DOI:
10.1039/D1GC00604E
(Paper)
Green Chem., 2021,
23, 3312-3321
Replacing DMF in solid-phase peptide synthesis: varying the composition of green binary solvent mixtures as a tool to mitigate common side-reactions†
Received
16th February 2021
, Accepted 31st March 2021
First published on 19th April 2021
Abstract
Significant efforts have been made in recent years to identify more environmentally benign and less hazardous alternatives to N,N-dimethylformamide (DMF) in solid-phase peptide synthesis (SPPS). Several greener solvents have been endorsed as suitable candidates yet finding a neat solvent that fully matches the qualities of DMF in SPPS has proven challenging. To this end, we recently demonstrated that green binary solvent mixtures are viable alternatives to DMF and showed that the polarity and viscosity profile of a binary solvent mixture can be used to predict its utility in SPPS. In this report, we systematically investigate how the composition of green binary solvent mixtures influences Fmoc-removal, peptide coupling and common side-reactions in SPPS and show that the purity profile is not impacted adversely in binary solvent mixtures when compared to DMF. Furthermore, we demonstrate that the sole variation of the composition of the binary solvent mixture during synthesis represents a novel and simple tool to mitigate certain side-reactions in SPPS, exemplified by suppression of Arg-lactamisation and aspartimide formation. When applied to the synthesis of the peptide therapeutic Bivalirudin on a 7.5 mmol scale, we showed that simply adjusting the solvent ratio in a single step of the synthesis significantly suppressed a problematic Arg-lactamisation side-reaction. These results underline that green binary solvent mixtures not only can replace DMF in SPPS but also provide novel solutions for mitigating common side-reactions in SPPS.
Introduction
Solid-phase peptide synthesis (SPPS) enables straightforward and reproducible synthesis of peptides with high purity and is generally the preferred method in industry for the production of therapeutic peptides.1,2 In SPPS, factors such as the choice of resin and the sequence and length of the target peptide have a direct impact on the generation and accumulation of side-products, and in extension the efficiency of the synthesis. Numerous side-reactions occurring at different stages of SPPS e.g. aspartimide and diketopiperazine (DKP) formation, arginine δ-lactamisation, and amino acid epimerisation/racemisation have been extensively studied and are known to negatively impact the overall peptide yield and purity.3,4 Strategies to improve synthesis efficiency typically include temporary backbone modification using designated protecting groups, and optimisation of reagent concentrations, reaction time and temperature.4–7 However, the influence of solvents on these side-reactions has not yet been systematically explored, likely due to the limited choice of solvents conventionally used in SPPS. N,N-Dimethylformamide (DMF), dichloromethane (DCM), and N-methyl-2-pyrrolidone (NMP) are the gold standard SPPS solvents in academic research environments and industrial production facilities alike, yet these solvents are to be restricted by the European Chemicals Agency (ECHA) due to their hazardous nature.8 In recent years, this has triggered considerable research into identifying greener and less toxic alternatives to especially DMF in SPPS.9–12 We have found that binary mixtures of green solvents (e.g. dimethyl sulfoxide (DMSO)/ethyl acetate (EtOAc), DMSO/1,3-dioxolane (DOL), and DMSO/2-methyl tetrahydrofuran (2-Me-THF) that display similar polarity and viscosity profiles to DMF are viable, less toxic solvent alternatives to DMF in SPPS.13 The binary solvent mixtures performed comparably to DMF in terms of synthetic yield and purity in the SPPS of a range of model peptides displaying a variety of synthetic challenges and sequence lengths (up to a 29-mer) at ambient temperature.13 In the present study, we put further emphasis on the composition of the binary solvent mixtures and systematically evaluate the influence of solvent polarity on commonly observed side-reactions in SPPS, including Arg-lactamisation, DKP formation, aspartimide formation, and amino acid racemisation. Furthermore, faced with numerous possible solvent combinations and the flexibility to vary the ratios of the individual solvents, we hypothesised that binary solvent mixtures could provide an unprecedented opportunity to customise the solvent properties to suit the separate steps in the SPPS cycle. We envisioned that such a ‘toolbox’ of useful binary solvent mixtures would provide increased control over coupling and Fmoc-removal reactions, while also limiting side-product formation. As a proof of concept, we herein demonstrate that varying the composition of green binary solvent mixtures can be used to mitigate aspartimide formation in the scorpion toxin II model system,14 and furthermore limit Arg-lactamisation in the fully automated SPPS of the anti-coagulant peptide therapeutic Bivalirudin.
Results and discussion
Varying the solvent polarity between coupling and Fmoc-removal impacts the purity of Bivalirudin
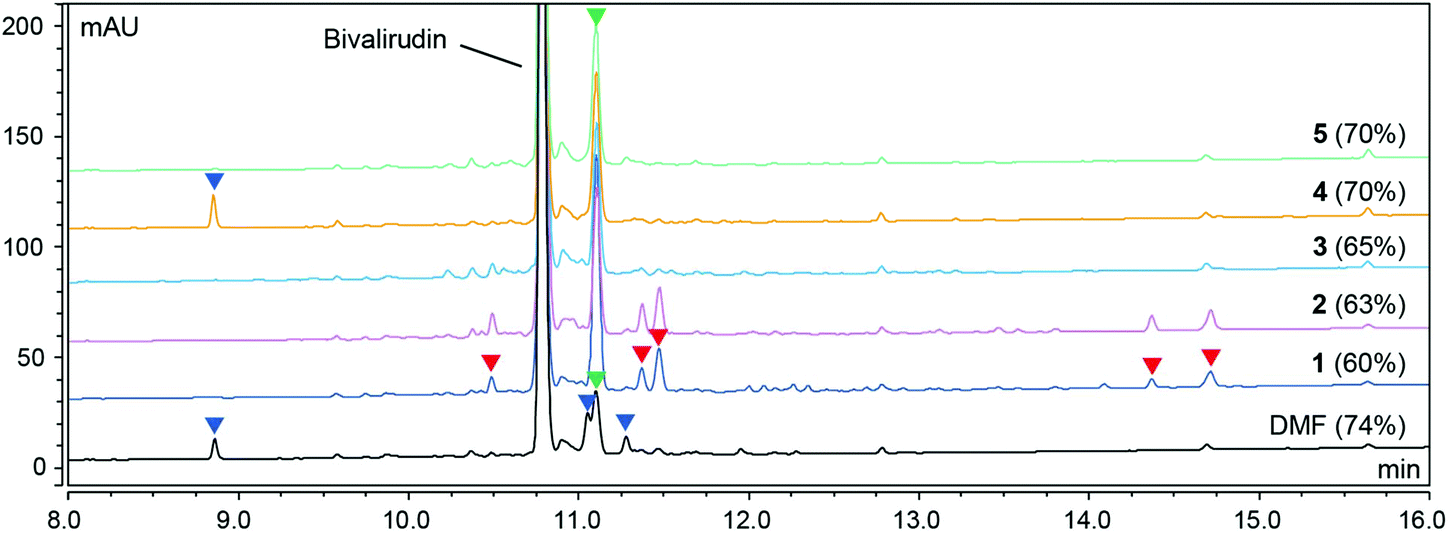
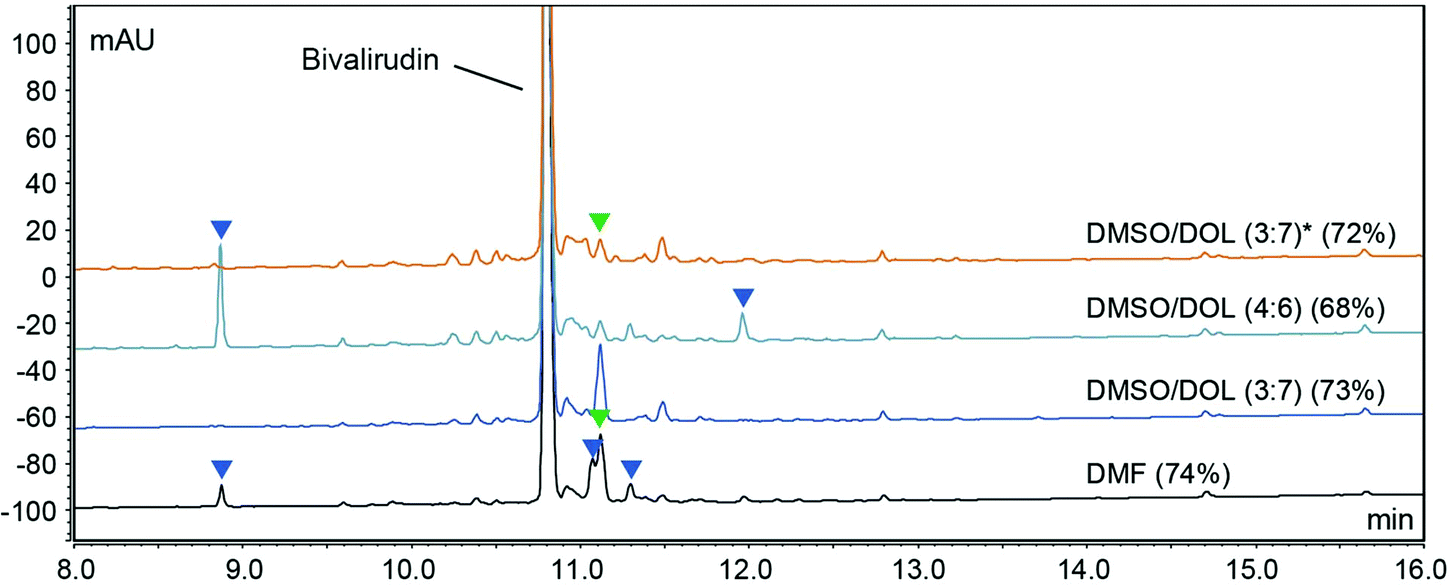
We recently demonstrated that the polarity and viscosity (PV) profile of a given solvent is a valuable indicator of its utility in SPPS. While green binary solvent mixtures displaying a similar PV profile to DMF proved to be viable alternatives for the SPPS of a range of synthetically challenging model peptides, we observed some sequence dependence and identified impurities arising from incomplete Fmoc-removal and amide bond formation (coupling) reactions, as well as side-reactions such as Arg-lactamisation.13 Importantly, solvents not only provide a medium to facilitate chemical reactions but also exert a significant influence on reagent properties and reaction rates on both molecular,15,16 and supramolecular level.17 We have demonstrated that solvent viscosity plays an important role for adequate resin swelling and efficient diffusion of reagents and by-products, and that solvent polarity is fundamental to govern Fmoc-removal and coupling reactions.13 Using reaction kinetics measurements we demonstrated that coupling reactions using diisopropylcarbodiimide (DIC)/OxymaPure are favoured in non-polar solvents, while Fmoc-removal reactions are favoured in polar solvents. The difference is a consequence of the respective reaction mechanisms and whether the stabilisation of charged intermediates is favourable for reaction progression (ESI Fig. S1 and S2†).18–21 Overall, DMF strikes a good balance that makes it useful in both coupling and Fmoc-removal reactions and therefore the gold standard solvent in SPPS. However, we acknowledged that the use of binary solvent mixtures enables a possible tailoring of the composition and ratio of the mixture to suit the application. Hence, we hypothesised that using less polar solvent mixtures for coupling reactions (albeit polar enough to adequately dissolve reagents and by-products), and more polar solvent mixtures for Fmoc-removal reactions (while ensuring that the solvent viscosity and resin swelling capacity was within a useful range) would increase the reaction control. A dynamic solvent viscosity of 0.5–1.1 mPa s is a guideline,13 but slightly higher solvent viscosities may be tolerated. To test the hypothesis, we decided to use the 20-mer peptide therapeutic Bivalirudin (H-fPRPGGGGNGDFEEIPEEYL-OH) as a model and all syntheses were carried out in the designated solvent mixture (and DMF as reference) on 7.5 mmol scale. We started by investigating a small set of binary solvent mixtures i.e. N-formyl morpholine (NFM)/DOL (entry 1, Table 1), N-butyl pyrrolidinone (NBP)/DOL (entry 2), DMSO/DOL (entry 3), DMSO/2-Me-THF (entry 4), and DMSO/EtOAc (entry 5) using ratios of relatively low polarity for coupling reactions, and ratios of higher polarity for Fmoc-removal (generally 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 and 4:6 respectively, Table 1). The Bivalirudin peptide synthesised in DMF and entries 1–5 gave varying purity profiles and the individual side-product peaks were carefully analysed by LC-MS (Fig. 1).
:8 and 4:6 respectively, Table 1). The Bivalirudin peptide synthesised in DMF and entries 1–5 gave varying purity profiles and the individual side-product peaks were carefully analysed by LC-MS (Fig. 1).
 |
| | Fig. 1 HPLC purity profiles (220 nm) of Bivalirudin (H-fPRPGGGGNGDFEEIPEEYL-OH) syntheses 1–5 and reference SPPS in DMF. The corresponding crude purity of Bivalirudin is indicated in parentheses. The syntheses were carried out on an automated peptide synthesiser (Sonata) using H-Leu-2-chlorotrityl resin (CTR) (0.81 mmol g−1 loading) on 7.5 mmol scale, after each coupling reaction the resin was capped using a basic solution of Ac2O (full experimental details in the ESI†). The side-products arising from poor Fmoc-removal (deletion peptides; red arrows), various incomplete coupling reactions (acetylated peptides; blue arrows), and incomplete coupling attributed to Arg-lactamisation (acetylated peptide; green arrows) are highlighted. | |
Table 1 Varying solvent polarity for the SPPS of Bivalirudin
| Entry |
Solvent for coupling reaction |
Polarity [ET(30)] (kcal mol−1) |
Solvent for Fmoc-removal |
Polarity [ET(30)] (kcal mol−1) |
|
NFM/DOL (4:6) and DMSO/EtOAc (6:4) both display dynamic viscosities above 1.1 mPa s at 20 °C (1.29 and 1.13 mPa s respectively) but are efficient solvent mixtures for Fmoc-removal reactions.13
|
| Reference |
DMF |
43.38 |
DMF |
43.38 |
| 1 |
NFM/DOL (2:8) |
40.89 |
NFM/DOL (4:6)a |
41.83 |
| 2 |
NBP/DOL (2:8) |
40.84 |
NBP/DOL (4:6) |
41.11 |
| 3 |
DMSO/DOL (2:8) |
42.21 |
DMSO/DOL (4:6) |
43.38 |
| 4 |
DMSO/2-Me-THF (2:8) |
41.94 |
DMSO/2-Me-THF (4:6) |
43.16 |
| 5 |
DMSO/EtOAc (1:9) |
41.29 |
DMSO/EtOAc (6:4)a |
44.26 |
| 6 |
NBP/DOL (4:6) |
41.11 |
NBP/DOL (4:6) |
41.11 |
| 7 |
NBP/DOL (4:6) |
41.11 |
DMSO/DOL (4:6) |
43.38 |
| 8 |
DMSO/DOL (4:6) |
43.38 |
DMSO/DOL (4:6) |
43.38 |
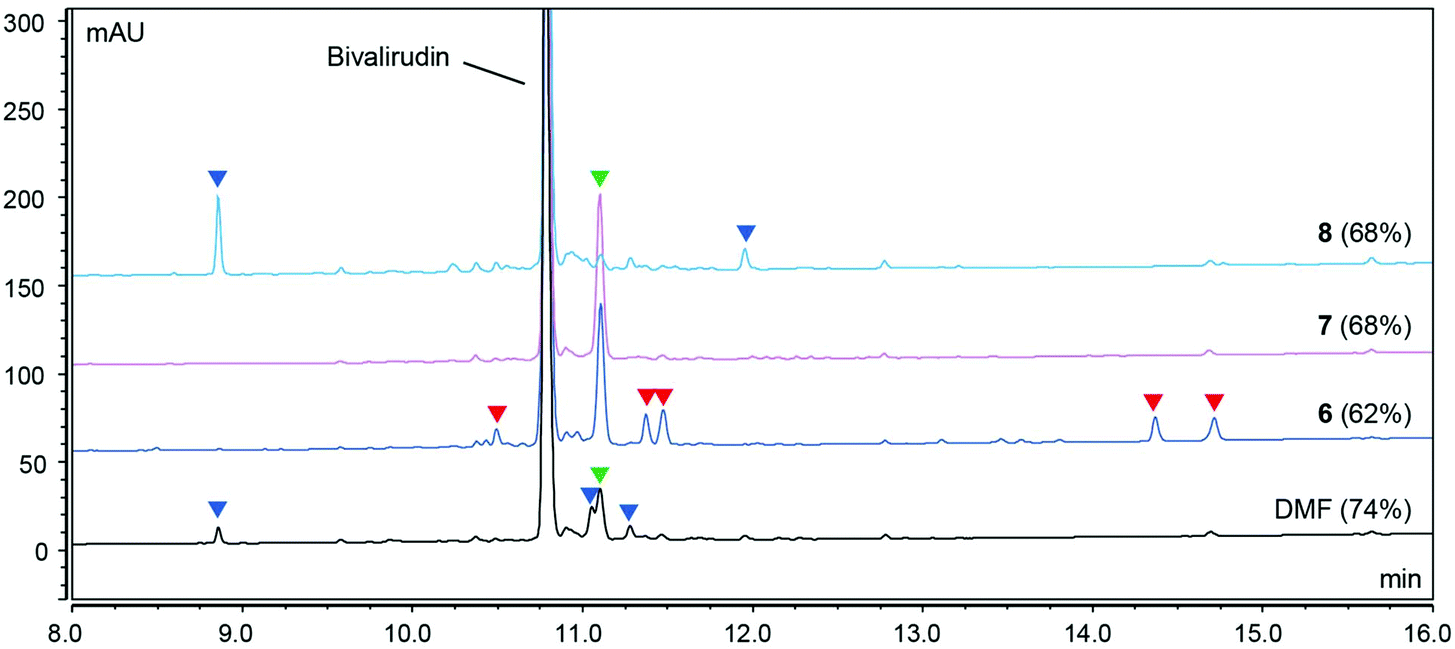
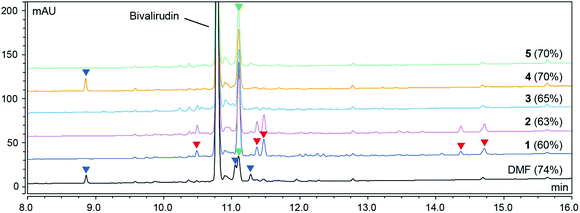
As shown in Fig. 1, the peptide purities obtained from the syntheses of Bivalirudin varied with the green binary solvent mixture. Among the mixtures used for Fmoc-removal, NFM/DOL (4:6) and NBP/DOL (4:6) displayed the lowest polarity (ET(30) < 42 kcal mol−1), and consequently a number of additional impurities due to incomplete Fmoc-removal were observed compared to the DMF control (entries 1 and 2, Fig. 1). Contrariwise, the solvent mixtures of higher polarity (ET(30) > 42 kcal mol−1, entries 3–5) did not give rise to these impurities, suggesting there might be a polarity threshold for sufficient Fmoc-removal in the synthesis of Bivalirudin. Moreover, out of the solvents used for the coupling reactions in entries 3–5, DMSO/EtOAc (1:9) and DMSO/2-Me-THF (2:8) (entries 4 and 5) displayed the lowest polarity ((ET(30) = 41.29 and 41.94 kcal mol−1 respectively), and also gave rise to the highest Bivalirudin purity (70%) of the combinations screened. However, in all binary solvent mixtures a significant increase of a side-product arising from incomplete coupling attributed to Arg-lactamisation was observed, and in addition, side-products arising from other incomplete couplings were observed in DMSO/2-Me-THF (entry 4) and DMF. We confirmed that the variation observed between the different solvent mixtures was not due to instability of the Fmoc-amino acids reagents, which overall showed good solubility and stability in the various solvent mixtures (ESI Table S1†). To corroborate the data and to further understand the correlation between stepwise modulation of the solvent mixture's polarity from coupling to Fmoc-removal, and the effects on crude peptide purity, we carried out three more syntheses of Bivalirudin using the same SPPS protocol (entries 6–8, Table 1 and Fig. 2).
 |
| | Fig. 2 HPLC purity profile (220 nm) of Bivalirudin (H-fPRPGGGGNGDFEEIPEEYL-OH) syntheses 6–8 (Table 1). The corresponding crude purity of Bivalirudin is shown in parenthesis. After each coupling reaction the resin was capped using a basic solution of Ac2O (full experimental details in the ESI†). The side-products arising from incomplete Fmoc-removal (deletion peptides; red arrows), various incomplete coupling reactions (acetylated peptides; blue arrows), and incomplete coupling attributed to Arg-lactamisation (acetylated peptide; green arrows) are highlighted. | |
In these syntheses we compared combinations of the relatively non-polar NBP/DOL (4:6) ((ET(30) = 41.11 kcal mol−1) and polar DMSO/DOL (4:6) ((ET(30) = 43.38 kcal mol−1) to further assess the impact of solvent polarity on coupling and Fmoc-removal and as a result the final crude purity profile. In entries 6 and 8 we used either NBP/DOL (4:6) or DMSO/DOL (4:6) for both coupling and Fmoc-removal, while we used NBP/DOL (4:6) for coupling reactions and DMSO/DOL (4:6) for Fmoc-removal in entry 7 (Fig. 2). We found that merely changing from a low polarity to high polarity mixture for Fmoc-removal reactions effectively eliminates the formation of side-products related to incomplete Fmoc-removal and increases the overall crude peptide purity (entry 6 vs. 7, Fig. 2). Furthermore, when performing both the coupling reactions and the Fmoc-removal at the same relatively high polarity (entry 8, Fig. 2), a significantly different purity profile was obtained. When using the lower polarity NBP/DOL (4:6) for coupling reactions, significant side-product formation due to incomplete arginine coupling (attributed to Arg-lactamisation) was observed (entry 6 and 7, Fig. 2). However, by switching to the polar DMSO/DOL (4:6), the side-product related to Arg-lactamisation was supressed (green arrow), but additional side-products attributed to other incomplete couplings were observed, with no overall change in peptide purity (entry 7 vs. 8, Fig. 2). Based on these findings it was clear that in addition to governing coupling and Fmoc-removal reactions, the solvent polarity could directly impact specific side-reactions. We therefore decided to delve deeper, and systematically investigate a number of fundamental SPPS-related side-reactions, including Arg-lactamisation, DKP formation, aspartimide formation as well as amino acid racemisation in selected green binary solvent mixtures.
Arginine-lactamisation
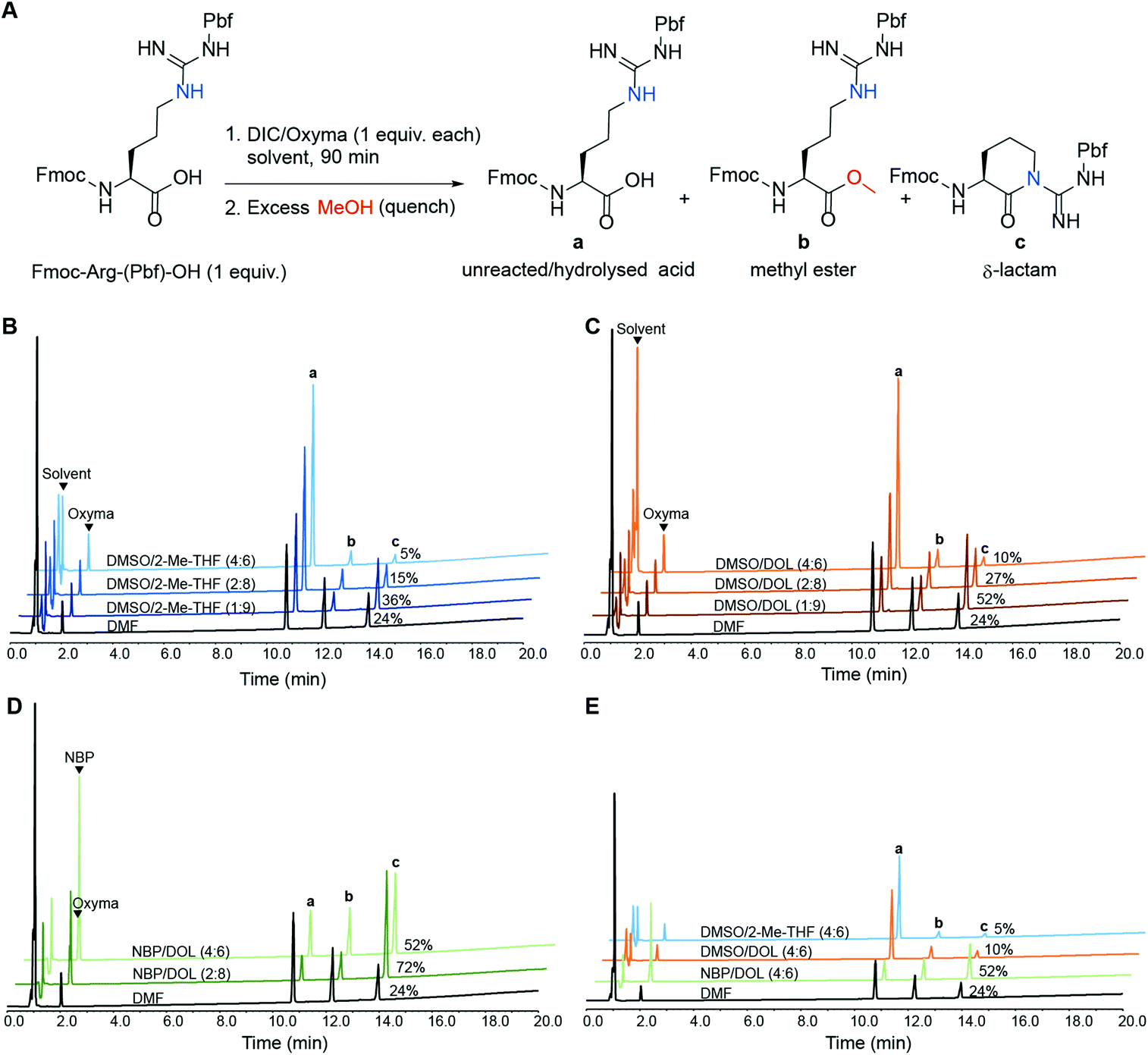
During coupling reactions arginine is susceptible to δ-lactam formation between its side-chain guanidine moiety and the activated carboxylic acid.22 This unwanted self-condensation effectively diminishes the reactive arginine species in the reaction mixture, risking formation of target peptides devoid of Arg (deletion peptides or acetylated truncated side-product peptides if capping is carried out after coupling). Side-product formation can be suppressed by increasing the number of equivalents of the starting amino acid reagent, or by performing iterative (e.g. double) couplings. However, this is not a cost-efficient approach, especially not for industrial-scale SPPS. Intramolecular cyclisation reactions such as Arg-lactamisation are often influenced by the solvent and are hard to avoid in SPPS solvents such as DMF and NMP, especially when a derivative mono-protected on the sidechain of Arg such as Fmoc-Arg(Pbf)-OH is employed.23,24 Furthermore, it was recently reported that the green solvent NBP increases the tendency for Arg-lactamisation in SPPS compared to DMF and a special protocol had to be developed to enable quantitative incorporation of arginine into the growing peptide chain.25 To this end, we sought to evaluate the influence of solvent polarity on Arg-lactamisation and we determined the extent of Arg-lactamisation in selected binary solvent mixtures. Fmoc-Arg(Pbf)-OH was treated with DIC/Oxyma (0.1 M each) in selected binary solvent mixtures in the absence of amine nucleophile for 90 min, then the mixture was quenched with excess MeOH to capture any activated species as the corresponding methyl ester (Fig. 3A).26,27 The binary solvent mixtures DMSO/2-Me-THF (1:9, 2:8 and 4:6), DMSO/DOL (1:9, 2:8 and 4:6) and NBP/DOL (2:8 and 4:6) were screened along with DMF as a reference, and the ratio between (a) Fmoc-Arg(Pbf)-OH, unreacted or arising from hydrolysis of the active ester, (b) Fmoc-Arg(Pbf)-OMe, arising from quenching the active ester with MeOH, and (c) arginine δ-lactam arising from Arg-lactamisation was determined by HPLC analysis (Fig. 3B–E and ESI Fig. S18†).
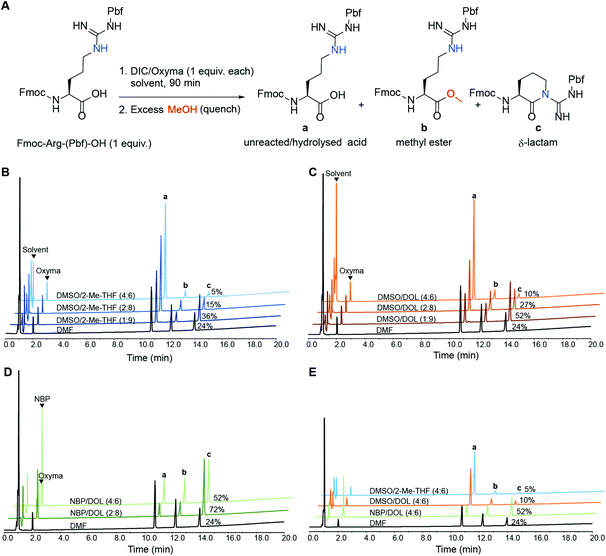 |
| | Fig. 3 (A) HPLC analysis (220 nm) for the activation of Fmoc-Arg(Pbf)-OH (1 mmol) using DIC/Oxyma (1 mmol each) in various binary solvent mixtures (10 mL) and the species present after quenching the reaction with excess MeOH: (a) unreacted/hydrolysed arginine, (b) methyl ester and (c) δ-lactam; the impact of solvent polarity on activation and subsequent Arg-lactamisation in binary mixtures of (B) DMSO/2-Me-THF, (C) DMSO/DOL and (D) NBP/DOL. The polarities of the investigated ratios of binary mixtures are in the order 4:6 > 2:8 > 1:9, and the percentage of undesired δ-lactam (peak c) in the corresponding solvent is indicated. (E) Comparison of the binary mixtures in panels B, C and D with the lowest prevalence of δ-lactam. | |
As demonstrated in Fig. 3, Arg-lactamisation was suppressed with increasing solvent polarity in all tested binary solvent combinations. DMSO/2-Me-THF (4:6) and DMSO/DOL (4:6) gave the least δ-lactam species (5 and 10% respectively), which was significantly lower than for DMF (24%). Interestingly, both DMSO/2-Me-THF (4:6) and DMSO/DOL (4:6) display polarities (ET(30) = 43.16 and 43.38 kcal mol−1 respectively) similar to DMF (43.38 kcal mol−1), indicating that solvent polarity alone cannot account for this discrepancy. Solvent combinations with significantly lower polarity than DMF gave considerably higher Arg-lactamisation, NBP/DOL (2:8; ET(30) = 40.84 kcal mol−1) giving the highest content of all solvents screened (72%). Noteworthy, the solvents giving the least formation of δ-lactam (DMSO/2-Me-THF (4:6) and DMSO/DOL (4:6)) also displayed the highest amounts of residual Fmoc-Arg(Pbf)-OH in the reaction mixture (Fig. 3B and C), which is likely due to hydrolysis of the active ester in the absence of an amine nucleophile rather than slow amino acid activation.28 Overall, binary solvent mixtures of higher polarity appeared advantageous for mitigating lactamisation during arginine couplings, which was supported by the synthesis of Bivalirudin (Fig. 2). In practice, by simply switching from NBP/DOL (4:6, ET(30) = 41.11 kcal mol−1) to the more polar solvent DMSO/DOL (4:6; ET(30) = 43.38 kcal mol−1) for the coupling reactions, the side-product arising from the Arg-lactamisation side-reaction could be reduced from 18% to 2% (entries 7 vs. 8, Fig. 2, vide supra).
Diketopiperazine (DKP) formation
DKP formation is a problematic side-reaction during both SPPS as well as during long-term peptide storage.29 The configuration, conformation and steric hindrance of the amino acids all play important roles, as DKP formation is especially prone to occur when a sterically unhindered amino acid (e.g. glycine) is followed by an amino acid more likely to adopt a cis-amide conformation (e.g. proline), or when combinations of L- and D-amino acids are used.30 It can be particularly pronounced when the dipeptide is attached to the solid support via an ester linkage (e.g. Wang linkers),31 and effectively leads to loss of peptide and lower yields. We investigated DKP formation in green binary solvent mixtures using the dipeptide Fmoc-Arg(Pbf)-Gly anchored on a Wang-PS-resin as a model sequence. The peptidyl resin was treated with base (piperidine or 4-methylpiperidine) in selected binary solvent mixtures for 10–30 min (Table 2). The percentage of DKP formation was then determined by HPLC, by quantifying the amount of DKP in solution using a standard curve for H-Arg(Pbf)-OH (assuming the same absorption coefficient for H-Arg(Pbf)-Gly-DKP and H-Arg(Pbf)-OH at 280 nm; ESI Fig. S4–S6 and Tables S2–S4†).
Table 2 DKP formation for Fmoc-Arg(Pbf)-Gly-O-Wang-PS resin in binary solvent mixtures and DMF
| Method |
A |
B |
C |
| Solvent/conditionsa |
Time = 30 min |
Time = 30 min |
Time = 10 min |
| Base = Pip |
Base = 4-Me-Pip |
Base = Pip |
|
Fmoc-Arg(Pbf)-Gly-O-Wang resin (30 mg) was allowed to swell in the candidate solvent (8 mL g−1 resin) for 60 min. The indicated base (2 mL g−1 resin) was added and the mixture was agitated for 10 or 30 min.
|
| DMF |
17% |
15% |
6% |
| DMSO/2-Me-THF (3:7) |
24% |
21% |
8% |
| DMSO/2-Me-THF (4:6) |
22% |
21% |
9% |
| DMSO/EtOAc (4:6) |
23% |
28% |
8% |
| DMSO/EtOAc (6:4) |
13% |
16% |
7% |
| DMSO/DOL (3:7) |
17% |
18% |
7% |
| DMSO/DOL (4:6) |
18% |
17% |
6% |
| NBP/DOL (4:6) |
20% |
17% |
6% |
| NFM/DOL (2:8) |
19% |
19% |
6% |
When using piperidine as base the DKP formation was generally on par with DMF or slightly higher in the binary solvent mixtures (Method A, Table 2). As anticipated, switching to the alternative base 4-methylpiperidine yielded similar results as when using piperidine (Method B) and was not deemed a useful alternative. Reducing the reaction time from 30 min to 10 min when using piperidine as base led to a pronounced decrease of DKP formation in all solvents (Method C). Fmoc-removal kinetics experiments in binary solvent mixtures have shown that effective Fmoc-removal can be accomplished in 5–10 min.13 Taken together with the data in Table 2, an Fmoc-removal protocol of 2 × 5–10 min might be useful for suppressing DKP formation in problematic peptide sequences. Interestingly, while DKP formation has been shown to be influenced by a number of factors such as pH, polarity, dipole moment and H-bond donor/acceptor capabilities,32 we did not observe any correlation between the polarity and the rate of DKP formation in the selected binary solvent mixtures.
Aspartimide formation
Aspartimide formation occurs through cyclisation of the Asp side-chain with the peptide backbone and can pose a significant challenge in peptide synthesis, as it accumulates throughout the synthesis with every iterative base treatment for Fmoc-removal.33–35 Furthermore, the aspartimide side-product subsequently can undergo epimerisation, hydrolysis to yield the α- or β-Asp isomers, or base-induced, ring-opening amidation to yield α- or β-piperidide side-products (ESI Fig. S3†). Aspartimide formation can be mitigated by the use of backbone amide protecting groups,36–39 bulky side-chain ester protecting groups for Asp,40 or alternatively by masking the side-chain carboxylate as a cyanosulfurylide (CSY).41 However, the polarity of the solvent is also believed to play a central role, and a recent report showed that aspartimide formation was reduced when switching from DMF to the less polar green solvent NBP.42 To shed more light on this finding, we investigated aspartimide formation in binary solvent mixtures of different polarity. To this end, we synthesised scorpion toxin II (Fmoc-Val-Lys(Boc)-Asp(OtBu)-Gly-Tyr(tBu)-Ile) on a Wang-PS resin, a well-studied model peptide that readily undergoes aspartimide formation.14,43,44 The peptide-Wang resin was treated with 20% piperidine (v/v) in binary solvent mixtures (and DMF as control) for 240 min to simulate 8 deprotection cycles (30 min each). The amounts of remaining peptide (α- and β-isomers combined), aspartimide and piperidide side-products were first determined by LC-MS, and subsequently the degree of α- and β-isomerisation was quantified by HPLC analysis. The results showed that more than 85% of the model peptide did not undergo aspartimide or piperidide formation in all solvents after base treatment (Table 3, ESI Tables S5 and S6†). By adding 0.1 M Oxyma to the deprotection solution,45 the peptide purity generally increased in all solvents, with approximately 90% of the peptide remaining in all binary solvent mixtures. The amount of side-product formation appeared to only correlate weakly with solvent polarity. For example, for a given binary solvent system the solvent mixture with the highest solvent polarity generally only resulted in a slight increase in aspartimide formation. When comparing across different types of binary solvent mixtures there was no clear correlation between solvent polarity and aspartimide formation, although a trend towards a higher peptide purity with the lowest polarity solvents was observed (e.g. NFM/DOL (2:8) and NBP/DOL (4:6)). In terms of α- and β-isomerisation, only the solvent systems DMF and DMSO/2-Me-THF (3:7) induced low levels of isomerisation to the β-isomer (0.3–0.4%, ESI Table S7†).
Table 3 Percentage of target peptide (α- and β-isomer combined), aspartimide and piperidide measured as peak area by LC-MS (210 nm)
| |
|
No additivesa |
0.1 M Oxyma Pureb |
| Solvent |
Solvent polaritycET(30) (kcal mol−1) |
Target peptide (%) |
Aspartimide (%) |
Piperidide (%) |
Target peptide (%) |
Aspartimide (%) |
Piperidide (%) |
|
The reaction was carried out in triplicate and an average value is reported.
The reaction was carried out as a single experiment.
Measured solvent polarity based on Reichardt polarity index.13
|
| DMF |
43.38 |
86 |
6 |
8 |
86 |
9 |
5 |
| DMSO/2-Me-THF (3:7) |
42.84 |
88 |
5 |
7 |
88 |
8 |
4 |
| DMSO/2-Me-THF (4:6) |
43.17 |
88 |
6 |
6 |
89 |
9 |
2 |
| DMSO/EtOAc (4:6) |
43.48 |
89 |
5 |
6 |
90 |
7 |
3 |
| DMSO/EtOAc (6:4) |
44.27 |
87 |
6 |
7 |
89 |
7 |
4 |
| DMSO/DOL (3:7) |
42.90 |
88 |
4 |
8 |
93 |
5 |
2 |
| DMSO/DOL (4:6) |
43.38 |
85 |
7 |
8 |
89 |
8 |
3 |
| NBP/DOL (4:6) |
41.11 |
90 |
3 |
7 |
92 |
4 |
4 |
| NFM/DOL (2:8) |
40.89 |
90 |
3 |
7 |
92 |
4 |
4 |
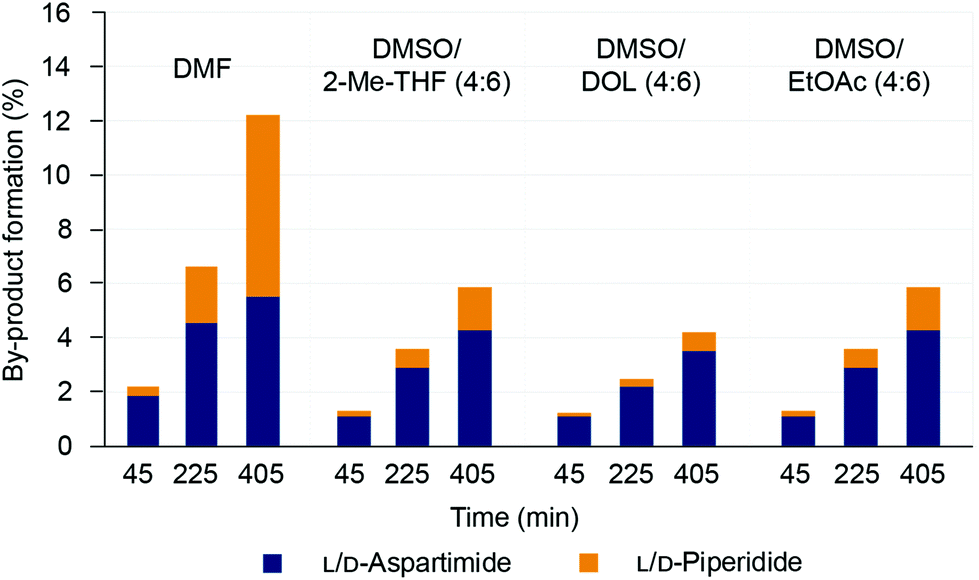
Although similar amounts of aspartimide formation were observed for the various solvents, there appeared to be some differences between the different solvent mixtures in their innate propensity for inducing aspartimide formation. We therefore decided to investigate a focused set of binary solvent mixtures displaying similar solvent polarities. DMSO/2-Me-THF (4:6), DMSO/DOL (4:6) and DMSO/EtOAc (4:6) all display similar polarities to DMF (ET(30) = 43.38 ± 0.21 kcal mol−1), and were used to evaluate aspartimide formation in the scorpion toxin II (H-Val-Lys(Boc)-Asp(OMpe)-Gly-Tyr(tBu)-Ile-Wang) model peptide (with the bulkier 3-methylpent-3-yl ester (OMpe)40 group for Asp side-chain protection). The peptide was treated with 20% piperidine (v/v) in the selected solvents and the extent of aspartimide formation was quantified by HPLC analysis of the crude peptides after 45, 225 and 405 minutes of base treatment (Fig. 4 and ESI Fig. S19†).
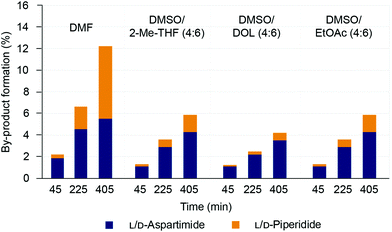 |
| | Fig. 4 Formation of L/D-aspartimide and L/D-piperidide (α/β) side-products in the Asp(OMpe)-protected model peptide Scorpion toxin II-Wang resin (100 mg), after treatment with 20% (v/v) piperidine in various solvents (1 mL) The amount of side-product formation was quantified by HPLC analysis (220 nm) of the crude peptides after 45, 225 and 405 min base treatment. | |
In general, prolonged base treatment led to increased D/L-aspartimide and piperidide side-product formation in all solvents screened (Fig. 4). However, the amount of side-product formation was significantly lower in the green binary solvent mixtures compared to DMF, especially in DMSO/DOL (4:6). The data suggests that solvent polarity is not the only decisive factor for aspartimide formation, and all the binary solvent mixtures screened showed an innate tendency to mitigate aspartimide formation. However, when evaluating mixtures of DMSO/DOL in different ratios (1:9 to 4:6) there was a clear trend of increased aspartimide formation with increasing solvent polarity (ESI Fig. S20 and S21†). Taken together, the results suggest that aspartimide formation can be mitigated by using binary solvent mixtures. Solvent polarity appeared to have an influence as binary solvent mixtures of lower polarity generally gave less aspartimide formation, although the composition of the binary mixture also seemed to play a role. Mixtures of DMSO and DOL were particularly successful in suppressing aspartimide formation.
Racemisation of amino acids
Amino acid racemisation is a common side-reaction in SPPS that can pose severe challenges in the downstream purification of peptides. The biological activity of a peptide therapeutic is directly correlated to its stereochemical integrity, and thus racemisation of even a single amino acid could have a detrimental impact on its activity.46 Amino acid racemisation primarily occurs during coupling reactions and proceeds mainly via oxazolone or enolization pathways.4,47 We initially set out to determine racemisation of the two particularly racemisation prone amino acids Cys and His studying Fmoc-Cys(Trt)-OH and Fmoc-His(Trt)-OH during activation and coupling with a H-Val-Gly-Rink Amide resin. However, due to the limited solubility of Fmoc-His(Trt)-OH in most binary solvent mixtures we decided to also evaluate racemisation of Fmoc-His(Boc)-OH, which was fully soluble in all solvents and has previously been shown to have a lower tendency to racemise.48 Fmoc-His(Trt)-OH showed 1–2% of epimerisation in DMF and DMSO/2-MeTHF (3:7), while less epimeric peptide was observed when using Fmoc-His(Boc)-OH, showing that it is a viable alternative to Fmoc-His(Trt)-OH for use in green binary solvent mixtures (Table 4). Furthermore, no apparent racemisation occurred during coupling of Fmoc-Cys(Trt)-OH in all tested solvents, which is in accordance with the observation that racemisation of Cys primarily occurs under basic reaction conditions and not in the slightly acidic conditions such as in DIC/Oxyma mediated couplings.49
Table 4 Racemisation of His and Cys during coupling to the H-Val-Gly-Rink Amide resin measured as peak area of the tripeptide (%) by HPLC (214 nm) of the cleaved and deprotected tripeptide
| Solvent |
His(Trt) |
His(Boc) |
Cys(Trt) |
|
Not soluble at a concentration of 0.2 M. n.d. = not determined.
|
| DMF |
1.4% |
<0.01% |
<0.01% |
| DMSO/2-MeTHF (3:7) |
2.0% |
0.4% |
<0.01% |
| DMSO/EtOAc (1:9) |
n.d.a |
<0.01% |
<0.01% |
| NBP/DOL (2:8) |
n.d.a |
<0.01% |
<0.01% |
| NBP/DOL (4:6) |
n.d.a |
<0.01% |
<0.01% |
| NFM/DOL (2:8) |
n.d.a |
<0.01% |
<0.01% |
| DMSO/DOL (1:9) |
n.d.a |
0.2% |
<0.01% |
Varying the solvent composition is a tool to mitigate common side-reactions in SPPS
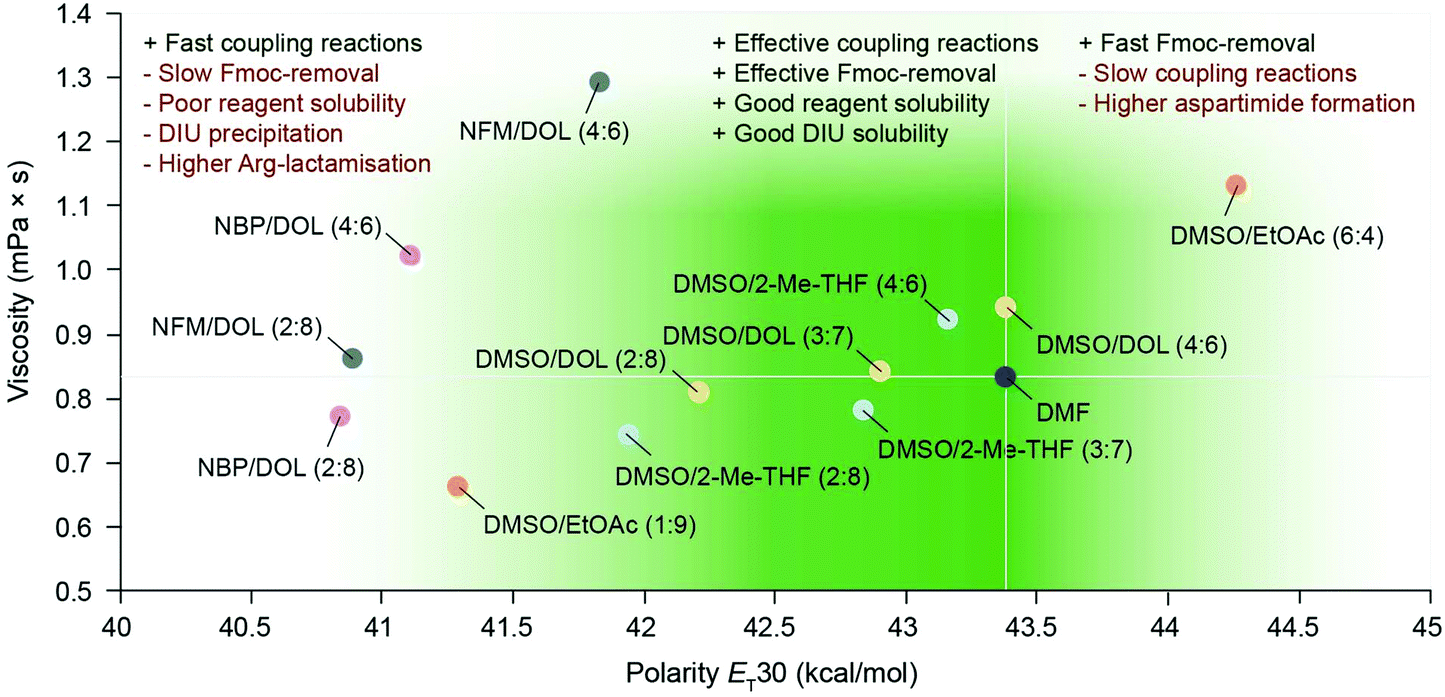
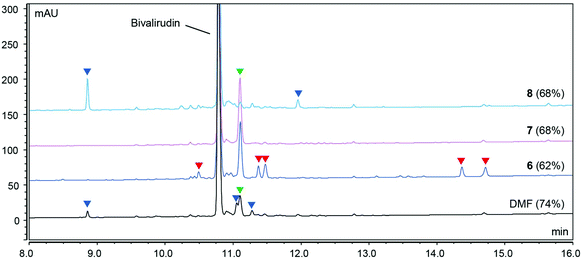
Gratifyingly, our findings demonstrated that green binary solvent mixtures do not generally lead to an increased propensity for common side-reactions in SPPS compared to DMF, and that they may in fact be used as a tool to mitigate common side-reactions. For example, Arg-lactamisation was suppressed by employing solvent mixtures of higher polarity (e.g. DMSO/2-Me-THF (4:6) and DMSO/DOL (4:6)), and aspartimide formation could be reduced when compared to DMF, particularly when the OMpe side chain protection group was employed. Therefore, optimisation of SPPS using green, binary solvent mixtures should entail careful consideration of the sequence of the target peptide and the potential specific side-reactions incurred and how these may be mitigated by solvent optimisation in critical steps of the synthesis. In Fig. 5 the learnings from the synthesis of Bivalirudin and our investigation of solvent impact on different side-reactions are summarised. As illustrated, green binary solvent mixtures displaying similar polarity and viscosity to DMF are characterised by effective Fmoc-removal and coupling reactions, good solubility of reagents and by-products, and strike a beneficial balance in mitigating common side-reactions such as Arg-lactamisation and aspartimide formation. Consequently, during synthesis optimisation the choice of solvent can be tailored to the application. We have previously reported that DMSO/DOL (3:7) and DMSO/2-Me-THF (3:7) represent excellent alternatives to DMF in SPPS.13 However, in the synthesis of Bivalirudin we found that Arg-lactamisation represented a major side-reaction in both DMF and DMSO/DOL (3:7) (Fig. 6). We leveraged our knowledge from the present study, and performed a final synthesis of Bivalirudin in DMSO/DOL (3:7) for both Fmoc-removal and coupling reactions, but importantly the coupling of [Arg3] was carried out in DMSO/DOL (4:6) that we have shown suppresses Arg-lactamisation. To our delight, this seemingly minor change resulted in substantial suppression of the major side-product related to Arg-lactamisation (green arrow) compared to our previous syntheses in DMF and DMSO/DOL (3:7) (Fig. 6). This result is significant in two ways; it demonstrates that varying the solvent composition based on polarity and viscosity can be used as a mild and facile tool to control common side-reactions in SPPS, and furthermore it underlines our findings that green binary solvent mixtures represent viable, less toxic alternatives to DMF in SPPS.
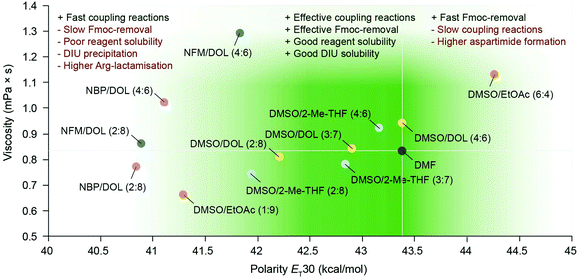 |
| | Fig. 5 The overall performance of binary solvent mixtures in SPPS correlated to the polarity (kcal mol−1) and viscosity (mPa s) of the solvents, benchmarked to DMF. The green gradient indicates empirically favourable viscosity and polarity ranges for useful solvent performance in SPPS. Binary solvent mixtures with the same individual components are shown in same colour. | |
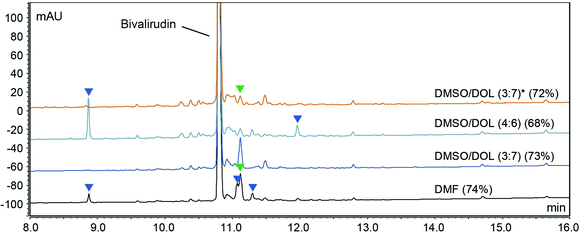 |
| | Fig. 6 Synthesis of Bivalirudin (H-fPRPGGGGNGDFEEIPEEYL-OH) on 7.5 mmol scale using DMSO/DOL (3:7)* where the single coupling of Arg3 was carried out in DMSO/DOL (4:6), compared to the syntheses carried out in DMF, DMSO/DOL (3:7) and DMSO/DOL (4:6). After each coupling reaction the resin was capped using a basic solution of Ac2O (full experimental details in the ESI†). The side-products arising from incomplete coupling reactions (blue arrows), and incomplete coupling attributed to Arg-lactamisation (green arrows) are highlighted. | |
Conclusion
In recent years significant progress has been made to identify more environmentally friendly and less hazardous solvent alternatives to DMF in SPPS. However, finding neat solvents that fulfil all the technical, regulatory and commercial criteria required for widespread adoption has proven challenging. We have demonstrated that binary solvent mixtures represent a way forward, as the polarity and viscosity profile of the binary mixture can be used to predict its utility in SPPS at ambient temperature. Binary mixtures with optimal polarity and viscosity efficiently dissolve reagents and by-products, swell peptide resins to adequate levels for effective flux of reagents, and significantly impact Fmoc-removal and peptide coupling reactions. As coupling reactions are generally promoted by non-polar solvents while Fmoc-removal reactions are more efficient in polar solvents, the use of binary solvent mixtures presents an opportunity to vary the composition of the solvent mixture to fine-tune the solvent characteristics to suit the application. In this report, we delve deeper into the effects of solvent polarity on coupling and Fmoc-removal reactions, and systematically evaluate its effects on the prevalence of side-reactions such as Arg-lactamisation, DKP and aspartimide formation and amino acid racemisation at ambient temperature. We show that utilising green binary solvent mixtures does not affect the purity profile in SPPS adversely and that it can in fact be used as a tool to suppress certain side-reactions as exemplified herein for Arg-lactamisation and aspartimide formation. Importantly, we demonstrate that solely varying the composition of the binary mixture represents a novel tool to control specific side-reactions in SPPS. This is significant as it expands the peptide chemist's ‘toolbox’ within the state-of-the-art Fmoc/tBu-based SPPS, which is the preferred approach in the pharmaceutical industry. In our findings, binary solvent mixtures of DMSO in combination with DOL, 2-Me-THF or EtOAc are suitable for efficient Fmoc-removals, whereas coupling reactions can be envisioned in these mixtures as well as in NFM/DOL and NBP/DOL. These results show that polarity and viscosity are key physicochemical parameters for solvent selection and that green binary solvent mixtures are attractive alternatives to DMF in SPPS. Based on the findings reported herein and in our previous publication13 we intend to apply the developed toolbox concept to our pipeline projects in order to assess its full potential for large-scale production of therapeutic peptides. Although some of the solvents identified in this work presently will incur a higher solvent cost, we anticipate significant improvement of the supply chain and concomitant cost reductions with increasing demand from us and others. Therefore, in addition to achieving our goal of reducing our environmental impact and ensuring future ECHA REACH compliance, our platform provides new tools for mitigating side-reactions and improving purity profiles of peptide APIs, with potential for ready implementation in peptide synthesis in both academia and industry.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
We thank Dr Sebastian Brandes, Dr Per Vedsø and Dr Rune Severinsen from Novo Nordisk for providing the necessary resources and support to realise the work presented herein. Dr Rolf H. Taaning, Dr Søren Bertelsen, Dr Brian Rasmussen and Dr Michael Raunkjær from Novo Nordisk are gratefully acknowledged for fruitful discussion during the start-up of the project that helped set the scope and direction. Marcela Castro is thanked for assistance with creating the graphical abstract. Dr Vincent Martin was funded by the Novo Nordisk R&D STAR Fellowship Programme. We acknowledge Dr Stefan Eissler from Bachem AG for diligently reviewing the manuscript internally. We thank and acknowledge the members of the research committee of Bachem AG for providing all necessary resources to accomplish the work presented here. Finally, we want to thank the unknown reviewers for their time reviewing this manuscript.
References
- A. A. Zompra, A. S. Galanis, O. Werbitzky and F. Albericio, Future Med. Chem., 2009, 1, 361–377 CrossRef CAS PubMed.
- T. Bruckdorfer, O. Marder and F. Albericio, Curr. Pharm. Biotechnol., 2004, 5, 29–43 CAS.
- M. D'Hondt, N. Bracke, L. Taevernier, B. Gevaert, F. Verbeke, E. Wynendaele and B. De Spiegeleer, J. Pharm. Biomed. Anal., 2014, 101, 2–30 CrossRef.
-
Y. Yang, Side Reactions in Peptide Synthesis, Elsevier, 2015 Search PubMed.
- D. M. M. Jaradat, Amino Acids, 2018, 50, 39–68 CrossRef CAS PubMed.
- A. Isidro-Llobet, M. Alvarez and F. Albericio, Chem. Rev., 2009, 109, 2455–2504 CrossRef CAS.
-
K. Akaji, G. Barker, L. F. Bonewald, J. Boydston, N. Chino, P. E. Dawson, A. Escherich, A. Friedler, H. F. Gaertner, C. Gilon, M. Goodman, M. T. W. Heam, R. W. Jack, A. M. Jansson, G. Jung, H. Kessler, T. A. Keideding, T. Kmura, Y. Kiso, E. Locardi, E. Lohof, G. Loidl, C. Mang, M. P. Meldal, R. B. Merrifield, L. Moroder, H.-J. Musiol, J. L. Offer, R. E. Offord, S. Pegorano, J. W. Perich, J. Rivier, T. T. Romoff, S. Sakakibara, P. W. Schiller, R. A. G. D. Silva, G. Siuzdak, S. Sperl, P. M. St. Hilaire, R. D. Süssmuth, E. J. Want, E. Weyher and R. W. Woody, Synthesis of Peptides and Peptidomimetics, in Houben-Weyl Methods of Organic Chemistry, ed. A. Felix, L. Moroder and C. Toniolo, Thieme, 4th edn supplement, 2004, vol. E 22b Search PubMed.
- The European Chemicals Agency, ECHA. http://echa.europa.eu. The restrictions under ECHA regulation number (EC) 1907/2006 and 276/2010 for DMF and DCM, respectively. Restrictions under commission regulation (EU) 2018/588 for NMP.
- F. Albericio, O. Al Musaimi and B. G. de la Torre, Green Chem., 2020, 22, 996–1018 RSC.
- V. Martin, P. H. G. Egelund, H. Johansson, S. Thordal Le Quement, F. Wojcik and D. Sejer Pedersen, RSC Adv., 2020, 10, 42457–42492 RSC.
- A. Isidro-Llobet, M. N. Kenworthy, S. Mukherjee, M. E. Kopach, K. Wegner, F. Gallou, A. G. Smith and F. Roschangar, J. Org. Chem., 2019, 84, 4615–4628 CrossRef CAS PubMed.
- K. G. Varnava and V. Sarojini, Chem. – Asian J., 2019, 14, 1088–1097 CrossRef CAS PubMed.
- V. Martin, S. Jadhav, P. H. G. Egelund, R. Liffert, H. Johansson Castro, T. Krüger, K. F. Haselmann, S. Thordal Le Quement, F. Albericio, F. Dettner, C. Lechner, R. Schönleber and D. Sejer Pedersen, Green Chem., 2021 10.1039/d1gc00603g.
- E. Nicolás, E. Pedroso and E. Girald, Tetrahedron Lett., 1989, 30, 497–500 CrossRef.
- C. Reichardt, Org. Process Res. Dev., 2007, 11, 105–113 CrossRef CAS.
- C. Reichardt, Pure Appl. Chem., 1982, 54, 1867–1884 CAS.
- K. V. Rao, M. F. J. Mabesoone, D. Miyajima, A. Nihonyanagi, E. W. Meijer and T. Aida, J. Am. Chem. Soc., 2020, 142, 598–605 CrossRef CAS PubMed.
- L. C. Chan and B. G. Cox, J. Org. Chem., 2007, 72, 8863–8869 CrossRef CAS PubMed.
- B. J. Balcom and N. O. Petersen, J. Org. Chem., 1989, 54, 1922–1927 CrossRef CAS.
- L. A. Carpino and G. Y. Han, J. Org. Chem., 1972, 37, 3404–3409 CrossRef CAS.
-
G. B. Fields, in Peptide Synthesis Protocols, ed. M. W. Pennington and B. M. Dunn, Humana Press, Totowa, NJ, 1995, pp. 17–27 Search PubMed.
- R. Paul, G. W. Anderson and F. M. Callahan, J. Org. Chem., 1961, 26, 3347–3350 CrossRef.
- M. H. Cezari and L. Juliano, Pept. Res., 1996, 9, 88–91 CAS.
- M. Alhassan, A. Kumar, J. Lopez, F. Albericio and B. G. de la Torre, Int. J. Mol. Sci., 2020, 21, 4464 CrossRef CAS PubMed.
- B. G. de la Torre, A. Kumar, M. Alhassan, C. Bucher, F. Albericio and J. Lopez, Green Chem., 2020, 22, 3162–3169 RSC.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed. Engl., 1978, 17, 522–524 CrossRef.
- M. Tsakos, E. S. Schaffert, L. L. Clement, N. L. Villadsen and T. B. Poulsen, Nat. Prod. Rep., 2015, 32, 605–632 RSC.
- V. Goldschmidt Gőz, A. Nagy, V. Farkas, E. Keszei and A. Perczel, RSC Adv., 2019, 9, 30720–30728 RSC.
-
M. Bodanszky, in Principles of Peptide Synthesis, Springer-Verlag Berlin Heidelberg, 1984, pp. 158–201 Search PubMed.
- P. M. Fischer, J. Pept. Sci., 2003, 9, 9–35 CrossRef CAS PubMed.
- J. Alsina, E. Giralt and F. Albericio, Tetrahedron Lett., 1996, 37, 4195–4198 CrossRef CAS.
- S. Capasso and L. Mazzarella, J. Chem. Soc., Perkin Trans. 2, 1999, 329–332 RSC.
- M. Mergler, F. Dick, B. Sax, C. Stahelin and T. Vorherr, J. Pept. Sci., 2003, 9, 518–526 CrossRef CAS PubMed.
- M. Mergler, F. Dick, B. Sax, P. Weiler and T. Vorherr, J. Pept. Sci., 2003, 9, 36–46 CrossRef CAS PubMed.
- J. P. Tam, M. W. Riemen and R. B. Merrifield, Pept. Res., 1988, 1, 6–18 CAS.
- A.-B. M. Abdel-Aal, G. Papageorgiou, R. Raz, M. Quibell, F. Burlina and J. Offer, J. Pept. Sci., 2016, 22, 360–367 CrossRef CAS PubMed.
- J. Offer, M. Quibell and T. Johnson, J. Chem. Soc., Perkin Trans. 1, 1996, 175–182 RSC.
- M. Quibell, D. Owen, L. C. Packman and T. Johnson, J. Chem. Soc., Chem. Commun., 1994, 2343–2344 RSC.
- V. Cardona, I. Eberle, S. Barthélémy, J. Beythien, B. Doerner, P. Schneeberger, J. Keyte and P. D. White, Int. J. Pept. Res. Ther., 2008, 14, 285–292 CrossRef CAS.
- R. Behrendt, S. Huber, R. Martí and P. White, J. Pept. Sci., 2015, 21, 680–687 CrossRef CAS PubMed.
- K. Neumann, J. Farnung, S. Baldauf and J. W. Bode, Nat. Commun., 2020, 11, 982 CrossRef CAS PubMed.
- A. Kumar, M. Alhassan, J. Lopez, F. Albericio and B. G. de la Torre, ChemSusChem, 2020, 13, 5288–5294 CrossRef CAS PubMed.
- J. L. Lauer, C. G. Fields and G. B. Fields, Lett. Pept. Sci., 1995, 1, 197–205 CrossRef CAS.
- R. Behrendt, S. Huber and P. White, J. Pept. Sci., 2016, 22, 92–97 CrossRef CAS PubMed.
- R. Subirós-Funosas, A. El-Faham and F. Albericio, Biopoylmers, 2012, 98, 89–97 CrossRef PubMed.
- D. L. Lee, J. P. S. Powers, K. Pflegerl, M. L. Vasil, R. E. W. Hancock and R. S. Hodges, J. Pept. Res., 2004, 63, 69–84 CrossRef CAS PubMed.
- A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS PubMed.
- N. Hartrampf, A. Saebi, M. Poskus, Z. P. Gates, A. J. Callahan, A. E. Cowfer, S. Hanna, S. Antilla, C. K. Schissel, A. J. Quartararo, X. Ye, A. J. Mijalis, M. D. Simon, A. Loas, S. Liu, C. Jessen, T. E. Nielsen and B. L. Pentelute, Science, 2020, 368, 980–987 CrossRef CAS PubMed.
- T. Kaiser, G. J. Nicholson, H. J. Kohlbau and W. Voelter, Tetrahedron Lett., 1996, 37, 1187–1190 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc00604e |
| ‡ These authors contributed equally as first authors. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Vincent
Martin‡
a,
Vincent
Martin‡