
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA more sustainable synthesis approach for cellulose acetate using the DBU/CO2 switchable solvent system†
Jonas
Wolfs
a and
Michael A. R.
Meier
 *ab
*ab
aInstitute of Organic Chemistry (IOC), Materialwissenschaftliches Zentrum MZE, Karlsruhe Institute of Technology (KIT), Straße am Forum 7, 76131 Karlsruhe, Germany. E-mail: m.a.r.meier@kit.edu; Web: http://www.meier-michael.com
bInstitute of Biological and Chemical Systems – Functional Molecular Systems (IBCS-FMS), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 18th May 2021
Abstract
Cellulose acetate is one of the most important cellulose derivatives and commercially mainly produced using the Acetic Acid Process, in which overstoichiometric amounts of acetic anhydride and concentrated acetic acid are used to obtain cellulose triacetate. A subsequent partial hydrolysis is necessary to achieve evenly substituted cellulose acetates with lower degrees of substitution. Homogeneous acetylations in ionic liquids or other cellulose dissolving solvent systems often offer milder conditions and the possibility of a one-step synthesis of cellulose acetates with lower degrees of substitution by simply adjusting the equivalents of the acetylation agent. Here, we show an efficient homogeneous cellulose acetylation process without the need of any additional catalyst or activation step using the DBU/CO2 switchable solvent system. Vinyl acetate was used as a more benign acetylation agent under mild conditions and straightforward recyclability of all employed components was demonstrated with high recycling ratios (87.0–98.9%). Less cellulose backbone degradation compared to a cellulose acetate sample synthesized by the Acetic Acid Process from the same cellulose source was shown by size exclusion chromatography (Mn = 35 kDa vs. 12 kDa), which resulted in improved mechanical properties of solvent casted foils. Other homogeneous procedures reported so far (e.g. in ionic liquids) reached lower degrees of substitution, needed additional catalysts, proved to be less advantageous in terms of recycling, or required more reactive acetylation agents. Our results thus demonstrate a cellulose acetylation method with full focus on sustainability, efficiency, and applicability, resulting in an E-factor of 1.92 for the overall process.
Introduction
The conversion of renewable resources and biomass into sustainable materials is an important step to reduce the dependence on depleting fossil resources, especially for polymeric materials. Cellulose, as the most abundant biopolymer with a natural production of 1.5 × 1012 tons p.a., has received substantial attention in this regard over the last few years.1 One of the most important technical derivatives of cellulose is cellulose acetate with its broad application possibilities in the field of films for optical devices, textile fibers, or cigarette filters.1–4 The commercial production is mainly carried out using the Acetic Acid Process, in which the cellulose is (initially) heterogeneously modified using an excess of acetic anhydride (Ac2O) in acetic acid (AcOH) with sulfuric acid as a catalyst.4–6 This results in cellulose triacetate with a degree of substitution (DS) between 2.8–3.0.3 Partial cleavage of the polymeric chain is unavoidable in this process, as the acetal moiety in the cellulose backbone is prone to hydrolysis under acidic conditions. Cellulose acetates with lower DS, especially cellulose diacetates (DS of 2.0–2.5), are obtained via a subsequent partial hydrolysis of the triacetate.3,4 The Acetic Acid Process uses overstoichiometric amounts of acetylating agent, concentrated acids, and needs two steps to obtain cellulose acetates with lower DS, generating large amounts of waste and compromising material properties due to molecular weight degradation. Directly produced cellulose diacetate using the Acetic Acid Process with lower equivalents of the acetylating agent results in an inhomogeneous distribution of the functional groups along the chain, causing irreproducible product characteristics, which makes it unsuitable for commercial processing.3,7,8Homogeneous modifications generally enable better control over the DS and regioselectivity in terms of primary vs. secondary hydroxyls as well as a homogeneous functionalization along the polymer chain, leading to more defined cellulose derivatives with tunable properties.9,10 Various solvent systems have been developed over the past years for homogeneous cellulose derivatization, including N,N-dimethylacetamide–lithium chloride (DMAc–LiCl) or dimethyl sulfoxide–tetrabutylammonium fluoride (DMSO–TBAF), but these solvents did not overcome the lab-scale synthesis stage due to their high cost and poor recyclability.11 Besides that, several ionic liquids (ILs) for the dissolution of cellulose were developed, which are considered “greener” solvents for cellulose dissolution and functionalization. However, ILs still suffer from limitations like their relatively expensive synthesis and easy contamination during the reaction, making their recovery more difficult.11,12
Based on the reversible nonpolar-to-polar switchable solvent developed by Jessop et al.,13 a new strategy for reversible cellulose dissolution using CO2 was almost simultaneously proposed by Xie et al.14 and Jérôme et al.15 in 2013. Two classes of cellulose dissolution systems were developed: the derivative and non-derivative approach. In the derivative approach, the hydroxyl groups of the cellulose are functionalized reversibly with CO2 in the presence of a superbase, e.g. 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) or 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), forming a cellulose carbonate complex, which is DMSO-soluble. The non-derivative approach includes an additional alcohol, like methanol or ethylene glycol, forming the carbonate complex with CO2. This ionic compound then acts as a solvent to solubilize cellulose with DMSO as a co-solvent. Upon removal of CO2 from the system, cellulose could be regenerated, and the solvent recovered for further solubilizations. Moreover, DMSO is categorized as a “usable” solvent by most sustainability focused solvent selection guides with only minor issues concerning reactivity and stability.16,17 With its non-volatility, ease of purification, and low toxicity, DMSO can be considered as a more sustainable solvent compared to DMAc or most ILs, which commonly need multi-step syntheses increasing the cost and environmental impact.
DBU is known as an efficient organocatalyst for various transformations, such as alkylation and transesterification reactions.18,19 With both being part of the solubilization process and acting as a transesterification catalyst, DBU has a dual role in this system requiring no additional catalyst for a successful acetylation. Despite its high efficiency and dual role, DBU remains the most critical component of this solvent system, but its problems are put into perspective considering the low amount required for the solubilization along with good recyclability.
A useful measure for the evaluation of the environmental impact of chemical processes is the E-factor, which is defined as the mass ratio of waste produced in a process taking the chemical yield, reagents, side products, solvent losses, and process aids into account.20 However, the E-factor does not take toxicity into account, which has a marked impact on the sustainability and safety as described by the twelve principles of green chemistry.21 Despite this limitation, the E-factor represents a rapid and easy indicator for the sustainability and efficiency of a given process and was thus applied here as the main sustainability metric.
For a truly sustainable production process from renewable resources, every component of the reaction needs to be considered. This aspect is often overlooked in the literature, as often only some reactants are derived from renewable resources. In the case of cellulose acetate production, especially the acetylation agent needs to be evaluated concerning its sustainability. Vinyl acetate is mainly produced on a large industrial scale by the ethylene-based process (ca. 70% of the total worldwide production).22 In this gas-phase process, ethylene reacts with acetic acid and oxygen in a palladium catalyst-containing fixed-bed reactor, giving vinyl acetate and water with a selectivity of 94% for ethylene and 98–99% for acetic acid.22,23 This makes the vinyl acetate production very efficient, but for an entirely sustainable process, ethylene and acetic acid also need to be originated from a sustainable resource. Industry chooses the cheapest way to produce these two compounds, which is mainly the carbonylation of methanol for acetic acid and steam cracking for ethylene.24,25 With methanol being mainly produced from syngas,26 the industrial synthesis of vinyl acetate today is entirely based on fossil resources. The potential for a greener synthesis of vinyl acetate exists with acetic acid and ethylene both being produced from bioethanol by fermentation (acetic acid) or dehydration (ethylene).24,25 In countries with cheap bioethanol availability, ethylene is produced in relevant amounts by dehydration already today.23 This renders vinyl acetate a promising candidate as a greener acetylating agent for cellulose acetate synthesis, especially also considering its lower toxicity and non-corrosiveness compared to acetic anhydride (LD50 oral, rat for vinyl acetate: 3470–3500 mg kg−1; LD50 oral, rat for acetic anhydride: 1780 mg kg−1)22,27 making its handling, transportation, and storage easier and safer.
In the last few years, several homogeneous cellulose acetylation methods were reported, such as the homogeneous esterification with acetic anhydride, vinyl acetate, and isopropenyl acetate in the ionic liquid [DBNH][OAc] (Ioncell) by Kilpeläinen et al.,28 the acetylation with acetic anhydride and acetyl chloride in 1-N-butyl-3-methylimidazolium chloride ([C4mim]+Cl−) by Heinze et al.29 or the acetylation in the DBU/CO2 switchable solvent system with acetic anhydride by Xie et al.30 Additionally, transesterification reactions in the DBU/CO2 switchable solvent system with different vinyl esters (i.e. vinyl benzoate, vinyl pivalate, vinyl laurate, vinyl 2-ethylhexyloate, vinyl octanoate, and vinyl palmitate) was shown by Liu et al. in 2018.31 However, vinyl acetate was not considered yet as an efficient acetylating agent using the DBU/CO2 switchable solvent system and detailed sustainability evaluations are missing in other publications.
Herein, we thus report an efficient and sustainable method for the homogeneous synthesis of cellulose acetate with tunable DS using vinyl acetate in the DBU/CO2 switchable solvent system with easy recyclability of the employed solvents and improved material properties due to less cellulose degradation.
Results and discussion
For the homogeneous acetylation investigated herein, cellulose was first dissolved using the DBU/CO2 switchable solvent system with the optimized conditions as reported by our group in 2018.32 Therefore, all cellulose samples were dried under vacuum for 24 h at 100 °C to remove water, possibly causing deviations in the exact ratio of cellulose to the acetylating agent and furthermore minimizing the risk of potential side reactions. The dried cellulose was first suspended in dry DMSO at 40 °C in the presence of DBU (3 eq. per anhydroglucose unit (AGU)) and then solubilized under stirring by bubbling CO2 into the suspension for 20 min. These very mild conditions and the rapid dissolution contribute to several benefits in terms of sustainability and efficiency compared to classic ionic liquids, which often need elevated temperatures and several hours for a complete dissolution of cellulose.29,33 For the derivatization reaction, a calculated amount of vinyl acetate was added slowly to the solution in order to prevent local precipitation of the cellulose. The temperature was then elevated to 60 °C ensuring a rapid reaction, but at the same time prevents the vinyl acetate (b.p. 72 °C) from evaporating (steps 1 and 2, solubilization and derivatization, in Scheme 1).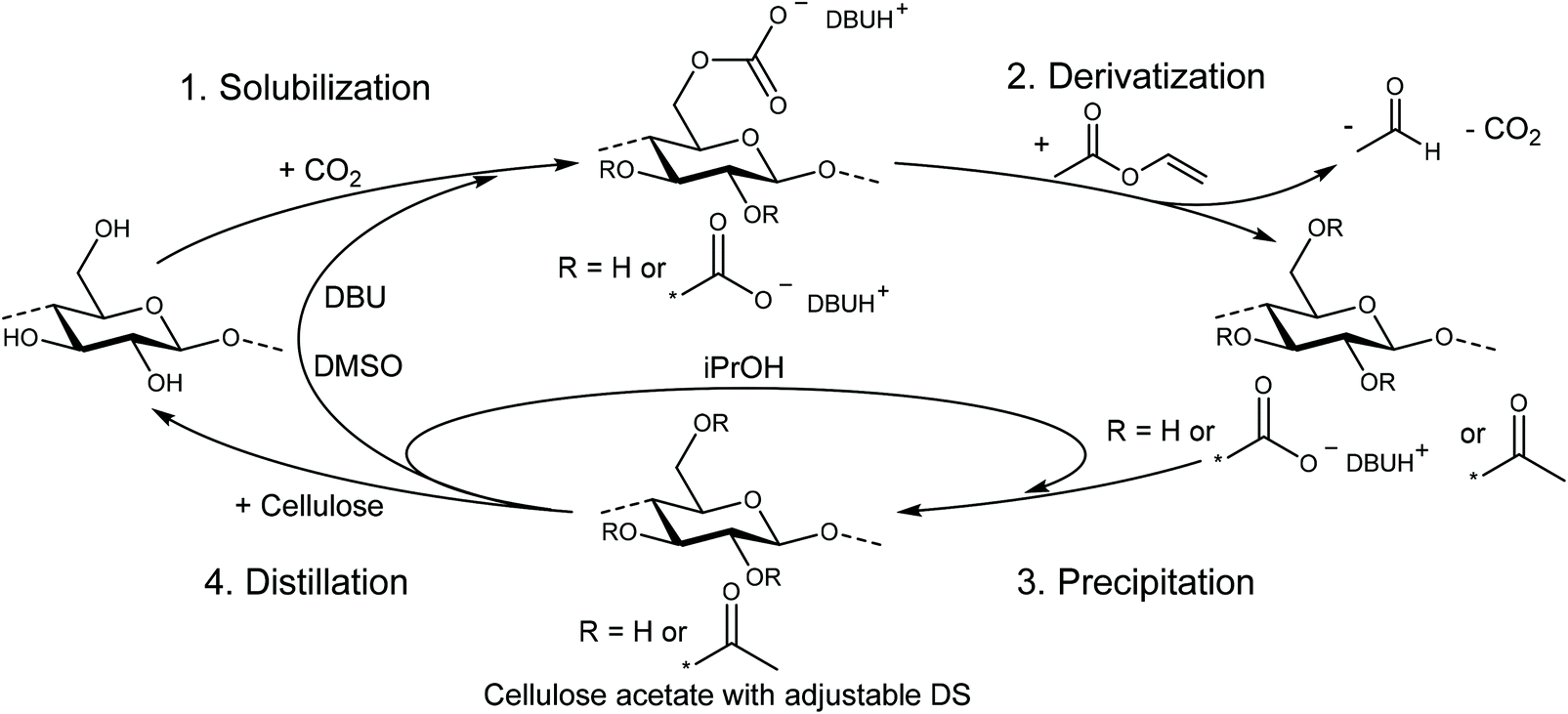 | ||
| Scheme 1 Dissolution and activation of cellulose in a DBU/CO2 switchable solvent system with subsequent acetylation using vinyl acetate and recycling of the solvents. | ||
The use of vinyl acetate has several benefits compared to acetic anhydride as an acetylating agent: first, the formed vinyl alcohol byproduct immediately tautomerizes to acetaldehyde, which evaporates due to its low boiling point (b.p. 20 °C), shifting the chemical equilibrium to the product side. This ensures high conversions, reducing the amount of reactant needed, and thus makes the synthesis more efficient. Second, the acetaldehyde does not react or form a salt with DBU, keeping the concentration and therefore the catalytic activity high, even at low temperatures. When anhydrides are used, the formed acid byproduct forms a salt with DBU, reducing its catalytic activity as reported by Liu et al.31 Third, the evaporation of acetaldehyde during the reaction facilitates the subsequent recycling process of the DMSO/DBU mixture. If anhydrides are used as acetylating agents, the formed DBU salt ([DBUH][OAc] in the case of acetic anhydride) commonly needs to be neutralized (with e.g. NaOH) and then extracted to recover the DBU.30 This additional step leads to generation of more waste, consequently increasing the environmental footprint and cost. Instead, the captured acetaldehyde can be used for other reactions (especially from larger scale acetylations with vinyl acetate), as acetaldehyde is a bulk chemical needed for many industrial processes. Moreover, as already described in the introduction, vinyl acetate is a more sustainable and less toxic alternative to acetic anhydride.
A linear correlation between the DS values determined via31P NMR analysis and the relation of integrals from the ATR-IR spectra was shown by Kilpeläinen et al.28 Therefore, ATR-IR spectroscopy was used to optimize the reaction time by monitoring the integrals of the decreasing O–H stretching vibration signal at 3390 cm−1, the increasing signal of the carbonyl stretching vibration at 1728 cm−1 and the increasing intensity of the C–O stretching vibration of the acetyl moiety at 1230 cm−1. All signals were normalized to the intensity of the C–O stretching vibration of the AGU at 1025 cm−1 for a relative comparison of the achieved DS, as the intensity of this signal is not significantly affected by the acetylation. It was observed that almost maximal conversion was reached after 240 min (Fig. 1). To prevent side reactions, which can potentially lower the DMSO/DBU recycling rate, all acetylations were thus terminated after 240 min.
 | ||
| Fig. 1 Normalized integrals of the characteristic vibrations over time in the homogeneous cellulose acetylation using vinyl acetate from the respective ATR-IR signals, referenced to the AGU C–O stretching vibration signal. | ||
Purification of the synthesized cellulose acetate samples was performed by precipitation from an anti-solvent. Methanol, ethanol, and isopropanol are commonly used antisolvents for cellulose acetate synthesized using a homogeneous approach, but optimization studies revealed significantly lower DS values when methanol or ethanol were used. A partial transesterification of cellulose acetate with methanol/ethanol catalyzed by DBU, which is still present in the precipitation mixture, was assumed. Precipitation of a reaction mixture (3 eq. vinyl acetate per AGU, 4 h at 60 °C) from methanol, ethanol, and isopropanol with subsequent stirring for 30 min revealed a lower DS for the samples precipitated from methanol (DS1H = 1.73) and ethanol (DS1H = 2.12) compared to the sample precipitated from isopropanol (DS1H = 2.43) as determined by 1H NMR spectroscopy (ESI Fig. S36†). To confirm a possible heterogeneous deacetylation by transesterification catalyzed by DBU, a purified cellulose acetate sample (DS1H = 2.43) was subsequently stirred in methanol, ethanol, and isopropanol with 3 eq. of DBU per AGU for 12 h at room temperature. The resulting suspension was filtered, the solids were analyzed by ATR-IR spectroscopy and the filtrate by 1H NMR spectroscopy, respectively. ATR-IR spectroscopy confirmed the deacetylation as the characteristic bands (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1728 cm−1, C–O at 1230 cm−1 and C–H at 1025 cm−1) of the acetyl moiety decreased and the O–H band at around 3390 cm−1 increased relative to the C–O vibration of the AGU at 1025 cm−1 (ESI Fig. S38†) when methanol or ethanol was used. For the sample stirred in isopropanol, no significant decrease of the acetyl signals or increase of the O–H signal was detected. The filtrates analyzed by 1H NMR spectroscopy showed the formation of the respective transesterification products (methyl acetate and ethyl acetate) when methanol or ethanol was used (ESI Fig. S37†), which confirms a transesterification and deacetylation of cellulose acetate in methanol and ethanol when DBU is present. In contrast, the formation of isopropyl acetate was not observed by 1H NMR spectroscopy when cellulose acetate was stirred in isopropanol in the presence of DBU.
O at 1728 cm−1, C–O at 1230 cm−1 and C–H at 1025 cm−1) of the acetyl moiety decreased and the O–H band at around 3390 cm−1 increased relative to the C–O vibration of the AGU at 1025 cm−1 (ESI Fig. S38†) when methanol or ethanol was used. For the sample stirred in isopropanol, no significant decrease of the acetyl signals or increase of the O–H signal was detected. The filtrates analyzed by 1H NMR spectroscopy showed the formation of the respective transesterification products (methyl acetate and ethyl acetate) when methanol or ethanol was used (ESI Fig. S37†), which confirms a transesterification and deacetylation of cellulose acetate in methanol and ethanol when DBU is present. In contrast, the formation of isopropyl acetate was not observed by 1H NMR spectroscopy when cellulose acetate was stirred in isopropanol in the presence of DBU.
Cellulose acetates with different DS values were synthesized by varying the equivalents of vinyl acetate using the optimized reaction and work-up procedure (CA-1–CA-7, Table 1 and Fig. 2). The DS of the prepared CAs was determined by reacting the free hydroxyl groups of the respective CA sample with the phosphorylating agent 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (2-Cl-TMDP) to yield phosphite esters, which can be detected quantitatively via31P NMR spectroscopy. By integration of these signals and comparison with the internal standard endo-N-hydroxy-5-norbornene-2,3-dicarboximide (e-HNDI-TMDP), the average DS can be calculated according to a procedure introduced by Kilpeläinen et al.34 (ESI eqn (2)†). Almost full conversion of the acetylating agent was observed for the synthesis of CAs with lower DS (entries CA-1 to CA-4, Table 1), whereas a 1.5-fold excess of vinyl acetate per hydroxyl group (4.5 eq. per AGU) was needed to obtain a per-O-acetylation of cellulose (entry CA-7, Table 1).
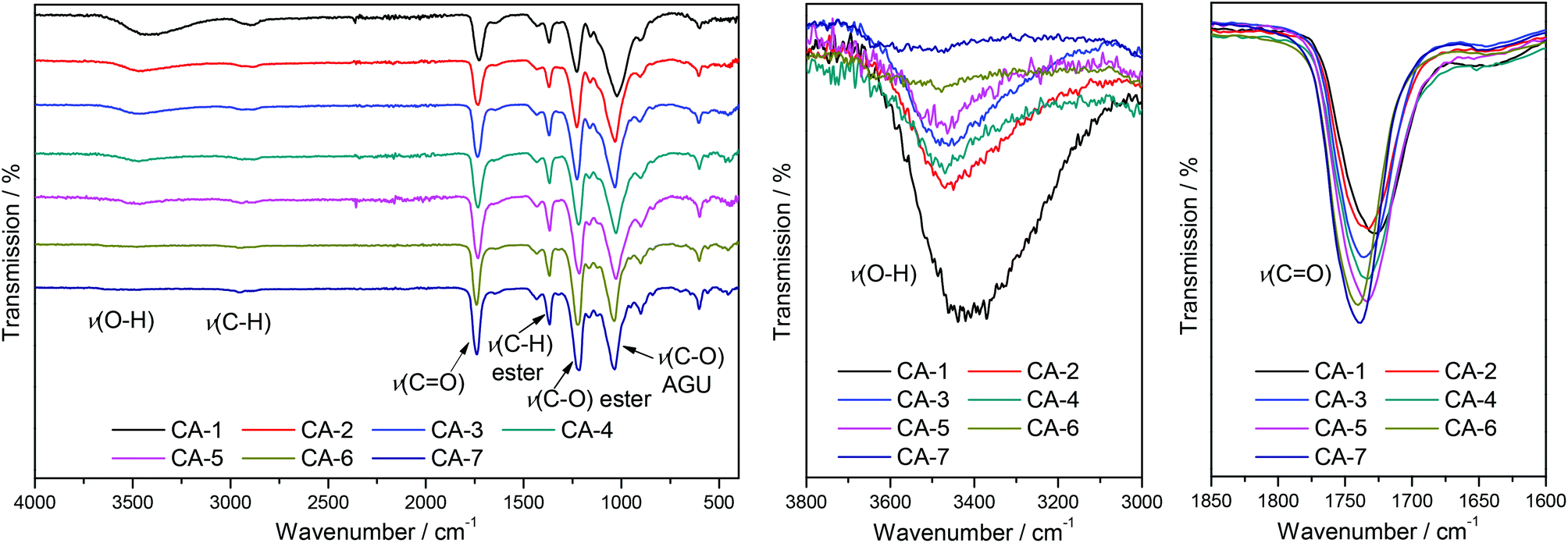 | ||
| Fig. 2 ATR-IR spectra (left) and expanded views of the O–H stretching vibration (middle) and CO stretching vibration (right) for CA-1–CA-7. | ||
| Sample | Cellulose typeb | Eq. VA | Yieldd | Conversiond | DS1H | DS31P | DS C6f | DS C3f | DS C2f |
|---|---|---|---|---|---|---|---|---|---|
| a Synthesized via a heterogeneous acetylation route analogously to the Acetic Acid Process. b MCC = micro crystalline cellulose, FP = filter paper. c Acetic anhydride used as acetylating agent. d Calculated based on the DS1H. e Quantitative conversion assumed based on a calculated conversion of 102%. f Calculated from 13C NMR method with peak deconvolution based on the DS31P. | |||||||||
| CA-1 | MCC | 1.0 | 91% | Quant.e | 1.02 | 1.04 | — | — | — |
| CA-2 | MCC | 1.5 | 91% | 97% | 1.45 | 1.47 | — | — | — |
| CA-3 | MCC | 2.0 | 91% | 85% | 1.70 | 1.73 | 0.80 | 0.71 | 0.22 |
| CA-4 | MCC | 2.5 | 84% | 87% | 2.17 | 2.18 | 0.90 | 0.87 | 0.40 |
| CA-5 | MCC | 3.0 | 92% | 79% | 2.36 | 2.50 | 0.89 | 0.95 | 0.67 |
| CA-6 | MCC | 3.5 | 98% | 74% | 2.59 | 2.52 | — | — | — |
| CA-7 | MCC | 4.5 | 99% | 65% | 2.94 | 2.97 | 0.98 | 0.97 | 1.02 |
| CA-FP | FP | 4.5 | 91% | 59% | 2.66 | 2.44 | — | — | — |
| CA-HET | MCC | 12.9c | 91% | 23% | 2.92 | 2.96 | — | — | — |
| CA-REC | MCC | 2.5 | 95% | 92% | 2.29 | 2.32 | — | — | — |
| CA-REC2 | MCC | 3.0 | 99% | 80% | 2.40 | 2.42 | — | — | — |
Compared to other homogeneous acetylation reactions reported so far (acetylation with isopropenyl acetate in 1-ethyl-3-methyl-imidazolium acetate35), less equivalents of acetylating agent was required to obtain peracetylation. Higher DS could be obtained with less acetylating agent (compared to acetylation with acetic anhydride in 1-allyl-3-methylimidazolium chloride33,36 or acetylation with acetic anhydride in 1,5-diazabicyclo(4.3.0)non-5-enium acetate37). Acetylation in the DBU/CO2 switchable solvent system using acetic anhydride30 generally reached lower DS with higher loadings of reactant (DS = 2.89, DS = 2.27, and DS = 1.78 for 5, 4, and 3 eq. per AGU) demonstrating the high efficiency of vinyl acetate as a mild and less toxic acetylating agent in this solvent system with the benefit of an easier solvent recycling due to the in situ evaporation of the formed acetaldehyde byproduct as already explained before. Kilpeläinen et al.28 demonstrated an acetylation of cellulose in the ionic liquid 1,5-diazabicyclo[4.3.0]non-5-ene acetate with vinyl acetate as acetylating agent under comparable reaction conditions, but the conversion remained low (DS = 1.58 with 3 eq. of VA).
The acetaldehyde side product formed during the acetylation reaction could be captured in a cold trap (−50 °C) when a slight flow of argon was applied, as confirmed by 1H NMR spectroscopy (ESI Fig. S42†). However, vinyl acetate was also captured in significant amounts due to its relatively high vapor pressure at the reaction temperature of 60 °C. On an industrial scale with the use of more advanced fractionating columns, this can certainly be prevented, and acetaldehyde could be captured in high purity for a use in other processes. This reduces the amount of waste produced, further increasing the sustainability and efficiency. To proof the versatility of this system, filter paper was also used as a cellulose source and acetylated successfully (CA-FP, Table 1).
A per-O-acetylated CA sample using the Acetic Acid Process was also synthesized (CA-HET) as a reference material in a modified protocol according to Malm et al.6 from the same cellulose source as in the homogeneous acetylation using the DBU/CO2 switchable solvent system. Size exclusion chromatography measurements of the two samples reveal significantly higher molecular weight for CA-7 synthesized using the DBU/CO2 switchable solvent system (Mn = 35 kDa) compared to CA-HET synthesized by the Acetic Acid Process (Mn = 12 kDa) as shown in Fig. 3a. This validates the distinctly milder acetylation conditions using this solvent system with less degradation of the cellulose backbone. Furthermore, significant deacetylation was observed for CA-HET after 2.5 months of storage in a closed screw cap vial (30.1% deacetylation, ESI Fig. S35†), which is caused by the literature-known vinegar syndrome.38,39 This degradation process is catalyzed by acids. Therefore, a thorough removal of residual acetic acid is crucial when using the Acetic Acid Process for the synthesis of cellulose triacetate. Despite profound washing and no detection of acetic acid in the 1H NMR spectrum, presumably trace amounts of acetic acid remained in the sample causing an accelerated degradation. Contrary, no deacetylation was observed for CA-7 in the same time frame (ESI Fig. S34†), highlighting another advantage of this procedure.
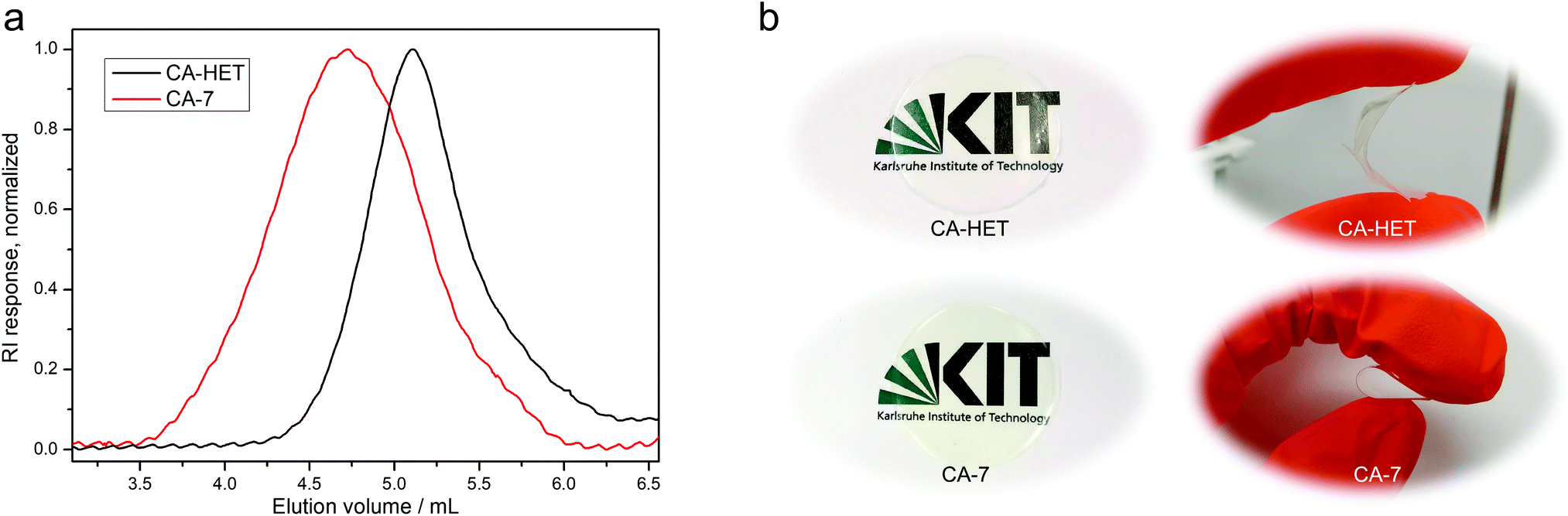 | ||
| Fig. 3 (a) Size exclusion chromatography traces of cellulose triacetate synthesized by the heterogeneous acetylation using acetic anhydride (CA-HET, DS31P = 2.96, Mn = 12 kDa) and the homogeneous route using vinyl acetate (CA-7, DS31P = 2.97, Mn = 35 kDa). Mn were extrapolated from a PMMA standard. (b) Photographs of CA-HET and CA-7 foils with bending tests of the material. | ||
Tensile strength measurements were performed for CA-5, CA-6, and CA-7, as their DS was high enough to solubilize in acetone or chloroform, which is a prerequisite to prepare cellulose acetate foils by a solvent casting technique (Table 2 and ESI Fig. S29†). A tensile strength measurement of CA-HET was planned as a comparison, but the foil was too brittle to be inserted into the instrument. The foils prepared from CA-7 and CA-HET were therefore compared by bending and CA-HET started to shatter at low deformation, while CA-7 could be bent by 180° without breaking as shown in Fig. 3b, which indicates considerably better mechanical properties of CA-7 compared to CA-HET, presumably due to its higher Mn. This was also observed by Sookne and Harris comparing the tensile strength of CA films with different number average degrees of polymerization 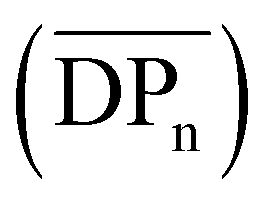 .40,41 In this report, the tensile strength of a CA film with
.40,41 In this report, the tensile strength of a CA film with 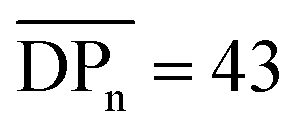 (corresponding to Mn = 12.4 kDa) could not be measured due to its high brittleness, which is in accordance to the results obtained for CA-HET. The tensile strength of a comparable sample to CA-7 with an Mn ≈ 36 kDa showed a tensile strength of approx. 62 MPa, which is higher, but in a comparable range to the ultimate tensile strength measured for CA-7 (42.9 ± 2.8 MPa). According to Sookne and Harris, tensile strength of CA films increase with higher
(corresponding to Mn = 12.4 kDa) could not be measured due to its high brittleness, which is in accordance to the results obtained for CA-HET. The tensile strength of a comparable sample to CA-7 with an Mn ≈ 36 kDa showed a tensile strength of approx. 62 MPa, which is higher, but in a comparable range to the ultimate tensile strength measured for CA-7 (42.9 ± 2.8 MPa). According to Sookne and Harris, tensile strength of CA films increase with higher 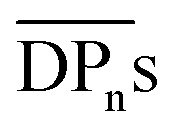 up to an asymptotic value for CAs with
up to an asymptotic value for CAs with 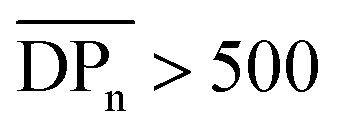 (corresponding to Mn > 144 kDa).40 Hence, CAs with higher Mn are desirable, which can be achieved by the herein introduced homogeneous acetylation due to its milder reaction conditions, leading to less degradation of the cellulose backbone compared to the Acetic Acid Process as shown in Fig. 3a. It has to be mentioned that in industrial acetylation processes, dissolving pulp is mainly used as cellulose source, which has a higher Mn compared to microcrystalline cellulose. Partial backbone degradation still leads to cellulose acetate with adequate Mn, but it remains a challenge for industrial acetylations to reach almost fully substituted cellulose acetate with a sufficiently high
(corresponding to Mn > 144 kDa).40 Hence, CAs with higher Mn are desirable, which can be achieved by the herein introduced homogeneous acetylation due to its milder reaction conditions, leading to less degradation of the cellulose backbone compared to the Acetic Acid Process as shown in Fig. 3a. It has to be mentioned that in industrial acetylation processes, dissolving pulp is mainly used as cellulose source, which has a higher Mn compared to microcrystalline cellulose. Partial backbone degradation still leads to cellulose acetate with adequate Mn, but it remains a challenge for industrial acetylations to reach almost fully substituted cellulose acetate with a sufficiently high 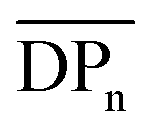 .5 The Young's moduli were determined from the linear region of the stress–strain curves. An average value from 3 measurements was determined by using a linear fit and the standard deviation was calculated (Table 2 and ESI Fig. S30–S32†).
.5 The Young's moduli were determined from the linear region of the stress–strain curves. An average value from 3 measurements was determined by using a linear fit and the standard deviation was calculated (Table 2 and ESI Fig. S30–S32†).
| Sample | T d,5%/°C | T d,50%/°C | T g/°C | Young's modulus/MPa | Ultimate tensile strength/MPa |
|---|---|---|---|---|---|
| CA-1 | 260.7 | 341.4 | — | — | — |
| CA-2 | 233.3 | 350.6 | — | — | — |
| CA-3 | 280.2 | 355.3 | — | — | — |
| CA-4 | 292.0 | 369.9 | 190.7 | — | — |
| CA-5 | 286.7 | 365.1 | 188.9 | 747.1 ± 89.3 | 19.5 ± 0.9 |
| CA-6 | 286.4 | 364.9 | 190.0 | 955.9 ± 31.0 | 22.9 ± 1.1 |
| CA-7 | 320.0 | 375.3 | 183.7 | 2005.5 ± 105.4 | 42.9 ± 2.8 |
The thermal stability of the CAs was determined by thermogravimetric analysis (TGA). The calculated results for Td,5% and Td,50% are summarized in Table 2. All samples showed a single major degradation step and an improvement in thermal stability was observed with an increase of the DS value. The onset degradation temperature Td,5% increased from 260.7 to 292.0 and 320.0 °C for the CAs with a DS31P of 1.04, 2.18, and 2.97, respectively. Analogously, the Td,50% also increased from 341.4 to 369.9 and 375.3 °C for the same CA samples. A Td,onset of 252 °C for a cellulose triacetate sample (DS = 2.92) and 224–243 °C for a CA (DS = 2.50) was observed by Kamide et al.,42 which is lower than the Td,5% observed for comparable CAs (CA-7 and CA-5, respectively), but shows the same trend with a higher Td for increasing DS values. Higher thermal degradation temperatures of CA-7 and CA-5 could be explained by the absence of any residual acetic acid, which possibly lowers the thermal stability of the material.
The three hydroxyl groups of cellulose at the C2, C3, and C6 position are known to differ in their reactivity, resulting in a distribution of the acetyl moiety among the three hydroxyl groups for CA samples with a DS < 3.36 The DS for those three individual functionalities was investigated via13C NMR spectroscopy in an inverse gated decoupling experiment by comparing the integral of the three signals at 169.49 ppm, 169.84 ppm and 170.78 ppm, which can be assigned to the carbonyl carbons of the acetyl moiety at C2, C3, and C6 position, respectively. Due to the overlap of the signals, a peak deconvolution method was used to determine the integrals of the three peaks as shown in Fig. 4. Based on the calculated individual DS values, a higher functionalization is obtained for the C6 and C3 positions compared to the C2 position for all investigated CA samples. Consequently, the observed order of reactivity for the acetylation is C6–OH > C3–OH > C2–OH. This result is similar to the homogeneous acetylation of cellulose with acetic anhydride in the ionic liquid 1-allyl-3-methylimidazolium chloride36 or in the solvent system DMAc–LiCl.43 In the commercial synthesis of partially substituted CA, the distribution of the acetyl moieties is different from that observed in these homogeneous acetylation reactions because of the deacetylation process: during the partial hydrolytic deacetylation of fully acetylated CA, the C6 position is preferably hydrolyzed, resulting in a lower individual DS at C6 relative to the DS at C3 and C2.8 Different substitution patterns obviously lead to deviations in properties of the material, especially the solubility. Despite the different substitution pattern, CA-4 (DS31P = 2.18), CA-5 (DS31P = 2.50) and CA-6 (DS31P = 2.52) were soluble in acetone, which is also observed for commercially produced cellulose diacetate (DS of 2.0–2.5) and is an important property for processability (e.g. foil casting) of the material.
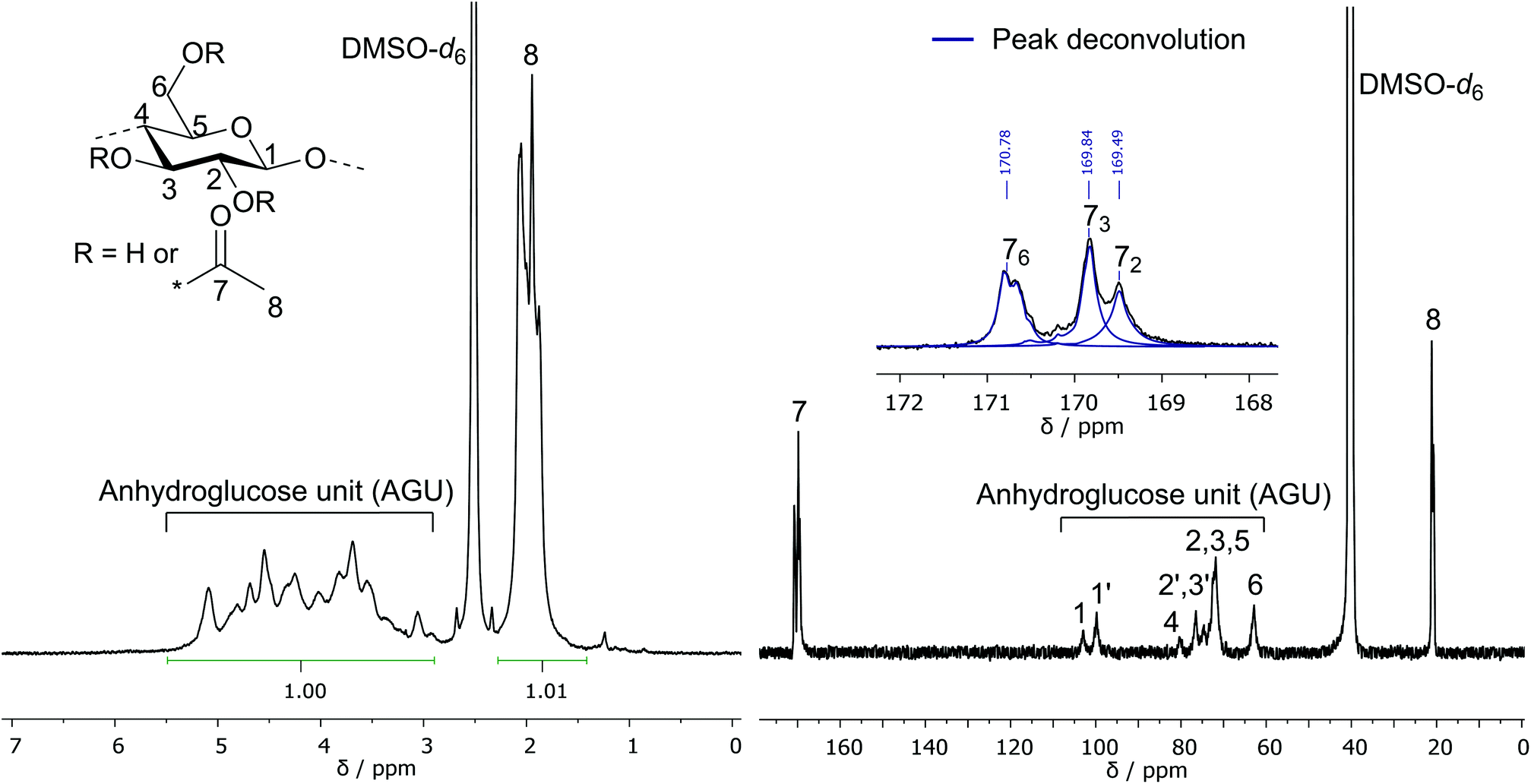 | ||
| Fig. 4 1H NMR (left, DMSO-d6 + TFA) and 13C NMR (right, DMSO-d6) spectra of partially acetylated cellulose acetate (CA-5, DS31P = 2.50). Peak deconvolution: blue lines in expanded view. | ||
For a sustainable homogeneous cellulose acetylation, recyclability of the solvent and antisolvent used for precipitation is crucial as this accounts most to the E-factor (Fig. 5b). To reduce the influence of sample loss during the process and thus ensuring a more precise recycling ratio determination, a larger batch (CA-REC, 4.00 g cellulose) was performed. After precipitation and filtration, a mixture of isopropanol, DMSO, and DBU remains to be recycled. Advantageous for the recycling steps is the low number of compounds, which need to be purified. Due to the almost quantitative conversion of vinyl acetate (92%) and the evaporation of acetaldehyde during the reaction, no side product needs to be removed. This enables an easier purification by fractional distillation as illustrated in Fig. 5a. In other procedures, i.e. when acetic anhydride is used as acetylating agent, the acetic acid byproduct forms a salt with DBU, complicating the recovery process.30,44,45 After recovery of isopropanol in a first fraction, an intermediate fraction containing isopropanol and DMSO (Fig. 5a) needed to be collected to ensure complete removal of the alcohol. This prevents a partial non-derivative dissolution mechanism with a possible transesterification side reaction of isopropanol with vinyl acetate in a second cycle. Transesterification of methanol with acetic anhydride as acetylation agent in the non-derivative dissolution approach was shown to occur by Liu et al.46 To facilitate the recycling process, DMSO and DBU was then distilled together in a third fraction and the molar composition of the obtained mixture was calculated by comparison of the integrals in the 1H NMR spectrum (ESI eqn (7) and (8), Fig. S39†). Even though minor impurities were detected in the recycled DMSO/DBU fraction, the solvent could be successfully reused in a second cycle for the homogeneous synthesis of CA-REC2 after adjusting the molar composition by the addition of a calculated amount of fresh DBU (67 eq. DMSO and 3 eq. DBU for 1 eq. AGU) without prior drying or considerable differences compared to a fresh batch. CA-REC2 with a DS1H of 2.40 was obtained using the recycled solvents, compared to a DS1H of 2.36 for CA-5 when fresh solvents were used (Table 1).
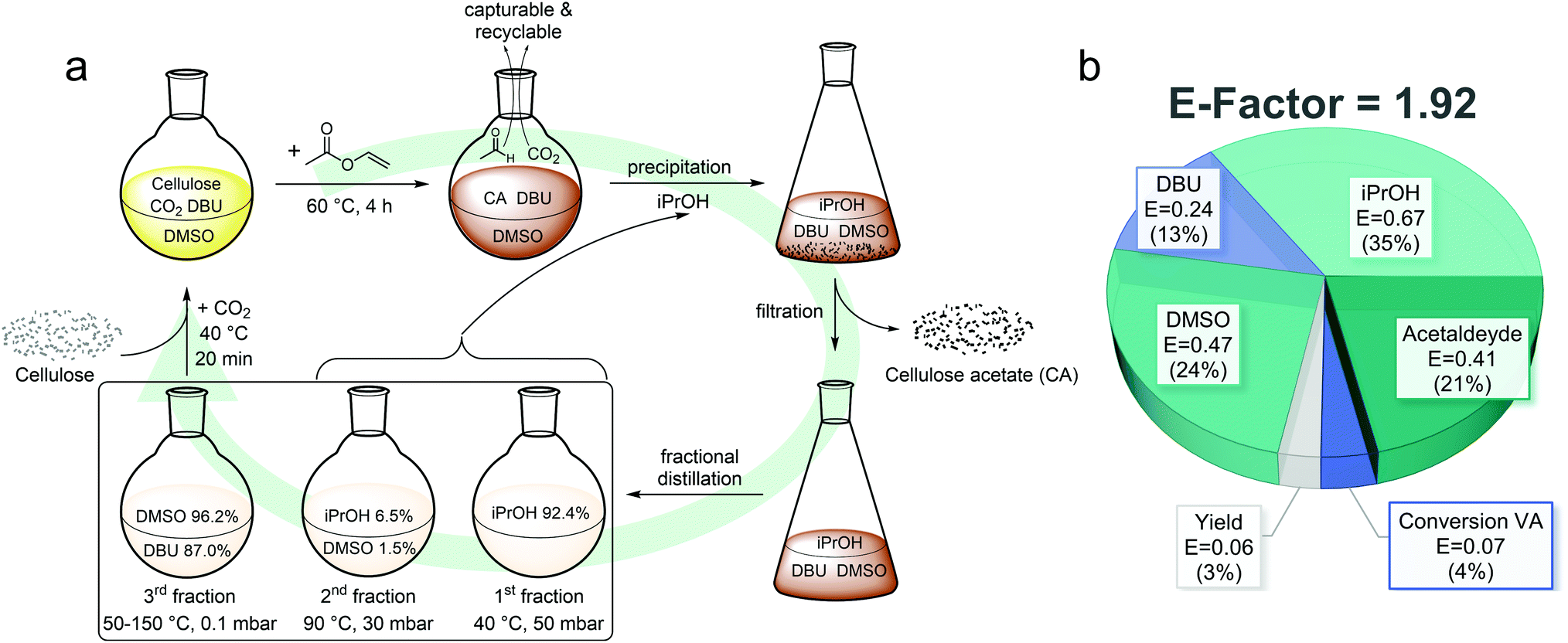 | ||
| Fig. 5 (a) Scheme for the homogeneous cellulose acetylation and recycling procedure. All recycling ratios given in % are related to the used starting material, respectively. (b) Pie chart of the partial E-factors for the synthesis of CA-REC (DS1H = 2.29) in the first recovery cycle with the respective contribution to the total E-factor in %. | ||
The distilled intermediate fraction consisting of isopropanol and DMSO was analyzed for its molar composition, analogously to the mixed DMSO/DBU fraction, by 1H NMR spectroscopy and then added to the pure isopropanol fraction as the low amount of DMSO does not affect the precipitation of cellulose acetate when used in a second synthesis and purification cycle. Including the recovered DMSO and isopropanol from the intermediate fraction, a total recycling ratio of 97.7% DMSO (96.2 + 1.5% from the intermediate fraction), 87.0% DBU and 98.9% iPrOH (92.4 + 6.5%) was achieved for the first cycle. This is close to the recovery ratio obtained by Xie et al. (92% DMSO and 91% DBU) for the acetylation of cellulose with acetic anhydride using the DBU/CO2 switchable solvent system.30 However, the recovery of DBU is significantly more complex when acetic anhydride is used, because the solution had to be extracted with ethyl acetate and neutralized with sodium hydroxide to remove the acetic acid byproduct, which generates more waste and consequently decreases the overall sustainability.
The CO2 used for solubilization was neglected for E-factor calculations due to its low impact and complex quantification. Furthermore, on larger scale, it could be recovered as it is released during the functionalization reaction. An E-factor of 1.92 was calculated for the overall process, which is in the range of the E-factor from industrial syntheses of bulk chemicals (<1–5) as reported by Sheldon.20 This E-factor is significantly lower (∼3 to almost 10 times) than for other reported cellulose acetate synthesis procedures.10 After calculation of the partial E-factor for every single component, it is obvious that the major contribution to the E-factor comes from iPrOH, as it contributed 35% to the total E-factor in the first cycle and 72% in the second cycle (Fig. 5b and ESI Fig. S41†). We assume that the loss of iPrOH can mainly be explained by evaporation during the work-up procedure due to its comparably high volatility. Working in a closed system using an industrial setup together with a recovery of the used CO2 and capturing of the acetaldehyde byproduct, an E-factor close to 1 should realistically be possible, which is why we see high potential in this procedure. In order to evaluate whether this process is also superior over the well-refined Acetic Acid Process on industrial scale, further research in pilot plants will certainly be necessary, but is outside the scope of this scientific manuscript. In particular, the one-step synthesis of cellulose diacetate (DS = 2.0–2.5), which typically can only be achieved in a two-step process, with a more benign acetylation agent, mild conditions, high reagent conversion, low degradation, and easy recycling process with high recovery ratios makes the presented process a very promising and more sustainable alternative to existing procedures.
Conclusion
A sustainable and homogeneous acetylation of cellulose with vinyl acetate was achieved under mild conditions in a CO2-based switchable solvent. After optimization of the reaction and work-up procedure by monitoring the reaction using Fourier-transform infrared spectroscopy, a set of CAs with different DS ranging from 1.04 to 2.97 were directly synthesized by varying the equivalents of vinyl acetate. The direct synthesis of cellulose diacetates (DS = 2.0–2.5) is advantageous over the commercially used Acetic Acid Process, in which cellulose triacetate needs to be partially hydrolyzed due to its otherwise inhomogeneous distribution of acetyl groups along the polymeric chain. Higher molecular weight of the synthesized cellulose acetate was maintained when the presented procedure with vinyl acetate was applied, if compared to a sample synthesized via the Acetic Acid Process from the same cellulose source (Mn = 35 kDa vs. Mn = 12 kDa). No additional catalyst was needed, since DBU was already part of the solvent system acting as the transesterification catalyst. In other homogeneous syntheses of cellulose acetate using acetic anhydride (or acetyl chloride) as acetylation agent, the byproduct acetic acid (or HCl) is generated, which forms a salt with DBU and therefore reduces its catalytic activity and complicates the recovery, leading to the generation of more waste. All employed solvents in this system were recovered by simple fractional distillation with recycling ratios of 98.9% iPrOH, 97.7% DMSO and 87.0% DBU with 95% CA yield and a vinyl acetate conversion of 92%, resulting in an E-factor of 1.92. A possible capture of the generated acetaldehyde during the functionalization was shown. Thermal properties of the synthesized products were investigated by thermogravimetric analysis and differential scanning calorimetry with degradation temperatures (Td,5%) ranging from 233.3 to 320.0 °C and glass transition temperature (Tg) between 183.7–190.7 °C. Foils from acetone soluble samples were prepared by solvent casting and the mechanical properties were analyzed via tensile strength measurements revealing elastic moduli (E) between 747.1–2005.5 MPa and ultimate tensile strength values between 19.5–42.9 MPa depending on the DS of the samples.Experimental part
Materials
All cellulose samples were dried at 100 °C under vacuum for 24 h prior to use. Microcrystalline cellulose (MCC) was purchased from Sigma-Aldrich and filter paper type MN 615 was purchased from Machery-Nagel. Diazabicyclo[5.4.0]undec-7-ene (DBU, >98%, TCI), dimethyl sulfoxide (DMSO, dry and stored over molecular sieve, Acros Organics), vinyl acetate (stabilized, Acros Organics), pyridine (Fisher Scientific), 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP, Sigma Aldrich), endo-N-hydroxy-5-norbornene-2,3-dicarboximide (Sigma Aldrich), glacial acetic acid (≥99%, Sigma Aldrich), acetic anhydride (≥99%, Carl Roth) and CO2 (99.995%, Air Liquide) were used without further purification. Deuterated solvents (DMSO-d6 and CDCl3) were purchased from Eurisotop. All other solvents were used in technical grade.General procedure for the homogeneous synthesis of cellulose acetate
In a round-bottom flask, cellulose (2.00 g, 12.33 mmol) was suspended in 60 mL DMSO followed by the dropwise addition of 1,8-diazabicyclo[5.4.0]undec-7-ene (5.5 mL, 37.00 mmol, 3.0 eq. per anhydroglucose unit). After applying a CO2 flow through the solution for 20 min at 40 °C, a clear solution was obtained and vinyl acetate (1.0–4.5 eq. per AGU, depending on the experiment) was added dropwise. The homogeneous solution was then heated to 60 °C and stirred for 4 h. Subsequently, the dark solution was added dropwise into 300 mL of isopropanol under vigorous stirring to precipitate cellulose acetate. It was then vacuum filtrated and washed with isopropanol (3 × 50 mL). The obtained precipitate was dried under high vacuum (0.02 mbar) for 24 h. In case of residual DMSO in the product, the material was stirred with 40 mL of isopropanol (or methanol) under reflux for 1–12 h, vacuum filtrated and dried again. The final product was obtained as a white or yellow powdery substance. Yields were calculated based on the DS1H (ESI eqn (1)†) and ranged from 84 to 99%. CA-1: Yield: 91% ATR-IR (cm−1): 3032–3675 ν(O–H), 2819–2991 ν(C–H), 1728 ν(CO), 1370 methyl ν(C–H), 1229 ester ν(C–O), 1023 AGU ν(C–O). 1H NMR (400 MHz, DMSO-d6) δH ppm: 2.78–5.30 (m, AGU, 7H), 1.69–2.27 (m, Ac6,2,3, 9H).
General procedure for the heterogeneous synthesis of cellulose acetate using the Acetic Acid Process
In a two-necked round-bottom flask equipped with a thermometer and a magnetic stirring bar, cellulose (2.00 g, 12.33 mmol) was suspended in 10 mL glacial acetic acid and stirred for 1 h at 40 °C. Afterward, a mixture of 0.1 mL concentrated sulfuric acid in 20 mL glacial acetic acid was added dropwise and stirred at 30 °C for another 20 min. Acetic anhydride (15 mL, 159 mmol, 12.9 eq. per anhydroglucose unit) was then added dropwise and the temperature was kept below 40 °C. After 1 h stirring, the reaction mixture was added dropwise to vigorously stirred water (500 mL) and the product precipitated as a white solid. It was then vacuum filtrated, washed with excess water until the filtrate was not acidic anymore and then dried under vacuum. The yield was calculated based on the DS1H (ESI eqn (1)†). CA-HET: Yield: 91% ATR-IR (cm−1): 2826–3048 ν(C–H), 1738 ν(CO), 1366 methyl ν(C–H), 1214 ester ν(C–O), 1032 AGU ν(C–O). 1H NMR (400 MHz, CDCl3) δH ppm: 5.07 (t, H3, 1H), 4.79 (t, H2, 1H), 4.28–4.50 (m, H1,6, 2H), 3.98–4.09 (m, H6, 1H), 3.71 (t, H4, 1H), 3.46–3.56 (m, H5, 1H), 2.12 (s, Ac6, 3H), 2.01 (s, Ac2, 3H), 1.94 (s, Ac3, 3H).
DS determination by 31P NMR method
DS were determined by derivatization of the CA samples using a phosphorylating agent according to the following procedure: an exact amount of 25 mg of each sample was dissolved in 1 mL of pyridine followed by the addition of 1 mL CDCl3. Next, 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (2-Cl-TMDP, 100 μL, 0.63 mmol) was added and the solution was stirred for 15 min. Then, the internal standard endo-N-hydroxy-5-norbornene-2,3-dicarboximide (150 μL, 123.21 mM in pyridine/CDCl3 = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, 0.0154 mmol) was added and the solution was stirred for further 10 min. Then, 0.6 mL of the solution was transferred to an NMR tube and a 31P NMR measurement was performed. The DS values were calculated according to ESI eqn (2)† as reported by Kilpeläinen et al.34
:2, 0.0154 mmol) was added and the solution was stirred for further 10 min. Then, 0.6 mL of the solution was transferred to an NMR tube and a 31P NMR measurement was performed. The DS values were calculated according to ESI eqn (2)† as reported by Kilpeläinen et al.34
Recycling process
After filtration, the product was dried under vacuum and the evaporated iPrOH was captured in a cold trap. The residual filtrate containing iPrOH, DMSO, and DBU was purified by distillation. First, iPrOH was recovered (50–90 mbar, 40 °C) and combined with the captured iPrOH in the cold trap. The second (intermediate) fraction was collected containing iPrOH and DMSO (30 mbar, 90 °C) to remove the iPrOH quantitatively. Then, the third fraction DMSO/DBU was distilled at 0.1 mbar and 50–150 °C oil bath temperature until dry. Recovery yield: 97.7% DMSO, 87.0% DBU, 98.9% iPrOH.Instrumentation
624 scans and a delay time d1 of 3 seconds at 298 K. The signals were referenced to the solvent peak of partly deuterated DMSO-d6 at 39.52 ppm. 31P NMR spectra were recorded using a Bruker Ascend instrument at 162 MHz with 1024 scans and a delay time d1 of 5 seconds at 298 K.
000 Å), PSS PFG Micro (25.0 × 0.46 cm, 1000 Å), and PSS PFG Micro (25.0 × 0.46 cm, 100 Å) and a differential RI detector. Hexafluoroisopropanol (HFIP) enriched with 0.1 wt% potassium trifluoroacetate (KTFAc) was used as eluent at a temperature of 35 °C and a flow rate of 0.4 mL min−1. The samples were prepared with a concentration of 1.0 mg mL−1 and 50 μL were injected into the system. A poly(methyl methacrylate) standard (Polymer Standard Service, Mp = 102–981 kDa) was used for extrapolation of the number average molar mass (Mn).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Katharina Elies for SEC measurements, Hans Weickenmeier for TGA measurements and Daniel Zimmermann for assistance during tensile strength measurements.References
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- R. C. Law, Macromol. Symp., 2004, 208, 255–266 CrossRef CAS.
- T. Heinze and T. Liebert, Celluloses and Polyoses/Hemicelluloses, Elsevier B.V., 2012, vol. 10 Search PubMed.
- S. Fischer, K. Thümmler, B. Volkert, K. Hettrich, I. Schmidt and K. Fischer, in Macromolecular Symposia, John Wiley & Sons, Ltd, 2008, vol. 262, pp. 89–96 Search PubMed.
- H. Steinmeier, Macromol. Symp., 2004, 208, 49–60 CrossRef CAS.
- C. J. Malm, L. J. Tanghe and B. C. Laird, Preparation of Cellulose Acetate ACTION OF SULFURIC ACID, UTC, 1946, vol. 38 Search PubMed.
- C. R. Fordyce and United States Patent, 2129052, 1938, 50–52.
- T. Heinze, O. El Seoud and A. Koschella, Cellulose Derivatives. Synthesis, Structure, and Properties, 2018 Search PubMed.
- T. Heinze and T. Liebert, Prog. Polym. Sci., 2001, 26, 1689–1762 CrossRef CAS.
- K. N. Onwukamike, S. Grelier, E. Grau, H. Cramail and M. A. R. Meier, ACS Sustainable Chem. Eng., 2019, 7, 1826–1840 CrossRef CAS.
- M. Gericke, P. Fardim and T. Heinze, Molecules, 2012, 17, 7458–7502 CrossRef PubMed.
- Z. Söyler, K. N. Onwukamike, S. Grelier, E. Grau, H. Cramail and M. A. R. Meier, Green Chem., 2018, 20, 214–224 RSC.
- P. G. Jessop, D. J. Heldebrant, X. Li, C. A. Eckert and C. L. Liotta, Nature, 2005, 436, 1102–1102 CrossRef CAS PubMed.
- H. Xie, X. Yu, Y. Yang and Z. K. Zhao, Green Chem., 2014, 16, 2422–2427 RSC.
- Q. Zhang, N. S. Oztekin, J. Barrault, K. De Oliveira Vigier and F. Jérôme, ChemSusChem, 2013, 6, 593–596 CrossRef CAS PubMed.
- R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC.
- I. Flores, J. Demarteau, A. J. Müller, A. Etxeberria, L. Irusta, F. Bergman, C. Koning and H. Sardon, Eur. Polym. J., 2018, 104, 170–176 CrossRef CAS.
- O. Kreye, L. C. Over, T. Nitsche, R. Z. Lange and M. A. R. Meier, Tetrahedron, 2015, 71, 293–300 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- F. Bienewald, E. Leibold, P. Tužina and G. Roscher, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2019, pp. 1–16 Search PubMed.
- H. J. Arpe, Industrielle Organische Chemie: Bedeutende Vor- und Zwischenprodukte, Wiley, 6th edn, 2007 Search PubMed.
- H. Zimmermann and R. Walzl, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2009, pp. 465–526 Search PubMed.
- C. Le Berre, P. Serp, P. Kalck and G. P. Torrence, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2014, pp. 1–34 Search PubMed.
- J. Ott, V. Gronemann, F. Pontzen, E. Fiedler, G. Grossmann, D. B. Kersebohm, G. Weiss and C. Witte, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2012 Search PubMed.
- H. Held, A. Rengstl and D. Mayer, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2000 Search PubMed.
- T. Kakko, A. W. T. King and I. Kilpeläinen, Cellulose, 2017, 24, 5341–5354 CrossRef CAS.
- S. Barthel and T. Heinze, Green Chem., 2006, 8, 301–306 RSC.
- Y. Yang, L. Song, C. Peng, E. Liu and H. Xie, Green Chem., 2015, 17, 2758–2763 RSC.
- H. Chen, F. Yang, J. Du, H. Xie, L. Zhang, Y. Guo, Q. Xu, Q. Zheng, N. Li and Y. Liu, Cellulose, 2018, 25, 6935–6945 CrossRef CAS.
- K. N. Onwukamike, T. Tassaing, S. Grelier, E. Grau, H. Cramail and M. A. R. Meier, ACS Sustainable Chem. Eng., 2018, 6, 1496–1503 CrossRef CAS.
- Y. Cao, J. Wu, T. Meng, J. Zhang, J. He, H. Li and Y. Zhang, Carbohydr. Polym., 2007, 69, 665–672 CrossRef CAS.
- A. W. T. King, J. Jalomäki, M. Granström, D. S. Argyropoulos, S. Heikkinen and I. Kilpeläinen, Anal. Methods, 2010, 2, 1499–1505 RSC.
- R. Kakuchi, M. Yamaguchi, T. Endo, Y. Shibata, K. Ninomiya, T. Ikai, K. Maeda and K. Takahashi, RSC Adv., 2015, 5, 72071–72074 RSC.
- J. Wu, J. Zhang, H. Zhang, J. He, Q. Ren and M. Guo, Biomacromolecules, 2004, 5, 266–268 CrossRef CAS PubMed.
- O. Jogunola, V. Eta, M. Hedenström, O. Sundman, T. Salmi and J. P. Mikkola, Carbohydr. Polym., 2016, 135, 341–348 CrossRef CAS PubMed.
- R. M. Robertson, W. C. Thomas, J. N. Suthar and D. M. Brown, Green Chem., 2012, 14, 2266–2272 RSC.
- N. S. Allen, M. Edge, J. H. Appleyard, T. S. Jewitt, C. V. Horie and D. Francis, Polym. Degrad. Stab., 1987, 19, 379–387 CrossRef CAS.
- A. M. Sookne and M. Harris, Ind. Eng. Chem., 1945, 37, 478–482 CrossRef CAS.
- A. M. Sookne and M. Harris, Mechanical Properties of Cellulose Acetate as Related to Molecular Chain Length, 1943, vol. 30 Search PubMed.
- K. Kamide and M. Saito, Polym. J., 1985, 17, 919–928 CrossRef CAS.
- O. A. El Seoud, G. A. Marson, G. T. Ciacco and E. Frollini, Macromol. Chem. Phys., 2000, 201, 882–889 CrossRef CAS.
- H. Hanabusa, E. I. Izgorodina, S. Suzuki, Y. Takeoka, M. Rikukawa and M. Yoshizawa-Fujita, Green Chem., 2018, 20, 1412–1422 RSC.
- A. W. T. King, J. Asikkala, I. Mutikainen, P. Järvi and I. Kilpeläinen, Angew. Chem., Int. Ed., 2011, 50, 6301–6305 CrossRef CAS PubMed.
- Y. Yang, H. Xie and E. Liu, Green Chem., 2014, 16, 3018–3023 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc01508g |
| This journal is © The Royal Society of Chemistry 2021 |