
One-step plasma-enabled catalytic carbon dioxide hydrogenation to higher hydrocarbons: significance of catalyst-bed configuration†
Jiajie
Wang
ab,
Mohammad S.
AlQahtani
 ac,
Xiaoxing
Wang
*a,
Sean D.
Knecht
d,
Sven G.
Bilén
de,
Chunshan
Song
*acf and
Wei
Chu
b
ac,
Xiaoxing
Wang
*a,
Sean D.
Knecht
d,
Sven G.
Bilén
de,
Chunshan
Song
*acf and
Wei
Chu
b
aClean Fuels & Catalysis Program, EMS Energy Institute, Department of Energy and Mineral Engineering, The Pennsylvania State University, University Park, PA 16802, USA. E-mail: xuw4@psu.edu; cxs23@psu.edu; chunshansong@cuhk.edu.hk
bDepartment of Chemical Engineering, Sichuan University, Sichuan, P.R. China
cDepartment of Chemical Engineering, The Pennsylvania State University, University Park, PA 16802, USA
dSchool of Engineering Design, Technology, and Professional Programs, The Pennsylvania State University, University Park, PA 16802, USA
eSchool of Electrical Engineering and Computer Science, The Pennsylvania State University, University Park, PA 16802, USA
fDepartment of Chemistry, Faculty of Science, The Chinese University of Hong Kong, Shatin, Hong Kong, China
First published on 10th February 2021
Abstract
Effectively converting CO2 into fuels and value-added chemicals remains a major challenge in catalysis, especially under mild conditions. In this study, we report a one-step plasma-enabled catalytic process for CO2 hydrogenation to C2+ hydrocarbons operated at low temperature and atmospheric pressure in a dielectric barrier discharge (DBD) packed-bed reactor. Plasma without catalyst produces mainly CO (over 80% selectivity), while CH4 becomes the main product when plasma is coupled with the alumina-supported Co catalyst. Interestingly, by simply changing the catalyst-bed configuration within the plasma discharge zone, more C2+ hydrocarbons are selectively produced. High C2+ hydrocarbons selectivity of 46% at ca. 74% CO2 conversion is achieved when operated at the furnace temperature of 25 °C and 10 W DBD plasma. The possible origin of C2+ formation and the significance of catalyst-bed configuration are discussed.
Transforming waste CO2 into chemicals and fuels offers not only a possible solution to mitigate anthropogenic CO2 emissions, but also an alternative to alleviate dependence on fossil fuels,1 supporting sustainable development and green chemistry.1–6 CO2 is thermodynamically stable (ΔG° = −393.5 kJ mol−1), thus preferably converted with a co-reactant having higher free Gibbs energy such as CH4 (ΔG° = −50.7 kJ mol−1) and H2 (ΔG° = 0 kJ mol−1).1 CO2 reaction with H2 is thermodynamically favorable;7 furthermore, H2 can be produced from H2O using renewable energy, which makes the overall process environmentally friendly.1
Much effort has been devoted to improving the performance of catalysts for producing higher hydrocarbons from CO2.8–11 C2+ hydrocarbons can be used as a liquid fuel or an entry platform (e.g., light olefins) for existing chemical production chains.12,13 In order to achieve high C2+ yield, high pressure (1.0–4.0 MPa) under the temperature of 200–400 °C is normally required.2 Another critical issue in thermal-catalytic processes is the catalyst deactivation caused by metal sintering and carbon deposition.14 Thus, achieving CO2 activation and carbon chain growth under mild conditions remains a great challenge.2,13
Non-thermal plasma (NTP) provides a unique medium for performing catalytic CO2 conversion at low temperatures owing to its non-equilibrium characteristics. Highly energetic electrons (1–10 eV) generated in NTP collide with gas molecules to produce highly reactive species (i.e., ions, radicals, excited atoms and excited molecules), enabling thermodynamically and/or kinetically unfavored reactions at low temperatures.15–17
Although various catalysts, such as nickel,18–20 cobalt,21 copper,7,22 platinum,7 transition metal oxide23 and molecular sieves,21 have been studied for plasma-catalytic CO2 hydrogenation during the past few decades,1,16,24 CO and CH4 are always the major products while the amount of C2+ is scarce. Only Lan et al. have achieved 13.7% selectivity of higher hydrocarbons over a Co/ZSM-5 catalyst.21 Hence, the selective production of long-chain hydrocarbons from CO2 hydrogenation using plasma-catalysis is still challenging.
Herein, we report a non-thermal plasma-driven catalytic process for one-step conversion of CO2 and H2 into higher hydrocarbons operated at low temperature and atmospheric pressure. We demonstrate the significance of catalyst-bed configuration within a dielectric barrier discharge (DBD) plasma discharge zone for the production of C2+ hydrocarbons on a γ-Al2O3 supported Co catalyst (the content of cobalt metal was fixed at 15 wt% based on the support, and termed as 15Co). To the best of our knowledge, it is the first report of plasma-enabled one-step CO2 hydrogenation into C2+ hydrocarbons with C2+ selectivity over 46% at ca. 74% CO2 conversion.
Fig. 1a shows the CO2 conversion and product selectivity in CO2 hydrogenation under different operation modes including catalyst alone, plasma alone, and plasma-catalyst operated at the furnace temperature of 25 °C (no external heating). As expected, both 15Co catalyst and alumina support are thermally inactive at room temperature. Plasma alone initiates CO2 hydrogenation at mild conditions with CO as the main product (over 80% selectivity), consistent with early reports.7,23 Packing alumina in the discharge zone does not seem to affect the plasma performance. Coupling 15Co catalyst with plasma significantly changes the product distribution, as the methane selectivity increases significantly from 3% to 45%, while CO selectivity drops down to 38%. Additionally, a small amount of C2+ hydrocarbons (ca. 3% selectivity) is produced. Part of the observed changes could be attributed to plasma-induced thermal effects as the temperature inside the catalyst bed increased to 200 °C (Table S3†).
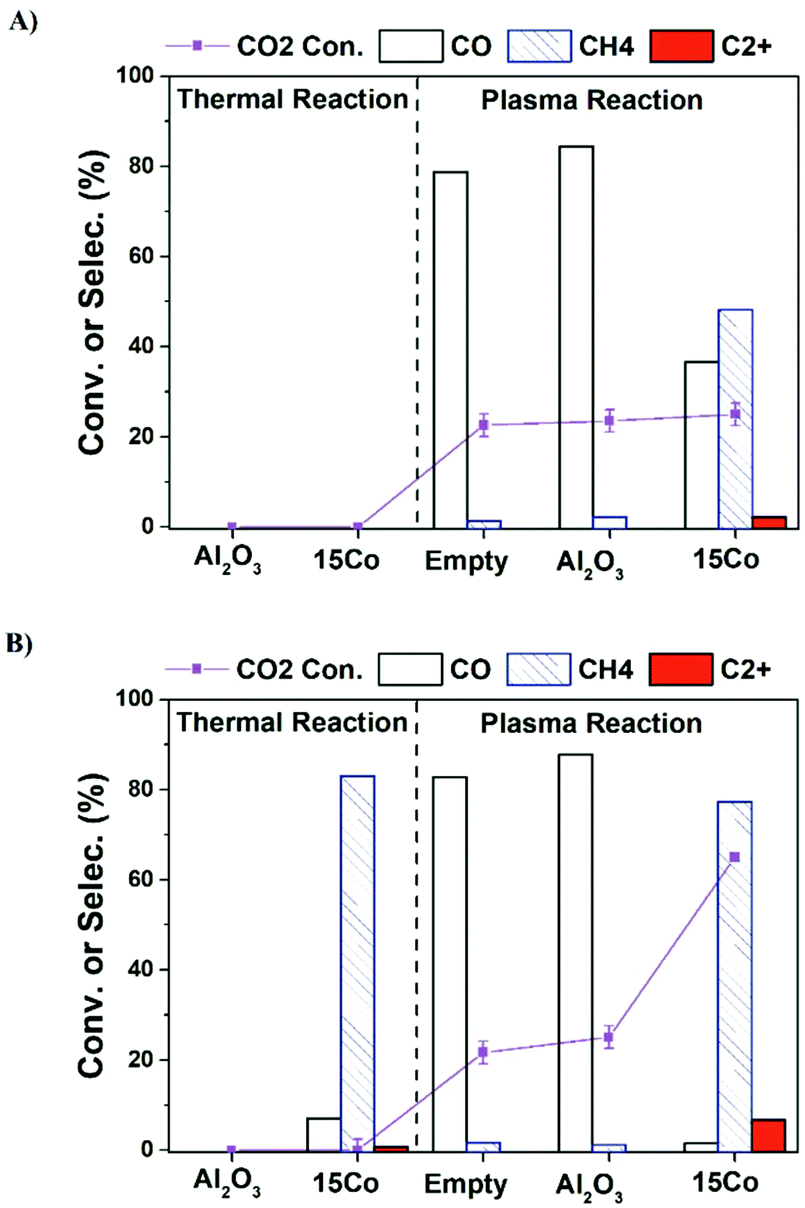 | ||
| Fig. 1 CO2 conversion and product selectivity in the three different modes including catalyst alone, plasma alone and plasma-catalyst operated (a) without external heating and (b) at furnace temperature of 250 °C. Reaction conditions: 20 v% CO2–60 v% H2 in Ar balance, H2/CO2 = 3, flow rate = 20 mL min−1, P = 1 atm, voltage = 13.6 kV, frequency = 23.5 kHz, plasma power = 4 W. | ||
Although C2+ hydrocarbons are successfully obtained when operated at the furnace temperature of 25 °C (Fig. 1a), its selectivity is low. In order to get a more distinct result in C2+ production for accurate comparison, we have further operated the reactions at the furnace temperature of 250 °C. Fig. 1b illustrates the influence of increasing the furnace temperature to 250 °C, which reveals the possibility of obtaining more C2+ hydrocarbons. Under thermal reaction conditions, about 45% of CO2 is converted over the 15Co catalyst, and methane is the main product (83% selectivity) along with traces of C2+ hydrocarbons (<1%). For an empty or alumina-packed reactor under plasma, both CO2 conversion and CO selectivity do not show appreciable changes after raising the operating temperature to 250 °C. In contrast, a significant increase in CO2 conversion (63%) and methane selectivity (81%) is observed over the 15Co catalyst under plasma. More importantly, the selectivity to C2+ hydrocarbons increases from 3% to 7%.
It should be pointed out that plasma can generate heat and thus cause the reactor temperature to rise. At the furnace temperature of 250 °C, unlike the thermal reactions where the reactor temperature is normally similar to the furnace temperature, the real reaction temperature with excited electrons could be much higher in the presence of plasma. Therefore, the reactor temperature was measured (within 10 s) by swiftly replacing the high-voltage (HV) electrode with a thermocouple in the center of the catalyst bed when the stable plasma-catalytic reaction is obtained. The measured reactor temperature was about 400 °C with the presence of plasma either empty or packed with alumina or the 15Co catalyst (Table S2†).
Recent work on plasma-assisted CO2 and CH4 reforming25–28 shows mainly the production of CO, H2 and oxygenates while only a small amount of higher hydrocarbons was reported. On the contrary, almost no oxygenates are detected in our system. Hence, we think the reaction process in our system is different from plasma-assisted dry reforming. We hypothesize that the formation of C2+ is mainly from plasma-driven activation and reaction of the produced methane. If the produced CH4 is effectively converted via electron impact fragmentations (R1, R2), which is otherwise thermodynamically unfavorable even at 400 °C (R3, R4), the formation of higher hydrocarbons could be enhanced. To test our hypothesis, we have designed a series of catalyst-bed configurations for CO2 hydrogenation to higher hydrocarbons under plasma (details of each configuration are given in the Table S1†). The obtained CO2 conversion and product selectivity are presented in Fig. 2. The reactor temperature was also measured for different bed-configurations at different pre-marked positions for a better assessment of the temperature distribution under DBD plasma. As seen in Fig. S3 and Table S2,† when the furnace temperature is 250 °C at the plasma applied voltage of 13.6 kV, the temperature inside the reactor reaches about 400 °C for all configurations. The temperature distribution is relatively uniform within the entire bed.
CH3 − H + e− ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ⇄ CH3˙ + H˙ + e− ⇄ CH3˙ + H˙ + e− | (1) |
| H − CH3 − H + 2e− ⇄ :CH2 + 2H˙ + 2e− | (2) |
| 2CH4 ⇄ C2H6 + H2 ΔG (400 °C) = 71 kJ mol−1 | (3) |
| 3CH4 ⇄ C3H8 + H2 ΔG (400 °C) = 159 kJ mol−1 | (4) |
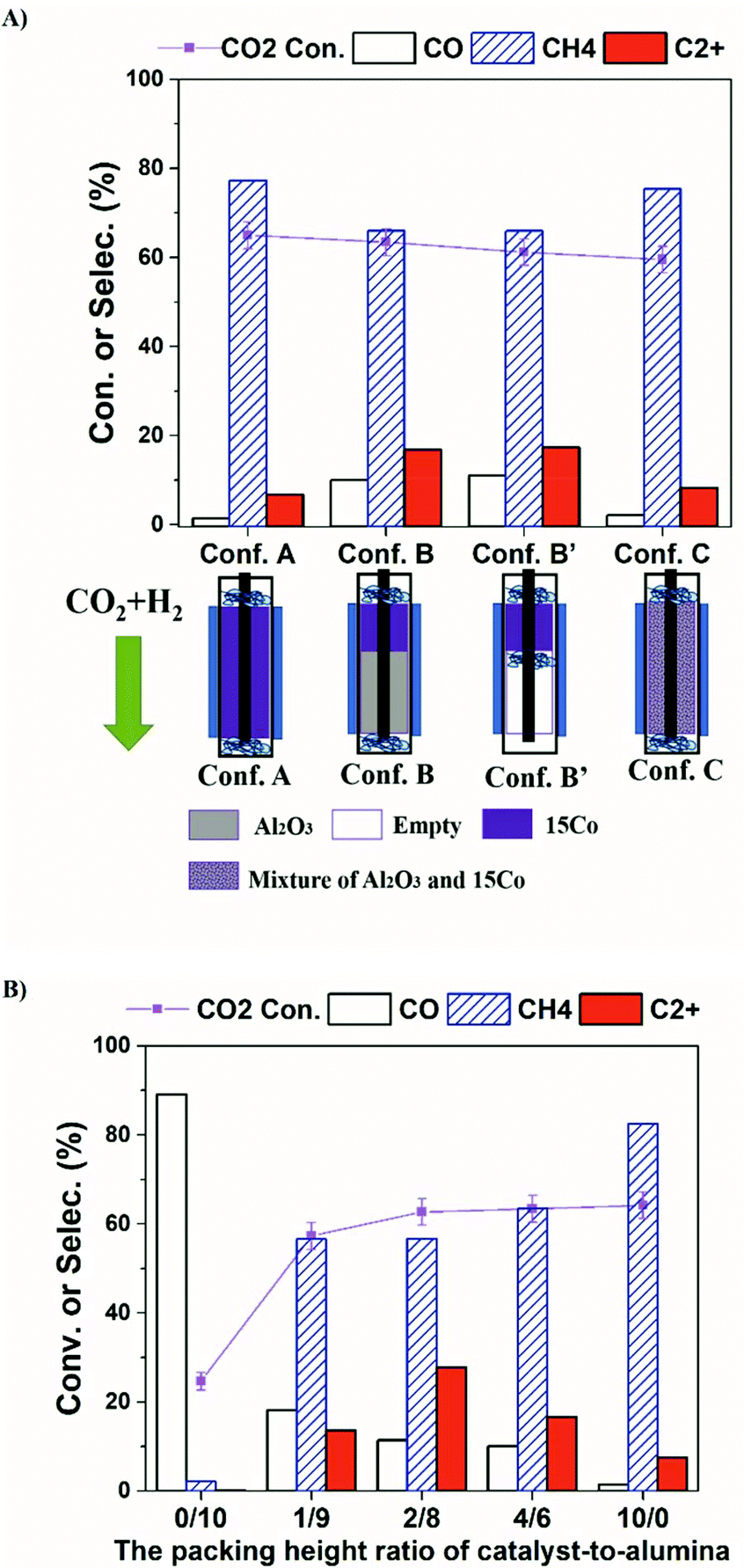 | ||
| Fig. 2 Influence of catalyst-bed configuration (a) and catalyst-to-alumina ratio over Conf. B (b) on the CO2 conversion and product distributions in the CO2 hydrogenation over 15Co catalyst coupled with DBD plasma at the furnace temperature of 250 °C. Reaction conditions: 20 v% CO2–60 v% H2 in Ar balance, H2/CO2 = 3, flow rate = 20 mL min−1, P = 1 atm, voltage = 13.6 kV, frequency = 23.5 kHz, plasma power = 4 W. | ||
In our DBD plasma reactor, gases flow vertically downward through the catalyst bed and the height of the discharge zone is fixed at 5 cm. The catalyst amount is first reduced from 1.25 g (fully packed, termed as Conf. A) to 0.50 g, occupying only the top 2 cm of the discharge zone, while the rest is packed with γ-Al2O3 particles (termed as Conf. B, and noted as the ratio of 4/6 in the packing-height for catalyst-to-support). Compared to the discharge zone fully packed with the catalyst (Conf. A), Conf. B significantly enhances the selectivity towards C2+ hydrocarbons from 7% to 17%, while the CO2 conversion stays the same (∼62%). For a fair comparison, the same amounts of the catalyst and alumina support as those for the Conf. B are physically and homogeneously mixed, then packed fully within the discharge zone, which is termed as Conf. C. Interestingly, methane again becomes the dominant product and the product distribution is very similar to that from the Conf. A, while the CO2 conversion does not change significantly. The results clearly demonstrate that the configuration of the catalyst-bed significantly influences the reactions in plasma-enhanced CO2 hydrogenation, resulting in the change in the product distributions. Compared to thermal catalysis at 400 °C, where only trace amount of C2+ hydrocarbons (1.4%) were formed (Table S4†), Conf. B seems to separate CH4 formation and C−C coupling into two different packing portions with complementary properties, indicating the critical role of plasma for the formation of C2+ products. CO2 is reduced to CH4 over 15Co catalyst with plasma, whereas C−C coupling occurs within the alumina-packed plasma portion. Consequently, identification of the thermal-catalysis and plasma-catalysis contribution on the overall CO2 hydrogenation to C2+ hydrocarbons over cobalt catalyst is crucial, which is under investigation in our laboratory.
The C2+ products are mainly C2–C5 paraffins along with some iso-paraffins and olefins, which are summarized in Table 1. Comparing Conf. B with Conf. B′ (which has a similar bed configuration as Conf. B except no alumina packed in the second part of the discharge zone), there is no significant difference in both CO2 conversion and products selectivity. No carbon deposition was observed on the reactor wall when using Conf. B′. In thermal catalytic reactions, acid sites on alumina usually work as active sites for isomerization or carbon deposition.29,30 Here, the Conf. B and Conf. B′ exhibit similar distributions in C2+ hydrocarbon product, iso/normal ratio (I/N), and olefin/paraffin ratio (O/P), indicating that the alumina-packing does not affect the methane conversion reactions, which occur in the gas phase and are driven by the plasma. Hence, we keep alumina packed for further study.
| Mode | Molar distributionb /% | Carbon balance/% | I/Nc | O/Pd | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C2= | C2 | C3= | C3 | i-C4 | C4= | n-C4 | i-C5 | C5= | n-C5 | ||||
| a Furnace temperature = 250 °C; P = 1 atm; 20 v% CO2–60 v% H2 in Ar balance; total flow = 20 ml min−1; voltage = 13.6 kV; frequency = 23.5 kHz; plasma power = 4 W. b Methane is not included here. c Iso-paraffins to normal paraffins molar ratio (C4–C5). d Olefins to paraffins molar ratio (C2–C5). | |||||||||||||
| Conf. B | 1.2 | 69.8 | 0.7 | 18.5 | 4.7 | 0.6 | 2.5 | 1.2 | 0.4 | 0.4 | 96 | 2.1 | 0.03 |
| Conf. B′ | 2.5 | 65.8 | 1.1 | 19.4 | 5.1 | 1.0 | 2.9 | 1.4 | 0.4 | 0.4 | 97 | 2.0 | 0.05 |
Fig. 2b illustrates the effect of the packing-height ratio between the 15Co catalyst and the alumina support in the discharge zone (Conf. B). Increasing the height of catalyst-packing increases the CO2 conversion from 58% to 63%, whereas the overall C2+ selectivity increases first, reaching 27.8% at the packing-height ratio of 2/8, then decreases. When less catalyst is used (i.e., at the packing-height ratio of 1/9), less CO2 is converted to methane. Consequently, the concentration of active CHx species generated from methane activation by plasma is relatively low, which limits the carbon-chain growth in the second part of the bed. Hence, the overall C2+ selectivity at 1/9 ratio is low. The CO2 molecules that have not been reduced to methane are further converted to CO in the alumina sector by plasma, leading to a higher CO selectivity. On the contrary, when more catalyst is packed (e.g., at the packing-height ratio of 4/6), the catalyst amount is sufficient to convert CO2 at near equilibrium level as evidenced by the similar CO2 conversion shown in Fig. 2b. However, the length of the alumina zone in plasma becomes relatively short, so does the residence time of plasma-activated CH4 molecules, leading to less probability for carbon-chain propagation to higher hydrocarbons. Thereby, the selectivity of CH4 increases while the C2+ selectivity decreases. As a result, the packing-height ratio of 2/8 gives the optimal C2+ production, for the investigated ratios.
It is worth mentioning that when 15Co catalyst is packed at the bottom 2 cm of the discharge zone, the performance is similar to those of Conf. A and Conf. C (Table S4†). Furthermore, when the total flow rate of the reactant gases increases, methane selectivity increases while the C2+ selectivity decreases as shown in Fig. S5.† The Lissajous figures in Fig. S6† display little change in the macro-scale plasma properties with different catalyst-bed configurations, suggesting that the dramatic change in C2+ production is mainly due to plasma-induced chemical reactions. These results suggest that both methane and CO are primary products, whereas C2+ hydrocarbons are secondary products forming from methane activation. It indicates that the generation of methane in the catalyst bed is essential for the production of higher hydrocarbons in the current plasma-catalyst system, likely through the reactions among the plasma-activated CHx species.7,31,32 Thus, we propose a possible C2+ formation mechanism in our system, which is described in Scheme 1. First, CO2 is selectively reduced to CH4 over 15Co catalyst under DBD plasma. Although CHx species are simultaneously generated by plasma on the catalyst, they are quickly terminated by plasma-activated H species, leading to trace amount of C2+ hydrocarbons, as observed when the catalyst is fully packed (Conf. A). Within the alumina-packed portion, the plasma-activated CHx species interact via gas-phase carbon-chain-growth reactions, generating more C2+ hydrocarbons. The proposed mechanism is supported by the fact that the distribution of C2+ products is very similar to that from the plasma methane conversion (Table S5†). However, spectroscopic analysis is needed to mechanistically understand reaction pathways, which is underway in our lab.
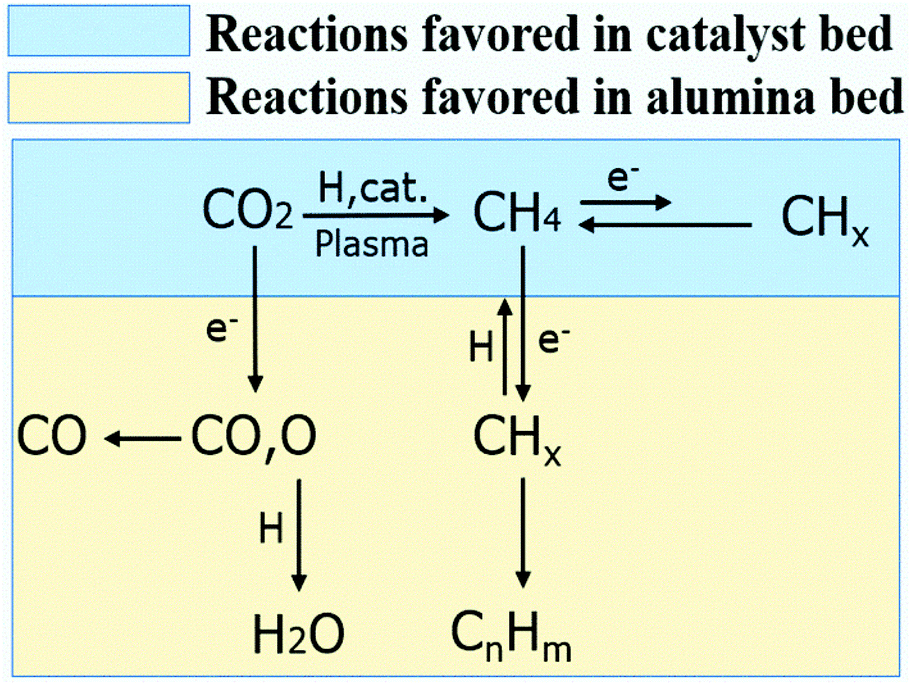 | ||
| Scheme 1 The possible reaction pathways for higher hydrocarbons formation in the plasma promoted low-temperature CO2 hydrogenation. | ||
The production of C2+ hydrocarbons can be further enhanced via increasing the input plasma power. As shown in Fig. 3, higher CO2 conversion is obtained over all cases by increasing plasma power to 10 W compared to the results in Fig. 1a (4 W). The selectivity to C2+ hydrocarbons is significantly improved. With Conf. B under a 10 W plasma at the furnace temperature of 25 °C (with no external heating), C2+ selectivity as high as 46.5% at CO2 conversion of 74% (i.e., C2+ hydrocarbons yield of 34.4%) is obtained. After 2 hours of reaction, we found the catalyst-bed temperature was about 400 °C. This is a result of inevitable heating effect of plasma and possibly the heat releasing from CO2 hydrogenation to hydrocarbons.18,20 Therefore, we have examined the thermal catalytic CO2 hydrogenation at 400 °C without plasma (Table S3†). Although the CO2 conversion is high (71%), the C2+ selectivity is only 1.4% while CH4 selectivity is 92%. Such a difference strongly substantiates the key role of plasma and catalyst-bed configuration in CO2 hydrogenation to higher hydrocarbons.
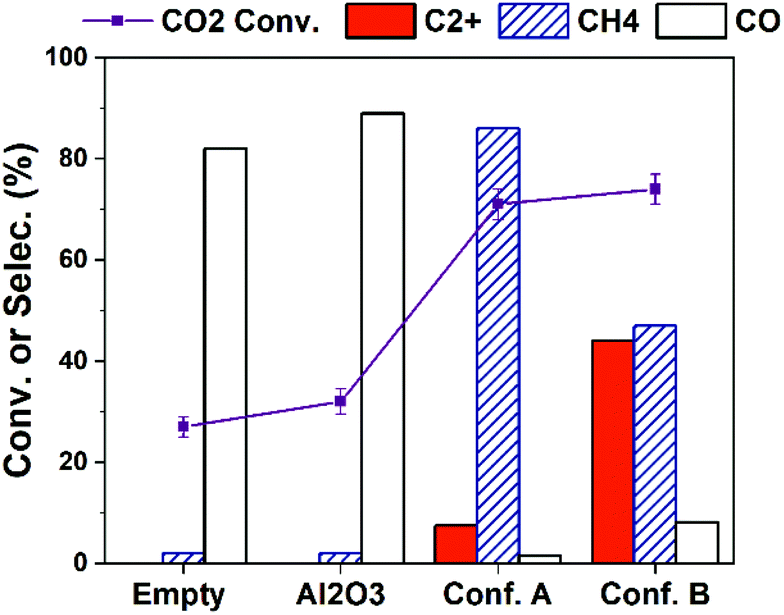 | ||
| Fig. 3 Influence of catalyst-bed configuration on the CO2 hydrogenation performance over 15Co catalyst under 10 W of DBD plasma at the furnace temperature of 25 °C. Reaction conditions: 20 v% CO2–60 v% H2 in Ar balance, H2/CO2 = 3, flow rate = 20 mL min−1, P = 1 atm, voltage = 18.5 kV, frequency = 23.5 kHz. | ||
In plasma methane conversion, carbon deposition is usually observed in the first few hours.26,33 On the other hand, metal sintering is a common problem of cobalt catalyst in CO2 hydrogenation.14 Hence, we have examined the stability of the plasma catalytic process (Conf. B). As shown in Fig. 4, the reaction reaches steady state in a fairly short time and is stable for at least 6 hours of operation with plasma. No tendency of deactivation is observed, which can be attributed to the existence of plasma-induced active H or O radicals to remove deposited carbon and the ability of plasma to enhance metal dispersion and/or prevent metal agglomeration.18,34,35
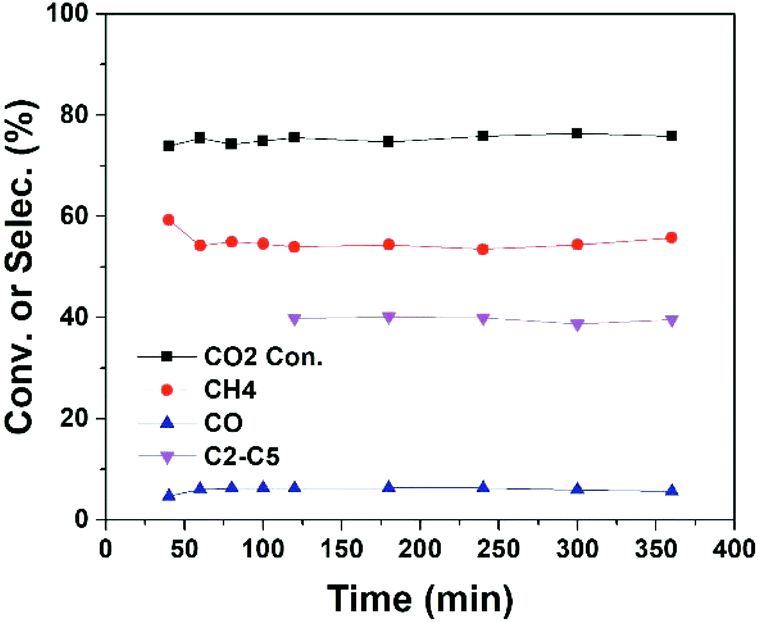 | ||
| Fig. 4 CO2 conversion and products selectivity as a function of time on stream (TOS) in CO2 hydrogenation over 15Co catalyst under 7 W of DBD plasma at the furnace temperature of 250 °C. Reaction conditions: 17 v% CO2–67 v% H2 in Ar balance, H2/CO2 = 4, flow rate = 20 mL min−1, P = 1 atm, voltage = 16.5 kV, frequency = 23.5 kHz. | ||
In conclusion, our work clearly demonstrates the significant impact of the catalyst-bed configuration on plasma-catalytic CO2 hydrogenation to higher hydrocarbons in one-step operated at low temperature and atmospheric pressure. With proper catalyst-bed configuration, high C2+ hydrocarbons selectivity of 46% at CO2 conversion of over 70% is achieved. C2+ hydrocarbons are likely formed through the plasma-driven gas-phase methane conversion. Thus, the key in optimizing the catalyst-bed configuration is the balance between methane formation and plasma reactions for carbon-chain-growth from methane. It should also be pointed out that the catalyst used in this work is a conventional alumina-supported Co catalyst for Fischer–Tropsch synthesis. With further optimization of Co catalyst and/or development of more effective catalysts, the plasma-promoted catalytic CO2 hydrogenation to higher hydrocarbons could be more promising. The present work may broaden the utilization of the plasma-catalyst synergy for effective CO2 conversion to higher hydrocarbons.
Author contributions
Jiajie Wang: conceptualization, methodology, investigation, writing – original draft, visualization. Mohammad S. AlQahtani: methodology, writing – review & editing, visualization. Xiaoxing Wang: conceptualization, methodology, resources, writing – original draft, review & editing, visualization, project administration, supervision, funding acquisition. Sean D. Knech: resources, writing – review & editing, visualization. Sven G. Bilén: resources, writing – review & editing, visualization. Chunshan Song: conceptualization, resources, writing – review & editing, visualization, supervision, funding acquisition. Wei Chu: writing – review & editing.Conflicts of interest
The authors claim no conflicts of interest.Acknowledgements
This work is financially supported by the Pennsylvania State University and EMS Energy Institute Seed Grant. J.W. also acknowledges the financial support from the Chinese Scholarship Council (CSC). M.S.Q. gratefully acknowledges the PhD scholarship from Saudi Aramco. The authors would like to thank Dr Na Liu and Dr Wenjia Wang for their helpful discussion and advices.References
- R. Snoeckx and A. Bogaerts, Chem. Soc. Rev., 2017, 46, 5805–5863 RSC.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- A. Sternberg, C. M. Jens and A. Bardow, Green Chem., 2017, 19, 2244–2259 RSC.
- H. Blanco and A. Faaij, Renewable Sustainable Energy Rev., 2018, 81, 1049–1086 CrossRef.
- Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707–3728 RSC.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono and A. Ahmad, Green Chem., 2015, 17, 2647–2663 RSC.
- L. Wang, Y. Yi, H. Guo and X. Tu, ACS Catal., 2017, 8, 90–100 CrossRef.
- W. Wang, X. Jiang, X. Wang and C. Song, Ind. Eng. Chem. Res., 2018, 57, 4535–4542 CrossRef CAS.
- R. Satthawong, N. Koizumi, C. Song and P. Prasassarakich, Top. Catal., 2013, 57, 588–594 CrossRef.
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Nat. Commun., 2017, 8, 15174 CrossRef.
- Z. Li, Y. Qu, J. Wang, H. Liu, M. Li, S. Miao and C. Li, Joule, 2019, 3, 570–583 CrossRef CAS.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711 RSC.
- Y. Gao, S. Liu, Z. Zhao, H. Tao and Z. Sun, Acta Phys.-Chim. Sin., 2018, 34, 858–872 CAS.
- W. Li, X. Nie, X. Jiang, A. Zhang, F. Ding, M. Liu, Z. Liu, X. Guo and C. Song, Appl. Catal., B, 2018, 220, 397–408 CrossRef CAS.
- A. Fridman, Plasma Chemistry, Cambridge University Press, 2008 Search PubMed.
- E. C. Neyts, K. K. Ostrikov, M. K. Sunkara and A. Bogaerts, Chem. Rev., 2015, 115, 13408–13446 CrossRef CAS.
- M. S. AlQahtani, S. D. Knecht, X. Wang, S. G. Bilén and C. Song, ACS Catal., 2020, 10, 5272–5277 CrossRef CAS.
- E. Jwa, S. B. Lee, H. W. Lee and Y. S. Mok, Fuel Process. Technol., 2013, 108, 89–93 CrossRef CAS.
- M. Nizio, A. Albarazi, S. Cavadias, J. Amouroux, M. E. Galvez and P. Da Costa, Int. J. Hydrogen Energy, 2016, 41, 11584–11592 CrossRef CAS.
- M. Mikhail, P. Da Costa, J. Amouroux, S. Cavadias, M. Tatoulian, S. Ognier and M. E. Gálvez, Catal. Sci. Technol., 2020, 10, 4532–4543 RSC.
- L. Lan, A. Wang and Y. Wang, Catal. Commun., 2019, 130, 105761 CrossRef.
- B. Eliasson, U. Kogelschatz, B. Xue and L. Zhou, Ind. Eng. Chem. Res., 1998, 37, 3350–3357 CrossRef CAS.
- Y. Zeng and X. Tu, IEEE Trans. Plasma Sci., 2016, 44, 405–411 CAS.
- A. H. Khoja, M. Tahir and N. A. S. Amin, Energy Convers. Manage., 2019, 183, 529–560 CrossRef CAS.
- L. Wang, Y. Yi, C. Wu, H. Guo and X. Tu, Angew. Chem., Int. Ed., 2017, 56, 13679–13683 CrossRef CAS.
- K. Krawczyk, M. Młotek, B. Ulejczyk and K. Schmidt-Szałowski, Fuel, 2014, 117, 608–617 CrossRef CAS.
- J. Kim, D. B. Go and J. C. Hicks, Phys. Chem. Chem. Phys., 2017, 19, 13010–13021 RSC.
- X. Tu and J. C. Whitehead, Appl. Catal., B, 2012, 125, 439–448 CrossRef CAS.
- J. E. Samad, J. Blanchard, C. Sayag, C. Louis and J. R. Regalbuto, J. Catal., 2016, 342, 203–212 CrossRef CAS.
- J. Ni, L. Chen, J. Lin and S. Kawi, Nano Energy, 2012, 1, 674–686 CrossRef CAS.
- A. Gómez-Ramírez, V. J. Rico, J. Cotrino, A. R. González-Elipe and R. M. Lambert, ACS Catal., 2013, 4, 402–408 CrossRef.
- L. Wang, S. Y. Liu, C. Xu and X. Tu, Green Chem., 2016, 18, 5658–5666 RSC.
- A. H. Khoja, M. Tahir and N. A. S. Amin, Energy Convers. Manage., 2017, 144, 262–274 CrossRef CAS.
- W. Chu, L. N. Wang, P. A. Chernavskii and A. Y. Khodakov, Angew. Chem., Int. Ed., 2008, 47, 5052–5055 CrossRef CAS.
- M. S. AlQahtani, X. Wang, J. L. Gray, S. D. Knecht, S. G. Bilén and C. Song, J. Catal., 2020, 391, 260–272 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc03779f |
| This journal is © The Royal Society of Chemistry 2021 |