
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterobimetallic complexes composed of bismuth and lithium carboxylates as polyurethane catalysts – alternatives to organotin compounds†
Emre
Levent
a,
Oliver
Sala
b,
Lukas F. B.
Wilm
a,
Pawel
Löwe
a and
Fabian
Dielmann
 *ac
*ac
aInstitut für Anorganische und Analytische Chemie, Westfälische Wilhelms-Universität Münster, Corrensstraße 30, 48149 Münster, Germany
bBASF SE, Digitalization of R&D, Carl-Bosch-Straße 38, 67056 Ludwigshafen am Rhein, Germany
cInstitut für Allgemeine, Anorganische und Theoretische Chemie, Leopold-Franzens-Universität Innsbruck, Innrain 80-82, 6020 Innsbruck, Austria. E-mail: fabian.dielmann@uibk.ac.at
First published on 25th March 2021
Abstract
Organotin compounds are important catalysts for the synthesis of polyurethanes consisting of aliphatic isocyanates and polyols. Due to their toxicity, however, it has been a long-standing goal to develop more environmentally benign catalysts. Bismuth octoates and neodecanoates are such well-known alternatives, but their catalytic activity is insufficient for many applications. Herein we show that the catalytic activity of bismuth carboxylates can be enhanced significantly by the addition of lithium carboxylates. Structural and spectroscopic results reveal the spontaneous formation of heterobimetallic complexes consisting of two bismuth and four lithium carboxylates that show dynamic behaviour in solution. The mechanism of the bismuth-catalyzed urethane reaction was elucidated in detail using quantum chemical calculations, paving the way for a rational catalyst design.
Introduction
Polyurethanes (PUs) represent one of the most important classes of polymer materials with numerous technical applications, including elastomers, rigid foams, soft foams, coatings and adhesives.1 These materials account for 7.9 wt% of the world polymer2 production due to their versatile applications at low cost. A key advantage of PU technology is the simple construction of material composites in a single processing step via a polyaddition reaction of diisocyanates and diols.1 Depending on the desired end product, the polymerization is controlled by task-specific catalysts.3,4 Tertiary amines and Lewis-acidic metal complexes are commonly employed to mediate this polyaddition reaction. Recent developments in organocatalytic PU formation include the use of amidines/guanidines,5 NHCs6–9 and organic acids.10–12 For coating applications involving aliphatic polyisocyanate components,13,14 catalysts with high catalytic activity at very low concentrations are required that accelerate the gelling reaction between isocyanate and alcohol, but do not promote the isocyanate–water reaction as this would lead to blistering. Further important catalyst properties are sufficient stability against light and air as well as tolerance against influences from other ingredients of the coating formulation. Organotin compounds, in particular dibutyltin dilaurate (DBTL), remain the most important catalysts in aliphatic urethane synthesis due to their remarkable catalytic activity and advantageous properties.4,15 However, owing to the toxicity of organotin compounds and the difficulties to remove them from the final products,16,17 it has been a long-standing goal to develop more environmentally benign alternative catalysts.18 In efforts to replace organotin catalysts, numerous Lewis-acidic metal complexes were identified as effective catalysts for the isocyanate-hydroxyl reaction,19–27 including some inorganic tin complexes with amino/alkoxide ligands that exhibit thermolatent behaviour.28–35 However, apart from DBTL, only a few metal complexes based on bismuth, aluminium, and zirconium are among the promising candidates for clear coat applications, as discoloration of the coating formulation is highly undesirable.21,26,27 While the practical use of Zr catalyst is hampered by their sensitivity towards hydrolysis and low tolerance against functional groups,27 bismuth carboxylates are recognized as non-toxic alternatives to DBTL catalysts.15,36–39 In addition, there is evidence that combinations of bismuth and zinc carboxylates or bismuth and lithium carboxylates can accelerate the drying time of polyurethane coating formulations.40 Herein, we investigate the influence of lithium carboxylates in the bismuth-catalyzed PU reaction.Results and discussion
Synthesis and solid-state structure of heterobimetallic carboxylate complexes
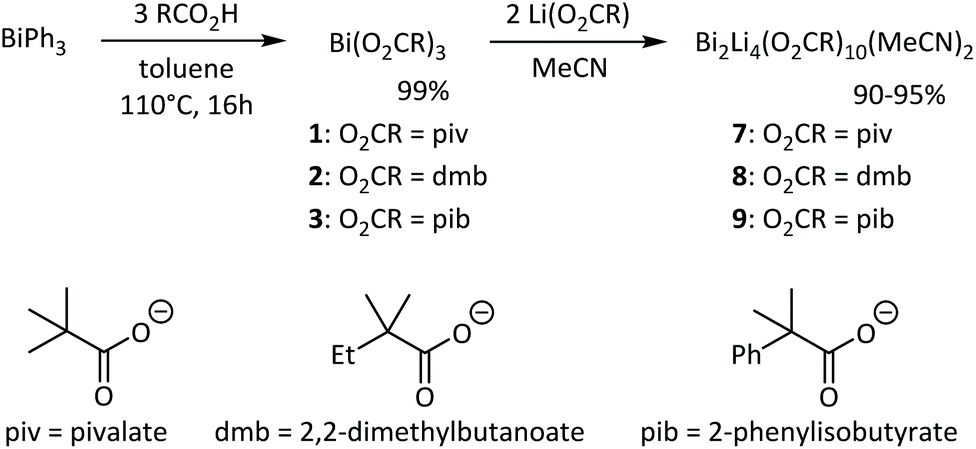
Based on the superior performance of bismuth neodecanoate among different carboxylates in PU synthesis,41 three alpha-branched carboxylates were chosen as model systems for the present study (Scheme 1). Various methods exist for the synthesis of bismuth carboxylates Bi(O2CR)3.42–44 The synthesis of bismuth carboxylates via salt metathesis reactions often results in salt contaminations due to the variable and high coordination number of bismuth(III) complexes.45 We therefore prepared bismuth pivalate Bi(piv)3 (1), bismuth 2,2-dimethylbutanoate Bi(dmb)3 (2) and bismuth 2-phenylisobutyrate Bi(pib)3 (3) according to the procedure reported by Andrews.44 Heating a mixture of BiPh3 and stochiometric amounts of carboxylic acid at 110 °C for 16 hours afforded bismuth carboxylates 1–3 in quantitative yield (Scheme 1). The progress of the reaction was monitored by 1H NMR spectroscopy based on the formation of benzene and the decrease in BiPh3 signals. Complexes 1–3 were obtained as white solids that are soluble in common polar and nonpolar solvents. In the solid-state structures of bismuth carboxylates, the bismuth centres are typically nine coordinate and comprise the carboxylates in chelating and bridging (μ2 and μ3) binding modes.46,47 Thus, polymeric structures are generally obtained with small substituents such as in Bi(O2CH)3, Bi(O2CCH3)3, Bi(O2CC6H5)3, and Bi(O2CC6H4NH2)3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 48–50 while the bulky pivalate ligands lead to the tetrameric complex [{Bi(O2CtBu)(μ2-O2CtBu)(μ3-O2CtBu)}4] in which the Bi atoms are connected by multiple Bi–O bonds formed by the bridging pivalate ligands.51,52 The almost identical steric bulk of the 2,2-dimethylbutanoate ligands and the observation that complex 2 is soluble in nonpolar solvents, suggests that 2 is also a tetramer in the solid state. The introduction of a phenyl group in 2-phenylisobutyrate further increases the steric bulk of the carboxylate ligand. An X-ray diffraction study of a single crystal grown from a saturated THF solution revealed that 3 is dimeric in the solid state (Fig. S57†). Each Bi atom is coordinated by three chelating pib ligands and one THF molecule. These monomers are connected by two pib ligands in tridentate bridging coordination mode forming a Bi2O2 four-membered ring with long intermolecular Bi–O bonds of 2.87 Å.
48–50 while the bulky pivalate ligands lead to the tetrameric complex [{Bi(O2CtBu)(μ2-O2CtBu)(μ3-O2CtBu)}4] in which the Bi atoms are connected by multiple Bi–O bonds formed by the bridging pivalate ligands.51,52 The almost identical steric bulk of the 2,2-dimethylbutanoate ligands and the observation that complex 2 is soluble in nonpolar solvents, suggests that 2 is also a tetramer in the solid state. The introduction of a phenyl group in 2-phenylisobutyrate further increases the steric bulk of the carboxylate ligand. An X-ray diffraction study of a single crystal grown from a saturated THF solution revealed that 3 is dimeric in the solid state (Fig. S57†). Each Bi atom is coordinated by three chelating pib ligands and one THF molecule. These monomers are connected by two pib ligands in tridentate bridging coordination mode forming a Bi2O2 four-membered ring with long intermolecular Bi–O bonds of 2.87 Å.
 | ||
| Scheme 1 Synthesis of Bi carboxylates and heterobimetallic complexes containing bismuth and lithium carboxylates. | ||
Next, we explored the influence of lithium carboxylates on the coordination behaviour of bismuth complexes 1–3. Heating a suspension of complex 1–3 and two equivalents of the corresponding lithium carboxylate in acetonitrile gave a clear solution, which indicates the formation of a heterobimetallic compound as Li(piv) is poorly soluble in acetonitrile. The heterobimetallic complexes 7–9 were readily obtained as white crystalline solid in excellent yield after allowing the reaction mixture to cool to room temperature (Scheme 1). Note that complexes 7–9 also crystallize from an equimolar mixture of bismuth and lithium carboxylates, which demonstrates the propensity for the formation of heterobimetallic complexes. The asymmetric and symmetric CO2 stretching frequencies in the IR spectra of complexes 1–3 and 7–9 appear at very similar frequencies (Table 1). The separation Δν of these bands can provide information about the coordination mode of the carboxylate ligand.53 For complexes 1–3 and 7–9 Δν values between 160 and 186 cm−1 are obtained, which are in the typical range of chelating and bridging carboxylates (150–200 cm−1).53
| Compound | ν AS(COO) | ν s(COO) | Δν |
|---|---|---|---|
| Bi(piv)3 (1) | 1538 | 1357 | 181 |
| Bi(dmb)3 (2) | 1527 | 1358 | 169 |
| Bi(pib)3 (3) | 1521 | 1360 | 160 |
| Bi2Li4(piv)10(MeCN)2 (7) | 1570, 1536 | 1409, 1360 | 161, 176 |
| Bi2Li4(dmb)10(MeCN)2 (8) | 1572, 1531 | 1399, 1357 | 173, 174 |
| Bi2Li4(pib)10(MeCN)2 (9) | 1575, 1544 | 1395, 1358 | 180, 186 |
Single crystals X-ray diffraction studies revealed the formation of isostructural complexes 7, 7′, 8, 9 and 9′ with the general formula [Bi2Li4(O2CR)2(μ-O2CR)4(μ3-O2CR)2(μ4-O2CR)2(MeCN)2], consisting of two bismuth carboxylates, four lithium carboxylates and two acetonitrile ligands (Fig. S58–S62†). The solid-state structure of the pivalate complex is depicted in Fig. 1. Compound 7′ and 9′ are two polymorphs containing one acetonitrile solvate molecules in the asymmetric unit (see the ESI† for details). Their geometrical parameters are very similar to those of 7 and 9, respectively, and thus are not included in the discussion. Selected bond lengths for the heterobimetallic complexes 7, 8 and 9 are summarized in Table 2. In the hexanuclear complexes 7–9, two Bi(O2CR)3 units are linked by a Li4(O2CR)4(MeCN)2 ladder-type structure with a crystallographic inversion centre located in the Li2(O2CR)2 six-membered ring. Each Bi is eight coordinate in a disordered square antiprismatic environment (Fig. 2) while the lithium adopts a tetrahedral geometry. For each Bi atom, four different types of carboxylate ligands can be distinguished. The first carboxylate asymmetrically chelates the Bi atom with rather short Bi–O distances of 2.20–2.63 Å. Two further carboxylates are chelating the Bi atom with a short (2.21–2.38 Å) and a long (2.72–3.00 Å) Bi–O bond. The oxygen atom of the latter is additionally coordinated by a Li atom. Finally, two carboxylates are bridging in μ3 (Bi–O8: 2.29–2.52 Å) and μ4 (Bi–O5: 2.69–2.90 Å) coordination mode. Note that well-defined heterometallic Bi–Li complexes have been described with alkoxide,54tert-butylamine55 and boraamidinate56 ligands. The latter Bi/Li complexes exhibit fluxional behaviour in solution.
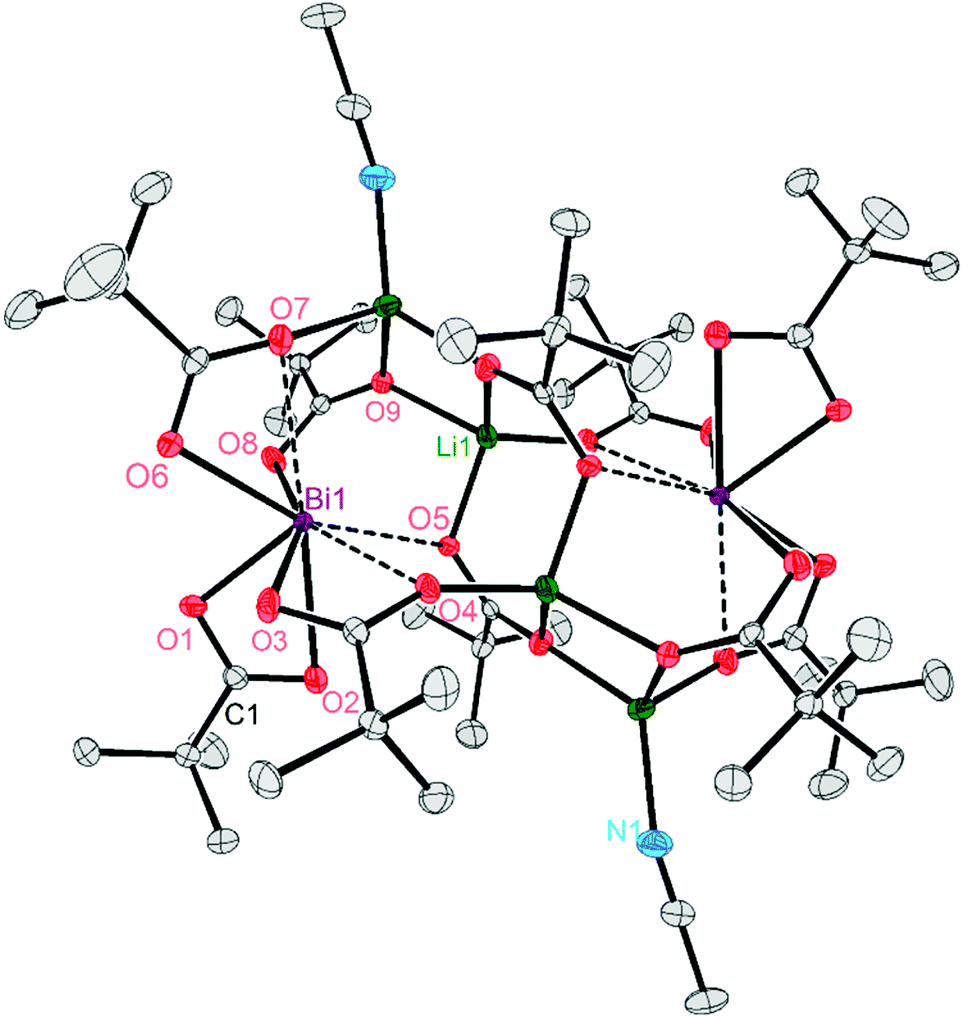 | ||
| Fig. 1 Molecular structure of the heterobimetallic cluster 7 in the solid state. Complexes 8 and 9 are isostructural and thus depicted in the ESI.† Hydrogen atoms are omitted for clarity. Thermal ellipsoids are set at 50% probability. | ||
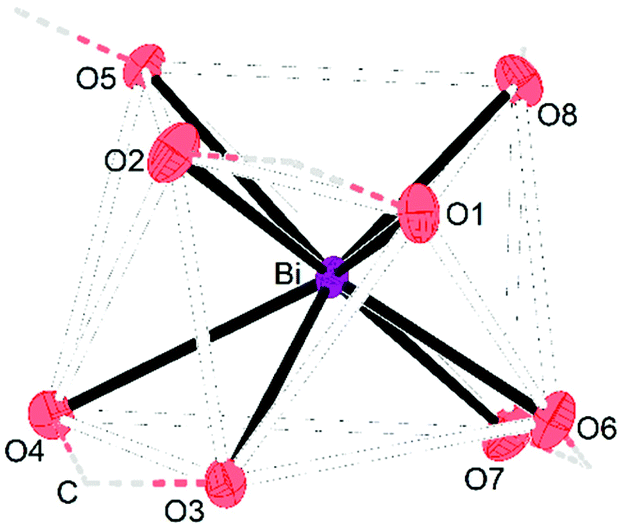 | ||
| Fig. 2 Section of the molecular structure of 7 showing the distorted square-planar coordination geometry of the Bi atom. Connection of O atoms via the C atom are illustrated by dashed lines. | ||
| Entry | Bi2Li4(piv)10 (MeCN)2 (7) | Bi2Li4(dmb)10 (MeCN)2 (8) | Bi2Li4(pib)10 (MeCN)2 (9) |
|---|---|---|---|
| C–O | 1.247(3)–1.287(3) | 1.233(1)–1.311(4) | 1.240(2)–1.287(2) |
| Li–O | 1.877(4)–2.000(9) | 1.849(3)–2.011(1) | 1.897(4)–2.040(4) |
| Bi⋯Li | 3.432(3) | 3.522(3) | 3.545(3) |
| Bi1–O1 | 2.236(1) | 2.256(4) | 2.204(1) |
| Bi1–O2 | 2.515(1) | 2.420(1) | 2.627(1) |
| Bi1–O3 | 2.355(1) | 2.375(2) | 2.318(1) |
| Bi1–O4 | 2.718(1) | 2.772(1) | 3.000(1) |
| Bi1–O5 | 2.687(1) | 2.889(1) | 2.802(1) |
| Bi1–O6 | 2.294(1) | 2.213(1) | 2.254(1) |
| Bi1–O7 | 2.781(1) | 2.930(1) | 2.737(1) |
| Bi1–O8 | 2.294(1) | 2.515(5) | 2.341(1) |
| Bi1–O9 | 3.163(1) | 3.355(9) | 3.181(1) |
NMR-study of heterobimetallic carboxylate complexes
To study the behavior of the Bi carboxylate complexes 1–3 and 7–9 in solution we performed 1H, 13C, 7Li and 1H DOSY NMR experiments. The 1H and 13C NMR spectra of Bi complexes 1–3 in CD2Cl2 show only one set of signals for the carboxylate ligands. The characteristic 13C signal of the carboxylate groups appears in the range of 190.0–187.6 ppm. By means of 1H DOSY NMR spectroscopy the hydrodynamic radius rH of 1 was determined, which is in good agreement with the radius of the tetramer in the solid state (Table 3). However, three sets of signals would be expected for the carboxylate ligands of the tetramer according to the solid-state structure of 1. Thus, the NMR data clearly indicates that in solution complexes 1–3 form dynamic aggregates.Dynamic behavior was also observed for the heterobimetallic complexes 7–9 in dichloromethane solutions, as indicated by the appearance of only one set of signals for the carboxylate ligands in the 1H or 13C NMR spectra. The carboxylate 13C NMR resonances of 7–9 (189.7–187.5 ppm) are significantly broadened and appear in the same range of those of 1–3. In the 7Li NMR spectra two signals were observed for complexes 8 (0.01 ppm, −0.5 ppm) and 9 (−0.1 ppm, −1.5 ppm) while 7 shows only one signal at −0.1 ppm. The hydrodynamic radius rH of 7 is significantly larger than that of 1 and agrees with the radius for the presence of dynamic aggregates in solutions of 7–9 was provided by low temperature 1H NMR experiments. Upon cooling a solution of 7 in CD2Cl2 to 193 K, the singlet for the methyl groups split into more than eight resonances, indicating the presence of several different aggregates rather than the discrete complex 7 in solution (Fig. S30†). Several signals for the methyl protons were also observed in the low temperature 1H NMR spectra of complexes 8 and 9 (Fig. S31 and S32†), which, however, can also be attributed to the presence of several rotamers.
It should be noted that Li(piv) is very poorly soluble in CD3CN, but dissolves quickly upon addition of Bi(piv)3. Mixtures of Bi(piv)3 and Li(piv) with 1:1 and 1:2 stoichiometry in CD3CN both show a singlet at 1.13 ppm in the 1H NMR spectra, which appears downfield compared to that of Bi(piv) (1.06) and downfield relative to that of Li(piv) (1.16 ppm). Collectively, the NMR studies show that the addition of Li carboxylates to bismuth carboxylates leads to the spontaneous formation of heterobimetallic complexes in solution. However, these species are not well defined, but rather constitute a highly dynamic mixture of aggregates with an average size comparable to that of 7–9.
Catalysis of the isocyanate/alcohol reaction
The catalytic performance of complexes 1–3 and 7–9 in the urethane reaction was examined using ethanol and the aliphatic isocyanate 2-ethylhexyl-(6-isocyanatohexyl)-carbamates (Desmodur LD). The latter was chosen as model for 1,6-hexamethylene diisocyanate (HDI) owing to its lower volatility and toxicity. The urethane reactions were performed at ambient temperature with 0.05 mol% catalyst loading using xylene as solvent (Scheme 2). DBTL was included in our study as a reference. Using ATR-IR spectroscopy the reaction progress was monitored by measuring the decreasing intensity of the isocyanate band at 2260 cm−1. The selectivity of the urethane reaction was verified by the simultaneous increase of the characteristic ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) (1692 cm−1) and ν(CHN) (1519 cm−1) frequencies of the alkyl urethane group.57
O) (1692 cm−1) and ν(CHN) (1519 cm−1) frequencies of the alkyl urethane group.57
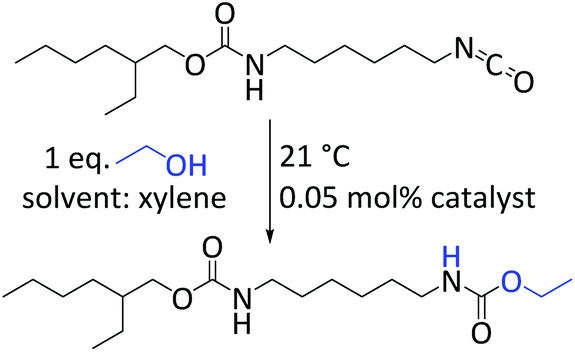 | ||
| Scheme 2 Catalysis conditions for isocyanate alcohol reaction. Catalyst concentration is based on the amount of bismuth. | ||
From these experiments, summarized in Fig. 3, the Bi carboxylates 1–3 show moderate catalytic activity and accelerate the urethane reaction significantly compared to the non-catalyzed reaction. Among the bismuth compounds the catalytic activity was found to increase with the steric bulk of the carboxylate ligands pib > dmb > piv. Strikingly, the heterobimetallic complexes 7–9 clearly emerge as superior catalysts compared to their lithium free counterparts and compound 8 was found to be almost as active as DBTL, leading to 90% conversion after only 15 minutes.
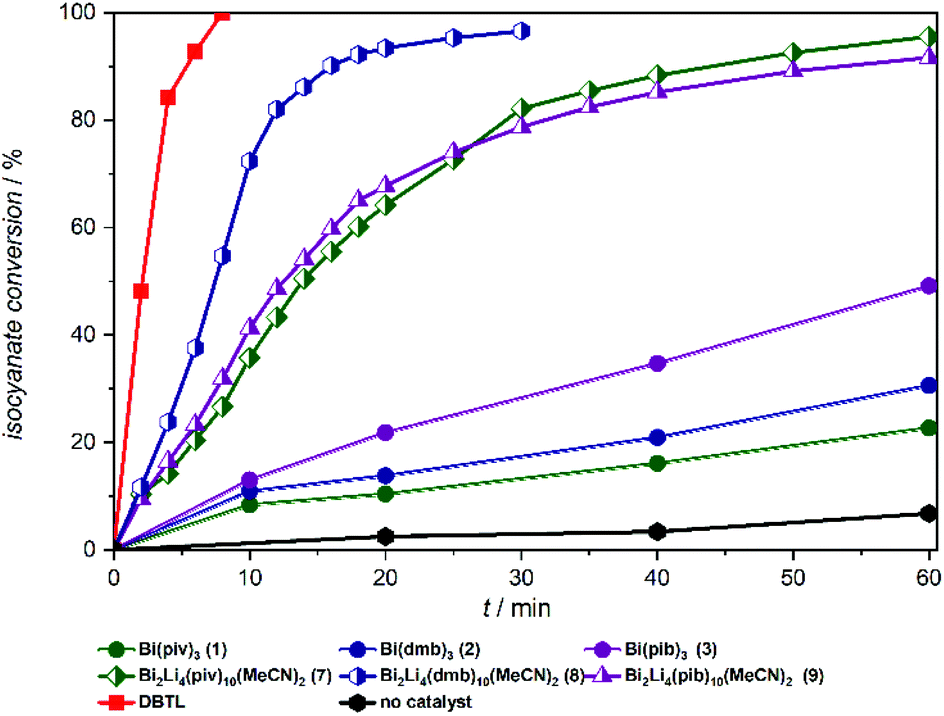 | ||
| Fig. 3 Lewis acid catalysed urethan reaction of Desmodur LD and ethanol in the presence of Bi complexes 1–3, Bi/Li complexes 7–9 and DBTL. | ||
We next wanted to explore the influence of lithium carboxylates on the catalytic performance of the bismuth complexes in more detail. Catalyst mixtures containing different amounts of bismuth and lithium carboxylates were prepared by adding the respective carboxylates to the xylene/ethanol mixture immediately before catalysis, one after the other (Fig. 4). Owing to the sufficient solubility of Li(pib) in xylene, the pib based catalyst system was chosen for this study. As expected, Li(pib) does not accelerate the isocyanate conversion at room temperature. However, the catalytic activity of Bi(pib)3 is slightly increased upon adding 0.5 equiv. of Li(pib), while the addition of two or more equiv. of Li(pib) resulted in reaction rates that are identical to that of complex 9, which suggests that complex 9 is the major species in these reaction mixtures and heterobimetallic compounds with higher Li content are not formed. Surprisingly, the best catalytic performance was observed using a 1:1 mixture of Bi(pib)3 and Li(pib). Collectively, these results reveal the superior performance of the 1:1 mixture while the stability of complex 9 might be responsible for the lower reaction rate with higher lithium content.
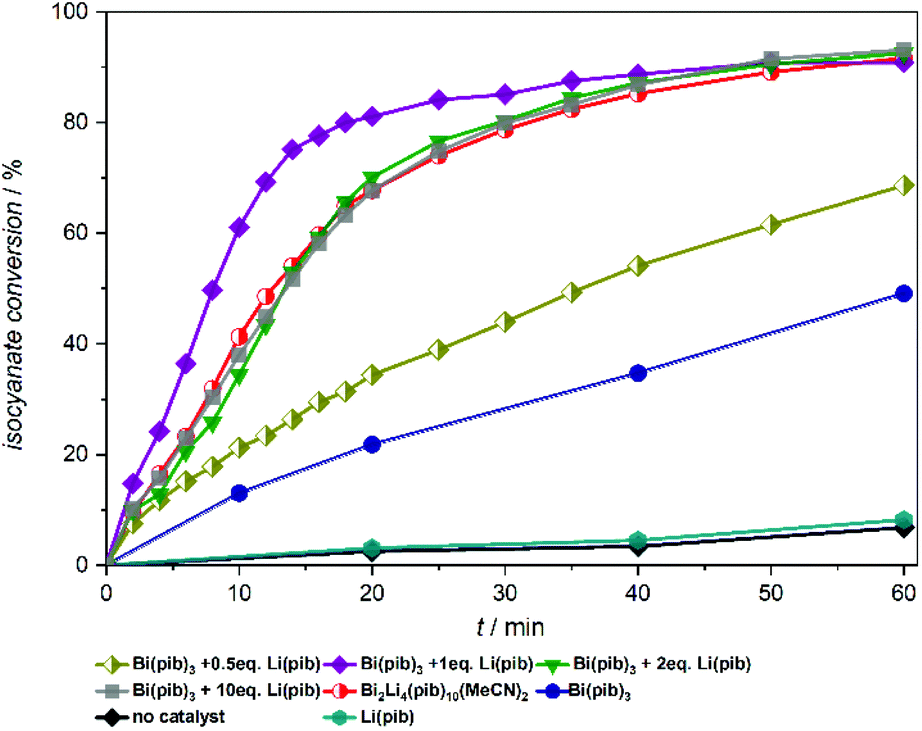 | ||
| Fig. 4 Lewis acid catalysed urethan reaction of Desmodur LD and ethanol in the presence of different amounts of Bi(pib)3 and Li(pib), and with complex 9 for comparison. | ||
Mechanistic investigation and density functional theory calculations
In an attempt to evaluate the reactivity of the urethane catalysts with alcohol and isocyanate functions, complexes 1 and 7 were mixed with stoichiometric amounts of ethanol or 1,6-hexamethylene diisocyanate (HDI) and the resulting 1H NMR spectra were compared to those of the individual compounds (Fig. S33–S36†). The 1H NMR spectrum of the mixtures of 1 or 7 with HDI show the resonances of the individual components and thus do not indicate a reaction. The same is observed for the mixture of 1 and ethanol. However, the CH2 resonance of ethanol in the mixture with 7 is significantly broadened and is not shifted to lower/higher frequency, suggesting that ethanol is involved in the dynamic formation of coordination compounds in solution, although the formation of free pivalic acid was not detected.58,59 Preliminary kinetic studies on the bismuth pivalate catalysed urethane reaction reveal a 0.4 order kinetics with respect to ethanol (see the ESI†).So far, mechanistic studies on metal complex catalyzed PU reaction have been performed mainly for organotin catalysts,58,60–65 leaving critical questions still unanswered. It remains for example unclear what factors determine the selectivity and speed of the isocyanate-hydroxyl reaction when catalyzed by tin, zirconium, bismuth, or zinc catalysts. We investigated the Bi-catalyzed urethane formation by means of quantum chemical computations, providing the basis for further mechanistic investigations. These computations confirm that bismuth carboxylates successfully catalyze urethane formation for both, aliphatic and aromatic isocyanates, speeding up the reaction by a factor of 104 to 105. Complex kinetics govern the bismuth pivalate catalysis, albeit partially following pseudo 0th-order kinetics (cf.Fig. 5). The turnover-limiting step in the catalytic cycle is the migratory insertion of the isocyanate into the Bi–O bond of the alkoxy ligand, finally forming the carbamate that remains coordinated to the metal center in a κ1- or κ2-fashion. The urethane product is released from the catalyst by protonation at the carbamate nitrogen- or carbonyl oxygen atom. Bismuth pivalate serves as model for gaining conceptional mechanistic insight into the Bi-catalysed urethane formation. Propyl isocyanate and ethyl alcohol serve as model substrates. Further information on the condensed phase representation and computational details in general can be found in the ESI.† After briefly addressing the uncatalyzed isocyanate-hydroxyl reaction we will take a closer look at its mechanistic spectrum in the context of bismuth catalysis.
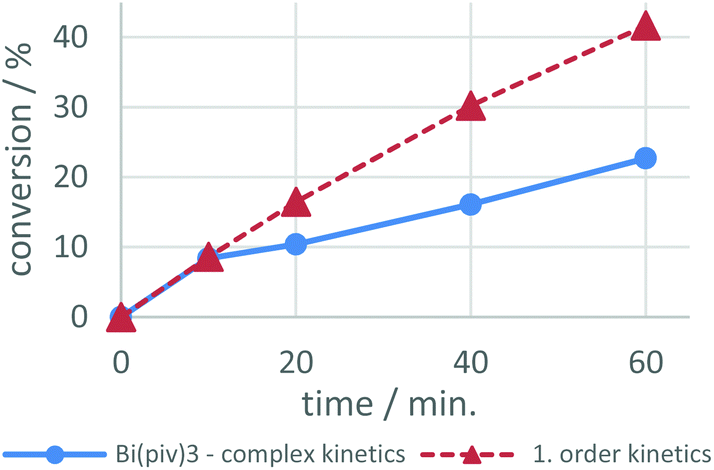 | ||
| Fig. 5 Isocyanate conversion over time utilizing Bi(piv)3. This curve initially follows pseudo first-order kinetics, deviating though after a while from the simulated first-order kinetics profile and appearing similar to 0th-order kinetics. We assume complex kinetics because of this change of kinetic profile. | ||
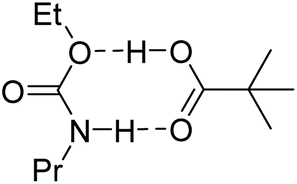
The uncatalyzed reaction between aliphatic alcohol and aliphatic or aromatic isocyanate requires a proton shuttle in the 6-membered ring transition-state (TS) structure to give urethane product in reasonable time (Scheme 3). In the absence of a suitable proton shuttle in the reaction mixture, the alcohol itself acts as such. This stoichiometry – two alcohols and one isocyanate – leads to pseudo second-order kinetics with a strong dependence on the concentration of the alcohol. The computed reaction barriers for our model system are 110 and 100 kJ mol−1 for aliphatic and aromatic isocyanates, respectively, in the infinite dilution scenario. The reaction rates are consistent with the experimental reaction times, which are in the range of days and hours, respectively, at ambient temperature. Note that the effective rate of the urethan bond forming reaction can be influenced by various phenomena such as catalysis, autocatalysis, solvent effects and molecular interactions.63,66–69
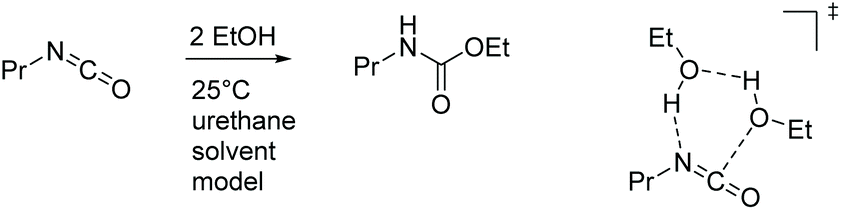 | ||
| Scheme 3 Uncatalyzed model reaction between propyl isocyanate and ethyl alcohol. A urethane species containing also an ester functionality represents the reaction medium. On the right side the 6-membered ring transition-state structure is shown. | ||
Computations reveal that cyclic, tetrameric bismuth tricarboxylate clusters form thermodynamically driven by a free energy difference of −40 and −18 kJ mol−1 at 25 °C and 60 °C, respectively (Fig. 6). Contrast this with the endergonic formation of di- and trimeric bismuth-carboxylate complexes, their free energy differences amounting to roughly +35 kJ mol−1, starting from the monomer at 60 °C. Because of the huge computational effort of using the tetramer as catalyst, we limit our discussion to the monomer. Consider that the concentration of Bi(piv)3 in solution is low (0.05 mol%) and that entropy (and diffusion) hamper the formation of clusters.
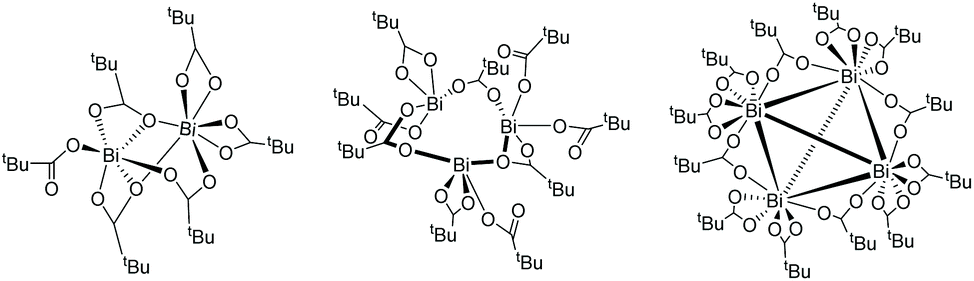 | ||
| Fig. 6 Computed Bi(piv)3-dimer, -trimer, and -tetramer structures. The highly symmetric Bi(piv)3-tetramer structure consists of Bi atoms in tetrahedral a geometry and agrees with the solid-state structure.42 | ||
Considering the multiple coordination forms of Bi, we first examined the alcohol–isocyanate coupling in more detail. In general, the coupling reaction is accelerated by facilitating the nucleophilic attack of the alcohol at the carbon atom of the isocyanate thereby lowering the activation barriers. Coordination of alcohol to bismuth lowers the alcohol's pKa, facilitating its deprotonation and enhancing its nucleophilic character. Several pathways of urethane formation have been considered with the insertion reaction either proceeding concertedly with protonated or unprotonated alcohol (coordinated to the catalyst's metal center) or following an outer-sphere type of mechanism (cf.Fig. 7). Clearly, the higher nucleophilicity of coordinated alkoxides in concert with the higher electrophilicity of the isocyanate carbon atom by interaction of the isocyanate nitrogen atom with a Lewis acid is most effective in lowering the reaction barrier. This reaction, by taking place on a metal center, is reminiscent of a 1,2-migratory insertion. Migratory insertion rates are strongly dependent on the solvent or reaction matrix.70 Consequently, the polarity of ligands (forming the local matrix around the Bi center) affects the rate of 1,2-migratory insertion.
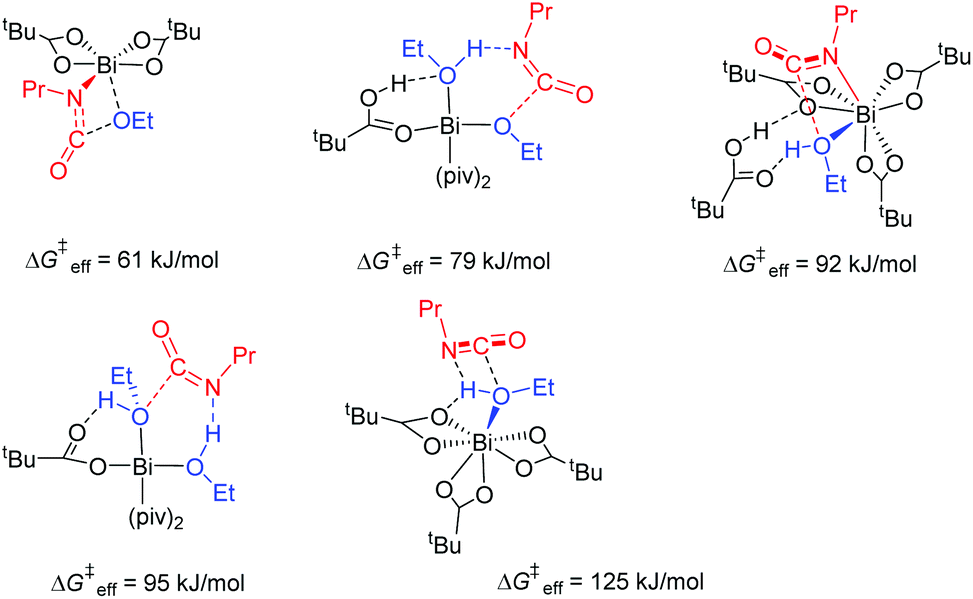 | ||
| Fig. 7 Potential modes of reaction between isocyanate and alcohol on the bismuth center. These modes are shown in increasing order of the effective barrier height. Concerted protonation of the forming carbamate can be ruled out. Protonated alcohol also is unlikely to react with isocyanate in a Bi complex. | ||
The initial ligand exchange most likely follows an associative mechanism because of the sterically accessible bismuth center in Bi(piv)3. There is no significant electronic stabilization of the pivalate's negative charge formed upon initial departure from the bismuth center, which additionally supports this mechanism. Conversely, conjugate base stabilization hints at a dissociative ligand exchange mechanism. Computations additionally confirm that the associative mechanism is dominant. Hence, the ligand exchange rate law is first order in the entering alcohol ligand. The expected large negative entropy of activation is the reason for an additional contribution to the solvent dependence of the hydroxyl-isocyanate reaction. It turns out that Bi(piv)3 prefers exchanging pivalate for alcohol only once (see Fig. 8 and 9). The concerted 1,2-migratory insertion of the alkoxide ligand is identified by quantum chemical computations as predominant reaction mechanism (compare the reaction barriers in Fig. 7).
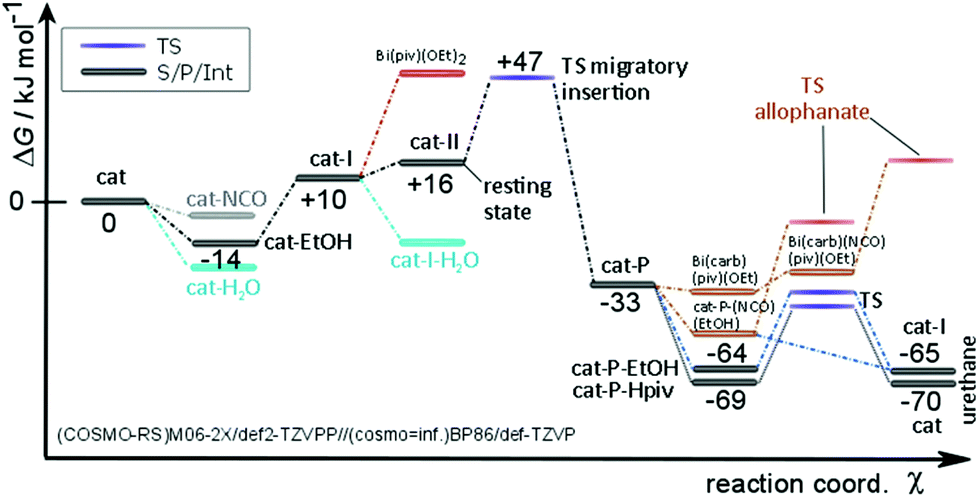 | ||
| Fig. 8 Energy diagram of the Bi(piv)3 catalytic cycle. Migratory insertion is turnover limiting. The resting state consists of Bi(piv)2(OEt)(PrNCO) (cat-II). The effective equilibria are shifted towards the resting state. | ||
 | ||
| Fig. 9 Catalytic cycle starting with Bi(piv)3 as catalyst precursor. EtO-Bi(piv)2 is the active form of the catalyst, its being regenerated by the alcohol component. Highest efficiency is achieved by following this catalytic cycle (heavy arrows). | ||
The efficiency of a catalytic cycle depends on the barrier of the turnover-limiting step but most importantly though on the absence of easily accessible off-cycle species with siphoning character. Favorable equilibria towards off-cycle species retard the catalytic cycle and open the door to the possible formation of side products such as allophanates (Fig. 9). Clearly, the Bi(piv)3 catalytic cycle consists of easily accessible off-cycle equilibria. These efficiency-lowering equilibria have been computationally identified (Fig. 8 and 9). Their presence additionally is supported by the overall kinetic profile, obtained by experiments, hinting at complex kinetics (cf.Fig. 5).
The kinetics of Bi(piv)3-catalysed urethane formation is mainly determined by which catalyst intermediate is regenerated after the last step of the catalytic cycle, that is the step releasing the product from the catalyst. The intermittent 0th-order kinetic character hints at a resting state located at Bi(piv)(OEt)(PrNCO) (cat-II). Up to this point we have not considered the computed energy diagram in Fig. 8. The relative energy levels of Bi(piv)3 (cat), Bi(piv)2(OEt) (cat-I), and cat-II seem to contradict this hypothesis. However, taking into account the uncertainty of quantum chemical computations in condensed phase, the energy level of cat-II might be lower than the one of cat-I. Seeing this, we should not be surprised to find cat-I being regenerated in the catalytic cycle. Computations show, however, that both the alcohol and free pivalic acid ligand can release the urethane product from the catalyst via proton transfer. Whether the alcohol or the carboxylic acid coordinates to the catalyst depends on their instantaneous concentration. Off-cycle equilibria become important in case of the regeneration of cat, retarding the efficiency of the catalytic cycle (Fig. 9). The most efficient catalytical path, having the least probability of off-cycle intermediates, follows the regeneration of cat-I. As the reaction proceeds, urethane increasingly forms and may immobilize the catalyst ligand pivalic acid, as outlined in Fig. 10. This immobilization redirects the catalytic cycle towards from cat being regenerated to the more efficient pathway regenerating only cat-I.
 | ||
| Fig. 10 Possible immobilization of pivalic acid as protonating agent for product release in the catalytic cycle. | ||
Computations reveal that protonation of carbamate is thermoneutral and, in addition, does not discriminate between carboxylic acid and alcohol as protonating agents in terms of kinetics (cf.Fig. 8). The corresponding barriers amount to 30 and 33 kJ mol−1, respectively. The alcohol and carboxylic acid concentrations determine – next to steric demand – which species favorably coordinates prior to protonating the carbamate to release the product. Let us have a closer look at the two scenarios. Firstly, when the alcohol protonates the carbamate (via associative ligand exchange), cat-I is regenerated, leading to the catalytic cycle consisting of less off-cycle equilibria (cf.Fig. 9). Secondly, when free carboxylic acid takes over protonation, catalyst precursor cat is regenerated, which appears to be the non-active form of the catalyst. In this latter scenario, ligand exchange with alcohol must take place, competing with off-cycle equilibria already at the beginning of the catalytic cycle or perhaps before forming the active catalyst species cat-I. Pre-equilibria including alcohol corroborate the experimental finding of the non-integer partial reaction order of the alcohol reagent in the rate law.
The selectivity of this catalyzed isocyanate-hydroxyl reaction depends on the height of the barrier of the turnover-limiting step compared to the ones that lead to side products. Formation of allophanate requires conquering a barrier of as much as 46 kJ mol−1, compared to 41 kJ mol−1 for the turnover-limiting migratory insertion. The extent of allophanate formation mainly depends on the relative height of the reaction barrier forming allophanates compared to that of carbamate protonation. As outlined in Fig. 8, isocyanate may coordinate to cat-P, competing with the coordination of alcohol (or pivalic acid). Latter coordination is clearly favored by 10–15 kJ mol−1 over that of isocyanate. Moreover, the barrier to form allophanate amounts to as much as 46 kJ mol−1 and, thus, lies higher than the ones leading to product release. These results corroborate the absence of allophanates in experiments.
Because the 1,2-migratory insertion is turnover limiting the resting state of the Bi(piv)3 catalyzed reaction is cat-II. Consequently, the barrier of the liberation of the product from the catalyst is not turnover limiting. Generally spoken, if computations result in this scenario, the overall kinetics most probably exhibits pseudo 0th-order. The case in which product release is turnover limiting also leads to pseudo 0th-order kinetics. But now the resting state consists of the catalyst-product (cat-P) intermediate, raising the probability of side-product formation. Let us assume now that the barrier of ligand exchange with alcohol is higher compared to the one of the insertion step. In this scenario, the reaction will follow pseudo 1st-order kinetics and the resting state would be the catalyst precursor species (cat).
Often humidity cannot be excluded during the isocyanate-hydroxyl reaction, depending on the application. Therefore, water may be present during synthesis, raising the question of the hydrolytic stability of Bi(piv)3. Water coordinates favorably to cat and cat-I (Fig. 8). However, the formation of bismuth hydroxides is unlikely because the reaction pathway is consistently uphill in energy. The computations further show that the barrier for the reaction of water with isocyanate is higher than that for the reaction with alcohol. This high barrier explains why Bi(piv)3 does not promote the isocyanate–water reaction. Apparently, water blocks the catalyst and removes it from the catalytic cycle by siphoning. Unfortunately, this bismuth-aqua complex is no longer available for catalyzing the hydroxyl-isocyanate reaction.
The mechanistic concepts can be applied (in a first approximation) to multi-nuclear bismuth complexes (e.g. to Bi–Li clusters). Preliminary quantum chemical computations reveal that in contrast to mononuclear bismuth catalysis, isocyanate ligand association is thermoneutral or even exergonic when lithium is involved in the catalyst complex. The substrate isocyanate then preferably coordinates to lithium, whereas the deprotonated alcohol substrate is bridging between lithium and bismuth. Presumably, the higher Lewis acidity of lithium compared to that of the bismuth species leads to a higher protonating potential of alcohols and free carboxylic acids, resulting in a more efficient release of product from the dinuclear complex.
Conclusions
In conclusion, a series of Bi carboxylates 1–3 and heterobimetallic complexes 7–9 have been synthesized and characterized. The isostructural clusters 7–9 consist of two bismuth carboxylates and four lithium carboxylates and are spontaneously formed upon mixing bismuth and lithium carboxylates in solution. 1H DOSY NMR experiments and variable temperature NMR studies reveal that solutions of 1–3 and 7–9 contain a dynamic mixture of aggregates with an average size comparable to that of their solid-state structures.Studies on the catalytic performance of the Bi compounds in the urethane forming reaction show that the Bi-catalyzed reaction can be significantly accelerated by using heterobimetallic mixtures of Bi and Li carboxylates, with 8 almost reaching the activity of DBTL without promoting the isocyanate–water reaction. Theoretical studies show that urethane formation utilizing Bi(piv)3 follows complex kinetics that exhibit intermittent pseudo 0th-order kinetics. The turnover-limiting step in the catalytic cycle is the migratory insertion of the isocyanate into the Bi–O bond of the alkoxy ligand, eventually forming the carbamate. The urethane product is released from the catalyst by proton transfer from either the alcohol substrate or the free pivalic acid ligand. Collectively, heterobimetallic complexes composed of bismuth and lithium carboxylates are a promising alternative to the toxic organotin-containing catalysts in PU synthesis. Successive studies on the synthesis of even more active bismuth catalysts are under investigation in our laboratories.
Author contributions
E.L. carried out the synthetic experiments and analysed the experimental data. O.S. performed the computational investigations. L.W. and P.L. performed the X-ray single-crystal structure analyses. F.D., E.L. and O.S. wrote the manuscript. F.D. directed the investigations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was generously provided by the BASF Coatings GmbH and BASF SE. Funding for F. D. was provided by the Emmy Noether Program of the DFG (DI 2054/1-1). We thank Prof. F. E. Hahn for his generous support. We thank Klaus Bergander for performing and evaluating the DOSY NMR experiments.References
- H.-W. Engels, H.-G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Angew. Chem., 2013, 125, 9596–9616 CrossRef.
- PlasticsEurope – Association of Plastic Manufacturers, available at: https://www.plasticseurope.org/de/resources/market-data.
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed.
- A. L. Silva and J. C. Bordado, Catal. Rev., 2004, 46, 31–51 CrossRef.
- J. Alsarraf, Y. A. Ammar, F. Robert, E. Cloutet, H. Cramail and Y. Landais, Macromolecules, 2012, 45, 2249–2256 CrossRef CAS.
- B. Bantu, G. M. Pawar, U. Decker, K. Wurst, A. M. Schmidt and M. R. Buchmeiser, Chem. Eur. J., 2009, 15, 3103–3109 CrossRef CAS PubMed.
- H.-J. Noh, T. Sadhasivam, M. Han, K. Lee, J.-Y. Kim and H.-Y. Jung, J. Catal., 2020, 382, 77–85 CrossRef CAS.
- O. Coutelier, M. El Ezzi, M. Destarac, F. Bonnette, T. Kato, A. Baceiredo, G. Sivasankarapillai, Y. Gnanou and D. Taton, Polym. Chem., 2012, 3, 605 RSC.
- E. Peris, Chem. Rev., 2018, 118, 9988–10031 CrossRef CAS PubMed.
- H. Sardon, A. C. Engler, J. M. W. Chan, J. M. García, D. J. Coady, A. Pascual, D. Mecerreyes, G. O. Jones, J. E. Rice, H. W. Horn and J. L. Hedrick, J. Am. Chem. Soc., 2013, 135, 16235–16241 CrossRef CAS PubMed.
- H. Sardon, A. Pascual, D. Mecerreyes, D. Taton, H. Cramail and J. L. Hedrick, Macromolecules, 2015, 48, 3153–3165 CrossRef CAS.
- F.-Z. Belmokaddem, J. Dagonneau, J. Lhomme, R. Blanc, A. Garduno-Alva, C. Maliverney, A. Baceiredo, E. Maerten, E. Fleury and F. Méchin, Des. Monomers Polym., 2016, 19, 347–360 CrossRef CAS.
- F. E. Golling, R. Pires, A. Hecking, J. Weikard, F. Richter, K. Danielmeier and D. Dijkstra, Polym. Int., 2019, 68, 848–855 CrossRef CAS.
- D. K. Chattopadhyay and K. V. S. N. Raju, Prog. Polym. Sci., 2007, 32, 352–418 Search PubMed.
- D. Guhl, in Polyurethanes for High Performance Coatings, European Coatings Conference, vol. 5, pp. 119–137.
- M. Hoch, J. Appl. Geochem., 2001, 16, 719–743 CrossRef CAS.
- Y. Yang and M. W. Urban, Angew. Chem., Int. Ed., 2014, 53, 12142–12147 CrossRef CAS PubMed.
- Y. Schellekens, B. van Trimpont, P.-J. Goelen, K. Binnemans, M. Smet, M.-A. Persoons and D. de Vos, Green Chem., 2014, 16, 4401–4407 RSC.
- Q. Han and M. W. Urban, J. Appl. Polym. Sci., 2002, 86, 2322–2329 CrossRef CAS.
- L. Rand, B. Thir, S. L. Reegen and K. C. Frisch, J. Appl. Polym. Sci., 1965, 9, 1787–1795 CrossRef CAS.
- W. J. Blank, Z. A. He and E. T. Hessell, Prog. Org. Coat., 1999, 35, 19–29 CrossRef CAS.
- S. Petrak, V. Shadurka and W. H. Binder, Prog. Org. Coat., 2009, 66, 296–305 CrossRef CAS.
- R. Gertzmann and C. Gürtler, Tetrahedron Lett., 2005, 46, 6659–6662 CrossRef CAS.
- I. Ahmad, J. H. Zaidi, R. Hussain and A. Munir, Polym. Int., 2007, 56, 1521–1529 CrossRef CAS.
- C. Korah Bina, K. G. Kannan and K. N. Ninan, J. Therm. Anal., 2004, 78, 753–760 CrossRef CAS.
- W. J. Blank, Macromol. Symp., 2002, 187, 261–270 CrossRef CAS.
- H. Sardon, L. Irusta and M. J. Fernández-Berridi, Prog. Org. Coat., 2009, 66, 291–295 CrossRef CAS.
- J. Krause, S. Reiter, S. Lindner, A. Schmidt, K. Jurkschat, M. Schürmann and G. Bradtmöller, WO Pat., 2009132784(A1), 2009 Search PubMed.
- K. Jurkschat, L. Lovkova-Berends, T. Zöller and M. Schürmann, WO Pat., 2011113926(A1), 2011 Search PubMed.
- F. Richter, K. Jurkschat, L. Lovkova-Berends and T. Zöller, EP Pat., 2772496(A1), 2013 Search PubMed.
- F. Richter, K. Jurkschat, M. Wagner, V. Abeyawarathan and M. Gock, EP Pat., 2990381(A1), 2014 Search PubMed.
- L. Iovkova-Berends, T. Berends, C. Dietz, G. Bradtmöller, D. Schollmeyer and K. Jurkschat, Eur. J. Inorg. Chem., 2011, 2011, 3632–3643 CrossRef CAS.
- L. Iovkova-Berends, T. Berends, T. Zöller, G. Bradtmöller, S. Herres-Pawlis and K. Jurkschat, Eur. J. Inorg. Chem., 2012, 2012, 3191–3199 CrossRef CAS.
- L. Iovkova-Berends, T. Berends, T. Zöller, D. Schollmeyer, G. Bradtmöller and K. Jurkschat, Eur. J. Inorg. Chem., 2012, 2012, 3463–3473 CrossRef CAS.
- B. Glowacki, R. Pallach, M. Lutter, F. Roesler, H. Alnasr, C. Thomas, D. Schollmeyer and K. Jurkschat, Chem. – Eur. J., 2018, 24, 19266–19279 CrossRef CAS PubMed.
- T. Vanbergen, I. Verlent, J. de Geeter, B. Haelterman, L. Claes and D. de Vos, ChemSusChem, 2020, 13, 3835–3843 CrossRef CAS PubMed.
- G. Yin, D. Zhao, X. Wang, Y. Ren, L. Zhang, X. Wu, S. Nie and Q. Li, RSC Adv., 2015, 5, 79070–79080 RSC.
- B. Xu, L. Li, K. Zhang, P. M. Macdonald, M. A. Winnik, R. Jenkins, D. Bassett, D. Wolf and O. Nuyken, Langmuir, 1997, 13, 6896–6902 CrossRef CAS.
- B. Jousseaume, C. Laporte, T. Toupance and J.-M. Bernard, Tetrahedron Lett., 2002, 43, 6305–6307 CrossRef CAS.
- P. Hoffmann and B. Schnier, EP Pat., 3247757(B1), 2016 Search PubMed.
- D. Guhl, FAPU, 2008, 49, 30–33 Search PubMed.
- J. D. Donaldson, J. F. Knifton and S. D. Ross, Spectrochim. Acta, Part A, 1965, 21, 275–277 CrossRef CAS.
- S. I. Troyanov and A. P. Pisarevsky, J. Chem. Soc., Chem. Commun., 1993, 335 RSC.
- P. C. Andrews, G. B. Deacon, P. C. Junk, I. Kumar and M. Silberstein, Dalton Trans., 2006, 4852–4858 RSC.
- G. G. Briand and N. Burford, in Main Chemistry Group, ed. A. H. Cowley and A. G. Sykes, Elsevier textbooks, s.l., 1st edn, 2000, pp. 285–357 Search PubMed.
- H. Suzuki, Elsevier Science, Amsterdam, New York, 2001 Search PubMed.
- G. G. Briand and N. Burford, in Advances in Inorganic Chemistry, Academic Press, 2000, vol. 50, pp. 285–357 Search PubMed.
- C.-I. Stålhandske, A. Kjekshus, S. Svensson, T. Holme, A. A. Lindberg, G. Jansen, B. Lamm and B. Samuelsson, Acta Chem. Scand., 1969, 23, 1525–1533 CrossRef.
- S. I. Troyanov and A. P. Pisarevskii, Koord. Khim., 1991, 909 CAS.
- L. Wrobel, T. Rüffer, M. Korb, H. Lang and M. Mehring, Eur. J. Inorg. Chem., 2017, 2017, 1032–1040 CrossRef CAS.
- T. Hatanpää, M. Vehkamäki, M. Ritala and M. Leskelä, Dalton Trans., 2010, 39, 3219–3226 RSC.
- S. I. Troyanov and A. P. Pisarevsky, J. Chem. Soc., Chem. Commun., 1993, 335 RSC.
- G. Deacon, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- M. Mehring, D. Mansfeld, S. Paalasmaa and M. Schürmann, Chem. Eur. J., 2006, 12, 1767–1781 CrossRef CAS PubMed.
- A. J. Edwards, M. A. Beswick, J. R. Galsworthy, M. A. Paver, P. R. Raithby, M.-A. Rennie, C. A. Russell, K. L. Verhorevoort and D. S. Wright, Inorg. Chim. Acta, 1996, 248, 9–14 CrossRef CAS.
- J. Konu, M. S. Balakrishna, T. Chivers and T. W. Swaddle, Inorg. Chem., 2007, 46, 2627–2636 CrossRef CAS PubMed.
- G. G. Suchkova and L. I. Maklakov, Vib. Spectrosc., 2009, 51, 333–339 CrossRef CAS.
- A. J. Bloodworth and A. G. Davies, Chem. Commun., 1965, 24 RSC.
- R. P. Houghton and A. W. Mulvaney, J. Organomet. Chem., 1996, 518, 21–27 CrossRef CAS.
- J. Otera, T. Yano and R. Okawara, Chem. Lett., 1985, 14, 901–904 CrossRef.
- M. V. Zabalov, R. P. Tiger and A. A. Berlin, Russ. Chem. Bull., 2012, 61, 518–527 CrossRef CAS.
- R. Devendra, N. R. Edmonds and T. Söhnel, J. Mol. Catal. A: Chem., 2013, 366, 126–139 CrossRef CAS.
- W. Cheikh, Z. B. Rózsa, C. O. Camacho López, P. Mizsey, B. Viskolcz, M. Szőri and Z. Fejes, Polymers, 2019, 11, 1543 CrossRef CAS PubMed.
- A.-C. Draye and J.-J. Tondeur, J. Mol. Catal. A: Chem., 1999, 138, 135–144 CrossRef CAS.
- A. Draye, J. Mol. Catal. A: Chem., 1999, 140, 31–40 CrossRef CAS.
- G. Raspoet, M. T. Nguyen, M. McGarraghy and A. F. Hegarty, J. Org. Chem., 1998, 63, 6878–6885 CrossRef CAS PubMed.
- W. Sultan and J. P. Busnel, J. Therm. Anal., 2006, 83, 355–359 CrossRef CAS.
- M. Muuronen, P. Deglmann and Ž. Tomović, J. Org. Chem., 2019, 84, 8202–8209 CrossRef CAS PubMed.
- C. O. C. López, Z. Fejes and B. Viskolcz, J. Flow Chem, 2019, 9, 199–204 CrossRef.
- J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461–1475 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2057615–2057620. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1gc00446h |
| This journal is © The Royal Society of Chemistry 2021 |