
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePaired electrolysis of 5-(hydroxymethyl)furfural in flow cells with a high-performance oxide-derived silver cathode†
Hengzhou
Liu‡
,
Ting-Han
Lee‡
,
Yifu
Chen
 ,
Eric W.
Cochran
* and
Wenzhen
Li
*
,
Eric W.
Cochran
* and
Wenzhen
Li
*
Department of Chemical and Biological Engineering, Iowa State University, 618 Bissell Road, Ames, IA 50011, USA. E-mail: wzli@iastate.edu; ecochran@iastate.edu
First published on 10th May 2021
Abstract
Electrocatalytic upgrading of biomass-derived feedstocks driven by renewable electricity offers a greener way to reduce the global carbon footprint associated with the production of value-added chemicals. In this respect, a key strategy is the electrocatalytic hydrogenation (ECH) reaction, which is typically paired with the anodic oxygen evolution reaction (OER) with sluggish kinetics, producing O2 with little value. Here we prepared an oxide-derived Ag (OD-Ag) electrode with high activity and up to 98.2% selectivity for the ECH of 5-(hydroxymethyl)furfural (HMF) to 2,5-bis(hydroxymethyl)furan (BHMF), and such efficient conversion was achieved in a three-electrode flow cell. The excellent BHMF selectivity was maintained over a broad potential range with long-term operational stability. We then considered the oxidation of HMF to 2,5-furandicarboxylic acid (FDCA) and hydrogen (to water) as more efficient and productive alternatives to the OER. In HMF-to-BHMF paired with 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO)-mediated HMF-to-FDCA conversion, a markedly reduced cell voltage from ∼7.5 V to ∼2.0 V was observed by transferring the electrolysis from the H-type cell to the flow cell, corresponding to more than four-fold increase in energy efficiency in operation at 10 mA. A combined faradaic efficiency of 163% was obtained for BHMF and FDCA. Alternatively, the anodic hydrogen oxidation reaction on platinum further reduced the cell voltage to only ∼0.85 V at 10 mA. These paired processes show the potential for integration of renewable electricity and carbon for green and economically feasible distributed chemical manufacturing.
Introduction
Electrocatalytic conversions of biomass-derived feedstocks, particularly driven by renewable electricity sources from wind and sunlight, have attracted enormous attention.1 Replacing fossil-based processes with renewable energy reduces the emission of greenhouse gases and helps mitigate their adverse impact on the environment.2 The driving force of electrochemical reactions is the electrode potential instead of thermal energy, with “clean” electrons as the reducing and oxidizing agents rather than toxic reductants and oxidants. However, in many circumstances, the complementary reaction at the counter electrode has sluggish kinetics and yields low-value products.3 For instance, the anodic four-electron oxygen evolution reaction (OER) is unfavorable both thermodynamically and kinetically, and produces O2 with little value. Indeed, it was found that the anodic OER consumes ∼90% of the full cell voltage and energy consumption in CO2 electro-reduction processes.4Pairing two desirable electrochemical half-reactions in one reactor can achieve the co-generation of valuable chemicals and fuels at both the cathode and anode, which is regarded as a promising approach to increase energy efficiency and economic feasibility.5 Ideally, a 200% theoretical faradaic efficiency (FE) to desired products can be obtained, doubling the conventional cells with only one desirable half-reaction. Moreover, pairing one electrochemical half-reaction more favorable than OER at the anode or hydrogen evolution reaction (HER) at the cathode can significantly lower the cell voltage, and hence, the overall energy consumption, as suggested by the recent techno-economic study.6 Some examples of paired electrolysis have been demonstrated, such as CO2 reduction coupled with organic oxidations4,7–11 or oxidative treatment of nitrogenous wastes,12 and co-electrolysis of two organics.13–16 To date, reports of paired electrolysis have mainly been proof-of-concept studies conducted in H-type reactors in order to evaluate electrolysis performances;13,17 however, the high energy cost and operational inefficiencies associated with the H-cells are critical barriers to further development of the economically feasible scale-up design of such processes.18 In this regard, continuous operation in a flow reactor can greatly reduce the ohmic loss, thus improving the energy efficiency. In addition, enhanced mass transport would be desirable for scale-up studies for economically feasible electrosynthesis.
5-(Hydroxymethyl)furfural (HMF) has been listed as one of the “top 10” biobased chemicals by the U.S. Department of Energy as a platform for value-added products.19,20 Its hydrogenation product 2,5-bis(hydroxymethyl)furan (BHMF) is an essential precursor for the production of polyesters and resins.21 Its oxidation product 2,5-furandicarboxylic acid (FDCA) is a feedstock to produce renewable polymers such as polyethylene 2,5-furandicarboxylate (PEF) as a promising alternative to polyethylene terephthalate (PET).22 Electrochemical conversions of HMF have been studied in recent years, such as its electrocatalytic hydrogenation (ECH) to BHMF on Ag-based catalysts,17,23 and electrocatalytic oxidation (ECO) to FDCA via a redox mediator24 or on Ni- and Co-based catalysts in alkaline electrolytes.25–28 Our previously reported Ag/C catalyst with nano-sized Ag showed higher activity than Ag foil, but the carbon support was found to interfere with the HMF reduction pathway, leading to the generation of undesirable dimeric product [5,5′-bis(hydroxymethyl)hydrofuroin, BHH] at higher overpotentials.17,23 Moreover, co-electrolysis by pairing ECH and ECO of HMF in one cell has rarely been reported, especially in flow electrolyzers with optimized cell voltage.
Herein, we demonstrated the significant enhancement in ECH activity for the HMF-to-BHMF reaction on an oxide-derived silver (OD-Ag) electrode compared to Ag foil, alongside 90+% BHMF selectivity in a wide potential range. The excellent performance on OD-Ag is due to its wave-like surface morphology, which provides more active sites and lowers the energy barriers for hydrogen and HMF adsorption. HMF conversion and BHMF selectivity were further improved in a three-electrode flow cell for long-term operations as compared with the H-type cell. Coupling the ECH (HMF-to-BHMF) reaction on OD-Ag with the ECO (HMF-to-FDCA) reaction mediated by (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO), a significantly reduced cell voltage from ∼7.5 V in the H-type cell to ∼2.0 V in the flow cell was achieved, corresponding to more than four-fold increase in energy efficiency at 10 mA. The total FE to BHMF and FDCA of up to 163% was obtained for the co-electrolysis of HMF. The same ECH reaction was also coupled with hydrogen oxidation reaction (HOR) on Pt/C, resulting in a cell voltage of only ∼0.85 V at 10 mA. Our paired electrolytic system will facilitate further advancements in coupled processes for efficient and sustainable utilization of bio-derived molecules to produce value-added chemicals with lower energy inputs.
Results and discussion
Synthesis and characterization of the OD-Ag electrocatalyst
OD-Ag was prepared by treating Ag foil with high-frequency (500 Hz) square-wave voltammetry (SWV) followed by constant-potential electrolysis at −1.3 VAg/AgCl (VAg/AgCl: V vs. Ag/AgCl, hereinafter). The color of the Ag foil periodically changes between white and black during the SWV operation, and a yellow surface was finally obtained after the constant-potential electrolysis (Fig. S1a†). These black and yellow layers were AgOx and OD-Ag, respectively, as confirmed by the cyclic voltammogram (CV) under the same alkaline system and the Pourbaix diagram of silver (Fig. S1b and c†).29 XRD patterns (Fig. 1a) confirmed the mono-constituent Ag0 in OD-Ag. XPS (Fig. 1b) exhibited the same binding energies of Ag 3d3/2 (374.1 eV) and 3d5/2 (368.0 eV) for OD-Ag and Ag foil, respectively. SEM imaging (Fig. 1c) shows that OD-Ag has a rough surface consisting of around 100 nm particles. AFM analysis (Fig. 1d) revealed that OD-Ag has a periodic wave-like morphology with ± 250 nm of surface depth, in contrast to the smooth surface of Ag foil (Fig. S2†). The above characterization results clearly demonstrate the successful preparation of the OD-Ag electrode. | ||
| Fig. 1 Physical characterization of OD-Ag electrocatalyst. (a) XRD patterns and (b) XPS Ag 3d spectra of Ag foil, freshly prepared OD-Ag, and OD-Ag after electrolysis. (c) SEM image of OD-Ag. (d) AFM image of OD-Ag. The inset graph is the height profile of a 6 μm section (the white horizontal line). | ||
ECH of HMF on OD-Ag in the H-type cell
It is known that metal electrodes subjected to oxidation–reduction treatment may possess nanostructured surfaces, which could enhance their electrocatalytic performances.30,31 Such enhancement is yet to be reported for HMF reduction.We first performed CV in the electrolyte with 20 mM HMF (Fig. 2a and b). Compared to Ag foil, OD-Ag showed 240 mV of a positive shift in the onset potential (defined as the potential at −1.0 mA cm−2 in the negative scan). The current density on OD-Ag was 2–3 times higher than that on Ag foil in the measured potential range. Besides, half-hour CA tests showed that OD-Ag delivered not only higher HMF conversion but also higher BHMF selectivity than Ag foil at potentials more negative than −1.25 VAg/AgCl (Fig. 2c): 90+% of BHMF selectivity was obtained on OD-Ag, in contrast to 67.0–82.9% of selectivity on Ag foil. At −1.25 VAg/AgCl, the selectivity to the dimeric product BHH was 10.2% on OD-Ag, much lower than that of 17.3% on Ag foil. In addition, the chemical state of Ag in OD-Ag was unchanged after ECH of HMF (Fig. 1a and b).
 | ||
| Fig. 2 ECH of HMF in the H-type cell. (a) Schematic illustration of the H-type cell for ECH of HMF. (b) Cyclic voltammograms of Ag foil and OD-Ag in 0.5 M borate buffer (pH 9.2) with 20 mM HMF. The geometric area of the electrode was 2 cm2 and the scan rate was 20 mV s−1. Onset potential was defined as the potential at −1.0 mA cm−2 in the negative scan. (c) Product selectivity and conversion of HMF in 0.5 M borate buffer (pH 9.2) with 20 mM HMF on Ag foil (left columns) and OD-Ag (right columns) at different applied potentials for half-hour electrolysis. | ||
Selectivity depicts the distribution of HMF reduction products based on the carbon balance, while FE is defined by the fraction of total charges to produce a certain product. As shown in Fig. 2c and Fig. S4,† the selectivity of BHMF increased in line with the increased FE of H2 (from HER) as the potential became more negative on OD-Ag. Also, the observed FE of H2 on OD-Ag outperformed that on Ag foil. Such parallel behavior could be attributed to the common reaction intermediate (Hads, generated from H+ + e− → Hads) of ECH and HER; thus, higher coverage of Hads on the electrode surface would increase the BHMF selectivity, and simultaneously sacrifice some charges for Hads–Hads combination to form H2.32 Apparently, binding of Hads is intrinsically more favorable on the surface of OD-Ag compared to Ag foil. Accordingly, the dimeric product BHH becomes less favorable on OD-Ag. As the mechanisms suggested in previous studies,33,34 BHH is formed by one adsorbed H coupled with one adsorbed HMF molecule, followed by rapid desorption as free radicals for dimerization before the addition of the second Hads to BHMF. The lower BHH selectivity on OD-Ag could be due to the appropriate binding energy of HMF and its reduction intermediates on the surface for two Hads addition to BHMF instead of one Hads addition to radicals. The above observations essentially stem from the wave-like morphology and higher surface area of OD-Ag, which reduce the energy barriers for the adsorption of both H and HMF.
ECH of HMF on OD-Ag in the three-electrode flow cell
A three-electrode flow electrolyzer was designed to reduce the limitation of mass transport in the continuous ECH of HMF. As shown in Fig. 3a, the catholyte was fed into a PTFE spacer with the Ag/AgCl reference electrode, and the anolyte was fed via a flow-field plate to a catalyst-supported gas diffusion electrode attached to the anion exchange membrane. | ||
| Fig. 3 ECH of HMF in the flow reactor. (a) Schematic illustration of the three-electrode flow cell, where Ag/AgCl was placed in the cathodic spacer to control the applied potential. ECH of HMF was performed on OD-Ag. The anodic reaction was OER. (b) Comparison of product selectivity and HMF conversion on different electrodes and cell configurations at −1.25 VAg/AgCl for half-hour electrolysis. (c) HMF conversion and (d) BHMF selectivity in three-electrode flow cell on Ag foil and OD-Ag at different potentials for 3-hour electrolysis. The samples were quantified in half-hour intervals. The electrolyte was 0.5 M borate buffer (pH 9.2) with 20 mM HMF. Error bars represent the standard deviation of the mean value from at least three measurements. The detailed experimental setup is shown in Fig. S3.† | ||
The ECH performances in different cell configurations are compared in Fig. 3b. Both HMF conversion and BHMF selectivity are higher in the flow cell, following the sequence: OD-Ag in flow cell > OD-Ag in H-type cell ≫ Ag foil in flow cell > Ag foil in H-type cell. In the flow cell, HMF conversion and BHMF selectivity were also higher on OD-Ag than that on Ag foil (HMF conversion: 28.9% vs. 22.1%; BHMF selectivity: 95.3% vs. 68.8% at −1.25 VAg/AgCl for half-hour electrolysis), showing the same trend as the measurements in the H-type cell. In addition, with the same electrode of OD-Ag for a half-hour electrolysis, HMF conversion in the flow cell has outperformed the performance in the H-type cell (28.9% vs. 24.0%), benefiting from the enhanced mass transport in the flow reactor.
The superior ECH activity and BHMF selectivity on OD-Ag can be maintained in a broad potential range. As shown in Fig. 3c and d, in 3-hour continuous measurements, the BHMF selectivity maintained 80+% in the potential range of −1.05 to −1.35 V. The high HMF conversion has achieved over 90% at potentials more negative than −1.15 V, which also revealed the superior capability of the flow reactor for continuous ECH of HMF with minimal external mass transport limitations. In addition, the consistent activity obtained on OD-Ag confirmed the high durability and robustness of the system under testing conditions, without significant performance drop issues.
ECH–ECO paired electrolysis in the flow cell
The above experiments were carried out with OER as the anodic reaction, which is known to be thermodynamically and kinetically sluggish. Thermodynamic calculations of the ECH–OER electrolytic system also suggest that the energy consumption is mainly attributed to OER (eqn (1)–(3)):Cathode:
 | (1) |
Anode:
| 2OH− → H2O + 0.5O2 + 2e−, ΔG° = 237.10 kJ mol−1 | (2) |
Overall:
| HMF + H2O → BHMF + 0.5O2, ΔG° = 214.10 kJ mol−1 | (3) |
Alternatively, by replacing the anodic OER with the ECO of HMF to a value-added product FDCA (denoted as ECH–ECO paired electrolysis), the required energy input can be largely brought down, as demonstrated by the decrease in the thermodynamic cell potential |E°| from 1.11 V to 0.33 V (Fig. 4a).
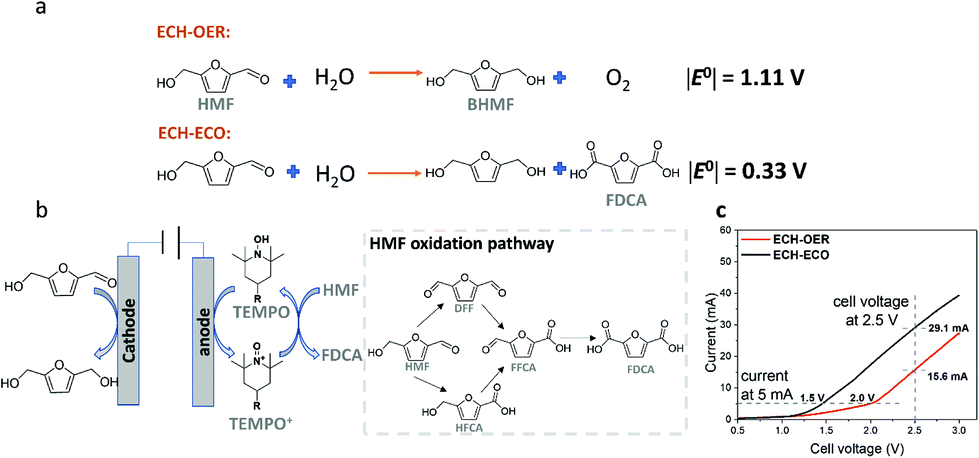 | ||
Fig. 4 Comparison of ECH–OER and ECH–ECO paired electrolytic system in the flow reactor. (a) Schematic illustration of ECH–ECO paired electrolytic system, with HMF reduction to BHMF at the OD-Ag cathode and TEMPO-mediated HMF oxidation at the anode. Two possible HMF oxidation pathways17,24 are shown in the dotted box. (b) Calculated thermodynamic cell potentials  of the ECH–OER system and the ECH–ECO system. (c) Linear sweep voltammograms of the ECH–OER and ECH–ECO systems in 0.5 M borate buffer (pH 9.2) with 20 mM HMF. The scan rate was 5 mV s−1. of the ECH–OER system and the ECH–ECO system. (c) Linear sweep voltammograms of the ECH–OER and ECH–ECO systems in 0.5 M borate buffer (pH 9.2) with 20 mM HMF. The scan rate was 5 mV s−1. | ||
To demonstrate the feasibility of the paired electrolysis experimentally, we first performed linear sweep voltammetry (LSV) analysis in the flow cell, with ECH of HMF catalyzed by OD-Ag and ECO of HMF mediated by TEMPO (Fig. 4b). The results for ECH–ECO paired electrolysis (Fig. 4c) exhibited a remarkable decrease in cell voltage compared to the ECH–OER system: 1.5 V vs. 2.0 V at 5 mA; 29.1 mA vs. 15.6 mA at 2.5 V.
To conduct bulk electrolysis in the ECH–ECO paired system, we first confirmed the facile kinetics and high HMF-to-FDCA selectivity mediated by TEMPO in the H-type cell. As shown in Table S2,† both constant-potential (in the range of 0.4–0.8 VAg/AgCl) and constant-current measurements (at 10 mA) achieved ∼100% HMF conversion with ∼100% selectivity and 90+% FE to FDCA.
The ECH–ECO paired electrolysis was then conducted in different cell configurations with ECH on OD-Ag and ECO mediated by TEMPO as the cathodic and anodic reactions, respectively. A schematic illustration of the flow cell configuration is shown in Fig. S5.† A substantial decrease in the steady-state cell voltage from ∼7.5 V to ∼2.0 V was observed when the electrolysis was conducted in the flow cell compared to the H-type cell (Fig. 5a), at a constant current of 10 mA. The reduction in voltage is in line with the decrease in the measured resistance between the cathode and anode from 483.2 Ω for the H-type cell to 39.3 Ω for the flow cell (Table S7†). No considerable performance drop was observed in three consecutive 1-hour measurements.
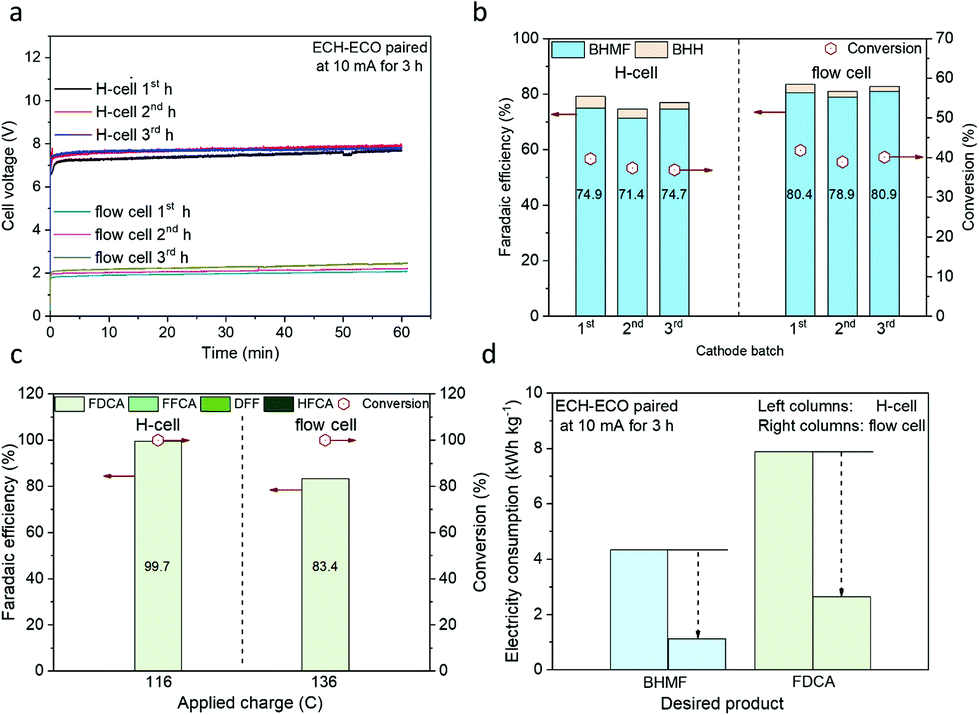 | ||
| Fig. 5 Comparison of ECH–ECO paired electrolysis in the flow cell and H-type cell at 10 mA. (a) Cell voltage profiles for three consecutive 1-hour electrolysis in the H-type cell and flow cell. (b) and (c) Faradaic efficiency and HMF conversion for cathodic and anodic reactions in the H-type cell (left columns) and flow cell (right columns). (d) Comparison of electricity consumption for BHMF and FDCA production in different cell configurations. The catholyte was 20 ml of 0.5 M borate buffer (pH 9.2) with 20 mM HMF, and the anolyte was 20 ml of 0.5 M borate buffer (pH 9.2) with 10 mM HMF and 7.5 mM TEMPO. The applied charge for the H-type cell test was 116 C, corresponding to the theoretical charge required for 100% conversion of HMF to FDCA. For the flow cell, 136 C was applied in order to achieve ∼100% conversion to FDCA. Fresh catholyte was replaced every hour during tests. More details and additional data can be found in Tables S3–S6.† | ||
Furthermore, the FE of BHMF was well maintained at ∼80% in the flow cell, slightly higher than the results in the H-type cell (Fig. 5b). The anodic FE was marginally lower in the flow cell compared to the H-type cell, as more charge (136 C) than the theoretical value (116 C) was required to fully convert HMF to FDCA (Fig. 5c). The additional charge consumption, presumably due to the side reaction OER, could be attributed to the imprecise control of the anodic potential. Alternatively, by controlling the applied potential between the reference electrode and anode at 0.6 V (which was selected due to the high performance of HMF-to-FDCA in the range of 0.4–0.8 VAg/AgCl from the H-type cell measurements) (Fig. S6†), the FE of HMF to FDCA was improved to 92.7% (Fig. 6). However, a decrease in the FE of BHMF on the cathode was observed as the potential between the cathode and reference electrode was no longer constant. Further optimization is needed to tackle the challenge of simultaneously controlling the applied potentials on both the cathode and anode within the desirable range, thereby maximizing the overall FE.
 | ||
| Fig. 6 ECH–ECO paired electrolysis in the three-electrode flow cell at a constant potential of 0.6 V between the reference electrode and anode. (a) Cell voltage profiles for ECH–ECO paired electrolysis in the H-type cell and flow cell at a constant potential of 0.6 VAg/AgCl between the reference electrode and anode. The cell voltage was measured with a multimeter. (b) Faradaic efficiency and HMF conversion for cathodic and anodic reactions in the H-type cell (left columns) and flow cell (right columns).The catholyte was 20 ml of 0.5 M borate buffer (pH 9.2) with 20 mM HMF, and the anolyte was 20 ml of 0.5 M borate buffer (pH 9.2) with 5 mM HMF and 7.5 mM TEMPO. The applied charge was 57.9 C, corresponding to the theoretical charge required for 100% conversion of HMF to FDCA. More details and additional data can be found in Tables S8 and S9.† | ||
No matter in constant current (10 mA) or constant half-cell potential (0.6 VAg/Agcl) operations, a combined FE of over 160% was always maintained in our flow reactor to the desired BHMF and FDCA. The selectivities to BHMF and FDCA were close to 90% and 100% on the cathode and anode, respectively (Tables S3–S6†). The electricity consumption (unit: kW h kg−1, normalized to the mass of the product) to the desired products is shown in Fig. 5d. Compared to the H-type cell, an over three-fold decrease of electricity consumption was obtained in the flow cell to produce BHMF and FDCA. The cell energy efficiency (ε) as a function of cell voltage was also calculated (detailed in Table S1†), which is 24.5% for the flow cell, exhibiting an over four-fold increase compared to the H-type cell (5.7%).
The versatility of the paired electrolysis was demonstrated by extending the substrate to furfural, another bio-derived chemical with an industrial production capacity of 400 ktons per year.35,36 By pairing the ECH of furfural-to-furfuryl alcohol (FA) on Pb foil with the ECO of furfural-to-furoic acid mediated by TEMPO, a total FE of 177% (90.6% to FA and 87.2% to 2-furoic acid) was obtained (Fig. S7 and Tables S10, 11†). In addition, the superior performance and excellent flexibility of our flow cell for paired electrolysis have approached or outperformed many state-of-the-art reaction systems, as shown in a detailed comparison in Table S12.†
ECH–HOR paired electrolysis in the flow cell
To further reduce the cell voltage, we performed ECH–HOR paired electrolysis with HMF reduction as the cathodic reaction and HOR as the anodic reaction (Fig. 7a). OD-Ag and Pt/C-coated carbon paper were used as the cathode and anode, respectively. As shown in the LSV curve (Fig. 7b), only 0.85 V of the total cell voltage is required to attain a current of 10 mA. The selectivity to BHMF was as high as 82.3% at the cell voltage of 0.90 V (Fig. 7c). The ECH–HOR paired electrolysis offers another sustainable pathway for BHMF production with low energy cost. The continuous fall of renewable electricity prices, along with further development of electrolyzers for H2 generation and conversion of bio-derived chemicals, warrants the potential feasibility of such electrochemical process in the future. | ||
| Fig. 7 ECH of HMF paired with the hydrogen oxidation reaction (HOR) in the flow cell. (a) Schematic illustration of the flow cell for ECH–HOR paired electrolysis. (b) Linear sweep voltammogram of paired electrolysis with a scan rate of 5 mV s−1. (c) Product selectivity and HMF conversion on OD-Ag at the cell voltage of 0.9 V for half-hour electrolysis. The cathode was OD-Ag and the anode was a Pt/C-loaded carbon paper (0.5 mgPt cm−2). The catholyte was 20 ml of 0.5 M borate buffer (pH 9.2) with 20 mM HMF. The flow rate of H2 was 100 ml min−1. | ||
Conclusions
High selectivity (90+%) of HMF reduction to BHMF was observed on OD-Ag, which significantly outperformed Ag foil. Such performance can be maintained in a wide potential range (−1.05 V to −1.35 V) and is stable for prolonged electrolysis. Substitution of the anodic OER by TEMPO-mediated HMF oxidation notably lowered the overall cell voltage, which was further reduced to 2.0 V (at 10 mA) in a flow cell. Such ECH–ECO paired electrolysis in the flow cell offered a combined FE of 163% to the valuable products (BHMF and FDCA) and demonstrated an over four-fold increase in energy efficiency. Using HOR as an alternative anodic reaction, the cell voltage was further reduced to 0.85 V, offering a potential renewable substitute of thermocatalytic hydrogenation of bio-based chemicals. The efficient conversion of HMF, alongside the concept of paired electrolysis developed in the present study, will open up sustainable, economically viable, and environmentally benign routes of harnessing renewable energy for efficient utilization of biomass-derived feedstocks for distributed manufacturing of valuable chemicals.Experimental section
Chemicals and materials
Sodium hydroxide (97%), potassium hydroxide (85%), furfural (99%), furfural alcohol (FA, 98%), sodium sulfate (99%), 5-(hydroxymethyl)furfural (HMF, 99%), TEMPO[(2,2,6,6-tetramethylpiperidin-1-yl)oxyl] (98%), 2-furoic acid (98%), 2,5-furandicarboxylic acid (FDCA, 97%), 5-hydroxymethyl-2-furancarboxylic acid (HFCA, 99%), and 2,5-diformylfuran (DFF, 97%) were purchased from Sigma-Aldrich. Acetonitrile (CH3CN, HPLC grade), platinum foil (0.025 mm thick, 99.9%), 2-propanol (99.9%), and boric acid (≥99%) were purchased from Fisher Scientific. 2,5-Bis(hydroxymethyl)furan (BHMF, 98%) was purchased from Ark Pharm, Inc. 5-Formyl-2-furoic acid (FFCA, 99%) was purchased from TCI. Silver foil (0.5 mm thick, 99.9985%) was purchased from Alfa Aesar. Teflon (0.002-inch thickness), silicon (0.01-inch thickness) gaskets, Nafion membrane, Toray carbon paper 030, and plain carbon cloth were purchased from the Fuel Cell Store. 40 wt% Pt on Vulcan XC-72 (Pt/C) was purchased from Premetek. A201 anion exchange membrane was purchased from Tokuyama Corp. H2 calibration gases (10 ppm, 100 ppm, 1000 ppm, 5000 ppm, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm, balance helium) were purchased from Cal Gas Direct. Deionized (DI) water (18.2 MΩ cm, Barnstead™ E-Pure™) was used for all experiments in this work. All electrochemical tests were performed by a Biologic SP-300 potentiostat with a ±2 A/±30 V booster.
000 ppm, balance helium) were purchased from Cal Gas Direct. Deionized (DI) water (18.2 MΩ cm, Barnstead™ E-Pure™) was used for all experiments in this work. All electrochemical tests were performed by a Biologic SP-300 potentiostat with a ±2 A/±30 V booster.
Preparation of the working electrode
OD-Ag was prepared in a standard three-electrode system by a modified electrochemical square-wave-voltammetry (SWV) method.37 A piece of polished and cleaned Ag foil was immersed in a 0.2 M NaOH electrolyte as the working electrode. Ag/AgCl (saturated KCl, Pine Research) and platinum foil served as the reference electrode and counter electrode, respectively. Cyclic voltammetry (CV) was applied from 0 to 1.2 VAg/AgCl with a scan rate of 20 mV s−1. A symmetric square-wave pulse potential was then applied on Ag foil from 0 to 1.0 VAg/AgCl at a frequency of 500 Hz for 3 h with both positive and negative scans. Subsequently, a constant potential (−1.3 VAg/AgCl) was applied for 10 min to obtain OD-Ag.To prepare Pt/C on carbon paper, catalyst ink was prepared by dispersing Pt/C in a mixture of DI water and 2-propanol (1:2 volume ratio) with added AS-4 ionomer by ultrasonication. The mass ratio of Pt/C and AS-4 ionomer was 4:1. The ink was then airbrushed onto the carbon paper to a final loading of around 0.5 mgPt cm−2.
Electrochemical measurements in the H-type cell
To perform ECH in an H-type cell, a three-electrode configuration was set up with Ag/AgCl as the reference electrode and Pt foil as the counter electrode. The geometric area of the working electrode was 2 cm2. Anode and cathode compartments were separated by a Tokuyama A201 anion-exchange membrane. The electrolyte was prepared in 0.5 M borate buffer solution (pH 9.2), and 20 ml of the electrolyte was used in each compartment. The resistance between the working and reference electrodes was determined by potentiostatic electrochemical impedance spectroscopy, and 90% iR-compensation was applied for all measurements.Cyclic voltammetry (CV), linear sweep voltammetry (LSV), and chronoamperometry (CA) tests were conducted under a constant Ar flow through the catholyte for deaeration and on-line analysis of evolved H2 by gas chromatography (GC). CV and LSV were carried out without magnetic stirring. During CA tests, the catholyte and anolyte were stirred by PTFE-coated magnetic bars (20 × 6 mm, Chemglass Life Sciences) at 350 rpm.
ECO (HMF oxidation) and paired electrolysis were performed in a similar configuration. A piece of carbon cloth (2 cm2) was used for TEMPO-mediated HMF oxidation. The concentration of TEMPO in the anolyte was 7.5 mM.
Electrochemical measurements in the flow cell
Flow cell electrolysis was performed by an in-house built system, which contains stainless-steel endplates, gold current collectors, PTFE and silicone gaskets, a PTFE flow chamber (cathode) with an Ag/AgCl reference electrode, an anion exchange membrane (Tokuyama A201), and a graphite flow-field plate (anode). The geometric area of the cathode and anode were 2 and 5 cm2, respectively. The catholyte and anolyte (both 20 ml) were circulated by a peristaltic pump (Masterflex® L/S®) at 50 ml min−1.Quantification methods
Liquid products were analyzed by High-Performance Liquid Chromatography (HPLC, Agilent Technologies, 1260 Infinity II LC System) equipped with a variable wavelength detector (Agilent 1260 Infinity Variable Wavelength Detector VL). Wavelengths of 225 nm and 260 nm were applied to quantify cathodic products (including HMF, BHMF, furfural, FA, and dimers) and anodic (including HMF, FDCA, HFCA, FFCA, and DFF) products, respectively. Detailed HPLC conditions were reported in our previous studies.17,32 For simplicity, two isomers of the dimeric product [5,5′-bis(hydroxymethyl)hydrofuroin, BHH] are reported together. For the quantification of furfural and 2-furoic acid, the same conditions were used for the analysis of HMF oxidation products except that the flow rate of the mobile phase was increased to 1.1 ml min−1, and the retention times were around 28.7 min and 18.6 min for furfural and 2-furoic acid, respectively.H2 was quantified by on-line GC (SRI Instrument 8610C MG#3) equipped with HaySep D and MolSieve 5 Å columns and a thermal conductivity detector. The calibration curve was established by analyzing the standard calibration gases with different concentrations (10–10000 ppm).
The GC program was started 2 min after the electrolysis was initiated, and a 6 min programmed cycle (including a 4 min running period and a 2 min cooling period) was repeated throughout the measurement.
The rate of H2 generation (r, mol s−1) for each cycle was calculated by eqn (4):
r = c × 10−6 × [p![[V with combining dot above]](https://www.rsc.org/images/entities/i_char_0056_0307.gif) × 10−6/(RT)] × 10−6/(RT)] | (4) |
is the volumetric flow rate of the inlet gas (12.5 mL min−1); p is the ambient pressure (p = 1.013 × 105 Pa); R is the gas constant (R = 8.314 J mol−1 K−1); and T is the room temperature (293.15 K). The total amount of H2 produced (mol) was calculated by integrating the plot of H2 production rate (mol s−1) vs. reaction time (s) with polynomial curve fitting.
Characterization of materials
X-ray diffraction (XRD) crystallography was performed using a Siemens D500 diffractometer operated with a Cu Kα source (λ = 1.5418 Å) at 45 kV and 30 mA and equipped with a diffracted beam monochromator (carbon). X-ray photoelectron spectroscopy (XPS) was performed using a Kratos Amicus/ESCA 3400 X-ray photoelectron spectrometer with Mg Kα X-ray (1253.7 eV). All XPS spectra were calibrated with the C 1s peak at 284.8 eV. Scanning Electron Microscopy-Energy Dispersive X-ray Spectroscopy (SEM-EDS) was conducted on a field-emission scanning electron microscope (FEI Quanta-250) equipped with a light-element X-ray detector and an Oxford Aztec energy-dispersive X-ray analysis system.Calculations
The conversion (X) and product selectivity (Si) were calculated by eqn (5) and (6):| X = (n0 − n)/n0 × 100% | (5) |
| Si = ni/(n0 − n) × 100% | (6) |
The FE of product i (FEi) was calculated by eqn (7):
| FEi = (niziF)/Q × 100% | (7) |
485 C mol−1); and Q is the total charge passed through the electrolytic cell.
Energy efficiency (ε) as a function of cell voltage (Vcell) was calculated by eqn (8):
 | (8) |
Author contributions
W.L. and E.W.C. conceived and supervised the project. H.L., T.L., and Y.C. performed all electrochemical measurements and physical characterization. All authors discussed the results and contributed to the manuscript writing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Science Foundation under grant No. CBET-1947435. We thank Drs Dapeng Jing, Curtis L. Mosher, and Warren E. Straszheim for their assistance with XPS, AFM, and SEM measurements. W. Li is grateful to his Herbert L. Stiles Faculty Fellowship and the IEC competitive fund (20-IEC-019).References
- T. Mai, M. M. Hand, S. F. Baldwin, R. H. Wiser, G. L. Brinkman, P. Denholm, D. J. Arent, G. Porro, D. Sandor and D. J. Hostick, IEEE Trans. Sustain. Energy, 2013, 5, 372–378 Search PubMed.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo and L. A. Hackett, Energy Environ. Sci., 2018, 11, 1062–1176 Search PubMed.
- E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302–308 Search PubMed.
- S. Verma, S. Lu and P. J. Kenis, Nat. Energy, 2019, 4, 466–474 Search PubMed.
- J. G. Ibanez, B. A. Frontana-Uribe and R. Vasquez-Medrano, J. Mex. Chem. Soc., 2016, 60, 247–260 Search PubMed.
- J. Na, B. Seo, J. Kim, C. W. Lee, H. Lee, Y. J. Hwang, B. K. Min, D. K. Lee, H.-S. Oh and U. Lee, Nat. Commun., 2019, 10, 1–13 Search PubMed.
- M. J. Llorente, B. H. Nguyen, C. P. Kubiak and K. D. Moeller, J. Am. Chem. Soc., 2016, 138, 15110–15113 Search PubMed.
- M. A. Bajada, S. Roy, J. Warnan, K. Abdiaziz, A. Wagner, M. M. Roessler and E. Reisner, Angew. Chem., 2020, 59, 15633–156419 Search PubMed.
- S. Choi, M. Balamurugan, K.-G. Lee, K. H. Cho, S. Park, H. Seo and K. T. Nam, J. Phys. Chem. Lett., 2020, 11, 2941–2948 Search PubMed.
- Q. Qin, T. Heil, J. Schmidt, M. Schmallegger, G. Gescheidt, M. Antonietti and M. Oschatz, ACS Appl. Energy Mater., 2019, 2, 8359–8365 Search PubMed.
- X. Wei, Y. Li, L. Chen and J. Shi, Angew. Chem., 2021, 60, 3148–3155 Search PubMed.
- X. V. Medvedeva, J. J. Medvedev, S. W. Tatarchuk, R. M. Choueiri and A. Klinkova, Green Chem., 2020, 22, 4456–4462 Search PubMed.
- R. S. Sherbo, R. S. Delima, V. A. Chiykowski, B. P. MacLeod and C. P. Berlinguette, Nat. Catal., 2018, 1, 501–507 Search PubMed.
- P. Zhang, X. Sheng, X. Chen, Z. Fang, J. Jiang, M. Wang, F. Li, L. Fan, Y. Ren and B. Zhang, Angew. Chem., Int. Ed., 2019, 58, 9155–9159 Search PubMed.
- S. Li, X. Sun, Z. Yao, X. Zhong, Y. Cao, Y. Liang, Z. Wei, S. Deng, G. Zhuang and X. Li, Adv. Funct. Mater., 2019, 29, 1904780 Search PubMed.
- X. Zhang, M. Han, G. Liu, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Appl. Catal., B, 2019, 244, 899–908 Search PubMed.
- X. H. Chadderdon, D. J. Chadderdon, T. Pfennig, B. H. Shanks and W. Li, Green Chem., 2019, 21, 6210–6219 Search PubMed.
- W. A. Smith, T. Burdyny, D. A. Vermaas and H. Geerlings, Joule, 2019, 3, 1822–1834 Search PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 Search PubMed.
- J. N. Chheda, Y. Román-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342–350 Search PubMed.
- C. Moreau, M. N. Belgacem and A. Gandini, Top. Catal., 2004, 27, 11–30 Search PubMed.
- S. Rajendran, R. Raghunathan, I. Hevus, R. Krishnan, A. Ugrinov, M. P. Sibi, D. C. Webster and J. Sivaguru, Angew. Chem., 2015, 127, 1175–1179 Search PubMed.
- J. J. Roylance, T. W. Kim and K.-S. Choi, ACS Catal., 2016, 6, 1840–1847 Search PubMed.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 Search PubMed.
- W.-J. Liu, L. Dang, Z. Xu, H.-Q. Yu, S. Jin and G. W. Huber, ACS Catal., 2018, 8, 5533–5541 Search PubMed.
- N. Zhang, Y. Zou, L. Tao, W. Chen, L. Zhou, Z. Liu, B. Zhou, G. Huang, H. Lin and S. Wang, Angew. Chem., 2019, 131, 16042–16050 Search PubMed.
- B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 Search PubMed.
- N. Jiang, B. You, R. Boonstra, I. M. Terrero Rodriguez and Y. Sun, ACS Energy Lett., 2016, 1, 386–390 Search PubMed.
- M. Hans, S. Mathews, F. Mücklich and M. Solioz, Biointerphases, 2016, 11, 018902 Search PubMed.
- J. E. Pander, D. Ren, Y. Huang, N. W. X. Loo, S. H. L. Hong and B. S. Yeo, ChemElectroChem, 2018, 5, 219–237 Search PubMed.
- N. Tian, Z.-Y. Zhou, N.-F. Yu, L.-Y. Wang and S.-G. Sun, J. Am. Chem. Soc., 2010, 132, 7580–7581 Search PubMed.
- X. H. Chadderdon, D. J. Chadderdon, J. E. Matthiesen, Y. Qiu, J. M. Carraher, J.-P. Tessonnier and W. Li, J. Am. Chem. Soc., 2017, 139, 14120–14128 Search PubMed.
- E. Andrews, J. A. Lopez-Ruiz, J. D. Egbert, K. Koh, U. Sanyal, M. Song, D. Li, A. J. Karkamkar, M. A. Derewinski and J. Holladay, ACS Sustainable Chem. Eng., 2020, 8, 4407–4418 Search PubMed.
- J. Anibal and B. Xu, ACS Catal., 2020, 10, 11643–11653 Search PubMed.
- R. Mariscal, P. Maireles-Torres, M. Ojeda, I. Sádaba and M. L. Granados, Energy Environ. Sci., 2016, 9, 1144–1189 Search PubMed.
- A. Banerjee, G. R. Dick, T. Yoshino and M. W. Kanan, Nature, 2016, 531, 215–219 Search PubMed.
- M. Ma, B. J. Trzesniewski, J. Xie and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 9748–9752 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc00988e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |