
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot route to convert technical lignin into versatile lignin esters for tailored bioplastics and sustainable materials†
Li-Yang
Liu
,
Siwei
Chen
,
Lun
Ji
,
Soo-Kyeong
Jang
and
Scott
Renneckar
 *
*
Department of Wood Science, The University of British Columbia, Vancouver, BC, Canada V6T 1R9. E-mail: scott.renneckar@ubc.ca
First published on 24th May 2021
Abstract
The valorization of lignin resources requires a functionalization route that satisfies the “greenness” and scale-up efficiency simultaneously. In this study, an efficient one-pot scalable method was created to convert kraft lignin into esterified lignin derivatives of controlled size, structure, and Tg. Specifically, the two-step reaction performed using a “one-pot method” involved the hydroxyethyl derivatization of phenolics and carboxylic acids on lignin with ethylene carbonate, followed by the direct esterification using an organic acid. The resulting homogeneous mixtures were fractionated by downward precipitation with water to obtain five different lignin fractions resulting in low process mass intensity. This one-pot route improved upon past work that involved recovery of the lignin after the first modification step. The one-pot result can esterify around 90% aliphatic hydroxyl groups in the hydroxyethyl derivatives. Analysis with NMR, GPC using multiple detectors, and DSC provided a greater view towards the structure and properties of technical lignin. 50–60% of the obtained esterified lignin derivatives have a polymeric characteristic with resulting Tg around 80 °C. The remaining 40%–50% of the technical lignin is oligomeric in size with more end group units and a corresponding Tg near or below room temperature. In solution, these fractions had conformation ranging from a rod-like structure to dense spheres, indicating a highly compact structure for the lignin. As a result, this route provided a starting material with a near-complete defined conformation and functionality to aid in-depth understanding of the structural features and properties of lignin-based materials. Integrating ethylene carbonate and organic acid as multi-functional reagents in this route can satisfy many sustainable chemistry requirements, delivering a clear path towards uniform esterified kraft lignin potentially available for various applications at the Mt level.
Introduction
Valorization of lignin alongside other biobased resources into new bioproducts (e.g., fuels, chemicals, and materials) is crucial to facilitate the economic conversion of biomass and shift towards a fossil-free society.1–4 Currently available on the market in North America and Europe is softwood lignin.5–8 As a consequence of the severity of the isolation process, softwood kraft lignin (SKL) has a heterogeneous chemical structure, various solubility, differing thermal stability, and broad distribution of molar mass.9 Further, the structural features and even the functional groups of lignin are dependent on their molecular weight.9–12 A way to obtain more homogeneous lignin is through fractionation. Gigli et al.13 and Sadeghifar et al.14 have reviewed the existing approaches (e.g., solvent fractionation and membrane fractionation) and the positive impacts on technical lignin valorization.However, the fractionation itself may not be enough to fully valorize the lignin, especially in preparing advanced polymeric materials. Additional structural changes have been observed during processing when fractionated lignin was blended with thermoplastics resulting in various responses when heated at elevated temperatures in the melt. Consequently, additional modification methods such as etherification or esterification are essential to improve their thermal stability or add more reactive functional groups, as highlighted in recent reviews.1,3,14–16 For example, researchers adopted a series of low boiling point solvents (e.g., ethylacetate, ethanol, methanol, acetone, and water) to refine kraft lignin as raw materials.17–22 Fractionated lignin was subsequently chemically modified to tailor properties of resulting lignin-based materials, such as polyurethane foams,23 polylactic acid (PLA),24 resin,25 and star-copolymers.18 These works described how the specific properties were affected by the molar mass and functional groups of fractionated lignin; the high molar mass lignin fractions can improve the resin's mechanical properties compared to original lignin.25 As a substrate for the core of a star-like copolymers using fractionated lignin, fractionated lignin created a spectrum of properties from a more ‘linear’-like compound to a densely grafted structure.18
Yet, several issues are still unresolved mechanisms on how fractionated lignin impact properties of lignin-based materials, such as the density of PUF,23 the tensile strength of PLA,24 and the stiffness of copolymers.18 This aspect is especially true as significant structural differences can arise during the derivatization of lignin. These potential changes will weaken the benefits of utilizing the fractionated lignin as starting materials to investigate the structure–property relationships for advancing lignin-based materials. The modification such as etherification of phenolic groups in lignin using epochlorihydrin,25 alkyl oxide,26 or organic carbonates27–31 always is accompanied by side reactions, causing unknown structural changes and increased molar mass with a significant change in the molecular weight distribution; in some cases, the molar mass and polydispersity will rise to over 20-fold of its original value.23,25–28,32–34 Also, these derivatization processes, such as esterification, utilized toxic and water-reactive reagent (e.g., halogenated compounds) with the harmful catalysts (e.g., pyridine) and solvents (e.g., DMF, THF).35–37 Beyond the “greenness” issues that cloud these chemicals, the multiple solvent mixtures lead to a more complex solvent parameter and additional difficulties to realize the separation of reagents mixture (solvents, reaction reagents, and catalysts) and the isolation of lignin from this mixture.
Actually, the “greenness” and efficiency for the above strategy of lignin fractionation and modification can have notable enhancements. Overcoming multiple challenges require integrative system thinking to use greener reaction reagents in a more efficient way.38 An emerging concept is the one-pot process for organic synthesis.39 In this process, the targeted products requiring several steps were synthesized in a single vessel. In so doing, isolating and purifying the intermediate chemicals can be omitted to reduce the cost regarding the amount of solvent, reagent, labor, waste, energy (heat), and time. The organic catalytic reactions in the pharmacetutical39,40 and biomass conversion41,42 are particularly suitable for the one-pot system.
For the valorization of lignin, a promising lignin modification route is the esterification of lignin resources. This route is capable of modifying their hydrogen bonding network, adding additional functional groups, and changing its physical properties, including solubility, glass transition temperature, and hydrophobicity.37,43–45 Consequently, it leads to a unique path in advancing lignin-based materials for various applications.3,16,30,32,45,46 In comparison with aforementioned process, researchers recently developed a series of novel and greener esterification approaches such as modification in multiphase emulsions,47 acidic ionic liquid,48 and supercritical conditions,49 and the one pot strategy. As highlighted above with the one pot study, Zhu and his collaborators performed this process during isolation50,51 enhancing the value of lignin while reducing processing steps. The approach can be done at temperatures lower than 100 °C, resulting in the selective esterification of the aliphatic hydroxyl groups (AlOH).50 Also, our group developed a highly efficient direct esterification (organic acid as solvent, reagent, and catalyst) to synthesize esterified lignin derivatives from technical lignin (degree of esterification = 90%).46 Due to the high selectivity toward AlOH of this route, a real-time monitoring hydroxyethyl reaction with ethylene carbonate was required as the first step to monitor the amount of AlOH in technical lignin and enhance their reactivity and uniformity.30,32 However, it is recognized that there are trade-offs when compared with other traditional approaches, such that using anhydrides can achieve high degrees of substitution at lower reaction temperatures and times, but also have lower atom economy values and potential toxicity of some of the catalysts or reagents.
Therefore, to further improve the greenness (e.g., lower process mass intensity) and avoid problems of traditional lignin fractionation and modification methods, we aimed at performing this two-step reaction with subsequent fractionation in a one-pot process in this study. The two-step reactions have a similar temperature (120–150 °C) so that a one-pot system can efficiently prevent waste heat requirements of cooling and reheating reactions. Another benefit of this one pot esterification process was the solvent mixture (organic acid/ethylene carbonate) can readily dissolve the derivatized lignin.46 The modification in advance enables us to obtain more uniform esterified lignin derivatives that have minimized lignin aggregates from hydrogen bonds,52 so that the resulting mixture does not require slow dissolution of aggregated structures. The addition of antisolvent such as water or alcohol to this solution can modulate the solvent parameters53 and realize the downward precipitation fractionation based on molecular weight. This process may be more efficient than the traditional fractionation process adopting unmodified lignin as starting materials if they are followed by chemical modification. The obtained lignin fractions with a specific degree of polymerization (DP) can then be used as building blocks, either as an “ideal” precursor for subsequent characterization to analyze the salient structural features, including chemical linkages, molecular weight, conformation, solubility, and glass transition temperature (Tg) or as a polymeric material. We anticipate these uniform esterified lignin fractions with well-characterized structures can highlight the mechanism for how the resulting properties of lignin polymeric materials arise and open a new path in sustainable materials research based on underutilized lignin resources.
Experimental section
Materials and chemicals
Softwood kraft lignin (West Fraser, Hinton, AB, Canada) was washed with distilled water, lyophilized, and further dried in the vacuum oven at 50 °C for 48 h before the modification and characterization. The functional groups of the starting softwood kraft lignin (aliphatic hydroxyl groups = 2.73 mmol g−1, aromatic hydroxyl groups = 4.77 mmol g−1, and carboxylic acid groups = 0.68 mmol g−1) were analyzed using quantitative 31P NMR analysis. The analysis procedures were described in our previous report.32Ethylene carbonate (99%, Alfa Aesar) was pre-dried in the vacuum oven at 50 °C for 48 h to remove the moisture. The propionic acid (99%, Arco Organics), sodium carbonate (anhydrous, Fischer Chemicals), dimethyl sulfoxide (DMSO, HPLC grade, Fischer Chemicals), and lithium bromide (LiBr, 99.995%, Alfa Aesar) were all purchased from Fischer Scientific and used without any treatment. Deuterium dimethyl sulfoxide (99.9% atom D%), chromium(III) acetylacetonate (99.99% trace metal basis) were obtained from Sigma Aldrich. Pyridine (anhydrous, 99.8%, Fisher Chemicals) was mixed with molecular sieves (3 Å) in advance. Deuterium chloroform was purchased from Cambridge Isotope Laboratories, Inc.
Three ingredients, two steps, and one-pot process
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, solvent and reagent) and Na2CO3 (0.25:1, catalyst) in the molar ratios to the sum of ArOH and COOH in a round-bottom flask. The mixture was loaded in an oil bath at 120 °C with continuous stirring. During the reaction, the produced CO2 was collected and measured by a self-designed real-time equipment to monitor the extent of the reaction.32
:2) to precipitate the lignin and centrifuge at 4000 rpm for 10 min. The collected solid residue was washed and dried to obtain lignin powders as lignin fraction 2 (F2). These procedures were repeated by adding 2/3 parts (v/v = 1:1), 1 part (v/v = 2:3), 3 part of water (v/v = 1:3) to obtain lignin fraction 3 (F3), lignin fraction 4 (F4), and lignin fraction 5 (F5), respectively (Fig. 1). These fractions were thoroughly washed with three ×200 ml distilled water and lyophilized to obtain lignin powders. After recovering F5, the supernatant was collected to recover propionic acid and other side products by a rotation evaporator. Note, room temperature was selected during precipitation, and temperature impacts were not considered in this study. This process was repeated six times (Table S1†). The corrected yield of each fraction was calculated as below.
:1, solvent and reagent) and Na2CO3 (0.25:1, catalyst) in the molar ratios to the sum of ArOH and COOH in a round-bottom flask. The mixture was loaded in an oil bath at 120 °C with continuous stirring. During the reaction, the produced CO2 was collected and measured by a self-designed real-time equipment to monitor the extent of the reaction.32
:2) to precipitate the lignin and centrifuge at 4000 rpm for 10 min. The collected solid residue was washed and dried to obtain lignin powders as lignin fraction 2 (F2). These procedures were repeated by adding 2/3 parts (v/v = 1:1), 1 part (v/v = 2:3), 3 part of water (v/v = 1:3) to obtain lignin fraction 3 (F3), lignin fraction 4 (F4), and lignin fraction 5 (F5), respectively (Fig. 1). These fractions were thoroughly washed with three ×200 ml distilled water and lyophilized to obtain lignin powders. After recovering F5, the supernatant was collected to recover propionic acid and other side products by a rotation evaporator. Note, room temperature was selected during precipitation, and temperature impacts were not considered in this study. This process was repeated six times (Table S1†). The corrected yield of each fraction was calculated as below.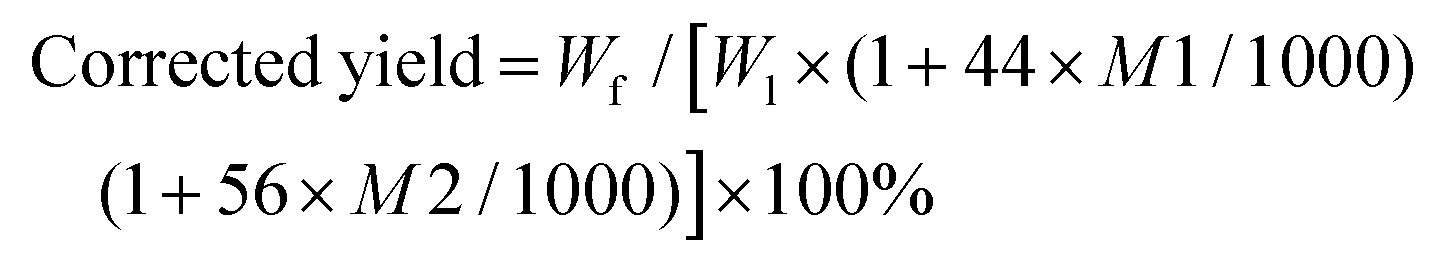 | (1) |
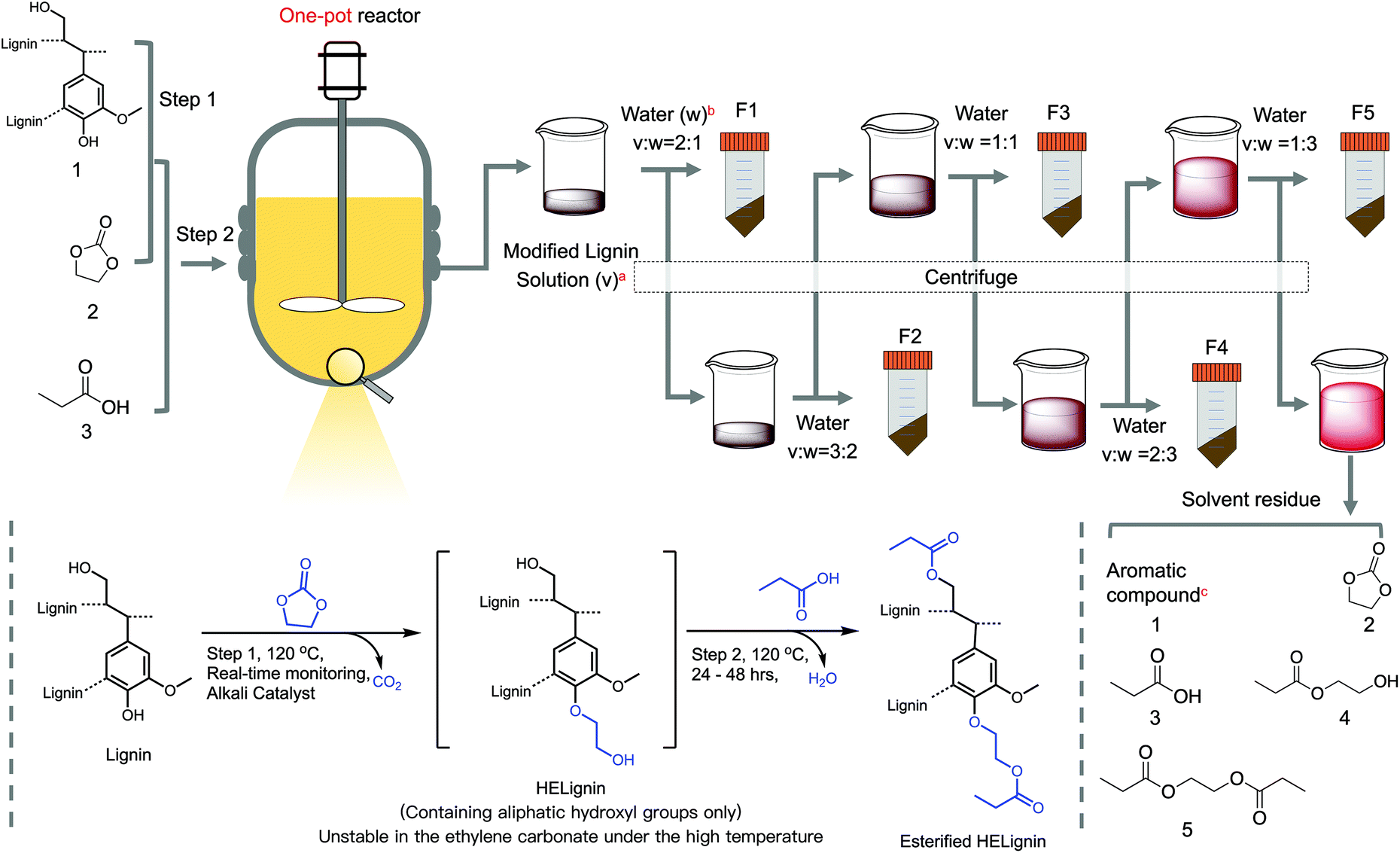 | ||
| Fig. 1 Scheme of one-pot process to synthesize esterified lignin derivatives with subsequent downward precipitation fractionation; athe modified lignin solution were centrifuged first (solid yield after centrifugation is less than 1%), bthe amount of water were calculated based on the volume of modified lignin solution mixtures, caromatic compounds were not defined as it is lower than the detection limit but caused visible reddish color of the solution. | ||
Nuclear magnetic resonance analysis
The sample preparation process for 1H, 13C, 31P, and HSQC NMR and related parameters were described in our previous studies. The 1H and 13C NMR spectra were acquired by Bruker Avance 300 MHz (Bruker Corp. Billerica, US) equipped with a BBO probe at 25 °C. 2D HSQC NMR spectra were analyzed by a 600 MHz NMR equipped with CryoProbe (Bruker AVANCE III, Bruker Corp. Billerica, US) at 25 °C. The obtained spectrum was processed using TopSpin 3.6.2 software.54GPC-MALS-IV-dRI analysis
10 mg lignin samples or standard polystyrene sulfonate (PSS) were dissolved into 1 ml degassed eluent (DMSO/LiBr, 0.5%) and stabilized at room temperature 48 h. For PSS, the solvent mixtures were heated up to 50 °C for two weeks in a closed vessel to obtain dissolved samples in the DMSO/LiBr.The lignin mixture and standard PSS samples were then analyzed using an Agilent 1100 series (Agilent Technologies, Santa Clara, US) system containing a pump, autosampler, two gel permeation columns (PolarGel M and Polar Gel L) at 35 °C with a flow rate 0.5 ml min−1. Three types of detectors (Wyatt Technology, Santa Barbara, CA, USA): Dawn HELEOS I Multi-angle static light scattering (MALS) detectors, ViscoStar viscometer (IV) detector, and Optilab T-rEX differential refractive index (dRI) detector. The temperature of all these detectors was set at 35 °C. Both the MALS detector and the dRI detector had a laser operating at 785 nm. The MALS detector was pre-installed with narrow bandpass filters (±10 nm) to minimize overlap of the fluorescence from lignin. The obtained molecular weight traces were recorded and analyzed using Astra 6.1 software.54
With molar mass ranging from the highest 751.5 kDa to the lowest 2.4 kDa, the PSS standard samples were analyzed and exported as the standard curve for the conventional (dRI traces) and universal (IV traces) calibration analysis by the ASTRA 6.1 software (Table S2†). The dRI traces were also adopted for the refractive index increment (dn/dc) value calculation, assuming 100% mass recovery and high purity for selected dRI peaks of each fraction in DMSO/LiBr (0.5% w/v).
In combination with dRI, the MALS detector traces recorded the intensity of scattered light at different angles (17 angles). Based on the Zimm's formalism (eqn (2)), the intensity of scattered light directly relates to the properties of lignin, including absolute molar mass (Fig. S1†).
| (K* × c)/R(θ) = 1/MP(θ) + 2A2c + … | (2) |
The specific viscosity obtained from the viscometer detector (IV) can be converted into the intrinsic viscosity for the Mark–Houwink–Sakurada model (eqn (3)) to describe the conformation and hydrodynamic radius (Rhv) of each lignin fraction.55
| log[η] = logK + α × logM | (3) |
Differential scanning calorimetry
Differential scanning calorimetry (DSC) was performed using a TA Q1000 (TA Corporation, US). 5 mg dried lignin powders were sealed into the aluminum pan. The heating procedure was a three-cycle process: ramp 20 °C min−1 to 105 °C and isothermal hold for 10 min as the first cycle, ramp 10 °C min−1 to −50 °C as the second cycle, and then ramp up to 220 °C as the third cycle. The glass transition temperature (Tg) of each lignin fraction was analyzed based on heat flow as a function of temperature in the third cycle.46Results and discussion
One-pot process and the “greenness” evaluation
The two-step reaction followed by the downward precipitation fractionation was illustrated in Fig. 1. In this system (Fig. 1), three ingredients, including lignin (compound 1), ethylene carbonate (EC, compound 2), and propionic acid (PA, compound 3), were reacted in the presence of a sodium carbonate catalyst as two steps to form the final esterified HElignin compound. In the first step, the hydroxyethyl reaction involved alkaline catalyzed lignin modification with EC to form hydroxyethyl derivatives, which has similar functionality with native lignin (e.g., containing over 80% aliphatic hydroxyl groups, AlOH) and enhanced thermal stability.30 The in situ monitoring process of CO2 was crucial to know the endpoint of this step since the excess time will cause more condensation and increase molar mass.32 Then, organic acid was added to neutralize the alkali (carbonate salts), quench the side-reaction between EC and hydroxyethyl lignin derivatives, and react with the aliphatic hydroxyl groups (newly formed and original) to form the esterified lignin derivatives. By using the in situ real-time process, there was enhanced reproducibility as the solid residue in the solvent mixture at the end of the reaction was less than 4% (Table S1†).As a result of near-complete solubilization, this homogeneous system is ideal for downward precipitation fractionation. Based on the Flory–Huggins equation, the interaction parameters χ of the solution under this stable state can be treated as χcritical ∝ 1/DP.53 This parameter is proportional to the solubility parameter differences between lignin and their solvent (δlignin–δsolvent).2 Adding antisolvent in a specific ratio with the solvent will gradually change the solvent parameter (δsolvent) and inflect the stable state, leading to the precipitation of polymer with a different average degree of polymerization (DP).17,22,53 The selected antisolvent should have good miscibility with the selected organic acid and ethylene carbonate.17,19 Hence, water was chosen in this case, while organic alcohol, such as ethanol, would be a suitable antisolvent for other types of organic acids considering their miscibility.46 As the EC and PA serve as solvents, the ratio of these two types of reagents will dominate the solvent solubility parameter and affect this system's reproducibility. Their ratio can affect the solubility parameter from these two aspects; the excess EC served as cosolvent can improve the lignin solubility in the organic acid and prompt the subsequent esterification reaction. A lower amount of EC would lead to a higher yield of solid residue (Table S1†). The reaction between EC and PA will also produce derivatized compounds, such as hydroxyethyl propionate (compound 3 and 4), based on the 1H NMR spectrum. This cosolvent will further change the solubility parameter (Fig. S2†).
Theoretically, we can tune lignin fractions as needed by changing the ratio of antisolvent/solvent. In this study, five fractions were isolated (Fig. 1), with a range of apparent yields from the lowest 11.3% (F4) to the highest 41.6% (F2). The sum of the yields of the modified lignin approached 120.6% related to the modified lignin to the unmodified starting material (Table 1). Based on the lignin content and its hydroxyl groups, each fraction's corrected yield was recalculated with a yield range from the lowest 7.6% (F4) to the highest 27.9% (F2). Consequently, over 10% of lignin was lost during the washing procedure, which can potentially be avoided by working on a larger-scale process. At the end of fractionation, the last supernatant was collected and concentrated via a rotation evaporator. The GC-MS results indicated (Fig. S3†) that the residual reddish solution was dominated by the solvent-related compounds, including EC (compound 2) and PA (compound 3), and their derived compounds: hydroxyethyl propionate (compound 4), and ethane-1,2-dilyl dipropionate (compound 5). Lignin residues were below the detection limit, indicating the lignin was thoroughly recovered in the precipitation process.
| Apparent yield/% | Corrected yield/% | dn/dc | M w/kDa | M n/kDa | Đ | DEa/% | DEb/% | |
|---|---|---|---|---|---|---|---|---|
|
a 13C NMR, degree of esterification = peak 7/(peak 8 + peak 7) based on Table S5.†
b 31P NMR, degree of esterification = (total OH + COOH in SKL − total OH + COOH in esterified lignin)/total OH + COOH in SKL; total OH = AlOH + ArOH (Table S5).†
|
||||||||
| Lignin-SKL | 0.1034 | 27.5 | 6.0 | 4.5 | ||||
| HELignin | 0.1000 | 166.2 | 9.8 | 16.9 | ||||
| F1 | 32.5 | 21.8 | 0.0995 | 740.0 ± 103.3 | 162.3 ± 38.9 | 4.6 | 87.6 | 89.1 |
| F2 | 41.6 | 27.9 | 0.1100 | 82.4 ± 8.0 | 35.9 ± 3.1 | 2.3 | 88.9 | 87.0 |
| F3 | 24.7 | 16.5 | 0.1092 | 14.7 ± 1.9 | 11.2 ± 1.9 | 1.3 | 91.3 | 88.7 |
| F4 | 11.3 | 7.6 | 0.0854 | 6.0 ± 1.1 | 3.3 ± 0.7 | 1.8 | 91.4 | 86.7 |
| F5 | 12.3 | 8.2 | 0.0863 | 2.2 ± 0.9 | 1.9 ± 0.8 | 1.8 | 91.0 | 85.1 |
To evaluate the greenness of this one-pot process, we adopted the process mass intensity (PMI = total mass in a process/mass of product)56 to compare the conventional methods (esterification of fractionated lignin) (Table S3†) and our one-pot process (Table S1†). Without considering the efficacy of reagent recovery and purification, the PMI for the typical multiple solvent fractionation process21,25 followed with the esterification using acyl chloride reagents will range from 146–287 g g−1 (Table S3†). This number will further increase depending on how many fractions require esterification. If all fractions were esterified, the total PMI would rise to 951 g g−1. In comparing with this traditional route, our one-pot process with subsequent esterified lignin fractions has much lower PMI as 67.1 g g−1 (Table S1†). Also, the amount of the solvent PA and EC can be optimized to lower their consumption. The previous study showed that the other lignins have enhanced solubility with EC.46,54 This simple solventless system using less-toxic and stable reagent can minimize the workload, cost, and energy consumption for the subsequent isolation and purification, resulting in decreased PMI-water and PMI-solvent. Isolating PA from water has been thoroughly studied by previous biochemists, demonstrating good recovery efficacy.57 Besides, the recovered ethylene glycol esters (Fig. S3†) as side products are useful to prepare polyester compounds via transesterification or further degraded to produce ethylene glycol.58 The collected high-purity CO2 during the one-pot modification process can be recycled and used to acidify the black liquor and coagulate the lignin, as demonstrated in our previous study.32 Since the consumption of CO2 occupied 50% cost of lignin recovery plant,6 our one-pot process can improve the overall economic efficiency of lignin functionalization. One of the drawbacks for this one-pot process is the harsher reaction conditions (e.g., higher temperature) may consume more energy. If renewable energy prices keep falling, these extra costs may not be such a great issue in the future.59
The molar mass of esterified lignin
Besides the greener metrics, the efficacy of the downward precipitation fractionation process to obtain uniform building blocks in this study was revealed in the molar mass analysis using gel permeation chromatography (GPC) combined with multi-angle light scattering (MALS), differential refractive index (dRI), and intrinsic viscosity (IV) detection (Fig. 2). The hydroxyethyl lignin (HELignin) and original lignin were also analyzed as comparisons. Because of some crosslinking during hydroxyalkylation, the modified sample had a broader multimodal molecular weight distribution and a larger molar mass than the parent sample32 (Fig. 2). | ||
| Fig. 2 GPC traces of lignin, HELignin, and different esterified lignin fractions (F1, F2, F3, F4, and F5) using different detectors including refractive index (dRI, a and d), light scattering (MALS, b and e), and viscometer (IV, c and f). | ||
After our one-pot process, a series of molar mass traces with narrower distribution for lignin was obtained. With increasing retention time, the molecular weight peak shifted to the right in order F1, F2, F3, F4, and F5, highlighting the efficiency of the fractionation process (Fig. 2 and 3). In the current study, the commercial PSS standard samples were pre-run to obtain the standard curve for both conventional calibration analysis (dRI) and universal calibration analysis (IV) (Table S2†). As expected, the fractionated lignins had a narrower molecular weight distribution noted by the polydispersity (Đ), with a significant difference in their molar mass between the fractions. Nonetheless, these calibration methods assumed that the lignin fraction and PSS standards had close molar mass, DP, and specific viscosity, when their retention time is the same. However, PSS and lignin have an entirely different structure (linear vs. branched) and the procedure underestimated the molar mass of lignin, as discussed below when compared to the PSS standards.
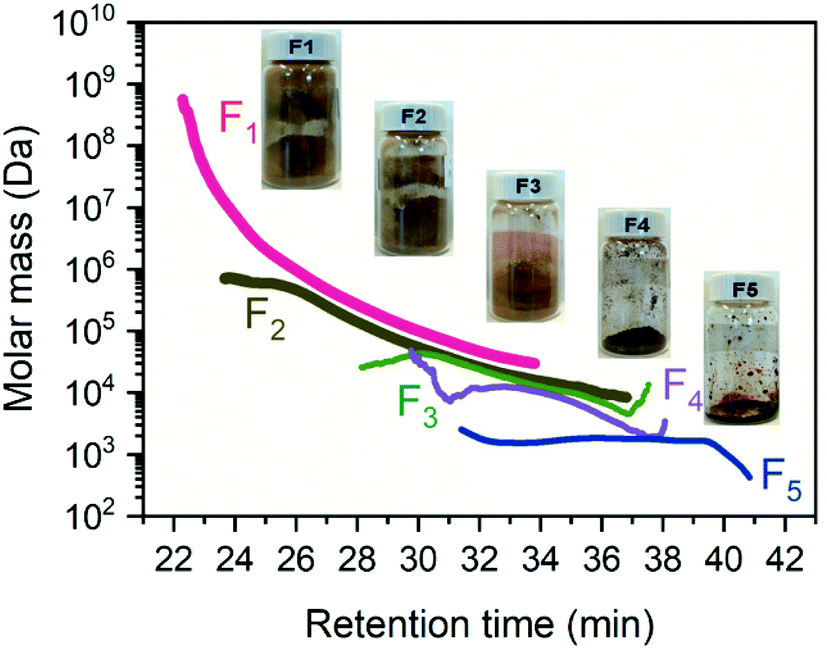 | ||
| Fig. 3 The molar mass distribution of different lignin fractions and sample images of recovered fractions. | ||
More advanced mean of molecular weight analysis uses multi-angle light scattering (MALS) to obtain the absolute molar mass of lignin. Based on the Zimm analysis, the intensity of scattered light (R(θ) scattered) has a direct correlation with the molar mass (M), sample concentration (c), and the specific refractive index increment (dn/dc) (Fig. S1†). This approach allows the measurement of the absolute weight-average molar mass of lignin without additional standard samples.55,60–62 One limitation of this technique when applied to lignin samples analysis is that the self-association of lignin from the hydrogen bonding and π–π interaction63,64 will lead to the formation of multimodal peaks and more noise within traces, causing overestimation of molecular weight analysis. For example, the LS signals of lignin and HELignin have multimodal peaks indicating the formation of aggregated lignin, though these lignin were acetylated in advance, and the LiBr salt was added in the eluent to help shield these interaction forces (Fig. 2).
Fig. 2 showed that the fractionation approach was a useful way to obtain Gaussian – distribution curves with higher signal/noise ratios and lower Đ for all detectors, even light scattering detectors. In line with previous studies,11,65 both the esterification and the fractionation can efficiently minimize the intermolecular aggregation arising from π–π stacking to hydrogen bonding. Further, the concentration of the smaller molecular weight lignin (less than 10 kDa) for GPC analysis can be enhanced to obtain more accurate molar mass and intrinsic viscosity for these fractions. The absolute molar mass of different lignin fractions ranged from 740.0 kDa to 2.2 kDa, which is higher than their relative molar mass (Table 1 and Table S4†). The F1 fraction had the largest molar mass, over 90-fold of the smallest F5, suggesting the heterogeneous features of lignin. Their color and state, from typical brownish-yellowish powder to dark liquid state, further provides obvious qualitative differences arising from their molar mass and structural difference (Fig. 3). The molar mass distribution of F1 and F2 have much larger molar masses and polydispersity (Đ) than F3, F4, and F5, suggesting the downward precipitation can be further optimized to narrow Đ. Both F3 and F4 fractions contain significant hooks in the molecular weight profiles (Fig. 3). This provides evidence of a branched structure within lignin,66 which is fundamentally different in structure than the PSS, commonly used for the relative molecular weight values. The F5 profile was essentially flat across the retention time, suggesting this low MW sample has a relatively uniform size with unique conformational features such as branching that cause a shift in retention time (Fig. 3).
The structural features of esterified lignin
The degree of esterification (DS) for each fraction was analyzed based on the 31P and 13C NMR spectra, respectively. Quantitative 31P NMR results indicated decreasing amount of hydroxyl groups, including AlOH, ArOH, and COOH groups for all esterified fractions (Table S5†). Previously, with the same lignin used in this study and corresponding amount of CO2 detected at the end point of the reaction, the total hydroxyl content of the first step was reported as 4.81 mmol g−1. In the fractionated, esterified samples total hydroxyl content was reduced to the range between 0.88 mmol g−1 to 1.22 mmol g−1 dependent upon fraction (Table S5†). The disappearance of hydroxyethyl carbon (peak 8) and corresponding downfield shift to the esterified derivative (peak 7) were also adopted to calculate the degree of esterification (Fig. 4).46,67 For all isolated fractions, the DS was near 90%, indicating the high robustness of our developed one-pot direct esterification process across all the various lignin fractions (Table 1). | ||
| Fig. 4 Part of HSQC NMR spectrum of F5 including the alkyl chain (a), oxygenated alkyl chain (b), and aromatic region (c), and 13C NMR spectrum of acetylated lignin (d), acetylated HELignin (e), and propionic esterified F5 lignin (f) and the major structural features including units, linkages, and functional groups of lignin during different stages. | ||
The chemical structure of lignin is vital in understanding the structure–property of lignin-based materials. For these esterified lignin fractions, their functional groups, chemical linkages, and fundamental units were analyzed with spectroscopic techniques. In association with the isolation and modification, lignin always has some structural changes.68 In the plant cell wall (softwood), phenyl propane units (e.g., coniferyl alcohol and p-coumaryl alcohol) were crosslinked, resulting in aryl ether linkages (β-O-4) and carbon–carbon linkages (phenylcoumaran β-5, and resionl β–β) (Fig. 4).69,70 During the subsequent alkali pulping process, the ether linkages in the native lignin were cleaved, accompanied by intermolecular condensation. The dominant detected linkages in the softwood kraft lignin were carbon–carbon linkages (e.g., 5–5, β-1 stilbene, and β-5 stilbene). The majority of hydroxyl groups in the kraft lignin will be changed from AlOH to ArOH groups (Fig. 4). These fundamental units also change slightly, partial cleavage of the γ carbon, intermolecular condensation, the oxidization of hydroxyl groups, and propyl chain C–C cleavage to create vanillin (V) or its derivatives: vanillic acid (VA) or aceto-vanillone (AV) (Fig. 4).68,71,72 In contrast, the hydroxyethyl reaction can enhance uniformity by modifying the ArOH group of technical lignin to AlOH.30,32 As side reactions, this process will also generate carbonate linkages (C1 and C2) as discussed in the previous studies32 (Fig. 5). The propionic acid will then selectively esterify the AlOH groups46 (Fig. 1).
 | ||
| Fig. 5 Correlation analysis between the molar mass (GPC analysis) and represented units in esterified lignin (13C NMR or 2D 13C–1H HSQC NMR) including native linkages (a); methoxy groups (MeO), guaiacyl units (G2), aromatic hydrogen (ArH), (b); kraft linkages (stilbene-1 and stilbene-5), (c); kraft units (vanillin, vanillic acid, and acetovanillone), (d); carbonyl units (ester), (e); and different carbonate linkages (carbonate 1), and oxygenated ethyl groups (f, peak 5 + 6 + 7), (f). | ||
As mentioned earlier, the fractionated lignin with decreased Đ can provide more accurate NMR quantification results.72 Interestingly, the spectra of 13C (Fig. S4†) and 2D HSQC NMR (Fig. S5†) of different lignin fractions were similar in light of their functional groups, units, and linkages. Hence, the lignin structure was relatively uniform from this perspective, but the number of different linkages and end units were diverse and heavily dependent on its molar mass (Fig. 5 and Table S5†). The native lignin units linkages (e.g., β-O-4, β–β, and β-5) were depleted as molecular weight decreased for the fractionated samples, in line with previous reports (Fig. 5a).72 Further, the lower amount of aromatic hydrogen (ArH, Fig. 5b) can reflect the number of condensed linkages (e.g., 5–5 and β-5). The amount of G2 signal was higher than the amounts of their methoxy groups, which indicated the demethylation occurred during the kraft pulping process.73 The harsh reaction conditions (strong alkali and high temperature) would significantly modify the lignin with low molar mass leading to more demethylation (larger differences between G2 and methoxyl groups, and a higher amount of ArH) (Fig. 5b), the formation of stilbene linkages (Fig. 5c), and vanillin-related compounds (Fig. 5d).71 The ethyl groups from the derivatization were correlated with the ArOH and COOH groups due to the selectivity of hydroxyethyl modification. Note that the number of total ester groups was higher than the number of ethyl groups because the original lignin contained AlOH groups, which were also esterified (Fig. 5f). As chain scission of β-O-4 bonds created phenolics during kraft pulping, the lower molar mass of lignin had a higher amount of esterified chain end units proportional to the total aromatic content on the polymer chain. It is in line with previous studies that lower molar mass lignin will contain more hydroxyl groups, especially phenolic groups.10,13,14,74 Conversely, the large molar mass lignin has more obvious condensation showing the decreased amount of ArH and enhanced carbonate linkages (Fig. 5b and e) as illustrated with the GPC analysis.
Conformation and hydrodynamic radius of esterified lignin
Though the NMR technique can provide detailed chemical insight on the kraft lignin, a considerable amount of unknown linkages will limit the understanding of the technical lignin structure and related physical character.72 A key in understanding the physical characteristics of polymer chains, such as their flexibility, entanglement, and multiplicity of interactions, is their conformation. For lignin, there have been several questions around what makes it suitable for certain applications. Hence a physical representation plays a central role for lignin-based materials, such as block copolymer melts and polymer blends.75–79The MALS signal provides for the detection of the radius of gyration (Rg) for the highest MW fraction and hydrodynamic radius (Rh(Q)) of the polymers (Fig. S6†).80 As tabulated in Table S6,† the Rg of F1 is almost 2.3 fold of its Rh(Q) value, indicating F1 has a shape factor of at least a prolate spheroid shape but most likely a rod-like structure.81 This conclusion is the same by Chakraborty et al., who utilized small angle neutron scattering (SANS) to study a fractionated kraft lignin from an aqueous lignin purification with hot acids (ALPHA) process, where the highest MW lignins were observed to be elongated rigid rod structures; the SANS data indicated a cylinder type structure as molar mass scaled with cylinder length, not diameter.82 This type of structure would also provide an explanation towards orientation and alignment of lignin in rapidly drawn fibers.83
For the other fractions, the physical limitation of using MALS detectors limits the ability to analyze the other four lignin fractions with the Rg of less than 10 nm. With knowledge of the molar mass and chemical structure, the conformation is also related to the intrinsic viscosity of lignin that can be described based on GPC combined with intrinsic viscosity detector (IV) and concentration detectors (dRI).55 After the one-pot process and fractional precipitation, the peaks with a high signal/noise level revealed more accurate intrinsic viscosity. The slope of MHS plot α value had a different range for the polymers signifying different conformation within solution (Fig. S7†). The lower the α value indicated a dense structure in the solution, which may arise from significant branching. The F1 and F2 lignin (large MW fractions) had α-values 0.35 and 0.31, indicating that these fractions are closer to a branched structure and not a random linear coil (Fig. 6a). The α-values of F1 were slightly higher than the results obtained by Glasser and co-workers,84 as we removed the bias impacts from low molar mass lignin. Other unique samples such single walled carbon nanotubes (SWNT) and graphene oxide have been analyzed for their MHS α-value. For analysis of single walled carbon nanotubes, an α-value ranged from a flexible rod (1.14) to a branched structure (0.37) down to almost a dense sphere of carbonaceous product was revealed (0.15) dependent upon the dispersion method. The branched structure (0.37) was related to the free radical grafting reaction bundling SWNTs or potentially strong entanglement.81 For graphene oxide MHS α parameter of 0.33 was reported related to a self-avoiding sheet with a moderate bending rigidity.85 In the current study, F3, F4, and F5 (lower MW fractions) had an α value approaching 0 and are essentially dense non-draining “particles” in solution without excluded volume (Fig. 6a). These significant differences in their conformation further highlight the importance of fractionation as the lignin derivatives would behave very differently in material applications.
 | ||
| Fig. 6 The slope of Mark–Houwink–Sakurada (MHS) plot of lignin (a) and the hydrodynamic radius (b) for both fractionated lignin and standard PSS samples with different molar mass. | ||
Since the intrinsic viscosity and absolute molar mass could be measured accurately, the Einstein–Simha relation was applied to analyze the hydrodynamic volume of each fraction. The hydrodynamic radius Rh(V) was calculated based on the assumption that lignin particles in the solvent are close to Einstein spheres (which for lower MW fractions may be accurate from the MHS parameter).55 With an increasing molar mass, each fraction's hydrodynamic radius increased from 1 nm to 10.2 nm (Fig. 6b). Zhao et al. obtained similar results, using the small-angle neutron scattering to analyze lignin aggregates in DMSO.63,64 Hence, the lignin molecules under theta solvent condition (the lignin aggregates were neglected here) are less than 10 nm (quantum dot size). As a comparison, the Rh(V) of PSS standard with a different molar mass was also calculated.(Table S2†) When the molar mass of esterified lignin and PSS were close, the Rh of PSS was much higher than related lignin samples. This difference was more evident for the large molar mass lignin when compare to the Rh of PSS (35 nm); the value was more than three-fold of the Rh(V) of F1 (10.2 nm) (Fig. 6b). As mentioned above, this significant difference leads to low accuracy of conventional calibration methods (dRI) and universal calibration methods (IV) to analyze the relative molar mass of lignin using non-fractionated samples.
Thermal properties of esterified lignin
Like other polymers, the Tg of lignin was noted to be affected by their chemical structure (e.g., backbone side groups, interaction), molar mass, and composition.16,78 By esterification, we can replace the intermolecular hydrogen bond with weaker dipole–dipole interactions derived from side propionate ethyl chains. Consequently, the different lignin fractions have a lower Tg with a range from 11 °C to 85 °C than unmodified lignin (160 °C) and hydroxyethyl lignin.35 The Tg of different lignin fractions increased as a function of molar mass, in line with other types of polymers (Fig. 7 and Fig. S8†). The decreased molar mass leads to higher proportion of propionate ethyl chains ends with a correlation that the concentration of chain ends ∝ 1/Mn.86 These chain ends (e.g., ester and oxygenated ethyl) can be viewed as colligative properties resulting in a significant impact on the density and thermal properties of lignin (Fig. 5f). For lignin fractions, a relation of Tg = 80.1–1.36 × 105/Mn was obtained to describe their correlation and imply an independent Tg from the molar mass when the number exceeds 1.36 × 105 Da (Fig. 7). Within this system, we can better control the thermal properties through selection of the molar mass of lignin. | ||
| Fig. 7 The glass transition temperature (Tg) of the different esterified lignin fractions and their correlation with the molar mass (Mn). | ||
The potential application of esterified lignin building blocks
In the long term, one of the most significant misunderstandings about the lignin resources is that “it can make everything”,87 because this statement lacks an essential condition that the application is highly dependent on the structure of lignin. The lignin resource, especially industrial technical lignin, has a well-known heterogeneous structure that limits their characterization and engineering toward high-performance materials. Based on their characterization, it is clear they do not behave like a random coil and should not be thought as one. The data suggests that technical lignin are fragmented particles that have potential to aggregate. Given their conformation, molar mass, chemical structure, Tg, and solubility (Table S7†), these different fractions should be targeted to specific applications.F1 and F2 with larger molar mass and rod-like structures most likely should be considered for applications where their asymmetric shape can be utilized in material properties like fiber spinning. Removing low molar mass lignin, these fractions showed an advance in making microfibres by electrospinning.88 After the esterification, the enhanced solubility of these lignin fraction in a variety of solvents, including acetone, THF, NMP, and ethyl acetate (Table S7†), can eliminate the need for high boiling point or toxic solvent (e.g., DMF) for solution processing89
It is hard to convert the F3, F4, and F5 themselves to high-performance thermoplastic materials due to their low molar mass and highly-branched structures (the conformation is close to densified spheres). However, these fractions could be valuable additives for preparing more sustainable materials as additives. These fractions contain structures that were close to the typical plasticizer (e.g., phthalates compounds) and contain both polar (aromatic backbone and carbonyls) and non-polar groups (propionate ethyl chain).79,90 The traditional plasticizer as “everywhere chemicals” can cause adverse health effects in children's brain development;86 thus their applications are regulated in North America, Europe, and Japan. Potentially, the esterified lignin fraction can be used as a plasticizer to enhance the thermal processibilities of conventional plastic materials. These lignin fractions can be used as UV light blockers in the plastic materials because they contain abundant UV chromophore functional groups (e.g., stilbene, Fig. 5c), as indicated by their darker color (Fig. 3).91 Besides, these fractions had excellent solubility in acetone, and THF (Table S7†) can be used to prepare colloidal lignin particles as their amphiphilic properties are critical for emulsions.92,93 These lower molar mass samples, especially F5 in liquid state at room temperature, are closer to lignin oligomers and soluble in solvents such as toluene (Table S7†).94 One approach to the utilization of these soluble materials is to exploit the small amount of residual hydroxyl groups as critical crosslinking sites in potential thermosetting adhesives when mixed with other reactive compounds.95 Further, this lower molecular weight sample would be an exciting starting material for further transformation to make more advanced chemicals.2,96 Another large quantitative applications would be the bitumen modifier. As natural cementitious material, lignin can modify bitumen pavements with enhanced lifespan, workability, compaction, fatigue and rutting resistance in asphalt roads at warm temperatures.97–99
On the other hand, we can better understand these lignin structures within a known absolute molar mass. Balakshin et al. revealed the conformation of spruce milled wood lignin. The study provided a model structure on this type of native lignin by analyzing the branching points, such as the condensed linkages (e.g., 5–5 or 4-O-5).100 The application of these developed characterization methods on these derivatized kraft lignin fractions would enable us to obtain an in-depth understanding of kraft lignin's structural features in the future.
Conclusions
This study developed a simple one-pot route to synthesize lignin esters with the controlled molar mass in a single reactor improving past research efforts in this direct esterification research area. This process satisfies both high greenness requirements (e.g., low PMI) and high efficacy (e.g., 90% degree of esterification). Our results revealed that their conformation, hydrodynamic radius (Rh), and glass transition temperature (Tg) are highly dependent on the absolute molar mass of lignin measured with multi-angle light scattering. Over 50% of lignin with polymeric characteristics have a more rod-like to extended branched structure and higher Tg; the other 50% of esterified lignin derivatives as oligomers have Tg around or below room temperature. These fractions are densified spheres and contain more derivatized functional groups because of the enhanced number of chain ends per molecule. As a consequence of understanding the structural differences, we can target the different lignin fractions in designing or engineering high-performance materials in different areas, from plastic materials to carbon fibers to bitumen replacement and colloidal particles.Author contributions
The manuscript was written through contributions of Li-Yang Liu and Scott Renneckar with input from all authors. Li-Yang Liu and Scott Renneckar designed the study. Li-Yang Liu, and Siwei Chen performed the experiments and characterization. Li-Yang Liu and Scott Renneckar analyzed the data. Lun Ji and Soo-Kyeong Jiang aided in interpreting the GPC and GC-MS results.Funding sources
The research was supported by the Alberta Innovates, Alberta Bio Future, Lignin Challenge 1.0 program project #BFL18010, the Paul and Edwina Heller Memorial Fund and the Canada Research Chairs program # 950-232330.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Thank you to Mark Okan in the UBC Chemistry Department for his help and suggestion on NMR analysis. Thank you to Muzaffer Karaaslan, MiJung Cho, and Qi Hua for their discussion and comments on this research. The research was supported in part by Alberta Innovates, Alberta Bio Future Lignin Challenge 1.0 (grant #BFL18010) and the Canada Research Chair (Tier 2) in Advanced Renewable Materials (grant #950-232330).Notes and references
- W. G. Glasser, Front. Chem., 2019, 7, 1–17 CrossRef PubMed.
- W. Schutyser, T. Renders, S. Van Den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116, 2275–2306 CrossRef CAS PubMed.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- Z. Hu, X. Du, J. Liu, H. M. Chang and H. Jameel, J. Wood Chem. Technol., 2016, 36, 432–446 CrossRef CAS.
- L. Kouisni, A. Gagné, K. Maki, P. Holt-Hindle and M. Paleologou, ACS Sustainable Chem. Eng., 2016, 4, 5152–5159 CrossRef CAS.
- P. Tomani, Cellul. Chem. Technol., 2010, 44, 53–58 CAS.
- I. Brodin, Chemical Properties and Thermal Behaviour of Kraft Lignins, 2009 Search PubMed.
- C. Crestini, H. Lange, M. Sette and D. S. Argyropoulos, Green Chem., 2017, 19, 4104–4121 RSC.
- C. Cui, R. Sun and D. S. Argyropoulos, ACS Sustainable Chem. Eng., 2014, 2, 959–968 CrossRef CAS.
- L. Ji, Characterization of lignin molar mass and molecular conformation by multi-angle light scattering, University of British Columbia, 2019 Search PubMed.
- H. Sadeghifar, T. Wells, R. K. Le, F. Sadeghifar, J. S. Yuan and A. J. Ragauskas, ACS Sustainable Chem. Eng., 2017, 5, 580–587 CrossRef CAS.
- M. Gigli and C. Crestini, Green Chem., 2020, 22, 4722–4746 RSC.
- H. Sadeghifar and A. J. Ragauskas, ACS Sustainable Chem. Eng., 2020, 8, 8086–8101 CrossRef CAS.
- D. Kai, M. J. Tan, P. L. Chee, Y. K. Chua, Y. L. Yap and X. J. Loh, Green Chem., 2016, 18, 1175–1200 RSC.
- C. Wang, S. S. Kelley and R. A. Venditti, ChemSusChem, 2016, 9, 770–783 CrossRef CAS PubMed.
- V. Passoni, C. Scarica, M. Levi, S. Turri and G. Griffini, ACS Sustainable Chem. Eng., 2016, 4, 2232–2242 CrossRef CAS.
- P. Olsén, M. Jawerth, M. Lawoko, M. Johansson and L. A. Berglund, Green Chem., 2019, 21, 2478–2486 RSC.
- H. Zhang, Y. Bai, B. Yu, X. Liu and F. Chen, Green Chem., 2017, 19, 5152–5162 RSC.
- A. S. Jääskeläinen, T. Liitiä, A. Mikkelson and T. Tamminen, Ind. Crops Prod., 2017, 103, 51–58 CrossRef.
- A. Tagami, C. Gioia, M. Lauberts, T. Budnyak, R. Moriana, M. E. Lindström and O. Sevastyanova, Ind. Crops Prod., 2019, 129, 123–134 CrossRef CAS.
- A. Duval, F. Vilaplana, C. Crestini and M. Lawoko, Holzforschung, 2016, 70, 11–20 CAS.
- B. Li, M. Zhou, W. Huo, D. Cai, P. Qin, H. Cao and T. Tan, Ind. Crops Prod., 2020, 143, 111887 CrossRef CAS.
- S. Y. Park, J. Y. Kim, H. J. Youn and J. W. Choi, Int. J. Biol. Macromol., 2019, 138, 1029–1034 CrossRef CAS PubMed.
- C. Gioia, G. Lo Re, M. Lawoko and L. Berglund, J. Am. Chem. Soc., 2018, 140, 4054–4061 CrossRef CAS PubMed.
- H. Nadji, C. Bruzzèse, M. N. Belgacem, A. Benaboura and A. Gandini, Macromol. Mater. Eng., 2005, 290, 1009–1016 CrossRef CAS.
- A. Duval and L. Avérous, ACS Sustainable Chem. Eng., 2017, 5, 7334–7343 CrossRef CAS.
- I. Kühnel, B. Saake and R. Lehnen, React. Funct. Polym., 2017, 120, 83–91 CrossRef.
- A. Duval and L. Avérous, ACS Sustainable Chem. Eng., 2016, 4, 3103–3112 CrossRef CAS.
- L. Y. Liu, M. Cho, N. Sathitsuksanoh, S. Chowdhury and S. Renneckar, ACS Sustainable Chem. Eng., 2018, 6, 12251–12260 CrossRef CAS.
- A. Duval, G. Layrac, A. van Zomeren, A. T. Smit, E. Pollet and L. Avérous, ChemSusChem, 2021, 14, 387–397 CrossRef CAS PubMed.
- L. Y. Liu, K. Bessler, S. Chen, M. Cho, Q. Hua and S. Renneckar, Eur. Polym. J., 2021, 142, 110082 CrossRef CAS.
- H. Sadeghifar, C. Cui and D. S. Argyropoulos, Ind. Eng. Chem. Res., 2012, 51, 16713–16720 CrossRef CAS.
- C. A. Cateto, M. F. Barreiro, A. E. Rodrigues and M. N. Belgacem, Ind. Eng. Chem. Res., 2009, 48, 2583–2589 CrossRef CAS.
- K. A. Y. Koivu, H. Sadeghifar, P. A. Nousiainen, D. S. Argyropoulos and J. Sipilä, ACS Sustainable Chem. Eng., 2016, 4, 5238–5247 CrossRef CAS.
- O. Gordobil, I. Egüés and J. Labidi, React. Funct. Polym., 2016, 104, 45–52 CrossRef CAS.
- L. Dehne, C. Vila, B. Saake and K. U. Schwarz, J. Appl. Polym. Sci., 2017, 134, 1–8 CrossRef.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- Y. Hayashi, Chem. Sci., 2016, 7, 866–880 RSC.
- L. Albrecht, H. Jiang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 8492–8509 CrossRef CAS PubMed.
- S. Tulaphol, M. A. Hossain, M. S. Rahaman, L. Y. Liu, T. K. Phung, S. Renneckar, N. Grisdanurak and N. Sathitsuksanoh, Energy Fuels, 2020, 34, 1764–1772 CrossRef CAS.
- J. Shi, J. M. Gladden, N. Sathitsuksanoh, P. Kambam, L. Sandoval, D. Mitra, S. Zhang, A. George, S. W. Singer, B. A. Simmons and S. Singh, Green Chem., 2013, 15, 2579–2589 RSC.
- E. L. Hult, K. Koivu, J. Asikkala, J. Ropponen, P. Wrigstedt, J. Sipilä and K. Poppius-Levlin, Holzforschung, 2013, 67, 899–905 CAS.
- S. Luo, J. Cao and A. G. McDonald, Ind. Crops Prod., 2017, 97, 281–291 CrossRef CAS.
- W. Thielemans and R. P. Wool, Biomacromolecules, 2005, 6, 1895–1905 CrossRef CAS PubMed.
- L. Y. Liu, Q. Hua and S. Renneckar, Green Chem., 2019, 21, 3682–3692 RSC.
- L. An, C. Si, G. Wang, C. S. Choi, Y. H. Yu, J. H. Bae, S. M. Lee and Y. S. Kim, BioResources, 2020, 15, 89–104 CAS.
- E. Husson, L. Hulin, C. Hadad, C. Boughanmi, T. Stevanovic and C. Sarazin, Front. Chem., 2019, 7, 1–13 CrossRef PubMed.
- N. Cachet, S. Camy, B. Benjelloun-Mlayah, J. S. Condoret and M. Delmas, Ind. Crops Prod., 2014, 58, 287–297 CrossRef CAS.
- C. Cai, K. Hirth, R. Gleisner, H. Lou, X. Qiu and J. Y. Zhu, Green Chem., 2020, 22, 1605–1617 RSC.
- C. Su, K. Hirth, Z. Liu, Y. Cao and J. Y. Zhu, Ind. Crops Prod., 2021, 159, 113017 CrossRef CAS.
- W. Zhao, L. P. Xiao, G. Song, R. C. Sun, L. He, S. Singh, B. A. Simmons and G. Cheng, Green Chem., 2017, 19, 3272–3281 RSC.
- F. Francuskiewicz, Polymer Fractionation, Springer Berlin Heidelberg, Wilminton, 1994 Search PubMed.
- L. Y. Liu, K. Bessler, S. Chen, M. Cho, Q. Hua and S. Renneckar, Data Brief, 2020, 33, 106512 CrossRef PubMed.
- S. Podzimek, Light Scattering, Size Exclusion Chromatography and Asymmetric Flow Field Flow Fractionation, 2011 Search PubMed.
- C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef CAS.
- A. Keshav, K. L. Wasewar and S. Chand, Ind. Eng. Chem. Res., 2008, 47, 6192–6196 CrossRef CAS.
- H. Yue, Y. Zhao, X. Ma and J. Gong, Chem. Soc. Rev., 2012, 41, 4218–4244 RSC.
- IRENA, Renewable Power Generation Costs in 2019 - Key Findings, 2020 Search PubMed.
- L. Wang, Y. Uraki, K. Koda, A. Gele, X. Zhou and F. Chen, Holzforschung, 2019, 73, 363–369 CrossRef CAS.
- G. Zinovyev, I. Sulaeva, S. Podzimek, D. Rössner, I. Kilpeläinen, I. Sumerskii, T. Rosenau and A. Potthast, ChemSusChem, 2018, 11, 3259–3268 CrossRef CAS PubMed.
- E. J. Siochi, T. C. Ward, M. A. Haney and B. Mahn, Macromolecules, 1990, 23, 1420–1429 CrossRef CAS.
- B. Cathala, B. Saake, O. Faix and B. Monties, J. Chromatogr. A, 2003, 1020, 229–239 CrossRef CAS PubMed.
- W. Zhao, B. Simmons, S. Singh, A. Ragauskas and G. Cheng, Green Chem., 2016, 18, 5693–5700 RSC.
- G. E. Fredheim, S. M. Braaten and B. E. Christensen, J. Chromatogr. A, 2002, 942, 191–199 CrossRef CAS PubMed.
- S. Podzimek, T. Vlcek and C. Johann, J. Appl. Polym. Sci., 2001, 81, 1588–1594 CrossRef CAS.
- M. Balakshin and E. Capanema, J. Wood Chem. Technol., 2015, 35, 220–237 CrossRef CAS.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. De Peinder, R. Boelens, D. S. Van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- J. Ralph, K. Lundquist, G. Brunow, F. Lu, H. Kim, P. F. Schatz, J. M. Marita, R. D. Hatfield, S. A. Ralph, J. H. Christensen and W. Boerjan, Phytochem. Rev., 2004, 3, 29–60 CrossRef CAS.
- J. Ralph, C. Lapierre and W. Boerjan, Curr. Opin. Biotechnol., 2019, 56, 240–249 CrossRef CAS PubMed.
- J. Gierer, Wood Sci. Technol., 1980, 14, 241–266 CrossRef CAS.
- C. S. Lancefield, H. J. Wienk, R. Boelens, B. M. Weckhuysen and P. C. A. Bruijnincx, Chem. Sci., 2018, 9, 6348–6360 RSC.
- M. Y. Balakshin and E. A. Capanema, RSC Adv., 2015, 5, 87187–87199 RSC.
- A. P. Dodd, J. F. Kadla and S. K. Straus, ACS Sustainable Chem. Eng., 2015, 3, 103–110 CrossRef CAS.
- O. Gordobil, I. Egüés, R. Llano-Ponte and J. Labidi, Polym. Degrad. Stab., 2014, 108, 330–338 CrossRef CAS.
- S. Kubo and R. D. Gilbert, Kadla in Natural Fibers, Biopolymers, and Biocomposites, 2004, 682 Search PubMed.
- A. Naseem, S. Tabasum, K. M. Zia, M. Zuber, M. Ali and A. Noreen, Int. J. Biol. Macromol., 2016, 93, 296–313 CrossRef CAS PubMed.
- S. Sen, S. Patil and D. S. Argyropoulos, Green Chem., 2015, 17, 4862–4887 RSC.
- D. Kun and B. Pukánszky, Eur. Polym. J., 2017, 93, 618–641 CrossRef CAS.
- L. Wang, K. Shigetomi, K. Koda, A. Gele and Y. Uraki, Holzforschung, 2020, 74, 551–558 CrossRef CAS.
- A. K. Brewer and A. M. Striegel, Analyst, 2011, 136, 515–519 RSC.
- S. Chakraborty, J. Ding, M. C. Thies and C. L. Kitchens, ACS Appl. Polym. Mater., 2019, 1, 2561–2565 CrossRef CAS.
- M. Cho, F. K. Ko and S. Renneckar, Biomacromolecules, 2019, 20(12), 4485–4493 CrossRef CAS PubMed.
- W. G. Glasser, V. Davé and C. E. Frazier, J. Wood Chem. Technol., 1993, 13, 545–559 CrossRef CAS.
- P. Li, S. Wang, F. Meng, Y. Wang, F. Guo, S. Rajendran, C. Gao, Z. Xu and Z. Xu, Macromolecules, 2020, 53, 10421–10430 CrossRef CAS.
- T. P. Lodge and P. C. Hiemenz, Polymer, 2020, 676 Search PubMed.
- S. Constant, C. S. Lancefield, B. M. Weckhuysen and P. C. A. Bruijnincx, ACS Sustainable Chem. Eng., 2017, 5, 965–972 CrossRef CAS.
- M. Cho, M. A. Karaaslan, S. Renneckar and F. Ko, J. Mater. Sci., 2017, 52, 9602–9614 CrossRef CAS.
- M. Zhang and A. A. Ogale, Carbon, 2014, 69, 626–629 CrossRef CAS.
- B. M. Elsiwi, O. Garcia-Valdez, H. C. Erythropel, R. L. Leask, J. A. Nicell and M. Maric, ACS Sustainable Chem. Eng., 2020, 8, 12409–12418 CrossRef CAS.
- H. Sadeghifar and A. Ragauskas, Polymers, 2020, 12, 1–10 CrossRef PubMed.
- Y. Qian, Y. Deng, X. Qiu, H. Li and D. Yang, Green Chem., 2014, 16, 2156–2163 RSC.
- M. H. Sipponen, H. Lange, C. Crestini, A. Henn and M. Österberg, ChemSusChem, 2019, 12, 2039–2054 CrossRef CAS PubMed.
- T. Renders, G. Van den Bossche, T. Vangeel, K. Van Aelst and B. Sels, Curr. Opin. Biotechnol., 2019, 56, 193–201 CrossRef CAS PubMed.
- S. Luo, J. Cao and A. G. McDonald, Ind. Crops Prod., 2018, 121, 169–179 CrossRef CAS.
- Z. Sun, B. Fridrich, A. De Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- E. Norgbey, J. Huang, V. Hirsch, W. Liu, M. Wang, O. Ripke, Y. Li, G. E. Takyi-Annan, D. Ewusi-Mensah, X. Wang, G. Treib, A. Rink, A. S. Nwankwegu, P. A. Opoku and P. N. Nkrumah, Constr. Build. Mater., 2020, 230, 116957 CrossRef CAS.
- K. B. Batista, R. P. L. Padilha, T. O. Castro, C. F. S. C. Silva, M. F. A. S. Araújo, L. F. M. Leite, V. M. D. Pasa and V. F. C. Lins, Ind. Crops Prod., 2018, 111, 107–116 CrossRef CAS.
- I. P. Pérez, A. R. Pasandín, J. C. Pais and P. A. Alves Pereira, J. Cleaner Prod., 2019, 220, 87–98 CrossRef.
- M. Balakshin, E. A. Capanema, X. Zhu, I. Sulaeva, A. Potthast, T. Rosenau and O. J. Rojas, Green Chem., 2020, 22, 3985–4001 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc01033f |
| This journal is © The Royal Society of Chemistry 2021 |