
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRoom temperature iron catalyzed transfer hydrogenation using n-butanol and poly(methylhydrosiloxane)†
Thomas G.
Linford-Wood
 ,
Nathan T.
Coles
and
Ruth L.
Webster
*
,
Nathan T.
Coles
and
Ruth L.
Webster
*
Department of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: r.l.webster@bath.ac.uk
First published on 11th March 2021
Abstract
Reduction of carbon–carbon double bonds is reported using a three-coordinate iron(II) β-diketiminate pre-catalyst. The reaction is believed to proceed via a formal transfer hydrogenation using poly(methylhydrosiloxane), PMHS, as the hydride donor and a bio-alcohol as the proton source. The reaction proceeds well using n-butanol and ethanol, with n-butanol being used for substrate scoping studies. Allyl arene substrates, styrenes and aliphatic substrates all undergo reduction at room temperature. Unfortunately, clean transfer of a deuterium atom using D-alcohol does not take place, indicating a complex catalytic mechanism. However, changing the deuterium source to D-aniline gives close to complete regioselectivity for mono-deuteration of the terminal position of the double bond. Finally, we demonstrate that efficient dehydrocoupling of alcohol and PMHS can be undertaken using the same pre-catalyst, giving high yields of H2 within 30 minutes at room temperature.
Introduction
The implementation and manipulation of carbon–carbon double bonds is of fundamental importance in organic synthesis. The reduction of unsaturated carbon–carbon bonds is one such transformation. This chemistry is dominated by transition metal-mediated reduction in the presence of H2.1,2 There are countless examples of homogeneous and heterogeneous catalysts undertaking the reduction of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, under a range of reaction conditions3,4 and H2 pressures. An alternative to the use of a gaseous reductant is the use of transfer hydrogenation (TH), which is traditionally undertaken using the conditions developed by Meerwein, Ponndorf and Verley in the 1920s.5,6 In order to develop the original chemistry for application in CC reduction, there have been many adaptations, including alternative catalysts7,8 to the traditional aluminium alkoxide system, chiral variants9 and a move away from isopropanol (IPA) as the hydrogen atom source in TH. Although H2 gas is impossible to compete with in terms of atom economy, the use of TH reagents,10–12 such as IPA or other alcohols, amines, boranes and silanes proffer the opportunity to develop chemistry that does not rely on gas handling, develop detailed mechanistic understanding13,14 that may not be viable if high pressures of H2 are required and, our own specific area of interest, developing chemistry that allows selective functionalization. In the case of TH, with an appropriate catalyst or substrate or TH reagent, regioselective mono-deuteration is feasible: this is simply not possible using D2. Since some of the earliest studies on Sn-catalyzed TH of activated functional groups (aldehydes and ketones) in refluxing EtOH, and alkene and nitro group reduction over Pd/C under similar conditions,15 much of the research in the area of TH mediated by alcohols and silanes is dominated by copper catalysis,16 in particular alkyne semi-hydrogenation,17,18 whereas iron-catalyzed transformations have relied on H2-release from a silane19 or formic acid.20
C bonds, under a range of reaction conditions3,4 and H2 pressures. An alternative to the use of a gaseous reductant is the use of transfer hydrogenation (TH), which is traditionally undertaken using the conditions developed by Meerwein, Ponndorf and Verley in the 1920s.5,6 In order to develop the original chemistry for application in CC reduction, there have been many adaptations, including alternative catalysts7,8 to the traditional aluminium alkoxide system, chiral variants9 and a move away from isopropanol (IPA) as the hydrogen atom source in TH. Although H2 gas is impossible to compete with in terms of atom economy, the use of TH reagents,10–12 such as IPA or other alcohols, amines, boranes and silanes proffer the opportunity to develop chemistry that does not rely on gas handling, develop detailed mechanistic understanding13,14 that may not be viable if high pressures of H2 are required and, our own specific area of interest, developing chemistry that allows selective functionalization. In the case of TH, with an appropriate catalyst or substrate or TH reagent, regioselective mono-deuteration is feasible: this is simply not possible using D2. Since some of the earliest studies on Sn-catalyzed TH of activated functional groups (aldehydes and ketones) in refluxing EtOH, and alkene and nitro group reduction over Pd/C under similar conditions,15 much of the research in the area of TH mediated by alcohols and silanes is dominated by copper catalysis,16 in particular alkyne semi-hydrogenation,17,18 whereas iron-catalyzed transformations have relied on H2-release from a silane19 or formic acid.20
We recently reported on the ability of a well-defined iron(II) β-diketiminate complex (1) to undertake transfer hydrogenation of unactivated carbon–carbon double bonds (Scheme 1a).23 The reaction is rapid and mild; proceeding smoothly at room temperature within minutes. Although an elegant method to reduce unsaturated systems and regioselectively introduce a single deuterium atom, our initial disclosure relies on benzene solvent (with C6D6 used for ease of in situ analysis), pinacol borane (HBpin) as a hydride source and an amine (such as PhNH2 or nBuNH2) as a proton source. We felt that the sustainability credentials could be readily improved by moving to the use of sustainable hydride and proton sources. Early indicators from our published work suggested that a change in proton source would be viable, since we had some positive TH results using o-allyl-phenol and propargyl alcohol as proton-containing substrates. Ideally, the reaction would also not rely on C6D6 as the reaction solvent. We herein report on our pursuit of a more benign transfer hydrogenation methodology.
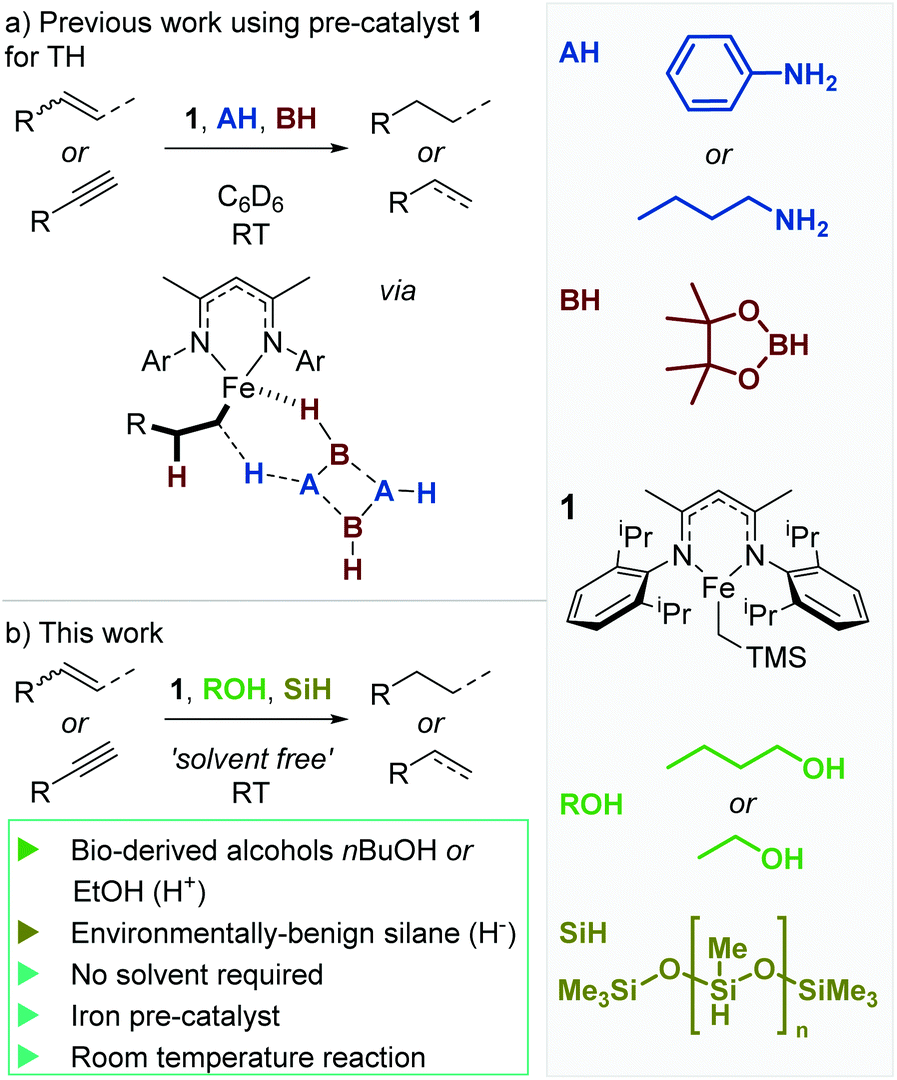 | ||
| Scheme 1 (a) Previous work on TH using pre-catalyst 1 was fast and mild, with reactions complete in minutes at RT. However, the chemistry relied on non-renewable reagents: HBpin and amines such as aniline or n-butylamine. (b) This work focuses on the development of a more sustainable and environmentally benign route to alkene TH using alcohols and poly(methylhydrosiloxane) (PMHS, a waste product from the silicone industry21,22). | ||
Results and discussion
We initiated our studies by optimizing the TH of allylbenzene using poly(methylhydrosiloxane) (PMHS) as the hydride donor (Table 1). PMHS has been used extensively with iron pre-catalysts to undertake hydrosilylation24 of carbonyl compounds, followed by hydrolysis to give the free alcohol,25–34 in the reduction of nitriles35 and amides36,37 in dehydroxylation38 or tandem hydrosilylation/isomerization.39 Enthaler has reported elegant examples of alkene synthesis via alkyne reduction mediated by silanes.40,41 However, application of PMHS in TH of carbon–carbon bonds42–45 is, to the best of our knowledge, unreported. Gratifyingly, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio of allylbenzene:PhNH2:PMHS gives good conversion and spectroscopic yield to propylbenzene at RT using 10 mol% 1 (entry 1). Increasing the loading of PMHS further increases the spectroscopic yield, with 3 equivalents of PMHS generating a quantitative amount of propylbenzene. In our search for a greener and safer alternative to C6D6, we found that key renewable solvents such as 2-methyl-THF and cyclopentyl methyl ether (CPME) lead to a drastic reduction in yield (entries 7 and 8). However, solvent free conditions, where PMHS is used to solubilize the other reagents, gives good yield of propylbenzene at both 10 mol% and 5 mol% loading of 1 (entries 9 and 10). Moving on to investigate more sustainable alternatives to PhNH2, we found that sugars are not suitable for the transformation; this may in part be due to lack of solubility in the reaction medium (entries 11 to 15). Pleasingly, nBuOH, which is biorenewable, gives 85% spectroscopic yield of propylbenzene in conjunction with 5 mol% 1 and PMHS at RT in 24 h. Notably iBuOH tends to be the key biofuel in the automotive industry and therefore this process would not impact demands for this fuel.46,47 Increasing the loading of nBuOH leads to complete loss of TH reactivity due to competing dehydrocoupling of the silane and alcohol, leading to H2 production (entries 20 and 21). H2 and Fe catalyst do not generate propylbenzene (vide infra). The catalyst is essential for TH to take place (entry 22). The proton source is necessary for efficient formation of propylbenzene (entry 23). 1 is sensitive due to its low coordinate environment but it is pleasing to note the ability of 1 to catalytically turnover alcohols, particularly when one considers the oxophilic nature of Fe(II).48
:1:1 ratio of allylbenzene:PhNH2:PMHS gives good conversion and spectroscopic yield to propylbenzene at RT using 10 mol% 1 (entry 1). Increasing the loading of PMHS further increases the spectroscopic yield, with 3 equivalents of PMHS generating a quantitative amount of propylbenzene. In our search for a greener and safer alternative to C6D6, we found that key renewable solvents such as 2-methyl-THF and cyclopentyl methyl ether (CPME) lead to a drastic reduction in yield (entries 7 and 8). However, solvent free conditions, where PMHS is used to solubilize the other reagents, gives good yield of propylbenzene at both 10 mol% and 5 mol% loading of 1 (entries 9 and 10). Moving on to investigate more sustainable alternatives to PhNH2, we found that sugars are not suitable for the transformation; this may in part be due to lack of solubility in the reaction medium (entries 11 to 15). Pleasingly, nBuOH, which is biorenewable, gives 85% spectroscopic yield of propylbenzene in conjunction with 5 mol% 1 and PMHS at RT in 24 h. Notably iBuOH tends to be the key biofuel in the automotive industry and therefore this process would not impact demands for this fuel.46,47 Increasing the loading of nBuOH leads to complete loss of TH reactivity due to competing dehydrocoupling of the silane and alcohol, leading to H2 production (entries 20 and 21). H2 and Fe catalyst do not generate propylbenzene (vide infra). The catalyst is essential for TH to take place (entry 22). The proton source is necessary for efficient formation of propylbenzene (entry 23). 1 is sensitive due to its low coordinate environment but it is pleasing to note the ability of 1 to catalytically turnover alcohols, particularly when one considers the oxophilic nature of Fe(II).48
|
|
|||||
|---|---|---|---|---|---|
| Entry | Proton source | PMHS (equiv.)/1 (mol%) | Solvent | Conv.a, % | Spec. yieldb, % |
| a Determined by 1H NMR spectroscopy based on consumption of allylbenzene. n.d. = not determined. b Determined by 1H NMR spectroscopy using 7.9 μL (0.1 mmol) DCE internal standard. c 16 h. d J-Young NMR-tube agitated at 10 rpm. e 4 equiv. nBuOH. f 3 equiv. nBuOH. g No catalyst. h 29% isomerization to β-methylstyrene also obtained. | |||||
| 1 | PhNH2 | 1/10 | C6D6 | 60 | 52 |
| 2 | PhNH2 | 2/10 | C6D6 | 85 | 76 |
| 3 | PhNH2 | 3/10 | C6D6 | >99 | >99 |
| 4 | PhNH2 | 3/5 | C6D6 | >99 | 99 |
| 5c | PhNH2 | 3/5 | C6D6 | >99 | 90 |
| 6 | PhNH2 | 3/5 | Tol-d8 | >99 | 82 |
| 7 | PhNH2 | 3/10 | 2-MeTHF | n.d. | 9 |
| 8 | PhNH2 | 3/5 | CPME | n.d. | 16 |
| 9 | PhNH2 | 3/10 | — | n.d. | 84 |
| 10 | PhNH2 | 3/5 | — | n.d. | 71 |
| 11d | D-(−)-Fructose | 1/10 | C6D6 | 39 | n.d. |
| 12d | D-(+)-Glucose | 1/10 | C6D6 | 2 | n.d. |
| 13d | Sucrose | 1/10 | C6D6 | 0 | n.d. |
| 14d | D-(−)-Fructose | 3/10 | C6D6 | 36 | 32 |
| 15d | D-(+)-Glucose | 3/10 | C6D6 | 6 | 4 |
| 16 | nBuOH | 3/5 | C6D6 | >99 | 70 |
| 17 | nBuOH | —/5 | PMHS | n.d. | 85 |
| 18 | EtOH | —/5 | PMHS | n.d. | 74 |
| 19 | MeOH | —/5 | PMHS | n.d. | 58 |
| 20e | nBuOH | 3/5 | — | n.d. | 0 |
| 21f | nBuOH | 1.5/5 | — | n.d. | 0 |
| 22g | nBuOH | —/— | PMHS | n.d. | 0 |
| 23h | — | —/5 | PMHS | n.d. | <5 |

Following this optimization process, we moved on to investigate substrate scope for this transformation (Scheme 2). It is worth noting that during the preparation of this manuscript an elegant Fe-catalyzed hydrogen atom transfer (HAT) method was published by Kattamuri and West.49 That work used PhSiH3 (2 equiv.), EtOH as the solvent, co-catalytic PhSH (10 mol%) in the presence of Fe(acac)3 (10 mol%) under very mild conditions. The transformation works well for a wide range of substrates, but struggles with styrenes, an area where our catalytic system operates well.
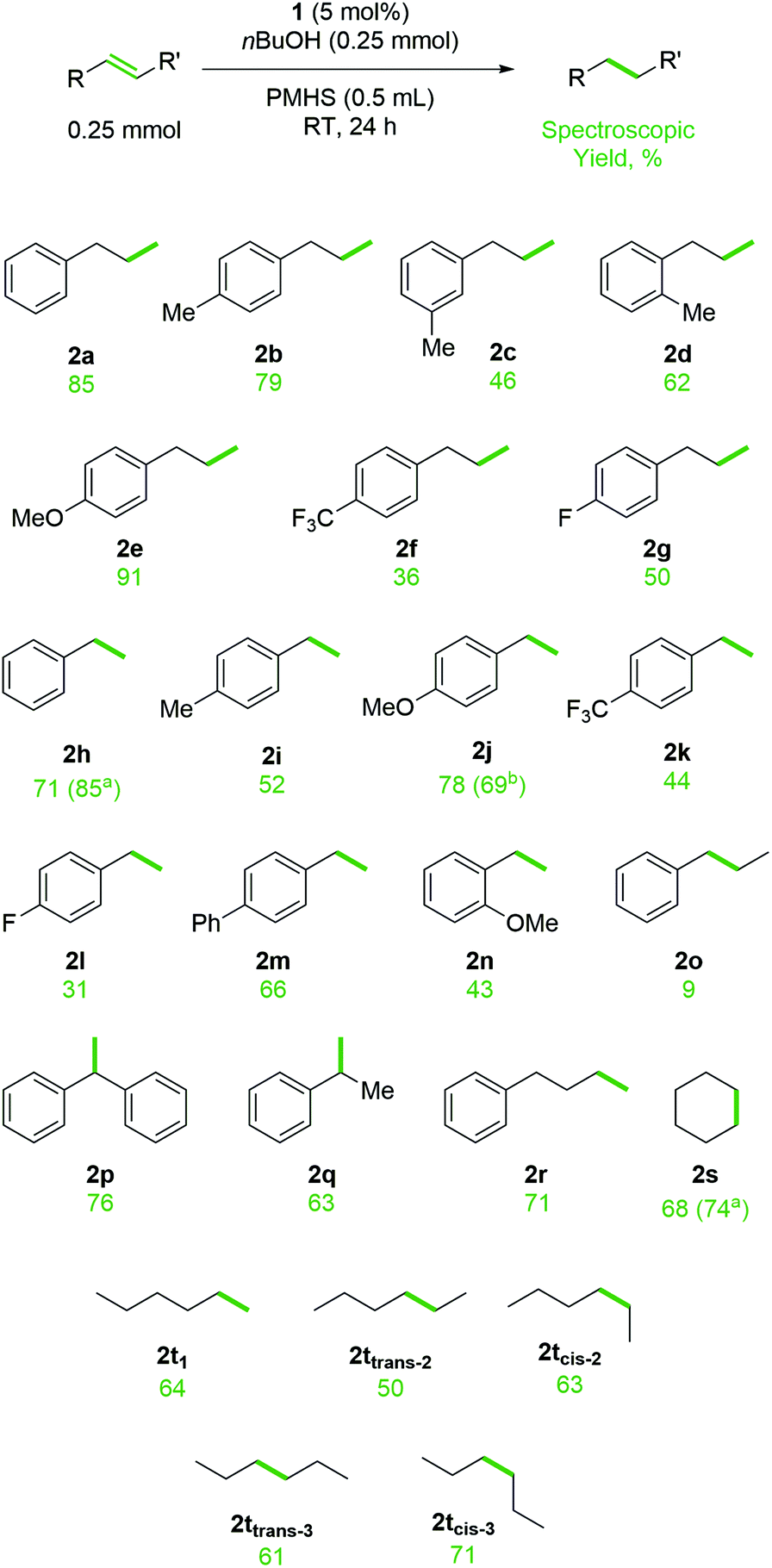 | ||
| Scheme 2 Substrate scope for TH of alkenes and alkynes using PMHS and nBuOH, catalyzed by 1. Values in brackets relate to a 2.5 mmol or b 0.5 mmol scale reactions and the isolated yield is reported. | ||
Allyl arenes respond well to the TH reaction, with unsubstituted, p-Me and p-OMe substrates (2a, 2b and 2e) all giving high yield of product. Steric hindrance and electron withdrawing groups do lower reactivity, as exemplified by the reduced yields observed at RT for m- and o-Me (2c and 2d) and p-CF3 and p-F substrates (2f and 2g). However, increasing the reaction temperature to 60 °C increases the yield of product, for example 68% 2c is obtained after 24 h. Pleasingly, styrene substrates, which appear to be more challenging substrates for Fe-catalyzed HAT chemistry and are, as yet, unproven substrates for iron-catalyzed silane/alcohol TH, react well under the standard reaction conditions. Spectroscopic yields ranging from 44% to 82% are obtained for unsubstituted, p-OMe, p-CF3 and p-Ph substrates (2h, 2j, 2k, 2m). There is no clear link between electronics and reactivity for substrates, with p-F styrene only giving 31% 2l. The effect of sterics are as expected, with o-OMe styrene giving 43% 2n. The transformation responds well to scale-up, with a 10-fold increase in scale (to 2.5 mmol) giving full conversion of styrene starting material and 85% isolated yield of 2h. Scale-up allows for facile isolation of the products. The by-product of the reaction is poly(butoxymethylsilane), a non-volatile compound that the reduced organic product is readily distilled away from. However, to demonstrate flexibility in isolation methods, scale-up synthesis of 2j is achieved with full conversion and 69% isolated yield via flash chromatography. trans-β-Methylstyrene is a poor substrate for TH, but 1,1-disubstituted styrenes are active giving 76% 2p and 63% 2q at RT. Unactivated alkenes, such as but-3-en-1-ylbenzene and cyclohexene give 71% and 68% of butylbenzene (2r) and cyclohexane (2s) respectively. Scale-up of the cyclohexene reduction (2.5 mmol) gives 74% isolated yield. Note the high isolated yield achieved from scale-up and distillation (2h and 2s) compared to the spectroscopic yield obtained on a small scale. Similarly, the isomers of hexene all give good yield of hexane (2t), this is in vast contrast to our previous report of TH using an amine and HBpin which only gave 28% and 48% conversion to product for cis-2-hexene and cis-3-hexene respectively.23 Unfortunately, internal alkynes do not react well with ≥90% starting material recovered. Substrates which carry an acidic proton, such as terminal alkynes and alkenes that are functionalized with carboxylic acids, do not react as desired and instead form bonds with PMHS (forming an (ethynyl)silane and silyl acetate respectively). No reaction is observed with alkenyl substrates such as allyl acetate, allyl 2,2,2-trifluoroacetate, cinnamyl chloride, pent-4-en-2-one, 1,2-epoxy-5-hexene: only unreacted starting material is observed. Acrylonitrile undergoes side-reactions, presumably forming poly(acrylonitrile). α,β-Unsaturated aldehydes, such as cinnamaldehyde, undergo preferential reduction of the carbonyl, forming cinnamyl alcohol in 65% yield.
Interestingly, compared to our previous studies using HBpin and nBuNH2, which undergoes reaction gelling that we use as a qualitative measure of reaction completion, which was subsequently linked to the formation of extended Lewis acid/base macrostructures,23 reactions using amines or alcohols with a silane do not gel. This is because the silane cannot form a Lewis acid adduct with the Lewis basic alcohol (or amine). The fact that the reactions take longer (hours using silane versus minutes using HBpin), there is opportunity for the silane to act as a reducing agent, taking Fe(II) to Fe(I) in situ and generating an η2-alkene complex,50,51 which then undertakes a competitive double bond isomerization process. We note that lower yielding allyl arene substrates show a higher percentage of isomerized product (i.e. trans-β-methylstyrenyl product). For example, in addition to 2c, the allyl-m-methylbenzene substrate also gives 26% isomerized product, likewise allyl-p-(trifluoromethyl)benzene produces 34% isomerized product as well as 36% 2f.52 It is important to note that no starting material is observed in the crude reaction mixtures where the reagent can be isomerized; there are clearly competing reaction processes taking place. High yielding reactions e.g. the formation of 2a and 2e show little or no isomerized product. Based on the lack of TH reactivity obtained using trans-β-methylstyrene (2o) there is no surprise that once isomerization takes place to form trans-β-methylstyrenyl compounds the TH process stalls. In short, we believe two competing catalytic cycles are viable with substrates that can be isomerized.
These results are also reflected in deuterium-labeling using nBuOD53,54 (Scheme 3a) where attempted mono-deuteration of 1-hexene gives a 60:40 ratio of hexane-2-d/hexane-3-d:hexane-1-d. All d-atoms were accounted for using an internal standard, thereby demonstrating high levels of deuteration for all substrates.55 Dropping the loading of PMHS to 1 equiv. gives almost identical levels of deuterium incorporation (compare Schemes 3a and b), but this comes at the expense of yield compared to that obtained using the standard reaction conditions.55 Our previous work using HBpin/PhND2 gave 8:92 (Scheme 3c). With PMHS and nBuOD allyl benzene gives 15:49:36 of (propyl-1-d):(propyl-2-d):(propyl-3-d)benzene while styrene gives a ratio of 37:63 (ethyl-1-d):(ethyl-2-d), the latter is not dissimilar to the results obtained using PhND2 and HBpin (24:76 ratio).23 Determined to provide selective mono-deuteration under sustainable conditions we continued to use PMHS as the hydride donor, but swapped D-alcohol for PhND2 (Scheme 3d). Pleasingly this system gives similar deuteration ratios to HBpin/PhND2 or a modest improvement. For example styrene is preferentially deuterated at the terminal position (17:83 (ethyl-1-d):(ethyl-2-d)benzene). Having noted these modest improvements in selectivity with a silane/amine system, we investigated the labeling selectivity obtained using Ph2SiH2 and PhND2 (Scheme 3e). Pleasingly we observe complete terminal deuteration with 1-hexene and allyl benzene, but once again styrene gives only moderate preference for terminal deuteration. In the case of styrene, we believe that electronics are influencing the regioselectivity, potentially by forming a mixture of linear and branched iron-alkyl species during the catalytic cycle. Finally, Ph2SiH2 and nBuOD (Scheme 3f) give similar results to those obtained using PMHS and PhND2 indicating that it is possible to obtain good terminal deuteration selectivity using one sustainable proton or hydride source but using both PMHS and nBuOD compromise the selectivity.
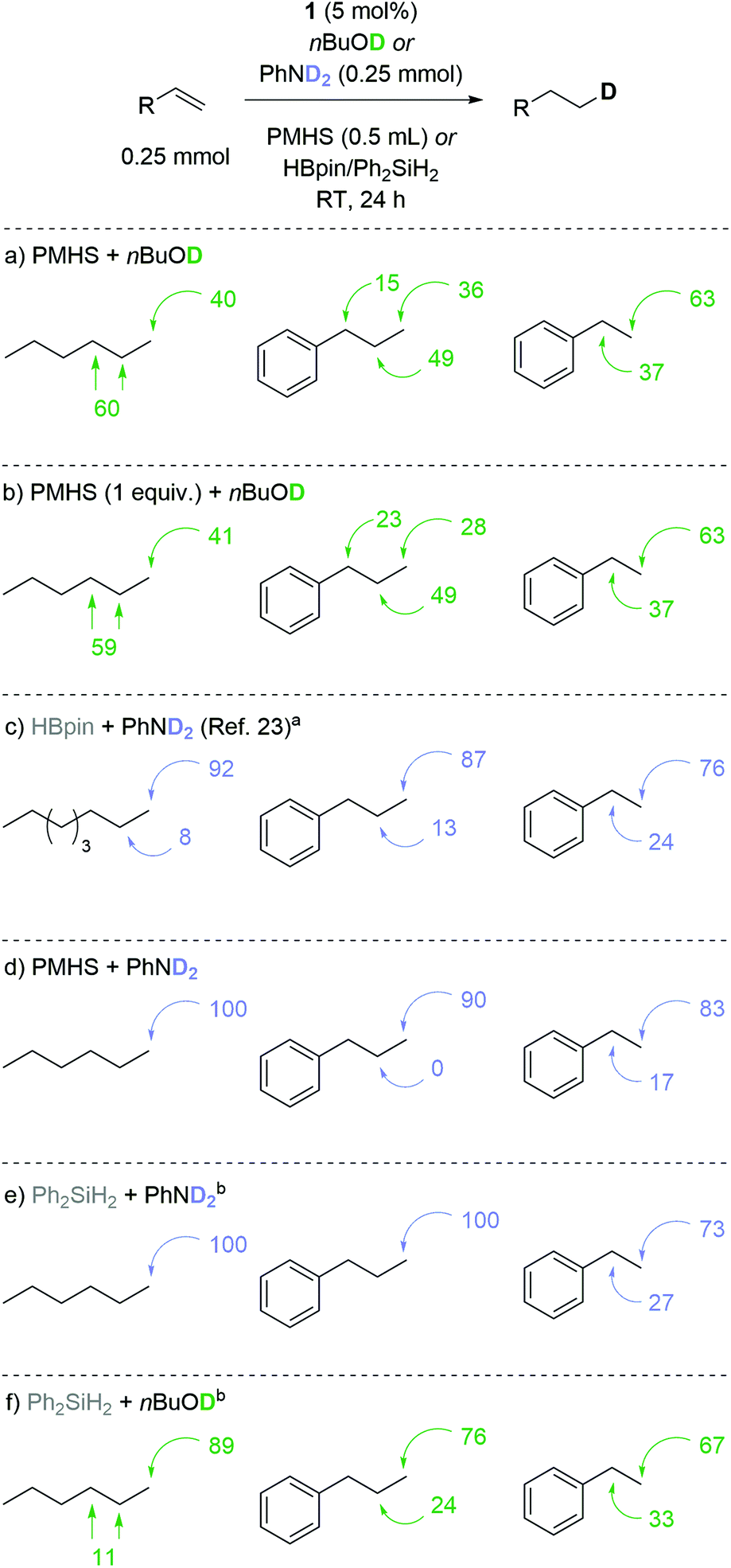 | ||
| Scheme 3 Mono-deuteration studies comparing the selectivity obtained using nBuOD and PhND2 using PMHS, HBpin or Ph2SiH2. a10 mol% 1, 0.26 mmol PhND2, 0.26 mmol HBpin, RT, 16 h. b 5 mol% 1, 0.26 mmol PhND2, 0.26 HBpin/Ph2SiH2, RT, 16 h. | ||
To further probe whether double bond reduction is taking place predominantly by a TH process (as opposed to hydrogenation by H2) we monitored gas evolution as a function of time for a standard reaction (Fig. 1a). As can be seen, a maximum 0.023 bar H2 is released for the standard reaction ( , 5 mol% 1, 0.25 mmol allyl benzene, 0.25 mmol nBuOH, 0.5 mL PMHS, RT). If dehydrocoupling of PMHS and nBuOH occurs with 100% yield of H2 then the maximum possible pressure of H2 in the reaction vessels used for TH is 0.645 bar. Even an attempted reduction of 0.25 mmol allyl benzene using 10 mol% 1 and 20 bar H2 in 0.5 mL C6D6 does not give any product. Attempted activation of 1 using 0.5 mL PMHS and reaction of allyl benzene with 2 bar H2 gives 5% 2a; this is the same result obtained in the absence of H2 (see Table 1, entry 23). The same reaction but using 0.25 mmol nBuOH instead of PMHS does not lead to the formation of 2a. To summarize the deuterium labelling and H2-mediated reduction results: the reaction is not a simple hydrogenation by H2.
, 5 mol% 1, 0.25 mmol allyl benzene, 0.25 mmol nBuOH, 0.5 mL PMHS, RT). If dehydrocoupling of PMHS and nBuOH occurs with 100% yield of H2 then the maximum possible pressure of H2 in the reaction vessels used for TH is 0.645 bar. Even an attempted reduction of 0.25 mmol allyl benzene using 10 mol% 1 and 20 bar H2 in 0.5 mL C6D6 does not give any product. Attempted activation of 1 using 0.5 mL PMHS and reaction of allyl benzene with 2 bar H2 gives 5% 2a; this is the same result obtained in the absence of H2 (see Table 1, entry 23). The same reaction but using 0.25 mmol nBuOH instead of PMHS does not lead to the formation of 2a. To summarize the deuterium labelling and H2-mediated reduction results: the reaction is not a simple hydrogenation by H2.
 | ||
Fig. 1 (a) H2 gas evolution during TH of allyl benzene using 1 equiv. nBuOH ( ) or 2 equiv. nBuOH ( ) or 2 equiv. nBuOH ( ); (b) dehydrocoupling studies using PMHS (0.5 mL) and 1 mmol of MeOH ( ); (b) dehydrocoupling studies using PMHS (0.5 mL) and 1 mmol of MeOH ( ), EtOH ( ), EtOH ( ) or nBuOH ( ) or nBuOH ( ), monitoring the change in H2 pressure as a function of time. ), monitoring the change in H2 pressure as a function of time. | ||
Dehydrocoupling becomes competitive with TH at higher alcohol loadings ( , 0.5 mmol nBuOH used, 0.083 bar released after 4 h, Fig. 1a). We therefore proceeded to explore the ability of 1 to dehydrocouple PMHS and alcohols in the absence of an alkene. Even though reactions have been scaled up, there is still a 10- to 25-fold increase in the quantity of gas released in these alkene-free reactions. This further supports the hypothesis that H2 release and uptake is not responsible for alkene reduction. A greater quantity of H2 is released from the reaction using 1 mmol MeOH as the proton source, which in turn is greater than 1 mmol EtOH and nBuOH (Fig. 1b). Based on the overall change in pressure, we calculate the number of mmol of H2 released is 0.88 (MeOH), 0.97 (EtOH) and 0.73 (nBuOH) which gives conversions of 88%, 97% and 73% for MeOH, EtOH and nBuOH respectively. Initial release of H2 from the nBuOH reaction is slower than those using EtOH and MeOH. The EtOH and nBuOH reactions are 90% complete after 15 minutes, whereas the MeOH reaction is 90% complete after 7 minutes. Two factors may be at play. Firstly, the steric bulk of nBuOH is limiting, where the reaction is more efficient with smaller proton sources and secondly the difference in pKa with MeOH and EtOH < nBuOH, the latter being closer in pKa to amines, which we know from our previous disclosure are very efficient TH reagents. This reflects in the reduced ability of MeOH to act as a TH reagent: dehydrocoupling is highly competitive during the TH of allylbenzene (Table 1, entry 19). Although the potential wt% of H2 that can be released is smaller than H3N·BH3, it is still interesting to note that up to 5.7 wt% can be released from the industrial by-product PMHS and a bio-alcohol using iron pre-catalyst 1.56
, 0.5 mmol nBuOH used, 0.083 bar released after 4 h, Fig. 1a). We therefore proceeded to explore the ability of 1 to dehydrocouple PMHS and alcohols in the absence of an alkene. Even though reactions have been scaled up, there is still a 10- to 25-fold increase in the quantity of gas released in these alkene-free reactions. This further supports the hypothesis that H2 release and uptake is not responsible for alkene reduction. A greater quantity of H2 is released from the reaction using 1 mmol MeOH as the proton source, which in turn is greater than 1 mmol EtOH and nBuOH (Fig. 1b). Based on the overall change in pressure, we calculate the number of mmol of H2 released is 0.88 (MeOH), 0.97 (EtOH) and 0.73 (nBuOH) which gives conversions of 88%, 97% and 73% for MeOH, EtOH and nBuOH respectively. Initial release of H2 from the nBuOH reaction is slower than those using EtOH and MeOH. The EtOH and nBuOH reactions are 90% complete after 15 minutes, whereas the MeOH reaction is 90% complete after 7 minutes. Two factors may be at play. Firstly, the steric bulk of nBuOH is limiting, where the reaction is more efficient with smaller proton sources and secondly the difference in pKa with MeOH and EtOH < nBuOH, the latter being closer in pKa to amines, which we know from our previous disclosure are very efficient TH reagents. This reflects in the reduced ability of MeOH to act as a TH reagent: dehydrocoupling is highly competitive during the TH of allylbenzene (Table 1, entry 19). Although the potential wt% of H2 that can be released is smaller than H3N·BH3, it is still interesting to note that up to 5.7 wt% can be released from the industrial by-product PMHS and a bio-alcohol using iron pre-catalyst 1.56
Finally, although detailed mechanistic studies have thus far been limited by the nature of the reaction (PMHS as a solvent, necessity for stirring and gas evolution) where any deviation from this may lead to kinetics results that are not representative of the reaction being undertaken, we postulate a catalytic cycle for TH that involves a completely redox-neutral process (Fig. 2). Activation of 1 involves formation of an iron-alkoxide complex,57,58 a σ-bond metathesis type process then takes place in a similar mode to that already reported by our group.23 The resultant iron hydride (which may exist as a dimer either on-cycle or off-cycle)59 then undergoes insertion, and finally protonolysis to release the product. The reaction using PhND2 as the deuterium source leads to higher selectivity for terminal deuteration. We, qualitatively, link this to the rate of protonolysis, with amine-mediated reactions going to completion faster than those mediated by alcohols. With this in mind, it may be that there is a change in the rate-limiting step when alcohols are used, so there is more opportunity to equilibrate between the branched and linear insertion product prior to a slower rate of protonolysis. The competing catalytic cycles (isomerization and dehydrocoupling) can become dominant under certain circumstances. For example, isomerization competes when there is a double bond that can isomerize to form a more stable, conjugated species. This isomerization process is aided by the slower rate of TH using PMHS/alcohol (compared to HBpin/amine) which facilitates access to the Fe(I) species necessary for isomerization.51 For dehydrocoupling, if TH is too challenging, or if the concentration of hydride and proton source become too great, then hydrogen release chemistry dominates and isomerization stalls.
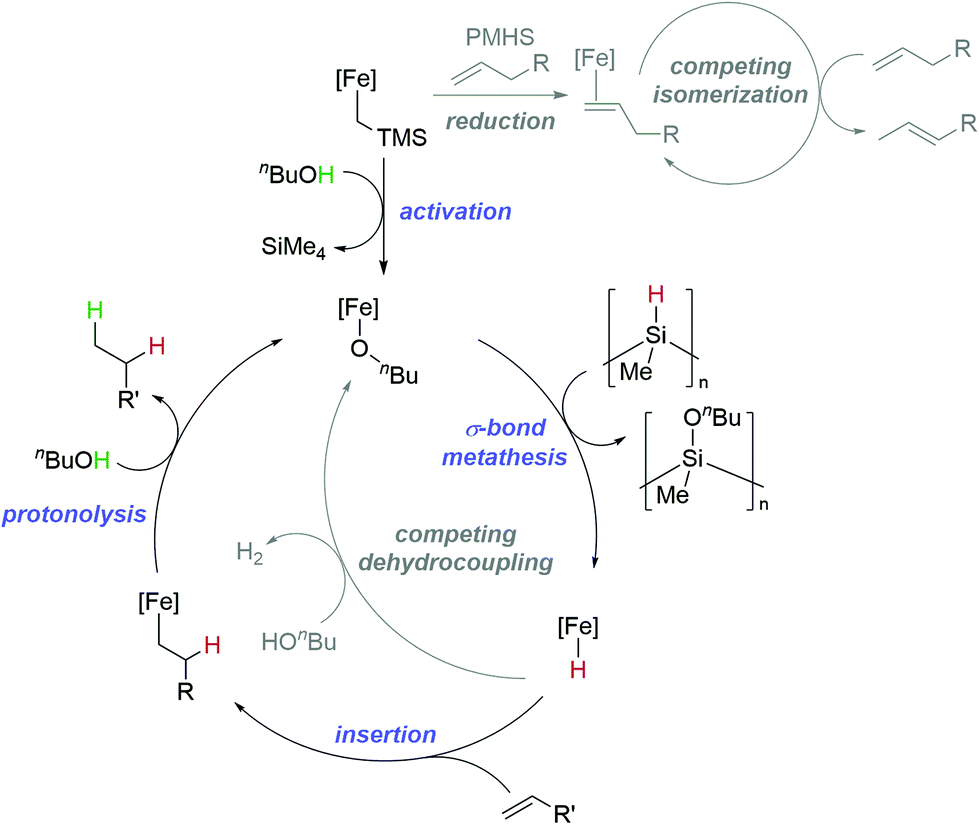 | ||
| Fig. 2 Postulated catalytic cycle for alkene TH, which is believed to proceed via a redox neutral Fe(II) cycle. Reagent dependent competing (redox neutral) dehydrocoupling cycles and (redox active) double bond isomerization are also at play and can affect TH outcomes. | ||
Conclusions
In summary, we have developed an iron-catalyzed TH process using the poly(silane) PMHS and a bio-alcohol. MeOH, EtOH and nBuOH all perform reasonably, but with a higher spectroscopic yield nBuOH was selected as the proton source. TH substrate scope included allylarenes, styrenes and unactivated alkenes. Deuteration using nBuOD proceeds with a reasonable level of selectivity but use of PhND2 as the D+ source gives a higher level of selectivity for the terminal position. Changing to use a less sustainable silane, PH2SiH2, does result in complete selectivity for terminal mono-deuteration of 1-hexene and allyl benzene but, irrespective of D+ or H− source, styrenes appear to suffer from lack of terminal (2-position) selectivity, likely linked to competing electronic effects. Double bond reduction is predominantly a TH process but excluding the alkene from the reaction mixture does allow for efficient dehydrocoupling to take place with 90% H2 released from PMHS reacting with EtOH in the presence of the iron catalyst. Further studies to understand the subtleties of the change in reaction mechanism postulated here, in comparison to previous published work is underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Danila Gasperini is thanked for some preliminary reaction optimization. The EPSRC is thanked for funding.References
- L. Červený, Catalytic hydrogenation, New York, Elsevier, New York, U.S.A, 1986 Search PubMed.
- P. A. Chaloner, M. A. Esteruelas, F. Joó and L. A. Oro, Homogeneous Hydrogenation, Dordrecht: Springer Netherlands, Imprint: Springer, 1st edn, 1994 Search PubMed.
- S. A. Green, S. W. M. Crossley, J. L. M. Matos, S. Vásquez-Céspedes, S. L. Shevick and R. A. Shenvi, Acc. Chem. Res., 2018, 51, 2628–2640 CrossRef CAS PubMed.
- C. S. G. Seo and R. H. Morris, Organometallics, 2019, 38, 47–65 CrossRef CAS.
- C. F. de Graauw, J. A. Peters, H. van Bekkum and J. Huskens, Synthesis, 1994, 1994, 1007–1017 CrossRef.
- J. S. Cha, Org. Process Res. Dev., 2006, 10, 1032–1053 CrossRef CAS.
- R. A. W. Johnstone, A. H. Wilby and I. D. Entwistle, Chem. Rev., 1985, 85, 129–170 CrossRef CAS.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- S. Gladiali and E. Alberico, Chem. Soc. Rev., 2006, 35, 226–236 RSC.
- S. Lau, D. Gasperini and R. L. Webster, Angew. Chem., Int. Ed. DOI:10.1002/anie.202010835.
- D. M. Sharma and B. Punji, Chem. – Asian J., 2020, 15, 690–708 CrossRef CAS PubMed.
- M. P. Doyle and C. C. McOsker, J. Org. Chem., 1978, 43, 693–696 CrossRef CAS.
- J. S. M. Samec, J.-E. Bäckvall, P. G. Andersson and P. Brandt, Chem. Soc. Rev., 2006, 35, 237–248 RSC.
- A. Comas-Vives, G. Ujaque and A. Lledós, in Adv. Inorg. Chem, ed. R. van Eldik and J. Harvey, Academic Press, 2010, vol. 62, pp. 231–260 Search PubMed.
- J. Lipowitz and S. A. Bowman, J. Org. Chem., 1973, 38, 162–165 CrossRef CAS.
- L. T. Brechmann and J. F. Teichert, Synthesis, 2020, 52, 2483–2496 CrossRef CAS.
- K. Semba, T. Fujihara, T. Xu, J. Terao and Y. Tsuji, Adv. Synth. Catal., 2012, 354, 1542–1550 CrossRef CAS.
- J. W. Hall, D. M. L. Unson, P. Brunel, L. R. Collins, M. K. Cybulski, M. F. Mahon and M. K. Whittlesey, Organometallics, 2018, 37, 3102–3110 CrossRef CAS.
- C. Johnson and M. Albrecht, Catal. Sci. Technol., 2018, 8, 2779–2783 RSC.
- G. Wienhöfer, F. A. Westerhaus, R. V. Jagadeesh, K. Junge, H. Junge and M. Beller, Chem. Commun., 2012, 48, 4827–4829 RSC.
- P. Döhlert and S. Enthaler, Catal. Lett., 2016, 146, 345–352 CrossRef.
- B. Tansel and S. C. Surita, Int. J. Environ. Sci. Technol., 2017, 14, 795–802 CrossRef CAS.
- M. Espinal-Viguri, S. E. Neale, N. T. Coles, S. A. Macgregor and R. L. Webster, J. Am. Chem. Soc., 2019, 141, 572–582 CrossRef CAS PubMed.
- H. Nagashima, Synlett, 2015, 866–890 CrossRef CAS.
- N. S. Shaikh, K. Junge and M. Beller, Org. Lett., 2007, 9, 5429–5432 CrossRef CAS PubMed.
- C. Dal Zotto, D. Virieux and J.-M. Campagne, Synlett, 2009, 276–278 CAS.
- D. Addis, N. Shaikh, S. Zhou, S. Das, K. Junge and M. Beller, Chem. – Asian J., 2010, 5, 1687–1691 CrossRef CAS PubMed.
- E. Buitrago, L. Zani and H. Adolfsson, Appl. Organomet. Chem., 2011, 25, 748–752 CrossRef CAS.
- K. Muller, A. Schubert, T. Jozak, A. Ahrens-Botzong, V. Schünemann and W. R. Thiel, ChemCatChem, 2011, 3, 887–892 CrossRef CAS.
- L. C. M. Castro, D. Bézier, J.-B. Sortais and C. Darcel, Adv. Synth. Catal., 2011, 353, 1279–1284 CrossRef CAS.
- A. P. Dieskau, J.-M. Begouin and B. Plietker, Eur. J. Org. Chem., 2011, 2011, 5291–5296 CrossRef CAS.
- E. Buitrago, F. Tinnis and H. Adolfsson, Adv. Synth. Catal., 2012, 354, 217–222 CrossRef CAS.
- J. Zheng, L. C. Misal Castro, T. Roisnel, C. Darcel and J.-B. Sortais, Inorg. Chim. Acta, 2012, 380, 301–307 CrossRef CAS.
- Q. Liang, N. J. Liu and D. Song, Dalton Trans., 2018, 47, 9889–9896 RSC.
- S. Das, H. S. Das, B. Singh, R. K. Haridasan, A. Das and S. K. Mandal, Inorg. Chem., 2019, 58, 11274–11278 CrossRef CAS PubMed.
- A. Volkov, E. Buitrago and H. Adolfsson, Eur. J. Org. Chem., 2013, 2013, 2066–2070 CrossRef CAS.
- S. Elangovan, S. Quintero-Duque, V. Dorcet, T. Roisnel, L. Norel, C. Darcel and J.-B. Sortais, Organometallics, 2015, 34, 4521–4528 CrossRef CAS.
- L. Y. Chan, J. S. K. Lim and S. Kim, Synlett, 2011, 2011, 2862–2866 CrossRef.
- H. Li, M. Achard, C. Bruneau, J.-B. Sortais and C. Darcel, RSC Adv., 2014, 4, 25892–25897 RSC.
- S. Enthaler, M. Haberberger and E. Irran, Chem. – Asian J., 2011, 6, 1613–1623 CrossRef CAS PubMed.
- M. Haberberger, E. Irran and S. Enthaler, Eur. J. Inorg. Chem., 2011, 2011, 2797–2802 CrossRef CAS.
- L. C. M. Castro, H. Q. Li, J. B. Sortais and C. Darcel, Green Chem., 2015, 17, 2283–2303 RSC.
- B. A. F. Le Bailly and S. P. Thomas, RSC Adv., 2011, 1, 1435–1445 RSC.
- D. Wei and C. Darcel, Chem. Rev., 2019, 119, 2550–2610 CrossRef CAS PubMed.
- R. Lopes and B. Royo, Isr. J. Chem., 2017, 57, 1151–1159 CrossRef CAS.
- C. Jin, M. Yao, H. Liu, C.-f. F. Lee and J. Ji, Renewable Sustainable Energy Rev., 2011, 15, 4080–4106 CrossRef CAS.
- T. L. Alleman, A. Singh, E. D. Christensen, E. Simmons and G. Johnston, Energy Fuels, 2020, 34, 8424–8431 CrossRef CAS.
- K. P. Kepp, Inorg. Chem., 2016, 55, 9461–9470 CrossRef CAS PubMed.
- P. V. Kattamuri and J. G. West, J. Am. Chem. Soc., 2020, 142, 19316–19326 CrossRef CAS PubMed.
- Y. Yu, J. M. Smith, C. J. Flaschenriem and P. L. Holland, Inorg. Chem., 2006, 45, 5742–5751 CrossRef CAS PubMed.
- C. R. Woof, D. J. Durand, N. Fey, E. Richards and R. L. Webster, Chem. – Eur. J. DOI:10.1002/chem.202004980.
- See ESI (Table S1)† for full details on levels of isomerization observed during TH.
- d 10-Butanol used.
- EtOD (0.25 mmol), PMHS (0.5 mL), 1 (5 mol%), RT, 24 h results in 12:50:38 (propyl-1-d):(propyl-2-d):(propyl-3-d)benzene; 38:62 (ethyl-1-d):(ethyl-2-d)benzene; 59:41 hexane-2-d/hexane-3-d:hexane-1-d.
- See ESI.†.
- 3200 g mol−1 PMHS will produce 2.6 wt% H2 when reacted with nBuOH; 4.1 wt% with EtOH; 5.7 wt% with MeOH.
- J. Vela, S. Vaddadi, T. R. Cundari, J. M. Smith, E. A. Gregory, R. J. Lachicotte, C. J. Flaschenriem and P. L. Holland, Organometallics, 2004, 23, 5226–5239 CrossRef CAS.
- K. P. Chiang, P. M. Barrett, F. Ding, J. M. Smith, S. Kingsley, W. W. Brennessel, M. M. Clark, R. J. Lachicotte and P. L. Holland, Inorg. Chem., 2009, 48, 5106–5116 CrossRef CAS PubMed.
- D. Gasperini, A. K. King, N. T. Coles, M. F. Mahon and R. L. Webster, ACS Catal., 2020, 10, 6102–6112 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc04175k |
| This journal is © The Royal Society of Chemistry 2021 |