
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Access to tough and transparent nanocomposites via Pickering emulsion polymerization using biocatalytic hybrid lignin nanoparticles as functional surfactants†
Adrian
Moreno
 ,
Mohammad
Morsali
,
Jinrong
Liu
and
Mika H.
Sipponen
*
,
Mohammad
Morsali
,
Jinrong
Liu
and
Mika H.
Sipponen
*
Department of Materials and Environmental Chemistry, Stockholm University, 10691, Stockholm, Sweden. E-mail: mika.sipponen@mmk.su.se
First published on 18th March 2021
Abstract
Weak interfacial binding of lignin within synthetic polymer composites results in unsatisfactory mechanical properties that limit their application prospects. In the present work, polystyrene (PS) and poly(butyl methacrylate) (PBMA) nanocomposites containing lignin nanoparticles (LNPs) are produced by simple melting of polymeric latex dispersions obtained from free radical polymerization of oil-in-water Pickering emulsions stabilized by hybrid LNPs coated with chitosan and glucose oxidase. Owing to the formation of viscous polymer melts, the hybrid LNPs ended up uniformly dispersed within the polymeric matrices, which gave the polymeric nanocomposites markedly improved tensile strength without sacrificing their elasticity in comparison to pure PS and PBMA. Consequently, the composites reinforced with 15 wt% of the hybrid particles showed improvement in toughness by a factor of 3.5 and 15 compared to those of the corresponding pristine PS and PBMA. In addition, the presence of the hybrid particles conferred the nanocomposites with commendable UV-blocking and antioxidant properties which are relevant for protective packaging and coating applications. Overall, our results show a new and green route with excellent material economy (overall mass yield up to 91%) to obtain strong and transparent polymeric nanocomposites reinforced with up to 30 wt% of LNPs, which is expected to attract renewed interest in lignin–polymer composites for a broad range of applications.
Introduction
Polymeric composites are multi-phase materials in which reinforcing fillers are integrated within a polymeric matrix, resulting in a synergistic effect on different material properties (e.g. mechanical, electrical, optical, magnetic and catalytic among others) that cannot be achieved from their constituent single components.1–4 With the widespread interest in the development of functional materials for a plethora of applications, polymeric composites with enhanced properties have attracted considerable attention during the last few years.5–8 In this context, the polymerization of monomers stabilized by solid particles in aqueous Pickering emulsions has emerged as one of the most popular and green strategies to generate different polymeric composites. To date, polymeric composites with improved mechanical, magnetic, and catalytic properties have been synthetized using Pickering emulsions stabilized by silica,9–11 clay,12,13 graphene,14,15 and magnetic nanoparticles.16,17The preparation of polymeric composites with a more favorable carbon footprint by using particulate stabilizers derived from lignocellulose raw materials remains less exploited, and hitherto limited to the use of cellulose nanocrystals (CNCs) and cellulose nanofibrils (CNFs).18–23 The use of lignin, the second most abundant natural polymer next to cellulose, as a reinforcing agent in composites has previously been seriously hindered due to the poor interfacial binding within the polymeric matrix. Consequently, aggregation of lignin in composites and phase separation in blends are common hurdles, especially with non-polar systems.24–30 To overcome this limitation, chemical functionalization of lignin by grafting polymer chains has classically been conducted.31–34 However, only minor positive effects or even detrimental impacts on the final mechanical properties of the materials have been reported,8,24,25 and therefore the development of sustainable alternative methodologies that avoid covalent functionalizations of lignin is still a challenge that needs to be addressed.
Nano- and microscale lignin particles have gained a great deal of interest in the preparation of lignin-based polymeric materials due to their spherical shape and colloidal stability compared to raw lignins.35–38 For instance, colloidal lignin nanoparticles (LNPs) can remain flowable at high concentrations (50 wt% or more) and act as reinforcing agents to improve the UV shielding and antioxidant properties of hydrophilic polymeric matrices such as CNF,39,40 poly(vinyl alcohol) (PVA),41,41,43 chitosan44 and polylactic acid (PLA).45 Besides the possibility to be used as a reinforcing agent, LNPs are also finding new applications as carriers for biologically active substances such as drugs, pesticides or enzymes.35,36,38,46,47 In addition, LNPs as anionic polyelectrolytes adsorb cationic polymers and proteins, and such coated particles are of interest for different applications ranging from catalytic transformations48,49 to sensors50,51 and water purification.52 These non-covalently modified LNPs have also been demonstrated as stabilizers for durable Pickering emulsions53,54 and vessels for the polymerization of hydrophobic monomers stabilized in water.55,56 However, only relatively low amounts of LNPs have been used (1–3 wt%), and none of the previous reports explored the extent that LNPs disperse within hydrophobic polymeric matrixes, and how this could affect the performance and the properties of the final composite material. Despite the anticipated potential of LNPs to improve the dispersibility within hydrophobic synthetic polymeric matrixes, the literature is lacking reports on methodologies to produce such hydrophobic polymeric particulate nanocomposites due to the considerable difficulty in efficiently dispersing LNPs (aqueous dispersion) in hydrophobic polymeric matrixes.
Recently, our research group reported a material-efficient method for the fabrication of biocatalyst-loaded LNPs and their application as functional surfactants for enzyme-degassed controlled radical polymerization in Pickering emulsions.57 This approach consists of a two-step adsorption immobilization process in which chitosan (chi) and glucose oxidase (GOx) were adsorbed onto LNPs to produce biocatalytic hybrid particles (GOx-chi-LNPs) capable of circumventing the oxygen inhibition of the polymerization process. We also found that immobilization of GOx increased the thermal stability of GOx from 55 °C to 75 °C, which was ascribed to the inherent ability of the particles to scavenge the hydrogen peroxide generated during the degassing step.
Inspired by the high thermal stability of the multifunctional emulsifiers, here we studied the application of biocatalyst-loaded LNPs (GOx-chi-LNPs) as particulate surfactants in free radical polymerization (FRP), a readily scalable polymerization technique. We demonstrate optimum conditions to produce stable hydrophobic monomer-in-water Pickering emulsions as well as their oxygen tolerant polymerization process catalyzed by GOx-chi-LNPs, using styrene and butyl methacrylate (BMA) as representative hydrophobic and industrially relevant monomers. Last but not least, we also pushed the limits of this system to produce hybrid polymer–GOx-chi-LNPs composites with a high content of GOx-chi-LNPs (up to 30 wt%) by a facile and green melting process of the obtained aqueous latex dispersions. The nanocomposites showed markedly enhanced mechanical properties compared to the raw polymeric materials and benchmarked to published data on lignin–polymer composites. Overall, our results demonstrate the potential of this approach to produce competitive and high performance polymeric composites with a favorable carbon footprint, while also equipping them with tailorable UV-shielding and antioxidant properties.
Results and discussion
Fabrication of biocatalyst loaded particles (GOx-chi-LNPs)
The preparation of biocatalyst loaded hybrid particles (GOx-chi-LNPs) was performed by following our recently reported methodology (Fig. 1a).57 Briefly, first negatively charged LNPs (approximate diameter 100 nm by DLS) were prepared from kraft lignin by solvent dilution in deionized water following previously reported methodologies with minor modifications.52 Afterwards, GOx-chi-LNPs were prepared by sequential adsorption of chitosan (100 mg g−1 LNPs) and GOx (10 mg g−1 chi-LNPs) onto LNPs, respectively. The DLS and zeta potential measurements confirmed the deposition of chitosan and then GOx on the nanoparticles based on a gradual increment in particle size and the reversal of zeta potential from negative to positive (Table S1†). After enzyme immobilization, GOx-chi-LNPs showed a high enzyme immobilization efficiency (88%) and a high retention of the enzyme activity (see Km and Vmax values in Table S2†).57 To evaluate the differences in particle size and morphology, negatively charged (LNPs) and positively charged (chi-LNPs and GOx-chi-LNPs) lignin particles were imaged by SEM. For all the colloidal lignin particles, well-defined and spherical particles could be observed (Fig. 1b–d) with a difference in size which is in agreement with the DLS data and attributed to the efficient adsorption/deposition of chitosan and GOx, respectively. The performance of GOx-chi-LNPs in the stabilization and enzyme-degassed FRP in Pickering emulsions will be discussed in the next sections.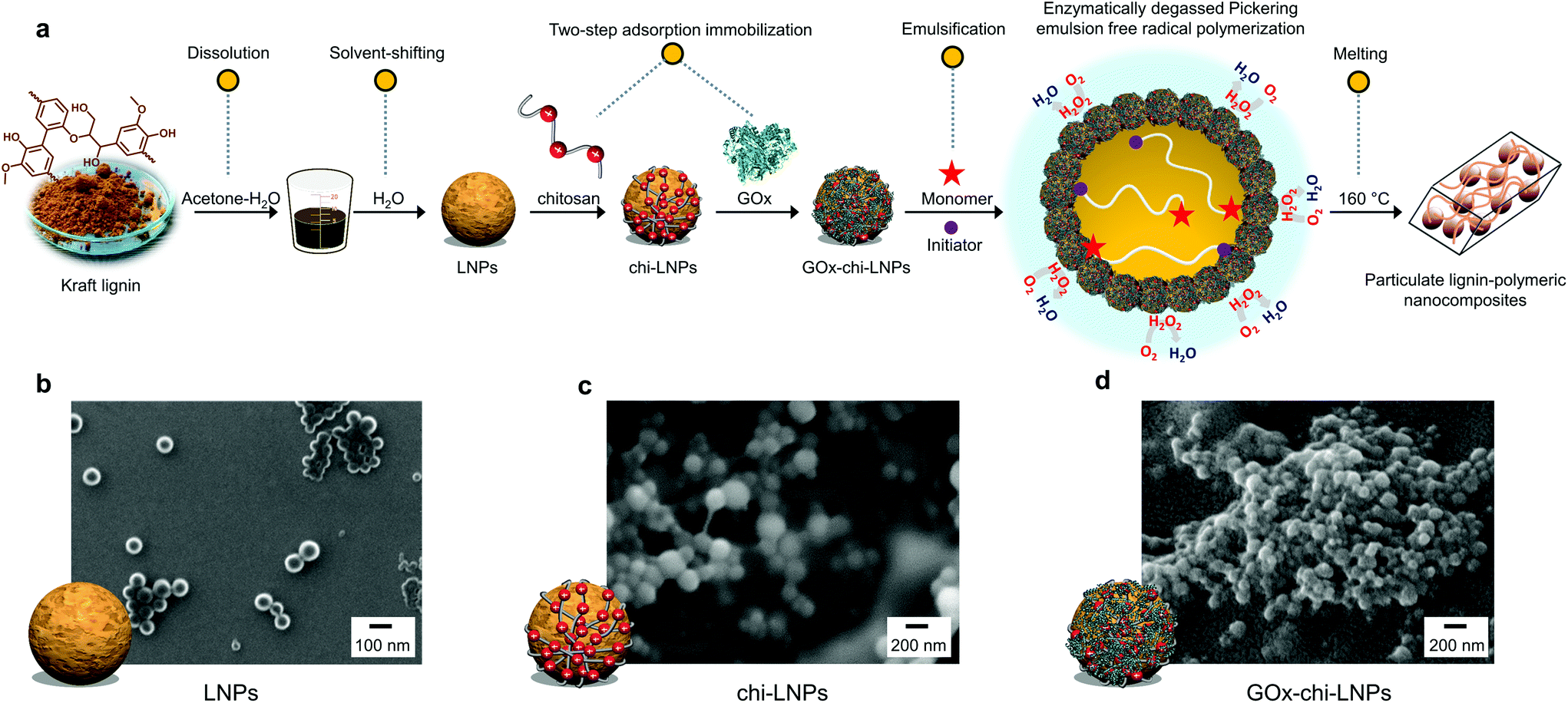 | ||
| Fig. 1 (a) Schematic illustration of the process for the preparation of biocatalyst loaded LNPs (GOx-chi-LNPs) and their application as functional surfactants in enzyme-degassed Pickering emulsion polymerization to produce particulate lignin–polymeric nanocomposites. SEM images of (b) LNPs produced by solvent exchange precipitation from pine kraft lignin, (c) chi-LNPs produced by the adsorption of chitosan (chi) onto LNPs and (d) GOx-chi-LNPs produced by the deposition of GOx onto chi-LNPs. | ||
Pickering emulsification and enzyme-degassed FRP of styrene in water
Considering the reported preferential use of LNPs and especially cationic LNPs as stabilizers for Pickering emulsions with a large number of hydrophobic compounds as oil phase,53,54 we decided to study GOx-chi-LNPs for the stabilization of styrene-based Pickering emulsions. The Pickering emulsions were formed by gradually adding styrene to a water suspension of GOx-chi-LNPs under sonication (final o/w ratio 20/80 v/v), following a previously described methodology by Zou et al.54 A set of emulsions by varying the concentration of GOx-chi-LNPs relative to the concentration of styrene (from 1 g L−1 to 20 g L−1 of GOx-chi-LNPs per L of styrene) were prepared and analyzed with the objective to determine the optimal conditions needed to obtain efficiently stabilized Pickering emulsions.All the emulsions were characterized after three hours from their preparation to allow for the stabilization of the system through the so-called limited coalescence process,58 which occurs when the particle concentration is low and a complete coverage of the oil–water interface is not produced during the agitation. To further assess the influence of the concentration of the GOx-chi-LNPs on the stability of the emulsion system, and to determine the minimum concentration of the particles to obtain a stable emulsion, the average reciprocal diameter of the emulsion droplets (inverse diameter of a droplet (1/D3,2), according to Sauter's nomenclature) is plotted in Fig. 2a.
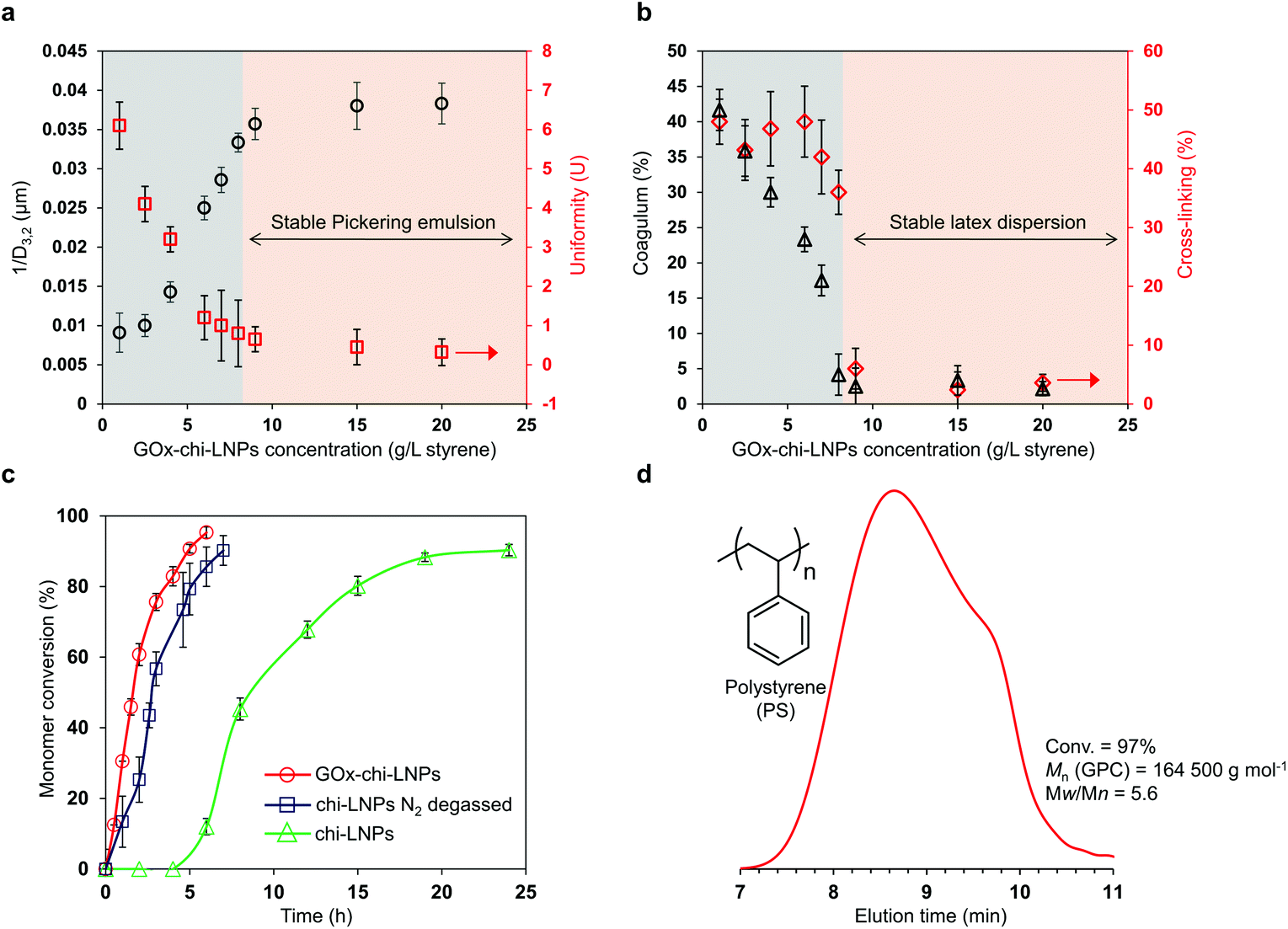 | ||
| Fig. 2 FRP and characterization of styrene-in-water Pickering emulsions stabilized by GOx-chi-LNPs. (a) Evolution of the average diameter (D3,2) and uniformity (U) of the emulsion droplets as a function of the concentration of GOx-chi-LNPs. (b) Evolution of the coagulum formation from the latex dispersions synthesized at 65 °C as a function of GOx-chi-LNP concentration. The % of cross-linked mass corresponds to the insoluble part after the filtration of coagulated latex dispersion. (c) Kinetic plots showing monomer conversion for the FRP of styrene-in-water Pickering emulsions using GOx-chi-LNPs (circles), chi-LNPs under inert conditions by degassing with N2 bubbling (squares), and chi-LNPs in the presence of oxygen (triangles). (d) GPC traces of polystyrene (PS) obtained after FRP-Pickering emulsion process using GOx-chi-LNPs (2.5 wt%) as emulsifiers. Error bars in (a, b, and c) represent one standard deviation (SD) from the mean values (n = 2). | ||
In agreement with the aforementioned limited coalescence mechanism, an increase in 1/D3,2 values can be observed when the concentration of GOx-chi-LNPs was gradually increased, until a critical concentration of 9 g GOx-chi-LNPs per L of styrene (1 wt%). This trait defines the previous value as the minimum concentration of GOx-chi-LNPs necessary to obtain a total coverage and stability of the oil–water interface to avoid coalescence of the emulsion. Such a transition could also be observed from the optical microscopy images through the vanishing of “macro” droplets of diameters 100–300 μm (Fig. S1a and b†) and the observation of oil droplets with a narrow dispersity associated with the uniformization of the droplet size and the absence of coalescence events at concentrations above 9 g L−1 (particles/styrene) (Fig. S1c and d†). Interestingly, even one week after the emulsification, phase separation or so-called creaming of large droplets was observed only for the preparations with lower concentrations of GOx-chi-LNPs (1–6 g L−1, particles/styrene) (Fig. S2a and b†) but not in the case of preparations with concentrations higher than 9 g L−1 (Fig. S2c†), which confirms a complete coverage of the oil phase during the emulsification process. In fact, above the optimum concentration value (9 g of GOx-chi-LNPs per L of styrene), all the emulsions remained visually stable (no evolution on droplet size) for one month indicating a comparable or higher stabilization behavior of the GOx-chi-LNPs compared to polysaccharide particles such as starch nanocrystals59 or CNCs.19
As we envisioned previously, the improved thermal stability of GOx immobilized onto chi-LNPs opens a possibility to apply these surfactants in polymerization processes such as FRP that occurs at elevated temperatures.57 Therefore, we decided to study the influence of the concentration of GOx-chi-LNPs on the colloidal quality of the latex dispersions obtained from the FRP of styrene-based Pickering emulsions using AIBN as a thermal initiator at 65 °C (Fig. 2b). Similar to the emulsion stability results, the critical concentration was observed at 9 g of GOx-chi-LNPs per L of styrene, at which GOx-chi-LNPs promote the formation of stable polymeric latexes after the emulsion polymerization process with a negligible extent of coagulation (lower than 5%). Only at low particle concentrations the formation of unstable latex dispersions by means of coarse and agglomerated polymer clumps could be observed (Fig. 2b and S3†). The observed critical concentration transition coincides with the previous one (compare Fig. 2a and b), and sets unequivocally the minimum concentration of particles (9 g L−1) to obtain a stable Pickering emulsion (average droplet diameter = 28.3 μm, uniformity parameter U = 0.4). In agreement with the literature,60–62 these results highlight the importance of ensuring a sufficient amount of the particulate emulsifier in order to promote a fast and complete coverage of the growing polymeric particles to avoid coagulation of the system. Noticeably, our system allows for increasing the concentration of GOx-chi-LNPs, opening an opportunity for the production of stable Pickering emulsions, and thereafter stable latex dispersions with a maximum content of 35 wt% of GOx-chi-LNPs relative to styrene. Above this concentration, the coagulation process occurred again during polymerization, leading to a high amount of cross-linked polymeric material (up to 60%) and unstable latex dispersions. This phenomenon could be associated with an increase in viscosity that counteracted the electrostatic repulsion of the cationic particles and resulted in the observed destabilization of the Pickering emulsions.63
Our polymerization experiments were conducted by adding glucose (0.14 M) as a substrate for GOx and without applying any external degassing procedures. In all cases, almost quantitative monomer conversion (>90%) was reached within 8 h, indicating an efficient enzyme catalyzed consumption of the dissolved oxygen during the whole polymerization process. However, it is important to note that additional analysis of the supernatant solution after purification of the latex dispersions via enzymatic tandem reaction using GOx and horseradish peroxidase (HRP) revealed residual glucose by an increase in UV-absorption at 500 nm owing to the formation of the oxidized dimer of o-dianisidine (Fig. S4†). The remaining amount of glucose after the polymerization reactions was 60% of the initial, which means that the ratio of glucose needed to produce 1 g of polymer is 44 mg, indicating the possibility to reduce the original amount when working with closed vials apart from the sampling. In addition, to evaluate in detail the real impact of GOx-chi-LNPs on the removal of oxygen during the polymerization process, styrene-in-water Pickering emulsions (20/80 v/v) stabilized with 22 g of lignin particles per L of styrene (2.5 wt%) were prepared using GOx-chi-LNPs and mere chitosan-coated LNPs (chi-LNPs) comparatively in the presence of AIBN as a radical initiator. The resulting emulsions were polymerized without degassing, and with N2 purging in sealed vials (Fig. 2c). In the case of the Pickering emulsions stabilized by GOx-chi-LNPs, no induction period was observed and after 6 h a near-complete conversion (95%) was determined (Fig. 2c, circles), while in the absence of GOx (chi-LNPs) there was a noticeable induction period (more than 4 h) and only 40% monomer conversion was determined after the next 8 h, requiring additional 20 h to reach 90% monomer conversion (Fig. 2c, triangles). This fact demonstrates the efficient in situ consumption of dissolved oxygen by GOx-chi-LNPs during the polymerization process even at high temperatures. A comparison with the performance of chi-LNPs without GOx but applying N2 purging for degassing with GOx-chi-LNPs showed a similar trend with complete conversion at 7 h without the presence of an induction period at the beginning of the reaction (Fig. 2c, squares). A slight decrease in the polymerization rate was nevertheless observed, likely due to the dissolution of oxygen during the sampling process with a consequent consumption of the propagating radicals. A GPC analysis of the polymerized latex dispersion stabilized by GOx-chi-LNPs was performed to prove the formation of high molecular weight polystyrene (PS) with an average molar mass (Mn) of 164 500 g mol−1 and a dispersity (Mw/Mn) of 5.9 (Fig. 2d). These results not only validate our previous findings on the use of GOx-chi-LNPs as efficient degassing stabilizers for Pickering emulsions and controlled radical polymerization processes57 but also demonstrate that these functional surfactants can be applied in industrially relevant FRP processes without the need for organic solvents or tedious and high cost degassing protocols (i.e. degassing with inert gas).
Analysis of the latex dispersions
The reaction mixtures from our Pickering emulsion polymerization experiments involving GOx-chi-LNPs as stabilizers were selected for further analysis of the nature of the obtained latex dispersion by SEM and optical light microscopy. Two different PS particles were produced, GOx-chi-LNP-coated PS microparticles by isolation of the particles by a simple centrifugation step and bare PS microparticles obtained by the extraction of GOx-chi-LNPs-coated particles with 35 wt% aqueous ammonium hydroxide solution to dissolve lignin (Fig. 3b, c and Fig. S5†). Based on the microscopic images of the GOx-chi-LNP-coated PS particles, the latex obtained under these polymerization conditions was composed of spherical PS microbeads, with a similar size to that of the initial monomer droplets (Fig. 3a) (average droplet diameter = 35.4 μm, uniformity parameter U = 0.5). The SEM images of GOx-chi-LNP-coated PS particles confirm a complete coverage through a thin layer of GOx-chi-LNPs (Fig. 3b and c), where the viscous nature of chitosan combined with the rigidity and radical scavenger nature of LNPs seems to be crucial to obtain a well-defined embedment of PS microparticles within the GOx-chi-LNPs, which could explain such a high emulsifier capacity and stability of these systems during the polymerization process. Interestingly, the presence of PS nanolatex particles about 500 nm in diameter (i.e. nanobeads, Fig. 3c, red marked sections) could also be detected on the surface of the PS microbeads. The origin of these PS nanolatex particles could be associated with the migration and subsequent stabilization with residual GOx-chi-LNPs of PS-oligomers from micrometric droplets to the aqueous phase due to the high internal pressure generated by the release of nitrogen coming from AIBN, as observed previously.18 On the other hand, the purified PS microparticles exhibited smooth surfaces, confirming the removal of the particulate coating (Fig. S5a†). However, it is important to mention that residual GOx-chi-LNPs could also be detected on the surface of the particles that went through the alkaline extraction process, revealing their original orientation at the oil–water interface (Fig. S5b†). The concentration of these residual particles was also evaluated by thermogravimetric analyses (TGA) performed before and after the treatment of the particles (GOx-chi-LNP-coated PS microparticles) with a basic solution. When analysed directly without alkaline extraction, TGA under nitrogen atmosphere revealed a clear and small amount of char (7 wt%) coming from GOx-chi-LNPs remaining at the end of the analysis for GOx-chi-LNP-coated PS microparticles, while in the case of purified PS microparticles a complete thermal decomposition could be observed (0.1 wt%, char residue) which confirmed the efficient purification of PS microparticles from GOx-chi-LNPs (Fig. S6†). The resulting latex dispersions stabilized by GOx-chi-LNPs were found to be stable for more than two months at room temperature. The stability of the PS latex dispersions was also investigated under different challenging conditions (Fig. 3d). First the effect of pH was examined; when decreased to pH 1, no coagulation took place and the optical microscopy image analysis did not reveal any significant changes in the particle size (Fig. 3d and e). However, when the pH was increased to 10, de-emulsification took place due to the charge neutralization of GOx-chi-LNPs at basic pH, causing aggregation and coagulation of the system (Fig. 3d). This de-emulsification process was found to be reversible upon restoration to the original pH 5, thus confirming the interesting pH-responsive behaviour of these systems, which could be exploited to recycle hybrid LNPs, albeit with the loss of enzyme activity (Fig. S7†). The stability against variation of temperature was also assessed. The latex dispersions were stored for 24 h in a fridge (5 °C) and freezer (−18 °C) without significant changes in the particle size, even in the case when the frozen sample was thawed at room temperature (Fig. 3d and e), demonstrating a higher thermal stability than that of the latex dispersions stabilized by conventional chemical surfactants or amphiphilic polymers.64,65 Ultimately, the stability to electrolyte addition was also proved and no significant changes in particle size or emulsion appearance were observed by saturating the dispersion system with sodium chloride (Fig. 3d and e).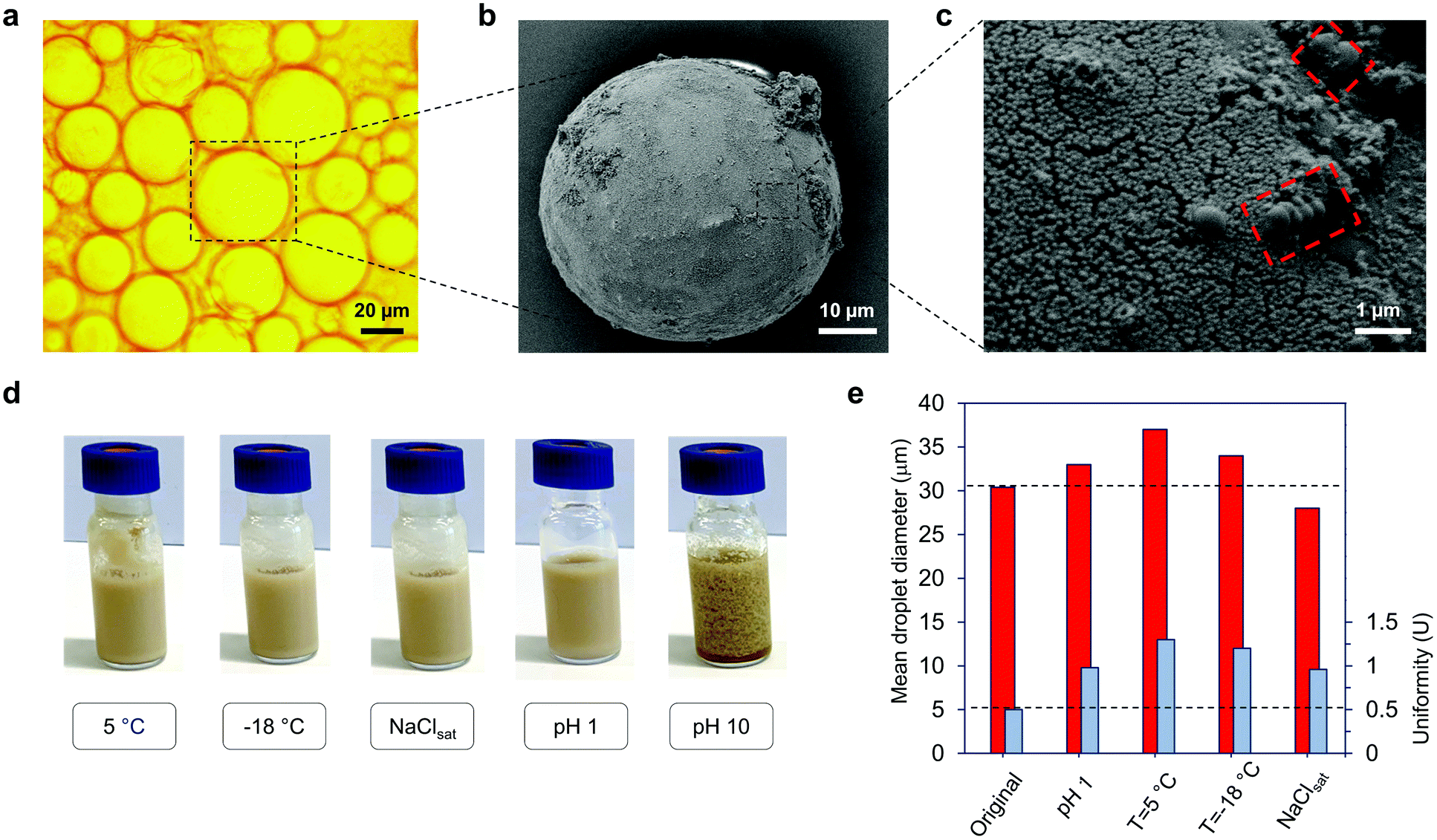 | ||
| Fig. 3 Characterization of PS latex dispersions. (a) Optical microscopic and (b and c) SEM images of the GOx-chi-LNP-coated PS microbeads after FRP at 65 °C (GOx-chi-LNPs, 2.5 wt%). (c) Magnification of (b) revealed the presence of GOx-chi-LNPs in the surface of PS particles. The red dashed section in (c) showed the presence of PS nanobeads on the surface of GOx-chi-LNP-coated PS microparticles. (d and e) Digital images and mean droplet evolution of PS latex dispersions under various challenging storage conditions. | ||
Elaboration and characterization of polymer–GOx-chi-LNP composites
Due to their excellent colloidal stability and uniformity, we envisioned that GOx-chi-LNP-coated PS microparticles could serve as precursors for generating uniform polymer–GOx-chi-LNP composites by taking advantage of the efficient coverage of GOx-chi-LNPs within the polymeric microparticles (Fig. 3c). Therefore, we performed a simple melting process of the PS microparticles at 160 °C and studied morphology of the GOx-chi-LNP particulate reinforced composites (Fig. 4a–c). | ||
| Fig. 4 Preparation of GOx-chi-LNP-reinforced PS composites. (a) Schematic illustrating the preparation of the composites by the melting process. (b) A digital image of GOx-chi-LNP-PS particles (GOx-chi-LNPs, 30 wt%) isolated from latex dispersion. (c) A digital image of PS-GOx-chi-LNP composite film obtained after the melting and pressing of particles shown on (b). The SEM micrographs of top and cross-sectional surfaces of PS-GOx-chi-LNP composite films at: (d) GOx-chi-LNPs 2.5 wt% (e) GOx-chi-LNPs 15 wt% and (f) GOx-chi-LNPs 30 wt%. Scale bars (1 μm). Red marked sections in (f) show the regions enriched with “GOx-chi-LNP-like clusters” in PS-GOx-chi-LNPs30 nanocomposite. | ||
To study the morphology of the particle reinforced composites, FRP of styrene was conducted under identical reaction conditions as those applied in the previous sections, but with higher contents of GOx-chi-LNPs (2.5, 15 and 30 wt%, respectively). After melting the aforementioned latex dispersions, uniform PS-GOx-chi-LNPs2.5, PS-GOx-chi-LNPs15 and PS-GOx-chi-LNPs30 composite films were obtained (Fig. 4c and S8†) and analyzed by SEM to gain insight into their morphologies. Delightfully, the SEM images of the surface and cross sections of PS-GOx-chi-LNPs2.5 and PS-GOx-chi-LNPs15 composites revealed that GOx-chi-LNPs were homogeneously dispersed within the polymeric matrixes without any micrometer sized agglomerates (Fig. 4d and e). One possible explanation for this well-defined particle dispersion could be the high viscosity of PS during the melting process, which likely restricted the diffusion of the particles and thus reduced their tendency to agglomerate based on a density difference compared to the matrix. A similar behavior could be observed in the case of PS-GOx-chi-LNPs30 composite film, albeit with the observation of minor agglomeration regions enriched in the “GOx-chi-LNP-like clusters” (Fig. 4f, red-marked sections) causing a less efficient coverage of the polymeric matrix within the particles.
This fact is not surprising since concentrations above 40 wt% of GOx-chi-LNPs during the Pickering emulsion polymerization destabilized the emulsions most likely through a particle saturation in the continuous phase of the Pickering emulsions. As stated before, lignin tends to generate aggregates by intermolecular interactions (π–π stacking), which consequently results in aggregation within composite matrixes.39 In this context, the use of spherical lignin particles with a high surface area to mass ratio has demonstrated the potential to enhance the interaction with hydrophilic polymeric matrixes and improve the mechanical and UV-shielding properties in various polymeric composites.39–42 However, the improvement of the properties is conditioned by the effective interfacial adhesion and uniform dispersibility of the particles within the polymer matrix, which is a non-trivial issue, especially in hydrophobic matrixes. Therefore, it is important to highlight that our approach not only successfully circumvented particle agglomeration when using the hybrid GOx-chi-LNPs as fillers but also allowed for the introduction of LNPs in hydrophobic polymeric systems, thereby overcoming a considerable challenge that has impeded the preparation of lignin–hydrophobic polymer composites during the last years.
Mechanical properties of polymer–GOx-chi-LNP composites
In order to evaluate the impact of GOx-chi-LNP as fillers on polymeric composites, uniaxial tensile testing was carried out to investigate their mechanical properties. Here, encouraged by the results obtained with PS, we decided to expand the polymer scope of our system by including poly(butyl methacrylate) (PBMA), another industrially relevant thermoplastic material (Fig. S9†). For comparison, six different types of samples for each polymer were prepared by varying systematically the amount of GOx-chi-LNPs as fillers from 2.5 wt% to 30 wt%. In all cases, optically transparent and uniform GOx-chi-LNP–polymeric films were obtained, with an increase in the brown color intensity as the amount of GOx-chi-LNPs increased (Fig. 5g). As a control, the mechanical properties of pure PS and PBMA isolated and purified from identical Pickering emulsion polymerization systems were also measured.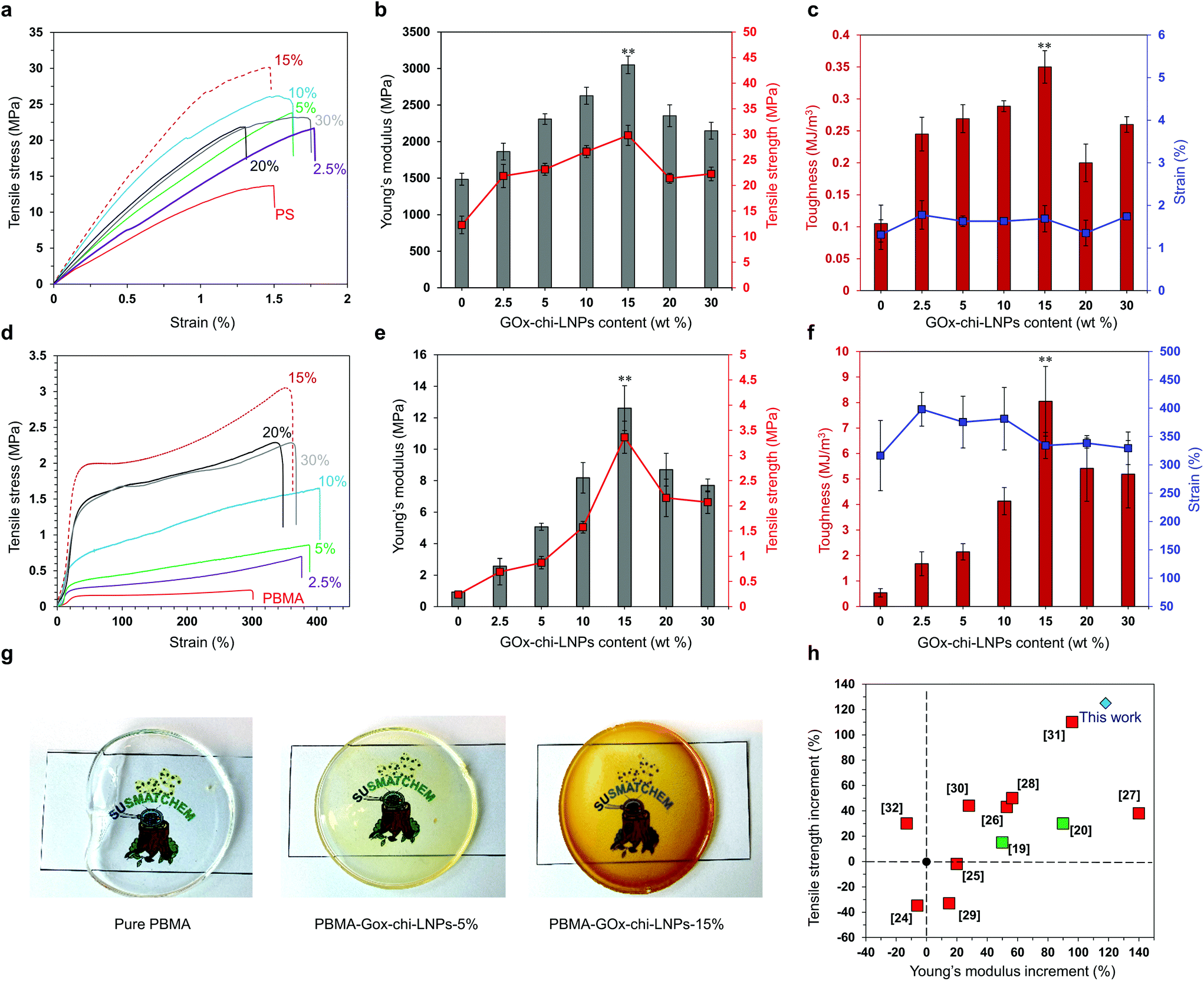 | ||
| Fig. 5 The mechanical properties of polymer–GOx-chi-LNP composites. (a) Tensile stress–strain curves of pure PS and PS-GOx-chi-LNP composites. (b) Young's modulus vs. tensile strength and (c) toughness vs. strain (%) for pure PS and PS-GOx-chi-LNP composites. (d) Tensile stress–strain curves of pure PBMA and PBMA-GOx-chi-LNP composites. (e) Young's modulus vs. tensile strength and (f) toughness vs. strain (%) for pure PBMA and PBMA-GOx-chi-LNP composites. In (b, c, e and f), the error bars represent one standard deviation (SD) from the mean values (n = 5) and asterisks (**) indicate the statistically significant differences (p < 0.05, one-way ANOVA with Tukey's HSD test). The details of ANOVA associated with b, c, e and f are given in the ESI Tables S5–S12.† (g) Digital images of pure PBMA and PBMA-GOx-chi-LNP composite films. (h) The influence of lignin on the mechanical properties of different PS–lignin composites; literature values are compared to the present work. | ||
In the case of composites with a polystyrene matrix, the particulate fillers caused a concentration-dependent augmentation of the mechanical properties compared to those of pure PS. As shown in Fig. 5a–c and Table S3,† at an optimum composition of 15 wt% loadings of GOx-chi-LNPs, the composite demonstrated the highest increments in Young's modulus and tensile strength (118% and 125%, respectively) relative to the values measured for pristine PS (Fig. 5a and b). Here, it is also important to highlight that the increase in tensile strength did not significantly affect the tensile strain of the composite materials, giving rise to a marked improvement in the toughness value (0.35 MJ m−3) of PS-GOx-chi-LNPs15 compared to that of pure PS (0.11 MJ m−3) (Fig. 5a and c). Remarkably, even in the presence of relatively lower amounts of GOx-chi-LNPs (2.5 wt% and 5 wt%), a notable increase in Young's modulus (25% and 55%, respectively) and tensile strength (77% and 88%, respectively) was observed compared to the values of pure PS (Fig. 5a–c). A further increase in the loading amount of the particulate fillers (i.e. 20 wt% and 30 wt%) did not further improve the mechanical properties of the composites; but despite their gradually decreasing mechanical strength, they still outperformed pure PS (Fig. 5a–c and Table S3†). A possible explanation for this effect could be related to the presence of the agglomerated regions of GOx-chi-LNPs at higher concentrations (i.e. 20% and 30%) that hindered the effective interaction between the particles and the polymeric matrix. A similar “bell-shape” trend in the mechanical performance was also found for PBMA-GOx-chi-LNP composites as a function of the amount of GOx-chi-LNPs (Fig. 5d–f and Table S4†). In this case, the positive reinforcing effect was even stronger than that observed for PS and likewise the maximum tensile strength and toughness values were obtained at a particulate concentration of 15 wt%. Upon comparing PBMA-GOx-chi-LNPs15 with pristine PBMA, we found an increase in Young's modulus value by a factor of 13 and an increase in the tensile strength by a factor of 14 with only subtle differences between elongation at break values. These results show an improvement in toughness by a factor of 15 (Fig. 5d–f and Table S4†).
Many previous works have reported detrimental and positive effects from the incorporation of lignin on polymeric matrixes, especially PS systems, but none of them have explored the potential of LNPs as reinforcing agents.24–32 In this context, it is important to highlight that our system surpasses the current state of the art PS–lignin composites without the need for the chemical functionalization of lignin (Fig. 5h, compare red squares with the blue diamond), and even the nanocellulose reinforced PS composites19,20 (Fig. 5h, compare green squares with blue diamond). The high mechanical performance of our polymer–GOx-chi-LNPs composites relies on the well-defined and uniform dispersion of the particles within the polymeric matrix as already discussed in the previous section, but also due to their favorable surface area to mass ratio and effective interaction within the polymeric matrix. We expect an irreversible denaturation of GOx during the melting process at high temperatures as reported elsewhere.66,67 This fact together with the low enzyme concentration (1 wt%) most probably resulted in the embedment of GOx residues within the chitosan layer. Therefore, enzyme impurities most likely do not impart major time-dependent changes in the properties of the composites. In this scenario, the main interactions within the polymeric matrix most probably took place through the chitosan deposited over LNPs. In this sense, in the case of the PS-GOx-chi-LNPs system, such interactions are based on the cationic–π-interactions between the primary amine groups of chitosan and the aromatic ring of styrene,68 while in the case of the PBMA-GOx-chi-LNPs those are mainly based on hydrogen bonding interactions,22 explaining the more pronounced reinforced effect observed for PBMA compared to PS. These non-covalent interactions would play a crucial role as sacrificial bonds able to form new interactions during the deformation process, which ultimately could explain the positive reinforcing effect in our polymeric composites (Fig. 6). Such a positive reinforcing effect of the particles can also be associated with the role of LNPs as ball bearing lubricating agents and promoting effective interaction between chitosan and the polymeric matrix due to a high active surface area. Such a ball bearing lubrication effect was earlier proposed for LNP-CNF composites39 but not for lignin–polymer composites because of the non-spherical lignins used in the prior studies.
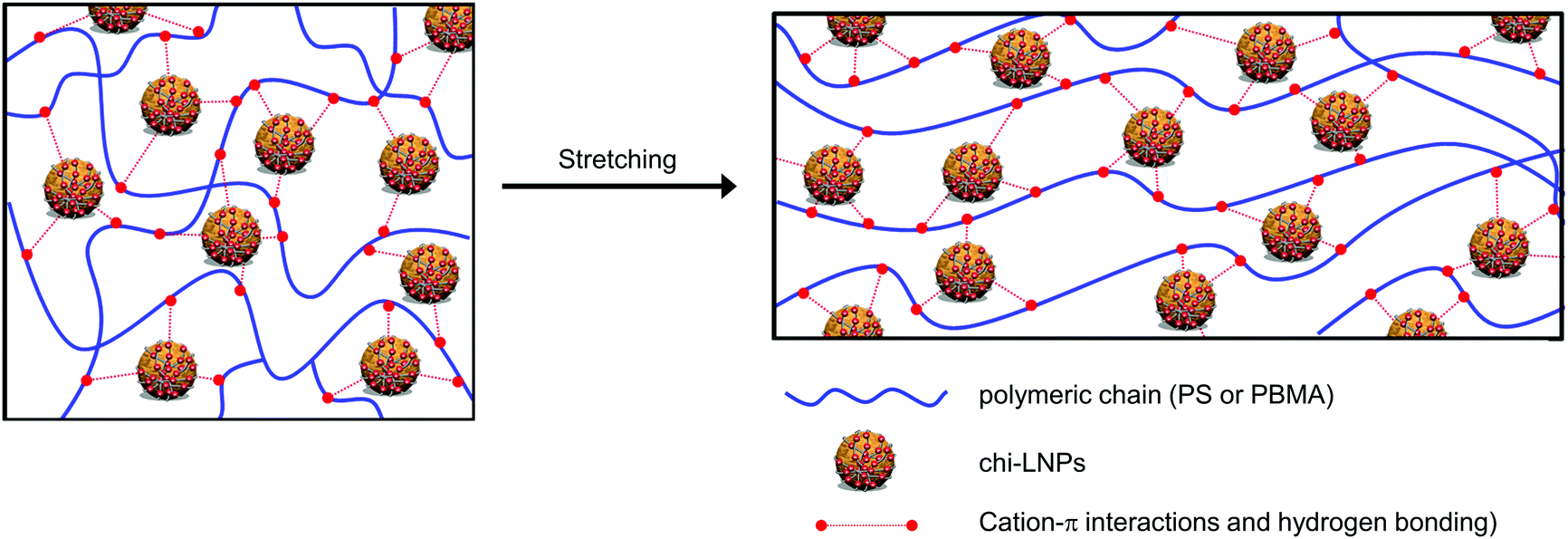 | ||
| Fig. 6 Schematic illustration of the proposed interactions between hybrid LNPs with polymeric chains before and after deformation in tensile testing. Note: image is not drawn to scale. | ||
UV-blocking and antioxidant activity of PBMA-GOx-chi-LNP composites
The inherent presence of phenolic hydroxyl groups in the aromatic rings of lignin equips the LNPs with well-known UV-blocking and antioxidant activity.69,70 In this sense, we also expected a beneficial performance from the introduction of GOx-chi-LNPs in our polymeric composites compared to pristine polymeric materials. It was of particular interest to determine the minimum amount of particulate fillers necessary to obtain efficient UV-blocking and antioxidant activity, and how that relates to the mechanical properties of the particulate nanocomposites. In this context, the UV-shielding potential of PBMA-GOx-chi-LNP-composite films was evaluated by the comparison of the UV-vis transmittance spectra of PBMA and the corresponding particulate reinforced composites (Fig. 7a). Regardless of the concentration of GOx-chi-LNPs within the polymeric matrixes, the PBMA-GOx-chi-LNP composite films revealed a strong absorption for both UV-B (280–315 nm) and UV-A (315–400 nm) regions compared to the absorption of only UV-C (200–280 nm) by the pristine PBMA (Fig. 7a). The transmittance values of the PBMA-GOx-chi-LNP composite films were close to zero in the entire UV wavelength region (200–400 nm), with the exception of PBMA-GOx-chi-LNPs2.5 showing a lower blockage in the UV-A region (16% transmittance). In the visible region (400–800 nm), the composite films showed a concentration-dependent optical transmittance. As expected from their color variation (Fig. 5g), increasing the loadings of GOx-chi-LNPs in the polymeric matrix decreased the optical transmittance of the films. However, it is important to highlight that a lower concentration of GOx-chi-LNP in the composites (i.e. 5 wt%) secured excellent UV-shielding behavior (3%), with only a moderate drop in visible light transmittance (40%). | ||
| Fig. 7 Optical and antioxidant properties of PBMA-GOx-chi-LNP composite films. (a) UV-vis light transmittance spectra of pure PBMA and PBMA-GOx-chi-LNP composite films. (b) Antioxidant activity kinetics of pure PBMA and PBMA-GOx-chi-LNP composite films. Reduction in the absorbance of ABTS˙+ radical solution was selected as reference. Error bars in (b) represent one standard deviation (SD) from the mean values (n = 2). | ||
The antioxidant activity of PBMA-GOx-chi-LNP composite films was evaluated by an ABTS˙+ radical scavenging assay that is adapted from a previous work.39 Kinetic experiments revealed a fast antioxidant activity within the first eight hours which thereafter slowly plateaued due to the depletion of ABTS˙+ and accessible GOx-chi-LNPs (Fig. 7b). The antioxidant activity of PBMA-GOx-chi-LNP composite films was estimated by monitoring and comparing the decrease in the UV-absorbance of ABTS˙+ solutions at different intervals of time and at different concentrations of PBMA-GOx-chi-LNPs. The results followed the same concentration-dependent trend as observed with UV-blocking activity (Fig. 7b). The antioxidant activity of pure PBMA was around 18% after 25 h while in the case of PBMA-GOx-chi-LNPs2.5 and PBMA-GOx-chi-LNPs5 antioxidant activities of 30% and 70% were determined, respectively. An increase in the amount of GOx-chi-LNPs to 10 wt% showed the quantitative ability of the composites to scavenge the ABTS˙+ radical (Fig. 7b and S10†). However, above this concentration threshold, no differences in the antioxidant activities were detected. This result could be associated with the increase of GOx-chi-LNPs in the inner part of the non-wettable polymeric matrix or even a layer by layer particle deposition on the surface of the hydrophobic polymer composites, since the antioxidant activity is postulated to occur mainly at the liquid–solid interface. Owing to its inherent radical scavenger ability, degradation of chitosan may occur upon exposure to active radicals, thereby affecting the mechanical properties of the composites. In order to investigate the stability of our composites as antioxidant materials, we performed additional tensile test assays on PBMA-GOx-chi-LNPs10 composite films after 24 h exposure (immersion) to ABTS˙+ radical solution (10 mM). The results showed a decrease in the tensile strength (31%) and toughness (37%) values compared to the values of the original sample before the antioxidant assay (Fig. S11†). This fact could be related to the partial degradation of chitosan, which affects the interaction with the polymeric matrix. However, regardless of the drop in mechanical strength, they still outperformed pure PBMA and no drastic drop in strength was observed. We speculate that these results can be attributed to two main factors: (i) lignin is the main radical scavenger and partially prevents the degradation of chitosan and (ii) as mentioned before the antioxidant process is expected to be limited to take place on the wettable surface of the composite film and therefore the inner part of the composite containing GOx-chi-LNPs is preserved. Finally, the effect of water should also be considered as the removal of some particles from the surface of the composite can take place during the immersion period.
Overall, our results are in agreement with previous reports that have demonstrated the benefit of combining lignin or LNPs with different polymeric matrixes to improve the aforementioned properties.39,49,71,72 However, it is important to highlight that the excellent UV-blocking and antioxidant activity performance of our polymer–GOx-chi-LNP composites not only envision potential application in a wide range of different areas (e.g. specific packaging materials) but are also expected to equip protection against the UV-degradation of the vinyl polymeric matrix at long-term exposure periods,73,74 and therefore preserve the mechanical properties which ultimately determine the lifetime of this type of materials.
Green metrics for biocatalytic Pickering emulsion polymerization process
As stated previously, the fabrication of lignin–polymer composites requires in most cases the chemical functionalization of lignin to improve its compatibility or increase its reactivity towards monomers/polymers.31–34 These classical chemical modifications usually require toxic chemical compounds such as anhydrides or acyl halides, together with the use of harmful organic solvents (dimethylformamide or tetrahydrofuran among others). Such steps are not necessarily environmentally benign when it comes to the preparation of lignin–polymer composites, and therefore limit scalability. In this sense, here it is important to highlight that our method avoids the use of chemical functionalization and uses water as the main solvent. To evaluate in detail the sustainability of our process, we analysed the green metrics through the calculation of the environmental factor (E-factor) according to Sheldon75 (eqn (1)), traditionally applied to give an idea of the waste generated during a chemical process.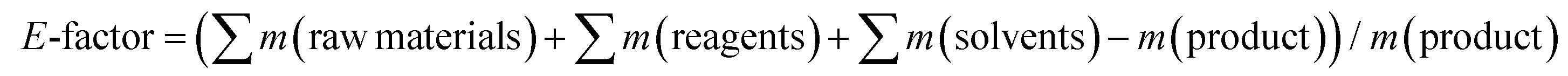 | (1) |
For comparative purposes, two representative procedures to obtain lignin–polymer composites, involving chemical functionalization of lignin were also analyzed (Fig. 8).31,32 Note that lignin–polymer composites obtained by directly blending lignin with polymers were excluded from this comparison, as, despite their likely lower E-factor values, the resulting composites have shown poor mechanical performance due to their incompatibility within the polymeric matrix.24,29 The two-step process involving the preparation of GOx-chi-LNPs and the Pickering emulsion polymerization had an overall E-factor of 0.2–0.5 for the first step (preparation of the hybrid particles) and 5–5.0 for the second step (polymerization). The entire two-step process gives an overall E-factor of 5.2–6.0, which is 80% lower than the alternative routes involving the chemical functionalization of lignin as depicted in Fig. 8. Noteworthy, the production of wastewater containing halogenated compounds or reactive anhydride species produced during chemical functionalization of lignin, which most probably would increase the E-factor values, was not taken into account in these calculations. Therefore, our route not only circumvents traditional problems in the preparation of lignin–polymer composites but also emerges as a promising alternative route that satisfies green chemistry principles.
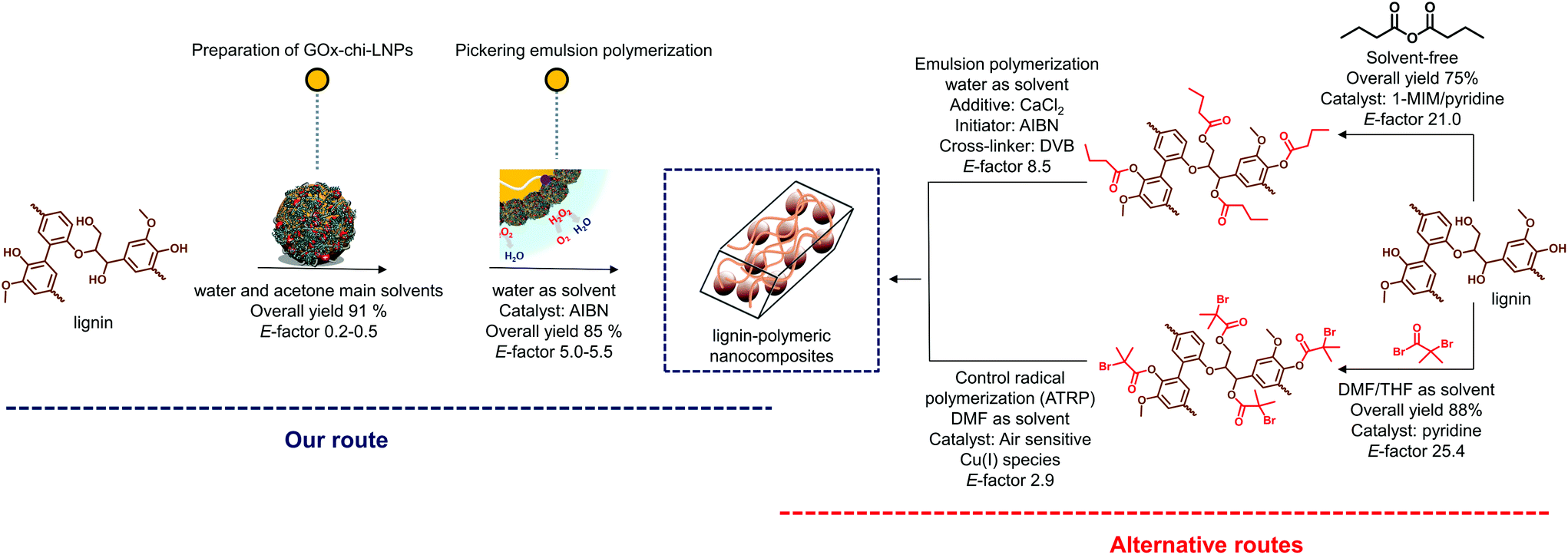 | ||
| Fig. 8 Green metrics of the biocatalytic Pickering emulsion polymerization process to produce lignin–polymer composites in comparison with alternative routes involving chemical functionalization of lignin. | ||
The recyclability and biodegradability of plastic polymers are crucial aspects of a circular economy. In this sense, it is important to note here that recycling of our lignin–polymer composites by means of separation of the synthetic polymer from hybrid GOx-chi-LNPs most probably could be a hard task, involving basic extraction to solubilize the bio-components. However, owing to the thermoplastic nature of vinyl polymers employed in this study, recycling by melting offers a possibility to reuse these materials without affecting the homogeneity of GOx-chi-LNPs, as we already demonstrated during the preparation of the films at high temperatures or by melt-processing an original specimen after tensile testing (Fig. S12†). The biodegradability of the system would remain a challenge since the incorporation of hybrid GOx-chi-LNPs would not affect the degradation of the synthetic part of the polymer. Hence, future works could benefit from this approach to open new avenues in the synthesis of other lignin–polymeric materials such as polyesters that would have a more sustainable impact through complete biodegradation in the environment.
Conclusions
In the present work, we have identified and exploited polymeric-latex dispersions produced by enzyme-degassed free radical polymerization in aqueous Pickering emulsion media stabilized by hybrid lignin particles (GOx-chi-LNPs) to prepare melt-casted hydrophobic polymeric composites incorporating hybrid LNPs as fillers. The kinetic polymerization experiments proved the efficient biocatalytic activity of the particles even at high temperatures, allowing for the polymerization reactions to proceed in the presence of air. An SEM analysis of GOx-chi-LNP–polymeric nanocomposites revealed the excellent dispersion of GOx-chi-LNP particles avoiding agglomeration even at a high particle loading of up to 30 wt%. It is worthy to note that our approach demonstrates unequivocally the possibility to prepare renewable-sourced particulate composites that significantly improve mechanical properties even at low and high contents of hybrid LNPs and also equip them with multifunctional properties such as UV-blocking and antioxidant activity that might be useful in specific packaging applications such as healthcare and medical devices. Overall, the approach presented in this work represents a new way to prepare lignin–hydrophobic polymer composites by overcoming the earlier challenges such as difficult blending of raw lignins or tedious functionalization steps to improve the compatibility of lignin within polymeric matrixes. Finally, we also hold a view that our system could be easily applied to the polymerization of bio-sourced vinyl monomers,76–78 and therefore will open and encourage renewed efforts to develop nanocomposites with multifunctional properties and a more favorable carbon footprint by exploiting the inherent beneficial properties of LNPs as building blocks.Experimental details
Materials
All lignin materials prepared in this work (LNPs, chi-LNPs and GOx-chi-LNPs) were prepared from BIOPIVA™ 100 pine kraft lignin (UPM, Finland), previously characterized.48 Chitosan from shrimp shells (>75% deacetylated, 50.000–190.000 Da) was purchased from Sigma Aldrich. Glucose oxidase (GOx) from Aspergillus niger was purchased from Sigma Aldrich as lyophilized powder and stored at −20 °C. All the chemicals and solvents were purchased from Sigma-Aldrich, Fischer and VWR and were used as received unless noted. Butyl methacrylate (BMA) and styrene (S) were passed through a short basic alumina column to remove the inhibitor before the polymerization reactions. Azobisisobutyronitrile (AIBN) was recrystallized in methanol before the polymerization reactions.Preparation of colloidal lignin nanoparticles (LNPs, chi-LNPs and GOx-chi-LNPs)
All colloidal lignin nanoparticles used in this work originated from the same batch as previously characterized and described.57 Briefly, the synthesis included the dissolution of kraft lignin in an acetone/water mixture (mass ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), removal of insoluble impurities by filtration, and production of LNPs by rapidly pouring deionized water to lignin solution followed by the rotary evaporation of acetone. The final aqueous dispersion of LNPs (0.8 wt%) was obtained after filtration with a lignin mass yield of 86%. The chitosan-coated LNPs (chi-LNPs) were prepared by adding the dispersion of LNPs under vigorous stirring into a 0.1 wt% chitosan solution. The ratio of chitosan to LNPs was 100 mg g−1. The GOx-chi-LNPs were produced by the addition of a GOx solution (sodium acetate buffer, 0.1 M; pH 5) to chi-LNP dispersion under orbital shaking at room temperature for 2 h. Then, GOx-chi-LNPs were recovered after three centrifugation/re-dispersion cycles (3000 rpm, 10 min), replacing each decanted supernatant with sodium acetate buffer (0.1 M; pH 5). The ratio of GOx to chi-CLPs was 10 mg g−1. The particle size and zeta potential of LNPs, chi-LNPs and GOx-chi-LNPs were measured using a Zetasizer Nano ZS (Malvern, UK). The zeta potential was determined using a dip cell probe. LNPs, chi-LNPs and GOx-chi-LNPs were diluted by a factor of 30 with deionized water and sodium acetate buffer (0.1 M; pH 4 and 5.5), respectively, before the analysis of particle size. The Scanning Electron Microscopy (SEM) images were recorded on a JEOL JSM-7401F (JEOL Ltd, Japan) operating at 2–5 kV. The colloidal dispersions of lignin nanoparticles (LNPs, chi-LNPs and GOx-chi-LNPs) were previously diluted by a factor of 1:40, followed by the deposition and evaporation of one droplet into a silicon wafer matrix for the SEM investigation.
:1), removal of insoluble impurities by filtration, and production of LNPs by rapidly pouring deionized water to lignin solution followed by the rotary evaporation of acetone. The final aqueous dispersion of LNPs (0.8 wt%) was obtained after filtration with a lignin mass yield of 86%. The chitosan-coated LNPs (chi-LNPs) were prepared by adding the dispersion of LNPs under vigorous stirring into a 0.1 wt% chitosan solution. The ratio of chitosan to LNPs was 100 mg g−1. The GOx-chi-LNPs were produced by the addition of a GOx solution (sodium acetate buffer, 0.1 M; pH 5) to chi-LNP dispersion under orbital shaking at room temperature for 2 h. Then, GOx-chi-LNPs were recovered after three centrifugation/re-dispersion cycles (3000 rpm, 10 min), replacing each decanted supernatant with sodium acetate buffer (0.1 M; pH 5). The ratio of GOx to chi-CLPs was 10 mg g−1. The particle size and zeta potential of LNPs, chi-LNPs and GOx-chi-LNPs were measured using a Zetasizer Nano ZS (Malvern, UK). The zeta potential was determined using a dip cell probe. LNPs, chi-LNPs and GOx-chi-LNPs were diluted by a factor of 30 with deionized water and sodium acetate buffer (0.1 M; pH 4 and 5.5), respectively, before the analysis of particle size. The Scanning Electron Microscopy (SEM) images were recorded on a JEOL JSM-7401F (JEOL Ltd, Japan) operating at 2–5 kV. The colloidal dispersions of lignin nanoparticles (LNPs, chi-LNPs and GOx-chi-LNPs) were previously diluted by a factor of 1:40, followed by the deposition and evaporation of one droplet into a silicon wafer matrix for the SEM investigation.
Preparation and polymerization of GOx-chi-LNP stabilized hydrophobic monomer-in-water Pickering emulsions
This procedure is representative of all the emulsions and polymerization experiments conducted herein. The emulsions were prepared by gradually adding a monomer (oil phase) containing the initiator (2 wt%) to a colloidal water dispersion of GOx-chi-LNPs containing glucose (200 mg, 0.14 M). The final fraction of oil/water was fixed at 20/80 v/v and the total volume of the emulsion was 10 mL. The final concentration of GOx-chi-LNPs was fixed depending on the experiment. Then emulsification was performed by sonication for 120 s in a BioBlock Vibra-Cell equipped with an ultrasonic tip with cooling in an ice bath (10 s on and 5 s off at 40% of amplitude power). Polymerizations were conducted by heating the Pickering emulsions stabilized by GOx-chi-LNPs at 65 °C in a thermostatic bath for 12 h. In the case of kinetic experiments, the samples were withdrawn periodically for monomer conversion by gravimetric analysis. After polymerization, the resulting lignin-coated polymeric microparticles were purified by three centrifugation/re-dispersion cycles, replacing each decanted supernatant with ammonium hydroxide solution (NH4OH, 35%), followed by drying of the purified polymer samples at 45 °C for 12 h prior to the GPC analysis. To purify the polymeric microparticles without removing the GOx-chi-LNPs from the microparticle surface, deionized water was used instead of the basic washing solution. An optical microscope analysis of the Pickering emulsions and latex dispersions was carried out using a Nikon (Alphaphot2) optical microscope. The emulsions and latex dispersions were previously diluted by adding one drop of the sample in 1 mL of deionized water. The ImageJ software was used to process the recorded images and calculate the Sauter diameter (D3,2) and uniformity (U) based on 50 measurements for each sample. The SEM images were obtained from the latex dispersions and polymeric microparticles, after drying the samples on a glass substrate and sputter-coating them with a thin layer of gold to prevent sample charging effects. The GPC analyses of the polymer samples were performed using an Agilent 1100 series system equipped with two gel columns (a guard column, 500 Å and PLgel 5 μm, 104 Å from Phenomenex) and an Agilent 1100 series refractive-index factor (RI) as a detector. THF (Fischer, HPLC grade) was used as an eluent at a flow rate of 1 mL min−1. The calibration curves for GPC analysis were obtained with polystyrene standards purchased from Polymer Standards Service (PSS) GmbH. The molecular weights were calculated using the universal calibration principle based on the Mark–Houwink parameters. The thermogravimetric analysis (TGA) of bare polymeric particles and GOx-chi-LNP coated polymeric particles was performed using a Discovery TG (TA Instruments, USA) under 50 mL min−1 nitrogen flow and at a heating rate of 10 °C min−1.Preparation of GOx-chi-LNP–polymer composites
This procedure is representative of all the GOx-chi-LNP nanocomposites produced herein. The preparation of PS-GOx-chi-LNPs2.5 is described as the representative example: a latex dispersion of PS (2 g approximately) stabilized with GOx-chi-LNPs was purified as described above and melted at 160 °C in 6 h. Then the temperature was gradually decreased to 50 °C. Afterwards, the obtained PS-GOx-chi-LNP composite film was stored at room temperature. For mechanical property analysis of GOx-chi-LNP–polymer composites, purified latex dispersions were placed in rectangular Teflon molds (0.5 × 3 cm) and subject to the same melting process described above in order to obtain regular and well-shaped rectangular specimens. The mechanical properties of the polymer–GOx-chi-LNP composites were measured using an Instron 5960 universal testing machine (Instron, USA) equipped with a 100 N loaded cell at a strain rate of 1 mm min−1 in the case of PS composites and 8 mm min−1 in the case of PBMA composites. The mechanical measurements were performed on rectangular shaped composites with dimensions of 0.5 cm × 3 cm using a gauge length of 1 cm. The specimens were conditioned 24 h prior to the measurement and measured at 50% relative humidity (RH) and 25 °C. The Young's modulus was calculated from the slope of the linear part of the stress–strain curve and the toughness was determined from the area under the stress–strain curve up to the fracture point. The results are reported as mean values ± standard deviation of a minimum of five samples.Optical and antioxidant properties of PBMA-GOx-chi-LNP composite films
The transmittance of PBMA and PBMA-GOx-chi-LNP composite films were recorded on a UV-vis spectrometer (PerkinElmer Lambda 19 UV/VIS/NIR spectrometer) in the range of 200–800 nm using air as background. The average thickness of the films was 500 μm. The antioxidant activity assay was adapted from Farooq et al.39 Briefly, a freshly prepared ABTS˙+ radical solution was diluted (1:60) until it reaches an absorbance of 0.8 at 734 nm at 25 °C. Then, rectangular specimens of PBMA-GOx-chi-LNP films (300–350 mg) were immersed in vials containing 3 mL of ABTS˙+ radical solution and agitated in a thermostatic orbital shaker at 25 °C. The antioxidant radical scavenger activity was tracked by monitoring the time evolution of the absorbance of the ABTS˙+ radical solution at 734 nm.
Author contributions
A. M. and M. H. S. conceived the idea and designed the experiments. A. M. performed the experiments and analyzed the data with inputs from M. H. S. J. L. assisted with mechanical characterization and SEM imaging. M. M. performed thermal analyses and designed the molding stage for mechanical testing. A. M. and M. H. S. co-wrote the manuscript. All the authors discussed the results commented on the manuscript and have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Vetenskapsrådet (grant number 2020-03752) and the Department of Materials and Environmental Chemistry (MMK) for financing this work. The authors also acknowledge Dr Kjell Jansson for helpful discussions and assistance with SEM imaging and Prof. Lennart Bergström and Dr Claudia Möckel for giving access to ultrasonication and GPC instrumentation, respectively. The authors thank Dr Konstantin Kriechbaum for his assistance with mechanical characterization.Notes and references
- R. Gangopadhyay and A. De, Chem. Mater., 2000, 12, 608–622 CrossRef CAS.
- J. Song, P. Huang, H. Duan and X. Chen, Acc. Chem. Res., 2015, 48, 2506–2515 CrossRef CAS PubMed.
- T. F. Jaramillo, S.-H. Baeck, B. R. Cuenya and E. W. McFarland, J. Am. Chem. Soc., 2003, 125, 7148–7149 CrossRef CAS PubMed.
- Z. Liu and Y. Hu, ACS Appl. Mater. Interfaces, 2016, 8, 21666–21673 CrossRef CAS PubMed.
- H. Wu, W. P. Fahy, S. Kim, H. Kim, N. Zhao, L. Pilato, A. Kafi, S. Bateman and J. H. Koo, Prog. Mater. Sci., 2020, 111, 100638 CrossRef CAS.
- D. K. Rajak, D. D. Pagar, R. Kumar and C. I. Pruncu, J. Mater. Res. Technol., 2019, 8, 6354–6374 CrossRef CAS.
- V. V. Kumar, G. Balaganesan, J. K. Y. Lee, R. E. Neisiany, S. Surendran and S. A. Ramakrishna, Polymer, 2019, 11, 644 Search PubMed.
- B. Ates, S. Koytepe, A. Ulu, C. Gurses and V. K. Thakur, Chem. Rev., 2020, 120, 9304–9362 CrossRef CAS PubMed.
- H. Ma, M. Luo, S. Sanyal, K. Rege and L. L. Dai, Materials, 2010, 3, 1186–1202 CrossRef CAS.
- K. Chen, S. Zhou, S. Yang and L. Wu, Adv. Funct. Mater., 2015, 25, 1035–1041 CrossRef CAS.
- X. Zhang, Y. Sun, Y. Mao, K. Chen, Z. Cao and D. Qi, RSC Adv., 2018, 8, 3910–3918 RSC.
- S. Cauvin, P. J. Colver and S. A. F. Bon, Macromolecules, 2005, 38, 7887–7889 CrossRef CAS.
- S. A. F. Bon and P. J. Colver, Langmuir, 2007, 23, 8316–8322 CrossRef CAS PubMed.
- X. Song, Y. Yang, J. Liu and H. Zhao, Langmuir, 2011, 27, 1186–1191 CrossRef CAS PubMed.
- G. Yin, Z. Zheng, H. Wang, Q. Du and H. Zhang, J. Colloid Interface Sci., 2013, 394, 192–198 CrossRef CAS PubMed.
- Y. J. Kim, Y. D. Liu, Y. Seo and H. J. Choi, Langmuir, 2013, 29, 4959–4965 CrossRef CAS PubMed.
- C. Wang, C. Zhang, Y. Li, Y. Chen and Z. Tong, React. Funct. Polym., 2009, 69, 750–754 CrossRef CAS.
- A. Werner, V. Schmitt, G. Sèbe and V. Héroguez, Polym. Chem., 2017, 8, 6064–6072 RSC.
- A. Werner, G. Sèbe and V. Héroguez, Polym. Chem., 2018, 9, 5043–5050 RSC.
- S. Fujisawa, E. Togawa and K. Kuroda, Biomacromolecules, 2017, 18, 266–271 CrossRef CAS PubMed.
- A. Werner, V. Schmitt, G. Sèbe and V. Héroguez, Biomacromolecules, 2019, 20, 490–501 CrossRef CAS PubMed.
- D. W. Kim, J. Shin and S. Q. Choi, Carbohydr. Polym., 2020, 247, 116762 CrossRef CAS PubMed.
- E. Limousin, R. Rafaniello, T. Schäfer, N. Ballard and J. M. Asua, Langmuir, 2020, 36, 2052–2062 CrossRef CAS PubMed.
- M. R. Barzegari, A. Alemdar, Y. Zhang and D. Rodrigue, Polym. Compos., 2012, 33, 353–361 CrossRef CAS.
- P. Pérez-Guerrero, J. Lisperguer, J. Navarretem and D. Rodrigue, BioResources, 2014, 9, 6514–6526 CrossRef.
- H. Jeong, J. Park, S. Kim, J. Lee and J. W. Cho, Fibers Polym., 2012, 13, 1310–1318 CrossRef CAS.
- V. Romhányi, D. Kun and B. Pukánszky, ACS Sustainable Chem. Eng., 2018, 6, 14323–14331 CrossRef.
- G. Szabó, V. Romhányi, D. Kun and B. Pukánszky, ACS Sustainable Chem. Eng., 2017, 5(1), 410–419 CrossRef.
- W. El-Zawawy, M. M. Ibrahim, M. N. Belgacem and A. Dufrense, Mater. Chem. Phys., 2011, 131, 348–357 CrossRef CAS.
- R. Pucciariello, V. Villani, C. Bonini, M. D′auria and T. Vetere, Polymer, 2004, 45, 4159–4169 CrossRef CAS.
- Z. Zhang, A. Mulyadi, X. Kuang, W. Liu, V. Li, P. Gogoi, X. Liu and Y. Deng, Polym. Eng. Sci., 2019, 59, 964–972 CrossRef CAS.
- S. L. Hilburg, A. E. Elder, H. Chung, R. L. Ferebee, M. R. Bockstaller and N. R. Washburn, Polymer, 2014, 4, 995–1003 CrossRef.
- K. S. Silmore, C. Gupta and N. R. Washburn, J. Colloid Interface Sci., 2016, 466, 91–100 CrossRef CAS PubMed.
- L. Dai, Y. Li, F. Kong, K. Liu, C. Si and Y. Ni, ACS Sustainable Chem. Eng., 2019, 7, 13497–13504 CrossRef CAS.
- M. Österberg, M. H. Sipponen, B. D. Mattos and O. J. Rojas, Green Chem., 2020, 22, 2712–2733 RSC.
- M. H. Sipponen, H. Lange, C. Crestini, A. Henn and M. Österberg, ChemSusChem, 2019, 12, 2039–2054 CrossRef CAS PubMed.
- A. Moreno and M. H. Sipponen, Mater. Horiz., 2020, 7, 2237–2257 RSC.
- S. Iravani and R. S. Varma, Green Chem., 2020, 22, 612–636 RSC.
- M. Farooq, T. Zou, G. Riviere, M. H. Sipponen and M. Österberg, Biomacromolecules, 2019, 20, 693–704 CrossRef CAS PubMed.
- O. Cusola, O. J. Rojas and M. B. Roncero, ACS Appl. Mater. Interfaces, 2019, 11, 45226–45236 CrossRef CAS PubMed.
- X. Wang, S. L. Ji, X. Q. Wang, H. Y. Bian, L. R. Lin, H. Q. Dai and H. Xiao, J. Mater. Chem. C, 2019, 7, 14159–14169 RSC.
- T. Ju, Z. Zhang, Y. Li, X. Miao and J. Ji, RSC Adv., 2019, 9, 24915–24921 RSC.
- P. Posoknistakul, C. Tangkrakul, P. Chaosuanphae, S. Deepentham, W. Techasawong, N. Phonphirunrot, S. Bairak, C. Sakdaronnarong and N. Laosiripojana, ACS Omega, 2020, 5, 20976–20982 CrossRef CAS PubMed.
- W. Yang, J. S. Owczarke, E. Fortunati, M. Kozanecki, A. Mazzaglia, G. M. Balestra, J. M. Kenny, L. Torre and D. Puglia, Ind. Crops Prod., 2016, 94, 800–811 CrossRef CAS.
- W. Wang, E. Fortunati, F. Dominici, G. Giovanale, A. Mazzaglia, G. M. Balestra, J. M. Kenny and D. Puglia, Eur. Polym. J., 2016, 79, 1–12 CrossRef.
- M. H. Sipponen, H. Lange, M. Ago and C. Crestini, ACS Sustainable Chem. Eng., 2018, 6, 9342–9351 CrossRef CAS PubMed.
- N. Chen, L. A. Dempere and Z. Tong, ACS Sustainable Chem. Eng., 2016, 4, 5204–5211 CrossRef CAS.
- M. H. Sipponen, M. Farooq, J. Koivisto, A. Pellis, J. Seitsonen and M. Österberg, Nat. Commun., 2018, 9, 2300 CrossRef PubMed.
- D. Piccinino, E. Capecchi, L. Botta, P. Bollella, R. Antiochia, M. Crucianelli and R. Saladino, Catal. Sci. Technol., 2019, 9, 4125–4134 RSC.
- E. Capecchi, D. Piccinino, E. Tomaino, B. M. Bizzarri, F. Polli, R. Antiochia, F. Mazzei and R. Saladino, RSC Adv., 2020, 10, 29031–29042 RSC.
- E. Capecchi, D. Piccinino, B. M. Bizzarri, D. Avitabile, C. Pelosi, C. Colantonio, G. Calabrò and R. Saladino, Biomacromolecules, 2019, 20, 1975–1988 CrossRef CAS PubMed.
- G. N. Rivière, A. Korpi, M. H. Sipponen, T. Zou, M. A. Kostiainen and M. Österberg, ACS Sustainable Chem. Eng., 2020, 8, 4167–4177 CrossRef PubMed.
- M. H. Sipponen, M. Smyth, T. Leskinen, L. S. Johansson and M. Österberg, Green Chem., 2017, 19, 5831–5840 RSC.
- T. Zou, M. H. Sipponen and M. Österberg, Front. Chem., 2019, 7, 370 CrossRef CAS PubMed.
- Z. Wei, Y. Yang, R. Yang and C. Wang, Green Chem., 2012, 14, 3230–3236 RSC.
- Y. Wang, X. Li, C. Shen, Z. Mao, H. Xu, Y. Zhong, X. Sui, X. Feng and B. Wang, Int. J. Biol. Macromol., 2020, 146, 1–8 CrossRef CAS PubMed.
- A. Moreno and M. H. Sipponen, Nat. Commun., 2020, 11, 5599 CrossRef CAS PubMed.
- S. Arditty, C. P. Whitby, B. P. Binks, V. Schmitt and F. Leal-Calderon, Eur. Phys. J. E, 2003, 11, 273–281 CrossRef CAS PubMed.
- S. B. Haaj, W. Thielemans, A. Magnin and S. Boufi, ACS Appl. Mater. Interfaces, 2014, 6, 8263–8273 CrossRef CAS PubMed.
- T. Chen, P. J. Colver and S. A. F. Bon, Adv. Mater., 2007, 19, 2286–2289 CrossRef CAS.
- P. J. Colver, C. A. L. Colard and S. A. F. Bon, J. Am. Chem. Soc., 2008, 130, 16850–16851 CrossRef CAS PubMed.
- A. Lotierzo and S. A. F. Bon, Polym. Chem., 2017, 8, 5100–5111 RSC.
- K. S. Mikkonen, Green Chem., 2020, 22, 1019–1037 RSC.
- B. M. Degner, C. Chung, V. Schlegel, R. Hutkins and D. J. Mcclements, Compr. Rev. Food Sci. Food Saf., 2014, 13, 98–113 CrossRef CAS PubMed.
- N. Bensabeh, A. Moreno, A. Roig, M. Rahimzadeh, K. Rahimi, J. C. Ronda, V. Cádiz, M. Galià, V. Percec, C. Rodriguez-Emmenegger and G. Lligadas, ACS Sustainable Chem. Eng., 2020, 8, 1276–1284 CrossRef CAS.
- G. Zoldák, A. Zubrik, A. Musatov, M. Stupák and E. Sedlák, J. Biol. Chem., 2004, 279, 47601–47609 CrossRef PubMed.
- N. Balistreri, D. Gaboriau, C. Jolivat and F. Launay, J. Mol. Catal. B: Enzym., 2016, 127, 26–33 CrossRef CAS.
- W. Jiang, X. Chen, B. Pan, Q. Zhang, L. Teng, Y. Chen and L. Liu, J. Hazard. Mater., 2014, 276, 295–301 CrossRef CAS PubMed.
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS.
- P. Figueiredo, K. Lintinen, J. T. Hirvonen, M. A. Kostiainen and H. A. Santos, Prog. Mater. Sci., 2018, 93, 233–269 CrossRef CAS.
- X. You, X. Wang, H. J. Zhang, K. Cui, A. Zhang, L. Wang, C. Yadav and X. Li, ACS Appl. Mater. Interfaces, 2020, 12, 39892–39901 CrossRef CAS PubMed.
- K. Kriechbaum and L. Bergström, Biomacromolecules, 2020, 21, 1720–1728 CrossRef CAS PubMed.
- G. Gomes de Castro Monsores, A. Oliveira da Silva, S. de Sant’ Ana Oliveira, J. G. Passos-Rodrigues and R. Pondé-Weber, J. Mater. Res. Technol., 2019, 8, 3713–3718 CrossRef.
- R. Shanti, A. N. Hadi, Y. S. Salim, S. Y. Chee, S. Ramesh and K. Ramesh, RSC Adv., 2017, 7, 112–120 RSC.
- R. A. Sheldon, Chem. Ind., 1992, 903 CAS.
- A. Moreno, N. Bensabeh, J. Parce, J. C. Ronda, V. Cádiz, M. Galià, L. Vares, G. Lligadas and V. Percec, Biomacromolecules, 2019, 20, 3200–3210 CrossRef CAS PubMed.
- N. Bensabeh, A. Moreno, A. Roig, O. R. Monaghan, J. C. Ronda, V. Cádiz, M. Galià, S. M. Howdle, G. Lligadas and V. Percec, Biomacromolecules, 2019, 20, 2135–2147 CrossRef CAS PubMed.
- C. Veith, F. Diot-Néant, S. A. Miller and F. Allais, Polym. Chem., 2020, 11, 7452–7470 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc00103e |
| This journal is © The Royal Society of Chemistry 2021 |