
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiobased aliphatic polyesters from a spirocyclic dicarboxylate monomer derived from levulinic acid†
Nitin G.
Valsange
 a,
Maria Nelly
Garcia Gonzalez
b,
Niklas
Warlin
a,
Smita V.
Mankar
a,
Nicola
Rehnberg
ac,
Stefan
Lundmark
d,
Baozhong
Zhang
*a and
Patric
Jannasch
*a
a,
Maria Nelly
Garcia Gonzalez
b,
Niklas
Warlin
a,
Smita V.
Mankar
a,
Nicola
Rehnberg
ac,
Stefan
Lundmark
d,
Baozhong
Zhang
*a and
Patric
Jannasch
*a
aCentre for Analysis and Synthesis, Department of Chemistry, Lund University, P.O. Box 124, SE-22100 Lund, Sweden. E-mail: patric.jannasch@chem.lu.se; baozhong.zhang@chem.lu.se
bEnvironmental and Energy Systems Studies, Department of Technology and Society, Lund University, P.O. Box 118, SE-221 00 Lund, Sweden
cBona Sweden AB, Box 210 74, 200 21 Malmö, Sweden
dPerstorp AB, Innovation, Perstorp Industrial Park, 284 80 Perstorp, Sweden
First published on 29th June 2021
Abstract
Levulinic acid derived from lignocellulose is an important biobased building block. Here, we report on the synthesis and polymerization of a rigid spirocyclic diester monomer to produce polyesters and copolyesters. The monomer was prepared via a one-step acid catalyzed ketalization involving ethyl levulinate and pentaerythritol by employing a straightforward, solvent-free, and readily scalable method which required no chromatographic purification. Still, careful removal of traces of water from the spiro-diester prior to polycondensations proved crucial to avoid side reactions. A preliminary life cycle assessment (LCA) in terms of greenhouse gas (GHG) emissions indicated that the corresponding spiro-diacid tended to be environmentally favourable, producing less CO2 emission than e.g., biobased succinic acid and adipic acid. A series of aliphatic polyesters with reasonably high molecular weights was subsequently prepared in melt and modified melt polycondensations of the spiro-diester with 1,4-butanediol, 1,6-hexanediol, neopentyl glycol and 1,4-cyclohexanedimethanol, respectively. The resulting fully amorphous polyesters showed glass transition temperatures in the range 12–49 °C and thermal stability up to 300 °C. Hot-pressed films of the polyesters based on neopentyl glycol and 1,4-cyclohexanedimethanol were transparent and mechanically strong, and dynamic melt rheology showed stable shear moduli over time to indicate good processability. In addition, the spiro-diester monomer was employed in copolycondensations with diethyl adipate and 1,4-butanediol and demonstrated good reactivity and stability. Hence, the results of the present study indicate that the spiro-diester based on levulinic acid is an effective monomer for the preparation of aliphatic polyesters and other condensation polymers.
1. Introduction
The development of biobased plastics from abundant and readily available sustainable sources has gained great momentum due to the serious environmental concerns of fossil-based plastics, such as depletion of fossil resources and generation of greenhouse gas emissions.1–3 However, the transition to biobased plastics cannot resolve the environmental problem of plastic littering since most biobased polymers on the market today are non-biodegradable (e.g. bio-PET).4 In this context, the development of biobased biodegradable polymers has received growing attention due to their unique characteristics of sustainability combined with biodegradability.5 Aliphatic polyesters are an important class of such polymers which can be conveniently produced using readily available biobased monomers and that are totally or partly biodegradable under certain conditions. In addition, they usually exhibit much better UV resistance compared to aromatic polyesters. Prominent examples of aliphatic polyesters are poly(L-lactic acid) (PLA), poly(butylene succinate) (PBS), and polyhydroxyalkanoates (PHAs).3,6 However, these polyesters are generally semicrystalline and have low glass transition temperatures (Tgs) and poor thermo-mechanical performance, which hinder their use in applications where high optical clarity, heat resistance, and impact strength are required. Therefore, the molecular design and synthesis of rigid amorphous polymers is an attractive pathway in the development of polymers with improved properties.The incorporation of rigid cyclic monomers into the polymer structure is an attractive pathway to increase the polymer chain stiffness of amorphous polymers. In this context, the potentially biobased diol 1,4-cyclohexanedimethanol (CHDM)7–9 and fossil-based diol cis/trans-2,2,4,4-tetramethyl-1,3-cyclobutanediol (CBDO)10,11 have received great attention. For example, CHDM has been utilized as a comonomer to produce a PET-like polyester (PETG) with a higher Tg and alkaline resistance than PET.12 In a similar way, CBDO has been used to prepare polyesters with high impact resistance and superior optical clarity.10 Furthermore, the Eastman Chemical company produces a high-performance amorphous polyester (Tritan™) using both CHDM and CBDO.13,14 Recently, a partially biobased rigid diol with a spirocyclic acetal structure has been introduced by Perstorp AB to produce a high-performance amorphous polyester, (Akestra™) with high transparency and heat resistance (Tg > 100 °C) for hot-filling applications.14,15 These industrial examples demonstrate the development of fully biobased rigid monomers for the production of high-performance amorphous polymers. In addition, various cyclic building blocks derived from lignocellulose have been exploited for the development of polymers with improved thermo-mechanical properties.16–18 Similarly, terpene-based cyclic monomers have also been investigated in polymerizations to obtain amorphous polyesters with Tg values up to 125 °C.19,20
Among various sustainable sources, sugar is an important source to produce valued monomers towards the development of new polymers. For example, furan dicarboxylic acid (FDCA) is the most well-known and commercially available sugar-based monomer today and has gained attention as a possible replacement to fossil-based terephthalic acid in PET production, yielding a 100% biobased poly(ethylene 2,5-furandicarboxylate) or PEF.21,22 Sugar-based cyclic building blocks have been extensively studied.23 For example, isosorbide is a commercially available sugar-based bicyclic rigid diol which has been used to produce polyesters with an increase in Tg values.24 Rigid bicyclic diacetalized structures were derived from sorbitol and mannitol and used in the preparation of polyesters.25–28 In addition, a galactaric acid-derived dicarboxylate was synthesized and used for polyester synthesis.29–31 Recently, furan-derived tricyclic diacid and tetracyclic anhydride monomers have been synthesized and investigated in melt and ring opening polymerizations to yield amorphous polyesters with Tg values up to 174 °C.32 In addition to rigid cyclic monomers, spirocyclic monomers have gained significant attention due to their ability to enhance thermal and mechanical properties.15 For example, a spiro-diol obtained through the acetalization reaction of fructose-derived 5-hydroxymethylfurfural (HMF) and biobased pentaerythritol was employed as a comonomer in the preparation of polyesters and poly(urethane-urea)s, which showed an increase in Tg values with the spiro-diol content.33 Various spiro-diols with ketal structures have been prepared from glycerol and cyclic diketones and used for the preparation of polyesters, polyurethanes, and polycarbonates.34 Recently, a new spiro-diol based on citric acid was reported to produce polycarbonates with Tgs up to 100 °C.35
Among sugar-based chemicals, levulinic acid (LA) has recently been recognized as a promising sustainable platform chemical that can conveniently be produced from hexoses (e.g. glucose and fructose), cellulose and lignocellulosic biomass.36–39 LA is a bifunctional molecule containing ketone and carboxylic acid groups, and has been used as a building block for the synthesis of polymer precursors such as levulinate esters, γ-valerolactone, acrylic acid, 1,4-pentadiol, angelica lactone, diphenolic acid, δ-aminolevulinic acid, and 5-hydroxylevulinic acid.36,40 Moreover, LA itself has also been directly used as a monomer. For instance, poly(ketal-ester)s with crosslinked structures were prepared from LA and pentaerythritol through concurrent ketalization and esterification reactions.41 The resulting polymer showed thermal stability up to 320 °C and good processability. However, the polymerization was difficult to control, because it mainly proceeded in a nonselective manner where esterification was favoured over ketalization. Recently, levulinate esters such as methyl levulinate and ethyl levulinate, which can be easily produced by esterification of LA, have become important precursors for the development of new monomers.42 For instance, a new diester monomer was synthesized by reductive amination between methyl levulinate and aspartic acid and was subsequently used to prepare polyesters containing pendent lactam units with Tg values of up to 45 °C.43 Using a similar pathway, diester and hydroxyl-ester monomers with cyclic amino units in the backbone were produced using methyl levulinate for poly(ester-co-amine) synthesis.44 The polymers were deemed suitable for biomedical applications, but the Tgs were rather low despite the cyclic units in the backbone.
The high selectivity of levulinate esters towards ketalization has enabled the development of new biobased ketal-ester building blocks for the production of polymers, solvents, lubricants and plasticizers.45,46 A patent reports on the preparation of plasticizers based on several rigid (spiro)cyclic ketal-esters from levulinate esters and various polyols.47 The (spiro)cyclic ketal-esters were also investigated in polycondensations with various diols. However, the polycondensations resulted in the formation of low molecular weight polyesters with high polydispersity. The latter may be related to a high degree of branching in polymers due to the formation of polyfunctional intermediates by the thermal cleavage of cyclic ketal units. In conclusion, the preparation of amorphous polyesters using (spiro)cyclic ketal-esters remains essentially unexplored. Hence, the utilization of these biobased rigid building blocks in polycondensations opens new possibilities for the production of high-performance polymers.
Here, we present a straightforward, high-yield and eco-friendly synthesis of a new biobased spirocyclic diester monomer (denoted as Monomer L, Scheme 1) using ethyl levulinate and biobased pentaerythritol. Pentaerythritol is a widely used building block in the polymer industry and is available in 100% renewable grade from Perstorp (Voxtar™ M100). The synthesis of the monomer included solvent-free reaction conditions and environmentally acceptable solvents/chemicals during the work-up and isolation procedures. The preliminary LCA results indicated a significantly lower CO2 emission for Monomer L production compared to biobased succinic acid and adipic acid. To explore the usefulness of Monomer L, it was employed in polycondensations with four different aliphatic diols (linear, branched and cyclic) to obtain biobased polyesters. We subsequently investigated the thermal and mechanical properties, as well as the processability of the obtained materials. In addition, Monomer L was further investigated in copolymerizations with diethyl adipate and 1,4-butanediol (1,4-BD) to produce a copolyester with an improved Tg value.
 | ||
| Scheme 1 Synthesis of the biobased Monomer L using a solvent-free acid catalyzed ketalization reaction. | ||
2. Experimental
2.1 Materials
Ethyl levulinate (99%), 1,4-butanediol (1,4-BD, >99%), 1,6-hexanediol (1,6-HD, 97%), neopentyl glycol (NPG, 99%), 1,4-cyclohexanedimethanol (1,4-CHDM, a mixture of cis and trans, 99%), diethyl adipate (Reagent plus 99%), dibutyltin(IV)oxide (DBTO, 98%), chloroform-d (99.8% atom D), and dimethyl sulfoxide (DMSO-d6, 99.9% atom D) were purchased from Sigma Aldrich. Sulfuric acid (H2SO4, 95%), sodium bicarbonate (99.7%), sodium sulfate (anhydrous, ≥99% ACS), ethyl acetate (≥99%), chloroform (99.1%, stabilized with 0.6% ethanol), heptane, toluene, xylene (a mixture of isomers, analytical grade, ACS), mesitylene, and methanol were purchased from VWR chemicals. Biobased pentaerythritol (Voxtar™, 99%) was kindly supplied by Perstorp AB. All chemicals and reagents were used as received.2.2 Monomer synthesis
Pentaerythritol (20.0 g, 147 mmol) and ethyl levulinate (116 g, 808 mmol) were added to a two-neck round bottom flask equipped with a magnetic stirrer and Dean-stark trap. The reaction mixture was heated to 120 °C, followed by addition of conc. H2SO4 (0.04 g, 0.4 mmol). After stirring for half an hour, a vacuum was applied to the reaction flask and the pressure was slowly reduced to 10 mbar. The reaction was maintained under these conditions for 4 h, whereafter no more liquid was collected in the Dean–Stark apparatus. Next, the reaction mixture was cooled to room temperature and the reactor was brought to atmospheric pressure. The contents of the flask were dissolved in ethyl acetate and washed with a saturated solution of NaHCO3, and then with water. The organic layer was dried over sodium sulfate, filtered, and concentrated on a rotary evaporator. Subsequently, the crude reaction mixture was subjected to high vacuum distillation at 130 °C to remove traces of ethyl levulinate. Finally, the pure product was isolated from the crude reaction mixture through extraction with heptane to collect Monomer L as a colorless viscous liquid (42.0 g, 74.0% yield).1H NMR (400.13 MHz, CDCl3, δ, ppm): 1.23 (t, 6H, J = 7.1 Hz, CH3CH2O–), 1.35 (s, 6H, CH3CCH2CH2), 2.04–1.95 (m, 4H, CH2CH2COO–), 2.45–2.36 (m, 4H, CH2COO–), 3.56 (s, 4H, CH2C(CH2)3), 3.74 (d, 2H, J = 11.9 Hz, CH2C(CH2)3), 3.92 (d, 2H, J = 11.8 Hz, CH2C(CH2)3), 4.10 (q, 4H, J = 7.1 Hz, –OCH2CH3).
13C NMR (100.61 MHz, CDCl3, δ, ppm): 14.15, 19.56, 28.34, 32.07, 33.78, 60.27, 63.70, 63.72, 98.92, 173.66.
Mass: HRMS (ESI+, m/z): exact mass calcd for C19H32O8+: 389.2181, found 389.2175.
2.3 Greenhouse gas (GHG) emissions
We carried out a preliminary and tentative evaluation by a Life Cycle Assessment (LCA) using literature data in order to verify that the production of biobased Monomer L followed an eco-friendly trend. The aim was also to identify both positive and negative aspects in the development of Monomer L, and to explore possible alternatives to decrease the environmental impact of the production of this new monomer. The impact category chosen was the total impact of greenhouse gas (GHG) emissions of Monomer L because it is one of the main environmental challenges of the present century. In addition, the data related to other impact categories in the literature are limited (like eutrophication that is also relevant in biomass derived products). The method that covers this impact category is the ‘Greenhouse Gas Protocol (GGP)’ (v1.01) method which is based on GWP100 (100-year timeframe) (IPCC, 2007).48 The characterization factors needed to quantify the amount of impact that the new monomer has in GHG emissions were taken from this GGP method.The system boundary was set up following a cradle-to-factory gate approach, comprising resource extraction, transportation to the raw monomer factory (i.e., levulinic acid (LA) and pentaerythritol) and production of Monomer L (before it is transported to the customer). The contributions of biobased LA, biobased pentaerythritol, ethanol and the contribution of the synthesis process were analyzed separately up to the step of Monomer L production. Any transportation of the raw monomer from one factory to the other for the Monomer L production was excluded. The GHG emission results on LA based on palm trees were obtained from Isoni's work49 after some assumptions. Data on biobased pentaerythritol (feedstock sugar beet pulp, commercially named Voxtar M100) were obtained from Perstorp AB.50 The synthesis simulations were obtained by an estimation from Ecoinvent database version 3.7. Likewise, the ethanol production based on rye was obtained from Ecoinvent database version 3.7. A comparative analysis of the diacid form of Monomer L was made with other biobased and conventional fossil-based monomers. The functional unit was defined as one kg of Monomer L. The geographical coverage was based on the European Union (EU) for all monomers except for LA as it is an Asian product and Bio_SA3 as it is USA. The LCA methodology is standardized in the ISO 14040-14044 series by the International Organization of Standardization.51,52
2.4 Monomer stability evaluation
Monomers with cyclic acetal and ketal structures are sensitive towards hydrolysis during polycondensations at high temperatures, often resulting in branching or crosslinking of the polymer products.17,33 Hence, it is important to study and verify the stability of the monomer prior to any condensation reaction. In the present case, the thermal stability of the monomer was investigated by 1H NMR spectroscopy. Monomer L samples were kept at defined temperatures for 5 h after different drying protocols before analysis to monitor any structural degradation. The drying protocols listed below were employed.2.5 Polyester synthesis
Monomer L was used in polycondensation with diols such as 1,4-BD, 1,6-HD, NPG, and 1,4-CHDM to obtain polyester PButL, PHexL, PNeoL, and PCycL, respectively.1H NMR (400.13 MHz, CDCl3, δ, ppm): 1.37 (s, 6H, CH3CCH2CH2), 1.69 (p, 4H, J = 3.3 Hz, CH2CH2O–), 2.05–1.96 (m, 4H, CH2CH2COO–), 2.48–2.39 (m, 4H, CH2COO–), 3.58 (s, 4H, CH2C(CH2)3), 3.76 (d, 2H, J = 11.8 Hz, CH2C(CH2)3), 3.94 (d, 2H, J = 11.8 Hz, CH2C(CH2)3), 4.08 (bt, 4H, –OCH2).
13C NMR (100.61 MHz, CDCl3, δ, ppm): 19.58, 25.24, 28.31, 32.10, 33.80, 63.73, 63.76, 63.85, 98.94, 173.63.
The polyesters PHexL, PNeoL, and PCycL were synthesized using a similar procedure to that described for PButL. However, in the synthesis of PNeoL a molar ratio of Monomer L to diol of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.25 was used, and the transesterification and polycondensation were carried out for 30 and 40 h, respectively.
:1.25 was used, and the transesterification and polycondensation were carried out for 30 and 40 h, respectively.
13C NMR (100.61 MHz, CDCl3, δ, ppm): 19.63, 25.57, 28.34, 28.50, 32.10, 33.74, 63.73, 63.76, 64.33, 98.95, 173.71
13C NMR (100.61 MHz, CDCl3, δ, ppm): 19.50, 21.72, 28.23, 32.06, 33.85, 34.65, 63.71, 63.75, 69.14, 98.90, 173.47
13C NMR (100.61 MHz, CDCl3, δ, ppm): 19.59, 25.29, 28.30, 28.82, 32.10, 33.80, 34.44, 37.00, 63.73, 63.77, 67.12, 69.32, 98.94, 173.69.
1H NMR (400.13 MHz, CDCl3, δ, ppm): 1.58–1.75 (m, 8H, CH2CH2O–, CH2CH2COO–), 2.25–2.38 (m, 4H, CH2COO–), 4.12–4.04 (m, 4H, –OCH2).
13C NMR (100.61 MHz, CDCl3, δ, ppm): 24.31, 25.24, 33.79, 63.78, 173.24.
A copolyester (PButLA-20) was synthesized using Monomer L (20 mol%), diethyl adipate and 1,4-BD.
1H NMR (400.13 MHz, CDCl3, δ, ppm): 1.37 (s, 6H, CH3CCH2CH2), 1.59–1.76 (m, 12H, CH2CH2COO–, CH2CH2O–), 2.04–1.95 (m, 4H, CH2CH2COO–), 2.25–2.38 (m, 4H, CH2CH2COO–), 2.48–2.39 (m, 4H, CH2COO–), 3.58 (s, 4H, CH2C(CH2)3), 3.76 (d, 2H, J = 11.9 Hz, CH2C(CH2)3), 3.94 (d, 2H, J = 11.8 Hz, CH2C(CH2)3), 4.08 (bt, 8H, –OCH2).
13C NMR (100.61 MHz, CDCl3, δ, ppm): 19.53, 24.32, 25.23, 25.25, 28.27, 32.07, 33.79, 33.82, 63.70, 63.73, 63.78, 63.80, 98.90, 173.23, 173.59.
2.6 Characterization
1H and 13C NMR measurements were performed on a Bruker DR X400 spectrometer at 400.13 MHz and 100.61 MHz, respectively, with the samples dissolved in chloroform-d or dimethyl sulfoxide-d6. Chemical shifts were reported as δ values (ppm). A Micromass QTOF mass spectrometer (ESI) was used for determining the exact mass of the monomer. The molecular weights of the polyesters were determined by size-exclusion chromatography (SEC) in THF. The SEC setup included three Shodex columns coupled in series (KF-805, -804, and -802.5) in a Shimadzu CTO-20A prominence column oven, a Shimadzu RID-20A refractive index detector, using the Shimadzu LabSolution software. All samples were run at 40 °C at an elution rate of 1 mL min−1. Calibration was done by using poly(ethylene oxide) standards (Mn = 3.86, 12.60, 49.64 and 96.1 kg mol−1). Thermogravimetric analysis (TGA) was performed on a TA Instruments model Q500 TGA system. The samples were heated from room temperature to 600 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere. Thermal decomposition temperatures (Tds) were determined at the maximum decomposition rates. Td,5 is the temperature taken at 5% weight loss, and the char yield is the remaining weight % at 600 °C. Differential scanning calorimetry (DSC) measurements were performed on a DSC Q2000 analyzer from TA Instruments. Glass transition temperatures (Tgs), melting temperatures (Tms) and heats of fusion (ΔHms) were determined from the second heating cycle where the samples were heated from −50 or −80 to 200 °C. The crystallization temperature (Tc) was measured from the DSC cooling curve. The polyesters PNeoL and PCycL were hot pressed into films (35 mm × 5 mm × 1 mm) for dynamic mechanical analysis (DMA). Hot pressing was performed at 100 and 110 °C, respectively, after which the samples were rapidly cooled to room temperature. DMA measurements were performed in tensile mode using a TA Instruments Q800 analyzer. The samples (17.5 mm between the clamps) were heated from −30 to 100 °C at a heating rate of 3 °C min−1 and a frequency of 1 Hz with a constant strain of 0.1%. Rheology measurements were carried out on an AR2000 ETC Advanced Rheometer from TA Instruments, using parallel plates with a diameter of 15 mm under air. Samples were prepared into discs of 15 mm diameter and 1 mm thickness by hot pressing the polyesters PNeoL and PCycL at 100 and 110 °C, respectively. Time sweep experiments were performed at 130 °C, with constant strain (1%) and oscillation (1 Hz) for 1500 s. Moreover, dynamic frequency sweep experiments were conducted on PCycL in the frequency range 0.1–100 Hz using constant strain (1%) at temperatures from 110 to 130 °C. The data obtained from the frequency sweeps were used in a time-temperature superposition to construct a master curve at 130 °C.3. Results and discussion
3.1 Monomer synthesis
Monomer L was synthesized using an acid-catalyzed double ketalization reaction of ethyl levulinate and pentaerythritol using a green chemistry protocol under solvent-free reaction conditions (Scheme 1). Here, a 5.5-fold excess of ethyl levulinate with respect to pentaerythritol was employed for the reaction, which was catalyzed by sulfuric acid at 120 °C. The use of excess ethyl levulinate is expected to favor the complete conversion to the ketal product without any undesirable transesterification products. In addition, an excess of ethyl levulinate facilitated the removal of the by-product water by forming an azeotrope that drove the ketalization to completion. Afterward, the mixture was dissolved in ethyl acetate and the acid catalyst was quenched by washing with a saturated aq. NaHCO3 solution. Complete quenching was essential because trace amounts of acid residues led to the slow decomposition of the monomer into the starting reactants. The 1H NMR spectrum of the crude reaction mixture revealed that it contained up to 95% of the desired Monomer L with a small percentage of monoketal as the impurity (structure shown in Scheme S3†). Up to 74% of the pure Monomer L was extracted from the viscous crude reaction mixture using heptane. Further extraction from the crude reaction mixture using more heptane gave Monomer L contaminated with small concentrations of the monoketal impurity. This straightforward and efficient purification procedure is attractive for upscaled production at industrial levels. Moreover, the green aspect of both the reaction (solvent-free) and purification procedures (no chromatographic separation) makes the monomer production eco-friendly.The structure and purity of Monomer L were confirmed by 1H NMR spectroscopy (Fig. 1). First, all the signals in the spectrum were unambiguously assigned. The signals of the ethyl ester groups appeared as a triplet at 1.23 (a) and quartet at 4.1 (b) ppm. Furthermore, the protons of the methyl groups attached to the spirocyclic unit gave rise to a singlet at 1.35 (e) ppm. The singlet at 3.56 (h) and two doublets at 3.74 (g) and 3.92 (f) ppm were attributed to the methylene protons of the spirocyclic unit. Surprisingly, the methylene protons attached to the ester group and the spirocyclic unit were observed as multiplets between 2.45 and 2.36 (c) and 2.04 and 1.95 (d) ppm, respectively (Fig. S4†), and not as triplets. This phenomenon is known as the second-order behavior in proton NMR spectra.53 It is mostly observed in rigid molecules wherein the hindered rotation around the C–C bond gives rise to the non-equal populations of the possible conformers, making the geminal protons magnetically non-equivalent. Thus, the hindered rotation of the C–C bond of the rigid spirocyclic and methylene units in Monomer L was the most likely reason for the second-order splitting of methylene protons. Still, when the spectrum was recorded in DMSO-d6 solution (Fig. S5†), the methylene protons (c and d) appeared as a first-order triplet, diminishing the appearance of the second-order multiplets due to the increased dynamic interconversion of conformers in polar solvent. The signal assignments were further confirmed by 13C and 2D NMR investigations (i.e., COSY, HMBC, and HSQC, Fig. S6–S9†).
 | ||
| Fig. 1 1H NMR spectrum of Monomer L in CDCl3. | ||
3.2 GHG emissions of Monomer L
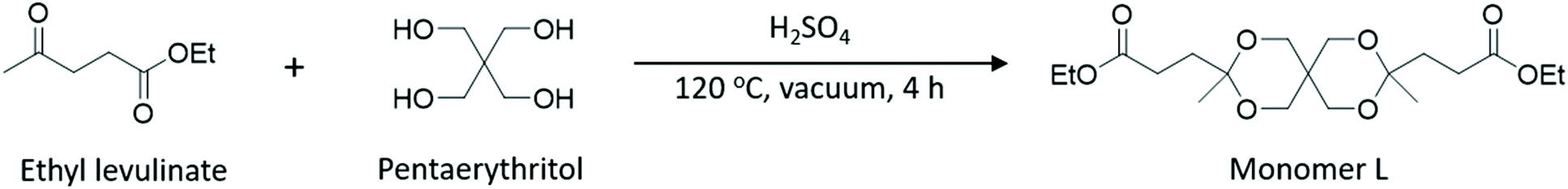
The evaluation of the total impact of GHG emissions by the LCA of Monomer L was done on the basis of a cradle-to-gate approach for the functional unit of 1.0 kg of Monomer L. As can be seen in Fig. 2a, the LCA of GHG emissions for Monomer L gave a value of 2.50 kg of CO2eq per kg of L, of which 1.45 kg of CO2eq per kg of L was derived from the LA (the largest impact). Moreover, 0.37 kg of CO2eq per kg of L originated from the biobased pentaerythritol and 0.35 kg of CO2eq per kg of L for ethanol. The synthetic processes, ketalization (“Synthesis 1”) and esterification (“Synthesis 2”) only contributed to the total emissions by small values, i.e., 0.24 and 0.1 kg CO2eq per kg L, respectively. In other words, 58% of the total GHG emissions was attributed to LA production, followed by pentaerythritol and ethanol productions with 15% and 14%, respectively.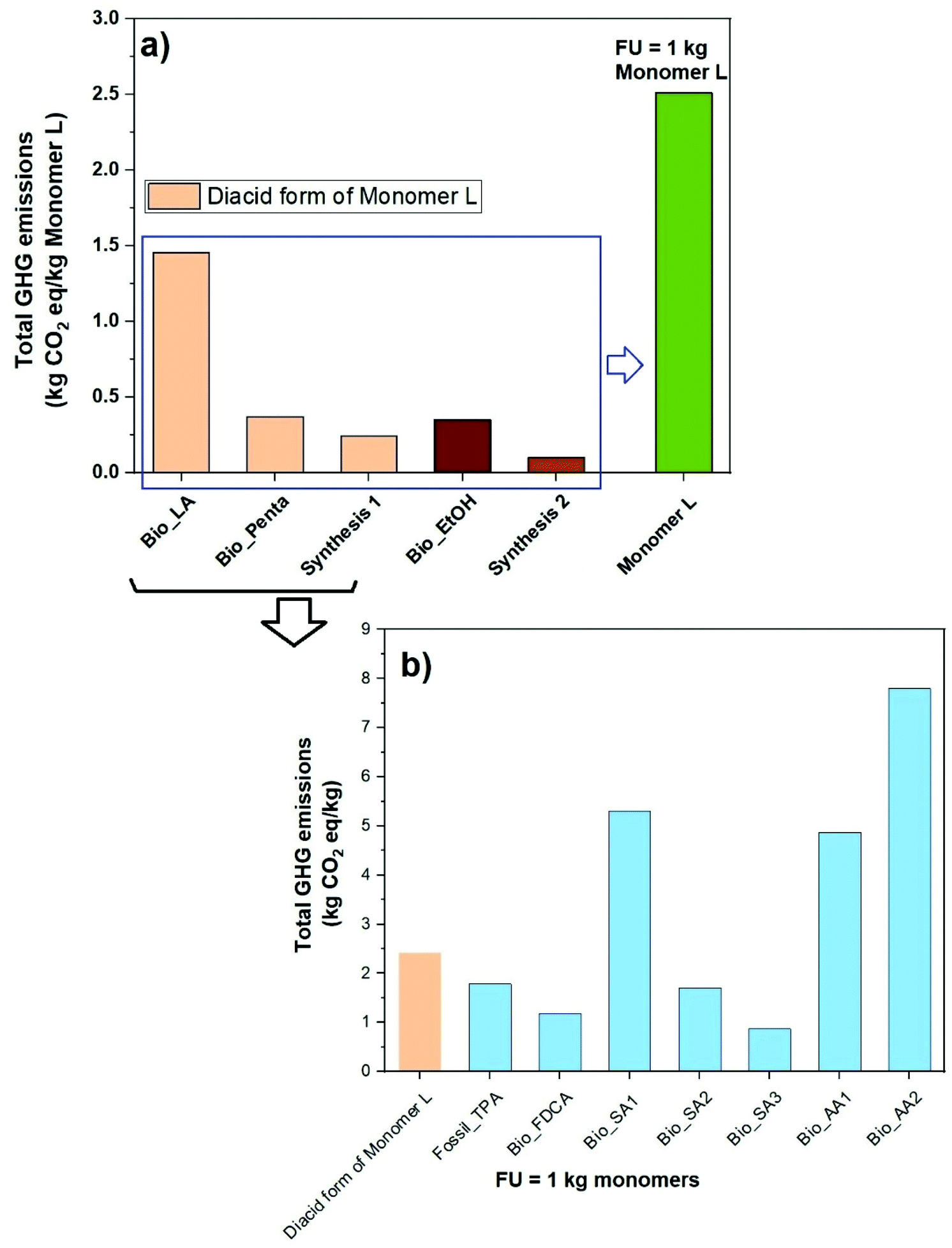 | ||
| Fig. 2 Evaluation of the total impact of GHG emissions of Monomer L (a), and the diacid form of Monomer L compared to other biobased and fossil-based conventional monomers (b). | ||
The diacid form of Monomer L was compared to Bio_SA1 (apple pomace-based),54 Bio_SA2 (based on dextrose from corn),55 Bio_SA3 (non-food crop feedstock),56 Bio_FDCA (beet sugar-based),57 Bio_AA1 (lignin feedstock),58 Bio_AA2 (from forest residues),59 and fossil-based terephthalic acid. As shown in Fig. 2b, the diacid form of Monomer L showed a value of 2.35 kg of CO2eq per kg, which is an average value in comparison with the other diacids. This initial result is quite promising for Monomer L. The assessment was only intended to provide preliminary first-hand information regarding the environmental impact of Monomer L based on literature data. Overall, the results suggest that the production of Monomer L tends to be environmentally favourable in terms of GHG emissions. As expected, the outcome of the analysis is very sensitive to the production of LA. With more favourable, yet realistic, input parameters such as source of biomass, location, type of process, efficiency of equipment, deforestation, etc., the results would be more advantageous. Therefore, in order to gain a deeper insight into the environmental impacts of Monomer L, more thorough LCA investigations and a full LCA development into the same geographical coverage will be needed.
3.3 Monomer stability and development of polycondensation conditions
As reported in the literature, cyclic acetal and ketal monomers are thermally and hydrolytically sensitive and degrade into multifunctional (polyhydroxy) products during polycondensations at high temperatures.26,60 This often leads to branching or crosslinking of the polymer, and may eventually lead to gel formation. Lower temperatures (typically ≤200 °C) are required in order to minimize these undesirable side reactions.25,27 Initially, conventional melt polycondensations (using high vacuum) of Monomer L with 1,6-HD were attempted at various temperatures between 130 and 160 °C (which may be compared with the decomposition temperature of the monomer, Td,5 = 201 °C) without any pre-drying of the monomer. However, these polycondensations led to partial or complete gel formation depending on the reaction time. It is reasonable to assume that the gelation was due to the degradation of ketal units, giving rise to additional hydroxyl groups that induce the formation of crosslinked structures. Because of this finding, studies of the monomer stability after different drying procedures were performed in order to investigate the degradation pathway and identify the conditions under which the monomer was stable. Firstly, the as-obtained monomer after the purification was heated under a nitrogen atmosphere for 5 h at temperatures between 90 and 170 °C at 10 °C intervals. The structural changes were subsequently studied by 1H NMR spectroscopy. Stacked spectra of the monomer samples after heating at various temperatures are shown in Fig. S10.† As seen, new signals emerged in the spectra, most of which overlapped with the monomer signals. The chemical shift and integrals of the isolated new signals at 2.19 (l), 2.56 (n), and 2.74 (m) and at 1.38 (e′), 3.62 (k), 3.71 (j), and 3.84 (i) revealed that they originated from ethyl levulinate (one of the starting reactants) and the monoketal ester, respectively (Fig. S10†). Thus, the partial hydrolysis of the ketal units of the monomer to form the monoketal ester and the starting ethyl levulinate was confirmed. As expected, the degree of hydrolysis increased with temperature, as indicated by the increased intensity of the corresponding signals of the degradation products. The degree of hydrolysis calculated from the 1H NMR spectra was in the range ∼5–20%, indicating the low thermal and hydrolytic stability of Monomer L under these conditions. Hence, the monomer was not suitable for use in polycondensation reactions before further treatment.We hypothesized that residual moisture was the main reason for the hydrolysis and low stability of the ketal unit. Hence, high vacuum drying of the monomer under stirring was carried out before heating under a nitrogen atmosphere prior to the NMR analysis. As seen in Fig. S11,† the 1H NMR spectra showed almost no degradation up to 130 °C, and only very slight degradation between 140 and 160 °C, when compared with the previous results on the as-obtained monomer sample. This strongly implied that hydrolysis due to the moisture present in the reaction mixture was responsible for the gel formation observed during the initial polycondensations, suggesting that moisture-free and inert conditions are critical for successful polymerizations. Subsequently, melt polycondensations of Monomer L with 1,6-HD using a high vacuum drying protocol was successfully carried out at 130 °C without any sign of gelation. However, high vacuum drying of the reaction mixture for a long time (3 h) is impractical for industrial production due to the low energy efficiency of the process and instead azeotropic distillation was performed. Hence, the stability of Monomer L was analyzed after azeotropic distillation using toluene and the results showed a very similar result to that observed after high vacuum drying with almost no thermal degradation up to 130 °C (Fig. 3). Hence, the removal of water by azeotropic distillation of the reaction mixture and polycondensation at a temperature of 130 °C were adopted for the successful preparation of polyesters without any gelation.
 | ||
| Fig. 3 1H NMR spectra of the monomer (pre-dried by azeotropic distillation) after stirring at different temperatures for 5 h. | ||
3.4 Polyester synthesis
Monomer L was polycondensed with the aliphatic diols 1,4-BD, 1,6-HD, NPG, and 1,4-CHDM, respectively, using dibutyltin oxide (DBTO) as a catalyst (0.5 mol% with respect to Monomer L) to obtain the corresponding polyesters (Scheme 2). A 10 mol% excess of 1,4-BD, 1,6-HD, and 1,4-CHDM and a 25 mol% excess of NPG with regard to Monomer L were used to compensate for the loss of the diols during the polymerization. In all cases, azeotropic distillation of the reaction mixture was carried out prior to the polycondensation. First, the melt polycondensation (conventional method using a high vacuum) of Monomer L with 1,6-HD and 1,4-CHDM, respectively, was investigated. In the first step, transesterification was achieved at 130 °C for 5 h under low nitrogen flow, and in the second step polycondensation was carried out under a high vacuum at 130 °C for 20 h. During the polycondensation of Monomer L with NPG, a significant loss via evaporation and sublimation of NPG was observed, which led to incomplete transesterification. Moreover, the transketalization of Monomer L with NPG was identified as a side reaction (details are given in ESI Note 1,† Schemes S1–S2 and Fig. S1–S3†). This side reaction was not observed for any of the other diols, and was presumably driven by the possibility of NPG to form a six-membered cyclic ketal structure. This observation was further supported in model reactions of Monomer L with a monofunctional alcohol (neopentyl alcohol or 1-butanol) in the presence of DBTO at 130 °C, i.e., under similar conditions to those used for the polycondensations. After driving off excess alcohol, the 1H NMR spectra showed a clean corresponding transesterification product with no sign of any transketalization product (Fig. S12 and S13†). Hence, the branching of the polyesters can most likely be attributed to the degradation of spirocyclic ketal structures by hydrolysis, as discussed in section 3.3. The transketalization of Monomer L with NPG was found to be reversible after an equimolar amount or slight excess of NPG was employed in the polycondensation. On the other hand, the large excess of NPG used to compensate for its loss favored the side reactions and restricted the reversible character. To avoid this issue, a modified melt polycondensation was investigated.61,62 In this procedure, the transesterification was completed in 30 h under continuous nitrogen flow using xylene (a mixture of isomers) as a solvent. The subsequent polycondensation was performed at 130 °C for 40 h using mesitylene as a solvent with increased nitrogen flow. The use of xylene in the transesterification helped to reduce the loss of NPG through evaporation. Still, a 25% molar excess was required to achieve complete transesterification. Mesitylene was added for the azeotropic removal of the excess diol during the polycondensation step, which efficiently increased the molecular weight of the resulting polymer (further details on the optimization of the polycondensation is found in ESI Note 2†). All the solvents driven off during the polyester synthesis may be recycled. In order to compare with the results obtained by the conventional melt condensation, the polyesters based on the other diols were also synthesized by the modified melt polycondensation method. The transesterification and polycondensation steps were performed at 130 °C for 5 h and 20 h, respectively. | ||
| Scheme 2 Synthesis of polyesters by a modified melt polycondensation of Monomer L and a variety of different aliphatic diols. | ||
After the polycondensations, the viscous reaction mixtures were dissolved in chloroform and the products were precipitated in methanol to give polyesters in 87–98% isolated yields (Table 1, photographs of polyester samples are shown in Fig. S14†). Notably, the use of chloroform can be avoided by instead using greener solvents such as ethyl acetate or dimethyl carbonate to dissolve the polyesters, but these solvents gave less distinct (but still handleable) precipitates. In a continuous industrial process, a polyester melt is typically fed directly into an extruder after the final polycondensation step, thus avoiding the use of solvents altogether. According to the SEC measurements, the polyesters PButL, PHexL, PNeoL, and PCycL reached reasonably high molecular weights with Mn = 18.3, 16.3, 17.1, and 10.0 kg mol−1, respectively, and polydispersities in the range Đ = 2.03–2.88, indicating only very limited branching apparently due to hydrolysis and ring-opening of the spirocyclic structure. However, the 1H NMR spectra showed no shifts related to branching except for PNeoL. This sample displayed small signals corresponding to the side product residues in the polymer (discussed in ESI Note 2†). Although the degree of branching is small and difficult to detect by 1H NMR spectroscopy because of the very low concentration of branch points, it can still result in Đ values significantly above the ideal value of 2. In fact, the shape of the SEC chromatograms (Fig. S15–S16†) indicated that the rather high Đ value in some cases was due to the presence of branched polymer chains. As can be seen, the chromatogram peaks indicated bimodal distributions with shoulders on the high molecular weight side (towards low retention volumes). These shoulders were most prominent for PButL and PNeoL, which showed the highest Đ values (2.88 and 2.73, respectively). Notably, polyester PCycL and PHexL synthesized by using the conventional melt polycondensation reached Mn = 9.02 and 17.0 kg mol−1, respectively, which was comparable with the samples produced via the modified method. This indicated that the modified melt polycondensation under nitrogen flow was equally efficient as the high-vacuum melt polycondensation to reach high molecular weights.
| Polyester | Melt polycondensation method | Isolated yielda (%) | M n (kg mol−1) | M w (kg mol−1) | Đ |
|---|---|---|---|---|---|
| a Calculated from the weight of the polyester obtained after purification. b Measured by SEC in THF against poly(ethylene oxide) standards. | |||||
| PButL | Modified | 92 | 18.3 | 52.7 | 2.88 |
| PHexL | Modified | 87 | 16.3 | 37.2 | 2.28 |
| Conventional | 94 | 17.0 | 37.1 | 2.18 | |
| PNeoL | Modified | 95 | 17.1 | 46.4 | 2.73 |
| PCycL | Modified | 98 | 10.0 | 20.4 | 2.03 |
| Conventional | 98 | 9.02 | 23.4 | 2.60 | |
| PButA | Modified | 91 | 5.14 | 9.53 | 1.85 |
| PButLA-20 | Modified | 89 | 20.5 | 52.4 | 2.55 |
The chemical structure of the polyesters was confirmed by 1H NMR spectroscopy and was further supported by 13C and 2D NMR investigations (i.e., COSY, HMBC and HMQC, Fig. S17–S32†). The stacked 1H NMR spectra of the polyesters, along with Monomer L, are depicted in Fig. 4. As expected, the polyesters showed broader 1H NMR signals in comparison with the monomer. The disappearance of the ethyl signals (a and b) of the ester group in the monomer confirmed the complete transesterification of Monomer L. Furthermore, the signals originating from the monomer unit, including the CH2 signals (c and d), the CH3 signal (e) and the CH2 signals of the ketal ring (f, g, and h), were observed at nearly identical chemical shifts as those of the monomer, clearly demonstrating the presence of the spirocyclic monomer residue in all the polyesters. In addition, the signals from the OCH2 (1) and CH/CH2/CH3 groups (2 and 3) of the aliphatic diol residues were also observed. It should be noted that in polyester PCycL, each signal from the cyclohexylenedimethylene units was split into two peaks because of the presence of a mixture of the two diastereoisomeric units (cis and trans) in 1,4-CHDM residues. The relative contents of these isomeric units in the polymer (30/70, cis/trans) were nearly identical to that in the monomer feed (28/72, cis/trans).
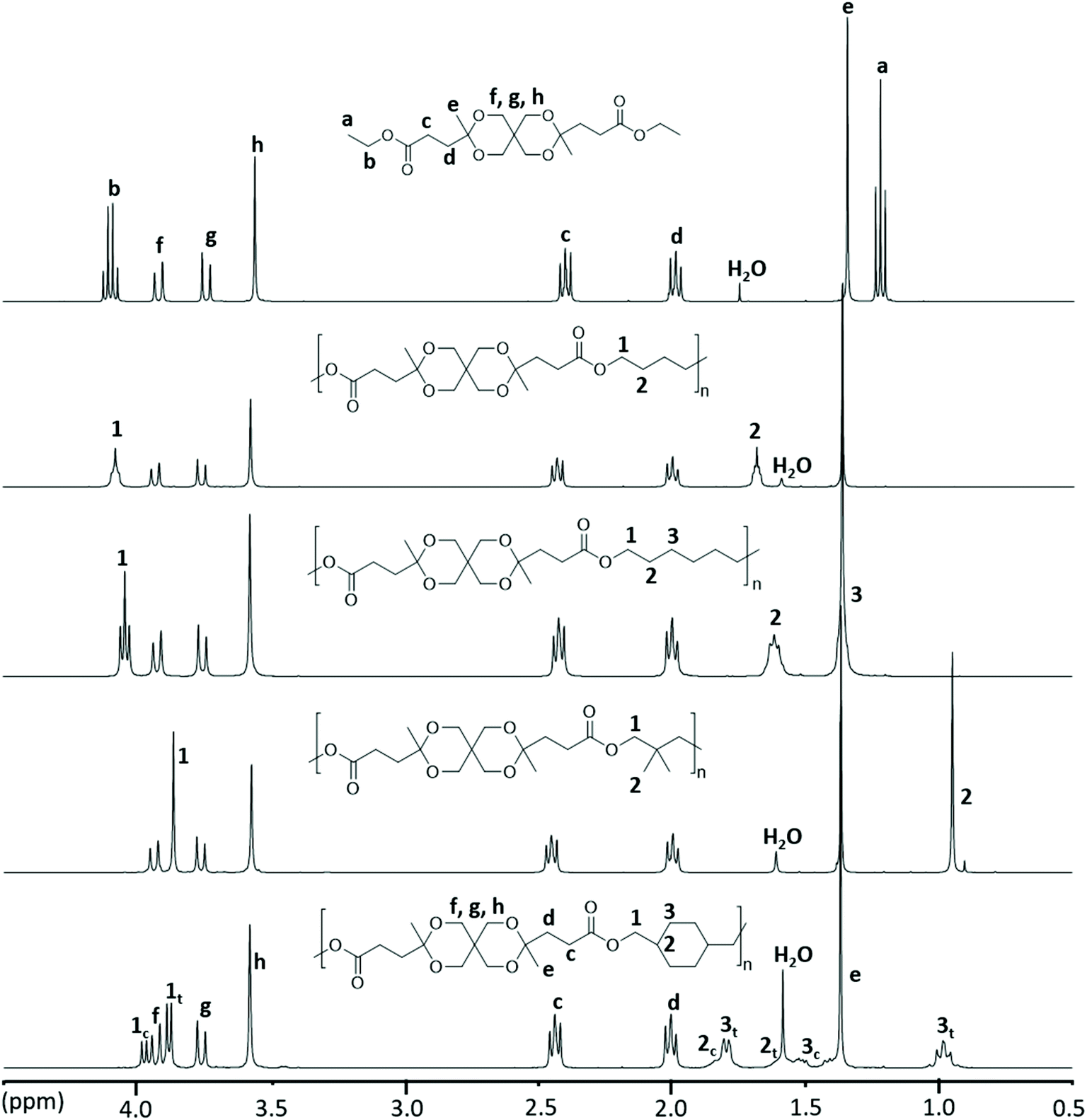 | ||
| Fig. 4 1H NMR spectra of polyesters produced via modified melt polycondensations. Data were recorded using CDCl3 solutions. | ||
3.5 Copolyester synthesis
Monomer L was further evaluated as a co-monomer to demonstrate its versatility and usefulness in copolymerizations to tune polymer properties. Poly(butylene adipate) (PButA) is a commercially available aliphatic polyester with a wide range of applications due to its mechanically flexible, semicrystalline and biodegradable properties and was selected as a starting point for the incorporation of Monomer L. Homopolyester PButA and a copolyester containing 20 mol% of Monomer L (PButLA-20) were produced via a modified melt polycondensation using a similar protocol to that described above for the homopolyesters (Scheme 3). The transesterification and polycondensation were performed at 130 °C for 5 and 20 h, respectively. After the polycondensations, the resulting viscous reaction mixtures were dissolved in chloroform and the products were precipitated in methanol to obtain PButA and PButLA-20 in 91 and 89% isolated yield, respectively. The Mn values of PButA and PButLA-20 were measured by SEC to be 5.14 and 20.5 kg mol−1, respectively. The Tg and Tm values of the PButA sample (further discussed below) were consistent with the literature values.63 Moreover, the 1H NMR spectrum of the polymer showed no end group signals (Fig. 5). | ||
| Scheme 3 Synthesis of copolyester PButLA-20 by the modified melt polycondensation of diethyl adipate, 1,4-butandiol and Monomer L. | ||
 | ||
| Fig. 5 1H NMR spectra of the PButA and PButL homopolyesters and copolyester PButLA-20 produced by the modified melt polycondensation. Data were recorded using CDCl3 solutions. | ||
The chemical structures of PButA and PButLA-20 were confirmed by 1H NMR, 13C NMR and 2D NMR (Fig. S33–S40†) spectroscopy. In the 1H NMR spectra of PButA (Fig. 5), the signals corresponding to the backbone polymers were clearly observed, including the CH2 signals of the adipate residue (signals 1 and 2) and the CH2 signals of the butylene chain segments (signals 3 and 4). Further signals of Monomer L residues (c–h), in addition to the signals from the PButA backbone, confirmed the structure of the copolyester PButLA-20. Finally, the Monomer L content in PButLA-20 was determined by comparing the integral of the adipate signal (1) with the signals (c and d) from the residues of Monomer L. In this way the Monomer L residue content was calculated to be 20%, i.e., precisely corresponding to the monomer feed. This demonstrated the excellent reactivity and stability of Monomer L in copolymerization, allowing for a high level of control of the polycondensation results.
3.6 Thermal properties
The thermal stability of the polyesters was evaluated by thermogravimetric analysis (TGA) under nitrogen. As shown in Fig. 6A–B and Table 2, all the polyesters showed two decomposition rate maxima (Td ∼350 °C and 420 °C). The first Td at ca. 350 °C had the highest rate, which corresponded to the thermal decomposition of polyester backbones. The second Td at ∼420 °C had a relatively low rate, which may be ascribed to the degradation-induced formation of a crosslinked structure during the TGA measurements at higher temperatures. This observation is consistent with previous reports.33 Furthermore, Td,5 (the temperature at 5% weight loss) was above 300 °C for all the polyesters, indicating the possibility for melt processing. The Td,5 values of the polyesters based on the non-cyclic diol units showed an increasing trend with an increase in the size of the diol units (i.e., PHexL > PButL > PNeoL). In addition, polyester PCycL with a cyclic diol unit showed a higher Td,5 compared to the polyesters containing non-cyclic diol units. This may be attributed to the higher structural rigidity of the cyclic diol unit. The char yields were relatively low, which is consistent with their aliphatic nature. In order to verify that no degradation occurred below Td,5, the samples of all the polyesters were first kept at 200 °C for 10 min in the TGA instrument before cooling to room temperature. All the thermally treated samples were perfectly soluble in chloroform-d and NMR analysis detected no structural change, thus demonstrating the high thermal stability of the present polyesters (Fig. S41†). | ||
| Fig. 6 (A) TGA thermograms and (B) first derivative curves of the polyesters recorded under a nitrogen atmosphere. | ||
| Polyester | T d, max (°C) | T d,5 (°C) | Char yielda (wt%) | T g (°C) | T m (°C) | T c (°C) | ΔHmb |
|---|---|---|---|---|---|---|---|
| a Obtained by TGA. b Determined by DSC. | |||||||
| PHexL | 356, 430 | 316 | 12 | 12 | — | — | — |
| PButL | 356, 425 | 315 | 15 | 25 | — | — | — |
| PNeoL | 351, 421 | 308 | 15 | 41 | — | — | — |
| PCycL | 352, 421 | 318 | 12 | 49 | — | — | — |
| PButA | 365 | 302 | 4 | −55 | 57 | 30 | 58.3 |
| PButLA-20 | 346, 435 | 307 | 8 | −40 | 31 | — | 0.61 |
The thermal transitions of the polymers were investigated by differential scanning calorimetry (DSC) and the data obtained are shown in Fig. 7 and Table 2. The glass transition of the fully amorphous polyesters was observed in the range Tg = 12–49 °C, and increased with increasing rigidity of the diol unit of the polymer. In the series, PCycL exhibited the highest Tg at 49 °C, which was attributed to the presence of the bulky cyclohexylenedimethylene units in the polymer chain, followed by PNeoL, PButL, and PHexL with Tg values of 41, 25 and 12 °C, respectively. The Tg values of polymers are in general influenced by the level of branching.64,65 and, as already mentioned, the present SEC results indicated limited branching. Still, at this low level the effect should be very small.
 | ||
| Fig. 7 DSC traces of the polyesters obtained during the second heating cycle (Tg values are indicated). | ||
The thermal properties of the copolyester PButLA-20 were investigated and compared with reference to the homopolyesters PButA and PButL. TGA measurements showed two decomposition rate maxima at Td ∼346 and 435 (°C) for PButLA-20, while the base polymer PButA only showed a single decomposition rate maximum at 365 °C (Table 2 and Fig. 8A and B). The second Td of PButLA-20 at ∼435 °C was similar to that observed for PButL and it exhibited a lower rate to that of PButL, which was consistent with the lower content of spirocyclic units in the copolymer chain. The Td,5 values were quite close for the two samples, 302 and 307 °C for PButA and PButLA-20, respectively. In addition, the Td,5 and char yield values for PButLA-20 were slightly higher in comparison with PButA, indicating enhanced thermal stability induced by the presence of Monomer L.
 | ||
| Fig. 8 TGA weight loss (A) and derivative weight loss (B), as well as DSC traces from the first cooling (C) and second heating of PButA, PButLA-20 and PButL (D). | ||
As expected, DSC analysis revealed changes in the Tg and Tm values of polyester PButA upon the incorporation of the spirocyclic monomer (Table 2 and Fig. 8C and D). PButA showed Tg = −55 °C and a crystalline melt interval between 52 and 61 °C with Tm = 57 °C. This indicated the presence of different populations of crystallites, which is consistent with the literature.66,67 In contrast, PButLA-20 showed an increased Tg value at −40 °C together with a minor melt endotherm at 31 °C. The melt enthalpy of PButLA-20 (ΔHm = 0.61 J g−1) was nearly 100 times lower than that of PButA (ΔHm = 58.3 J g−1), indicating an almost insignificant degree of crystallinity of the former sample. The observed decrease of the Tm value and melt enthalpy of PButLA-20 was attributed to the irregular structure of the statistical copolymer.
3.7 Film formation
Polymer films were prepared by casting from solution. Homogeneous polymer solutions in chloroform (100 mg mL−1) were spread evenly onto Teflon crucibles, followed by solvent evaporation at room temperature under a glass conical funnel overnight. PButL, PHexL, PNeoL and PButLA-20 formed smooth films without significant wrinkles, cracks or bubbles. However, only PNeoL produced a stable and free-standing film, while the other two polymers gave soft sticky films because their Tg values were close to room temperature. PCycL formed films with cracks upon casting, most likely due to its relatively low molecular weight. As can be seen in Fig. S42,† the film formed by PNeoL was transparent, tough and mechanically flexible. In addition, the Tgs of PNeoL and PCycL were above the minimum value typically required for amorphous polymers in powder coating applications (usually 40–45 °C).68–70 Therefore, these two polyesters were selected for dynamic mechanical and melt rheological analysis.3.8 Dynamic mechanical and rheological properties
The dynamic mechanical properties of polyesters PNeoL and PCycL were studied by DMA analysis of hot-pressed samples (Fig. S43†). Polyester PButL, PHexL and PButLA-20 could not be analyzed due to their soft and sticky character, and the dynamic mechanical properties of PButA have been reported previously.71,72As shown in Fig. 9A–C and Table 3, the storage modulus (E′) values at the glassy plateau (20 °C) of the polyesters PNeoL and PCycL were 2.36 and 1.91 GPa, respectively. Hence, PCycL showed a lower storage modulus than PNeoL. This observation may be explained by the lower molecular weight of PCycL (10 kg mol−1) compared to PNeoL (17 kg mol−1). The Tg values were taken at the local maximum of E′′ at the glass transition (Fig. 9B, E′′max in Table 3). Consequently, the Tg values were 43 and 48 °C for PNeoL and PCycL, respectively. These values were in good agreement with the corresponding data obtained by DSC, showing a good correlation between the two methods. The tanδ curve, which indicates the variation of the damping behavior, showed a local maximum (tanδmax, Table 3) due to the energy dissipation originating from a sharp increase of the segmental mobility of the polymer in the Tg region. However, after this region, the tanδ curve showed a quick and steady increase with temperature for both PCycL and PNeoL, respectively, without the presence of any well-defined rubbery plateau. This indicated a rapid transition into the viscous flow region. This observation may be attributed to the absence of crystallinity and a limited degree of chain entanglements in the two polymer samples due to their relatively low Mn values.
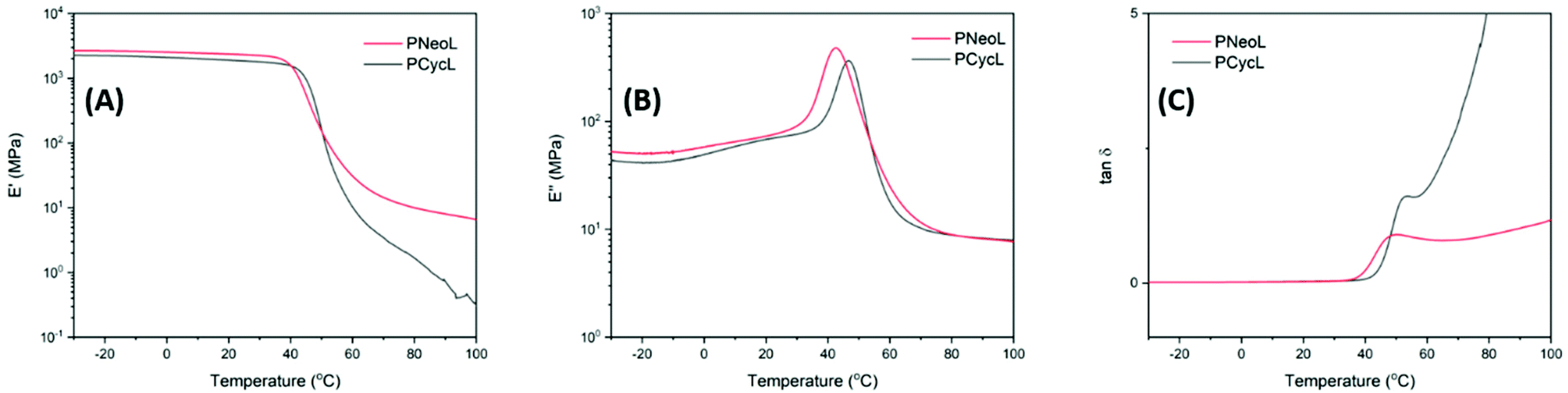 | ||
| Fig. 9 DMA data showing the (A) storage modulus, (B) loss modulus, and (C) tan δ of polyester PNeoL and PCycL (1 Hz, 0.1% strain). | ||
Dynamic rheology measurements were carried out to assess the stability, melt processability and flow characteristics of the polyesters in the melt state. Rheological properties measured at low strains and strain rates in the linear viscoelastic region are sensitive to molecular weight and structure, and changes in the modulus may thus indicate degradation by, e.g., polymer chain scission or crosslinking reactions. High precision time sweep experiments were performed in oscillatory shear mode on PNeoL and PCycL by using hot-pressed sample discs (Fig. S43†) in a parallel plate arrangement. Sweeps were carried out at 130 °C for 25 min at a frequency of 1 Hz using a constant strain of 1%. This ensured that the measurements were performed within the linear viscoelastic region. Fig. 10A and B show the melt shear storage modulus (G′), shear loss modulus (G′′) and phase shift (δ) as a function of time at 130 °C for PNeoL and PCycL, respectively. As can be seen, for both polyesters, these parameters were almost constant throughout the measurements. This observation indicated the high thermal stability of the polyesters under low shear in the melt state without any noticeable signs of polymer chain degradation. Furthermore, 1H NMR analysis of the samples directly after the rheological measurements revealed no structural changes in relation to the data obtained before the experiments (Fig. S44–S45†). This further indicated the absence of any significant degradation. The polyester PNeoL exhibited a higher value of G′ than PCycL at 130 °C, which was most probably related to its higher molecular weight. Overall, the time sweep experiments demonstrated a very stable rheological behavior of the polyesters during the measurements in the melt state.
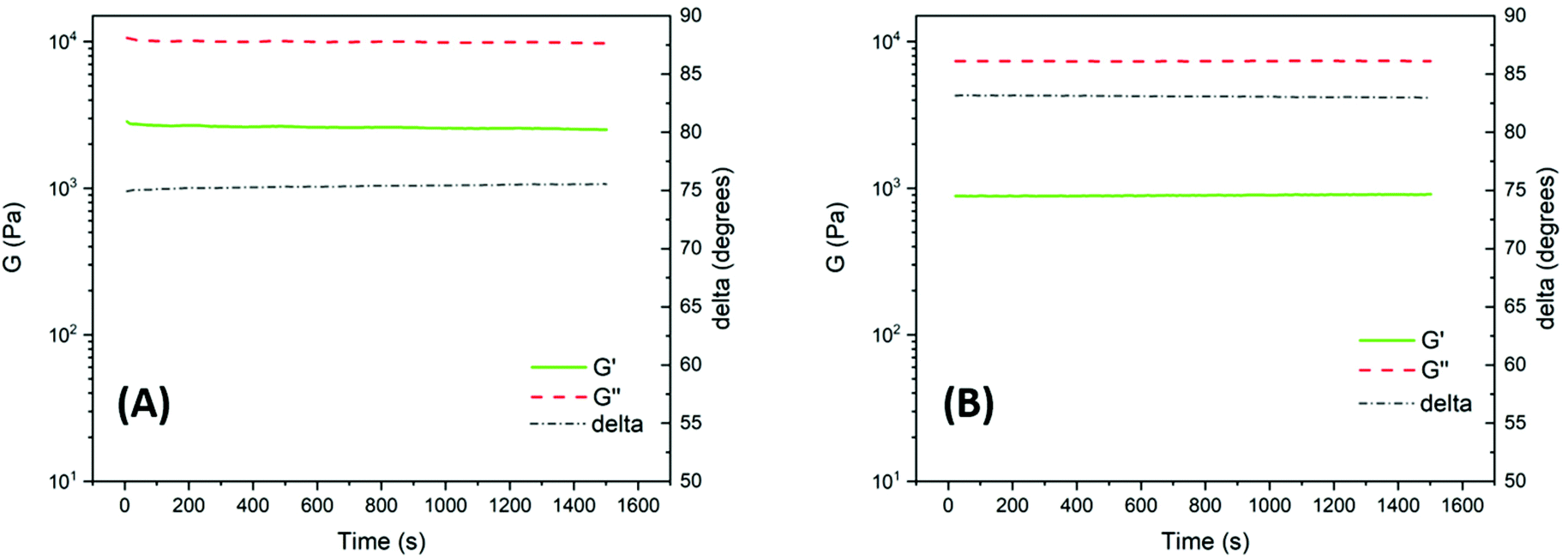 | ||
| Fig. 10 Time sweep traces showing the shear storage modulus (G′), shear loss modulus (G′′) and phase shift (delta) for PNeoL (A) and PCycL (B) at 130 °C (1 Hz, 1% strain). | ||
In order to further investigate the shear flow characteristics, PCycL was selected as a representative sample for frequency dependent dynamic rheological measurements. The frequency dependence of the rheological properties was measured at 110, 115, 120, 125 and 130 °C in the frequency range 0.1–100 Hz at a constant strain of 1%. Time–temperature superposition (TTS) was applied to the data in order to create a master curve covering the flow behavior of the polymer melt over a broader frequency window. Fig. 11A shows the master curves for G′ and G′′ as a function frequency at the chosen reference temperature (130 °C). At 330 Hz, the master curves of PCycL displayed a transition from the melt flow region, where G′′ dominated over G′, to a rubbery plateau where G′ increased above G′′. Furthermore, the shift factors (aT) versus temperature for PCycL obeyed the Williams–Landel–Ferry (WLF) equation:
 | (1) |
 | (2) |
| Cg2 = C2 + (Tg − Tref) | (3) |
 | ||
| Fig. 11 (A) Storage G′ and loss G′′ modulus master curves of PCycL at Tr = 130 °C, and (B) temperature dependence of the shift factor (squares) and fit of the Williams–Landel–Ferry equation (dashed curve, C1 = 3.156, C2 = 107.7 K). | ||
The calculated values of C1g and C2g were 12.8 and 26.7 K, respectively, which are close to the reported values for other non-aggregating polymers, displaying C1g and C2g values of 16.79 ± 5.43 and 51.6 ± 28.1 K, respectively.73,74
4. Conclusions
An eco-friendly, sugar-based spiro-diester monomer was prepared by a straightforward reaction of biobased ethyl levulinate and pentaerythritol. The synthesis of Monomer L was carried out with an environmentally friendly pathway and the product was straightforward to scale-up and purify. However, the melt polycondensation was challenging due to the hydrothermal sensitivity of Monomer L. However, after a thorough investigation of the thermal stability of Monomer L, suitable polymerization conditions were identified. Using optimized drying and polycondensation conditions, the spiro-diester was successfully polymerized with various potentially biobased diols to produce a series of aliphatic polyesters with reasonably high molecular weights. The fully amorphous polyesters exhibited sufficient thermal stability with Tds in the range 308–318 °C and Tgs in the range 12–49 °C, which were significantly higher than the corresponding polyesters of the diols and alternative aliphatic dicarboxylates. Both hot-pressed and solvent cast films of the polyesters based on NPG and 1,4-CHDM were flexible and mechanically strong with high transparency, which suggests their potential use in coating applications. Moreover, melt rheological experiments revealed sufficient thermo-rheological stability of these polyesters for melt processing.Conflicts of interest
Perstorp AB markets the biobased pentaerythritol (Voxtar™) used in the synthesis of Monomer L. There are no other conflicts to declare.Acknowledgements
This work was financially supported by the Mistra Foundation (Swedish Foundation for Strategic Environmental Research) through the “STEPS” project (No. 2016/1489). The research was also supported by grants from the Royal Physiographic Society in Lund. We thank Siim Laanesoo from the University of Tartu, Estonia, and Dr. Prakash P. Wadgaonkar and Amol Ichake from the CSIR-National Chemical Laboratory, Pune, for their valuable help with the SEC analysis.References
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed
.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed
- J. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef CAS PubMed
- C. Vilela, A. F. Sousa, A. C. Fonseca, A. C. Serra, J. F. J. Coelho, C. S. R. Freire and A. J. D. Silvestre, Polym. Chem., 2014, 5, 3119–3141 RSC
- D. Garlotta, J. Polym. Environ., 2019, 9, 63–84 CrossRef
- A. Celli, P. Marchese, L. Sisti, D. Dumand, S. Sullalti and G. Totaro, Polym. Int., 2013, 62, 1210–1217 CrossRef CAS
- S. R. Turner, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5847–5852 CrossRef CAS
- Y. Hu, Z. Zhao, Y. Liu, G. Li, A. Wang, Y. Cong, T. Zhang, F. Wang and N. Li, Angew. Chem., Int. Ed., 2018, 57, 6901–6905 CrossRef CAS
- D. R. Kelsey, B. M. Scardino, J. S. Grebowicz and H. H. Chuah, Macromolecules, 2000, 33, 5810–5818 CrossRef CAS
- N. C. Hoppens, T. W. Hudnall, A. Foster and C. J. Booth, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3473–3478 CrossRef CAS
- T. Chen, W. Zhang and J. Zhang, Polym. Degrad. Stab., 2015, 120, 232–243 CrossRef CAS
-
E. U. Elam, J. C. Martin and G. Russell, US Pat., 3313777, 1967 Search PubMed
- R. Hatti-Kaul, L. J. Nilsson, B. Zhang, N. Rehnberg and S. Lundmark, Trends Biotechnol., 2020, 38, 50–67 CrossRef CAS PubMed
- A. Hufendiek, S. Lingier and F. E. Du Prez, Polym. Chem., 2019, 10, 9–33 RSC
- S. Muñoz-Guerra, C. Lavilla, C. Japu and A. Martínez De Ilarduya, Green Chem., 2014, 16, 1716–1739 RSC
- S. V. Mankar, M. N. Garcia Gonzalez, N. Warlin, N. G. Valsange, N. Rehnberg, S. Lundmark, P. Jannasch and B. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 19090–19103 CrossRef CAS
- C. Lavilla and S. Muñoz-Guerra, Green Chem., 2013, 15, 144–151 RSC
- O. Nsengiyumva and S. A. Miller, Green Chem., 2019, 21, 973–978 RSC
- C. Pang, X. Jiang, Y. Yu, L. Chen, J. Ma and H. Gao, ACS Macro Lett., 2019, 8, 1442–1448 CrossRef CAS
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G. J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961–5983 RSC
- A. Gandini, D. Coelho, M. Gomes, B. Reis and A. Silvestre, J. Mater. Chem., 2009, 19, 8656–8664 RSC
- J. A. Galbis, M. D. G. García-Martín, M. V. De Paz and E. Galbis, Chem. Rev., 2016, 116, 1600–1636 CrossRef CAS PubMed
- F. Fenouillot, A. Rousseau, G. Colomines, R. Saint-Loup and J. P. Pascault, Prog. Polym. Sci., 2010, 35, 578–622 CrossRef CAS
- C. Japu, A. Alla, A. Martínez De Ilarduya, M. G. García-Martín, E. Benito, J. A. Galbis and S. Muñoz-Guerra, Polym. Chem., 2012, 3, 2092–2101 RSC
- E. Zakharova, A. Alla, A. Martínez De Ilarduya and S. Muñoz-Guerra, RSC Adv., 2015, 5, 46395–46404 RSC
- C. Lavilla, A. Alla, A. Martínez De Ilarduya and S. Muñoz-Guerra, Biomacromolecules, 2013, 14, 781–793 CrossRef CAS PubMed
- C. Lavilla, A. M. De Ilarduya, A. Alla, M. G. García-Martín, J. A. Galbis and S. Muñoz-Guerra, Macromolecules, 2012, 45, 8257–8266 CrossRef CAS
- C. Lavilla and S. Muñoz-Guerra, Polym. Degrad. Stab., 2012, 97, 1762–1771 CrossRef CAS
- C. Lavilla, A. Alla, A. M. De Ilarduya, E. Benito, M. G. García-Martín, J. A. Galbis and S. Muñoz-Guerra, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1591–1604 CrossRef CAS
- C. Lavilla, A. Alla, A. Martínez De Ilarduya, E. Benito, M. G. García-Martín, J. A. Galbis and S. Muñoz-Guerra, Biomacromolecules, 2011, 12, 2642–2652 CrossRef CAS PubMed
- H. Zhang, G. Zhou, M. Jiang, H. Zhang, H. Wang, Y. Wu and R. Wang, Macromolecules, 2020, 53, 5475–5486 CrossRef CAS
- N. Warlin, M. N. Garcia Gonzalez, S. Mankar, N. G. Valsange, M. Sayed, S. H. Pyo, N. Rehnberg, S. Lundmark, R. Hatti-Kaul, P. Jannasch and B. Zhang, Green Chem., 2019, 21, 6667–6684 RSC
- S. Lingier, Y. Spiesschaert, B. Dhanis, S. De Wildeman and F. E. Du Prez, Macromolecules, 2017, 50, 5346–5352 CrossRef CAS
- O. Bonjour, I. Liblikas, T. Pehk, T. Khai-Nghi, K. Rissanen, L. Vares, P. Jannasch and P. Jannasch, Green Chem., 2020, 22, 3940–3951 RSC
- F. D. Pileidis and M. M. Titirici, ChemSusChem, 2016, 9, 562–582 CrossRef CAS PubMed
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–555 RSC
- S. H. Pyo, S. J. Glaser, N. Rehnberg and R. Hatti-Kaul, ACS Omega, 2020, 5, 14275–14282 CrossRef CAS PubMed
- S. K. Bisen, P. S. Niphadkar, S. U. Nandanwar, I. Simakova and V. V. Bokade, Environ. Prog. Sustainable Energy, 2021, 40, 1–9 Search PubMed
- J. J. Bozell, L. Moens, D. C. Elliott, Y. Wang, G. G. Neuenscwander, S. W. Fitzpatrick, R. J. Bilski and J. L. Jarnefeld, Resour., Conserv. Recycl., 2000, 28, 227–239 CrossRef
- A. S. Amarasekara, U. Ha and N. C. Okorie, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 955–958 CrossRef CAS
- G. C. Hayes and C. R. Becer, Polym. Chem., 2020, 11, 4068–4077 RSC
- Y. Bernhard, S. Pellegrini, T. Bousquet, A. Favrelle, L. Pelinski, F. Cazaux, V. Gaucher, P. Gerbaux and P. Zinck, ChemSusChem, 2019, 12, 3370–3376 CrossRef CAS PubMed
- Y. Bernhard, L. Pagies, S. Pellegrini, T. Bousquet, A. Favrelle, L. Pelinski, P. Gerbaux and P. Zinck, Green Chem., 2019, 21, 123–128 RSC
- C. Leibig, B. Mullen, T. Mullen, L. Rieth and V. Badarinarayana, ACS Symp. Ser., 2011, 1063, 111–116 CrossRef CAS
- B. D. Mullen, V. Badarinarayana, M. Santos-Martinez and S. Selifonov, Top. Catal., 2010, 53, 1235–1240 CrossRef CAS
-
S. Selifonov, S. D. Rothstein, D. A. Wicks, B. D. Mullen, T. J. Mullen, J. D. Pratt, C. T. Williams, C. Wu and N. Zhou, US Pat., 8546519, 2013 Search PubMed
- IPPC Intergovernmental Panel on Climate, IPCC Fourth Assessment Report: Climate Change, 2007.
- V. Isoni, D. Kumbang, P. N. Sharratt and H. H. Khoo, J. Environ. Manage., 2018, 214, 267–275 CrossRef CAS PubMed
-
Perstorp AB
, Proof of Sustainability for Perstorp'sPro- Environment Polyols, 2007 Search PubMed
- ISO 14044:2006 – Environmental management – Life cycle assessment – Requirements and guidelines, 2006.
- ISO 14040:2006 – Environmental management – Life cycle assessment – Principles and framework, 2006.
- P. J. Stevenson, Org. Biomol. Chem., 2011, 9, 2078–2084 RSC
- S. González-García, L. Argiz, P. Míguez and B. Gullón, Chem. Eng. J., 2018, 350, 982–991 CrossRef
- B. Cok, I. Tsiropoulos, A. L. Roes and M. K. Patel, Biofuels, Bioprod. Biorefining, 2012, 6, 246–256 CrossRef
- H. I. Moussa, A. Elkamel and S. B. Young, J. Clean. Prod., 2016, 139, 761–769 CrossRef CAS
- M. N. García González, P. Börjesson, M. Levi and S. Turri, J. Polym. Environ., 2018, 26, 3626–3637 CrossRef
- A. Corona, M. J. Biddy, D. R. Vardon, M. Birkved, M. Z. Hauschild and G. T. Beckham, Green Chem., 2018, 20, 3857–3866 RSC
- R. Aryapratama and M. Janssen, J. Clean. Prod., 2017, 164, 434–443 CrossRef CAS
- E. Zakharova, A. M. De Ilarduya, S. León and S. Muñoz-Guerra, Des. Monomers Polym., 2017, 20, 157–166 CrossRef CAS PubMed
- P. Wang, C. R. Arza and B. Zhang, Polym. Chem., 2018, 9, 4706–4710 RSC
- C. R. Arza, P. Wang, J. Linares-Pastén and B. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 2314–2323 CrossRef CAS
- Z. Florjańczyk, A. Jóźwiak, A. Kundys, A. Plichta, M. Dbowski, G. Rokicki, P. Parzuchowski, P. Lisowska and A. Zychewicz, Polym. Degrad. Stab., 2012, 97, 1852–1860 CrossRef
- X. Zhu, Y. Zhou and D. Yan, J. Polym. Sci. Part B: Polym. Phys., 2011, 49, 1277–1286 CrossRef CAS
- A. Khalyavina, L. Häußler and A. Lederer, Polymer, 2012, 53, 1049–1053 CrossRef CAS
- C. Berti, A. Celli, P. Marchese, E. Marianucci, S. Sullalti and G. Barbiroli, Macromol. Chem. Phys., 2010, 211, 1559–1571 CrossRef CAS
- Z. Gan, K. Kuwabara, H. Abe, T. Iwata and Y. Doi, Biomacromolecules, 2004, 5, 371–378 CrossRef CAS PubMed
- E. Gubbels, J. P. Drijfhout, C. Posthuma-Van Tent, L. Jasinska-Walc, B. A. J. Noordover and C. E. Koning, Prog. Org. Coatings, 2014, 77, 277–284 CrossRef CAS
-
B. A. J. Noordover, R. Duchateau, C. E. Koning and R. A. T. M. van Benthem, in Proceedings of the 241st ACS National Meeting - Biobased monomers, polymers and materials, Anaheim, USA, 2011, pp. 1–3
- B. A. J. Noordover, V. G. van Staalduinen, R. Duchateau, C. E. Koning, R. A. T. M. van Benthem, M. Mak, A. Heise, A. E. Frissen and J. van Haveren, Biomacromolecules, 2006, 7, 3406–3416 CrossRef CAS PubMed
- C. W. Chen, T. S. Hsu, K. W. Huang and S. P. Rwei, Polymers, 2020, 12, 1160 CrossRef CAS PubMed
- B. Wu, Y. Xu, Z. Bu, L. Wu, B. G. Li and P. Dubois, Polymers, 2014, 55, 3648–3655 CrossRef CAS
-
J. D. Ferry, Viscoelastic properties of polymers, 1980 Search PubMed
- S. T. Hemp, M. Zhang, M. Tamami and T. E. Long, Polym. Chem., 2013, 4, 3582–3590 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc00724f |
| This journal is © The Royal Society of Chemistry 2021 |