
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Extractive in situ product removal for the application of naturally produced L-alanine as an amine donor in enzymatic metaraminol production†
Kevin
Mack‡
abc,
Moritz
Doeker‡
cd,
Laura
Grabowski
abc,
Andreas
Jupke
cd and
Dörte
Rother
 *abc
*abc
aForschungszentrum Jülich GmbH IBG-1, Wilhelm-Johnen-Straße, 52425 Jülich, Germany. E-mail: do.rother@fz-juelich.de; Fax: +49 2461 61-3870; Tel: +49 2461 61-6772
bRWTH Aachen University ABBt, Worringer Weg 1, 52056 Aachen, Germany
cBioeconomy Science Center (BioSC), Forschungszentrum Jülich, Jülich, Germany
dRWTH Aachen University AVT, Forckenbeckstraße 51, 52056 Aachen, Germany
First published on 7th June 2021
Abstract
Vicinal amino alcohols such as metaraminol find direct application in pharmaceuticals and serve as building blocks for fine chemicals. The amine transaminase enzyme can facilitate the stereoselective production of such amino alcohols, which have two chiral centers, e.g. by transamination from 2-hydroxy ketones in the presence of an amine donor. The feasibility of enzymatic metaraminol production has already been demonstrated with the amine donor isopropylamine. This amine donor has the drawback of an unfavorable reaction equilibrium for the target reaction and being crude oil-based. Therefore, we substituted isopropylamine with the bio-based amine donor L-alanine. As the transamination reaction is also thermodynamically limited when utilizing L-alanine, in situ liquid–liquid extraction of metaraminol can solve this drawback and was implemented to increase the conversions and initiate downstream processing steps. We investigated a suitable solvent–enzyme combination and determined a distinct operational window in terms of reaction conditions for combining the enzymatic metaraminol production with product extraction in a smart process concept. This study thus presents a powerful example for the use of the bio-based amine donor L-alanine in combination with efficient process intensification of biocatalytic drug synthesis by means of in situ product removal.
1 Introduction
The requirement of stereopure chiral compounds in pharmaceutical, fine chemical and agricultural industries has led to growing interest in one-step and multi-step biocatalysis since enzymes can combine high conversion rates with excellent stereoselectivity.1 In this context, one important group of enzymes are transaminases, which catalyze the transamination of ketones and aldehydes.2 This can be achieved by chiral resolution as well as in asymmetric synthesis, to obtain chiral amines and amino alcohols from simple precursor molecules at moderate operating conditions. By definition, chiral resolution can only yield a theoretical maximum of 50% product with the correct stereoselective conformation, while the other 50% is converted to an unwanted side product. Therefore, asymmetric synthesis is often the preferred reaction mode, even if an excess of amine donor is necessary. The amino alcohol metaraminol ((1R,2S)-2-amino-1-(3-hydroxyphenyl)propan-1-ol) is an active pharmaceutical ingredient for hypotension treatment3 and can moreover serve as a precursor of more complex, bioactive compounds like tetrahydroisoquinolines.4 The stereoselective production of metaraminol can be achieved within an enzymatic cascade starting from 3-hydroxybenzaldehyde and acetaldehyde or pyruvate, using a thiamine diphosphate depending enzyme for carboligation of the two aldehydes towards the 2-hydroxy ketone and the further transamination of this intermediate to the 3-hydroxylated amino alcohol metaraminol by a transaminase, highlighted in Fig. 1. Within this two-step reaction the amine donor choice greatly influences the atom efficiency, while Erdmann et al.4 are leveraging on isopropylamine for the amine donor choice. In theory, enhanced atom efficiency could be achieved by applying alanine and recycling the subsequent coproduct pyruvate, when suitable means of shifting the equilibrium are found. This hinges on the application of alanine as the amine donor in transaminase reactions, which additionally can be gained from renewable resources.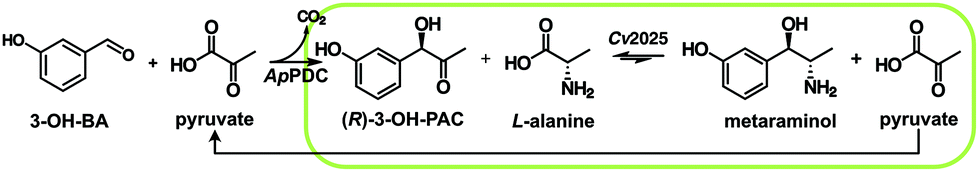 | ||
| Fig. 1 Reaction equation towards metaraminol, starting from 3-OH-BA with (R)-3-OH-PAC as intermediate, with the ApPDC and Cv2025 amine transaminase as catalysts yielding metaraminol and pyruvate and possible recycling of pyruvate for the first reaction step. Highlighted in green is the observed reaction step. ApPDC = pyruvate decarboxylase from Acetobacter pasteurianus, Cv2025 = transaminase from Chromobacterium violaceum, 3-OH-BA = 3-hydroxybenzaldehyde, and (R)-3-OH-PAC = 3-hydroxy phenylacetylcarbinol. | ||
Recently, efforts to integrate precursors from renewable feedstock into (chemo-) enzymatic cascades have been made to establish fully bio-based conversion routes to high value chemicals and pharmaceuticals.5 Fortunately, the large substrate spectrum of transaminases, partly exploited by extensive protein engineering advances, supports the use of sustainable catalysts for these applications and enlarges the possible product spectrum.6,7 Nonetheless, enzyme-catalyzed transamination often exhibits an unfavorable reaction equilibrium resulting in low product titers.8,9 To overcome these limitations, intensive prediction studies have taken place and showed a mild shift of the equilibrium while adjusting the pH and temperature of the reaction.10 Many other transaminase reactions make use of large amounts of the cheap, oil-based amine donor isopropylamine (IPA), producing acetone as the side product. Acetone can be removed via evaporation during the reaction, thus realizing an equilibrium shift to higher product titers,11 while ultimately reducing the overall atom efficiency of the process and leading to excess organic solvent waste. A combination of shifting the equilibrium while simultaneously removing the desired amine product from the reaction phase would be desired. In fact, several drawbacks are evident while using IPA as an amine donor: while there can be little to no activity of wild type amine transaminases towards IPA,12,13 also undesired side reactions can arise from the basicity of the amine donor.14 Furthermore, IPA is highly toxic, derived from fossil resources and complicates the downstream processing, which stands in the way of a sustainable production process with little residual, polluting waste streams. Another amine donor can be α-methylbenzylamine, which has the drawback of being a chiral compound and therefore entails higher costs, sharing the problem of atom efficiency as well.15 Alternative amine donors are the natural products of transaminases. These can be amino acids such as L-alanine, which is non-toxic and was shown to be a favorable co-substrate for the transaminase catalyzed transamination of pro-chiral precursors.16 In addition, L-alanine can be produced in microbial transformations from bio-based sugars. However, the major challenge of utilizing L-alanine for transamination reactions remain very low conversions due to a thermodynamic limitation when stoichiometric amounts are applied. To overcome this limitation, measures to shift the reaction equilibrium, predominantly by in situ product removal (ISPR), are a possible solution.
Established ISPR strategies are in situ product crystallization17 or supported liquid membrane product extraction,18 which were both shown with IPA as an amine donor. Common challenges of in situ product crystallization are (i) the choice of a suitable precipitation agent and (ii) the subsequent product recovery from the precipitate. Supported liquid membranes on the other hand suffer from membrane fouling when the crude cell extract is applied. Further strategies to shift the reaction equilibria involve the enzymatic depletion of the co-product using an additional enzymatic reaction, as shown for the in situ product removal of pyruvate.19,20 However, co-product removal also lowers the atom efficiency of the process and the required auxiliary enzymes heavily increase the overall operating costs and system complexity. Moreover, such strategies do not serve product purification purposes or alleviate possible product inhibition effects. In addition, none of the mentioned ISPR strategies have been applied to enzymatic cascades targeting metaraminol. Hence, alternative methods for a robust, efficient, and scalable product recovery are sought. One such alternative method is liquid–liquid extraction (LLE), which was already shown to be applicable to some enzymatic systems.21 In this contribution, we show that the integration of in situ LLE in the biocatalytic production of metaraminol allows for enhanced process productivity when an amine donor from renewable resources is applied.
2 Materials and methods
2.1 Chemicals
The following chemicals were purchased as given: (1R,2S)-3-(2-amino-1-hydroxypropyl)phenol (metaraminol bitartrate ≥99% purity, TRC, CA), thiamine pyrophosphate (ThDP; AppliChem, DE), acetophenone (AlfaAesar, DE), 2-amino-2-(hydroxymethyl)propane-1,3-diol (Tris; AlfaAesar, DE), L(+)-tartaric acid (Merck, DE), orthophthalic aldehyde/mercaptoethanol reagent (Maisch, DE), phosphoric acid (Fluka, DE), magnesium chloride hexahydrate (MgCl2·6H2O, Fluka, DE), L-alanine (Fluka, DE), α-methylbenzylamine (Sigma-Aldrich, DE), isopropylamine (IPA, Sigma-Aldrich, DE), pyridoxal-5-phosphate (PLP, Sigma-Aldrich, DE), boric acid (Sigma-Aldrich, DE), disodium tetraborate decahydrate (Merck, DE), disodium sulfate (VWR, DE), HCl (Carl Roth, DE), pyruvic acid sodium salt (Carl Roth, DE), potassium dihydrogen phosphate (KH2PO4, Carl Roth, DE), di-potassium hydrogen phosphate (K2HPO4, Carl Roth, DE), 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES, Carl Roth, DE), sodium hydroxide (NaOH, Carl Roth, DE), 3-(trimethylsilyl)propanoic acid (TMSP, Carl Roth, DE), trifluoroacetic acid (TFA, Carl Roth, DE), diethylamine (DEA, Carl Roth, DE), 1-octanol (Merck, DE), and 1-decanol (Merck, DE).2.2 LC/DAD quantification
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) : water containing 0.1% diethylamine (DEA) and 0.075% trifluoroacetic acid (TFA) and centrifuged for 3 min at 14000g. The supernatant was measured by UHPLC analysis (Agilent 1290 Infinity II, DAD 220 nm, Agilent Zorbax Eclipse Plus C18 column, 2 × 100 mm, 1.8 μm, with Zorbax RRHD Eclipse Plus C18 guard column, 2.1 mm, 1.8 μm, 20 °C, 5 μL injection volume). The flow rate was 0.5 mL min−1 and the elution was performed with 10% ACN 90% water + 0.1% DEA and 0.075% TFA in a gradient from 10 to 90% ACN in 5 min. The retention time of (R)-3-OH-PAC was 1.9 min. Metaraminol quantification was performed using an isocratic method with 15% ACN and a flow rate of 0.3 mL min−1 on the same system. The retention time of metaraminol was 0.9 min (for typical chromatograms see Fig. S5.4 and S5.5 of the ESI†).
: water containing 0.1% diethylamine (DEA) and 0.075% trifluoroacetic acid (TFA) and centrifuged for 3 min at 14000g. The supernatant was measured by UHPLC analysis (Agilent 1290 Infinity II, DAD 220 nm, Agilent Zorbax Eclipse Plus C18 column, 2 × 100 mm, 1.8 μm, with Zorbax RRHD Eclipse Plus C18 guard column, 2.1 mm, 1.8 μm, 20 °C, 5 μL injection volume). The flow rate was 0.5 mL min−1 and the elution was performed with 10% ACN 90% water + 0.1% DEA and 0.075% TFA in a gradient from 10 to 90% ACN in 5 min. The retention time of (R)-3-OH-PAC was 1.9 min. Metaraminol quantification was performed using an isocratic method with 15% ACN and a flow rate of 0.3 mL min−1 on the same system. The retention time of metaraminol was 0.9 min (for typical chromatograms see Fig. S5.4 and S5.5 of the ESI†).
2.3 Titration curves
The pKa values for metaraminol bitartrate and (R)-3-OH-PAC were determined in automatic titrations with 0.1 M NaOH (Titroline 7000, SI Analytics; Mainz, DE). The titrant was added in linear steps of 0.0250 mL at 25 °C and the pH change was measured by a combination pH electrode (InLab Micro, Mettler Toledo; Columbus, US).2.4 Biocatalyst production and formulation
The amine transaminase from Chromobacterium violaceum ATCC 12472 (Cv2025) was produced in Escherichia coli BL21 (DE3) cells in shake-flask cultivation using auto-induction medium (gene and protein sequences, plasmid maps and transformation and production details are given in the ESI†). Afterwards, the cells were harvested by centrifugation at 4 °C and 7000 rpm for 30 min. Cell disruption was performed by ultrasonication. The enzyme was purified by immobilized-metal affinity chromatography (IMAC) making use of a C-terminal histidine-tag (the protocol is specified in the ESI†). The purified enzyme was obtained in 10 mM Tris buffer pH 7.5 + 0.2 mM PLP and subsequently lyophilized at −80 °C and 0.2 mbar. Long-term storage took place at −20 °C. Whole cell biocatalysts were cultivated, harvested and lyophilized as stated above but no cell disruption and enzyme purification took place.2.5 Initial activity and long-term initial activity determination of Cv2025 in the presence of organic solvents
For the initial solvent screening (section 3.1), transamination reactions using IPA as an amine donor were performed with 10% (v/v) of organic solvents in a total reaction volume of 200 μL (20 mM (R)-3-OH-PAC, 0.2 mM PLP, 100 mM IPA in 100 mM HEPES buffer pH 7.5 containing 0.1 mM ThDP and 2.5 mM MgCl2). The reaction was started by the addition of 5 mg mL−1 purified Cv2025 from a 57 mg mL−1 enzyme stock solution in the reaction buffer. The enzyme concentration of the stock solution was determined via the Bradford assay using bovine serum albumin (BSA) as a standard.23 The reactions were incubated in 1.5 mL glass vials with sealed screw caps at 30 °C and 700 rpm.For the long-term initial activity assays (section 3.2), purified and lyophilized Cv2025 was incubated in 50 mM HEPES buffer pH 7.5, which was pre-saturated with the respective organic solvents. Pre-saturation was accomplished by mixing the respective solvent with the buffer and incubating this mixture for 24 h. After that, the organic solvent was discarded and only a monophasic buffer, with trace amounts of soluble organic solvent, remained and was applied subsequently. The enzyme concentration was determined via the Bradford assay. After distinct incubation times, the enzyme initial activity was measured in a quartz glass cuvette (d = 1 cm) at 25 °C by acetophenone production at 300 nm using a UV-Vis spectrophotometer (final concentrations in the assay: 50 mM HEPES buffer pH 7.5, 0.1 mM PLP, 2.5 mM α-methylbenzylamine, 5 mM pyruvate, 0.1 mg mL−1 enzyme). Under the stated conditions, one Unit (U) is defined as the amount of catalyst producing one μmol acetophenone per minute. This standard assay for transaminases was first published elsewhere.24
2.6 Liquid–liquid batch extraction of metaraminol and (R)-3-OH-PAC without enzyme addition
Solutions of 1 g L−1 metaraminol bitartrate were buffered in 12.5 mM sodium tetraborate decahydrate and 50 mM phosphoric acid. Both buffers are allowed to reach the pH range which is described as follows: The pH of the aqueous phases was adjusted to pH 7–10.5 in 0.25 increments with NaOH. The organic phase (1-decanol) was saturated with distilled water prior to the extraction to eliminate the volume changes of organic and aqueous phases due to cross solubility. 1 mL of the aqueous solution and 0.5 mL of the saturated organic phase were mixed for 16 h at room temperature and subsequently separated via centrifugation at 3000 rpm for 10 minutes. The organic phase was transferred to a vial containing 1 mL of 0.1 M HCl and was shaken for 16 h for back extraction. Metaraminol concentrations in the aqueous phase before and after extraction as well as after back extraction were determined via HPLC (Agilent 1200). Likewise, 0.5 mL solutions of 8 g L−1 (R)-3-OH-PAC in 50 mM potassium phosphate-borate buffer were adjusted to pH 7–10.5 in 0.5 increments with 4 M KOH. The initial aqueous concentration was determined by HPLC measurements before the addition of 0.5 mL of water-saturated 1-decanol and then incubation by rotating for 24 h. Liquid–liquid batch extraction experiments were performed in duplicate. To evaluate the extraction results, the overall extraction yield E is used as defined in eqn (1):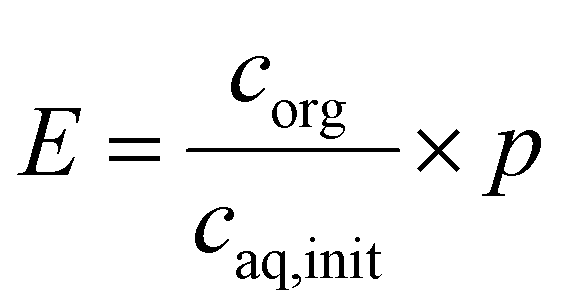 | (1) |
2.7 Integration of liquid–liquid extraction with enzymatic production of metaraminol and scale-up
Transaminations with integrated physical product extraction were performed in a 20 mL scale reaction phase. If not stated otherwise, the aqueous reaction phase contained 50 mM (R)-3-OH-PAC, 250 mM L-alanine and 1 mM PLP in 50 mM borate (boric acid–sodium hydroxide) buffer pH 9. The organic phase (1-decanol) was added in a 2:1 aqueous:organic ratio, after both phases were pre-saturated with water or organic solvent, respectively. The reaction was started by the addition of 10 mg mL−1 Cv2025 (lyophilized, whole cells) and incubated at 30 °C in an overhead shaker.
For repeated batch mode experiments, the organic phase was back extracted after 4, 6 and 24 h and the samples were taken from the aqueous phases as described before. Larger reactions (40 mL) were performed in a 100 mL stirred-tank reactor (EasyMax 402, Mettler Toledo; Columbus, US) at 30 °C and with the addition of pH control by automatic titration of 1 M NaOH. For product recovery, the organic extraction phase was transferred to an acidic aqueous phase (50 mM tartaric acid, 25 °C, stirred at 600 rpm for 1 h). To aid in phase separation, biphasic systems were transferred to Falcon reaction tubes (50 mL) and centrifuged (4000 rpm, 10 °C, 10 min). Afterwards, the samples were taken from the aqueous reaction and back extraction phases and analyzed separately via HPLC analysis. Reference reactions without organic overlay were performed under identical process conditions at pH 7.0 (40 mL reaction phase, 40 mM (R)-3-OH-PAC, 250 mM L-alanine, 1 mM PLP, 100 mM HEPES, 10 mg mL−1 lyophilized, whole cells Cv2025).
2.8 Definition of biocatalytic performance indicators
The enzymatic conversion was calculated as shown in eqn (2), with [metaraminol,t] and [3OHPAC,t] as the aqueous concentrations of the product and substrate of the transamination reaction at the time of sampling: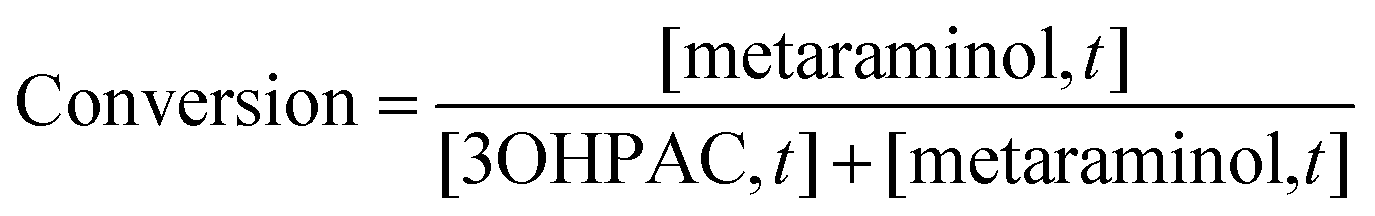 | (2) |
The metaraminol yield was defined as the total amount of produced metaraminol from the supplied amount of substrate (R)-3-OH-PAC as shown in the following equation:
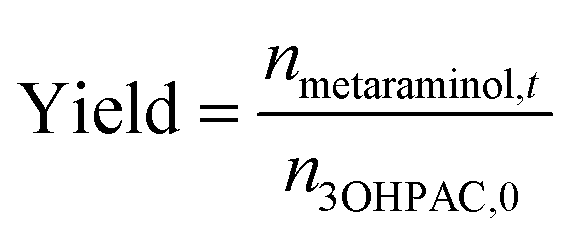 | (3) |
The selectivity (eqn (5)) of the reaction step was used to assess the amount of unwanted (chemical) side reactions as follows:
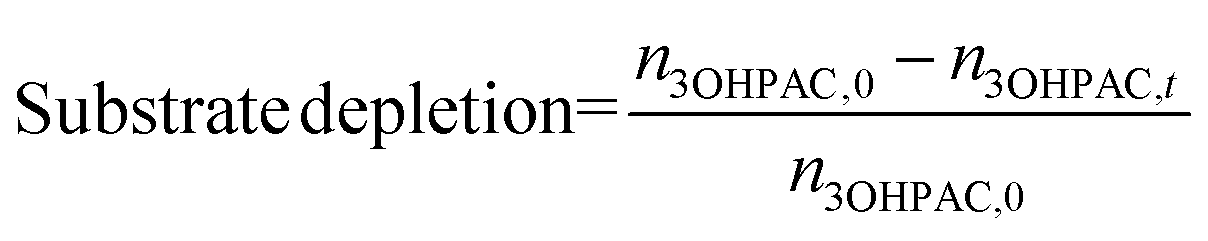 | (4) |
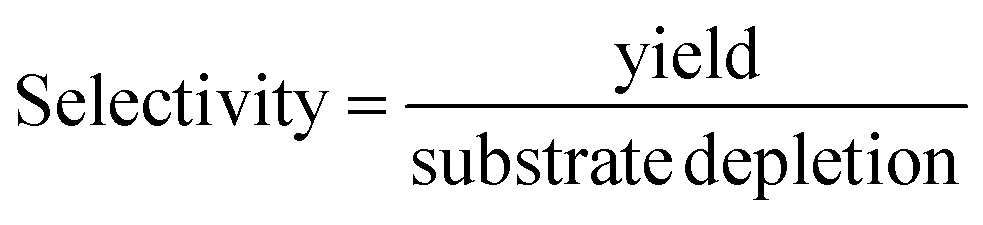 | (5) |
3 Results and discussion
3.1 Determination of enzyme initial activity in different organic solvents
Due to a hydroxyl substituent in the meta position, (R)-3-OH-PAC is a challenging substrate for most amine transaminases. Therefore, the enzyme selection was limited to the transaminase Cv2025, as this enzyme showed not only activity but also the desired (S)-stereoselectivity for the conversion to (1R,2S)-metaraminol in previous studies.4 The reaction scheme is shown in Fig. 1.For the efficient conversion of (R)-3-OH-PAC to metaraminol, Cv2025 transaminase must further exhibit high tolerance to organic solvents to enable in situ extraction within the enzymatic reaction system. The use of solvents within an enzymatic reaction can influence the activity, the stability or even the (stereo)selectivity of the enzyme.25 To validate this organic solvent tolerance, we determined the activity of Cv2025 transaminase for different organic solvents (Fig. 2). For this purpose, the reaction towards metaraminol was investigated using IPA as an amine donor. The use of IPA in excess allowed the exclusion of the limiting reaction equilibrium for transamination with L-alanine.
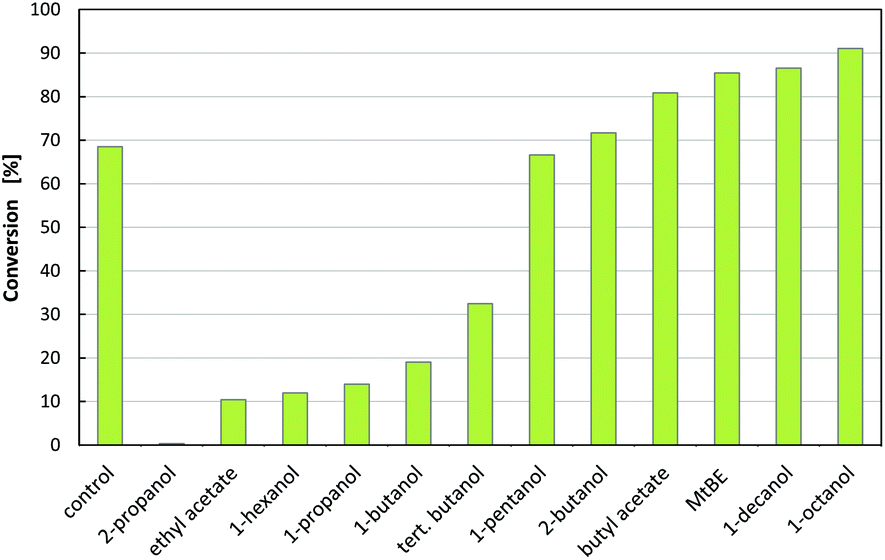 | ||
| Fig. 2 Conversion of (R)-3-OH-PAC after 3.5 h with 12 different organic solvents at 10% solvent addition using 100 mM IPA as an amine donor and 5 mg mL−1 purified Cv2025 (20 mM (R)-3-OH-PAC, 100 mM HEPES + 0.2 mM PLP). | ||
To obtain an economically efficient extraction process, organic solvent losses must be minimized. One main reason for the loss of organic solvents is the cross solubility in the aqueous phase, as the recovery of the dissolved solvent is rarely possible. Therefore, extraction systems with low water solubility are required. The degree of cross solubility of different solvents may be compared via the logP value, which describes the logarithmic distribution coefficient of a compound in a biphasic system of 1-octanol and water (logc1-octanol/cwater). In the initial solvent screening, high cross solubility was observed for 2-propanol, ethyl acetate, 1-propanol, tert-butanol and 1-butanol, which are not suitable for the extraction. Furthermore, these 5 solvents and 1-hexanol lead to inactivation of the catalyst. The remaining 6 solvents showed similar or better conversion of (R)-3-OH-PAC to metaraminol when compared to the reference without solvent addition. From the best performing solvents with respect to conversion in the screening, 1-decanol and 1-octanol exhibit a low distribution towards water.26 As both solvents also showed the highest conversions in the enzymatic system, without observable interface inactivation, both were chosen for further experiments.
3.2 Investigation of long-term initial enzyme activity in the presence of organic saturated aqueous phases
A critical aspect when determining the applicability of an in situ product extraction approach within an enzymatic reaction is the long-term compatibility of the biocatalyst with the organic solvent. From the initial solvent screening (section 3.1), even increased short-term activity (within the 3.5 h detection time) and therewith no detectable negative effect on the activity of the enzyme in the presence of a water–organic interface were shown. In addition, initial enzyme activity assays, after long-term organic solvent incubation, were performed to determine the possible effects of the selected solvents 1-decanol and 1-octanol on the Cv2025 performance. Here, repeated initial rate determinations after incubation in buffer solutions (saturated with the organic phase) were done using a standard photometer assay for transaminase activity.24 This allowed the exclusion of the putative detrimental effects of co-dissolved amounts of organic solvent on the enzyme in the aqueous reaction phase. For 1-octanol and 1-decanol, almost full activity was retained after 13 days of incubation, where activity in pure aqueous phases dropped over time (Fig. 3). Therefore, the addition of 1-decanol or 1-octanol not only has no negative effects but is even beneficial for the enzymatic long-term initial activity. Conclusively, with respect to biocatalyst suitability, both solvents are candidates for in situ extraction-enhanced transaminase reactions. Since 1-decanol enabled slightly higher initial activities to be reached, it was chosen for the following experiments.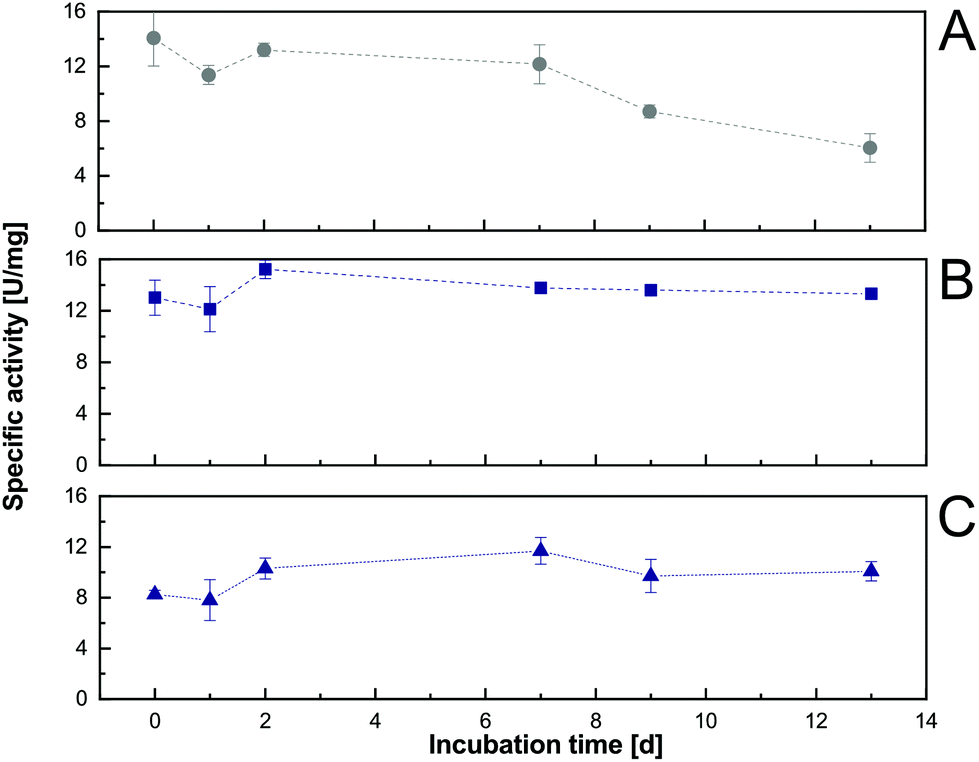 | ||
| Fig. 3 Long-term initial activity curves of Cv2025 over 13 days without organic solvents (A) and in the presence of 1-decanol (B), and 1-octanol (C) at 20 °C. Error bars show the standard deviation of three technical replicates. | ||
3.3 Characterization of metaraminol protonation states
Developing an efficient strategy for liquid–liquid extraction of metaraminol is, besides suitability with the biocatalyst's performance, highly dependent on identifying matching extraction conditions tailored to the target molecule. It is therefore inevitable to consider the molecular structure of metaraminol and characterize its functional groups which influence the extraction behavior. Metaraminol is a primary amino alcohol with a hydroxyl group in the meta-position. Protonation or dissociation of the functional groups strongly affects the hydrophobicity and thus affinity towards an organic solvent phase and solubility in it. As two functional groups can be protonated in metaraminol, the amino group and the hydroxyl functionality at the aromatic ring, two pKa values are expected.The dissociation behavior and respective pKa values were analyzed by titration experiments with commercial metaraminol bitartrate. The pKa values of tartaric acid were found to be 3.3 and 4.4, whereas the pKa values of metaraminol were approximated to 8.9 and 10.2 (Fig. 4) and confirmed in three separate experiments. The results for metaraminol are within the expected pKa range for the amino and phenolic groups, which varies slightly from a previously published pKa value of 8.6.27 Unfortunately, the publication does not clarify the method for pKa determination, which could explain the deviation from our findings.
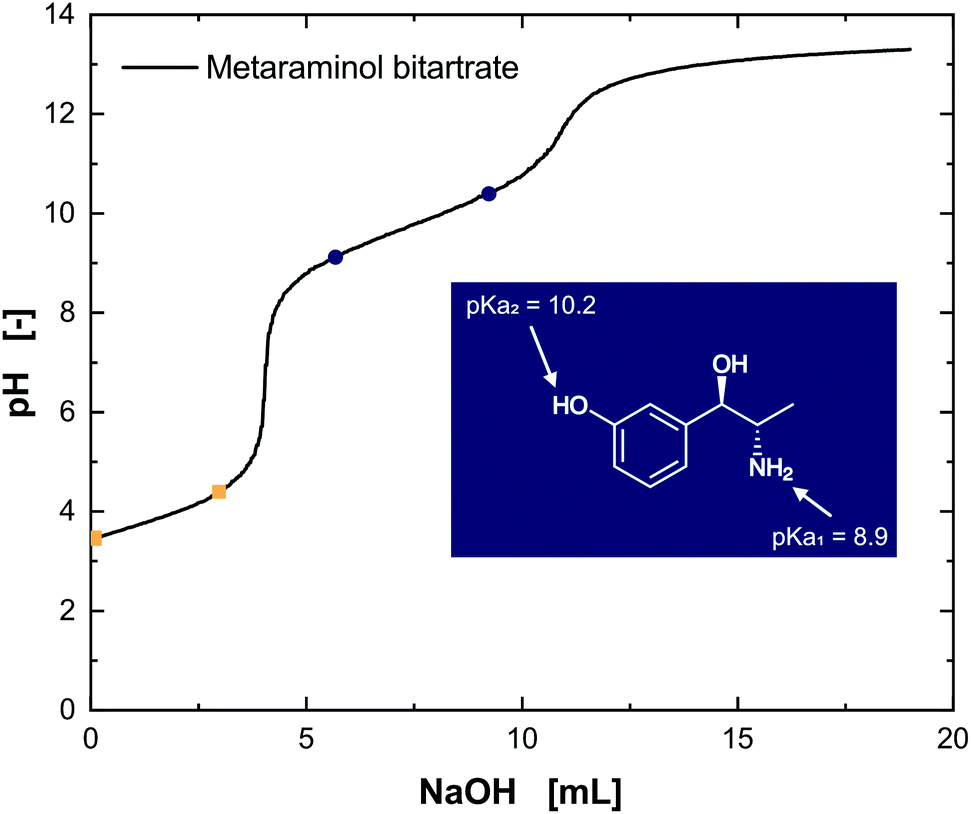 | ||
| Fig. 4 Titration of metaraminol bitartrate with NaOH. The pKa values for the tartaric acid (yellow, pKa1 = 3.3, pKa2 = 4.4) and metaraminol (blue, pKa1 = 8.9, pKa2 = 10.2) are highlighted. | ||
The assignment of the determined pKa values to the functional groups can be difficult. Nevertheless, a comparison with well-characterized compounds such as norephedrine, having only the amino group and therewith one pKa value, allows the matching of the ascertained pKa1 value of 8.9 to the amino group and conclusively the pKa2 = 10.2 to the hydroxyl group. The isoelectric point (IEP) of metaraminol was determined to be pH 9.55. Thus, three different dissociation states of metaraminol can be defined. At low pH values, metaraminol is mostly present in its cationic form, between both pKa values it is mostly net neutral and above the upper pKa metaraminol is present in its anionic form. These results are in line with the experimental data available for similar amino alcohols.28 The titration of the substrate (R)-3-OH-PAC resulted in a pKa value of 9.6, corresponding to the dissociation of the hydroxyl group at the aromatic ring (Fig. 5).
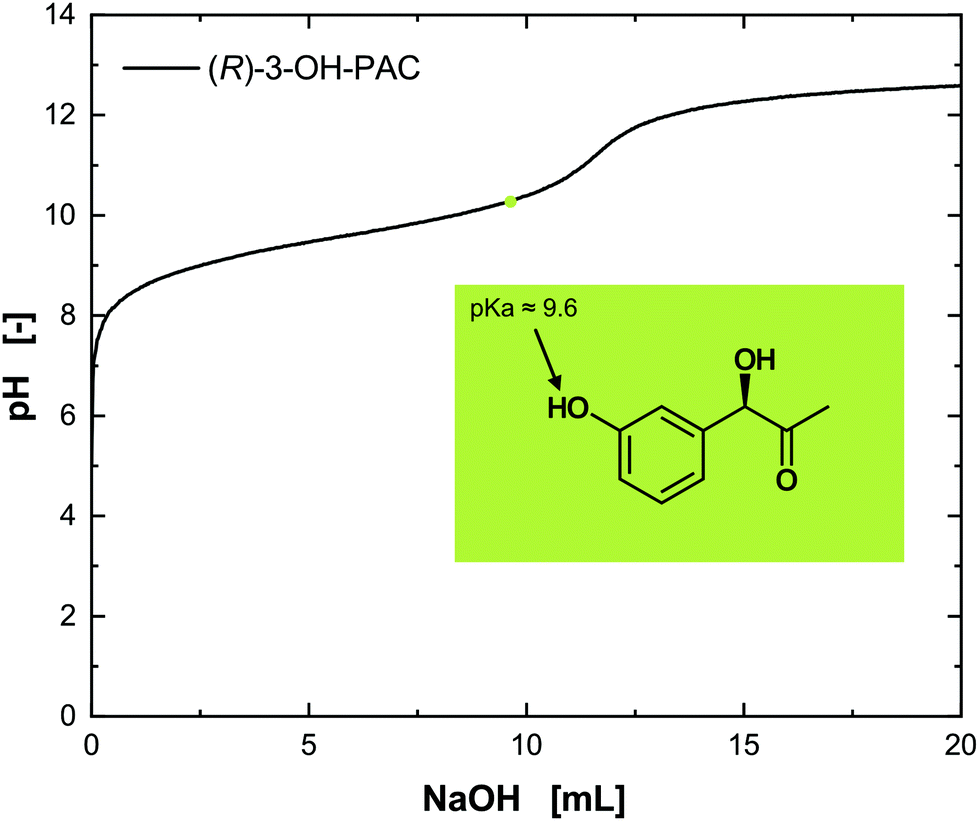 | ||
| Fig. 5 Titration of (R)-3-OH-PAC with NaOH. The pKa value for the hydroxy group can be estimated at pH 9.6. | ||
As a consequence, the protonation states of metaraminol and (R)-3-OH-PAC should be reflected by pH-dependent phase distributions in physical extraction experiments, which were investigated and verified in the following pH dependent liquid–liquid extraction studies.
3.4 Characterization of liquid–liquid extraction and back extraction in pure substance systems without enzyme addition
To characterize the distribution of substrates and products between the aqueous and the organic phase, extraction experiments using 1-decanol as extraction solvent were performed. For simplicity, only pure substances were investigated at different pH values in the range of 7.0–10.5. The experiments show that L-alanine and pyruvate could not be extracted into the 1-decanol phase at any pH (not shown), which is in line with comparable investigations.21,29 For metaraminol (Fig. 6) and (R)-3-OH-PAC (Fig. 7), a pH dependent phase distribution could be observed.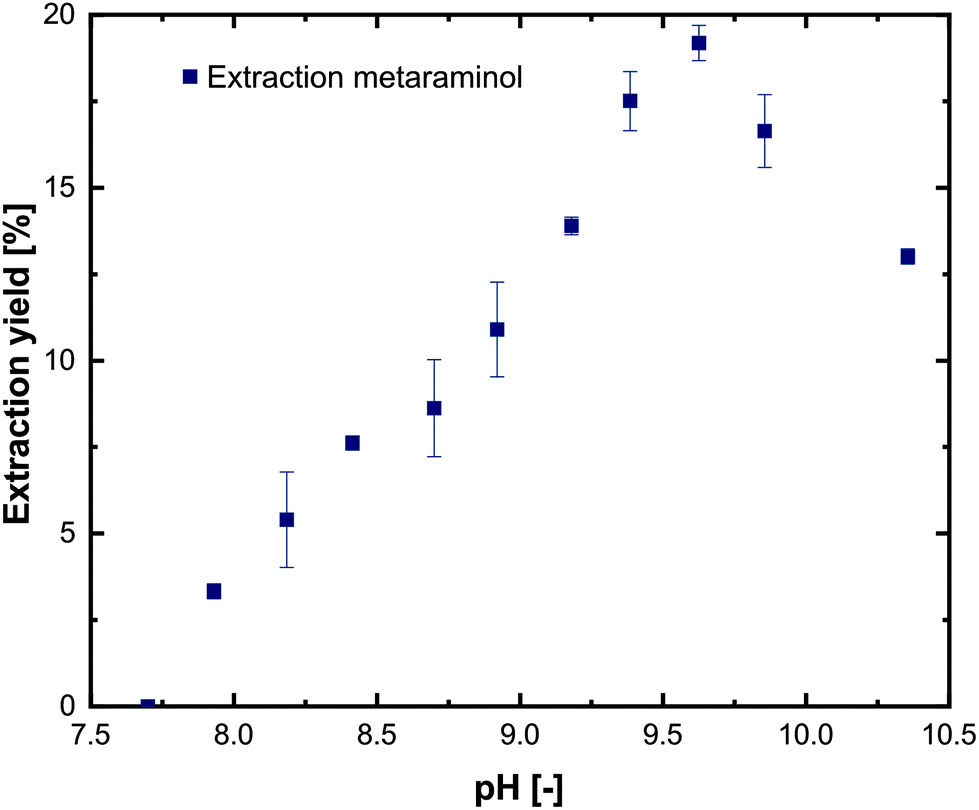 | ||
| Fig. 6 Extraction yield of metaraminol at different pH values using 1-decanol as the extraction solvent. Error bars show the range of two technical replicates. | ||
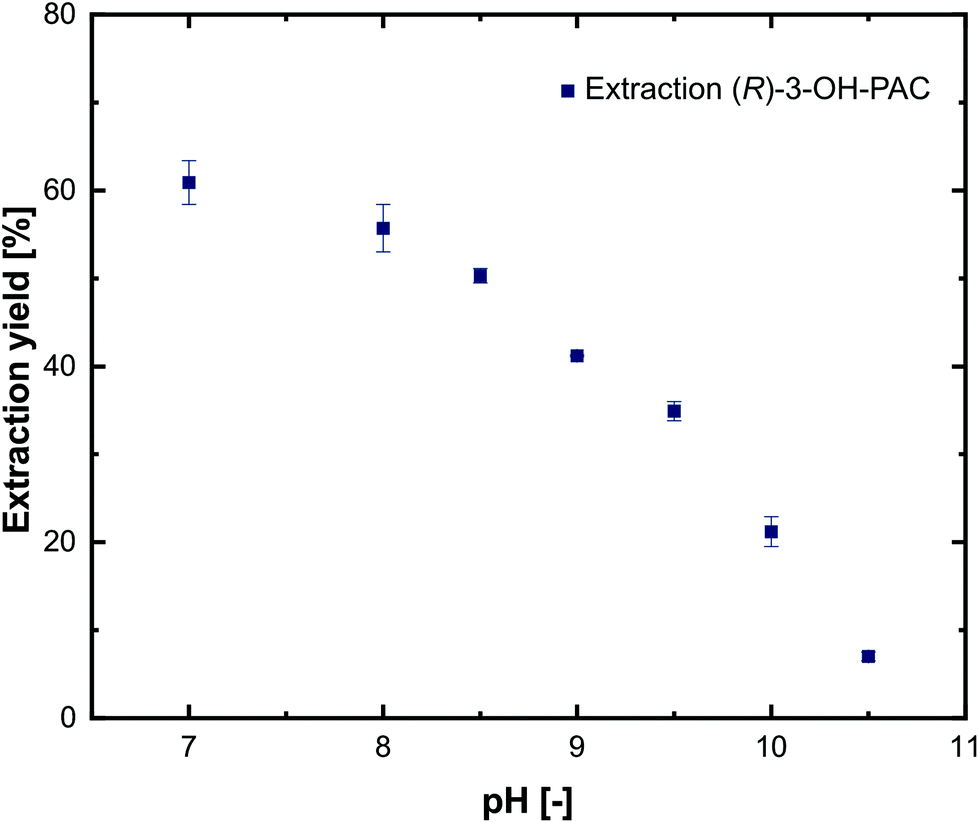 | ||
| Fig. 7 Extraction yield of (R)-3-OH-PAC from aqueous solutions at different pH values using 1-decanol as the extraction solvent. Error bars indicate the range of two independent measurements. | ||
The pH dependent extraction behavior of metaraminol is shown in Fig. 6. At pH levels below 7.75 negligible distribution to the organic phase was observed. Here, metaraminol is mostly present in its cationic form, which shows very low affinity for an aliphatic organic phase and thus low concentration in the organic phase was expected. Above pH 7.9, an increase of the distribution of metaraminol towards the organic phase can be observed until the maximal extraction yield was reached at pH 9.6. This increase in the extraction yield in the organic phase is due to the shift of the metaraminol species towards the neutral state, which has zero net charge and can thus be physically extracted. The experimentally verified results thus coincide with the estimated isoelectric point of metaraminol. Here, the highest measured extraction yield for metaraminol was 18%. A further increase of pH leads to a decrease of the neutral metaraminol species in favor of the anionic metaraminol species. This anionic species shows again lower affinity for the organic phase. Therefore, physical extraction of metaraminol is only possible around the isoelectric point and thus limits the operating pH range of the enzymatic reaction. Recovery of metaraminol from the organic phase can be realized by contacting the loaded organic phase with an acidic aqueous phase. The low pH of the back extraction phase causes protonation of metaraminol to its cationic form, while being transferred to the acidic aqueous phase. In all experiments, the determined recovery rate of metaraminol by back extraction was between 80% and 95%. Any discrepancies to complete full back extraction are due to metaraminol losses during handling of the samples and HPLC detection accuracy limits.
Similar to the results for the extraction behavior of metaraminol, dissociation states of (R)-3-OH-PAC influence the distribution between the organic and aqueous phases. The liquid–liquid extraction results for (R)-3-OH-PAC in the pH range of 7.0–10.5 are shown in Fig. 7. A maximal extraction yield of 60% was observed at pH 7.0, which decreased with higher pH values. Thus, significant amounts of (R)-3-OH-PAC will be present in the organic phase during the enzymatic reaction. This allows the organic phase to function as a reservoir for (R)-3-OH-PAC with a constant refeed, when (R)-3-OH-PAC is consumed in the aqueous reaction phase, while simultaneously trapping metaraminol. During back extraction, the remaining (R)-3-OH-PAC will also distribute into the acidic aqueous phase. This may be countered by a continuous operation mode where the equilibrium shift of the reaction towards metaraminol will constantly lower (R)-3-OH-PAC concentrations in the organic phase and thus also lower concentrations present in the acidic aqueous phase. We concluded that a pH of around 9.6 and back extraction in an acidic phase would be the optimal setup for the physical extraction of metaraminol. A schematic view of the setup with the in situ extraction including the distribution of (R)-3-OH-PAC and metaraminol is shown in Fig. 8.
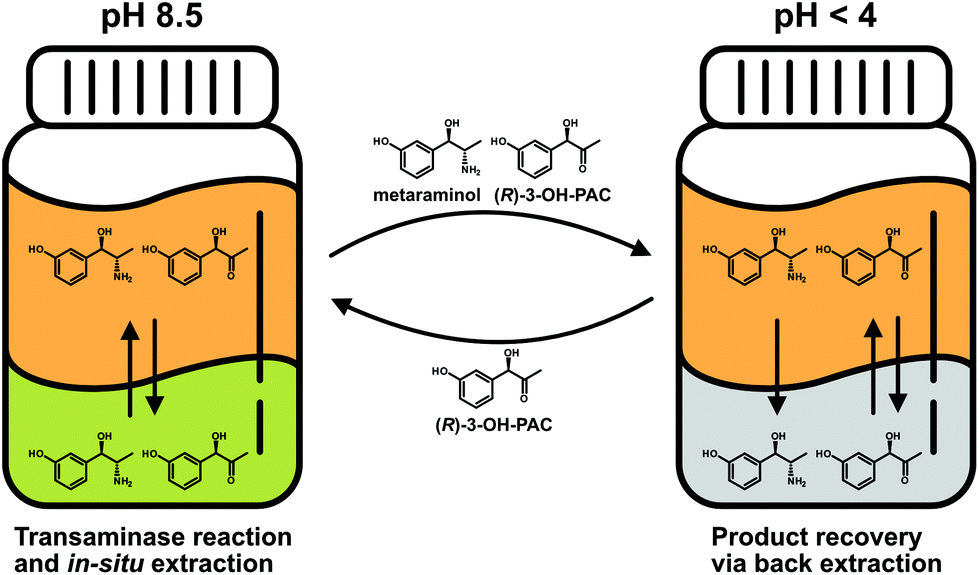 | ||
| Fig. 8 Schematic view of the enzymatic transamination reaction coupled with ISPR. Shown in green is the reaction phase containing the enzymatic catalyst at pH 8.5, orange is the organic phase and grey is the aqueous back extraction phase at a pH below 4.0. | ||
3.5 Integration of the ISPR concept with the enzymatic production of metaraminol
Based on the results obtained from single characterization steps, the next step was to integrate all process parameters for the in situ extraction of metaraminol into the enzymatic transamination reaction using L-alanine as an amine donor. As the reaction does not influence the aqueous pH, no pH control is required. Still buffer was applied to the reaction system to allow facilitated pH adjustment. While with Tris buffer purified enzymes were applied in the initial characterization experiments to assess the compatibility of the ISPR strategy with Cv2025, lyophilized whole cells, which were produced in a phosphate buffer containing medium, were applied instead of the purified enzymes.Here, borate and phosphate buffers were used to ensure the buffer capacity in the alkaline pH range, which is known to be suitable for transaminase reaction systems.18 Since pH values ≥ pH 9.0 showed lower activity of Cv2025 (refer to Fig. S1.1 and S1.2 in the ESI†), pH 8.5–9.0 was considered a suitable compromise for metaraminol production and extraction. Specifically with regard to an industrial scope, whole cell biocatalysts are favored over purified enzymes because of easier handling and substantial cost savings.30
The batch ISPR experiments were scaled up to biphasic systems (60 mL total volume) and performed in a stirred reactor, as this facilitated pH control and high mixing to ensure optimal mass transfer between the organic extraction phase and the aqueous reaction phase. Higher organic phase ratios were used when compared to the long-term initial enzyme activity experiments. These higher organic phase ratios were required for efficient extraction of metaraminol and have no influence on the stability of the enzymatic catalyst, as in both cases the maximal cross solubility of the organic solvent in the aqueous phase was reached and sufficient organic phase remained to ensure the formation of a two-phasic liquid system.31
For comparison, two control experiments at pH 7.0 and 9.0 without ISPR were performed prior to the ISPR assays. Then the ISPR setup at pH 9.0 with a single extraction step was tested as well as the ISPR setup at pH 8.5 with repeated batches. The results of the investigation are shown in Fig. 9.
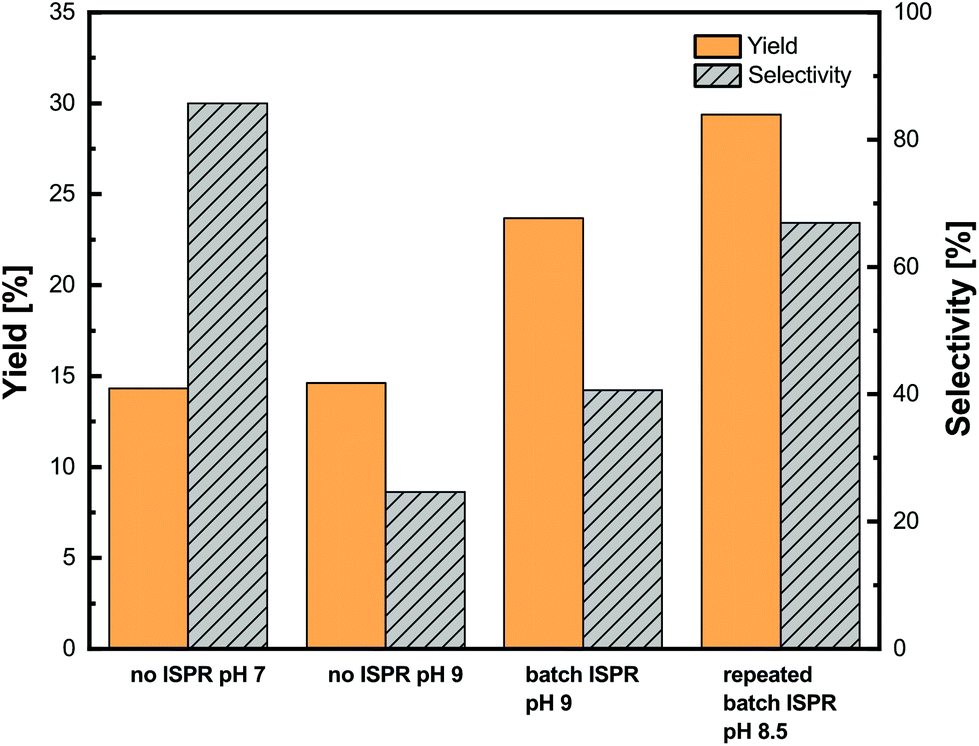 | ||
| Fig. 9 Integration of in situ product removal of metaraminol with the enzymatic transamination reaction. The total metaraminol yield was analyzed from the aqueous and organic phases upon back extraction after 24 h. | ||
The reaction setup at pH 7.0 without ISPR showed a low product yield of 14%, which was to be expected since the reaction equilibrium with L-alanine is largely on the substrate side. Furthermore, a good selectivity of above 85% was observed. An increase of pH to 9.0 without integration of the ISPR resulted in the same yield as that measured at pH 7.0 while the selectivity dropped to 25%. One explanation for this sharp drop in selectivity may be the formation of imines (see the ESI, Fig. S5.7†), as these side products can form when a constant alkaline pH is applied.32 By integrating ISPR with the enzymatic synthesis at pH 9.0, an increase of the metaraminol yield from 14 to 24% was achieved. This increase is due to the extraction of the metaraminol to the organic phase and the accompanying shift of the reaction equilibrium via ISPR. In addition, an increase of selectivity from 25% to over 40% compared to the enzymatic experiment at pH 9.0 without ISPR was observed. This indicates that both selectivity and yield can be improved by the presented ISPR technique. Nevertheless, the yield and selectivity observed at pH 9.0, with one stage batch extraction, are not sufficient for efficient production of metaraminol.
To further increase the yield and selectivity, we chose to slightly adapt the operating pH to 8.5. This reduction of the operating pH leads to an increase in selectivity at the cost of a lower single stage extraction yield. To cope with the reduction of the single stage extraction yield and to increase the overall achievable yield, three repeated batch extractions were performed after 4, 6 and 24 hours of the experiments. As can be seen in Fig. 9, the operation in the repeated batch mode allowed even higher yields of up to 29%, which is double the equilibrium yield without ISPR. Furthermore, the repeated batch operation at pH 8.5 allowed for a significant increase in the selectivity of the reaction to up to 68%, which is already close to the 85% selectivity achieved at pH 7.0. During all experiments, the reaction mixtures were carefully monitored for precipitation in the aqueous and organic phases, and we could not observe any precipitation in the reaction mixtures. Concluding this investigation, these results suggest that a continuous extraction and back extraction process within metaraminol production should be established to further increase the yield and selectivity. In this regard, Matassa et al. have demonstrated the successful application of membrane modules to increase the extraction yield and retaining of the biocatalyst.33 Furthermore, the accumulation of pyruvate in the reaction medium, which did not significantly influence the activity and stability of the enzymatic catalyst, can be recycled. Another advantage of a continuous process is the possibility of a substrate feeding strategy to further increase the overall productivity.
Aside from the clear optimization potential, the integration of extraction-based ISPR served three main purposes in the presented application. Firstly, the reaction equilibrium was shifted towards the product side and the biocatalyst yield was increased. Secondly, product isolation and recovery were implemented, either initiating complete downstream processing of the product or providing metaraminol in an acidic aqueous phase for subsequent chemoenzymatic modifications. Finally, applying a biphasic reaction system enabled in situ substrate supply via the organic phase, which can be especially beneficial for substrates with low solubility in aqueous reaction phases.34,35 The presented concept is expected to be transferable to the biocatalytic synthesis of a range of amino alcohols and chiral amines, although the operational conditions such as the applicable pH range and the optimal solvent must be determined for each biocatalyst and substrate system individually. Following the results of this investigation, a more detailed solvent screening, based on thermodynamic modelling, would be an interesting target, as this would further clarify the influence of different solvents and provide additional solvent candidates. For this, approaches based on COSMO-RS or PC-SAFT could be used.36,37 Furthermore, as this publication focuses on standard organic solvents, alternative solvents such as deep eutectic solvents can be the target of further investigations. These alternative solvents are known to influence the enzyme performance and might be useful to further optimize the enzymatic reaction system.38
4 Conclusions
Biocatalytic production of chiral amines offers great potential to gain valuable, chiral products from renewable resources under environmentally favorable conditions. Metaraminol is an active pharmaceutical ingredient for hypotension treatment, also serving as a potential precursor for a variety of bioactive compounds such as tetrahydroisoquinolines. The reported transaminations commonly rely on petrochemically gained amine donor molecules. Bio-based L-alanine can serve as a more sustainable alternative, while simultaneously producing pyruvate which is applicable in a separate reaction, but bearing the challenge that an unfavorable reaction equilibrium limits conversion. Therefore, in situ liquid–liquid extraction was investigated to increase conversions with L-alanine as an amine donor and to implement downstream processing of enzymatically produced metaraminol. In a comprehensive solvent screening, 1-decanol and 1-octanol were determined as suitable solvents for in situ removal of metaraminol, maintaining high transaminase activity and additionally stabilizing the enzyme. Extraction studies with metaraminol and (R)-3-OH-PAC further revealed a reciprocal pH dependency for the physical extraction of both compounds. In this instance, the two pKa values of metaraminol were experimentally determined as 8.9 and 10.2 through titration.By integrating the ISPR concept with the enzymatic reaction, the utilization of L-alanine as a bio-based amine donor was enabled by circumventing the unfavorable reaction equilibrium, accomplishing increased conversions and efficient product recovery by back extraction. As extraction efficiencies remained limited in the batch mode, a repeated batch mode was investigated, achieving a maximum metaraminol yield of 29% compared to 14% conversion without ISPR. It is expected that a continuous setup might be a good strategy to further enhance the overall process performance. Additionally, thermodynamic modelling, such as that conducted by Voges et al. in 2017 for another transaminase reaction, could further improve the shown concept.10 Conclusively, an operational window for simultaneous transamination and product extraction was defined within an economically and ecologically feasible framework.
The use of biocatalysts combined with the defined ISPR concept facilitates the use of highly stereoselective conversion routes under mild reaction conditions and is independent of harmful auxiliaries, thus avoiding toxic and polluting waste products and simultaneously facilitating a first purification step of metaraminol. Also the recycling of reaction components such as organic solvents further increases the greenness of the overall reaction. The utilized co-substrate L-alanine can be derived from renewable feedstock and is deaminated in the enzymatic reaction to the co-product pyruvate. In a preceding reaction step within the enzymatic cascade to metaraminol, pyruvate can be carboligated with 3-OH-benzaldehyde to yield (R)-3-OH-PAC as was shown for a similar reaction for the synthesis of nor(pseudo)ephedrines.31 By recycling pyruvate from the ISPR concept, the two-step enzyme cascade to metaraminol can accomplish a high atom economy. This concept is not only limited to the presented reaction but can also be applied within similar transaminase reactions contributing to an overall sustainable value chain to chiral amines. Simultaneously, we wanted to create awareness that the reaction setup and downstream processing go hand in hand and should be set up in conjunction to avoid unfavorable component choices and reaction conditions. In this way, the presented concept complies with the major key principles of green chemistry, highlighting the unique capability of integrated bioprocessing towards greener pharmaceutical and fine chemical manufacturing.
Author contributions
Enzyme long-term activity and initial activity experiments: KM and LG. Physical extraction studies in pure substance systems: MD and LG. ISPR in the enzymatic system: LG. The manuscript was written by KM, MD, and LG. Project conception: AJ and DR. All authors revised and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was conducted in the frame of the Bioeconomy Science Center focus lab project “HyImPact-Hybrid processes for Important Precursors and Active pharmaceutical ingredients”. The scientific activities of the Bioeconomy Science Center (BioSC) were financially supported by the Ministry of Culture and Science from North Rhine-Westphalia within the framework of the NRW strategic project BioSC (No. 313/323-400-002 13). We sincerely thank Helen Hailes and John Ward, University College London, for obtaining the plasmids for the expression of the amine transaminase from Chromobacterium violaceum (Cv2025).References
- P. D. de María, G. de Gonzalo and A. Alcántara, Catalysts, 2019, 9, 802 CrossRef.
- J. Ward and R. Wohlgemuth, Curr. Org. Chem., 2010, 14, 1914–1927 CrossRef CAS.
- F. F. de Aragão, P. W. de Aragão, C. A. de Souza Martins, N. S. Filho and E. de Souza Barcelos Barroqueiro, Braz. J. Anesthesiol., 2014, 64, 299–306 CrossRef PubMed.
- V. Erdmann, B. R. Lichman, J. Zhao, R. C. Simon, W. Kroutil, J. M. Ward, H. C. Hailes and D. Rother, Angew. Chem., 2017, 129, 12677–12681 CrossRef.
- F. Dumeignil, M. Guehl, A. Gimbernat, M. Capron, N. L. Ferreira, R. Froidevaux, J.-S. Girardon, R. Wojcieszak, P. Dhulster and D. Delcroix, Catal. Sci. Technol., 2018, 8, 5708–5734 RSC.
- F. G. Mutti, C. S. Fuchs, D. Pressnitz, J. H. Sattler and W. Kroutil, Adv. Synth. Catal., 2011, 353, 3227–3233 CrossRef CAS.
- S. Schätzle, F. Steffen-Munsberg, A. Thontowi, M. Höhne, K. Robins and U. T. Bornscheuer, Adv. Synth. Catal., 2011, 353, 2439–2445 CrossRef.
- F. Guo and P. Berglund, Green Chem., 2017, 19, 333–360 RSC.
- M. Voges, F. Schmidt, D. Wolff, G. Sadowski and C. Held, Fluid Phase Equilib., 2016, 422, 87–98 CrossRef CAS.
- M. Voges, R. Abu, M. T. Gundersen, C. Held, J. M. Woodley and G. Sadowski, Org. Process Res. Dev., 2017, 21, 976–986 CrossRef CAS.
- E.-S. Park, J.-Y. Dong and J.-S. Shin, Org. Biomol. Chem., 2013, 11, 6929 RSC.
- P. Kelefiotis-Stratidakis, T. Tyrikos-Ergas and I. V. Pavlidis, Org. Biomol. Chem., 2019, 17, 1634–1642 RSC.
- S.-W. Han and J.-S. Shin, Biosci., Biotechnol., Biochem., 2014, 78, 1788–1790 CrossRef CAS PubMed.
- S. A. Kelly, S. Pohle, S. Wharry, S. Mix, C. C. Allen, T. S. Moody and B. F. Gilmore, Chem. Rev., 2017, 118, 349–367 CrossRef PubMed.
- P. Tufvesson, M. Nordblad, U. Krühne, M. Schürmann, A. Vogel, R. Wohlgemuth and J. M. Woodley, Org. Process Res. Dev., 2015, 19, 652–660 CrossRef CAS.
- U. Kaulmann, K. Smithies, M. E. Smith, H. C. Hailes and J. M. Ward, Enzyme Microb. Technol., 2007, 41, 628–637 CrossRef CAS.
- D. Hülsewede, J.-N. Dohm and J. von Langermann, Adv. Synth. Catal., 2019, 361(11), 2727–2733 Search PubMed.
- G. Rehn, P. Adlercreutz and C. Grey, J. Biotechnol., 2014, 179, 50–55 CrossRef CAS PubMed.
- J.-S. Shin and B.-G. Kim, Production of chiral Amines with ω-Transaminase, ACS Symposium Series, American Chemical Society, 2001, ch. 16, pp. 248–262 Search PubMed.
- T. Börner, G. Rehn, C. Grey and P. Adlercreutz, Org. Process Res. Dev., 2015, 19, 793–799 CrossRef.
- M. Voges, I. V. Prikhodko, S. Prill, M. Hübner, G. Sadowski and C. Held, J. Chem. Eng. Data, 2017, 62, 52–61 CrossRef CAS.
- A. Bednarz, A. C. Spieß and A. Pfennig, J. Chem. Technol. Biotechnol., 2017, 92, 1817–1824 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- S. Schätzle, M. Höhne, E. Redestad, K. Robins and U. T. Bornscheuer, Anal. Chem., 2009, 81, 8244–8248 CrossRef PubMed.
- T. Gerhards, U. Mackfeld, M. Bocola, E. von Lieres, W. Wiechert, M. Pohl and D. Rother, Adv. Synth. Catal., 2012, 354, 2805–2820 CrossRef CAS PubMed.
- I. P. R. Grundtvig, S. Heintz, U. Krühne, K. V. Gernaey, P. Adlercreutz, J. D. Hayler, A. S. Wells and J. M. Woodley, Biotechnol. Adv., 2018, 36, 1801–1814 CrossRef.
- D. W. Newton and R. B. Kluza, Drug Intell. Clin. Pharm., 1978, 12, 546–554 CrossRef CAS.
- S. Riegelman, L. Strait and E. Fischer, J. Pharm. Sci., 1962, 51, 129–133 CrossRef CAS PubMed.
- N. A. Bowden, J. P. Sanders and M. E. Bruins, J. Chem. Eng. Data, 2018, 63, 488–497 CrossRef CAS PubMed.
- P. Tufvesson, J. Lima-Ramos, N. A. Haque, K. V. Gernaey and J. M. Woodley, Org. Process Res. Dev., 2013, 17, 1233–1238 CrossRef CAS.
- T. Sehl, H. C. Hailes, J. M. Ward, R. Wardenga, E. von Lieres, H. Offermann, R. Westphal, M. Pohl and D. Rother, Angew. Chem., Int. Ed., 2013, 52, 6772–6775 CrossRef CAS PubMed.
- C. Godoy-Alcántar, A. K. Yatsimirsky and J.-M. Lehn, J. Phys. Org. Chem., 2005, 18, 979–985 CrossRef.
- C. Matassa, A. Romani, D. Ormerod, U. T. Bornscheuer, M. Höhne and Y. Satyawali, J. Chem. Technol. Biotechnol., 2019, 95, 604–613 CrossRef.
- K. Castiglione, Y. Fu, I. Polte, S. Leupold, A. Meo and D. Weuster-Botz, Biochem. Eng. J., 2017, 117, 102–111 CrossRef CAS.
- K. Schmölzer, K. Mädje, B. Nidetzky and R. Kratzer, Bioresour. Technol., 2012, 108, 216–223 CrossRef PubMed.
- H. Veith, M. Voges, C. Held and J. Albert, ACS Omega, 2017, 2, 8982–8989 CrossRef CAS PubMed.
- M. Knierbein, M. Voges and C. Held, Org. Process Res. Dev., 2020, 24, 1052–1062 CrossRef CAS.
- M. M. C. H. van Schie, J.-D. Spöring, M. Bocola, P. Domínguez de María and D. Rother, Green Chem., 2021, 23, 3191–3206 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/D1GC00852H |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |