
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective lignin fractionation using CO2-expanded 2-methyltetrahydrofuran (2-MTHF)†
Dennis
Weidener
ab,
Holger
Klose
 ab,
William
Graf von Westarp
bc,
Andreas
Jupke
cb,
Walter
Leitner
de,
Pablo
Domínguez de María
*f and
Philipp M.
Grande
*ab
ab,
William
Graf von Westarp
bc,
Andreas
Jupke
cb,
Walter
Leitner
de,
Pablo
Domínguez de María
*f and
Philipp M.
Grande
*ab
aInstitute of Bio- and Geosciences, Plant Sciences (IBG-2), Forschungszentrum Jülich, 52425 Jülich, Germany. E-mail: p.grande@fz-juelich.de
bBioeconomy Science Center (BioSC), c/o Forschungszentrum Jülich, 52425 Jülich, Germany
cFluid Process Engineering (AVT.FVT), RWTH Aachen University, Forckenbeckstraße 51, 52074 Aachen, Germany
dInstitute of Technical and Macromolecular Chemistry (ITMC), RWTH Aachen University, Worringer Weg 1, 52074 Aachen, Germany
eMax-Planck-Institute for Chemical Energy Conversion, Stiftstraße 34-36, 45470 Mülheim an der Ruhr, Germany
fSustainable Momentum, SL Av. Ansite 3, 4-6, 35011 Las Palmas de Gran Canaria, Canary Islands, Spain. E-mail: dominguez@sustainable-momentum.net
First published on 9th August 2021
Abstract
Upon CO2 pressurization, 2-methyltetrahydrofuran (2-MTHF) expands. The expanded phase exerts different solvation properties, triggering fractional lignin precipitation. Depending on the actual CO2 pressure, fractions of lignin with a gradient of different sizes and chemical properties are selectively precipitated. A range of raw materials and applications may be thus reached.
Introduction
Lignin can play a central role in future lignocellulosic biorefineries, as it accounts for an important proportion of the biomass (20–40%), and holds promising uses ranging from innovative biomaterials to the production of aromatic chemicals.1 Hence, an adequate valorization of lignin appears mandatory to cope with the tight economic numbers of biorefineries, and the establishment of value-added lignin-based solutions is thus of utmost importance. Particularly, the generation of purified, narrowly-distributed, and well-defined lignin fractions results in an attractive path for research, as it potentially broadens the market options and economic margins of biorefining processes. To that end, the establishment of selective, environmentally-friendly, straightforward and cost-efficient methods for lignin recovery and fractionation are obviously of interest.1Lignin is typically obtained through so-called pretreatment processes, in which lignocellulose is fractionated in its three main components, hemicellulose, cellulose and lignin. Different pretreatment methods have been successfully reported.2 Among them, acid-catalyzed processes trigger the (more or less selective) depolymerization of the polysaccharide fraction(s), disentangling the lignocellulose and rendering different raw materials. In this area, we developed the OrganoCat pretreatment,3 which aims at using biogenic resources at mild processing conditions, to secure that degradation of raw materials is significantly reduced. The OrganoCat pretreatment uses an organic carboxylic acid as catalyst (namely oxalic acid or 2,5-furandicarboxylic acid, FDCA), and forms a biphasic system with water and 2-methyltetrahydrofuran (2-MTHF), a solvent that can be derived from biogenic resources.4 In the OrganoCat process conditions (e.g. 140–160 °C, 1–3 h), the acid selectively depolymerizes the hemicelluloses to render xylose and other sugars, while cellulose and lignin are obtained as macromolecules. Cellulose remains suspended in the aqueous phase, and lignin is in situ extracted to the 2-MTHF phase. Subsequently, lignin must be separated from the 2-MTHF-rich phase for further valorization. Apart from distillation, several alternatives have been recently proposed by some of us, such as using liquid–liquid extraction with concentrated NaOH aqueous solutions,5 or adding antisolvents like n-pentane or n-hexane to trigger the precipitation of lignin from 2-MTHF.6 Other fractional precipitation methods for lignins include solvent/anti-solvent mixtures (e.g. acetone/hexane, ethanol/water), sequential acid precipitation, ultrafiltration or the addition of sulfate and surfactants.7
This work explores the use of the gas-expanded liquid (GXL) concept for the lignin precipitation from the 2-MTHF phase. GXLs are formed when a compressible gas (e.g. CO2) is pressurized on solvents that can solubilize it to a given extent.8 Most notably, the chemo-physical properties (e.g. solvation properties) change in the resulting gas-expanded solvent phase,9 which can be reversed upon degassing. As the solvation properties of the GXL changes, the precipitation of the dissolved lignin (as macromolecule) can be triggered, and importantly, depending on the actual CO2 pressure, different fractions (with narrowed molecular size distributions, chemical bond proportions, etc.) could be selectively obtained. Despite its potential, only few references have assessed the use of GXL for lignin precipitation, e.g. using mixtures of acetic acid/water and CO2,10 or using methanol, for the extraction of vanillin and other value-added chemicals.11
In general, using gas-expanded (biorefinery-made) biogenic solvents might become an excellent alternative to create novel bioeconomy value chains. It enables using solvents generated within a biorefinery to valorize complex and diverse natural polymeric fractions (e.g. separations, narrowing polymer distributions, elimination of non-desired fractions, etc.).8 Particularly, if successful, the use of a CO2 expanded 2-MTHF phase would provide several advantages, such as obtaining different lignin fractions, while establishing a straightforward solvent reuse, as 2-MTHF can be recovered by depressurization. Despite this potential, CO2 expanded phases with 2-MTHF have only been reported for the highly selective catalytic decarbonylation of furans,12 as well as in biocatalysis, showing that the expanded phase with 2-MTHF can improve the enantioselectivity of several enzymes.13 Herein, we report on the potential of CO2-expanded 2-MTHF for biorefineries and lignin.
Experimental
Materials
Beech wood was obtained from local suppliers, the particle size was generated by a cutting mill with a 1 mm sieve and dried at 50 °C until constant weight (ca. 24 h). 2-MTHF and oxalic acid were obtained from Sigma-Aldrich and Carl Roth and were used without further modification.Exemplary procedure for lignocellulose fractionation via OrganoCat to obtain lignin
In a 300 mL high pressure reactor, 12 g beech wood lignocellulose and 120 mL of 0.1 M oxalic acid and 120 mL 2-MTHF were suspended. The reactor was heated to 140 °C for 3 h. After cooling of the reactor to room temperature, the liquid phases where separated by decantation and the aqueous phase was filtered to isolate the cellulose enriched pulp. The lignin solution was extracted with aqueous CaCl2 solution to remove oxalic acid from the solution. Lignin was obtained by evaporation of 2-MTHF and analyzed using NMR and SEC.Lignin precipitation using CO2-expanded 2-MTHF
All experiments were conducted in triplicates. A solution of 10 wt% lignin in 2-MTHF was prepared by addition of OrganoCat lignin to 2-MTHF. 5 mL of lignin solution in 2-MTHF was placed inside a 20 mL stainless steel autoclave with a glass inlet and equipped with a dip tube (see Fig. 1). Under stirring at room temperature, CO2 pressure was applied to the autoclave and the mixture was stirred for 15 min to saturate 2-MTHF with CO2 and reach precipitation equilibrium of lignin. The autoclave was removed from the CO2 line and the solution was let out slowly through the dip tube, while reducing the pressure to ambient pressure. The precipitated lignin inside the glass inlet was dissolved using acetone and the solvent was evaporated afterwards, to yield the precipitated fraction of the lignin. The lignin solution, let off through the dip tube, was placed inside the autoclave again and pressurized with CO2 at a higher pressure. The described procedure was repeated consecutively from 10 to 50 bar pressure, to provide several fractions of precipitated lignin, leaving residual lignin in solution (referred to as dissolved fraction). The dissolved lignin was obtained via solvent evaporation. | ||
| Fig. 1 Device to pressurize CO2 onto a lignin solution in 2-MTHF. The device consists of a high-pressure steel reactor (4), equipped with a dip tube (5), pressure gage (3) and connected to a CO2 gas line with adjustable pressure (1). | ||
Lignin analysis (NMR spectroscopy)
NMR measurements where conducted on a Bruker AS400 (400 MHz) Spectrometer. Approximately 5 to 20 mg of lignin where dissolved in 0.5 mL of DMSO-d6. 1H–13C-HSQC (measurement time 3:40 h) measurements were used to evaluate the type of linkages present in the respective lignin fractions of the reactions. The corresponding signals of each substructure were integrated and compared to each other.Lignin analysis (31P-NMR spectroscopy)
NMR measurements where conducted on a Bruker AS400 (400 MHz) Spectrometer. Following a procedure described by Pu et al.,15 20 mg of lignin where dissolved in 0.3 mL pyridine and 0.2 mL of deuterated chloroform. Chromium acetylacetonate was added (1 mM) together with 5 mg of cyclohexanol as internal standard. 100 μL of 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP) were added and the solution was mixed for 10 min. Afterwards the mixture was measured using quantitative 31P-NMR spectroscopy (25 s pulse delay, 128 scans).Size exclusion chromatography
The molecular size distribution of lignin samples was investigated using size exclusion chromatography (SEC). Measurements were performed using a Agilent Series 1200 SEC device equipped with a RID detector (Optilab Rex 837, Wyatt Technology) and a SEC column system consisting of a precolumn PSS PolarSil, 8 × 50 mm, with a particle size of 5 μm and three columns PSS PolarSil Linear S, 8 × 300 mm, particle size 5 μm downstream. SEC measurements were conducted using N,N-dimethylformamide with 0.1 M lithium chloride as eluent, a column temperature of 45 °C and a flow rate of 1 mL min−1. Samples of lignin with concentrations of 20 g L−1 were dissolved in the eluent. The injection volume used was 10 μL. The calibration was conducted using the PSS ReadyCal-Kit Poly(sterene) standard for a molecular weight range between 266 to 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da. The eluent was identical as for the lignin samples. The SEC data was analyzed using PSS WinGPC software.
000 Da. The eluent was identical as for the lignin samples. The SEC data was analyzed using PSS WinGPC software.
Results and discussion
OrganoCat lignin was produced as described previously,3 and was dissolved in 2-methyltetrahydrofuran (2-MTHF) at a concentration of 10 wt%. A device comprising a high-pressure reactor equipped with a dip tube (solvent outlet), manometer and connected to a CO2 line with adjustable pressure was built (Fig. 1).In a first set of experiments, the formation of the CO2-expanded phase with 2-MTHF was assessed, as well as the capacity of the created phase to trigger lignin precipitation, upon consecutively increasing pressures of CO2 and removing the precipitate after each step. The precipitated lignin was collected, and dried until constant weight to determine the yield (Fig. 2).
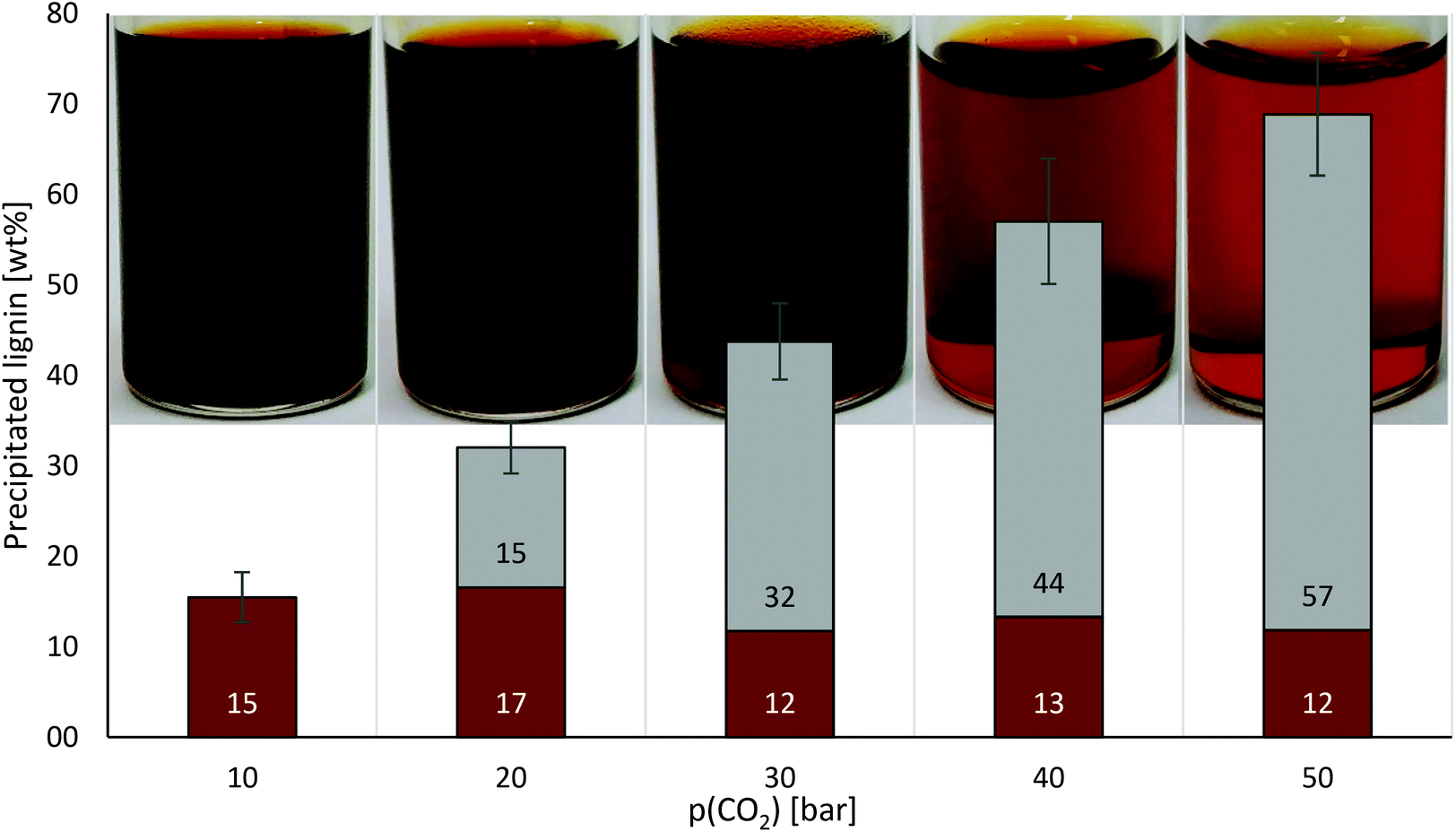 | ||
| Fig. 2 Consecutive precipitation of lignin at room temperature upon increasing pressures of CO2 in 2-MTHF, forming an expanded phase. The vials show samples of the supernatant after each precipitation. Experiments were conducted in triplicates. 10 wt% lignin in 2-MTHF stock solution, room temperature, solubility equilibrium reached after 15 min. Precipitated lignin at indicated pressures is given in wt% of the initial amount of lignin (brown bars); accumulated lignin from former precipitations (grey bars). | ||
Along the formation of the expanded phase, lignin precipitation started at a CO2 pressure of 10 bar. Starting from a stock solution with 10 wt% lignin, based on previous work on anti-solvent (e.g. n-pentane) precipitation of OrganoCat lignin from 2-MTHF,6 the concentration was reduced to 8.5 wt% at 10 bar and declined linearly (R2 = 0.9953) with enhancing CO2 pressures to 3.1 wt% at 50 bar. At 50 bar, approx. 70% of the lignin was obtained as precipitate, while 28% of the lignin remained dissolved. Results are comparable to those obtained in CO2-expanded phases formed with acetic acid–water mixtures,10 but with 2-MTHF a truly water-free solution is intended (what can provide benefits for future biorefinery plants, e.g. leading to simplified downstream processing units). Remarkably, compared to other lignins (e.g. Kraft derivatives), the OrganoCat lignin displays a lower and more homogeneous molecular weight distribution, in the range of 1000–3000 Da.3,14 This may result in more difficult-to-separate derivatives, but notably the concept works successfully at low pressures. It may be expected that at higher pressures of CO2, higher yields or selectivity could be achieved. Exploring even supercritical CO2 conditions may also become an important future research alternative for biorefineries, as this may show utility for the separation of other biomolecules as well. A judicious consideration on (higher) energy costs and achieved benefits should be considered as well.
Stimulated by the results, which provide different quantities of precipitated lignin, in a subsequent set of experiments, the qualities of the obtained precipitated lignins were assessed. Fig. 3 depicts the lignin unit proportions obtained in the different fractions, namely p-hydxoxy-phenyl (H), guaiacyl (G) and syringyl (S) units (blue bars, left axis) and the amount of linkages per 100 monomer units (yellow bars, right axis). Interestingly, the remaining dissolved lignin seems to contain a higher amount of H units (proportion of up to approx. 16%), whereas in the different precipitation steps (10–50 bar) almost no H units were observed. This suggests that in OrganoCat lignin from beech wood, H units are more present in low weight-average molecular weight (Mw) fractions and monomers (which tend to remain dissolved, and not precipitated). Conversely, S and G units followed a constant pattern at different pressures (approx. 40–55% proportion each), suggesting a statistic distribution of these moieties in all lignin fractions with higher Mw. This is in agreement with similar findings, fractionating lignin with anti-solvent precipitation.6 Albeit more research is needed to understand these precipitation patterns – including lignin from different plant origins –, overall, the results suggest that it is possible to obtain lignin fractions with different monomer contents, from a given pretreatment process by gas-expanding the 2-MTHF phase. Novel tailored applications, depending on the needs and lignin compositions, might be envisaged. It is worth mentioning that, with the same pretreatment, more than one lignin raw material for future valorization can be obtained, broadening the versatility and the viability of the biorefinery.
 | ||
| Fig. 3 Linkage and monomer unit composition in OrganoCat lignin and lignin fractions, generated by applying different CO2 pressures. Experiments were conducted in triplicates. Blue bars: Proportion of lignin units in the different fractions obtained. Yellow bars: Proportion of some lignin moieties in the different fractions. Green bars: Monomeric, dimeric and oligomeric structures. Data obtained using 2D-1H–13C-HSQC NMR; 10 wt% lignin in 2-MTHF stock solution, room temperature, solubility equilibrium reached after 15 min. | ||
Subsequently, we evaluated the proportion of some relevant chemical linkages present in lignin, namely β–β, β-O-4, β-5 moieties (Fig. 3, yellow bars), as well as the presence of some dissolved monomeric, dimeric and oligomeric structures (Fig. 3, green bars). Remarkably, lignin fractions with a rather high β-O-4 content were precipitated at low CO2 pressures (10–20 bar), whereas at higher CO2 pressures the β-O-4 content tended to be gradually lower, showing that it is possible to enhance the proportion of that moiety in pretreated lignins by means of expanded phases. Thus, the precipitated fraction obtained at 10 bar led to a 2-fold higher β-O-4 proportion than the starting material (raw lignin). The ratio of the other moieties – β–β and β-5 – remained constant along the fractions. Finally, monomers and small oligomers were not significantly precipitated at increasing pressures – as expected, given their low molecular weight – and were then accumulated in the final fraction in the 2-MTHF, which was not precipitated at the applied pressures. The presence of proportions of β–β and β-O-4 linkages in the final fraction (dissolved in 2-MTHF, not precipitated) suggests that there are relatively small oligomers with high proportion of these bonds present. Analysis of hydroxyl functions via31P-NMR in the different lignin fractions (see ESI, Table S3†) confirm the constant monomer ratios and the declining aliphatic hydroxyl functions at elevated pressures. Interestingly, in the dissolved lignin at 50 bar, a slightly higher amount of COOH groups was detected (see ESI, Table S3†), which might be caused by small amounts of residual oxalic acid from the OrganoCat process staying dissolved at the applied pressure.
To further characterize the lignin fractions, size exclusion chromatography (SEC) studies were performed. Results related to weight-average molecular weight (Mw), number average molecular weight (Mn) and polydispersity index (PDI) are depicted in Fig. 4. With rising CO2 pressure, Mw and Mn decrease gradually, consistent with the fact that the gradual precipitation of lignin fractions depends on the molecular size. The PDI of the fractions is lower than of the complete OrganoCat lignin, leading to a narrowing of the molecular size distribution. The fraction precipitated at 10 bar has a high content in β-O-4 and S and G units, with a Mw of approx. 4600 g mol−1, whereas the precipitated fraction at 50 bar has a low β-O-4 content, with a Mw of approx. 1040 g mol−1. The non-precipitated, dissolved lignin at 50 bar CO2 pressure displays an even more distinct decrease in the molecular weight (580 g mol−1, approx. 4-fold decrease compared to the complete OrganoCat lignin), indicating the smallest lignin fragments in the range of mono- and dimers. Being a rather homogeneous fraction, this might prove valuable to the chemical industry (e.g. hydrogenation of the aromatics16), and catalytic approaches could be focused there, rather than in crude lignin, where a broad weight distribution is present.
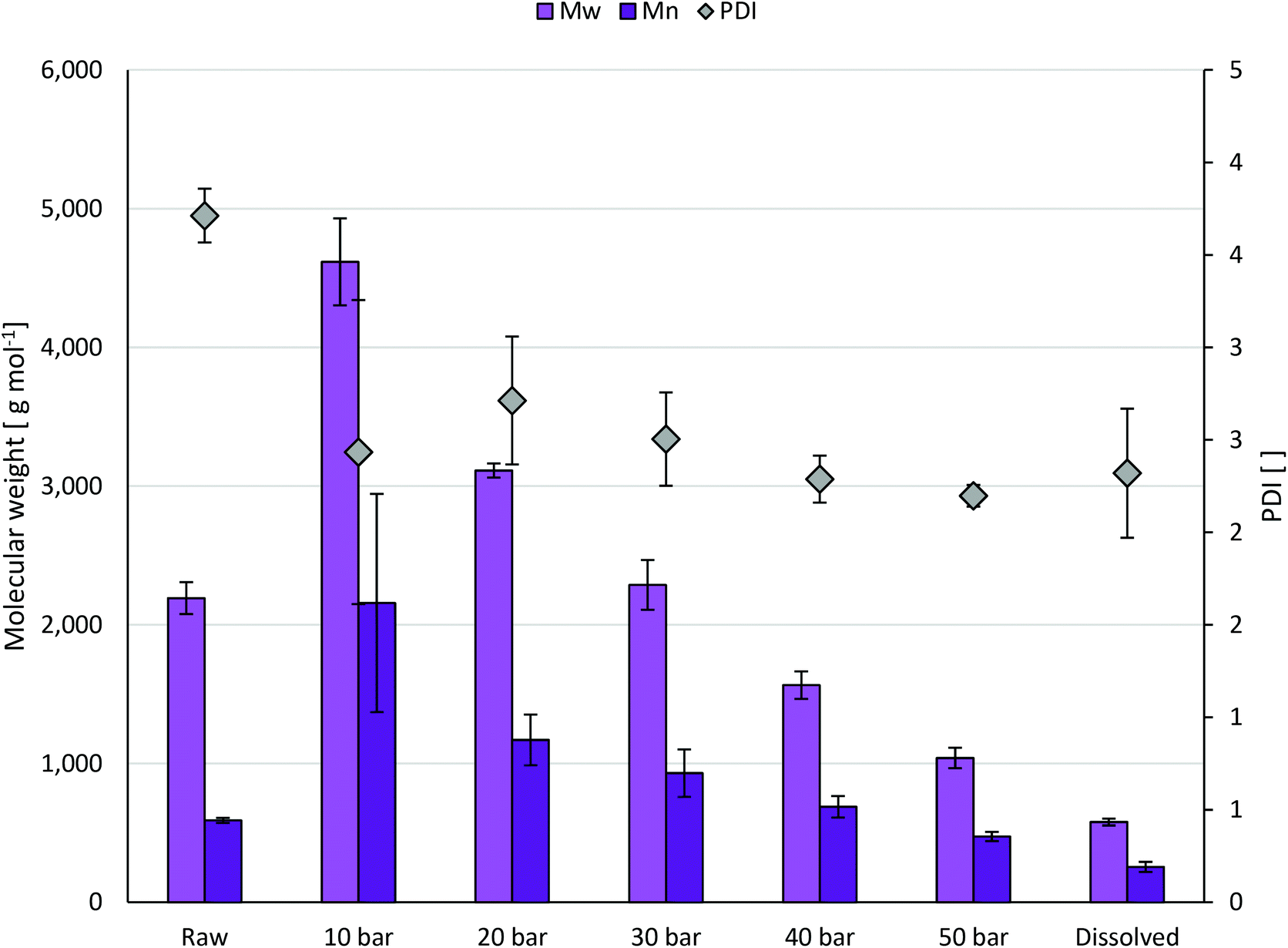 | ||
| Fig. 4 Size exclusion chromatography (SEC) data related to Mw (light purple bars), Mn (dark purple bars) and PDI (grey diamonds) of the different fractions gained at different applied CO2 pressures. “Raw” is referring to the feed solution and “Dissolved” covers remaining unprecipitated lignin fractions. Experiments were conducted as triplicates. 10 wt% lignin in 2-MTHF feed solution, room temperature, solubility equilibrium reached after 15 min. | ||
To benchmark the potential of GXL using CO2 for lignin fractionation with other techniques, economic and environmental aspects need to be considered. The use of CO2 does not generate significant amount of waste, and can be recycled once used, as well as the 2-MTHF fraction (e.g. for another OrganoCat cycle). Other approaches for lignin fractionation involve the use of petroleum-based solvents,6 which incorporate a higher environmental impact, although they can be, in principle, recovered and reused as well. In some other cases, water is used as anti-solvent,10 which implies consumption of large quantities of water, and the generation of wastewater for which a treatment should be incorporated. With respect to economics, it must be noted that the herein presented approach does not reach supercritical ranges, and thus limited pressure of CO2 is added (up to 50 bar). It is expected that the different lignin fractions will provide options for several markets, thus compensating the costs associated with CO2 storage, energy consumption, and needed devices and reactors.17 The lignin fractionation is performed at room temperature, what may save energy costs significantly. Overall, the use of CO2 for extractions has been used industrially (e.g. decaffeination), and gives promising prognosis that economics may be reached. However, careful optimizations and assessments must be made.
Conclusions
The use of biogenic solvents to create CO2-expanded phases may enable very promising applications for biorefineries. The expanded phases trigger the Mw-selective precipitation of polymeric materials with different amounts of β-O-4 linkages, facilitating its downstream, while allowing a straightforward solvent recovery at the same time. Herein, the generation of a CO2-expanded 2-MTHF phase has been used to precipitate and fractionate OrganoCat lignin in several products with different properties. Qualitatively, the composition of the different fractions, in terms of size distribution and linkages seem to be influenced by the CO2 pressure. Smaller oligomers, including mono- and dimeric lignin fragments remain dissolved, potentially allowing for their direct, subsequent conversion to high-value chemicals. More research is needed to understand the findings observed, and how applications of the fractions can be achieved. The herein reported proof-of-concept should serve as a basis to trigger research groups and industries to undertake assessment on this and other biogenic solvents for gas-expanded phases. Uses in future biorefineries may be rather broad and extending the concept to other macromolecular raw materials appears foreseeable.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed as part of the Cluster of Excellence “Fuel Science Center”, which is funded by the Excellence Initiative of the German Research Foundation to promote science and research at German universities, as well as part of the Bioeconomy Science Center (BioSC), supported in the projects AP3 Focus Lab and LIFT. The scientific activities of the Bioeconomy Science Center were financially supported by the Ministry of Innovation, Science and Research within the framework of the NRW Strategieprojekt BioSC (no. 313/323-400-002 13). PDdM thanks Prof. Andrés Alcántara for many fruitful scientific discussions related to biocatalysis and sustainable solvents, and in particular to 2-MTHF.References
- (a) Y. Liao, S. F. Koelewijn, G. van den Bossche, J. van Aelst, S. van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. van Aelst, M. Maesen, H. Matsushima, J. M. Thevelein, K. van Acker, B. Lagrain, D. Verboekend and B. F. Sels, Science, 2020, 367, 1385–1390 CrossRef CAS PubMed; (b) M. Mascal, ChemSusChem, 2020, 13, 274–277 CrossRef CAS PubMed; (c) E. Paone, T. Tabanelli and F. Mauriello, Curr. Opin. Green Sustainable Chem., 2020, 24, 1–6 CrossRef; (d) T. Renders, G. van den Bossche, T. Vangeel, K. van Aelst and B. F. Sels, Curr. Opin. Biotechnol., 2019, 56, 193–201 CrossRef CAS PubMed; (e) Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–618 CrossRef CAS PubMed; (f) R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS; (g) Industrial Renewables – A Practical Viewpoint, ed. P. Domínguez de María, John Wiley & Sons, Hoboken (NJ), 2016 Search PubMed.
- (a) C. G. Yoo, X. Meng, Y. Pu and A. J. Ragauskas, Bioresour. Technol., 2020, 301, 122784 CrossRef CAS PubMed; (b) Y. M. Questell-Santiago, M. V. Galkin, K. Barta and J. S. Luterbacher, Nat. Rev. Chem., 2020, 4, 311–330 CrossRef CAS; (c) P. Domínguez de María, P. M. Grande and W. Leitner, Chem. Ing. Tech., 2015, 87, 1686–1695 CrossRef.
- (a) P. M. Grande, D. Weidener, S. Dietrich, M. Dama, M. Bellof, R. Maas, M. Pauly, W. Leitner, H. Klose and P. Domínguez de María, ACS Omega, 2019, 4, 14451–14457 CrossRef CAS; (b) D. Weidener, H. Klose, W. Leitner, U. Schurr, B. Usadel, P. Domínguez de María and P. M. Grande, ChemSusChem, 2018, 11, 2051–2056 CrossRef CAS PubMed; (c) T. Damm, P. M. Grande, N. D. Jablonowski, B. Thiele, U. Disko, U. Mann, U. Schurr, W. Leitner, B. Usadel, P. Domínguez de María and H. Klose, Bioresour. Technol., 2017, 244, 889–896 CrossRef CAS PubMed; (d) P. M. Grande, J. Viell, N. Theyssen, W. Marquardt, P. Domínguez de María and W. Leitner, Green Chem., 2015, 17, 3533–3539 RSC; (e) T. vom Stein, P. M. Grande, H. Kayser, F. Sibilla, W. Leitner and P. Domínguez de María, Green Chem., 2011, 13, 1772–1777 RSC.
- (a) A. R. Alcántara and P. Domínguez de María, Curr. Green Chem., 2018, 5, 86–102 CrossRef; (b) V. Pace, P. Hoyos, L. Castoldi, P. Domínguez de María and A. R. Alcántara, ChemSusChem, 2012, 5, 1369–1379 CrossRef CAS.
- S. Stiefel, D. Di Marino, A. Eggert, I. R. Kühnrich, M. Schmidt, P. M. Grande, W. Leitner, A. Jupke and M. Wessling, Green Chem., 2017, 19, 93–97 RSC.
- (a) D. Weidener, A. Holtz, H. Klose, A. Jupke, W. Leitner and P. M. Grande, Molecules, 2020, 25, 3330 CrossRef CAS PubMed; (b) A. Holtz, D. Weidener, W. Leitner, H. Klose, P. M. Grande and A. Jupke, Sep. Purif. Technol., 2019, 236, 116295 CrossRef.
- (a) C. Cui, R. Sun and D. S. Argyropoulos, ACS Sustainable Chem. Eng., 2014, 2, 959–968 CrossRef CAS; (b) Y. Xu, K. Li and M. Zhang, Colloids Surf., A, 2007, 301, 255–263 CrossRef CAS; (c) M. A. Gilarranz, F. Rodriguez, M. Oliet and J. A. Revenga, Sep. Purif. Technol., 1998, 33, 1359–1377 CAS; (d) T. V. Lourençon, F. A. Hansel, T. A. da Silva, L. P. Ramos, G. I. B. de Muniz and W. L. E. Magalhães, Sep. Purif. Technol., 2015, 154, 82–88 CrossRef; (e) A. García, A. Toledano, L. Serrano, I. Egüés, M. González, F. Marín and J. Labidi, Sep. Purif. Technol., 2009, 68, 193–198 CrossRef; (f) A. Toledano, L. Serrano, A. Garcia, I. Mondragon and J. Labidi, Chem. Eng. J., 2010, 157, 93–99 CrossRef CAS; (g) M. Norgren and S. Mackin, Ind. Eng. Chem. Res., 2009, 48, 5098–5104 CrossRef CAS.
- (a) U. Hintermair, W. Leitner and P. Jessop, Expanded Liquid Phases in Catalysis: Gas-expanded Liquids and Liquid-Supercritical Fluid Biphasic Systems, in Handbook of Green Chemistry, ed. P. Anastas, Wiley-VCH, Weinheim, 2010, ch. 4, pp. 103–188 Search PubMed; (b) M. Aigner, A. Echtermeyer, S. Kaminski, J. Viell, K. Leonhard, A. Mitsos and A. Jupke, J. Chem. Eng. Data, 2020, 65, 993–1004 CrossRef CAS.
- E. Granero-Fernandez, C. Lacaze-Dufaure, J. S. Condoret, V. Gerbaud and Y. Medina-Gonzalez, Ind. Eng. Chem. Res., 2019, 58, 18942–18964 CrossRef CAS.
- A. S. Klett, A. M. Payne, T. Phongpreecha, D. B. Hodge and M. C. Thies, Ind. Eng. Chem. Res., 2017, 56, 9778–9782 CrossRef CAS.
- C. Eckert, C. Liotta, A. Ragauskas, J. Hallett, C. Kitchens, E. Hill and L. Draucker, Green Chem., 2007, 9, 545–548 RSC.
- F. M. A. Geilen, T. vom Stein, B. Engendahl, S. Winterle, M. A. Liauw, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2011, 50, 6831–6834 CrossRef CAS PubMed.
- (a) H. N. Hoang, Y. Nagashima, S. Mori, H. Kagechika and T. Matsuda, Tetrahedron, 2017, 73, 2984–2989 CrossRef CAS; (b) H. N. Hoang, E. Granero-Fernández, S. Yamada, S. Mori, H. Kagechika, Y. Medina-González and T. Matsuda, ACS Sustainable Chem. Eng., 2017, 5, 11051–11059 CrossRef CAS.
- L. Wiermans, H. Schumacher, C. M. Klaassen and P. Domínguez de María, RSC Adv., 2015, 5, 4009–4018 RSC.
- Y. Pu, S. Cao and A. J. Ragauskas, Energy Environ. Sci., 2011, 4, 3154 RSC.
- A. Bjelić, B. Likozar and M. Grilc, Chem. Eng. J., 2020, 399, 125712 CrossRef.
- (a) I. M. Prado, G. H. C. Prado, J. M. Prado and M. A. A. Meireles, Food Bioprod. Process., 2013, 91, 656–664 CrossRef CAS; (b) J. A. Rocha-Uribe, J. I. Novelo-Pérez and C. Araceli Ruiz-Mercado, J. Supercrit. Fluids, 2014, 93, 38–41 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc01651b |
| This journal is © The Royal Society of Chemistry 2021 |