- E. S. Stevens, A. Klamczynski and G. M. Glenn, eXPRESS Polym. Lett., 2010, 4, 311–320 CrossRef CAS.
- A. Sarwono, Z. Man, M. A. Bustam, K. Azizi and M. Azizli, Appl. Mech. Mater., 2015, 699, 204–209 Search PubMed.
- S. Baumberger, in Chemical Modification, Properties, and Usage of Lignin, ed. T. Q. Hu, Kluwer Academic/Plenum Publishers, 2002, pp. 1–19 Search PubMed.
- S. Baumberger, C. Lapierre and B. Monties, J. Agric. Food Chem., 1998, 46, 2234–2240 CrossRef CAS.
- K. Kaewtatip and J. Thongmee, Mater. Des., 2013, 49, 701–704 CrossRef CAS.
- N. Izaguirre, O. Gordobil, E. Robles and J. Labidi, Int. J. Biol. Macromol., 2020, 155, 447–455 CrossRef CAS PubMed.
- K. Ravishankar, M. Venkatesan, R. P. Desingh, A. Mahalingam, B. Sadhasivam, R. Subramaniyam and R. Dhamodharan, Mater. Sci. Eng., C, 2019, 102, 447–457 CrossRef CAS PubMed.
- T. Zou, M. H. Sipponen and M. Österberg, Front. Chem., 2019, 7, 2300 Search PubMed.
- M. H. Nguyen, I. C. Hwang and H. J. Park, J. Photochem. Photobiol., B, 2013, 125, 194–201 CrossRef CAS PubMed.
- E. Capecchi, D. Piccinino, B. M. Bizzarri, D. Avitabile, C. Pelosi, C. Colantonio and G. Calabro, Biomacromolecules, 2019, 20, 1975–1988 CrossRef CAS PubMed.
- A. B. Albadarin, M. N. Collins, M. Naushad, S. Shirazian, G. Walker and C. Mangwandi, Chem. Eng. J., 2017, 307, 264–272 CrossRef CAS.
- V. Nair, A. Panigrahy and R. Vinu, Chem. Eng. J., 2014, 254, 491–502 CrossRef CAS.
- L. Chen, C. Tang, N. Ning, C. Wang, Q. Fu and Q. Zhang, Chin. J. Polym. Sci., 2009, 27, 739–746 CrossRef CAS.
- K. Crouvisier-Urion, P. R. Bodart, P. Winckler, J. Raya, R. D. Gougeon, P. Cayot, S. Domenek, F. Debeaufort and T. Karbowiak, ACS Sustainable Chem. Eng., 2016, 4, 6371–6381 CrossRef CAS.
- K. Crouvisier-Urion, A. Lagorce-Tachon, C. Lauquin, P. Winckler, W. Tongdeesoontorn, S. Domenek, F. Debeaufort and T. Karbowiak, Food Chem., 2017, 236, 120–126 CrossRef CAS PubMed.
- H. Luo, Q. Shen, F. Ye, Y. F. Cheng, M. Mezgebe and R. J. Qin, Mater. Sci. Eng., C, 2012, 32, 2001–2006 CrossRef CAS PubMed.
- S. Rai, P. K. Dutta and G. K. Mehrotra, J. Polym. Mater., 2017, 34, 171–183 CAS.
- W. Wang, B. Shan, L. Zhu, C. Xie, C. Liu and F. Cui, Carbohydr. Polym., 2018, 187, 35–42 CrossRef CAS PubMed.
- Y. Zhang, M. Jiang, Y. Zhang, Q. Cao, X. Wang, Y. Han, G. Sun, Y. Li and J. Zhou, Mater. Sci. Eng., C, 2019, 104, 110002 CrossRef CAS PubMed.
- W. Zhu, J. Lu and L. Dai, Part. Part. Syst. Charact., 2018, 35, 1–8 CAS.
- R. Tian, Q. Liu, W. Zhang and Y. Zhang, J. Inorg. Organomet. Polym. Mater., 2018, 28, 2545–2553 CrossRef CAS.
- M. Yan, W. Huang and Z. Li, Int. J. Biol. Macromol., 2019, 136, 927–935 CrossRef CAS PubMed.
- K. Wang, L. S. Loo and K. L. Goh, Mater. Res. Express, 2016, 3, 035301 CrossRef.
- M. Schreiber, S. Vivekanandhan, P. Cooke, A. K. Mohanty and M. Misra, J. Mater. Sci., 2014, 49, 7949–7958 CrossRef CAS.
- M. M. Al-Rashed, S. Niknezhad and S. C. Jana, Macromol. Chem. Phys., 2019, 220, 1–8 CrossRef.
- S. Kim, M. M. Fernandes, T. Matamá, A. Loureiro, A. C. Gomes and A. Cavaco-Paulo, Colloids Surf., B, 2013, 103, 1–8 CrossRef CAS PubMed.
- R. P. Pandey, K. Rasool, P. A. Rasheed, T. Gomez, M. Pasha, S. A. Mansour, O. S. Lee and K. A. Mahmoud, Green Chem., 2020, 22, 678–687 RSC.
- X. Ji and M. Guo, Int. J. Adhes. Adhes., 2018, 82, 8–13 CrossRef CAS.
- E. Pasquier, B. D. Mattos, N. Belgacem, J. Bras and O. J. Rojas, Biomacromolecules, 2021, 22, 880–889 CrossRef CAS PubMed.
- B. Song, H. Liang, R. Sun, P. Peng, Y. Jiang and D. She, Int. J. Biol. Macromol., 2020, 144, 219–230 CrossRef CAS PubMed.
- S. P. Raman, P. Gurikov and I. Smirnova, J. Supercrit. Fluids, 2015, 106, 23–33 CrossRef CAS.
- M. H. Sipponen, M. Farooq, J. Koivisto, A. Pellis, J. Seitsonen and M. Österberg, Nat. Commun., 2018, 9, 1–7 CrossRef CAS PubMed.
- S. Fertahi, I. Bertrand, M. Ilsouk, A. Oukarroum, M. B. Amjoud, Y. Zeroual and A. Barakat, Int. J. Biol. Macromol., 2020, 143, 153–162 CrossRef CAS PubMed.
- L. Musilová, A. Kovalcik, P. Smolka, A. Mina, P. Humpolí, R. Vícha and P. Poní, Carbohydr. Polym., 2018, 181, 394–403 CrossRef PubMed.
- I. E. Raschip, O. M. Paduraru-Mocanu, L. E. Nita and M. V. Dinu, J. Appl. Polym. Sci., 2020, 137, e49111 CrossRef.
- S. Sathawong, W. Sridach and K. Techato, J. Polym. Environ., 2018, 26, 3307–3315 CrossRef CAS.
- X. Song, F. Chen and S. Liu, BioResources, 2016, 11, 6378–6392 CAS.
- S. Shankar, J. P. Reddy and J. Rhim, Int. J. Biol. Macromol., 2015, 81, 267–273 CrossRef CAS PubMed.
- F. Flores-céspedes, M. Villafranca-sánchez and M. Fernández-pérez, Int. J. Biol. Macromol., 2020, 153, 883–891 CrossRef PubMed.
- I. E. Raschip, G. E. Hitruc, C. Vasile and M. Popescu, Int. J. Biol. Macromol., 2013, 54, 230–237 CrossRef CAS PubMed.
- S. Sathawong, W. Sridach and K. Techato, J. Environ. Chem. Eng., 2018, 6, 5879–5888 CrossRef CAS.
- H. Bradford Vickery, The History of the Discovery of the Amino Acids II. A Review of Amino Acids Described Since 1931 as Components of Native Proteins, in Advances in Protein Chemistry, ed. C.B. Anfinsen Jr., J. T. Edsall and F. M. Richards, Academic Press, 1972, vol. 26, pp. 81–171 Search PubMed.
- S. Bleakley and M. Hayes, Foods, 2017, 6, 33 CrossRef PubMed.
- T. D. Sutherland, M. G. Huson and T. D. Rapson, J. Struct. Biol., 2018, 201, 76–83 CrossRef CAS PubMed.
