
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA predictive toolset for the identification of effective lignocellulosic pretreatment solvents: a case study of solvents tailored for lignin extraction†
Ezinne C.
Achinivu‡
ab,
Mood
Mohan‡
 ab,
Hemant
Choudhary
ab,
Lalitendu
Das
ab,
Kaixuan
Huang
acd,
Harsha D.
Magurudeniya
ab,
Venkataramana R.
Pidatala
ac,
Anthe
George
ab,
Blake A.
Simmons
ac and
John M.
Gladden
*ab
ab,
Hemant
Choudhary
ab,
Lalitendu
Das
ab,
Kaixuan
Huang
acd,
Harsha D.
Magurudeniya
ab,
Venkataramana R.
Pidatala
ac,
Anthe
George
ab,
Blake A.
Simmons
ac and
John M.
Gladden
*ab
aDeconstruction Division, Joint BioEnergy Institute, 5885 Hollis Street, Emeryville, California 94608, USA. E-mail: jmgladden@lbl.gov; jmgladd@sandia.gov
bDepartment of Biomass Science and Conversion Technology, Sandia National Laboratories, 7011 East Avenue, Livermore, California 94551, USA
cBiological Systems and Engineering Division, Lawrence Berkeley National Laboratory, 1 Cyclotron Road, Berkeley, California 94720, USA
dKey Laboratory of Forestry Genetics & Biotechnology (Nanjing Forestry University), Ministry of Education, Nanjing 210037, People's Republic of China
First published on 2nd September 2021
Abstract
Pretreatment of lignocellulosic biomass is essential for efficient conversion into biofuels and bioproducts. The present study develops a predictive toolset to computationally identify solvents that can efficiently dissolve lignin and therefore can be used to extract it from lignocellulose during pretreatment, a process known to reduce recalcitrance to enzymatic deconstruction and increase conversion efficiency. Two approaches were taken to examine the potential of eleven organic solvents to solubilize lignin, Hansen solubility parameters (HSP) and activity coefficients and excess enthalpies of solvent/lignin mixtures predicted by COSMO-RS (COnductor like Screening MOdel for Real Solvents). The screening revealed that diethylenetriamine was the most effective solvent, promoting the highest lignin removal (79.2%) and fermentable sugar yields (>72%). Therefore, a COSMO-RS-based predictive model for the lignin removal as a function of number and type of amines was developed. Among the fitted models, the non-linear regression model predicts the lignin solubility more accurately than the linear model. Experimental results demonstrated a >65% lignin removal and >70% of sugar yield from several amine-based solvents tested, which aligned very well with the model's prediction. Finally, to help understand the dissolution mechanism of lignin by these solvents, quantum theory of atoms in molecules (QTAIM) and quantum chemical calculations (interaction energies and natural bond orbital (NBO) analysis) was performed and suggest that amines exhibit strong electrostatic interactions and hydrogen bonding strengths with lignin leading to higher lignin removal. Together, these computational tools provide an effective approach for rapidly identifying solvents that are tailored for effective biomass pretreatment.
1. Introduction
Lignocellulosic biomass is one of the primary natural resources that can be utilized as a renewable source of key intermediates to produce bioenergy, chemicals, and biomaterials.1,2 However, the distinctive physical and chemical diversity of its major constituents (cellulose, hemicellulose, and lignin), along with its complex microstructure and chemical linkages make it difficult to be effectively processed or fractionated into its various constituents. This key hurdle must be overcome for the development of sustainable biorefineries and a robust bio-based economy.1,3 To effectively accomplish this, the cross-linked matrix of lignin and hemicelluloses that surrounds the cellulose fibers must be disrupted.4 In particular, the extraction of lignin, the component that predominantly makes the biomass recalcitrant to deconstruction, is necessary to enable the efficient conversion of lignocellulosic intermediates into valuable products (fuels and/or chemicals).1,4–7There are several different pretreatment strategies that have been investigated for the separation of pure lignin and amongst them four chemical industrial processes are noteworthy: sulfite, kraft, soda and organosolv pretreatments.6,8 Out of these methods, the organosolv fractionation process has been widely accepted as one of the most promising techniques for biomass fractionation due to its comparatively low environmental impact, high delignification efficiency, and the diversity of products that are released.1,9,10 Organosolv pulping or fractionation is one of the methods of biomass fractionation that can produce high-quality cellulosic biofuels (via monomeric sugar fermentation), along with a high purity lignin. Unlike other pretreatment methods, the organosolv process is sulfur free, thereby producing lignin streams with a high level of purity for subsequent valorization.6,9,11 Additionally, this approach is particularly appealing because of the possibility of recovery and recycling of the organic solvent.12–14 In a typical organosolv process, an organic solvent is used to pretreat lignocellulosic biomass with or without the addition of external catalysts.13,14 Organic solvents such as short alkyl chain aliphatic alcohols (e.g., methanol, ethanol), polyols (e.g., glycerol, ethylene glycol, triethylene glycol), amines, alkanolamines, organic acids, acetone, dioxanes, and phenols have been widely used for the organosolv process.12,15 In most cases, the biomass pretreated by organic solvents is very susceptible to hydrolysis (via enzymes) and can be readily deconstructed to yield monomeric sugars.12,14,16 Cheng et al. studied the ability of 12 organic solvents including alcohols, alcohol ethers, lactones, and alkanolamines, to fractionate poplar and rice straw and reported at least 70% delignification.17 Zhao et al. also reported >90% conversion of the polysaccharides (cellulose/hemicelluloses) for alcohol-pretreated biomasses,18 and, Qin et al. reported that ethylenediamine can be applied to corn stover, resulting in glucose and xylose yields of 92% and 70% respectively after enzymatic digestion.19
Despite the promise for the organosolv processes, the near limitless possibilities for solvent selection have not been fully explored within the context of a robust multi-product biorefinery. Solvents like alcohols and diols have dominated the organosolv literature,9,15,20 yet many other possible solvents may exist with better performance and/or recyclability. The identification of these solvents would be greatly accelerated by the development of a computational toolset that could predict lignin solubilization and be systematic and efficient. Nevertheless, researchers still require guidelines to be established for the choice of successful solvent systems to become methodical. These guidelines or design rules should offer insights into the key chemical functionalities within a solvent that promote lignin dissolution, as well as the structural and conformational variations within a solvent group that can affect it. Lastly, it would be ideal if this toolset could aid in revealing the mechanistic factors that control lignin dissolution, which would help further refine the design/development of new and effective solvent systems for lignin.
Alongside experimental studies, molecular simulations have also been employed to understand the dissolution mechanism of biomass and its components. Researchers have adopted quantum chemical (QC) and molecular dynamics (MD) simulations, which provide fundamental insights of the molecular systems (e.g., lignin and ionic liquids). Solubility parameters such as Hildebrand21–23 and Hansen solubility parameters (HSP)17,24–27 and the COSMO-RS (COnductor like Screening MOdel for Real Solvents) model have been widely used to design and develop effective solvents for biomass delignification.28–31 Balaji et al.28 and Casas et al.29,30 screened various ionic liquids (ILs) to understand the lignin dissolution ability by predicting Hildebrand solubility parameters and thermodynamic parameters namely excess enthalpy and activity coefficient using COSMO-RS. Casas reported that the strong exothermic behavior of excess enthalpy and lower activity coefficients are beneficial for higher lignin dissolution.29,30 However, in both studies, only lignin's monomeric structures were employed as a model component. Later, Zhang et al.32 and Ji et al.33 performed quantum chemical (density functional theory) simulations to reveal the mechanism of lignin dissolution in imidazolium-based ionic liquids. It has been reported that the stronger H-bonding interaction between lignin and IL is responsible for the greater ability to dissolve lignin. These molecular simulation techniques can help in identifying new potential effective solvents for biomass pretreatment. However, the dissolution mechanism of lignin from lignocellulosic biomass using molecular solvents and the development of a predictive model for lignin removal has not yet been fully addressed. There is still a need to develop a predictive model for lignin removal, which can describe the solubility, while exploring the relationship between lignin dissolution and pretreatment effectiveness.
The present study attempts to develop both predictive models to identify the best solvents for lignin dissolution and multiscale simulation approaches tailored to provide mechanistic insights into how these solvents interact with lignin. First, HSP were used to screen a wide range of molecular solvents and identify ones that may be effective at lignin extraction from lignocellulose, which were then tested experimentally to determine the accuracy of the HSP predictions. Next, COSMO-RS calculations were performed to examine the same solvents and study the solvent/lignin mixture's thermodynamic properties such as excess enthalpy, activity coefficient, and sigma potentials. The excess enthalpy and activity coefficients were then used as a method to rank these solvents’ ability to dissolve lignin, and this approach was compared to using HSP and found to have better predictability. The initial screening revealed that solvents containing amine were effective at dissolving lignin, so a broader class of amine-based solvents were screened using excess enthalpy and activity coefficients of solvent/lignin mixtures, and several amines were tested experimentally for their ability to extract lignin from biomass and promote efficient enzymatic saccharification of the lignocellulosic polysaccharides. This data was used to validate a COSMO-RS-based predictive model that was developed for the lignin removal as a function of number and type of amines, which can be used for future screening efforts of amine-based solvents. Finally, to gain a deeper mechanistic understanding of how these solvents act to dissolve lignin, quantum chemical simulations are performed to study the solvent's interactions with lignin. Quantum theory of atom in the molecule (QTAIM), reduced density gradient (RDG), and natural bonding orbital (NBO) analysis were also carried out to investigate the strength and nature of H-bonding present in the lignin/molecular solvents. This analysis provided key insights into lignin dissolution and revealed that H-bonding between solvent and lignin is a major driver of lignin dissolution. The predictive toolset developed in this study combined with the mechanistic insights into lignin dissolution lay a strong foundation for rapidly identifying effective solvents for biomass pretreatment and developing cost-effective lignocellulosic conversion technologies.
2. Results and discussion
2.1. Screening of molecular solvents for lignin dissolution
Lignin is a complex and random polymer that is held together with strong bonds, such as ether linkages (carbon–oxygen) and carbon–carbon bonds, as well as, weak inter- and intra-molecular forces, such as hydrogen bonds.34 The complete (or near-complete) dissolution of lignin and its removal from lignocellulosic biomass is challenging owing to the scarcity of effective, efficient, economic, and environmentally benign solvents. Therefore, methods that enable rapid identification of effective lignin solvents could be instrumental in expanding the list of available solvents and identifying those that can be incorporated into cost-effective lignocellulose conversion technologies.Hansen solubility parameters (HSP) could possibly be used to provide an expedient route to identifying a short list of good potential solvents for lignin dissolution. HSP values for many molecular solvents have been determined and these values are readily available. In addition, Hansen and Björkman report relative energy difference (RED) values for many solvents compared to lignin.27 RED values can be used to estimate a solvent's ability to dissolve a solute. If the RED value is less than 1, then the affinity between the solute and the solvent is said to be higher and will result in a higher dissolution capacity. If the RED is greater than 1, the affinity between the solvent and solute is lower, resulting in poor dissolution. These RED values were used as an initial screen to identify a short list of solvents with RED values less than 1 that could be tested experimentally for lignin dissolution. The solvents were intentionally selected to have different molecular functionalities to maximize the chemical space covered. Functional groups included amines (diethylenetriamine), lactams (2-pyrrolidone), alcohols (dipropylene glycol, benzyl alcohol, furfuryl alcohol, guaiacol, and 2-ethoxyethanol), ethers (2-ethoxyethanol and dipropylene glycol), esters (isobutyl acetate and trimethyl phosphate), and aromatics (benzyl alcohol, furfuryl alcohol, guaiacol, and furfural) (Fig. 1A).
 | ||
| Fig. 1 Chemical structures of (A) the organic solvents screened (B) the lignin model used in this study for COSMO-RS calculations. | ||
While the Hansen and Björkman reported HSP and RED values for extracted woody lignin (14.9 for polar (δp), 16.9 for hydrogen-bonded (δh), and 21.9 for dispersion (δd) contributor) are readily available, they are also based on kraft lignin extracted from pine trees during paper pulping, which is unlikely to have the same properties as intact lignin within plant biomass.27,35 In addition, it should be noted that the reported HSP values cannot be assumed to be universal for all lignin samples as there is an extensive chemical diversity that exists between the lignins from different biomass sources. Considering these issues, we sought to identify an alternate set of lignin HSP that could be used to calculate lignin RED values to accurately rank the selected solvent's ability to dissolve lignin. Thielemans and Wool have reported the HSP values for lignin as δp = 13.7, δh = 11.7, and δd = 16.7.36 In their model, the solubility behavior of the modified lignin was described using the Flory–Huggins solubility theory, combined with the group contribution model developed by Hoy.36–38 This is one of the more practical lignin models available because it has contributions for a large number of functional groups, and accounts for a variety of structural features, which is important for a complex polymer like lignin.
To develop a more accurate set of RED values for lignin solvents, the Thielemans and Wool reported HSPs were used to calculate a new set of RED values for the same solvents identified by Hansen and Björkman using COSMOquick. In the new set of RED values, diethylenetriamine and trimethylphosphate have the lowest RED values and are expected to be the most suitable solvents for delignification, while the other solvents are expected to extract little to no lignin (Table 1). To validate these predictions, the grassy crop sorghum was pretreated with the solvents listed in Table 1 at 140 °C for 3 h at 20 wt% solids loading. Pretreatment with diethylenetriamine resulted in the highest lignin extraction (79.2%) as predicted, but pretreatment with trimethylphosphate resulted in an unexpectedly low-level lignin extraction (28.5%; Table 1). None of the other solvents were able to extract high levels of lignin from sorghum. Therefore, the calculated RED values do not appear to be very predictive for lignin extraction from lignocellulose, and screening solvents with these values will likely result in many false positives.
| Lignin/solvents | Lignin removal (%) | Hansen solubility parameters | REDa | REDb | |||
|---|---|---|---|---|---|---|---|
| δ D | δ P | δ H | δ T | ||||
| a Taken from Thielemans and Wool.36 b Hansen and Björkman26,34 reported RED values for lignin. | |||||||
| Lignina | — | 16.7 | 13.7 | 11.7 | 24.57 | — | — |
| Diethylenetriamine | 79.20 | 16.7 | 13.3 | 14.3 | 25.70 | 0.192 | 0.785 |
| Trimethylphosphate | 30.32 | 16.7 | 15.9 | 10.2 | 25.21 | 0.194 | 0.913 |
| 2-Pyrrolidone | 36.28 | 18.2 | 12.0 | 9.0 | 23.58 | 0.320 | 0.599 |
| 2-Ethoxyethanol | 28.51 | 16.2 | 9.2 | 14.3 | 23.49 | 0.386 | 0.925 |
| Dipropylene glycol | 24.07 | 16.5 | 10.6 | 17.7 | 26.42 | 0.494 | 0.831 |
| Furfuryl alcohol | 22.37 | 17.4 | 7.6 | 15.1 | 24.26 | 0.520 | 0.821 |
| Guaiacol | 26.68 | 18.0 | 7.0 | 12.0 | 22.74 | 0.525 | 0.761 |
| Furfural | 12.72 | 18.6 | 14.9 | 5.1 | 24.37 | 0.563 | 0.989 |
| Benzyl alcohol | 19.82 | 18.4 | 6.3 | 13.7 | 23.79 | 0.612 | 0.800 |
| Aniline | 16.75 | 20.1 | 5.8 | 11.2 | 23.73 | 0.762 | 0.897 |
| Isobutyl acetate | 18.29 | 15.1 | 3.7 | 6.3 | 16.77 | 0.862 | 1.470 |
It is unclear why the HSP values are not very predictive for lignin dissolution, but one explanation is that HSP values are used to measure the intermolecular affinity between solvent and solute but do not account for their intramolecular affinities, which can affect their behavior. Therefore, to better understand both the inter- and intramolecular interactions in a lignin/solvent mixture, COSMO-RS calculations were performed to study the mixture's thermodynamic properties such as excess enthalpy, activity coefficient, and sigma potentials. Typically, monomeric and dimeric structures of lignin have been used as lignin models to perform these molecular simulations.28–30 However, the monomeric and dimer structures of lignin do not directly represent the lignin molecule due to the absence of many different linkages present in lignin. Therefore, to obtain more realistic results, a lignin structure was generated based on the G/S ratio of grassy biomass and built by joining all the major lignin linkages (β-O-4, β–β, 4-O-5, α-O-4, and β-5) present in the native lignin (Fig. 1B). As mentioned earlier, lignin is a heterogeneous macromolecule, therefore, it is not possible to create a single lignin structure that can fully capture that heterogeneity or represent all lignins. However, many insights can be gained by simply ensuring coverage of the typical linkages found in lignin for the biomass used for pretreatment, which is this study is the grass sorghum.
Two thermodynamic properties may be useful in predicting lignin dissolution in a solvent, excess enthalpy (HE) and logarithmic activity coefficients (ln(γ)). The HE is a useful thermodynamic property for measuring the difference in the strength of interactions between dissimilar species (i.e., lignin–solvents) in the mixture. While the ln(γ) values are often used as a quantitative descriptor for the dissolution power of a solvent. In the literature, ln(γ) has been reported as the dominating parameter in deciding the capability of a solvent and has also been successfully employed in previous studies to predict the solubility of cellulose in ILs.29,39,40 Studies have reported that both HE and ln(γ) parameters are good indicators of cellulose and lignin solubility in a solvent.29,30,41 Therefore, both HE and ln(γ) parameters were calculated for the model grass lignin in the same set of solvents screened by HSP to determine if they can be used to accurately predict lignin dissolution (Fig. 2). The solvent diethylenetriamine was determined to possess significantly lower HE and ln(γ) values (i.e., more negative) than the other solvents, including trimethylphosphate, which had a similar RED value as diethylenetriamine and was therefore predicted to be a good lignin solvent. The COSMO-RS predicted results are much more consistent with the experimental lignin removal than the HSP RED values (Table 1), suggesting the use of COSMO-RS to predict HE and ln(γ) parameters of lignin in solvents is a more realistic method to determine a solvents’ ability to extract lignin from lignocellulose.
 | ||
| Fig. 2 COSMO-RS predicted excess enthalpy and logarithmic activity coefficients of lignin in molecular solvents. | ||
The goal of lignin extraction from lignocellulose is to increase the efficiency of enzymatic digestion of the plant polysaccharides. Therefore, enzymatic hydrolysis of sorghum pretreated with these solvents was performed using commercial enzyme cocktails (Fig. 3). The pretreated sorghum was first washed to remove the solvent to prevent interference with enzymatic digestion and the sugar yields were calculated (eqn (5)) based on the recovered solids (Fig. S1 and S2†). These results indicate that there is a direct correlation between low HE and ln(γ) parameters of lignin in molecular solvents and saccharification efficiency. Diethylenetriamine had the lowest HE and ln(γ) parameters and promoted the highest lignin removal and highest glucose and xylose yields of 72.6% and 78.6%, respectively (Fig. 3). All other solvents investigated were unable to extract significant quantities of lignin (≤36%) and had low sugar yields (≤18% glucose and ≤13% xylose), indicating that amine solvents pretreat biomass more effectively than the other functional group categories investigated. The direct correlation between lignin removal efficacy during pretreatment and saccharification efficiency has been observed in many other studies in the literature.17,18,42 Overall, these data suggest that a general rule can be postulated that solvents that enable high lignin solubility and subsequent plant polysaccharide digestibility will have a HE value for lignin of ≤−1.5.
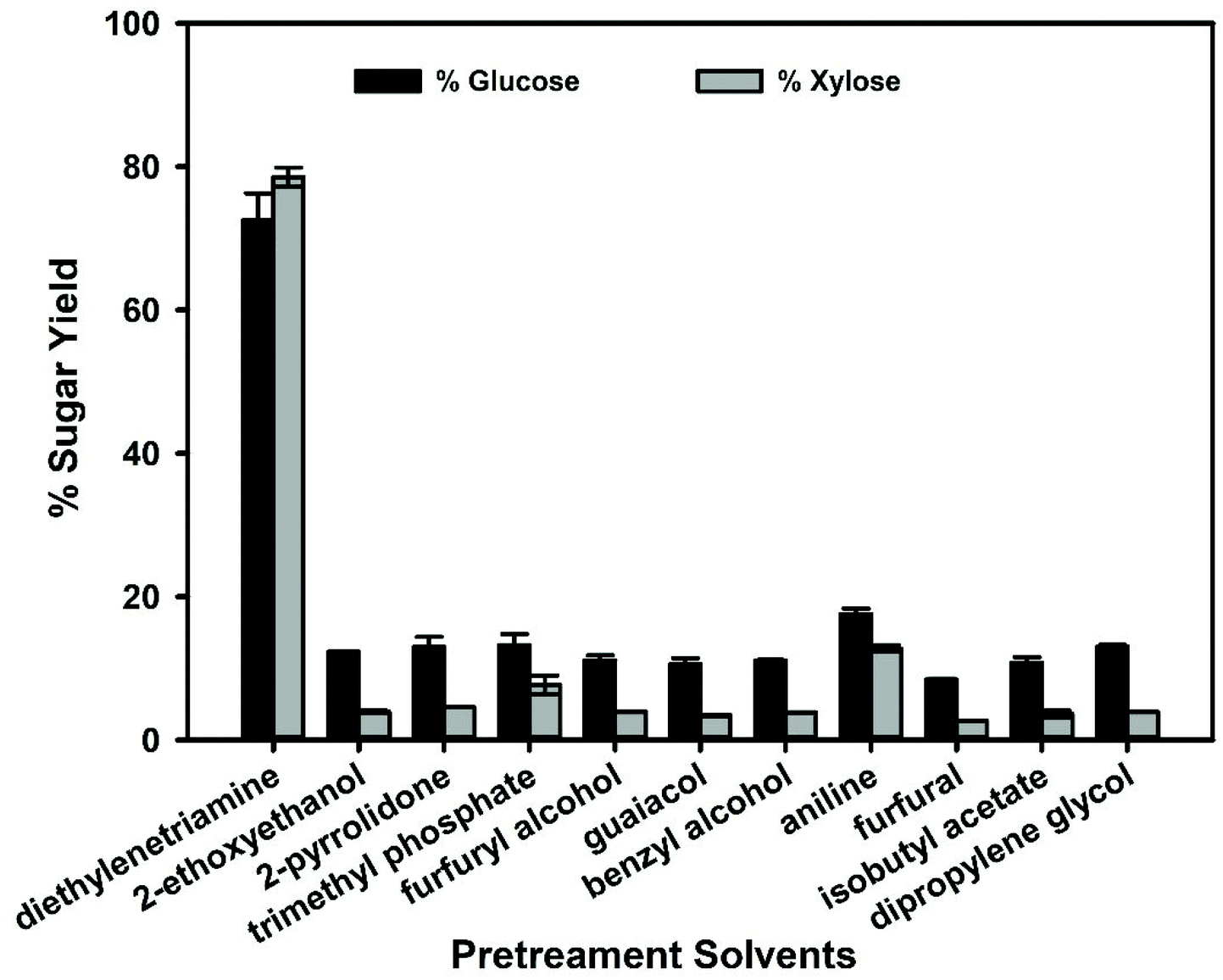 | ||
| Fig. 3 Glucose and xylose yields of pretreated sorghum after enzymatic hydrolysis. | ||
To better understand the experimental observations, sigma (σ)-potentials of the isolated molecules (solvents and lignin) were predicted using COSMO-RS. The σ-potential is a measure of the affinity of the system to a surface of polarity σ, which provides insights into a solvent's interactions with itself and with lignin. The σ-potential is divided into three regions: H-bond acceptor (σ > +0.01 e Å−2), H-bond donor (σ < −0.01 e Å−2), and non-polar (−0.01 e Å−2 < σ > +0.01 e Å−2) regions. Fig. S3a† depicts the σ-potentials of lignin and molecular solvents. On the negative side of screening charge density (SCD: σ > −0.01 e Å−2), the σ-potential (μ(σ)) value of diethylenetriamine is more negative than the other solvents, which implies that diethylenetriamine has more affinity to interact with the H-bond donor surfaces (blue color in Fig. S3b†) and has higher H-bond basicity, both of which would promote greater lignin solubility. In contrast, the μ(σ) value is positive in the region of large positive screening charge density values (σ > +0.01 e Å−2), which reflects diethylenetriamine's lack of H-bond donor surfaces (Fig. S3b†). Thus, the intramolecular interaction in diethylenetriamine is very weak, which enables the high interacting strength with the lignin. These results indicate that diethylenetriamine and potentially other amine-based solvents have an excellent ability to dissolve lignin from lignocellulosic biomass.
2.2. Development of COSMO-RS-based predictive model for lignin solubility in amines
The initial solvent screen identified diethylenetriamine as an effective pretreatment solvent. This prompted a more in-depth analysis of amine-based solvents, including a broad class of amines with one or more amine functional groups. A diverse set of amines was selected to help develop a deeper understanding of the key structural features in the amines that contribute to lignin extraction and to enable the development of an effective predictive model for amine-based dissolution of lignin. The new set of solvents was selected that vary in the number, type, and position of amine groups, as well as the carbon chain length of the molecule. The amines selected include 1,3-diaminopropane, 1,4-diaminobutane, 1,5-diaminopentane, 1,2-diaminopropane, spermidine, spermine, 2,2-dimethyl-1,3-propanediamine, ethylenediamine, and pentylamine. They were compared to diethylenetriamine as the baseline (see Fig. 4). | ||
| Fig. 4 Chemical structures of the amines evaluated in this study. | ||
Fig. 6 shows the COSMO-RS predicted HE and ln(γ) of lignin in these amine containing solvents. In the previous section, we established a general rule for lignin solubility and biomass digestibility as HE value ≤−1.5. Since all the selected amines have values below this cutoff, there is a strong indication that they will all be effective solvents for lignin extraction, except possibly 2,2-dimethyl-1,3-propanediamine with a borderline HE value of −1.57 (Fig. 5). Spermine and spermidine have the lowest HE and ln(γ), indicating that they may be able to extract and solubilize the greatest amount of lignin. As the number of carbon and amine groups increases, the HE and ln(γ) are predicted to be more negative. The polyamines (compounds with >2 amine groups) have HE ≤ −3.8 and are predicted to have the highest lignin solubility capacity, while the diamines, are expected to have an intermediate lignin dissolution ability −3.8 ≤ HE ≤ −2.5. Finally, the branched diamine (2,2-dimethyl-1,3-propanediamine) and monoamine (1-aminopentane) with −2.5 < HE < −1.5 are expected to have the lowest lignin extraction capacity.
 | ||
| Fig. 5 COSMO-RS predicted excess enthalpy and logarithmic activity coefficients of lignin. | ||
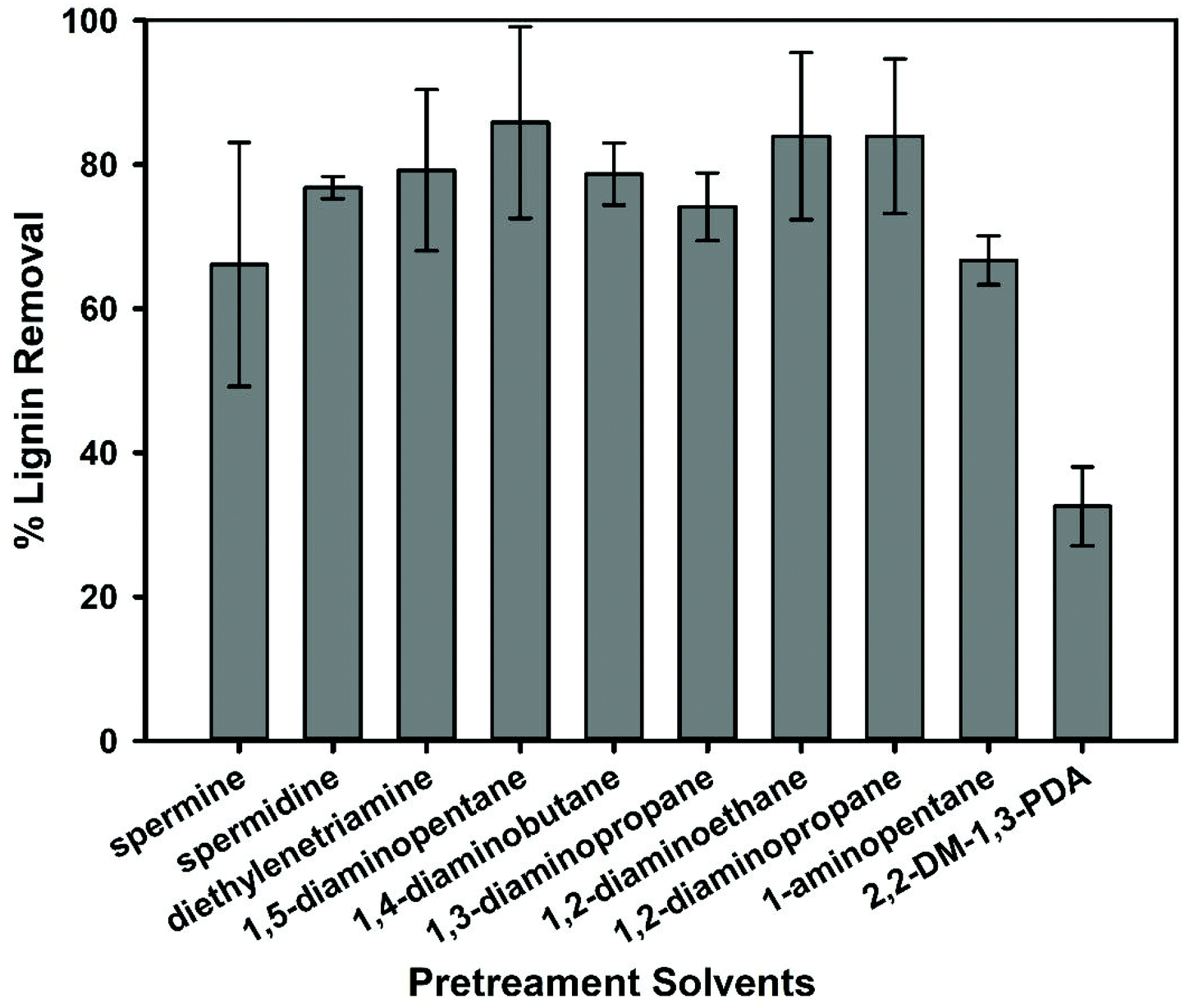 | ||
| Fig. 6 Solubility of lignin measured after biomass pretreatment in amines. Pretreatment conditions: 20% solids loading, 140 °C, and 3 h of reaction time. | ||
To confirm these predictions, biomass pretreatment experiments using these solvents were performed in a similar manner as the prior round of screening. Overall, the results indicate that all the poly and diamines were effective solvents. However, an examination of the experimental averages of lignin removal suggest that the polyamines do not actually have the highest lignin extraction capacity (66.1%–79.2%), but rather the diamines (74.1%–85.8%) (Fig. 6). This is not completely unexpected as both spermine and spermidine have a higher viscosity (η) than the diamines (Table S1†). Increases in a solvent's viscosity can limit effective mass transfer, which is known to have a negative effect on a pretreatment solvent's dissolution power.43–46 Therefore, solvents with low HE values should also be cross checked for high viscosity when screening. In addition to viscosity, the basicity and polarity of the solvent is another indicator of their ability to dissolve lignin. For example, 2,2-dimethyl-1,3-propanediamine has a lower viscosity than spermine and spermidine but only extracted a relatively low amount of lignin (32.6%). This is likely due to the lower polarity and basicity of this solvent (see Fig. S4†). Therefore, viscosity, polarity and basicity are important parameters to consider when attempting to predict dissolution. Since viscosities do have a notable impact, we demonstrated that we could predict the amine solvent viscosities using the COSMO-RS model and validated the predictions using the available experimental viscosity data for diethylenetriamine (Fig. S5†).47 This means COSMO-RS can be used to predict HE, ln(γ), and η to facilitate identification of good lignin solvents.
Using these COSMO-RS-predicted quantities, three different lignin solubility prediction models (linear and non-linear; eqn (1)–(3)) were developed for the amines and then validated. To develop a predictive model, the parameters excess enthalpy (correlated to the interactions), activity coefficient (related to the dissolution capability), viscosity (associated with the mass transfer rate), and the dissociation constant (pKa) related to the strength of acid/base were considered. The following equations were developed to predict the dissolution of lignin, but excluded pentylamine, which was used to validate the models.
Non-linear model 1:
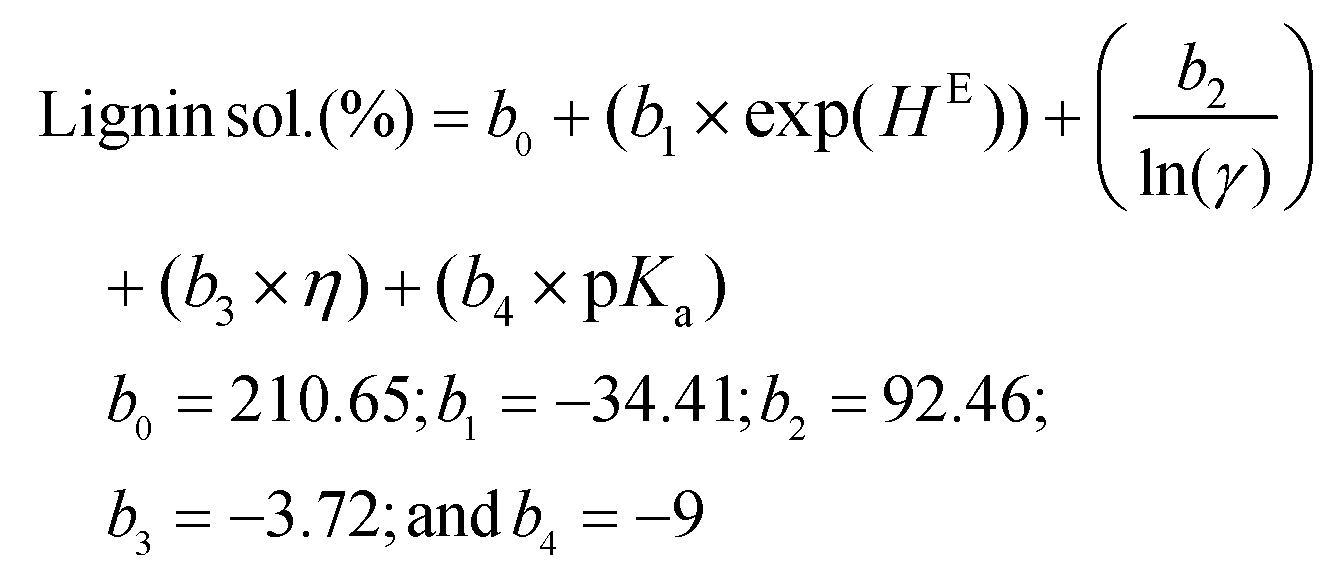 | (1) |
Non-linear model 2:
 | (2) |
Linear model 3:
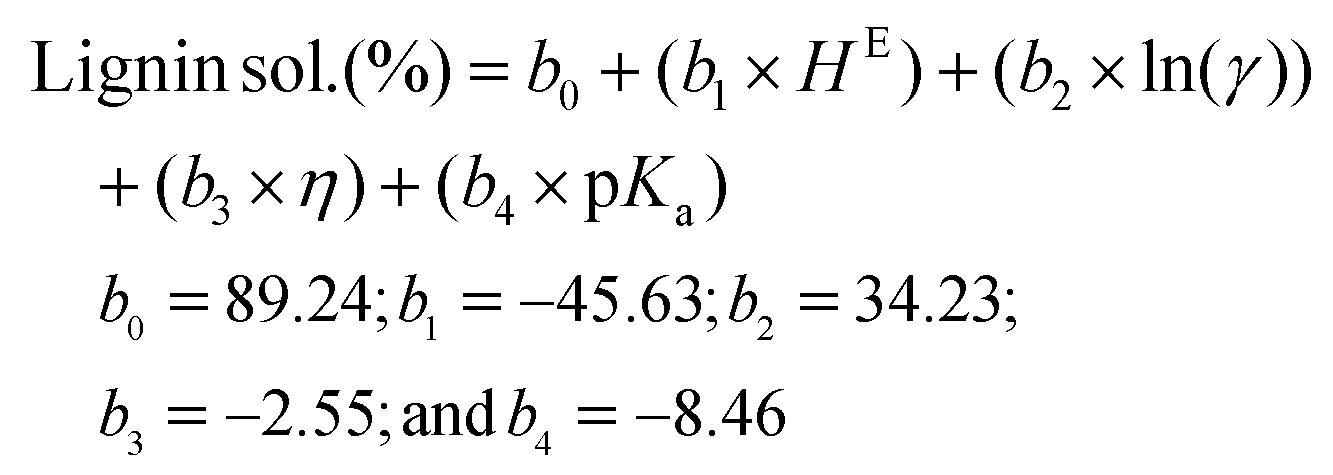 | (3) |
Here, the b0, b1, b2, b3, and b4 are the fit coefficients (i.e., constants). Experimental and predicted lignin solubility for amines based on these models are shown in Fig. 7 (model 1) and Fig. S6† (models 2 and 3). To assess the potential performance of the developed model equations, they were used to predict the solubility of lignin for pentylamine, which was excluded from the original training set. The predicted lignin solubility for models 1, 2, and 3 is 60.2%, 58.9%, and 53.1%, respectively while the experimental lignin solubility in pentylamine is 66.7 ± 3.4%. Further, the predictive models were also evaluated with lignin solubility data from the literature where Miscanthus biomass was pretreated with ethylenediamine at higher temperature (180 °C).48 The experimental lignin solubility was reported as ∼71 ± 4%, while the predicted solubility for models 1, 2, and 3 is 65.1%, 53.5%, and 96.1%, respectively. These data indicate that the non-linear model (1) predicts the lignin solubility more accurately than the linear models. This reveals that the relationship between the solvent type and lignin dissolution capacity (within the realm of biomass pretreatment) is not a simple linear relationship. When factors such as mass transfer and chemical reactivity are coupled, non-linear relationships have been more suitable at describing the experimental results. However, these developed lignin solubilities non-linear model could be applicable for amines only when HE ≤ 0.2 and ln(γ) ≤ −0.75.
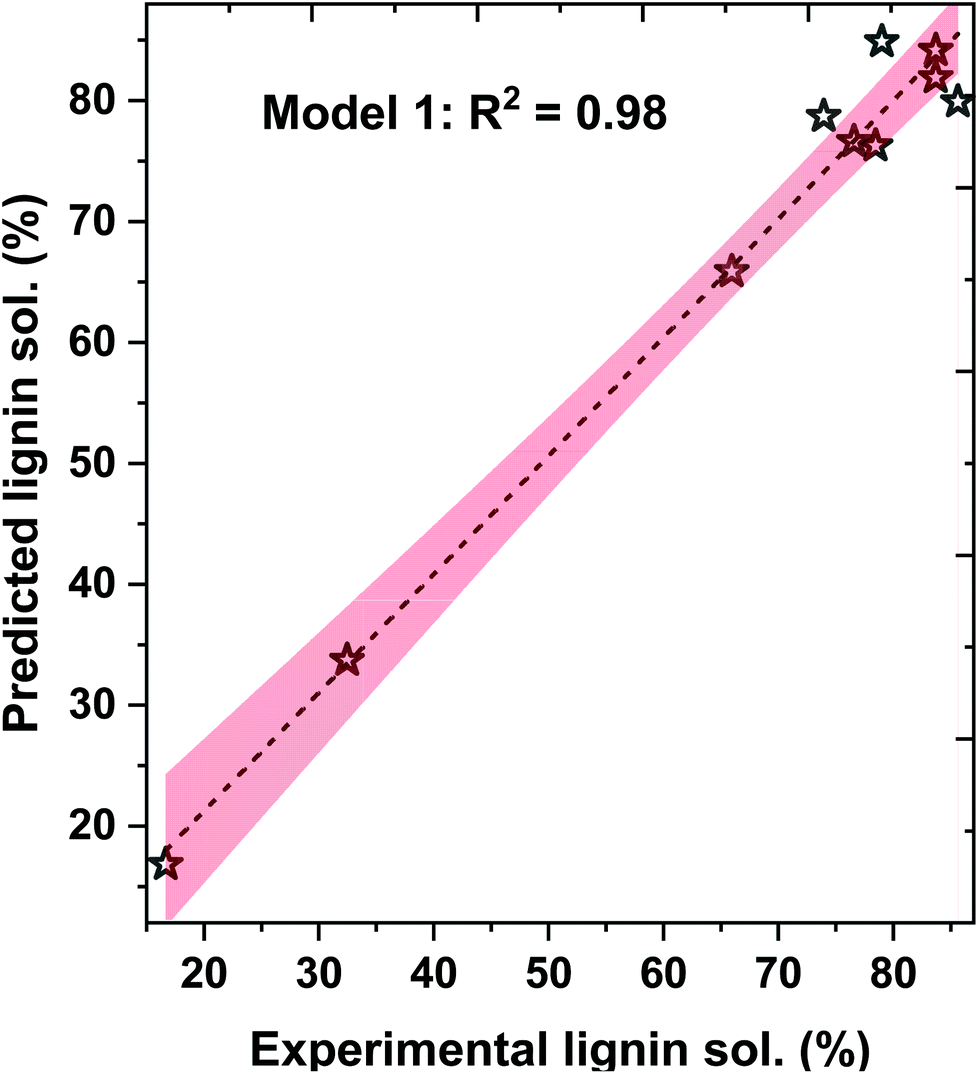 | ||
| Fig. 7 Experimental data and COSMO-RS-based model 1 predicted lignin solubility for amines with 95% confidence error band. | ||
2.3. Pretreatment with amines and enzymatic saccharification of residual biomass
As mentioned earlier, high lignin extraction has been directly correlated with an increase in enzymatic saccharification efficiency of pretreated lignocellulose. Therefore, we tested whether this correlation also holds for biomass pretreated with the new set of polyamines. Typically, efforts to extract lignin from biomass are not completely selective and some portions of polysaccharides are also extracted, so it is important to measure the solid recovery and composition post-pretreatment to fully understand the extent of this non-selective extraction and its impact on biomass deconstruction. For the set of amines studied, the solid recovery after pretreatment ranged from 61.5%–68.1% (Fig. S7†). The composition of the residual biomass was relatively similar across the different pretreatments, with an average loss of 27.1 ± 2.3% cellulose and 30.2 ± 1.9% hemicellulose (xylan and arabinan) during pretreatment (Fig. S7 and 8†). In many cases, lignin extraction is accompanied by some extent of hemicellulose/cellulose removal, so these results are not surprising. In an actual biorefinery, these extracted polysaccharides would be returned to the saccharification reaction after solvent recovery. Nevertheless, all amines investigated in this study afforded >70% glucose yields, with the highest glucose yield of ∼90% (Fig. 8). The glucose yields with these amines could be organized in the following order: 1,2-diaminopropane > 1,5-diaminopentane > 2,2-dimethyl-1,3-propanediamine > ethylenediamine > spermidine > 1,4-diaminobutane > pentylamine > 1,3-diaminopropane > diethylenetriamine > spermine. Similarly, >65% xylose yields were attained with these amines, with the highest xylose yield of ∼92% obtained also for 1,2-diaminopropane. The xylose yields followed the similar order as of glucose (Fig. 8). | ||
| Fig. 8 Glucose and xylose yields after enzymatic hydrolysis of pretreated sorghum with amines. | ||
The efficiency of sugar release for most amines was as expected based on the extent of lignin removal. Interestingly, 2,2-dimethyl-1,3-propanediamine extracted relatively low amounts of lignin (32.6%) but still permitted high sugar yields. This was very unexpected and does not fit well with previously reported studies of pretreatment solvents that selectively extract lignin. However, while lignin extraction can drive increases in saccharification efficiency, it is not the only factor that influences the release of sugars from biomass. Lignocellulose is complicated, and there are many possible outcomes of solvent-based pretreatment that can impact enzymatic sugar release, such polysaccharide extraction, or modification of macrostructure of biomass to increase the accessible surface area for enzymatic hydrolysis, etc. This is an interesting observation and suggests that amines are potentially acting to reduce the recalcitrance to enzymatic digestion by other mechanisms than lignin dissolution. Since this study is focused on lignin extraction, those other possibilities will be a focus area of future studies.
2.4. Cellulose structure and allomorphs
To study the structural changes in the polymers that occur during pretreatment, PXRD was utilized to determine the polymorph of cellulose and the proportions of crystalline and non-crystalline components in the biomass sample. The untreated sorghum displays diffraction patterns characteristic to the cellulose I polymorph, which is typical for native cellulose that is found in untreated lignocellulosic biomass49–51 (Fig. 9 and S9†) with a crystallinity content of 83.3%. Note: This is higher than that previously reported for sorghum (∼50–55%)52 due to the different radiation source that is used in these measurements (cobalt vs. copper). Nevertheless, we can compare the results qualitatively to determine the impact of pretreatment on the biomass. When 1,2-diaminoethane was utilized to pretreat the biomass, a change in the polymorph was observed. The identified peaks in the diffractogram match what has been previously reported as cellulose III49,50,53 and depicted a lower crystallinity index (70.3%) (Fig. 9 and S10†). This observation is in line with previous reports associated with amine pretreatment. Qin et al. showed that using 1,2-diaminoethane (also known as ethylenediamine) resulted in biomass modification to cellulose III especially when high temperatures are used followed by soaking and washing in ethanol (unlike water).51 On the other hand, the biomass recovered after pretreatment with the polyamine-spermine displayed mixed polymorphism. The diffractogram shows peaks analogous to both cellulose I and III, although most of the crystalline peaks are from cellulose I polymorph (Fig. 9 and S11†). Nevertheless, the recovered biomass has the highest amorphous character with a crystallinity index of 65.7%. | ||
| Fig. 9 X-ray diffraction profiles for untreated and treated sorghum including the relative percentage of each polymorph and crystallinity index (*measured by method or Segal et al.).59 | ||
The recovered biomass (after pretreatment) was subjected to saccharification and yielded an average sugar (glucose/xylose) of 68.6% and 85.4% for spermine and ethylenediamine, respectively. Although ethylenediamine pretreated biomass had a slightly higher crystallinity index, it permitted higher sugar yields. This indicates that while the level of crystallinity is known to impact biomass digestibility, sugar yields are impacted by a combination of factors, such as delignification. However, in conjunction with polymorph transformation, amine pretreatment can significantly reduce the crystallinity index, which can positively impact the cellulose digestibility. In their previous work, researchers reveal that ethylenediamine molecules penetrate the hydrophilic edges of the stacked sheets and enlarge cellulose III volume in the (010) direction.51 These changes have been reported to increase the enzymatic saccharification rate by 5 times,54 while other studies found that initial rates of digestion were strongly correlated with amorphous content, not the allomorph type.55,56
While celluloses having a higher amorphous content are typically easier to enzymatically digest, the accessibility of the plant cell-wall to the various glycoside hydrolases is also a very important factor in determining hydrolysis rate. While enzyme accessibility could be affected by crystallinity, it is also known to be affected by the lignin and hemicellulose contents/distribution, the particle size, and the porosity of the biomass. Since the lignin and hemicellulose removal for ethylenediamine were higher (83.9% and 32.5%) than that of spermine (66.1% and 29.7%), the greater sugar yields from the ethylenediamine pretreated biomass appear to also be influenced by their enhanced removal, potentially providing the enzymes greater access to the polysaccharides. It is well known that lignin plays a more important role than cellulose crystallinity on the digestibility of lignocellulose,57 and both the chemistry and physical barrier lignin provides can reduce the level of enzymatic hydrolysis.58 Therefore, lignin solubilization and removal remains the main focus area for optimization in this study.
2.5. Understanding the mechanistic behavior of lignin dissolution
To better understand the dissolution mechanism of lignin in the molecular solvents, quantum chemical (QC) simulations were performed. QC calculation is a prevailing computational technique for investigating the underlying molecular interactions of solvent/solute systems. These calculations can be used to investigate different types of interactions between solvents and solutes and help researchers to understand which interactions drive a solvent's ability to dissolve a solute. It is computationally difficult to conduct QC calculations with large macromolecules such as polymeric type lignin structure, so to simplify our analysis, we chose a representative lignin dimer, guaiacyl glycerol-β-guaiacyl ether (GGE) as a model molecule since it has the most common β-O-4 monomer–monomer linkage found in lignin. These complex solvent/solute systems can adopt many different conformations, which complicates analysis, so the most stable energy conformer structures of lignin GGE-molecular solvents were obtained and used to help understand several aspects of their interactions in more detail. According to Boltzmann distribution, the conformers with the lowest energy represent the maximum proportion of distribution. The lowest energy conformers are used to construct the initial structures for the geometry optimization between lignin–amines and lignin–organic solvents. All the lignin–amine/organic solvent conformers with relative energies are provided in the ESI (Fig. S12–S20 and Tables S2–S10†) and the most stable conformers are depicted in Fig. 10 and Fig. S21.† All the optimized conformers show that the ideal interaction site for the lignin–amine interaction is the α-OH, γ-OH, and phenol-OH group of lignin. In addition, interaction energies, non-covalent interactions, critical bonding topological characteristics, and an assessment of the strength of the electron donor–acceptor orbital interactions have also been performed for only the most stable lowest energy conformers. | ||
| Fig. 10 Optimized geometries for lignin GGE–amines (a) spermidine, (b) 1,5-diaminopentane, (c) diethylenetriamine, (d) 1,3-diaminopropane, and (e) 2,2-dimethyl-1,3-propanediamine. The H-bonds are indicated by dotted lines, the bond lengths are in Angstrom (Å) and given with corresponding atom numbers. The color scheme used for different atoms is C (gray), O (red), N (blue), and H (white), respectively. | ||
On a fundamental level, the total interaction energy of a solvent with lignin is decomposed into four chemically meaningful contributors: electrostatic, exchange-repulsion, induction, and dispersion (see Fig. 11). The electrostatic energy corresponds to the classic electrostatic interaction between the promoted fragments as they are brought into their positions in the final complexes, the term exchange repulsion accounts for Pauli repulsion between closed-shell fragments and is perpetually positive. The induction term, sometimes referred to as the orbital interaction or the polarization energy, arises from the orbital relaxation and the orbital mixing between the fragments (charge transfer). Dispersion energy which represents the amount of energy required to promote the fragments from their equilibrium geometry to the structure they will take up in the combined molecule. The stronger the interaction energy (more negative magnitude) between lignin and molecular solvents, the higher the anticipated lignin dissolution capacity. Analysis of these four contributors illustrates that the electrostatic interaction is the dominating attractive component between lignin and the amine, while dispersion and induction energies play a minor role in stabilizing the lignin–amine complexes. In the case of non-amine organic solvent systems, dispersion interactions are almost equal to the electrostatic interactions. Also, the induction interactions are significant in lignin–amine complexes relative to those calculated for the non-amine organic solvent complexes. This higher induction energy in lignin–amine complexes indicates that a substantial charge transfer occurred between lignin and amines. In terms of magnitude, all the energy components are greater in the lignin–amine complexes than the lignin–organic solvent complexes. The order of attractive interactions in lignin–amine complexes is electrostatic > dispersion > induction. Overall, the QC calculated interaction energies are in good agreement with the COSMO-RS predicted interactions and lignin solubility. The QC and COSMO-RS results can be used to suggest the solubility of lignin is lower in these types of organic solvents due to the weaker interactions.
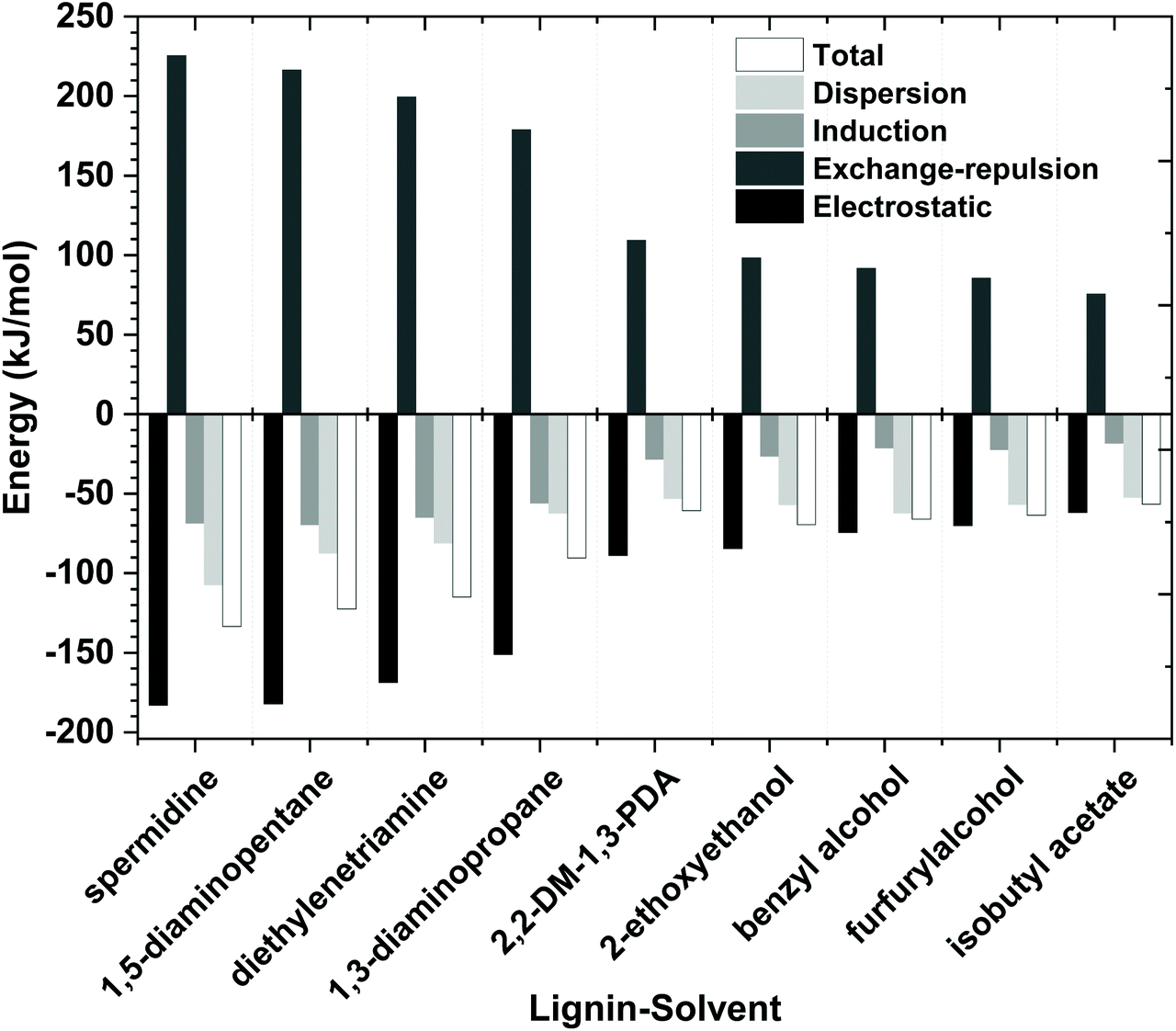 | ||
| Fig. 11 Functional group intramolecular symmetry adapted perturbation theory (FI-SAPT) decomposition of the non-bonded interaction energies between lignin and molecular solvents. | ||
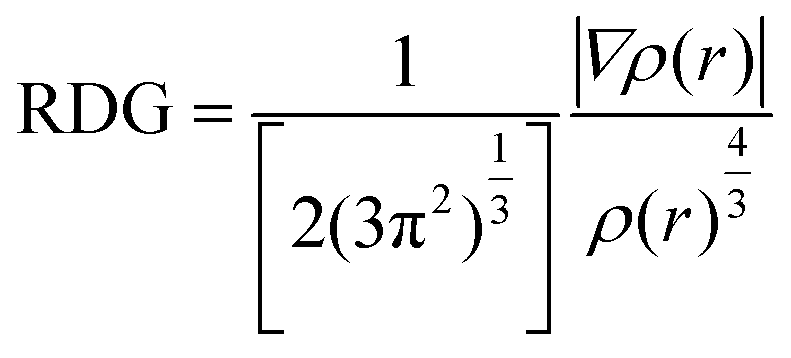 | (4) |
Regions where the RDG and electron densities are low representing the non-covalent interactions. Therefore, the isosurface of RDG at lower electron densities was used to visualize the position and nature of NCIs in 3D space. This is done by plotting the RDG vs. sign of second Hessian eigenvalue (λ2) multiplied with the electron density (ρ(r)) (sign(λ2). ρ(r)) in a scatter plot.
Two representative solvents, spermidine (amine) and furfuryl alcohol (non-amine), were selected to explore this analysis in detail (Fig. 12a and c) and plots of the remaining solvents are provided in Fig. S22.† Scanning across sign(λ2). ρ(r) from positive to negative values, there are several spikes in RDG scatter plot that correspond to the steric repulsion (red color), van der Waal (green color) interaction, and hydrogen bonding (blue color). In Fig. 12b and d, the interactions are visualized, and colored surfaces correspond to the respective colors in the respective NCI scatter plots. Examination of the NCI plots show that the amine-based solvents have spikes in the negative region of sign(λ2). ρ(r) that are more negative (O–H⋯N: −0.032 < sign(λ2). ρ(r) < −0.044) than the non-amine organic solvents (O–H⋯O: sign(λ2). ρ(r) > −0.03), which indicates that the strength of the H-bond interactions (blue region) is much stronger between lignin and amines. On the other hand, in the attractive region, multiple spikes are observed for lignin–amine interactions which are consistent with the geometrical analysis. Also, in the lignin–amine systems, the major steric repulsions (red color) occurred within the lignin molecule while it occurred between the lignin and solvent in the lignin–organic solvents, weakening their interaction and potentially explaining the lower lignin solubility observed in non-amine solvents (Fig. 12b and d).
 | ||
| Fig. 12 RDG scatter and NCI plots (isovalue 0.5 a.u.) of (a) lignin-spermidine (RDG), (b) lignin-spermidine (NCI), (c) lignin-furfuryl alcohol (RDG), and (d) lignin-furfuryl alcohol (NCI). The RDG/NCI plots are colored on a blue-green-red scale according to values of sign(λ2) ρ(r), ranging from −0.045 to 0.025. Blue indicates strong attractive interactions, green indicates the vdW interaction, and red indicated steric repulsions. The color scheme used for different atoms is C (cyan), O (red), N (blue), and H (white), respectively. | ||
Table 2 reports the ρ(r), ∇2ρ(r), and HBCP(r) of hydrogen bond critical points for the lignin–amine (O–H⋯N) and lignin–organic solvent (O–H⋯O) systems. For the lignin–amine system, the values of ρ(r) are in the range of 0.036–0.044 a.u., which is higher than the Koch and Popelier proposed range for electron densities for the hydrogen bond. Whereas, in the case of lignin–organic solvents, the values of ρ(r) and ∇2ρ(r) lies within the Koch and Popelier proposed ranges (0.002–0.035 a.u. and 0.014–0.139 a.u.). From these electron densities, the O–H⋯N bond between lignin and the amine solvent is predicted to be stronger than the O–H⋯O bond between lignin and non-amine solvents. The Laplacian electron densities at the BCP show positive values for both lignin–amines/organic solvent systems, implying that the characteristics of H-bonding interactions are non-covalent. Further examining energy densities (HBCP(r)), they are negative for O–H⋯N and positive for O–H⋯O at the BCP, indicating the amine–lignin interactions are more covalent in nature while the non-amine solvents have more non-covalent or weak interactions. These calculations (ρ(r), ∇2ρ(r), and HBCP(r)) help in explain why the organic solvents exhibit lower interactions with lignin.
| Amine/organic solvent | H-Bond | Distance (Å) | ρ(r) | ∇ 2 ρ(r) | H BCP(r) | E HB (kJ mol−1) |
|---|---|---|---|---|---|---|
H
BCP(r) = GBCP(r) + VBCP(r); 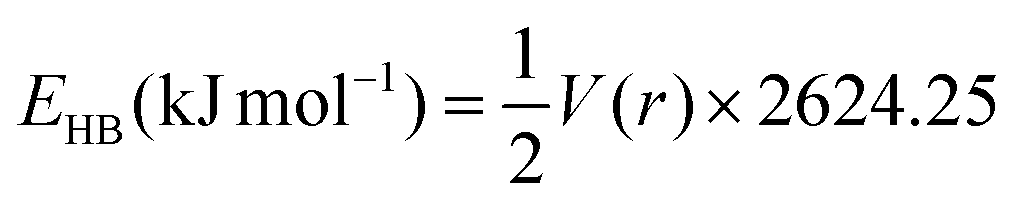 ; GBCP(r) is the Lagrangian kinetic energy density; VBCP(r) is the potential energy density. ; GBCP(r) is the Lagrangian kinetic energy density; VBCP(r) is the potential energy density. |
||||||
| Spermidine | O23–H42⋯N43 | 1.80 | 0.0436 | 0.0957 | −0.0059 | −46.84 |
| O13–H32⋯N47 | 1.87 | 0.0382 | 0.093 | −0.0032 | −38.84 | |
| N52–H64⋯O8 | 2.28 | 0.0128 | 0.0435 | 0.0013 | −10.76 | |
| 1,5-Diaminopentane | O23–H43⋯N44 | 1.79 | 0.045 | 0.0957 | −0.0067 | −48.94 |
| O13–H32⋯N50 | 1.85 | 0.0393 | 0.0947 | −0.0037 | −40.68 | |
| N44–H52⋯O20 | 2.38 | 0.0124 | 0.0424 | 0.0011 | −10.89 | |
| Diethylenetriamine | O11–H31⋯N50 | 1.86 | 0.038 | 0.095 | −0.0032 | −39.49 |
| O23–H43⋯N44 | 1.79 | 0.045 | 0.099 | −0.0062 | −48.94 | |
| 1,3-Diaminopropane | O23–H43⋯N48 | 1.79 | 0.0449 | 0.0994 | −0.0065 | −49.73 |
| O13–H32⋯N44 | 1.92 | 0.0334 | 0.0898 | −0.0011 | −32.28 | |
| N44–H55⋯O20 | 2.37 | 0.0124 | 0.0441 | 0.0014 | −10.89 | |
| 2,2-Dimethyl-1,3-propaneamine | O11–H31⋯N44 | 1.90 | 0.0347 | 0.0895 | −0.0018 | −34.12 |
| 2-Ethoxy ethanol | O11–H31⋯O46 | 1.88 | 0.030 | 0.101 | 0.0008 | −31.10 |
| O57–H59⋯O20 | 2.01 | 0.020 | 0.078 | 0.0023 | −19.42 | |
| Benzyl alcohol | O51–H59⋯O13 | 1.87 | 0.0306 | 0.1049 | 0.0008 | −32.28 |
| Furfuryl alcohol | O50–H56⋯O11 | 1.82 | 0.033 | 0.1184 | 0.0006 | −37.13 |
| Isobutyl acetate | O23–H43⋯O46 | 1.92 | 0.0247 | 0.0953 | 0.0023 | −25.19 |
Additionally, hydrogen bonding energies (EHB) are also calculated using the potential energy densities VBCP(r). The hydrogen bonding energy of O–H⋯N (lignin–amine) is much stronger than the O–H⋯O bond energy. It is worthwhile to mention that 2,2-dimethyl-1,3-propanediamine showed similar electronic properties (ρ(r), ∇2ρ(r), and EHB) as the organic solvents, which is consistent with the low solubility of lignin observed with this solvent. In addition to the discussions, there is a strong correlation between Hessian second eigenvalue (λ2) and EHB values (Fig. S23†). The lower the λ2 value, the stronger the hydrogen bonding energy. Overall, the electronic properties clearly indicate that the amine solvents are highly effective solvents for the lignin removal and have much stronger hydrogen bonding energies than the organic solvents.
| Amine/organic solvent | H-Bond interaction | π-Stacking interaction | ||||
|---|---|---|---|---|---|---|
| Donor | Acceptor | E (2)* (kJ mol−1) | Donor | Acceptor | E (2)* (kJ mol−1) | |
| Spermidine | LP (1) N43 | σ* O23–H42 | 101.87 | π C18–C21 | σ* C45–H65 | 1.46 |
| LP (1) N47 | σ* O13–H32 | 71.39 | π C2–C5 | σ* C50–H59 | 1.42 | |
| LP (2) O8 | σ* N52–H64 | 5.31 | ||||
| 1,5-Diaminopentane | LP (1) N44 | σ* O23–H43 | 107.73 | π C18–C21 | σ* C46–H56 | 0.71 |
| LP (1) N50 | σ* O13–H32 | 86.32 | ||||
| Diethylenetriamine | LP (1) N44 | σ* O23–H43 | 101.96 | π C12–C17 | σ* C49–H60 | 1.30 |
| LP (1) N50 | σ* O11–H31 | 80.34 | π C19–C21 | σ* C47–H57 | 2.38 | |
| 1,3-Diaminopropane | LP (1) N48 | σ* O23–H43 | 102.92 | π C19–C21 | σ* C46–H51 | 1.09 |
| LP (1) N44 | σ* O13–H32 | 66.92 | π C16–C18 | σ* C45–H50 | 0.59 | |
| 2,2-Dimethyl-1,3-propaneamine | LP (1) N44 | σ* O11–H31 | 69.92 | π C16–C18 | σ* C52–H64 | 2.51 |
| LP (1) N48 | σ* C22–H42 | 8.53 | ||||
| 2-Ethoxy ethanol | LP (1) O46 | σ* O11–H31 | 13.68 | π C12–C17 | σ* C45–H49 | 0.88 |
| LP (2) O46 | σ* O11–H31 | 30.28 | ||||
| LP (1) O20 | σ* O57–H59 | 8.11 | ||||
| LP (1) O20 | σ* O57–H59 | 10.66 | ||||
| Benzyl alcohol | LP (1) O13 | σ* O51–H59 | 8.28 | π C4–C9 | π* C47–C48 | 1.17 |
| LP (2) O13 | σ* O51–H59 | 41.23 | π C47–C48 | σ* C14–H35 | 1.21 | |
| Furfuryl alcohol | LP (1) O11 | σ* O50–H56 | 13.01 | π C4–C9 | π* C45–C46 | 1.13 |
| LP (2) O11 | σ* O50–H56 | 39.02 | π C47–C48 | π* C4–C9 | 1.17 | |
| Isobutyl acetate | LP (1) O46 | σ* O23–H43 | 17.90 | π C16–C18 | σ* C50–H60 | 1.42 |
| LP (2) O46 | σ* O23–H43 | 11.33 | ||||
In addition, the π–π and CH–π stacking interactions in the lignin–solvent complexes were also examined, where applicable. π–π interactions are observed in the benzyl alcohol and furfuryl alcohol lignin complexes whereas CH–π stacking interactions were observed in the isobutyl acetate, 2-ethoxyethanol, and amine–lignin complexes. Overall, the strength of CH–π interactions was predicted to be relatively stronger than π–π. However, compared to LP (O) → σ* orbital energies, the CH–π stacking interactions are not significant and therefore less relevant to lignin dissolution. Overall, the QC calculations indicate that hydrogen bonding interactions are playing a vital role in the dissolution of lignin. Organic solvents also exhibit significant hydrogen bonding energies, but due to the strong steric repulsions and weaker polarity, the net result is lower lignin solubility.
3. Materials and methods
3.1. Materials
In this study, Sorghum (Sorghum bicolor) biomass was obtained from Idaho National Labs (Idaho Falls, Idaho, USA) and used as a raw material. The acquired biomass samples were milled (Thomas-Wiley Model 4, Swedesboro, NJ) and sieved to attain a homogeneous particle size of 2 mm, and finally oven-dried (40 °C, 24 h). The resulting biomass was then placed in an airtight plastic bag and stored in a cool dry place until further use. The following solvents were purchased from Sigma Aldrich (St Louis, MO) and used as received: 2,2-dimethyl-1,3-propanediamine (purity: 99%), ethylenediamine (≥99), spermidine (≥99%), spermine (≥99%), 1,2-diaminopropane (99%), 1,3-diaminopropane (≥99%), 1,4-diaminobutane (99%), 1,5-diaminopentane (95%), diethylenetriamine (99%), pentylamine (99%). 2-Ethoxyethanol (≥99%), 2-pyrrolidone (≥99%), trimethyl phosphate (97%), furfuryl alcohol (98%), guaiacol (≥98%), benzyl alcohol (≥99%), isobutyl acetate (99%), aniline, (≥99.5%), furfural (99%), dipropylene glycol (99%), citric acid (ACS reagent ≥99.5%), sodium citrate tribasic dihydrate (ACS reagent, ≥99.0%) and sodium azide. Sulfuric acid (72% and 95–98%) was purchased from VWR, and sugar standards glucose (≥99.5%), xylose (≥99%), and arabinose (≥98%) were procured from Sigma-Aldrich for high-performance liquid chromatography (HPLC) analysis. Commercial cellulase (Cellic® CTec3) and hemicellulase (Cellic® HTec3) mixtures were provided by Novozymes, North America (Franklinton, NC).3.2. Pretreatment of sorghum biomass
The pretreatment of biomass sorghum was carried out using the conventional method that involves early separation (or washing) to remove the solvent after pretreatment. In a typical experiment, 1 g of the biomass was mixed with the 4 g of solvent and loaded into an ace pressure tube (50 mL, Ace Glass Inc., Vineland, NJ) and mixed well prior to the experiments. The pretreatment experiments were performed at 140 °C for 3 h of reaction time at a solid loading of 20 wt%. Post pretreatment, 25 mL of ethanol was added to the slurry before being transferred to a 50 mL Falcon tube and centrifuged at 4500 rpm to separate solids from liquid. The recovered solid was further washed with a mixture of ethanol and water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to remove any residual organic solvents. Finally, the recovered solid fractions were lyophilized before enzymatic hydrolysis (EH) and compositional analysis (CA). All the experiments were performed in duplicate, and the average values are reported here. The solid recovery (%SR) after pretreatment was calculated based on the following equation.
:1) to remove any residual organic solvents. Finally, the recovered solid fractions were lyophilized before enzymatic hydrolysis (EH) and compositional analysis (CA). All the experiments were performed in duplicate, and the average values are reported here. The solid recovery (%SR) after pretreatment was calculated based on the following equation. | (5) |
3.3. Enzymatic hydrolysis
The enzymatic saccharification of pretreated and untreated biomass was carried out using commercially available enzymes, Cellic® Ctec3 and Htec3 (9:1 v/v) from Novozymes, at 50 °C in a rotary incubator (Enviro-Genie, Scientific Industries, Inc.). All reactions were performed at 5 wt% biomass loading in a 15 mL centrifuge tube (using 0.25 g of the pretreated or untreated biomass). The pH of the mixture was adjusted to 5 with 50 mM sodium citrate buffer supplemented with 0.02 wt% sodium azide to prevent microbial contamination. The total reaction volume included a total protein content of 10 mg per g biomass. The amount of sugars released was analyzed on an Agilent HPLC 1260 infinity system (Santa Clara, California, United States) equipped with a Bio-Rad Aminex HPX-87H column (300 × 7.8 mm2) and a refractive index detector. An aqueous solution of sulfuric acid (4 mM) was used as the eluent (0.6 mL min−1, column temperature 60 °C). All enzymatic saccharification was conducted in duplicate. The sugar yield was calculated as an overall process yield using the formula below (eqn (6)), which accounts for sugars/oligosaccharides lost during pretreatment/washing. | (6) |
3.4. Compositional analysis
The compositional analysis of pretreated and untreated biomass sorghum was performed to determine the glucan, xylan, lignin and ash content following the two-step acid hydrolysis procedure previously described by NREL.62 In summary, 300 mg of the dry extractive-free biomass was exposed to 3 mL of 72% w/w H2SO4 and incubated at 30 °C for 1 h. Subsequently, the mixture was taken through secondary hydrolysis at 4% w/w H2SO4 at 121 °C for 1 h. After the two-step acid hydrolysis, the hydrolysates were filtered using medium porosity filtering crucibles. The filtrates were spectrophotometrically analyzed for the acid-insoluble lignin (ASL) (NanoDrop 2000, Thermo Fisher Scientific, Waltham, MA) using the absorbance at 240 nm. Additionally, monomeric sugars (glucose and xylose) were determined by HPLC using an Agilent 1200 series instrument equipped with a refractive index detector and Bio-Rad Aminex HPX-87H column, coupled with a guard column assembly. Product separation was obtained at 60 °C with 4 mM H2SO4 as a mobile phase at a flow rate of 0.6 mL min−1. Finally, the Klason lignin (acid-insoluble lignin – ASL) was determined gravimetrically by subtracting the weight of the oven-dried residual solids (105 °C) and the ash content (575 °C). All compositional analyses were conducted in duplicate. The amount of lignin removed can be calculated using the formula below (eqn (7)). Note: %lignin = %AIL + %ASL.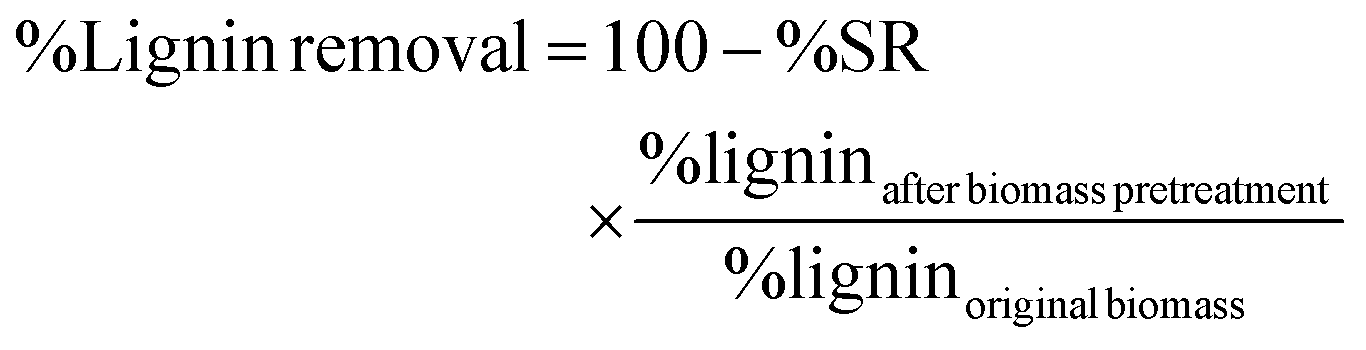 | (7) |
3.5. Structural characterization (P-XRD analyses)
The powder X-ray diffraction (P-XRD) analysis were performed to study the crystallinity behavior of cellulose before and after biomass pretreatment. The cellulose allomorph and crystallinity index was measured using in the 2θ range from 5 to 60° and an exposure time of 300 s with a voltage of 40 kV and current of 20 mA using Co-Kα radiation (λ = 1.7891 nm). It is important to mention that the diffraction angle was converted to the analogous Cu-Kα (λ = 1.5418 nm) for peak identification and analysis using previously assigned spectra. The peak deconvolution of the resulting diffractogram was performed using software PeakFit (SeaSolve Software Inc.). Gaussian/Lorentzian functions were applied in curve fitting analysis and iterations were repeated until the maximum F number was obtained. In all cases, the F number was >10000, which corresponds to a R2 value >0.99. Estimation of the content of cellulose I, cellulose III, and amorphous cellulose in the cellulosic samples was established by using the peak areas of cellulose I, cellulose III, and amorphous cellulose, respectively. According to previously defined diffractogram, the Bragg angles of peak (110), (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0), (020), and (004) belonging to cellulose I are ∼[14.8°, 16.3°, 22.3°, and 34.5°], respectively. The Bragg angles of peak (110) and (020) belonging to cellulose III are 11.3° and 20.0°, respectively. The Bragg angle of the amorphous peak is around 19.5–20.5°.49–51 The crystallinity index was also calculated according to the method of Segal et al., where the ratio of the height of the 002 peak (I002) and the height of the minimum (IAM) between the 002 and the 101 peaks.50,59
0), (020), and (004) belonging to cellulose I are ∼[14.8°, 16.3°, 22.3°, and 34.5°], respectively. The Bragg angles of peak (110) and (020) belonging to cellulose III are 11.3° and 20.0°, respectively. The Bragg angle of the amorphous peak is around 19.5–20.5°.49–51 The crystallinity index was also calculated according to the method of Segal et al., where the ratio of the height of the 002 peak (I002) and the height of the minimum (IAM) between the 002 and the 101 peaks.50,59
3.6. Computational details
After a successful geometry optimization step, further, the COSMO file was generated using the BVP86/TZVP/DGA1 level of theory.41,64,65 The ideal screening charges on the molecular surface were computed using the same level of theory i.e., BVP86 through the “scrf = COSMORS” keyword.66,67 The generated COSMO files were then used as an input in the COSMOtherm (version 19.0.1, COSMOlogic, Leverkusen, Germany) package.68,69 BP_TZVP_19 parametrization was used to predict the sigma potentials, viscosity, excess enthalpy, and logarithmic activity coefficients of the isolated and mixture of molecular systems. In COSMO-RS calculations, the molar fraction of lignin was set as 0.2, whereas the molar fraction of solvents was set to 0.8 to mimic the experimental pretreatment setup.
The excess enthalpy of a binary mixture can be predicted by using the following expression (eqn (8)):65
| HEM = ∑xiHEi = ∑xi[H(i,mixure) − H(i,pure)] | (8) |
| HEM = HEM(misfit) + HEM(H-bond) + HEM(vdW) | (9) |
The activity coefficient of component i is associated with the chemical potential μi and expressed as70 (eqn (10)):
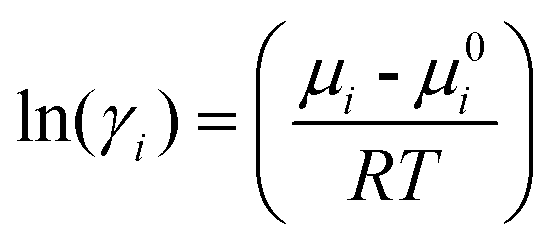 | (10) |
From QC calculations, the interaction energy (ΔEtotal) is calculated by following the eqn (11).39,73
| I.E. (kJ mol−1) = Ecomplex − ∑(Eisolated molecules) | (11) |
| ΔEtotal (kJ mol−1) = ΔEelec + ΔEexch + ΔEind + ΔEdisp | (12) |
NBO analysis was employed to understand the strength of the electron donor–acceptor interactions involved in the system. The electron donor i–j acceptor delocalized stabilization energies (E(2)*) were estimated from the second-order perturbation approach and are expressed in the eqn (13) below.74,75
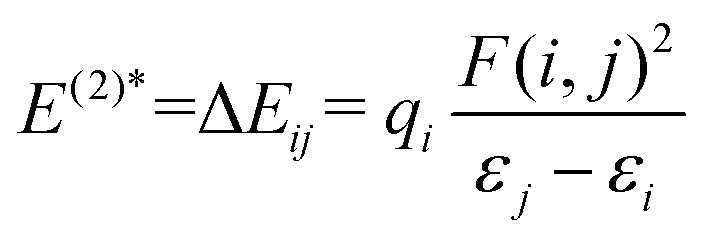 | (13) |
In addition to the NBO analysis, QTAIM76 analysis at the bond critical point (BCP) was performed to understand the strength (electron density, ρ(r)), characterization (Laplacian energy density (∇2ρ(r)), and nature (energy density H(r)) of the H-bond presented in lignin-molecular solvent systems using AllAIM (version 19.10.12) software.77 The H-bond energy (EHB) was calculated using Espinosa's equation: EHB = 1/2 × VBCP(r), in which VBCP(r) is the potential energy density at the BCP of the measured H-bond.78 Further, to examine the nature of intermolecular interactions in the complex systems, reduced density gradient non-covalent interactions (RDG-NCI) were analyzed using Multiwfn79 and VMD80 packages.
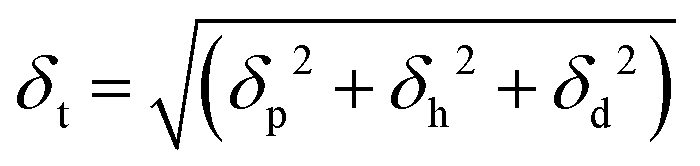 | (14) |
In addition to δt, Hansen also proposed a parameter called relative energy difference (RED) that correlates the interaction between a solute and a solvent. The RED is defined as the ratio between the radius of interaction (Ra) to the 3D sphere radius of the solute (R0) as shown in the below eqn (15) and (16).27,35,84,85
 | (15) |
 | (16) |
If the RED < 1, then the affinity of the solvent towards the solute is said to be higher. While If the RED > 1, the affinity between the solvent and solute is lower.
4. Conclusions
The current work demonstrates an effective framework for (1) discovering and predicting high performing solvents for the dissolution of lignin and (2) understanding the mechanistic factors that control the lignin dissolution capacity of a solvent. This framework relies on multiscale simulation approaches to develop a predictive model to identify potential solvents for lignocellulosic biomass pretreatment, which are then experimentally verified. To initiate the development of this framework, several molecular solvents were screened using HSP and COSMO-RS models and amines were predicted to be effective solvents. Next, amines with different chemical functionalities were experimentally tested and shown to promote higher lignin removal and fermentable sugar yields than several non-amine solvents. The solvents 1,5-diaminopentane and 1,2-diaminopropane were found to be the most effective at biomass pretreatment, extracting >82% of lignin from biomass and enabling >88% yields of fermentable sugars. COSMO-RS was then used to develop a predictive model for lignin removal based on the several influential quantities: HE, ln(γ), η, and pKa. Comparison of the predictive model and experimental results shows that they are in excellent agreement, with a deviation of less than 10%.Once effective lignin solvents were identified, QC calculations and QTAIM analysis were employed to understand the mechanism that drive the lignin solvent interactions and determine why the amines are more effective lignin extraction solvents than the other non-amine solvents examined in this study. QC and QTAIM analysis indicate that amines that form multiple strong H-bond interactions with lignin can extract high amounts of lignin from biomass. The use of computational platforms to both develop predictive models to identify effective pretreatment solvents and to then gain deeper insights into the mechanism of lignin dissolution by these solvents will lead to the rapid expansion of the list of solvents that can be used for efficient lignocellulose pretreatment and deconstruction. There are numerous considerations that must be made to effectively integrate a pretreatment technology into a biorefinery, including effectiveness on a broad range of feedstocks, fractionation of lignocellulose components, solvent cost, solvent recycling, generation of biomass-derived enzyme and microbe inhibitors, etc. Recommendations for subsequent research will study these additional components of process development and the expansive list of pretreatment solvents identified though the predictive framework established in this study will provide researchers and industry more options to consider in the development of highly efficient, low-cost lignocellulose conversion technologies.
Conflicts of interest
The authors declare the following competing financial interest(s): EA, MM, HC, LD, KH, HDM, VRP, BAS, and JMG are named inventors on at least one related patent application. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.Acknowledgements
This work was part of the DOE Joint BioEnergy Institute (http://www.jbei.org) supported by the US Department of Energy, Office of Science, Office of Biological and Environmental Research, through contract DE-AC02-05CH11231 between Lawrence Berkeley National Laboratory and the US Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. XRD characterization was conducted with the help of Dr Tevye Kuykendall at the Molecular Foundry in Lawrence Berkeley National Laboratory.Notes and references
- M. Galbe and O. Wallberg, Biotechnol. Biofuels, 2019, 12, 294 CrossRef PubMed.
- M. C. Y. Chang, Curr. Opin. Chem. Biol., 2007, 11, 677–684 CrossRef CAS PubMed.
- S. I. Mussatto and G. M. Dragone, in Biomass fractionation technologies for a lignocellulosic feedstock based biorefinery, Elsevier, 2016, pp. 1–22 Search PubMed.
- M. V. Rodionova, R. S. Poudyal, I. Tiwari, R. A. Voloshin, S. K. Zharmukhamedov, H. G. Nam, B. K. Zayadan, B. D. Bruce, H. J. M. Hou and S. I. Allakhverdiev, Int. J. Hydrogen Energy, 2017, 42, 8450–8461 CrossRef CAS.
- E. C. Achinivu, R. M. Howard, G. Li, H. Gracz and W. A. Henderson, Green Chem., 2014, 16, 1114–1119 RSC.
- Z. Strassberger, S. Tanase and G. Rothenberg, RSC Adv., 2014, 4, 25310–25318 RSC.
- B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116, 2275–2306 CrossRef CAS PubMed.
- P. Kumar, D. M. Barrett, M. J. Delwiche and P. Stroeve, Ind. Eng. Chem. Res., 2009, 48, 3713–3729 CrossRef CAS.
- L. Yao, C. Chen, C. G. Yoo, X. Meng, M. Li, Y. Pu, A. J. Ragauskas, C. Dong and H. Yang, ACS Sustainable Chem. Eng., 2018, 6, 14767–14773 CrossRef CAS.
- J. Kautto, M. J. Realff, A. Ragauskas and T. Kässi, BioResources, 2014, 9, 6041–6072 CrossRef.
- C. Tang, J. Shan, Y. Chen, L. Zhong, T. Shen, C. Zhu and H. Ying, Bioresour. Technol., 2017, 232, 222–228 CrossRef CAS PubMed.
- Z. Zhang, M. D. Harrison, D. W. Rackemann, W. O. S. Doherty and I. M. O'Hara, Green Chem., 2016, 18, 360–381 RSC.
- M. N. Borand and F. Karaosmanoğlu, J. Renewable Sustainable Energy, 2018, 10, 033104 CrossRef.
- X. Zhao, S. Li, R. Wu and D. Liu, Biofuels, Bioprod. Biorefin., 2017, 11, 567–590 CrossRef CAS.
- A. Rodríguez, E. Espinosa, J. Domínguez-Robles, R. Sánchez, I. Bascón and A. Rosal, in Pulp and paper processing, ed. S. N. Kazi, InTech, 2018 Search PubMed.
- H. Chen, J. Zhao, T. Hu, X. Zhao and D. Liu, Appl. Energy, 2015, 150, 224–232 CrossRef CAS.
- F. Cheng, T. Ouyang, J. Sun, T. Jiang and J. Luo, BioResources, 2019, 14, 486–499 CAS.
- X. Zhao, K. Cheng and D. Liu, Appl. Microbiol. Biotechnol., 2009, 82, 815–827 CrossRef CAS PubMed.
- L. Qin, X. Li, J.-Q. Zhu, W.-C. Li, H. Xu, Q.-M. Guan, M.-T. Zhang, B.-Z. Li and Y.-J. Yuan, Ind. Crops Prod., 2017, 102, 51–57 CrossRef CAS.
- F. Cheng, X. Zhao and Y. Hu, Bioresour. Technol., 2018, 249, 969–975 CrossRef CAS PubMed.
- D. T. Balogh, A. A. S. Curvelo and R. A. M. C. De Groote, Holzforschung, 1992, 46, 343–348 CrossRef CAS.
- Y. Ye, Y. Liu and J. Chang, BioResources, 2014, 9, 3417–3427 CrossRef.
- J. Quesada-Medina, F. J. López-Cremades and P. Olivares-Carrillo, Bioresour. Technol., 2010, 101, 8252–8260 CrossRef CAS PubMed.
- L. Soh and M. J. Eckelman, ACS Sustainable Chem. Eng., 2016, 4, 5821–5837 CrossRef CAS.
- T. T. Kwok, K. R. Bright, M. J. Realff and A. S. Bommarius, BioResources, 2019, 14, 5988–6003 CAS.
- L. P. Novo and A. A. S. Curvelo, Ind. Eng. Chem. Res., 2019, 58, 14520–14527 CrossRef CAS.
- C. M. Hansen, Hansen Solubility Parameters: A User's Handbook, Crc Press, Boca Raton, 2nd edn, 2007 Search PubMed.
- C. Balaji, T. Banerjee and V. V. Goud, J. Solution Chem., 2012, 41, 1610–1630 CrossRef CAS.
- A. Casas, J. Palomar, M. V. Alonso, M. Oliet, S. Omar and F. Rodriguez, Ind. Crops Prod., 2012, 37, 155–163 CrossRef CAS.
- A. Casas, S. Omar, J. Palomar, M. Oliet, M. V. Alonso and F. Rodriguez, RSC Adv., 2013, 3, 3453–3460 RSC.
- Q. Zhang, X. Tan, W. Wang, Q. Yu, Q. Wang, C. Miao, Y. Guo, X. Zhuang and Z. Yuan, ACS Sustainable Chem. Eng., 2019, 7, 8678–8686 CrossRef CAS.
- Y. Zhang, H. He, K. Dong, M. Fan and S. Zhang, RSC Adv., 2017, 7, 12670–12681 RSC.
- W. Ji, Z. Ding, J. Liu, Q. Song, X. Xia, H. Gao, H. Wang and W. Gu, Energy Fuels, 2012, 26, 6393–6403 CrossRef CAS.
- R. Vanholme, B. Demedts, K. Morreel, J. Ralph and W. Boerjan, Plant Physiol., 2010, 153, 895–905 CrossRef CAS PubMed.
- C. M. Hansen and A. Björkman, Holzforschung, 1998, 52, 335–344 CrossRef CAS.
- W. Thielemans and R. P. Wool, Biomacromolecules, 2005, 6, 1895–1905 CrossRef CAS PubMed.
- K. L. Hoy, J. Coated Fabr., 1989, 19, 53–67 CrossRef.
- D. W. van Krevelen and K. te Nijenhuis, Properties of Polymers, 4th edn, 2009 Search PubMed.
- M. Mohan, P. Viswanath, T. Banerjee and V. V. Goud, Mol. Phys., 2018, 116, 1–21 CrossRef.
- J. Kahlen, K. Masuch and K. Leonhard, Green Chem., 2010, 12, 2172–2181 RSC.
- Y.-R. Liu, K. Thomsen, Y. Nie, S.-J. Zhang and A. S. Meyer, Green Chem., 2016, 18, 6246–6254 RSC.
- Y. Liu, J. Zheng, J. Xiao, X. He, K. Zhang, S. Yuan, Z. Peng, Z. Chen and X. Lin, ACS Omega, 2019, 4, 19829–19839 CrossRef CAS PubMed.
- D. Song, A. F. Seibert and G. T. Rochelle, Energy Procedia, 2014, 63, 1268–1286 CrossRef CAS.
- D. Song, A. F. Seibert and G. T. Rochelle, Energy Procedia, 2017, 114, 2713–2727 CrossRef CAS.
- X. Zhao, R. Wu and D. Liu, Bioresour. Technol., 2018, 261, 52–61 CrossRef CAS PubMed.
- J. P. F. Simão, M. G. V. S. Carvalho and C. M. S. G. Baptista, Chem. Eng. J., 2011, 170, 264–269 CrossRef.
- A. Hartono and H. F. Svendsen, J. Chem. Thermodyn., 2009, 41, 973–979 CrossRef CAS.
- S. Padmanabhan, P. Schwyter, Z. Liu, G. Poon, A. T. Bell and J. M. Prausnitz, 3 Biotech., 2016, 6, 23 CrossRef PubMed.
- E. L. Hult, T. Iversen and J. Sugiyama, Cellulose, 2003, 10, 103–110 CrossRef CAS.
- S. Park, J. O. Baker, M. E. Himmel, P. A. Parilla and D. K. Johnson, Biotechnol. Biofuels, 2010, 3, 10 CrossRef PubMed.
- L. Qin, W.-C. Li, J.-Q. Zhu, J.-N. Liang, B.-Z. Li and Y.-J. Yuan, Biotechnol. Biofuels, 2015, 8, 174 CrossRef PubMed.
- Ismojo, R. Hadiwibowo, E. Yuanita, E. Kustiyah and M. Chalid, Macromol. Symp., 2020, 391, 1900129 CrossRef CAS.
- J. P. Vandenbrink, R. N. Hilten, K. C. Das, A. H. Paterson and F. A. Feltus, BioEnergy Res., 2012, 5, 387–397 CrossRef CAS.
- S. P. S. Chundawat, G. Bellesia, N. Uppugundla, L. da Costa Sousa, D. Gao, A. M. Cheh, U. P. Agarwal, C. M. Bianchetti, G. N. Phillips, P. Langan, V. Balan, S. Gnanakaran and B. E. Dale, J. Am. Chem. Soc., 2011, 133, 11163–11174 CrossRef CAS PubMed.
- A. Mittal, R. Katahira, M. E. Himmel and D. K. Johnson, Biotechnol. Biofuels, 2011, 4, 41 CrossRef CAS PubMed.
- T. Cui, J. Li, Z. Yan, M. Yu and S. Li, Biotechnol. Biofuels, 2014, 7, 134 CrossRef PubMed.
- Z.-H. Liu, L. Qin, B.-Z. Li and Y.-J. Yuan, ACS Sustainable Chem. Eng., 2015, 3, 140–146 CrossRef CAS.
- Z. Yu, K.-S. Gwak, T. Treasure, H. Jameel, H. Chang and S. Park, ChemSusChem, 2014, 7, 1942–1950 CrossRef CAS PubMed.
- L. Segal, J. J. Creely, A. E. Martin and C. M. Conrad, Text. Res. J., 1959, 29, 786–794 CrossRef CAS.
- U. Koch and P. L. Popelier, J. Phys. Chem., 1995, 99, 9747–9754 CrossRef CAS.
- P. L. A. Popelier, J. Phys. Chem. A, 1998, 102, 1873–1878 CrossRef CAS.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter and D. Templeton, NREL, 2008, TP-510-42618.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- M. Mohan, T. Banerjee and V. V. Goud, J. Chem. Eng. Data, 2016, 61, 2923–2932 CrossRef CAS.
- M. Gonzalez-Miquel, M. Massel, A. DeSilva, J. Palomar, F. Rodriguez and J. F. Brennecke, J. Phys. Chem. B, 2014, 118, 11512–11522 CrossRef CAS PubMed.
- M. Mohan, V. V. Goud and T. Banerjee, Fluid Phase Equilib., 2015, 395, 33–43 CrossRef CAS.
- R. Anantharaj and T. Banerjee, Ind. Eng. Chem. Res., 2010, 49, 8705–8725 CrossRef CAS.
- Y. Li and Y. Jin, Renewable Energy, 2015, 77, 550–557 CrossRef CAS.
- F. Eckert and A. Klamt, AIChE J., 2002, 48, 369–385 CrossRef CAS.
- K. A. Kurnia, S. P. Pinho and J. A. P. Coutinho, Ind. Eng. Chem. Res., 2014, 53, 12466–12475 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- Z.-D. Ding, Z. Chi, W.-X. Gu, S.-M. Gu, J.-H. Liu and H.-J. Wang, Carbohydr. Polym., 2012, 89, 7–16 CrossRef CAS PubMed.
- M. Mohan, P. K. Naik, T. Banerjee, V. V. Goud and S. Paul, Fluid Phase Equilib., 2017, 448, 168–177 CrossRef CAS.
- J. Guo, D. Zhang, C. Duan and C. Liu, Carbohydr. Res., 2010, 345, 2201–2205 CrossRef CAS PubMed.
- R. F. W. Bader, A Quantum Theory, 1990 Search PubMed.
- T. A. Keith, TK Gristmill Software, Overland Park, KS, USA, 2019 Search PubMed.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS , 27.
- C. Loschen and A. Klamt, Ind. Eng. Chem. Res., 2012, 51, 14303–14308 CrossRef CAS.
- A. Niederquell, N. Wyttenbach and M. Kuentz, Int. J. Pharm., 2018, 546, 137–144 CrossRef CAS PubMed.
- M. Diedenhofen, F. Eckert, A. Hellweg, H. C. Steffen, U. Huniar, A. Klamt, C. Loschen, L. Koch, J. Reinisch, J. Schwobel, M. Suray, S. Terzi, K. Wichmann and T. R. Rosenbaum, COSMOquick, Version 1.7, COSMOlogic GmbH & Co. KG, Leverkusen, Germany, 2018 Search PubMed.
- H. Yu, J. Hu and J. Chang, Ind. Eng. Chem. Res., 2011, 50, 7513–7519 CrossRef CAS.
- C. M. Hansen, J. Paint Technol., 1967, 39, 104–117 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: COSMO-RS calculated chemical potentials of investigated molecules, viscosity validation, lignin developed predictive model plots, and quantum-based calculated intermolecular geometries, QTAIM properties, biomass compositional analyses, and the coordinates (XYZ) of most stable conformation structural information. See DOI: 10.1039/d1gc01186c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |