
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mizoroki–Heck type reactions and synthesis of 1,4-dicarbonyl compounds by heterogeneous organic semiconductor photocatalysis†
Jagadish
Khamrai
 a,
Saikat
Das
a,
Aleksandr
Savateev
b,
Markus
Antonietti
b and
Burkhard
König
*a
a,
Saikat
Das
a,
Aleksandr
Savateev
b,
Markus
Antonietti
b and
Burkhard
König
*a
aFakultät für Chemie und Pharmazie, Universität Regensburg, 93040 Regensburg, Germany. E-mail: burkhard.koenig@ur.de
bDepartment of Colloid Chemistry, Max Planck Institute of Colloids and Interfaces, Research Campus Golm, 14424 Potsdam, Germany
First published on 2nd March 2021
Abstract
We report the synthesis of 1,4-dicarbonyl compounds and substituted alkenes (Mizoroki–Heck type coupling) starting from secondary and tertiary alkyl halides and vinyl acetate or styrene derivatives using visible-light photocatalysis. The protocol uses mesoporous graphitic carbon nitride (mpg-CN) as a heterogeneous organic semiconductor photocatalyst and Ni(II) salts as Lewis acid catalysts. Detailed post-characterization of the heterogeneous material has been carried out to support the proposed catalytic cycle. Apart from high functional-group tolerance, mild reaction conditions, scalability as well as easy recovery and reuse of the mpg-CN photocatalyst provide a practical solution to these widespread transformations in terms of sustainability and efficiency and this methodology is recommended for applications in academic and industrial synthesis.
Introduction
Highly substituted 1,4-dicarbonyl motifs are found in a wide variety of biologically active natural products and pharmaceutical agents,1 and the development of novel methods for their synthesis continues to be an active research area.2 Classical methods for the synthesis of such compounds involve the Michael type addition of acyl anion synthons to α,β-unsaturated carbonyl compounds or nucleophilic substitution between enolates and α-haloketones.3 However, these reactions lack general applicability due to limited functional group tolerance caused by the harsh reaction conditions and reactive intermediates.4 Lately, alternative routes such as oxidative cross-coupling of enolates,5 enamines,7 and enol silanes6 have been reported, but most of them suffer from the use of stoichiometric amounts of toxic oxidants as well as competing homocoupling reactions.8 Very recently, the application of visible light photocatalysis has offered the synthesis of such compounds under milder reaction conditions. Among them, visible light induced generation of acyl radicals followed by Giese-type addition to α,β-unsaturated carbonyl compounds is an effective synthetic route to 1,4-dicarbonyl compounds.9–12 Along these lines, direct construction of 1,4-dicarbonyls via α-photoalkylation of β-ketocarbonyls by merging ruthenium-based homogeneous photoredox catalysis and primary amine catalysis has also been reported (Scheme 1a).13–15 However, most of these methods suffer from the use of expensive transition metal photocatalysts, which limits their practical application specially in larger scale synthesis. On the other hand, Mizoroki–Heck reaction is one of the most useful cross-coupling reactions in organic synthesis which is mostly catalyzed by expensive transition metals, such as palladium.16,17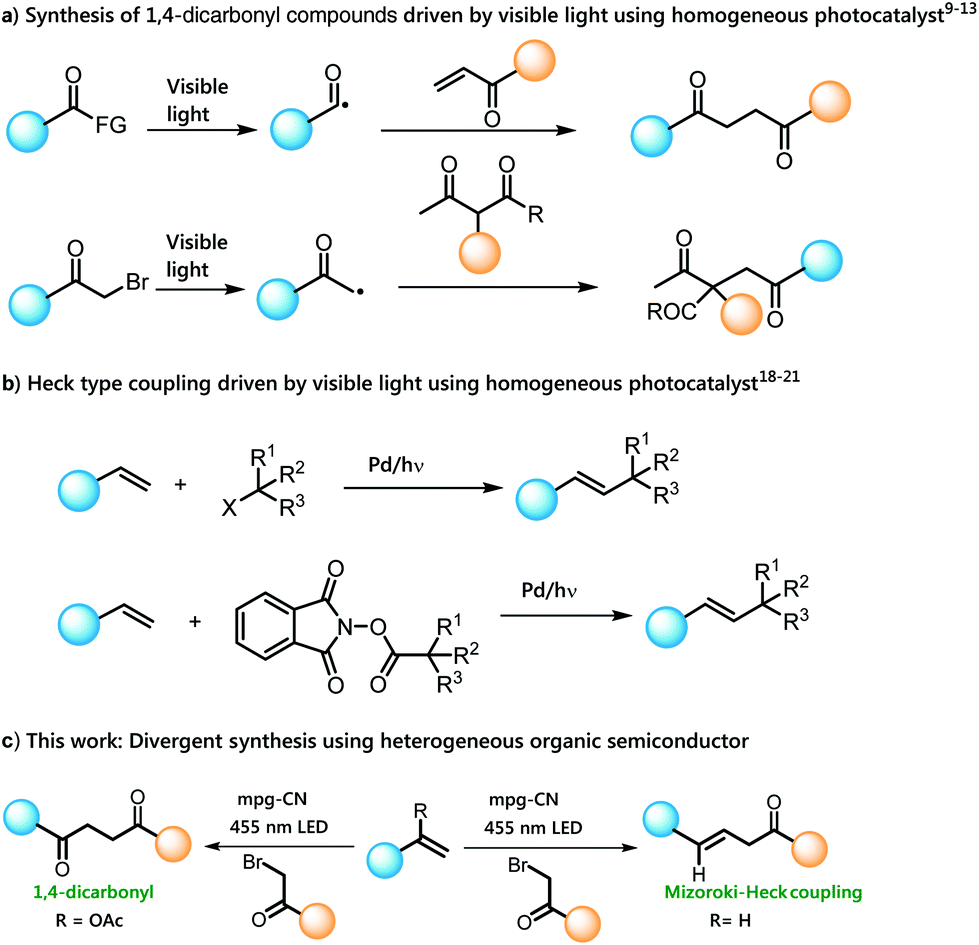 | ||
| Scheme 1 Mizoroki–Heck type coupling and synthesis of the 1,4-dicarbonyl compound using organic semiconductor visible light photoredox catalysis. | ||
Many applications have been found in visible light photocatalysis complementing classical methods for the preparation of substituted olefins, for example addition of alkyl radicals from alkyl halides18,19 or N-hydroxyphthalimide ester21 precursors to styrene derivatives using photoexcited palladium(0) complexes20 under visible light irradiation (Scheme 1b). However, most of these methods are constrained by the use of expensive metal catalysts and molecular photocatalysts. We describe a divergent, practicable and sustainable route for the synthesis of 1,4-dicarbonyl and Mizoroki–Heck type of cross coupling products using heterogeneous organic semiconductor photocatalysis namely the use of mesoporous graphitic carbon nitride (mpg-CN).
We recently reported mpg-CN as a purely organic semiconductor photocatalyst capable of performing many organic transformations under diverse reaction conditions.22,24 mpg-CN can easily be synthesized using inexpensive starting materials, and the polymeric material is stable towards reactive radicals or nucleophiles. It possesses a suitable bandgap between the valence band maxima (VBM) and conduction band minima (CBM),23 which allows the use of photoexcited mpg-CN for controlled oxidation and reduction of many substrates. Very recently, the use of carbon nitride-based heterogeneous photocatalysts in several synthetic transformations has been demonstrated by Seeberger and Reisner groups.25,26 However, intermolecular addition of radicals to olefins using a heterogeneous photocatalyst has not been thoroughly explored until today. Herein we report the use of an mpg-CN organic semiconductor as a sustainable heterogeneous photocatalyst to promote the reaction between alkyl halides and vinyl acetate or styrene derivatives under visible light for the synthesis of 1,4-dicarbonyl compounds and the products of Mizoroki–Heck type reactions, respectively (Scheme 1c).
Results and discussion
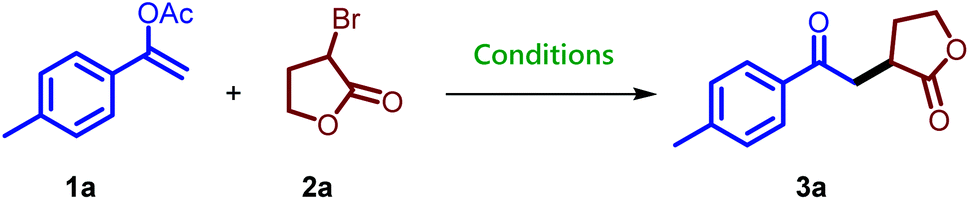
We explored the reactivity of mpg-CN as a heterogeneous organic semiconductor first by the synthesis of 1,4-dicarbonyl compounds. For this purpose, we chose vinyl acetate, easily synthesized from stable and commercially available starting materials and different alkyl bromides. We began our investigation with 1-(p-tolyl)vinyl acetate (1a) and α-bromo-γ-butyrolactone (1.5 equivalent) (2a) as model substrates and mpg-CN as a heterogeneous photocatalyst in the presence of 2,6-lutidine as a base. When the reaction mixture in DMF was irradiated using a 455 ± 15 nm (Imax = 1000 mA, 1.12 W) blue LED for 24 h under nitrogen, the corresponding 1,4-dicarbonyl product 3a was obtained in 29% GC yield (Table 1, entry 1). The use of a catalytic amount of NiBr2 increased the product yield to 79% (entry 2). Control reactions in the absence of light or mpg-CN confirmed their essential roles in the photocatalytic reaction (entries 3 and 4). While 1.25 mol% of NiBr2 decreased the yield to 43% (entry 5), the use of 1.25 mol% NiBr2·glyme provided the best result and the desired product was obtained in 83% isolated yield (entry 6, GC yield: 86%). When the reaction was performed using recovered mpg-CN without adding any Ni source, compound 3a was formed in 74% yield (entry 7), which indicates that the amount of Ni deposited on the surface of the mpg-CN is sufficient to accelerate the reaction in the 2nd cycle.|
|
|||
|---|---|---|---|
| Entry | Photocatalyst | Additive | Yieldb |
| a The reaction was performed using 0.2 mmol of 1-(p-tolyl)vinyl acetate, 1.25 equiv. of 2,6-lutidine as a base and 1 ml of DMF as a solvent under 455 nm blue LED irradiation for 24 h. b GC yields were determined using 1,3,5-trimethoxybenzene as an internal standard. c Isolated yield. d Without 2,6-lutidine. e DMSO was used as the solvent. f DMA was used as the solvent. g Dioxane was used as the solvent. h ACN was used as the solvent. i K-PHI (prepared from 5-aminotetrazole in the LiCl/KCl eutectic mixture using mechanochemical pretreatment of reagents). j K-PHI (prepared from 5-aminotetrazole in the LiCl/KCl eutectic mixture using 0.5 wt% of K-PHI nanoparticles as nucleation seeds). | |||
| 1 | mpg-CN | — | 29% |
| 2 | mpg-CN | NiBr2 (5 mol%) | 79% |
| 3 | mpg-CN | NiBr2 (5 mol%) | 0% (in the dark) |
| 4 | — | NiBr2 (5 mol%) | 1% (without mpg-CN) |
| 5 | mpg-CN | NiBr2 (1.25 mol%) | 43% |
| 6 | mpg-CN | NiBr 2 ·glyme (1.25 mol%) | 86% (83%) |
| 7 | Recovered mpg-CN | — | 74% |
| 8 | mpg-CN | NiBr2·glyme (1.25 mol%) | 59% (under air) |
| 9 | mpg-CN | NiBr2·glyme (1.25 mol%) | 49%d |
| 10 | mpg-CN | NiBr2·glyme (1.25 mol%) | 70%e |
| 11 | mpg-CN | NiBr2·glyme (1.25 mol%) | 83%f |
| 12 | mpg-CN | NiBr2·glyme (1.25 mol%) | 37%g |
| 13 | mpg-CN | NiBr2·glyme (1.25 mol%) | 72%h |
| 14 | mpg-CN | Cu(OTf)2 (1.25 mol%) | 38% |
| 15 | mpg-CN | In(OTf)3 (1.25 mol%) | 43% |
| 16 | mpg-CN | Sc(OTf)3 (1.25 mol%) | 40% |
| 17 | mpg-CN | Zn(OTf)2 (1.25 mol%) | 42% |
| 18 | mpg-CN | AlCl3 (5 mol%) | 38% |
| 19 | mpg-CN | Y(OTf)3 (1.25 mol%) | 30% |
| 20 | Na-PHI | NiBr2·glyme (1.25 mol%) | 31% |
| 21 | K-PHIi | NiBr2·glyme (1.25 mol%) | 38% |
| 22 | CN-ATZ-NaK | NiBr2·glyme (1.25 mol%) | 44% |
| 23 | K-PHIj | NiBr2·glyme (1.25 mol%) | 22% |
| 24 | Mn-PHI | NiBr2·glyme (1.25 mol%) | 69% |
| 25 | H-PHI | NiBr2·glyme (1.25 mol%) | 59% |
In the 3rd cycle, 46% product formation was observed without adding Ni, indicating that the Lewis acid is necessary to drive the reaction. The reaction becomes sluggish when performed in the presence of air (entry 8). Moreover, the reaction performed in the absence of 2,6-lutidine provides a diminished product yield27 (entry 9) along with the formation of 4-methyl acetophenone as a major side product. This is due to the generation of HBr during the reaction, which deprotects the acetate group leading to the formation of keto carbonyl compounds. Other commonly used solvents such as DMSO, dioxane, and ACN resulted in lower yield of the desired product compared to DMF except DMA (entries 10–13). Other Lewis acids as additives failed to improve the yield and the formation of 4-methyl acetophenone as a major side product was observed (entries 14–19). Finally, the use of other modified carbon nitrides such as Na-PHI,28a K-PHI,28b CN-ATZ-NaK,28c K-PHI,28d Mn-PHI,28e and H-PHI28e did not increase the product yield compared to mpg-CN (entries 20–25).
With the optimized reaction conditions in hand, we explored the scope of this reaction using different vinyl acetates as substrates as shown in Scheme 2.
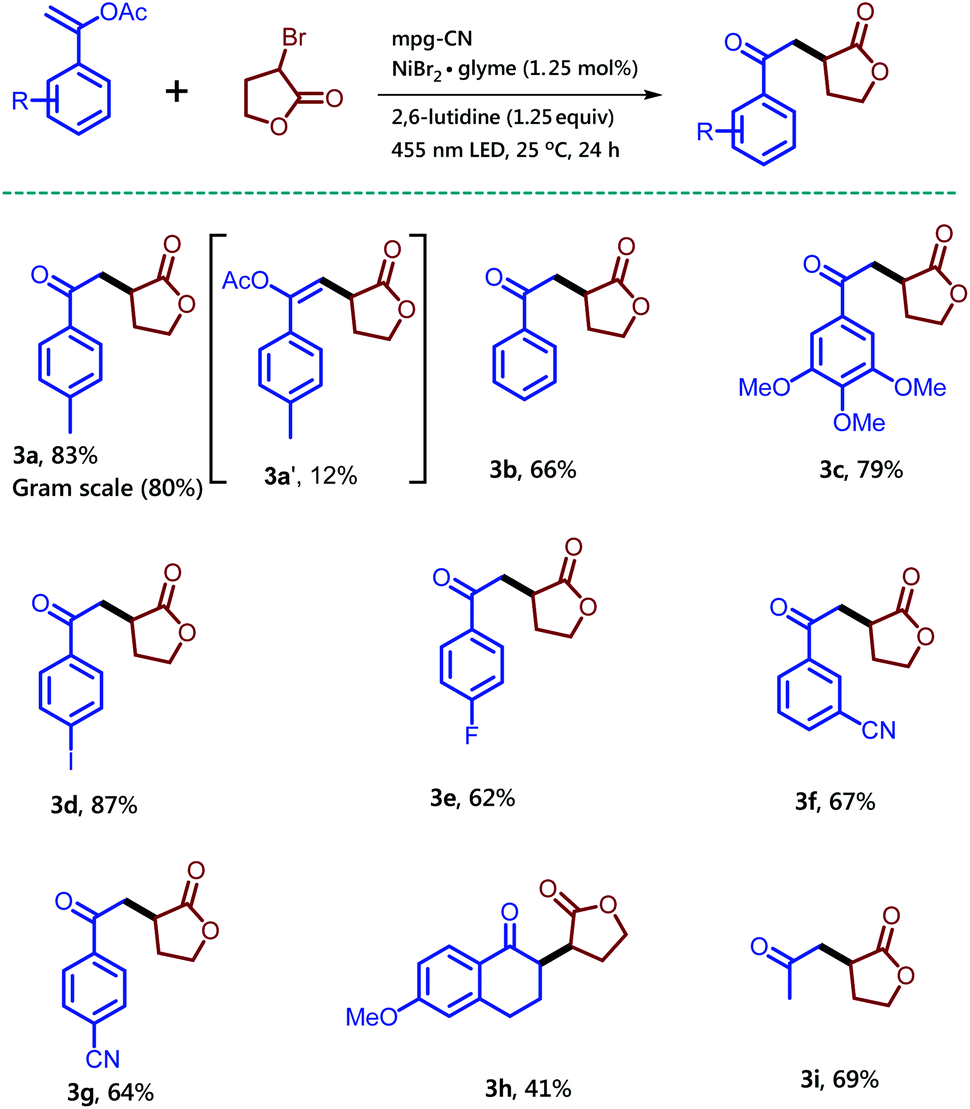 | ||
| Scheme 2 Synthesis of 1,4-dicarbonyl compounds using various vinyl acetate derivatives and α-bromo-γ-butyrolactone in the presence of mpg-CN as a heterogeneous photocatalyst. Standard conditions: Vinyl acetate (0.2 mmol, 1.0 equiv.), α-bromo-γ-butyrolactone (49.5 mg, 0.3 mmol, 1.5 equiv.), mpg-CN (10.0 mg), NiBr2·glyme (0.7 mg, 0.0025 mmol, 0.0125 equiv.), 2,6-lutidine (29 μL, 0.25 mmol, 1.25 equiv.) in DMF (1 mL), under a nitrogen atmosphere for 24 h. Similar conditions were used for gram scale reactions on a 6 mmol scale; for more details, see the ESI.† | ||
Notably, the side product 3a′ was formed in 12% isolated yield along with the desired compound 3a. Unsubstituted vinyl acetate gave the desired product 3b in 66% isolated yield. Enol-acetate functionalized with a highly electron rich aromatic ring delivered product 3c in 79% isolated yield. Moreover, the halogen substituent on the aromatic-ring did not alter the outcome of the reaction. Iodo and fluoro substituents at the para-position of the aromatic ring provided the desired products 3d and 3e in 87% and 62% isolated yields, respectively. Electron withdrawing substituents on the aromatic ring are tolerated well as shown by a cyano substituent at meta and para positions, which gave the corresponding products 3f and 3g in 67% and 64% isolated yields, respectively. This reaction is not only useful for the enol-acetates substituted with an aromatic ring, but also for the aliphatic enol-acetate. Successful examples of this type 3h and 3i were obtained by the reaction among dihydronaphthalene acetate, isopropenyl acetate and α-bromo-γ-butyrolactone, respectively.
After screening several enol-acetates we switched our focus to the exploration of the utilization of different alkyl bromides (Scheme 3). We chose the 1-(p-tolyl)vinyl acetate (1a) as a model substrate to study the reaction of other alkyl bromides.
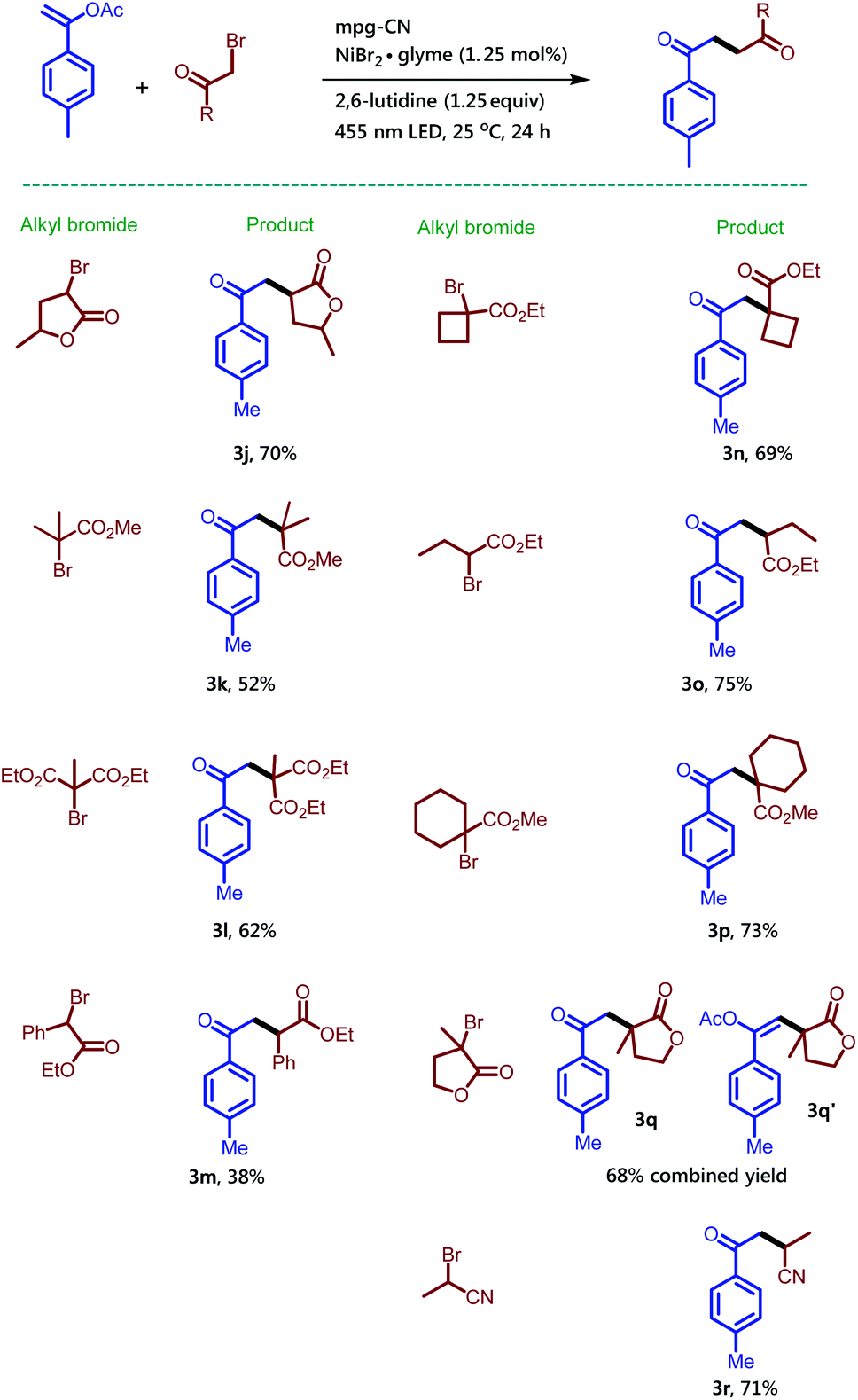 | ||
| Scheme 3 Synthesis of 1,4-dicarbonyl compounds using various alkyl bromides in the presence of mpg-CN as a heterogeneous photocatalyst. Standard conditions: 1-(p-tolyl)vinyl acetate (35.2 mg, 0.2 mmol, 1.0 equiv.), alkyl bromide (0.3 mmol, 1.5 equiv.), mpg-CN (10.0 mg), NiBr2·glyme (0.7 mg, 0.0025 mmol, 0.0125 equiv.), and 2,6-lutidine (29 μL, 0.25 mmol, 1.25 equiv.) in DMF (1 mL), under a nitrogen atmosphere for 24 h. | ||
Treatment of α-bromo-γ-butyrolactone containing a methyl substituent with 1a under the standard reaction conditions afforded the desired product 3j in 70% isolated yield. Methyl-2-bromo-2-methyl propanoate also provided the corresponding compound 3k in 52% isolated yield, while diethyl-2-bromo-2-methylmalonate converted to compound 3l in 62% isolated yield. Here it is noteworthy to mention that particularly for this bromide substrate a considerable amount of background reaction was observed in GC when the reaction was performed without a catalyst but in the presence of light. Ethyl-2-bromo-2-phenylacetate gave compound 3m in low yield along with the recovered starting material. The synthesis of 1,4-dicarbonyl compounds bearing all-carbon quaternary stereocenters is another commendable feature of the reaction. For example, bromo-compounds bearing a cyclobutane or cyclohexane ring delivered 1,4-dicarbonyl products 3n and 3p. Similarly, the all-carbon quaternary stereocenters also formed when 3-bromo-3-methyldihydrofuran-2(3H)-one was treated with 1a to provide an inseparable mixture of 3q and 3q′ combined in 68% isolated yield. Interestingly, 2-bromo-propionitrile also takes part in the reaction and gives the desired compound 3r bearing a keto and a cyano group, useful for follow-up chemistry.
Interestingly, addition of the alkyl radicals to styrene derivatives instead of vinyl acetate produced Mizoroki–Heck type coupling products under the previously optimized reaction conditions.
Under standard reaction conditions the reaction between 4-methoxystyrene and α-bromo-γ-butyrolactone (2a) yielded an E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :Z mixture of the desired compound 5a in an almost 1
:Z mixture of the desired compound 5a in an almost 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, while in the absence of a base the desired compound 5a was obtained in 77% isolated yield as a single regioisomer. The reaction conditions for Mizoroki–Heck type coupling required only mixing of styrene, alkyl bromide (2.0 equiv.), and 1.25 mol% NiBr2·glyme in the presence of mpg-CN and irradiation under nitrogen for 48 h. Variation of the alkyl bromides afforded the corresponding products 5b–5j in moderate to good isolated yields. The reaction of α-bromo-γ-butyrolactone with 4-acetoxystyrene and 4-fluorostyrene gave the expected products (5k–5l) demonstrating the applicability of the protocol. This reaction remained effective for a gram-scale protocol as shown by the preparation of 5g and 5h in Scheme 4.
:1 ratio, while in the absence of a base the desired compound 5a was obtained in 77% isolated yield as a single regioisomer. The reaction conditions for Mizoroki–Heck type coupling required only mixing of styrene, alkyl bromide (2.0 equiv.), and 1.25 mol% NiBr2·glyme in the presence of mpg-CN and irradiation under nitrogen for 48 h. Variation of the alkyl bromides afforded the corresponding products 5b–5j in moderate to good isolated yields. The reaction of α-bromo-γ-butyrolactone with 4-acetoxystyrene and 4-fluorostyrene gave the expected products (5k–5l) demonstrating the applicability of the protocol. This reaction remained effective for a gram-scale protocol as shown by the preparation of 5g and 5h in Scheme 4.
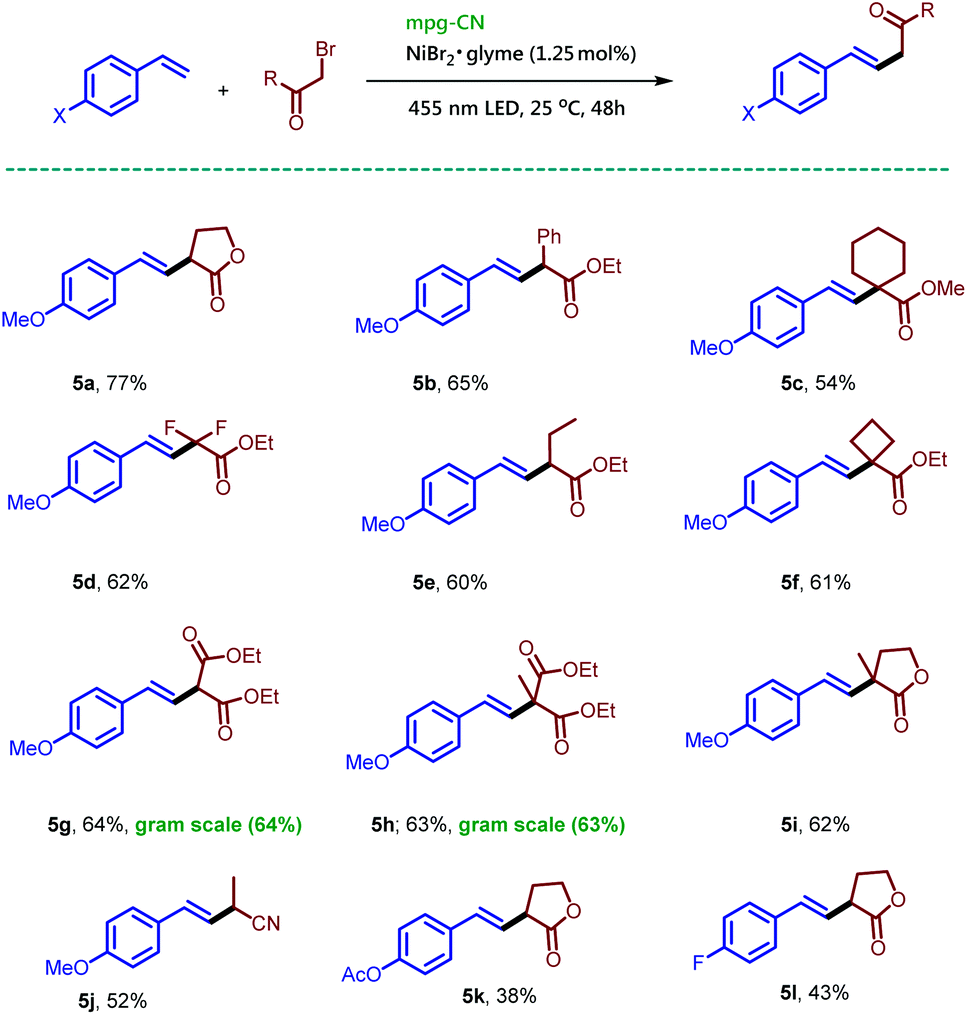 | ||
| Scheme 4 Mizoroki–Heck type reactions of olefins and alkyl bromides using mpg-CN as a heterogeneous photocatalyst. Standard conditions: 4-methoxystyrene (26.8 mg, 0.2 mmol, 1.0 equiv.), alkyl bromide (0.4 mmol, 2.0 equiv.), mpg-CN (10.0 mg), NiBr2·glyme (0.7 mg, 0.0025 mmol, 0.0125 equiv.), DMF (1 mL), under a nitrogen atmosphere for 48 h. Similar conditions were used for gram scale reactions on a 6 mmol scale; for more details, see the ESI.† | ||
Apart from operational simplicity, one of the main advantages of our protocol is the use of the heterogeneous mpg-CN photocatalyst, which can be easily recovered from the reaction mixture, even from gram-scale reactions, via simple filtration or centrifugation. The heterogeneous nature and the remarkable stability of mpg-CN under the photochemical reaction conditions allow the reuse of the material several times without the loss of the photocatalyst reactivity or decrease of the yield of the desired product. As shown in Fig. 1 the photocatalyst can be recycled at least six times, and the rates of the photochemical reactions remain the same over four catalytic cycles.
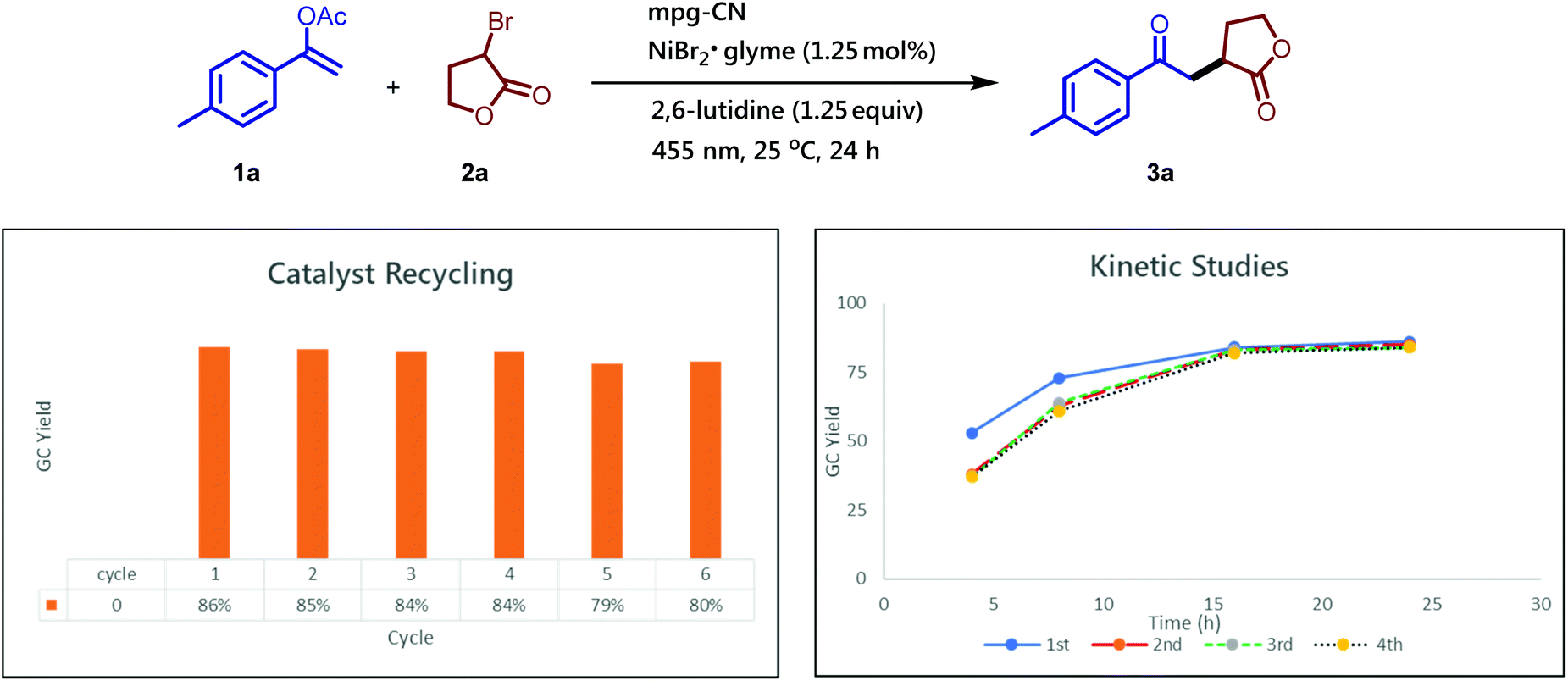 | ||
| Fig. 1 Catalyst recycling (for six catalytic cycles) and assessment of the reaction rates (for four catalytic cycles). Standard conditions: 1-(p-tolyl)vinyl acetate (35.2 mg, 0.2 mmol, 1.0 equiv.), α-bromo-γ-butyrolactone (49.5 mg, 0.3 mmol, 1.5 equiv.), mpg-CN (10.0 mg), NiBr2·glyme (0.7 mg, 0.0025 mmol, 0.0125 equiv.), and 2,6-lutidine (29 μL, 0.25 mmol, 1.25 equiv.) in DMF (1 mL), under a nitrogen atmosphere. GC yields were determined using 1,3,5-trimethoxybenzene as an internal standard. | ||
After the photocatalytic experiments, mpg-CN was washed with different solvents and water, and characterized by the same set of techniques used for fresh mpg-CN (see the ESI† for details). Thus, the position and intensity of all peaks observed in the attenuated total reflectance Fourier-transform infrared (ATR FT-IR) spectrum of mpg-CN revealed that the bulk chemical structure of the photocatalyst had not changed (Fig. S2†). Energy Dispersive X-Ray Analysis (EDX) and X-ray photoelectron spectroscopy (XPS) revealed slightly enhanced oxygen content (Table S1, S2 and Fig. S3b†) that we explained by partial hydrolysis of terminal amino-groups on the surface of mpg-CN during work-up washing with water. Inductively coupled plasma optical emission spectrometry (ICP-OES) revealed that mpg-CN after the photocatalytic tests contained 0.0268 ± 0.00033 wt% of Ni (Table S3,† entry 2). The detection limit of nickel by ICP-OES was determined to be >0.0013 ± 0.00001 wt% (Table S3,† entry 1). In agreement with the results of ICP-OES, Ni 2p3/2 XPS showed a distinct signal of nickel (Fig. S13b†). Due to its low intensity, a precise assignment of oxidation states is not possible. However, considering the presence of the complementary signal in high-resolution Br 3d XPS (Fig. S14b†), it likely arises from Ni(II) coordinated by the lone pairs of nitrogen rather than agglomerated Ni(0) particles. Such Ni(II) species could be considered a precatalyst. Taking into account the loading of NiBr2·glyme (0.77 mg) and mpg-CN (10 mg) in a typical experiment, <1% of nickel was retained in the mpg-CN after catalyst recovery. Given that our synthetic protocol does not require explicitly added ligands, the mpg-CN framework due to abundant nitrogen atoms, might indeed act as a polydentate chelating ligand stabilizing the Ni(II) precatalyst. In agreement with the hypothesis, Ni in the recovered mpg-CN is represented by atomically dispersed species rather than Ni(0) agglomerates. The powder X-Ray diffraction (PXRD) pattern that is similar to the PXRD pattern of fresh mpg-CN and does not show peaks that could be assigned to Ni(0) (Fig. S8†). Furthermore, analysis of transmission electron microscopy (TEM) images confirmed that the mesoporous structure of mpg-CN is retained after the photocatalytic synthesis (Fig. S12d†). High resolution TEM (HR-TEM, Fig. S12d†) and high angle annular darkfield scanning transmission electron microscopy (HAADF-STEM) (Fig. S12e†) also revealed the absence of Ni(0) agglomerates. HAADF-STEM in high resolution confirmed the presence of single heavy atom clusters that could be ascribed to Ni (Fig. S12e†) further supporting our hypothesis that Ni atoms are stabilized by chelation by the mpg-CN framework. However, the observed single atom clusters could alternatively represent Si derived from the template. The sensitivity of the technique is too low to support or refute this hypothesis. It should be noted that the deposition of Ni black (1.4–12.6 wt%) on the carbon nitride photocatalyst has been observed by Pieber et al. in dual nickel/photoredox C–N coupling.26a,29 We explain the difference in nickel content in the recovered photocatalyst found in this work (∼0.03 wt%) and earlier reports (up to 13 wt%) mainly by the different structures of the carbon nitride photocatalysts, covalent mpg-CN (used in this work) and ionic CN-OA-m30 (used by Pieber et al.). Ionic carbon nitrides contain N-metal moieties in their structure that can undergo ion exchange.31,28e In the context of dual Ni/photoredox catalysis, at the first step, covalent carbon nitrides form a chelating 16 electron nickel precatalyst complex (Fig. S15a†), while ionic carbon nitrides coordinate nickel atoms via transmetalation and form a more reactive 14 electron Ni(II)-amide complex (Fig. S15b†).
Therefore, the difference in the electronic configurations of the Ni(II) complexes coordinated by carbon nitrides defines their reactivity and tendency to form nickel black. Although the exact mechanistic picture of this transformation remains to be elucidated, we depict a working hypothesis in Fig. 2 based on the experimental results. The exact role of the nickel(II) salt is not clear; we speculate that the nickel salt acts as a Lewis acid to coordinate with the ester group of the alkyl bromide and thereby lowers the reduction potential of the alkyl bromide. Alternatively, it may be possible that it could activate the double bond by coordinating with the olefin as product formation was observed in the presence of other Lewis acids. However, reactions involving non-activated alkyl bromides did not yield any product, which ruled out a typical cross-coupling mechanism. Based on these observations we believe that mpg-CN under photochemical illumination generates two-dimensional surface redox centers as electron–hole pairs. The photogenerated electron effectively reduces the alkyl bromide and generates the alkyl radical. The alkyl radical adds to the double bond of vinyl acetate and the resulting radical is oxidized by the photogenerated hole delivering the carbocation. Successive loss of the acetyl group presumably supported by nucleophiles present in the reaction medium such as bromide anions or the solvent yields the corresponding 1,4-dicarbonyl compounds, whereas the proton loss in the case of olefins results in a Mizoroki–Heck type cross-coupling product.
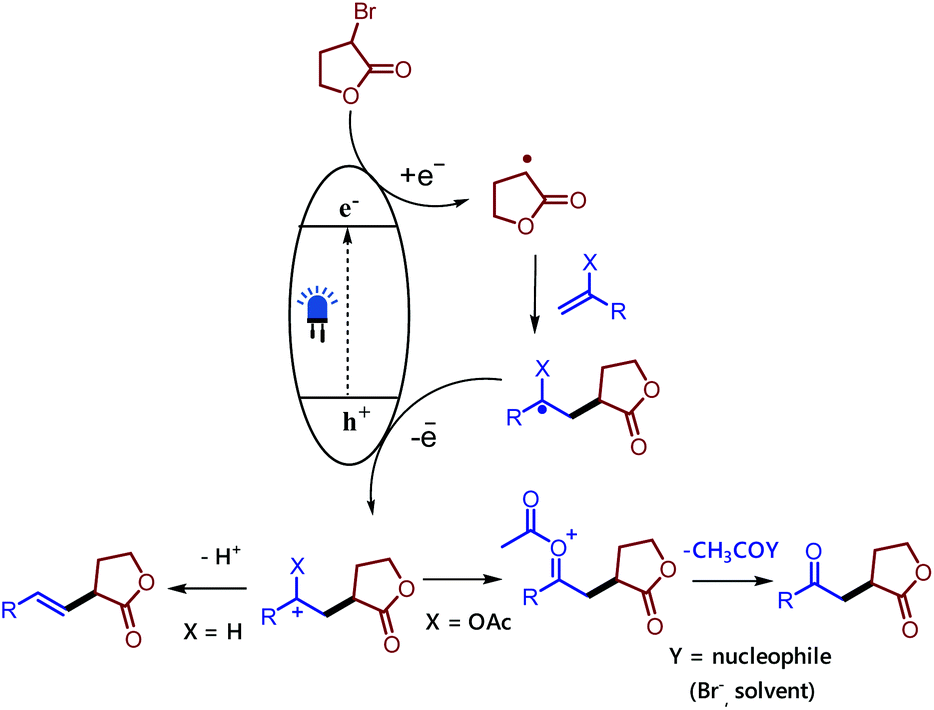 | ||
| Fig. 2 Plausible mechanism for the reaction involving a mpg-CN semiconductor. | ||
Conclusions
The present study illustrates the potential of mesoporous graphitic carbon nitride (mpg-CN) as a heterogeneous photocatalyst in the synthesis of 1,4-dicarbonyl compounds and Mizoroki–Heck type cross-coupling reactions from simple starting materials such as alkyl bromides and vinyl acetates or olefins. The protocol, based on a purely organic semiconductor photocatalyst, cheap and Earth abundant Lewis acid catalyst, provides a powerful alternative to conventional homogeneous catalysis. As the aforementioned transformations are extremely valuable and essential methods in organic synthesis, our protocol involving facile recovery and reuse of the mpg-CN photocatalyst overcomes some of its shortcomings in terms of sustainability and efficiency and the method is recommended for application in academia and industry with great environmental benefits.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the German Science Foundation (DFG, KO 1537/18-1). This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (grant agreement no. 741623). We thank Dr Rudolf Vasold for GC-MS measurements and Regina Hoheisel for CV measurements. The authors also thank Miss Jiamei Liu at the Instrument Analysis Center of Xi'an Jiaotong University for her assistance with XPS analysis, Bolortuya Badamdorj for acquiring TEM images and Dr. Nadezda V. Tarakina for fruitful discussion of TEM data interpretation.References
-
(a) M. Whittaker, C. D. Floyd, P. Brown and A. J. H. Gearing, Chem. Rev., 1999, 99, 2735 CrossRef CAS
; (b) T. Eicher, S. Hauptmann and A. Speicher, Chemistry of Heterocycles, Wiley–VCH, 2003 CrossRef
-
(a) J. Yang, F. Mei, S. Fu and Y. Gu, Green Chem., 2018, 20, 1367 RSC
- H. Setter, Angew. Chem., Int. Ed., 1976, 15, 639 CrossRef
-
(a) M. C. Myers, A. R. Bharadwaj, B. C. Milgram and K. A. Scheidt, J. Am. Chem. Soc., 2005, 127, 14675 CrossRef CAS
-
(a) P. S. Baran and M. P. DeMartino, Angew. Chem., Int. Ed., 2006, 45, 7083 CrossRef CAS
-
(a) C. T. Avetta, L. C. Konkol, C. N. Taylor, K. C. Dugan, C. L. Stern and R. J. Thomson, Org. Lett., 2008, 10, 5621 CrossRef CAS
-
(a) H.-Y. Jang, J.-B. Hong and D. W. C. MacMillan, J. Am. Chem. Soc., 2007, 129, 7004 CrossRef CAS
-
(a) C. Liu, Y. Deng, J. Wang, Y. Yang, S. Tang and A. Lei, Angew. Chem., Int. Ed., 2011, 50, 7337 CrossRef CAS
- G. Goti, B. Bieszczad, A. Vega-PeÇaloza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 1213 CrossRef CAS
- T. Morack, C. Mück-Lichtenfeld and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 1208 CrossRef CAS
- G.-Z. Wang, R. Shang, W.-M. Cheng and Y. Fu, Org. Lett., 2015, 17, 4830 CrossRef CAS
- C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991 CrossRef CAS
- Y. Zhu, L. Zhang and S. Luo, J. Am. Chem. Soc., 2014, 136, 14642 CrossRef CAS
- S. Mazzanti, B. Kurpil, B. Pieber, M. Antonietti and A. Savateev, Nat. Commun., 2020, 11, 1387 CrossRef CAS
-
(a) Q. Liu, R. Wang, H. Song, Y. Liu and Q. Wang, Adv. Synth. Catal., 2020, 362, 4391 CrossRef CAS
-
(a) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef CAS
-
(a) S. Iyer, C. Ramesh, A. Sarkar and P. P. Wadgaonkar, Tetrahedron Lett., 1997, 38, 8113 CrossRef CAS
-
(a) D. Kurandina, M. Parasram and V. Gevorgyan, Angew. Chem., Int. Ed., 2017, 56, 14212 CrossRef CAS
-
(a) G.-Z. Wang, R. Shang, W.-M. Cheng and Y. Fu, J. Am. Chem. Soc., 2017, 139, 18307 CrossRef CAS
- G.-Z. Wang, R. Shang and Y. Fu, Org. Lett., 2018, 20, 888 CrossRef CAS
-
(a) W. J. Zhou, G. M. Cao, G. Shen, X. Y. Zhu, Y. Y. Gui, J. H. Ye, L. Sun, L. L. Liao, J. Li and D. G. Yu, Angew. Chem., Int. Ed., 2017, 56, 15683–15687 CrossRef CAS
-
(a) I. Ghosh, J. Khamrai, A. Savateev, N. Shlapakov, M. Antonietti and B. Konig, Science, 2019, 365, 360 CrossRef CAS
-
(a) X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS
- J. Khamrai, I. Ghosh, A. Savateev, M. Antonietti and B. König, ACS Catal., 2020, 10, 3526 CrossRef CAS
- M. Woźnica, N. Chaoui, S. Taabache and S. Blechert, Chem. – Eur. J., 2014, 20, 14624 CrossRef
-
(a) B. Pieber, J. A. Malik, C. Cavedon, S. Gisbertz, A. Savateev, D. Cruz, T. Heil, G. G. Zhang and P. H. Seeberger, Angew. Chem., Int. Ed., 2019, 58, 9575 CrossRef CAS
- Due to the presence of surface primary and secondary amino groups (Fig. S5†), mpg-CN in addition to its role as a photocatalyst may facilitate deprotonation of the intermediate. Indeed, carbon nitride materials have been studied as a solid base catalyst, for example, in Knoevenagel condensations.
(a) F. Su, M. Antonietti and X. Wang, Catal. Sci. Technol., 2012, 2, 1005–1009 RSC
-
(a) Z. Chen, A. Savateev, S. Pronkin, V. Papaefthimiou, C. Wolff, M. G. Willinger, E. Willinger, D. Neher, M. Antonietti and D. Dontsova, Adv. Mater., 2017, 29, 1700555 CrossRef
- S. Gisbertz, S. Reischauer and B. Pieber, Nat. Catal., 2020, 3, 611 CrossRef CAS
- G. Zhang, G. Li, Z.-A. Lan, L. Lin, A. Savateev, T. Heil, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2017, 56, 13445 CrossRef CAS
- A. Savateev and M. Antonietti, ChemCatChem, 2019, 11, 6166 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc03792c |
| This journal is © The Royal Society of Chemistry 2021 |