- M. Zubair and A. Ullah, Crit. Rev. Food Sci. Nutr., 2020, 60, 406–434 CrossRef CAS PubMed.
- J. Gómez-Estaca, R. Gavara, R. Catalá and P. Hernández-Muñoz, Packag. Technol. Sci., 2016, 29, 203–224 CrossRef.
- S. B. Ross-Murphy, Imaging Sci. J., 1997, 45, 205–209 CrossRef CAS.
- S. Karnnet, P. Potiyaraj and V. Pimpan, Polym. Degrad. Stab., 2005, 90, 106–110 CrossRef CAS.
- R. Núñez-Flores, B. Giménez, F. Fernández-Martín, M. E. López-Caballero, M. P. Montero and M. C. Gómez-Guillén, Food Hydrocolloids, 2013, 30, 163–172 CrossRef.
- S. M. Ojagh, R. Núñez-Flores, M. E. López-Caballero, M. P. Montero and M. C. Gómez-Guillén, Food Chem., 2011, 125, 595–606 CrossRef CAS.
- R. Núñez-Flores, B. Giménez, F. Fernández-Martín, M. E. López-Caballero, M. P. Montero and M. C. Gómez-Guillén, Food Hydrocolloids, 2012, 27, 60–71 CrossRef.
- J. A. Belgodere, S. A. Zamin, R. M. Kalinoski, C. E. Astete, J. C. Penrod, K. M. Hamel, B. C. Lynn, J. S. Rudra, J. Shi and J. P. Jung, ACS Appl. Bio Mater., 2019, 2, 3562–3572 CrossRef CAS.
- K. R. Aadil, A. Barapatre and H. Jha, Bioresour. Bioprocess., 2016, 3, 27 CrossRef.
- M. J. Mehta and A. Kumar, Chem. – Eur. J., 2019, 25, 1269–1274 CrossRef CAS PubMed.
- B. L. Gadilohar and G. S. Shankarling, J. Mol. Liq., 2017, 227, 234–261 CrossRef CAS.
- M. R. Lumadue, F. S. Cannon and N. R. Brown, Fuel, 2012, 97, 869–875 CrossRef CAS.
- C. Nieto-Delgado, F. S. Cannon, P. D. Paulsen, J. C. Furness, R. C. Voigt and J. R. Pagnotti, Fuel, 2014, 121, 39–47 CrossRef CAS.
- Z. Zhao, F. S. Cannon and C. Nieto-Delgado, Carbon, 2019, 154, 254–265 CrossRef CAS.
- Z. Zhao, F. S. Cannon, C. Nieto-Delgado and L. Pena, Carbon, 2016, 108, 303–317 CrossRef CAS.
- H. Tian, G. Guo, X. Fu, Y. Yao, L. Yuan and A. Xiang, Int. J. Biol. Macromol., 2018, 120, 475–490 CrossRef CAS PubMed.
- N. Kitabatake, M. Tahara and E. Doi, Agric. Biol. Chem., 1990, 54, 2205–2212 CAS.
- E. Mohammad Zadeh, S. F. O’Keefe and Y.-T. Kim, J. Food Sci., 2019, 84, 1420–1426 CrossRef CAS PubMed.
- P. Chen, L. Zhang, S. Peng and B. Liao, J. Appl. Polym. Sci., 2006, 101, 334–341 CrossRef CAS.
- S. Pradyawong, G. Qi, N. Li, X. S. Sun and D. Wang, Int. J. Adhes. Adhes., 2017, 75, 66–73 CrossRef CAS.
- Z. Xiao, Y. Li, X. Wu, G. Qi, N. Li, K. Zhang, D. Wang and X. S. Sun, Ind. Crops Prod., 2013, 50, 501–509 CrossRef CAS.
- J. Xin, P. Zhang, M. P. Wolcott, J. Zhang, W. C. Hiscox and X. Zhang, J. Polym. Environ., 2017, 25, 599–605 CrossRef CAS.
- C. Salas, M. Ago, L. A. Lucia and O. J. Rojas, React. Funct. Polym., 2014, 85, 221–227 CrossRef CAS.
- M. Ago, K. Okajima, J. E. Jakes, S. Park and O. J. Rojas, Biomacromolecules, 2012, 13, 918–926 CrossRef CAS PubMed.
- C. Salas, O. J. Rojas, L. A. Lucia, M. A. Hubbe and J. Genzer, ACS Appl. Mater. Interfaces, 2013, 5, 199–206 CrossRef CAS PubMed.
- W. Yang, J. M. Kenny and D. Puglia, Ind. Crops Prod., 2015, 74, 348–356 CrossRef CAS.
- M. Oliviero, R. Rizvi, L. Verdolotti, S. Iannace, H. E. Naguib, E. Di Maio, H. C. Neitzert and G. Landi, Adv. Funct. Mater., 2017, 27, 1605142 CrossRef.
- W. J. Grigsby, S. M. Scott, M. I. Plowman-Holmes, P. G. Middlewood and K. Recabar, Acta Biomater., 2020, 104, 95–103 CrossRef CAS PubMed.
- J. Yang, Y. Wang, J. Luo and L. Chen, ACS Omega, 2018, 3, 4647–4656 CrossRef CAS PubMed.
- S. Yang, T.-Q. Yuan, Q. Shi and R.-C. Sun, in Application of Lignin in Thermoplastic Materials, Green Chemistry and Chemical Engineering. Encyclopedia of Sustainability Science and Technology Series, ed. B. Han and T. Wu, Springer New York, New York, NY, 2019, pp. 405–426 Search PubMed.
- D. Kun and B. Pukánszky, Eur. Polym. J., 2017, 93, 618–641 CrossRef CAS.
- S. I. Falkehag, Appl. Polym. Symp., 1975, 28, 247–257 CAS.
- C. Pouteau, S. Baumberger, B. Cathala and P. Dole, 2004, 327, 935–943.
- J. Yang, Y. C. Ching and C. H. Chuah, Polymers, 2019, 11, 751 CrossRef CAS PubMed.
- S. Sen, S. Patil and D. S. Argyropoulos, Green Chem., 2015, 17, 4862–4887 RSC.
- H. Liu and H. Chung, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3515–3528 CrossRef CAS.
- W. Ren, X. Pan, G. Wang, W. Cheng and Y. Liu, Green Chem., 2016, 18, 5008–5014 RSC.
- Y. Sun, L. Yang, X. Lu and C. He, J. Mater. Chem. A, 2015, 3, 3699–3709 RSC.
- S. Luo, J. Cao and A. G. McDonald, ACS Sustainable Chem. Eng., 2016, 4, 3465–3476 CrossRef CAS.
- S. S. Panesar, S. Jacob, M. Misra and A. K. Mohanty, Ind. Crops Prod., 2013, 46, 191–196 CrossRef CAS.
- Y.-L. Chung, J. V. Olsson, R. J. Li, C. W. Frank, R. M. Waymouth, S. L. Billington and E. S. Sattely, ACS Sustainable Chem. Eng., 2013, 1, 1231–1238 CrossRef CAS.
- C. E. Astete, J. U. De Mel, S. Gupta, Y. Noh, M. Bleuel, G. J. Schneider and C. M. Sabliov, ACS Omega, 2020, 5, 9892–9902 CrossRef CAS PubMed.
- D. Kai, K. Zhang, S. S. Liow and X. J. Loh, ACS Appl. Bio Mater., 2019, 2, 127–134 CrossRef CAS.
- M. Abdollahi, R. Bairami Habashi and M. Mohsenpour, Ind. Crops Prod., 2019, 130, 547–557 CrossRef CAS.
- H. Chung and N. R. Washburn, Green Mater., 2013, 1, 137–160 CrossRef.
- Y. Li and S. Sarkanen, Macromolecules, 2002, 35, 9707–9715 CrossRef CAS.
- Y. Teramoto, S.-H. Lee and T. Endo, Polym. J., 2009, 41, 219–227 CrossRef CAS.
- M. S. Ganewatta, H. N. Lokupitiya and C. Tang, Polymers, 2019, 11, 1176 CrossRef PubMed.
- H. Y. Liu, F. Q. Chen, R. B. Guo, G. Zhang and J. Qu, J. Polym. Eng., 2015, 35, 829–837 CAS.
- H. Nitz, H. Semke and R. Mülhaupt, Macromol. Mater. Eng., 2001, 286, 737–743 CrossRef CAS.
- H. Nitz, H. Semke, R. Landers and R. Mülhaupt, J. Appl. Polym. Sci., 2001, 81, 1972–1984 CrossRef CAS.
- A. Kumar, V. R. Tumu, S. Ray Chowdhury and S. V. S. Ramana Reddy, Int. J. Biol. Macromol., 2019, 121, 588–600 CrossRef CAS PubMed.
- R. Muthuraj, M. Hajee, A. R. Horrocks and B. K. Kandola, Int. J. Biol. Macromol., 2019, 132, 439–450 CrossRef CAS PubMed.
- A. Pascu, M. C. Vasile, C. Agafitei, G. E. Gazacu and G. Stoleriu, in Monomers, Oligomers, Polymers, Composites and Nanocomposites Research: Synthesis, Properties and Applications, 2010, pp. 265–282 Search PubMed.
- A. Leszczyńska, J. Njuguna, K. Pielichowski and J. R. Banerjee, Thermochim. Acta, 2007, 453, 75–96 CrossRef.
- V. Romhányi, D. Kun and B. Pukánszky, ACS Sustainable Chem. Eng., 2018, 6, 14323–14331 CrossRef.
- A. M. Borrero-López, L. Wang, C. Valencia, J. M. Franco and O. J. Rojas, Compos. Sci. Technol., 2021, 203, 108602 CrossRef.
- N. Laibach, A. Hillebrand, R. M. Twyman, D. Prüfer and C. S. Gronover, Plant J., 2015, 82, 609–620 CrossRef CAS PubMed.
- C. L. Swanson, R. A. Buchanan and F. H. Otey, J. Appl. Polym. Sci., 1979, 23, 743–748 CrossRef CAS.
- A. Kato, Y. Ikeda and S. Kohjiya, Polym.-Plast. Technol. Eng., 2018, 57, 1418–1429 CrossRef CAS.
- T. Nishi, H. Nukaga, S. Fujinami and K. Nakajima, Chin. J. Polym. Sci., 2007, 25, 35–41 CrossRef CAS.
- J. J. Keilen and A. Pollak, Rubber Chem. Technol., 1947, 20, 1099–1108 CrossRef.
- D. Barana, S. D. Ali, A. Salanti, M. Orlandi, L. Castellani, T. Hanel and L. Zoia, ACS Sustainable Chem. Eng., 2016, 4, 5258–5267 CrossRef CAS.
- Y. Ikeda, T. Phakkeeree, P. Junkong, H. Yokohama, P. Phinyocheep, R. Kitano and A. Kato, RSC Adv., 2017, 7, 5222–5231 RSC.
- K. Roy, S. C. Debnath and P. Potiyaraj, J. Polym. Environ., 2020, 28, 367–387 CrossRef CAS.
- N. A. Mohamad Aini, N. Othman, M. H. Hussin, K. Sahakaro and N. Hayeemasae, Front. Mater., 2020, 6, 329 CrossRef.
- O. Gordobil, I. Egüés, R. Llano-Ponte and J. Labidi, Polym. Degrad. Stab., 2014, 108, 330–338 CrossRef CAS.
- M. A. Abdelwahab, S. Taylor, M. Misra and A. K. Mohanty, Macromol. Mater. Eng., 2015, 300, 299–311 CrossRef CAS.
- F. Bertini, M. Canetti, A. Cacciamani, G. Elegir, M. Orlandi and L. Zoia, Polym. Degrad. Stab., 2012, 97, 1979–1987 CrossRef CAS.
- S. Shankar and J.-W. Rhim, Food Hydrocolloids, 2017, 71, 76–84 CrossRef CAS.
- S. Shankar, J.-W. Rhim and K. Won, Int. J. Biol. Macromol., 2018, 107, 1724–1731 CrossRef CAS PubMed.
- A. Grząbka-Zasadzińska, Ł. Klapiszewski, K. Bula, T. Jesionowski and S. Borysiak, J. Therm. Anal. Calorim., 2016, 126, 263–275 CrossRef.
- A. Grząbka-Zasadzińska, Ł. Klapiszewski, S. Borysiak and T. Jesionowski, Materials, 2018, 11, 2257 CrossRef PubMed.
- R. Zhang, X. Xiao, Q. Tai, H. Huang, J. Yang and Y. Hu, High Perform. Polym., 2012, 24, 738–746 CrossRef.
- J. Fal, K. Bulanda, J. Traciak, J. Sobczak, R. Kuzioła, K. M. Grąz, G. Budzik, M. Oleksy and G. Żyła, Molecules, 2020, 25, 1354 CrossRef CAS PubMed.
- D. Yang, S. Wang, R. Zhong, W. Liu and X. Qiu, Front. Chem. Sci. Eng., 2019, 13, 59–69 CrossRef CAS.
- W. Wu, T. Liu, X. Deng, Q. Sun, X. Cao, Y. Feng, B. Wang, V. A. L. Roy and R. K. Y. Li, Int. J. Biol. Macromol., 2019, 126, 1030–1036 CrossRef CAS PubMed.
- Ł. Klapiszewski, A. Grząbka-Zasadzińska, S. Borysiak and T. Jesionowski, Polym. Test., 2019, 79, 106058 CrossRef.
- Ł. Klapiszewski, K. Bula, A. Dobrowolska, K. Czaczyk and T. Jesionowski, Polym. Test., 2019, 73, 51–59 CrossRef.
- Ł. Klapiszewski, M. Nowacka, G. Milczarek and T. Jesionowski, Carbohydr. Polym., 2013, 94, 345–355 CrossRef PubMed.
- A. Grząbka-Zasadzińska, Ł. Klapiszewski, T. Jesionowski and S. Borysiak, Molecules, 2020, 25, 864 CrossRef PubMed.
- E. Konował, A. Modrzejewska-Sikorska, M. Motylenko, Ł. Klapiszewski, M. Wysokowski, V. V. Bazhenov, D. Rafaja, H. Ehrlich, G. Milczarek and T. Jesionowski, Int. J. Biol. Macromol., 2016, 85, 74–81 CrossRef PubMed.
- M. Hu, Z. Chen, S. Luo, X. Yang, R. Ye, M. Zheng and P. Chen, Wood Sci. Technol., 2019, 53, 649–664 CrossRef CAS.
- F. Zhou, X. Feng, J. Yu and X. Jiang, Environ. Sci. Pollut. Res., 2018, 25, 15651–15661 CrossRef CAS PubMed.
- X. Li, Y. Wang, B. Wang, X. Feng, Z. Mao and X. Sui, Int. J. Biol. Macromol., 2020, 144, 624–631 CrossRef CAS PubMed.
- K. R. Aadil, S. I. Mussatto and H. Jha, Int. J. Biol. Macromol., 2018, 120, 763–767 CrossRef CAS PubMed.
- R. P. de Oliveira Santos, B. V. M. Rodrigues, D. M. dos Santos, S. P. Campana-Filho, A. C. Ruvolo-Filho and E. Frollini, Polym. Test., 2017, 60, 422–431 CrossRef.
- M. Ago, J. E. Jakes, L.-S. Johansson, S. Park and O. J. Rojas, ACS Appl. Mater. Interfaces, 2012, 4, 6849–6856 CrossRef CAS PubMed.
- R. P. de Oliveira Santos, L. A. Ramos and E. Frollini, Cellulose, 2019, 26, 617–630 CrossRef CAS.
- M. Ago, J. E. Jakes and O. J. Rojas, ACS Appl. Mater. Interfaces, 2013, 5, 11768–11776 CrossRef CAS PubMed.
- Y. Li, M. Wu, B. Wang, Y. Wu, M. Ma and X. Zhang, ACS Sustainable Chem. Eng., 2016, 4, 5523–5532 CrossRef CAS.
- J. Geng, F. Gu and J. Chang, J. Hazard. Mater., 2019, 375, 174–181 CrossRef CAS PubMed.
- M. Nasrollahzadeh, N. S. S. Bidgoli, Z. Issaabadi, Z. Ghavamifar, T. Baran and R. Luque, Int. J. Biol. Macromol., 2020, 148, 265–275 CrossRef CAS PubMed.
- S. Li, D. Ogunkoya, T. Fang, J. Willoughby and O. J. Rojas, J. Colloid Interface Sci., 2016, 482, 27–38 CrossRef CAS PubMed.
- L. Bai, L. G. Greca, W. Xiang, J. Lehtonen, S. Huan, R. W. N. Nugroho, B. L. Tardy and O. J. Rojas, Langmuir, 2019, 35, 571–588 CrossRef CAS PubMed.
- S. Li, J. A. Willoughby and O. J. Rojas, ChemSusChem, 2016, 9, 2460–2469 CrossRef CAS PubMed.
- M. J. Zhou, S. Z. Zhou, X. C. Pang, K. R. Li and X. G. Qiao, Colloids Surf., A, 2020, 585, 124040 CrossRef CAS.
- A. Jędrzak, T. Rębiś, M. Kuznowicz and T. Jesionowski, Int. J. Biol. Macromol., 2019, 127, 677–682 CrossRef PubMed.
- Q. Zhang, M. Li, C. Guo, Z. Jia, G. Wan, S. Wang and D. Min, Nanomaterials, 2019, 9, 210 CrossRef CAS PubMed.
- K. A. Mahmoud and M. Zourob, Analyst, 2013, 138, 2712–2719 RSC.
- Z. Zhou, X. Peng, L. Zhong, X. Li and R. Sun, Polymers, 2018, 10, 724 CrossRef PubMed.
- R. Dong, D. Zheng, D. Yang and X. Qiu, Bioresour. Technol., 2019, 294, 122133 CrossRef CAS PubMed.
- X. Li, Y. He, H. Sui and L. He, Nanomaterials, 2018, 8, 162 CrossRef PubMed.
- X. Liu, X. He, J. Zhang, J. Yang, X. Xiang, Z. Ma, L. Liu and E. Zong, RSC Adv., 2020, 10, 1249–1260 RSC.
- P. Li, W. Lv and S. Ai, J. Exp. Nanosci., 2016, 11, 18–27 CrossRef CAS.
- X. Chen, D.-H. Kuo, D. Lu, Y. Hou and Y.-R. Kuo, Microporous Mesoporous Mater., 2016, 223, 145–151 CrossRef CAS.
- M. Morsella, M. Giammatteo, L. Arrizza, L. Tonucci, M. Bressan and N. d'Alessandro, RSC Adv., 2015, 5, 57453–57461 RSC.
- X. Wang, Y. Zhang, C. Hao, F. Feng, H. Yin and N. Si, Ind. Eng. Chem. Res., 2014, 53, 6585–6592 CrossRef CAS.
- J. M. Gutiérrez-Hernández, A. Escalante, R. N. Murillo-Vázquez, E. Delgado, F. J. González and G. Toríz, J. Photochem. Photobiol., B, 2016, 163, 156–161 CrossRef PubMed.
- J. Liu and R. H. Hurt, Environ. Sci. Technol., 2010, 44, 2169–2175 CrossRef CAS PubMed.
- H. Liang, W. Wang, Y. Huang, S. Zhang, H. Wei and H. Xu, J. Phys. Chem. C, 2010, 114, 7427–7431 CrossRef CAS.
- S. Hu and Y.-L. Hsieh, Int. J. Biol. Macromol., 2016, 82, 856–862 CrossRef CAS PubMed.
- Z. Shen, Y. Luo, Q. Wang, X. Wang and R. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 16147–16155 CrossRef CAS PubMed.
- M. Ahamed, M. S. AlSalhi and M. K. J. Siddiqui, Clin. Chim. Acta, 2010, 411, 1841–1848 CrossRef CAS PubMed.
- A. P. Richter, J. S. Brown, B. Bharti, A. Wang, S. Gangwal, K. Houck, E. A. Cohen Hubal, V. N. Paunov, S. D. Stoyanov and O. D. Velev, Nat. Nanotechnol., 2015, 10, 817–823 CrossRef CAS PubMed.
- K. Lintinen, S. Luiro, P. Figueiredo, E. Sakarinen, Z. Mousavi, J. Seitsonen, G. N. S. Rivière, U. Mattinen, M. Niemelä, P. Tammela, M. Österberg, L.-S. Johansson, J. Bobacka, H. A. Santos and M. A. Kostiainen, ACS Sustainable Chem. Eng., 2019, 7, 15297–15303 CrossRef CAS.
- Y. Xue, X. Qiu, Z. Liu and Y. Li, ACS Sustainable Chem. Eng., 2018, 6, 7695–7703 CrossRef CAS.
- S. Chandna, N. S. Thakur, Y. N. Reddy, R. Kaur and J. Bhaumik, ACS Biomater. Sci. Eng., 2019, 5, 3212–3227 CrossRef CAS PubMed.
- S. Chen, G. Wang, W. Sui, A. M. Parvez, L. Dai and C. Si, Ind. Crops Prod., 2020, 145, 112164 CrossRef CAS.
- Ł. Klapiszewski, T. Rzemieniecki, M. Krawczyk, D. Malina, M. Norman, J. Zdarta, I. Majchrzak, A. Dobrowolska, K. Czaczyk and T. Jesionowski, Colloids Surf., B, 2015, 134, 220–228 CrossRef PubMed.
- J. B. Edel, A. A. Kornyshev, A. R. Kucernak and M. Urbakh, Chem. Soc. Rev., 2016, 45, 1581–1596 RSC.
- C. Gao, X. Wang, S. Zhai and Q. An, Int. J. Biol. Macromol., 2019, 134, 202–209 CrossRef CAS PubMed.
- D. Gan, W. Xing, L. Jiang, J. Fang, C. Zhao, F. Ren, L. Fang, K. Wang and X. Lu, Nat. Commun., 2019, 10, 1487 CrossRef PubMed.
- N. Elahi, M. Kamali and M. H. Baghersad, Talanta, 2018, 184, 537–556 CrossRef CAS PubMed.
- N. Bumbudsanpharoke and S. Ko, Int. J. Biol. Macromol., 2018, 107, 1782–1791 CrossRef CAS PubMed.
- D. M. Rocca, J. P. Vanegas, K. Fournier, M. C. Becerra, J. C. Scaiano and A. E. Lanterna, RSC Adv., 2018, 8, 40454–40463 RSC.
- G. Han, X. Li, J. Li, X. Wang, Y. S. Zhang and R. Sun, ACS Omega, 2017, 2, 4938–4945 CrossRef CAS PubMed.
- F. Coccia, L. Tonucci, D. Bosco, M. Bressan and N. d'Alessandro, Green Chem., 2012, 14, 1073–1078 RSC.
- F. Coccia, L. Tonucci, N. d'Alessandro, P. D'Ambrosio and M. Bressan, Inorg. Chim. Acta, 2013, 399, 12–18 CrossRef CAS.
- A. Aiello, J. R. Morey, K. J. T. Livi, H. C. DeLong, H. ElBidweihy, P. C. Trulove and D. P. Durkin, J. Magn. Magn. Mater., 2020, 497, 165964 CrossRef CAS.
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167–R181 CrossRef CAS.
- K. Lintinen, M. Latikka, M. H. Sipponen, R. H. A. Ras, M. Österberg and M. A. Kostiainen, RSC Adv., 2016, 6, 31790–31796 RSC.
- K. Akhtar, F. Ali, S. Sohni, T. Kamal, A. M. Asiri, E. M. Bakhsh and S. B. Khan, Environ. Sci. Pollut. Res., 2020, 27, 823–836 CrossRef CAS PubMed.
- M. J. Rak, T. Friščić and A. Moores, RSC Adv., 2016, 6, 58365–58370 RSC.
- M. J. Rak, T. Friščić and A. Moores, Faraday Discuss., 2014, 170, 155–167 RSC.
- W. Gindl-Altmutter, J. Köhnke, C. Unterweger, N. Gierlinger, J. Keckes, J. Zalesak and O. J. Rojas, Composites, Part A, 2019, 121, 175–179 CrossRef CAS.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- P.-C. Ma, N. A. Siddiqui, G. Marom and J.-K. Kim, Composites, Part A, 2010, 41, 1345–1367 CrossRef.
- P. Estellé, S. Halelfadl and T. Maré, Int. Commun. Heat Mass Transfer, 2014, 57, 8–12 CrossRef.
- G. Milczarek and M. Nowicki, Mater. Res. Bull., 2013, 48, 4032–4038 CrossRef CAS.
- E.-S. Lee, Y.-O. Kim, Y.-M. Ha, D. Lim, J. Y. Hwang, J. Kim, M. Park, J. W. Cho and Y. C. Jung, Eur. Polym. J., 2018, 105, 79–84 CrossRef CAS.
- H. Neelamegan, D.-K. Yang, G.-J. Lee, S. Anandan and J. J. Wu, Mater. Today Commun., 2020, 22, 100746 CrossRef CAS.
- X. Geng, Y. Zhang, L. Jiao, L. Yang, J. Hamel, N. Giummarella, G. Henriksson, L. Zhang and H. Zhu, ACS Sustainable Chem. Eng., 2017, 5, 3553–3561 CrossRef CAS.
- W. Xiaoying, Y. Weijie and S. Runcang, Method of preparing graphene with lignin solution, CN105236391A, 2015.
- S.-K. Kim, Y. K. Kim, H. Lee, S. B. Lee and H. S. Park, ChemSusChem, 2014, 7, 1094–1101 CrossRef CAS PubMed.
- K. Song, I. Ganguly, I. Eastin and A. B. Dichiara, Int. J. Mol. Sci., 2017, 18, 2368 CrossRef PubMed.
- M. Si, J. Zhang, Y. He, Z. Yang, X. Yan, M. Liu, S. Zhuo, S. Wang, X. Min, C. Gao, L. Chai and Y. Shi, Green Chem., 2018, 20, 3414–3419 RSC.
- E. Lizundia, T.-D. Nguyen, R. J. Winnick and M. J. MacLachlan, J. Mater. Chem. C, 2021, 9, 796–817 RSC.
- T.-D. Nguyen, J. Li, E. Lizundia, M. Niederberger, W. Y. Hamad and M. J. MacLachlan, Adv. Funct. Mater., 2019, 29, 1904639 CrossRef.
- H. Li, Y. Zhao, S. Liu, P. Li, D. Yuan and C. He, Microporous Mesoporous Mater., 2020, 297, 109960 CrossRef CAS.
- H. Qin, S. Kang, Y. Wang, H. Liu, Z. Ni, Y. Huang, Y. Li and X. Li, ACS Sustainable Chem. Eng., 2016, 4, 1240–1247 CrossRef CAS.
- H. Qin, B. Wang, C. Zhang, B. Zhu, Y. Zhou and Q. Zhou, Catal. Commun., 2017, 96, 28–31 CrossRef CAS.
- Y. Ma, D. Zheng, Z. Mo, R. Dong and X. Qiu, Colloids Surf., A, 2018, 559, 226–234 CrossRef CAS.
- Z. Dai, P. Ren, Q. Cao, X. Gao, W. He, Y. Xiao, Y. Jin and F. Ren, J. Polym. Res., 2020, 27, 108 CrossRef CAS.
- B. Zhang, D. Yang, X. Qiu, Y. Qian, H. Wang, C. Yi and D. Zhang, Carbon, 2020, 162, 256–266 CrossRef CAS.
- H. Wang, X. Qiu, R. Zhong, F. Fu, Y. Qian and D. Yang, Mater. Chem. Phys., 2017, 199, 193–202 CrossRef CAS.
- C. D. Tran, H. C. Ho, J. K. Keum, J. Chen, N. C. Gallego and A. K. Naskar, Energy Technol., 2017, 5, 1927–1935 CrossRef CAS.
- Z. Zhou, F. Chen, T. Kuang, L. Chang, J. Yang, P. Fan, Z. Zhao and M. Zhong, Electrochim. Acta, 2018, 274, 288–297 CrossRef CAS.
- R. Wang, G. Xia, W. Zhong, L. Chen, L. Chen, Y. Wang, Y. Min and K. Li, Green Chem., 2019, 21, 3343–3352 RSC.
- G. Han, Q. Jiang, W. Ye, C. Liu and X. Wang, Sci. Rep., 2019, 9, 7300 CrossRef PubMed.
- B. Luo, M. Chi, Q. Zhang, M. Li, C. Chen, X. Wang, S. Wang and D. Min, Nanomaterials, 2019, 9, 1681 CrossRef CAS PubMed.
- M. Ago, M. Borghei, J. S. Haataja and O. J. Rojas, RSC Adv., 2016, 6, 85802–85810 RSC.
- M. H. Sipponen, M. Smyth, T. Leskinen, L.-S. Johansson and M. Österberg, Green Chem., 2017, 19, 5831–5840 RSC.
- E. Capecchi, D. Piccinino, B. M. Bizzarri, L. Botta and M. Crucianelli, Catalysts, 2020, 10, 313 CrossRef CAS.
- T. Leskinen, J. Witos, J. J. Valle-Delgado, K. S. Lintinen, M. A. Kostiainen, S. K. Wiedmer, M. Österberg and M.-L. Mattinen, Biomacromolecules, 2017, 18, 2767–2776 CrossRef CAS PubMed.
- E. Capecchi, D. Piccinino, I. Delfino, P. Bollella, R. Antiochia and R. Saladino, Nanomaterials, 2018, 8, 438 CrossRef PubMed.
- G. N. Rivière, A. Korpi, M. H. Sipponen, T. Zou, M. A. Kostiainen and M. Österberg, ACS Sustainable Chem. Eng., 2020, 8, 4167–4177 CrossRef PubMed.
- T. Zou, M. H. Sipponen, A. Henn and M. Österberg, ACS Nano, 2021, 15, 4811–4823 CrossRef CAS PubMed.
- A. Moreno, J. Liu, R. Gueret, S. E. Hadi, L. Bergström, A. Slabon and M. H. Sipponen, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202106743.
- A. M. Guerra and M. Sipponen, Nat. Commun., 2020, 11, 5599 CrossRef PubMed.
- A. Moreno, M. Morsali, J. Liu and M. H. Sipponen, Green Chem., 2021, 23, 3001–3014 RSC.
- A. Pereira, I. C. Hoeger, A. Ferrer, J. Rencoret, J. C. del Rio, K. Kruus, J. Rahikainen, M. Kellock, A. Gutiérrez and O. J. Rojas, Biomacromolecules, 2017, 18, 1322–1332 CrossRef CAS PubMed.
- P. Figueiredo, M. H. Sipponen, K. Lintinen, A. Correia, A. Kiriazis, J. Yli-Kauhaluoma, M. Österberg, A. George, J. Hirvonen, M. A. Kostiainen and H. A. Santos, Small, 2019, 15, 1901427 CrossRef PubMed.
- M. Delgado-Aguilar, I. González, Q. Tarrés, M. À. Pèlach, M. Alcalà and P. Mutjé, Ind. Crops Prod., 2016, 86, 295–300 CrossRef CAS.
- G. Albornoz-Palma, D. Ching, O. Valerio, R. T. Mendonça and M. Pereira, Cellulose, 2020, 27, 10631–10647 CrossRef CAS.
- M. J. Selig, S. Viamajala, S. R. Decker, M. P. Tucker, M. E. Himmel and T. B. Vinzant, Biotechnol. Prog., 2007, 23, 1333–1339 CrossRef CAS PubMed.
- B. S. Donohoe, S. R. Decker, M. P. Tucker, M. E. Himmel and T. B. Vinzant, Biotechnol. Bioeng., 2008, 101, 913–925 CrossRef CAS PubMed.
- S. V. Pingali, V. S. Urban, W. T. Heller, J. McGaughey, H. O'Neill, M. Foston, D. A. Myles, A. Ragauskas and B. R. Evans, Biomacromolecules, 2010, 11, 2329–2335 CrossRef CAS PubMed.
- Y. Xu, K. Li and M. Zhang, Colloids Surf., A, 2007, 301, 255–263 CrossRef CAS.
- A. Bjorkman, Ind. Eng. Chem., 1957, 49, 1395–1398 CrossRef CAS.
- A. Barakat, C. Gaillard, D. Lairez, L. Saulnier, B. Chabbert and B. Cathala, Biomacromolecules, 2008, 9, 487–493 CrossRef CAS PubMed.
- P. Sannigrahi, D. H. Kim, S. Jung and A. Ragauskas, Energy Environ. Sci., 2011, 4, 1306–1310 RSC.
- R. Kumar, F. Hu, P. Sannigrahi, S. Jung, A. J. Ragauskas and C. E. Wyman, Biotechnol. Bioeng., 2013, 110, 737–753 CrossRef CAS PubMed.
- J. L. Rahikainen, R. Martin-Sampedro, H. Heikkinen, S. Rovio, K. Marjamaa, T. Tamminen, O. J. Rojas and K. Kruus, Bioresour. Technol., 2013, 133, 270–278 CrossRef CAS PubMed.
- C. Fritz, A. Ferrer, C. Salas, H. Jameel and O. J. Rojas, Biomacromolecules, 2015, 16, 3878–3888 CrossRef CAS PubMed.
- R. Martín-Sampedro, J. L. Rahikainen, L.-S. Johansson, K. Marjamaa, J. Laine, K. Kruus and O. J. Rojas, Biomacromolecules, 2013, 14, 1231–1239 CrossRef PubMed.
- H. Bian, L. Chen, H. Dai and J. Y. Zhu, Carbohydr. Polym., 2017, 167, 167–176 CrossRef CAS PubMed.
- X. Liu, Y. Li, C. M. Ewulonu, J. Ralph, F. Xu, Q. Zhang, M. Wu and Y. Huang, ACS Sustainable Chem. Eng., 2019, 7, 14135–14142 CrossRef CAS.
- J. Jiang, N. C. Carrillo-Enríquez, H. Oguzlu, X. Han, R. Bi, M. Song, J. N. Saddler, R.-C. Sun and F. Jiang, ACS Sustainable Chem. Eng., 2020, 8, 7182–7191 CrossRef CAS.
- A. Tripathi, A. Ferrer, S. A. Khan and O. J. Rojas, ACS Sustainable Chem. Eng., 2017, 5, 2483–2492 CrossRef CAS.
- A. Kumagai, S.-H. Lee and T. Endo, Biotechnol. Bioeng., 2016, 113, 1441–1447 CrossRef CAS PubMed.
- C. I. K. Diop, M. Tajvidi, M. A. Bilodeau, D. W. Bousfield and J. F. Hunt, Cellulose, 2017, 24, 3037–3050 CrossRef CAS.
- F. Araya, E. Troncoso, R. T. Mendonça and J. Freer, Biotechnol. Bioeng., 2015, 112, 1783–1791 CrossRef CAS PubMed.
- D. Tian, J. Hu, J. Bao, R. P. Chandra, J. N. Saddler and C. Lu, Biotechnol. Biofuels, 2017, 10, 192 CrossRef PubMed.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941 RSC.
- D. Maldas and B. V. Kokta, Compos. Interfaces, 1993, 1, 87–108 CrossRef CAS.
- Y. Peng, S. S. Nair, H. Chen, N. Yan and J. Cao, ACS Sustainable Chem. Eng., 2018, 6, 11078–11086 CrossRef CAS.
- D. Ballner, S. Herzele, J. Keckes, M. Edler, T. Griesser, B. Saake, F. Liebner, A. Potthast, C. Paulik and W. Gindl-Altmutter, ACS Appl. Mater. Interfaces, 2016, 8, 13520–13525 CrossRef CAS PubMed.
- Z. Wang, P. Gnanasekar, S. Sudhakaran Nair, R. Farnood, S. Yi and N. Yan, ACS Sustainable Chem. Eng., 2020, 8, 11215–11223 CrossRef CAS.
- A. Gupta, W. Simmons, G. T. Schueneman, D. Hylton and E. A. Mintz, ACS Sustainable Chem. Eng., 2017, 5, 1711–1720 CrossRef CAS.
- A. Šutka, M. Antsov, M. Järvekülg, M. Visnapuu, I. Heinmaa, U. Mäeorg, S. Vlassov and A. Šutka, J. Appl. Polym. Sci., 2016, 134, 44361 Search PubMed.
- S. Herzele, S. Veigel, F. Liebner, T. Zimmermann and W. Gindl-Altmutter, Ind. Crops Prod., 2016, 93, 302–308 CrossRef CAS.
- C. Zhang, S. S. Nair, H. Chen, N. Yan, R. Farnood and F. Li, Carbohydr. Polym., 2020, 230, 115626 CrossRef CAS PubMed.
- S. Iwamoto, S. Yamamoto, S.-H. Lee and T. Endo, Cellulose, 2014, 21, 1573–1580 CrossRef CAS.
- Y. Kojima, A. Kawabata, H. Kobori, S. Suzuki, H. Ito, R. Makise and M. Okamoto, J. Wood Sci., 2016, 62, 518–525 CrossRef CAS.
- Y. Zhong, L. Gu, S. Wang, Y. Jin and H. Xiao, Ind. Eng. Chem. Res., 2019, 58, 20323–20330 CrossRef CAS.
- A. H. Tayeb, M. Tajvidi and D. Bousfield, Molecules, 2020, 25, 1344 CrossRef CAS PubMed.
- M. Visanko, J. A. Sirviö, P. Piltonen, R. Sliz, H. Liimatainen and M. Illikainen, Cellulose, 2017, 24, 4173–4187 CrossRef CAS.
- A. Winter, L. Andorfer, S. Herzele, T. Zimmermann, B. Saake, M. Edler, T. Griesser, J. Konnerth and W. Gindl-Altmutter, J. Mater. Sci., 2017, 52, 60–72 CrossRef CAS.
- H. Yang, B. Yu, X. Xu, S. Bourbigot, H. Wang and P. Song, Green Chem., 2020, 22, 2129–2161 RSC.
- V. Carretier, J. Delcroix, M. F. Pucci, P. Rublon and J.-M. Lopez-Cuesta, Materials, 2020, 13, 2450 CrossRef CAS PubMed.
- W. Hu, Y. Zhang, Y. Qi, H. Wang, B. Liu, Q. Zhao, J. Zhang, J. Duan, L. Zhang, Z. Sun and B. Liu, Macromol. Mater. Eng., 2020, 305, 1900840 CrossRef CAS.
- N. Mandlekar, A. Cayla, F. Rault, S. Giraud, F. Salaün and J. Guan, Polymers, 2019, 11, 180 CrossRef PubMed.
- A. Shukla, V. Sharma, S. Basak and S. W. Ali, Cellulose, 2019, 26, 8191–8208 CrossRef CAS.
- R. K. Sharma, J. B. Wooten, V. L. Baliga, X. Lin, W. G. Chan and M. R. Hajaligol, Fuel, 2004, 83, 1469–1482 CrossRef CAS.
- N. Mandlekar, A. Cayla, F. Rault, S. Giraud, F. Salaün, G. Malucelli and J. Guan, Ind. Eng. Chem. Res., 2017, 56, 13704–13714 CrossRef CAS.
- A. Cayla, F. Rault, S. Giraud, F. Salaün, V. Fierro and A. Celzard, Polymers, 2016, 8, 331 CrossRef PubMed.
- L. Costes, F. Laoutid, S. Brohez, C. Delvosalle and P. Dubois, Eur. Polym. J., 2017, 94, 270–285 CrossRef CAS.
- A. F. Baldissera, M. R. Silveira, A. C. Dornelles and C. A. Ferreira, J. Coat. Technol. Res., 2020, 17, 1297–1307 CrossRef CAS.
- Y. Wang, Y. Zhang, B. Liu, Q. Zhao, Y. Qi, Y. Wang, Z. Sun, B. Liu, N. Zhang, W. Hu and H. Xie, Compos. Commun., 2020, 21, 100382 CrossRef.
- J. Zhang, E. Fleury, Y. Chen and M. A. Brook, RSC Adv., 2015, 5, 103907–103914 RSC.
- X. Wang, S. L. Ji, X. Q. Wang, H. Y. Bian, L. R. Lin, H. Q. Dai and H. Xiao, J. Mater. Chem. C, 2019, 7, 14159–14169 RSC.
- L. Costes, F. Laoutid, M. Aguedo, A. Richel, S. Brohez, C. Delvosalle and P. Dubois, Eur. Polym. J., 2016, 84, 652–667 CrossRef CAS.
- R. Zhang, X. Xiao, Q. Tai, H. Huang and Y. Hu, Polym. Eng. Sci., 2012, 2620–2626 CrossRef CAS.
- A. M. Youssef and S. M. El-Sayed, Carbohydr. Polym., 2018, 193, 19–27 CrossRef CAS PubMed.
- W. Yang, Y. Weng, D. Puglia, G. Qi, W. Dong, J. M. Kenny and P. Ma, Int. J. Biol. Macromol., 2020, 144, 102–110 CrossRef CAS PubMed.
- W. Yang, E. Fortunati, F. Dominici, G. Giovanale, A. Mazzaglia, G. M. Balestra, J. M. Kenny and D. Puglia, Int. J. Biol. Macromol., 2016, 89, 360–368 CrossRef CAS PubMed.
- W. Yang, J. S. Owczarek, E. Fortunati, M. Kozanecki, A. Mazzaglia, G. M. Balestra, J. M. Kenny, L. Torre and D. Puglia, Ind. Crops Prod., 2016, 94, 800–811 CrossRef CAS.
- Q. Xing, P. Buono, D. Ruch, P. Dubois, L. Wu and W.-J. Wang, ACS Sustainable Chem. Eng., 2019, 7, 4147–4157 CrossRef CAS.
- H. Wang, W. Lin, X. Qiu, F. Fu, R. Zhong, W. Liu and D. Yang, ACS Sustainable Chem. Eng., 2018, 6, 3696–3705 CrossRef CAS.
- X. Wang, S.-L. Ji, X.-Q. Wang, H.-Y. Bian, L.-R. Lin, H.-Q. Dai and H. Xiao, J. Mater. Chem. C, 2019, 7, 14159–14169 RSC.
- V. Sinisi, P. Pelagatti, M. Carcelli, A. Migliori, L. Mantovani, L. Righi, G. Leonardi, S. Pietarinen, C. Hubsch and D. Rogolino, ACS Sustainable Chem. Eng., 2019, 7, 3213–3221 CrossRef CAS.
- A. García, G. Spigno and J. Labidi, Ind. Crops Prod., 2017, 104, 242–252 CrossRef.
- S. Falsini, I. Clemente, A. Papini, C. Tani, S. Schiff, M. C. Salvatici, R. Petruccelli, C. Benelli, C. Giordano, C. Gonnelli and S. Ristori, ACS Sustainable Chem. Eng., 2019, 7, 19935–19942 CrossRef CAS.
- J. Fischer, S. J. Beckers, D. Yiamsawas, E. Thines, K. Landfester and F. R. Wurm, Adv. Sci., 2019, 6, 1802315 CrossRef PubMed.
- T. O. Machado, S. J. Beckers, J. Fischer, B. Müller, C. Sayer, P. H. H. de Araújo, K. Landfester and F. R. Wurm, Biomacromolecules, 2020, 21, 2755–2763 CrossRef CAS PubMed.
- S. Peil, S. J. Beckers, J. Fischer and F. R. Wurm, Mater. Today Bio, 2020, 7, 100061 CrossRef CAS PubMed.
- M. H. Sipponen, H. Lange, C. Crestini, A. Henn and M. Österberg, ChemSusChem, 2019, 12, 2039–2054 CrossRef CAS PubMed.
- R. Weiss, E. Ghitti, M. Sumetzberger-Hasinger, G. M. Guebitz and G. S. Nyanhongo, ACS Omega, 2020, 5, 4322–4329 CrossRef CAS PubMed.
- D. Legras-Lecarpentier, K. Stadler, R. Weiss, G. M. Guebitz and G. S. Nyanhongo, ACS Sustainable Chem. Eng., 2019, 7, 12621–12628 CAS.
- J. Luo, D. Zhang, T. Jing, G. Liu, H. Cao, B. Li, Y. Hou and F. Liu, Chem. Eng. J., 2020, 394, 124854 CrossRef CAS.
- Y. Sun, Y. Ma, G. Fang, S Ren and Y. Fu, BioResources, 2016, 11, 2361–2371 CAS.
- F. Flores-Céspedes, C. I. Figueredo-Flores, I. Daza-Fernández, F. Vidal-Peña, M. Villafranca-Sánchez and M. Fernández-Pérez, J. Agric. Food Chem., 2012, 60, 1042–1051 CrossRef PubMed.
- J. Chen, X. Fan, L. Zhang, X. Chen, S. Sun and R.-C. Sun, ChemSusChem, 2020, 13, 4356–4366 CrossRef CAS PubMed.
- Y. Fu, M. Qin, Z. Wang and Z. Li, Pap. Biomater., 2018, 42–53 CAS.
- W. T. Jiang, L. Su, M. Yu, L. Fang, L. Liu, L. J. Xu and G. Z. Fang, Chem. Ind. For. Prod., 2015, 35, 27–32 CAS.
- W. T. Jiang, M. Yu, L. Fang, L. Liu, L. Xu, L. Su and G. Z. Fang, J. Beijing For. Univ., 2014, 36, 165–170 Search PubMed.
- Y. Ma, R. Wang, G. Z. Fang and D. Li, Trans. Chin. Soc. Agric. Eng., 2012, 28, 208–214 CAS.
- E. Lizundia and D. Kundu, Adv. Funct. Mater., 2020, 31, 2005646 CrossRef.
- M. Zhao, J. Wang, C. Chong, X. Yu, L. Wang and Z. Shi, RSC Adv., 2015, 5, 101115–101120 RSC.
- M.-J. Uddin, P. K. Alaboina, L. Zhang and S.-J. Cho, Mater. Sci. Eng., B, 2017, 223, 84–90 CrossRef CAS.
- D. Yuan, W. Manalastas Jr., L. Zhang, J. J. Chan, S. Meng, Y. Chen and M. Srinivasan, ChemSusChem, 2019, 12, 4889–4900 CrossRef CAS PubMed.
- Z. Zhang, S. Yi, Y. Wei, H. Bian, R. Wang and Y. Min, Polymers, 2019, 11, 1946 CrossRef CAS PubMed.
- S. Wang, L. Zhang, A. Wang, X. Liu, J. Chen, Z. Wang, Q. Zeng, H. Zhou, X. Jiang and L. Zhang, ACS Sustainable Chem. Eng., 2018, 6, 14460–14469 CrossRef CAS.
- K. Yu, T. Liu, Q. Zheng, X. Wang, W. Liu, J. Liang and C. Liang, J. Porous Mater., 2020, 27, 875–882 CrossRef CAS.
- N. Casado, M. Hilder, C. Pozo-Gonzalo, M. Forsyth and D. Mecerreyes, ChemSusChem, 2017, 10, 1783–1791 CrossRef CAS PubMed.
- Y. Zhang, Y. Zhu, J. Zhang, S. Sun, C. Wang, M. Chen and J. Zeng, Energy Technol., 2020, 8, 1900694 CrossRef CAS.
- G. Milczarek and O. Inganäs, Science, 2012, 335, 1468–1471 CrossRef CAS PubMed.
- F. N. Ajjan, N. Casado, T. Rębiś, A. Elfwing, N. Solin, D. Mecerreyes and O. Inganäs, J. Mater. Chem. A, 2016, 4, 1838–1847 RSC.
- S. Jha, S. Mehta, Y. Chen, P. Renner, S. S. Sankar, D. Parkinson, S. Kundu and H. Liang, J. Mater. Chem. C, 2020, 8, 3418–3430 RSC.
- L. Wang, M. Ago, M. Borghei, A. Ishaq, A. C. Papageorgiou, M. Lundahl and O. J. Rojas, ACS Sustainable Chem. Eng., 2019, 7, 6013–6022 CrossRef CAS PubMed.
- K. Zhang, M. Liu, T. Zhang, X. Min, Z. Wang, L. Chai and Y. Shi, J. Mater. Chem. A, 2019, 7, 26838–26848 RSC.
- F. Fu, D. Yang, H. Wang, Y. Qian, F. Yuan, J. Zhong and X. Qiu, ACS Sustainable Chem. Eng., 2019, 7, 16419–16427 CrossRef CAS.
- L. Cui, C. Cheng, F. Peng, Y. Yang, Y. Li, M. Jia and X. Jin, New J. Chem., 2019, 43, 14084–14092 RSC.
- A. L. Cottrill, A. T. Liu, Y. Kunai, V. B. Koman, A. Kaplan, S. G. Mahajan, P. Liu, A. R. Toland and M. S. Strano, Nat. Commun., 2018, 9, 664 CrossRef PubMed.
- M. H. Sipponen, A. Henn, P. Penttilä and M. Österberg, Chem. Eng. J., 2020, 393, 124711 CrossRef CAS.
- J. Gong, K. Sumathy, Q. Qiao and Z. Zhou, Renewable Sustainable Energy Rev., 2017, 68, 234–246 CrossRef CAS.
- M. Aoyagi and M. Funaoka, J. Photochem. Photobiol., A, 2006, 181, 114–119 CrossRef CAS.
- Y. Zhao, Y. Liu, C. Tong, J. Ru, B. Geng, Z. Ma, H. Liu and L. Wang, J. Mater. Sci., 2018, 53, 7637–7647 CrossRef CAS.
- Y. Wu, J. Wang, X. Qiu, R. Yang, H. Lou, X. Bao and Y. Li, ACS Appl. Mater. Interfaces, 2016, 8, 12377–12383 CrossRef CAS PubMed.
- G. Jaganathan, K. Manivannan, S. Lakshmanan and M. A. Sithique, Sustainable Chem. Pharm., 2018, 10, 27–32 CrossRef.
- T. Abudula, L. Gzara, G. Simonetti, A. Alshahrie, N. Salah, P. Morganti, A. Chianese, A. Fallahi, A. Tamayol, S. A. Bencherif and A. Memic, Materials, 2018, 11, 451 CrossRef PubMed.
- J. Domínguez-Robles, E. Larrañeta, M. L. Fong, N. K. Martin, N. J. Irwin, P. Mutjé, Q. Tarrés and M. Delgado-Aguilar, Int. J. Biol. Macromol., 2020, 145, 92–99 CrossRef PubMed.
- D. Kai, W. Ren, L. Tian, P. L. Chee, Y. Liu, S. Ramakrishna and X. J. Loh, ACS Sustainable Chem. Eng., 2016, 4, 5268–5276 CrossRef CAS.
- H. K. Zhao, X. Y. Wei, Y. M. Xie and Q. H. Feng, Int. J. Biol. Macromol., 2019, 137, 1161–1168 CrossRef CAS PubMed.
- I. Spiridon and C. E. Tanase, Int. J. Biol. Macromol., 2018, 114, 855–863 CrossRef CAS PubMed.
- X. Zhang, M. Morits, C. Jonkergouw, A. Ora, J. J. Valle-delgado, M. Farooq, R. Ajdary, S. Huan, M. Linder, O. Rojas, M. H. Sipponen and O. Monika, Biomacromolecules, 2020, 21, 1875–1885 CrossRef CAS PubMed.
- D. Kai, K. Zhang, L. Jiang, H. Z. Wong, Z. Li, Z. Zhang and X. J. Loh, ACS Sustainable Chem. Eng., 2017, 5, 6016–6025 CrossRef CAS.
- D. Kai, H. M. Chong, L. P. Chow, L. Jiang, Q. Lin, K. Zhang, H. Zhang, Z. Zhang and X. J. Loh, Compos. Sci. Technol., 2018, 158, 26–33 CrossRef CAS.
- J. Pérez, J. Muñoz-Dorado, T. de la Rubia and J. Martínez, Int. Microbiol., 2002, 5, 53–63 CrossRef PubMed.
- M. Thevenot, M.-F. Dignac and C. Rumpel, Soil Biol. Biochem., 2010, 42, 1200–1211 CrossRef CAS.
- Y. Cao, S. S. Chen, S. Zhang, Y. S. Ok, B. M. Matsagar, K. C.-W. Wu and D. C. W. Tsang, Bioresour. Technol., 2019, 291, 121878 CrossRef CAS PubMed.
- C. Abbati De Assis, L. G. Greca, M. Ago, M. Y. Balakshin, H. Jameel, R. Gonzalez and O. J. Rojas, ACS Sustainable Chem. Eng., 2018, 6, 11853–11868 CrossRef CAS PubMed.
- S. Yang, T. Q. Yuan, Q. Shi and R. C. Sun, in Green Chemistry and Chemical Engineering. Encyclopedia of Sustainability Science and Technology Series, ed. B. Han and T. Wu, Springer, 2019, 405–426 Search PubMed.
- I. Spiridon, K. Leluk, A. M. Resmerita and R. N. Darie, Composites, Part B, 2015, 69, 342–349 CrossRef CAS.
- R. Zhang, X. Xiao, Q. Tai, H. Huang, J. Yang and Y. Hu, J. Appl. Polym. Sci., 2013, 127, 4967–4973 CrossRef CAS.
- E. Bernier, C. Lavigne and P. Y. Robidoux, Int. J. Life Cycle Assess., 2013, 18, 520–528 CrossRef CAS.
- J. B. Guinée, R. Heijungs, G. Huppes, A. Zamagni, P. Masoni, R. Buonamici, T. Ekvall and T. Rydberg, Environ. Sci. Technol., 2011, 45, 90–96 CrossRef PubMed.
- M. Iturrondobeitia, O. Akizu-Gardoki, R. Minguez and E. Lizundia, ACS Sustainable Chem. Eng., 2021, 9, 7139–7153 CrossRef CAS.
- F. Hermansson, M. Janssen and M. Svanström, Int. J. Life Cycle Assess., 2020, 25, 1620–1632 CrossRef.
- C. Culbertson, T. Treasure, R. Venditti, H. Jameel and R. Gonzalez, Nord. Pulp Pap. Res. J., 2016, 31, 30–40 CrossRef CAS.
- P. Yadav, D. Athanassiadis, I. Antonopoulou, U. Rova, P. Christakopoulos, M. Tysklind and L. Matsakas, J. Cleaner Prod., 2021, 279, 123515 CrossRef CAS.
- A. Alsabri and S. G. Al-Ghamdi, Energy Rep., 2020, 6, 364–370 CrossRef.
- K. W. Meereboer, M. Misra and A. K. Mohanty, Green Chem., 2020, 22, 5519–5558 RSC.
- J. Turk, P. Oven, I. Poljanšek, A. Lešek, F. Knez and K. Malovrh Rebec, J. Cleaner Prod., 2020, 247, 119107 CrossRef CAS.
- C. W. Johnson, D. Salvachúa, N. A. Rorrer, B. A. Black, D. R. Vardon, P. C. St. John, N. S. Cleveland, G. Dominick, J. R. Elmore, N. Grundl, P. Khanna, C. R. Martinez, W. E. Michener, D. J. Peterson, K. J. Ramirez, P. Singh, T. A. VanderWall, A. N. Wilson, X. Yi, M. J. Biddy, Y. J. Bomble, A. M. Guss and G. T. Beckham, Joule, 2019, 3, 1523–1537 CrossRef CAS.
- J. Miller, Lignin 2021: A Pivotal Year, 2021 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |