
Synthesis of an Fe–Pd bimetallic catalyst for N-alkylation of amines with alcohols via a hydrogen auto-transfer methodology†
Peng-yu
Wu
,
Guo-ping
Lu
 and
Chun
Cai
*
and
Chun
Cai
*
School of Chemical Engineering, Nanjing University of Science & Technology, Xiaolingwei 200, Nanjing 210094, P. R. China. E-mail: c.cai@njust.edu.cn
First published on 9th December 2020
Abstract
Hydrogen auto-transfer (HAT) or borrowing hydrogen (BH) methodology which combines dehydrogenation, intermediate reaction and hydrogenation, is recognized as an excellent strategy for one-pot synthesis from an economic and environmental point of view. Although much effort has been made on the development of catalysts for HAT reactions, harsh conditions, external base or large amounts of noble metals are still required in most reported catalysis systems, and thus the exploration of a highly efficient and recyclable heterogeneous catalyst remains meaningful. In this work, a novel bimetallic catalyst, Fe10Pd1/NC500 derived from bimetallic MOF NH2-MIL-101(Fe10Pd1), has been prepared, and the catalyst exhibits superior catalytic performance for the N-alkylation of amines with alcohols via a hydrogen auto-transfer methodology. High yields of the desired products were achieved at 120 °C with an alcohol/amine molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and required no external additive or solvent. A distinct enhancement in catalytic performance is observed when compared with monometallic catalysts, which can be ascribed to the “synergistic effects” inside the bimetallic alloys. The N-doped carbon support has been revealed to provide the necessary basicity which avoids the requirement of an external base. Moreover, a wide substrate range and remarkable reusability have been shown by Fe10Pd1/NC500, and this work highlights new possibilities for bimetallic catalysts applied in sustainable chemistry.
:1 and required no external additive or solvent. A distinct enhancement in catalytic performance is observed when compared with monometallic catalysts, which can be ascribed to the “synergistic effects” inside the bimetallic alloys. The N-doped carbon support has been revealed to provide the necessary basicity which avoids the requirement of an external base. Moreover, a wide substrate range and remarkable reusability have been shown by Fe10Pd1/NC500, and this work highlights new possibilities for bimetallic catalysts applied in sustainable chemistry.
Introduction
Hydrogen auto-transfer methodology (HAT), also known as the borrowing hydrogen (BH) approach, which combines dehydrogenation, intermediate reaction and hydrogenation is recognized as a green and practical strategy for one-pot synthesis without tedious isolation processes. With the hydrogen atoms from a donor reactant stored by the catalyst and released in the final hydrogenation step, HAT methodology avoids the use of an external hydrogen source and shows both atom-efficiency and step-efficiency.1,2 For instance, the N-alkylation of amines with alcohols via a HAT methodology using alcohols as both the alkylation reagents and hydrogen donors, produces water as the only by-product (Scheme 1).3–6 Since the early reports by Grigg's group7 and Watanabe's group,8 a variety of organometallic homogeneous catalysts have been developed for HAT methodology reactions. Early developments were mediated by complexes of Ru and Ir, and recently the focus is on the Earth-abundant metals.9–11 Although notable progress has been achieved, homogeneous catalysis still requires tedious catalyst preparation under inert conditions and external additives, and the catalysts could hardly be separated and reused.12,13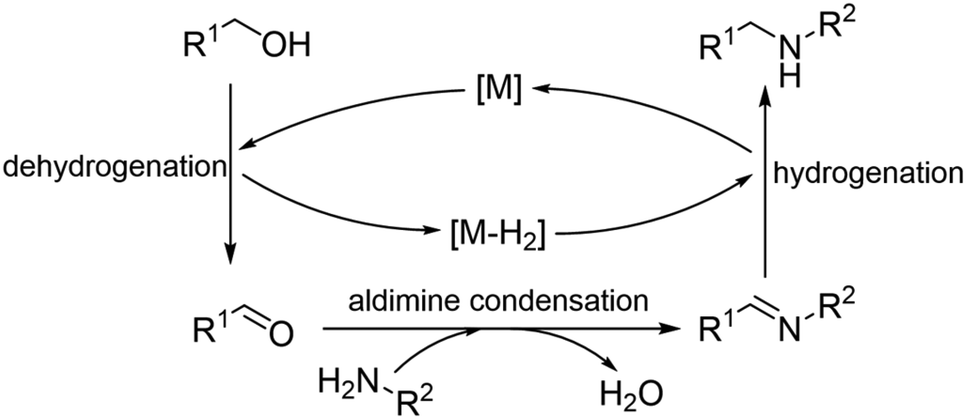 | ||
| Scheme 1 N-Alkylation of amines with alcohols via a HAT methodology. | ||
Early attempts with heterogeneous catalysis were driven by the advances in homogeneous catalysis and suffered from poor selectivity, limited substrate scope, relatively high reaction temperature (>200 °C) or the requirement of H2.2,14,15 Currently, much effort has been made on accomplishing the HAT reaction with an efficient and reusable heterogeneous catalyst under relatively mild conditions, with metals such as Ru,5,16 Pt,17 Pd,18,19 and Cu20,21 having been successfully applied in the HAT methodology. Nonetheless, an external base or a large amount of noble metals was required in most methodologies reported in the last few years (Table S1†). Iron, one of the most Earth-abundant metals with high biocompatibility, has been studied as a catalyst in various hydrogenation and dehydrogenation reactions.22,23 However, the application of Fe-based catalysts in HAT reactions is still a young field of research. The pioneering work on Fe catalysed N-alkylation of amines following the HAT concept was reported by Ramón and Yus and co-workers,24 in which commercial magnetite was used as the catalyst, and the reaction proceeded slowly (7 days) in the presence of 2 equiv. of t-BuOK. Namitharan et al.25 recently reported direct N-alkylation of amines with alcohols following a HAT concept, wherein nano-Fe2O3 was successfully explored as a catalyst, but the addition of KOH was still necessary for the reaction. As we know, by alloying two different metals, bimetallic catalysts always show excellent catalytic performance in hydrogen transfer reactions due to their unique electronic states and structure.26–28 On account of the various reaction steps involved in the HAT methodology, a multifunctional bimetallic catalyst may be preferable. Recently, Obora and co-workers6 employed Ti–Pd alloys as the catalysts for the N-alkylation of amines with alcohols via the HAT methodology. The catalytic activity of the Ti-based alloys was significantly improved by the incorporation of hydrogen-active Pd species. Inspired by that, we envisaged that alloying Pd species with an Fe-based heterogeneous catalyst could enhance the catalytic activity in HAT reactions, which may avoid the necessity of a base or other additives.
The synthesis and application of metal–organic frameworks (MOFs) has drawn a lot of attention in the past decade.29,30 But poor stability and blockage of active sites restrict the applications of MOFs in catalysis. After pyrolysis or etching of the MOFs, the obtained catalysts could achieve exposed active sites and high surface area and usually show good stability in organic reactions.31 Moreover, some MOF-derived catalysts were reported to attain improved basicity from the N-doped carbon (NC) support and performed well in hydrogen transfer and alcohol activation without the addition of an external base.32,33 With the incorporation of a second metal species and the resulting synergistic effects, the bimetallic MOFs and bimetallic MOF-derived catalysts show improved catalytic performance compared to their monometallic counterparts, and it has been proved to be an efficient method for the preparation of bimetallic catalysts.34
Herein, a novel bimetallic catalyst, Fe10Pd1/NC500, was successfully prepared by the pyrolysis of the bimetallic MOF NH2-MIL-101(Fe10Pd1), and the N-alkylation of amines with alcohols via a HAT methodology was studied as the model reaction, for its great value in pharmaceuticals, agrochemicals and material chemistry.15,35 To our delight, the N-doped carbon (NC) supported bimetallic catalyst Fe10Pd1/NC500 showed excellent catalytic performance in the N-alkylation of amines with alcohols. High yields of N-alkylamines were achieved under solvent-free conditions without any additive, which could be attributed to the cooperation of the oxophilic Fe, hydrogen-active Pd and basic N-doped carbon support. Furthermore, the catalytic protocol exhibits a broad substrate scope as well as excellent stability during the reaction process, and no significant loss in catalytic activity was observed after eight catalytic cycles.
Experimental section
Catalysts preparation
The synthesis of NH2-MIL-101(Fe) was similar to the synthesis of NH2-MIL-101(Fe10Pd1), except that no PdCl2 was added. The synthesis of MIL-101(Fe10Pd1) also followed the preparation procedure of NH2-MIL-101(Fe10Pd1) while using 500.0 mg (3 mmol) terephthalic acid instead of 2-aminoterephthalic acid. NH2-MIL-101(Al) was synthesized following the preparation procedure of NH2-MIL-101(Fe10Pd1) while using 1207.2 mg (5.0 mmol) of AlCl3·6H2O as the metal precursor.
N-Doped carbon derived from cellulose was prepared by the following procedure. Typically, 4.0 g of cellulose and 2.0 g of dicyandiamide was dropped into a 100 mL Teflon-lined autoclave. Then 50 mL deionized water was added, followed by ultrasound treatment at 550 W for 30 min. The mixture was treated in an oven for 4 h at 180 °C. The resulting solid was separated by centrifugation, washed with deionized water and dried under vacuum. Next, the solid was ground into a powder and then annealed in a tubular furnace under a nitrogen atmosphere at 800 °C (ramp rate: 5 °C min−1) for 2 h. The obtained product was designated as NC(cellulose).
The Pd nanoparticles or Fe10Pd1 nanoparticles were immobilized by the impregnation–reduction method. In a typical procedure, 100 mg of NC500 was dispersed in 50 mL of deionized water with subsequent addition of Na2PdCl4 solution (0.1 M, 0.3 mL) under stirring. After 30 min, NaBH4 solution (30 mg NaBH4 dissolved in 1 mL H2O) was added dropwise into the mixture under a nitrogen atmosphere with continuous stirring for 1 h. Finally, the catalyst was collected by centrifugation, washed with deionized water three times and dried under vacuum at 50 °C for 12 h. The obtained black powder was the Pd/NC500 catalyst. Fe10Pd1/AC and Fe10Pd1/NC(cellulose) were prepared in the same way except using 81.0 mg of FeCl3·6H2O and Na2PdCl4 solution (0.1 M, 0.3 mL) as the metal precursors and commercial activated carbon (AC) or NC(cellulose) as the catalyst supports.
Characterization
XRD analysis was performed on a Shimadzu X-ray diffractometer (XRD-6000) with Cu Kα irradiation. The scanning electron microscopy (SEM) images were obtained using a Hitachi S-4800 apparatus on a sample powder previously dried and sputter-coated with a thin layer of gold. The transmission electron microscopy (TEM) images were recorded using a PHILIPS Tecnai 12 microscope operating at 120 kV. High resolution transmission electron microscopy (HRTEM) was performed on a Philips-FEI Tecnai G2 F20 operating at 300 kV. The CO2-TPD data were obtained on a Micromeritics AutoChem II S3 2920 instrument. X-ray photoelectron spectroscopy (XPS) was performed on an ESCALAB 250Xi spectrometer, using an Al Kα X-ray source (1486.6 eV of photons) and calibrated by setting the C 1s peak to 284.80 eV. Inductively coupled plasma mass spectrometry (ICP-MS) was analyzed on an Optima 7300 DV. The BET surface area and pore size measurements were performed with N2 adsorption/desorption isotherms at 77 K on a Micromeritics ASAP 2020 instrument. Before measurements, the samples were degassed at 80 °C for 12 h.Catalytic reaction
In a typical reaction procedure, 3 mmol amine, 6 mmol alcohol and 20 mg of catalyst were placed in a 25 mL sealed tube, and then the tube was purged with N2 gas, and heated at 120 °C for 16 h (or other mentioned conditions). After the reaction was completed, the sealed tube was cooled down to room temperature, and the solution was diluted by ethyl acetate and analyzed by gas chromatography (GC) and GC-mass spectroscopy (GC-MS).Recyclability test
In order to investigate the recyclability of Fe10Pd1/NC500, the catalyst was collected using a magnet after the reaction, washed four times with methanol, and dried under vacuum at room temperature, and then reused for another reaction cycle.Results and discussion
Characterization of Fe10Pd1/NC500
Fig. 1 illustrates the synthesis strategy of Fe10Pd1/NC500, and the SEM image (Fig. 2a) shows the octahedral morphology of NH2-MIL-101(Fe10Pd1). After the pyrolysis of the bimetallic MOF at 500 °C, porous Fe10Pd1/NC500 was obtained, which showed magnetic properties that enabled the catalyst to be easily separated from the reaction solution (Fig. S1†). As measured by ICP-MS (Table S2†), the contents of Fe and Pd in Fe10Pd1/NC500 are 16.0 wt% and 3.2 wt%. The molar ratio of Fe to Pd is 9.6:1, which is close to the theoretical ratio of Fe to Pd (10:1). The SEM image (Fig. 2b) and TEM image (Fig. 3a) show the morphology and structure of the as-synthesized Fe10Pd1/NC500. The obtained catalyst maintains the octahedral frame of NH2-MIL-101(Fe10Pd1), and nanoparticles (NPs) were formed and exposed during the pyrolysis process and dispersed homogeneously on the surface of the catalyst with an average diameter of 8.5 nm (±0.5 nm). More information about the structure of the NPs is revealed by the high-resolution TEM (HRTEM) image (Fig. 3b) and the lattice fringe with an interplanar spacing of 0.212 nm which corresponds to the (111) plane of the Fe–Pd alloy.37 The structure of Fe10Pd1/NC500 is more evident from the results of STEM, and from the corresponding elemental mapping (Fig. 3c–g), carbon and nitrogen show a uniform distribution throughout the as-obtained catalyst, thus illustrating the homogeneous surface of the N-doped carbon material. The distribution of Pd matches well with the distribution of Fe, further indicating the formation of Fe–Pd alloy NPs.
 | ||
| Fig. 1 Schematic illustration of the preparation process of Fe10Pd1/NC500. | ||
 | ||
| Fig. 2 SEM images of (a) NH2-MIL-101(Fe10Pd1) and (b) Fe10Pd1/NC500. | ||
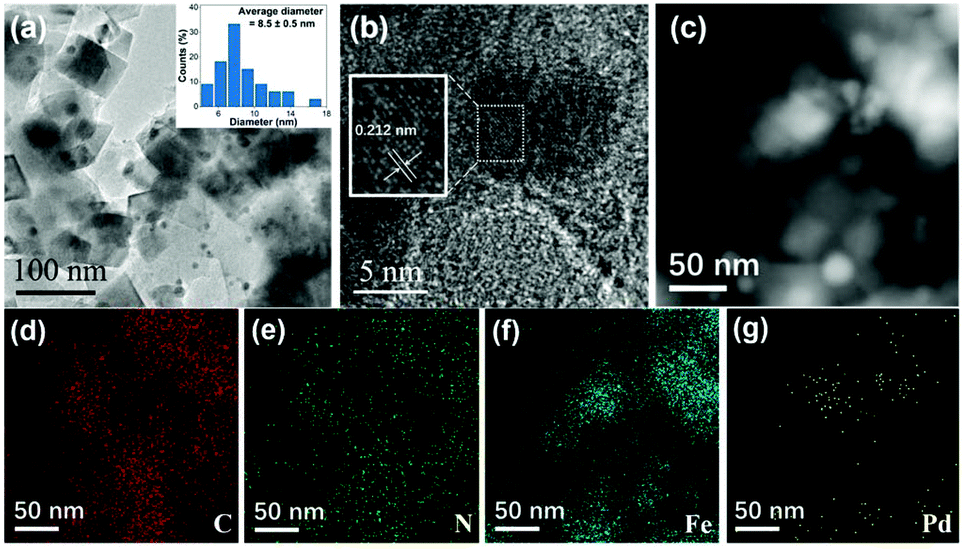 | ||
| Fig. 3 (a) TEM image of Fe10Pd1/NC500. (b) HRTEM image of Fe10Pd1/NC500. (c) STEM image of Fe10Pd1/NC500. Elemental mapping images of C (d), N (e), Fe (f) and Pd (g) for Fe10Pd1/NC500. | ||
The crystallite phases of the prepared MOFs and catalysts were analysed by X-ray diffraction analysis (XRD). As shown in Fig. 4, the XRD pattern of NH2-MIL-101(Fe10Pd1) was consistent with the XRD pattern of NH2-MIL-101(Fe), indicating that the uniform incorporation of a small amount of Pd does not obviously affect the crystallographic structures of the NH2-MIL-101-type MOFs. The XRD pattern of the obtained Fe10Pd1/NC500 displayed peaks at around 35.5° and 40.7°, indexed to Fe3O4 (311) and FePd (111),38,39 which confirmed the formation Fe–Pd alloy NPs during the pyrolysis of NH2-MIL-101(Fe10Pd1). The graphitization degree of the carbon matrix in the Fe10Pd1/NC500 was investigated by Raman spectra (Fig. S2†); the D-band (defect) and G-band (graphitic) can be observed at about 1350 cm−1 and 1582 cm−1, and the peak intensity ratio of the D-band to the G-band (ID/IG) is 1.15, which suggests a favorable graphitic feature and therefore high electronic conductivity of Fe10Pd1/NC500.
 | ||
| Fig. 4 XRD patterns of NH2-MIL-101(Fe), NH2-MIL-101(Fe10Pd1) and Fe10Pd1/NC500. | ||
To ascertain the detailed electronic structure and chemical states of Fe10Pd1/NC500, X-ray photoelectron spectroscopy (XPS) was employed. The survey spectrum of Fe10Pd1/NC500 (Fig. S3†) shows the existence of C, N, O, Fe and Pd that are from the precursor bimetallic MOF NH2-MIL-101(Fe10Pd1). As shown in Fig. 5a, the C 1s spectra could be deconvoluted into four peaks at 284.5 eV, 285.5 eV, 286.3 eV and 289.4 eV, corresponding to the C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, C–N and O–CO functional groups.40 The N 1s spectra (Fig. 5b) could be deconvoluted into five peaks, indicating five types of nitrogen species: pyridinic N at 398.3 eV, Fe–Nx at 399.4 eV, pyrrolic N at 400.4 eV, graphitic N at 401.3 eV and oxidized N at 404.8 eV.39,41 These spectra combined with the element maps (Fig. 3d and e) evidence that the nitrogen atoms have been well embedded in the carbon matrix. Moreover, pyridinic N and pyrrolic N are generally recognized as basic sites,42 and a high density of these sites enhances the basicity of Fe10Pd1/NC500. The XPS spectra of the metals were also carefully analysed. In the Fe 2p spectra (Fig. 5c), the Fe 2p3/2 band contains a divalent Fe peak at 710.4 eV and a trivalent Fe peak at 712.9 eV while the Fe 2p1/2 band contains a divalent Fe peak at 724.8 eV and a trivalent Fe peak at 730.7 eV, and the peak at 719.1 eV can be assigned to metallic Fe.43 The existence of metallic Fe proved the reduction of Fe2+ precursors during the pyrolysis of NH2-MIL-101(Fe10Pd1). Also, the reduction of Pd2+ precursors was proved by the Pd 3d spectra (Fig. 5d): metallic Pd peaks located at 335.4 eV (Pd 3d5/2) and 340.6 eV (Pd 3d3/2) are observed and divalent Pd (peaks at 337.5 eV and 342.4 eV) may come from the oxidation of metallic Pd in air. Compared with the Pd 3d spectra of the monometallic catalyst Pd/NC500 (Fig. S4†), the content of Pd0 in Fe10Pd1/NC500 was higher than that in Pd/NC500, and a shift (about 0.3 eV) of the Pd 3d5/2 peak in Fe10Pd1/NC500 towards lower binding energy was observed, indicating that alloying Fe with Pd induced the electron transfer from Fe to Pd. The electronic effect inside the bimetallic NPs came from the different electronegativities of Pd (2.20) and Fe (1.80), and prevented the fast oxidation of nano Pd in air and improved the durability of Fe10Pd1/NC500.44
N, C–N and O–CO functional groups.40 The N 1s spectra (Fig. 5b) could be deconvoluted into five peaks, indicating five types of nitrogen species: pyridinic N at 398.3 eV, Fe–Nx at 399.4 eV, pyrrolic N at 400.4 eV, graphitic N at 401.3 eV and oxidized N at 404.8 eV.39,41 These spectra combined with the element maps (Fig. 3d and e) evidence that the nitrogen atoms have been well embedded in the carbon matrix. Moreover, pyridinic N and pyrrolic N are generally recognized as basic sites,42 and a high density of these sites enhances the basicity of Fe10Pd1/NC500. The XPS spectra of the metals were also carefully analysed. In the Fe 2p spectra (Fig. 5c), the Fe 2p3/2 band contains a divalent Fe peak at 710.4 eV and a trivalent Fe peak at 712.9 eV while the Fe 2p1/2 band contains a divalent Fe peak at 724.8 eV and a trivalent Fe peak at 730.7 eV, and the peak at 719.1 eV can be assigned to metallic Fe.43 The existence of metallic Fe proved the reduction of Fe2+ precursors during the pyrolysis of NH2-MIL-101(Fe10Pd1). Also, the reduction of Pd2+ precursors was proved by the Pd 3d spectra (Fig. 5d): metallic Pd peaks located at 335.4 eV (Pd 3d5/2) and 340.6 eV (Pd 3d3/2) are observed and divalent Pd (peaks at 337.5 eV and 342.4 eV) may come from the oxidation of metallic Pd in air. Compared with the Pd 3d spectra of the monometallic catalyst Pd/NC500 (Fig. S4†), the content of Pd0 in Fe10Pd1/NC500 was higher than that in Pd/NC500, and a shift (about 0.3 eV) of the Pd 3d5/2 peak in Fe10Pd1/NC500 towards lower binding energy was observed, indicating that alloying Fe with Pd induced the electron transfer from Fe to Pd. The electronic effect inside the bimetallic NPs came from the different electronegativities of Pd (2.20) and Fe (1.80), and prevented the fast oxidation of nano Pd in air and improved the durability of Fe10Pd1/NC500.44
 | ||
| Fig. 5 XPS spectra of the (a) C 1s region, (b) N 1s region, (c) Fe 2p region, and (d) Pd 3d region of Fe10Pd1/NC500. | ||
Surface area and pore structure are important parameters for a catalyst, and the porous nature of Fe10Pd1/NC500 was investigated by N2 adsorption–desorption isotherms (Fig. 6). Based on N2 sorption isotherms, the Brunauer–Emmett–Teller surface area (SBET) of Fe10Pd1/NC500 is 192 m2 g−1. The pore size distribution analysis reveals that Fe10Pd1/NC500 possesses a complex pore structure including both micropores and mesopores. The high surface area and complex pore structure of Fe10Pd1/NC500 could benefit the exposure of the active sites and the absorption of the reactant molecules, resulting in an efficient catalytic performance.
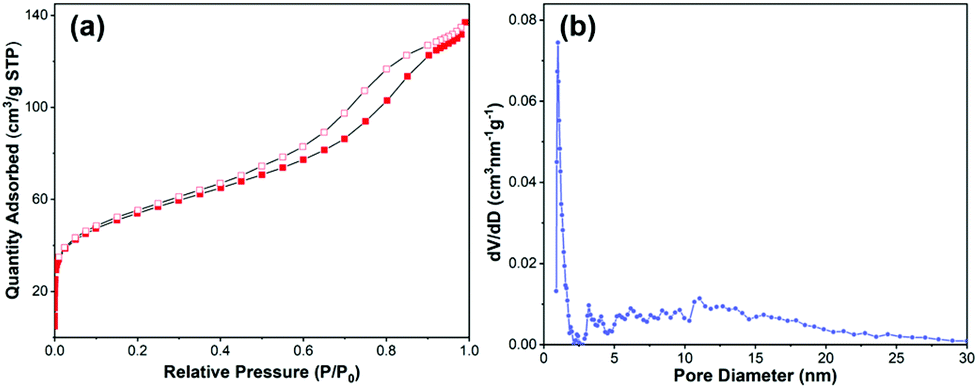 | ||
| Fig. 6 (a) N2 adsorption/desorption isotherms of Fe10Pd1/NC500, (b) the corresponding pore size distributions. | ||
Catalytic performance
The as-prepared catalysts were employed for the N-alkylation of amines with alcohols. Benzyl alcohol 1a and aniline 2a were chosen as the model substrates, and 2a was converted only to N-benzylaniline 3a or N-benzylideneaniline 4a in our tests. A small amount of benzaldehyde 5a, which was generated from the dehydrogenation of 1a, was detected in the reaction mixtures. Firstly, the bimetallic MOF NH2-MIL-101(Fe10Pd1) was directly used as the catalyst (Table 1, entry 1), a quite low conversion of 2a was obtained, and no desired product 3a was detected, the poor catalytic activity could be because the metal sites in the MOF were unreduced (Fig. S4† for XPS spectra). Moreover, the bimetallic MOF disintegrated under the reaction conditions, which signifies that NH2-MIL-101(Fe10Pd1) is not a suitable catalyst for the HAT reaction. To our delight, Fe10Pd1/NC500 showed a good catalytic performance at 110 °C (Table 1, entry 2), when the reaction temperature was increased to 120 °C (Table 1, entry 3), and all 2a was converted and the selectivity for the desired 3a was up to 98%. Based on the ICP-MS results (Table S2,† entry 1), when 20 mg Fe10Pd1/NC500 was loaded in the reaction, the ratios of Fe and Pd to 2a were 1.9 mol% and 0.2 mol%, respectively, suggesting a low dosage of the noble metal Pd. When the reaction was carried out in air (Table 1, entry 4), most of the obtained product was N-benzylideneaniline 4a, which could imply that the activated H atoms combined with the oxygen in air instead of hydrogenating the intermediate imine. The stoichiometry of the reactants may have an impact on the reaction process, as shown in entries 5 and 6: both the conversion of 2a and selectivity for 3a were slightly decreased when the amount of 1a was less than 2 equiv.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | T (°C) | Conv.b (%) | Yield (%) | 5a | |
| 3a | 4a | |||||
| a Reaction conditions: 1a (6 mmol), 2a (3 mmol), catalysts 20 mg, N2 atmosphere, 16 h. Conversions and yields were determined by GC with hexadecane as an internal standard. b Conversion of 2a. c Content of 5a in reaction solution. d Reacting in air. e 3 mmol of 1a. f 4.5 mmol of 1a. g 20 mg (2 mol% Fe to 2a). h 20 mg (0.2 mol% Pd to 2a). i 20 mg Fe/NC500 and 20 mg Pd/NC500. j 30 mg (2.7 mol% Fe and 0.2 mol% Pd to 2a). k 11 mg (1 mol% Fe and 0.2 mol% Pd to 2a). l 20 mg (1.8 mol% Fe and 0.2 mol% Pd to 2a). m 20 mg (1.7 mol% Fe and 0.2 mol% Pd to 2a). n 20 mg (1.6 mol% Fe and 0.2 mol% Pd to 2a). o 20 mg Fe10Pd1/C500, 3 mmol of t-BuOK added. | ||||||
| 1 | NH2-MIL-101(Fe10Pd1) | 120 | 7 | 0 | 7 | <1 |
| 2 | Fe10Pd1/NC500 | 110 | 87 | 74 | 13 | 6 |
| 3 | Fe10Pd1/NC500 | 120 | 100 | 98 | 2 | 9 |
| 4d | Fe10Pd1/NC500 | 120 | 82 | 15 | 67 | 13 |
| 5e | Fe10Pd1/NC500 | 120 | 74 | 65 | 9 | 2 |
| 6f | Fe10Pd1/NC500 | 120 | 87 | 81 | 6 | 4 |
| 7g | Fe/NC500 | 120 | 12 | 5 | 7 | <1 |
| 8h | Pd/NC500 | 120 | 97 | 58 | 39 | 11 |
| 9i | Fe/NC500 + Pd/NC500 | 120 | 95 | 61 | 34 | 8 |
| 10j | Fe15Pd1/NC500 | 120 | 70 | 59 | 11 | 2 |
| 11k | Fe5Pd1/NC500 | 120 | 85 | 63 | 22 | 14 |
| 12l | Fe10Pd1/AC | 120 | 34 | 12 | 22 | 2 |
| 13m | Fe10Pd1/NC(cellulose) | 120 | 76 | 55 | 21 | 8 |
| 14n | Fe10Pd1/C500 | 120 | 61 | 13 | 48 | 5 |
| 15o | Fe10Pd1/C500 | 120 | 90 | 84 | 6 | 16 |
| 16 | Fe10Pd1/NC400 | 120 | 56 | 42 | 14 | 10 |
| 17 | Fe10Pd1/NC600 | 120 | 89 | 51 | 38 | 5 |

For comparison, a series of catalysts was synthesized and tested (Table S2† for metal contents in different catalysts). Monometallic catalyst Fe/NC500 was prepared by pyrolysis of NH2-MIL-101(Fe), and a poor reaction activity was observed (Table 1, entry 7). Besides, almost no benzaldehyde 5a (<1%) was detected in the reaction solution of Fe/NC500 while 9% was detected in the solution of Fe10Pd1/NC500 (Table 1, entry 3), suggesting that hydrogen-active Pd sites in Fe10Pd1/NC500 play an important role in the dehydrogenation of alcohol, which is the first step of the HAT reaction. Pure Pd NPs were supported on the NH2-MIL-101(Al)-derived N-doped carbon by an impregnation–reduction method, and the obtained Pd/NC500 afforded good conversion of 2a but low selectivity for 3a, which may be attributed to the lack of Fe sites. The oxophilic Fe sites were reported to enhance the interaction of the CO groups with the catalyst surface,45,46 which facilitated the intermediate reaction step. In addition, the electron transfer from Fe to Pd, which was revealed by XPS results, made the Pd sites more electron-rich, accelerating the hydrogenation process and causing the better selectivity in the HAT reaction.37 These synergistic effects were verified by the results of bimetallic Fe10Pd1/NC500 and monometallic catalysts (Table 1, entries 3 and 7, 8). A physical mixture of Fe/NC500 and Pd/NC500 does not lead to a significant enhancement in catalytic performance (Table 1, entry 9), demonstrating that the alloying of Fe and Pd accounts for the bimetallic synergistic effects. The compositions of bimetallic catalysts could have a strong influence on their catalytic efficiency, and bimetallic catalysts prepared from MOFs with different Fe/Pd ratios were tested (Table 1, entries 10 and 11). Keeping the Pd load in the reaction constant (0.2 mol%), a loss in catalytic activity was observed when the Pd load in the catalyst decreased (Fe15Pd1/NC500), which may be attributed to the reduction of the Pd site density; a decrease in selectivity was observed when Fe5Pd1/NC500 was used (1 mol% Fe loaded in the reaction), and it could be concluded that an Fe/Pd ratio of 10:1 was the optimal ratio in this reaction.
Next, Fe–Pd NPs were immobilized by the impregnation–reduction method on different supports such as activated carbon (AC) and N-doped carbon derived from cellulose, and the obtained catalysts were applied in the reaction under the optimized conditions (Table 1, entries 12 and 13). Much lower yields of 3a were afforded by these reference catalysts, and the poor activity could be attributed to the weaker metal–support interaction in catalysts obtained from the impregnation–reduction method as compared to those from the pyrolysis method, and more metal sites were leached in Fe10Pd1/NC(cellulose) than in Fe10Pd1/NC500 (Table S2,† entries 2 and 9) during the reaction. The above results also indicated that the MOF-derived N-doped carbon support may play a direct role in improving the catalytic performance of Fe10Pd1/NC500. To elucidate the effect of the MOF-derived N-doped carbon support, comparative experiments were conducted. Fe10Pd1/C500 without N atoms embedded in the carbon matrix was prepared by the pyrolysis of MIL-101(Fe10Pd1) at 500 °C, and the catalyst showed poor conversion of 2a and quite low selectivity for 3a (Table 1, entry 14). When 3 mmol of t-BuOK was added, an obvious growth in the yield of 3a was achieved (Table 1, entry 15), and this result proved the promoting role of the base in the N-alkylation of amines with alcohols. Since no external base is used in the reaction with Fe10Pd1/NC500, the N sites in the catalyst were believed to provide the required basicity.
Different pyrolysis temperatures of the MOF may cause changes in the catalyst surface composition and therefore impact the catalytic performance. When pyrolyzed at 400 °C, the obtained Fe10Pd1/NC400 displayed a poor catalytic activity (Table 1, entry 16). Fe10Pd1/NC600 which was obtained at a higher pyrolysis temperature, successfully converted most 2a, but the selectivity for the desired 3a is poor (Table 1, entry 17). The atomic contents of N, Fe and Pd on the surfaces of catalysts were determined by the XPS results. As listed in Table S3,† the atomic contents of N, Fe and Pd on the surface of Fe10Pd1/NC400 are lower than in Fe10Pd1/NC500, revealing that the exposure of active sites is insufficient at a lower temperature. Although the atomic contents of metals on the surface of Fe10Pd1/NC600 are almost equal to the contents on Fe10Pd1/NC500, the atomic content of N on the surface of Fe10Pd1/NC600 is relatively low, which is probably due to the partial disintegration of pyridinic N and pyrrolic N at 600 °C. The Lewis basicity of Fe10Pd1/C500, Fe10Pd1/NC400, Fe10Pd1/NC500 and Fe10Pd1/NC600 were evaluated by CO2-TPD (Fig. 7). No intense peak was observed in the profile of Fe10Pd1/C500 illustrating that the catalyst has quite a weak Lewis basicity. In the profiles of catalysts derived from NH2-MIL-101(Fe10Pd1), the intense peaks at 550–650 °C suggest that the catalysts possess strong Lewis basicity. The intensity of the basic peak reduced in the following order: Fe10Pd1/NC500 > Fe10Pd1/NC400 > Fe10Pd1/NC600 > Fe10Pd1/C500, which showed a positive correlation with the atomic contents of N on the surface of the catalysts (Table S3†). Furthermore, a shift of the basic peak at high temperature can be observed by comparing the profiles of Fe10Pd1/NC400 and Fe10Pd1/NC500, which indicates that the dopant of the N atoms leads to a stronger Lewis basicity at a higher pyrolysis temperature.33 The above results further confirmed that the N sites in the catalyst support provide the necessary basicity and consequently avoid the need for an external base for Fe10Pd1/NC500 in HAT reactions.
 | ||
| Fig. 7 CO2-TPD profiles of Fe10Pd1/C500, Fe10Pd1/NC400, Fe10Pd1/NC500 and Fe10Pd1/NC600. | ||
A model reaction was conducted under the optimized conditions (Table 1, entry 3) and tracked by GC to gain preliminary insights into the reaction mechanism. As showed in Fig. 8, benzaldehyde 5a was detected in the reaction mixture, the yield of N-benzylideneaniline 4a increased first and then decreased with the increase in the reaction time. These results identified 5a and 4a as the intermediates in the reaction, which is in accord with the HAT methodology: dehydrogenation of 1a occurs first, then the intermediate reaction between 5a and 2a forms the intermediate imine 4a, and finally 4a is hydrogenated to give the desired product 3a.
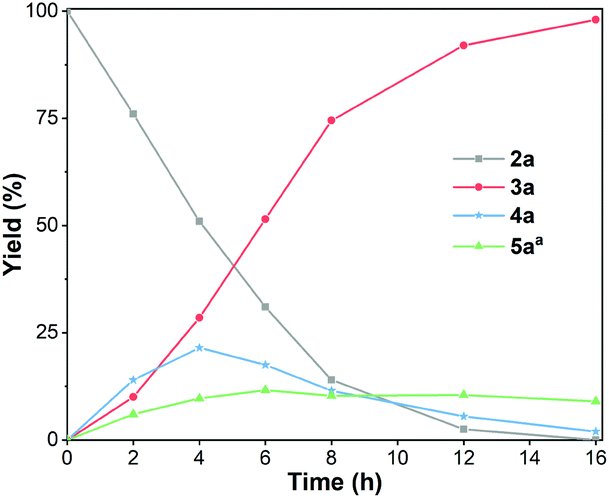 | ||
| Fig. 8 Time-course monitoring of the reaction. Reaction conditions: 1a (6 mmol), 2a (3 mmol), Fe10Pd1/NC500 20 mg, N2 atmosphere, 120 °C. Yields were determined by GC with hexadecane as an internal standard. aContent of 5a in reaction solution. | ||
With the optimized reaction conditions established, the substrate scope of the N-alkylation of amines with alcohols was examined, and the reactions were performed with 0.2 mol% (Pd to amines) Fe10Pd1/NC500 loading under solvent-free conditions (Table 2). Excellent yields of corresponding N-alkylamines were obtained upon reaction with p-toluidine (3b) and m-toluidine (3c), and the steric hindrance effect may cause the relatively low yield of 3d. Benzylic amines were tested and the desired products were produced in acceptable yields (3e–3f). Gratifyingly, for the aniline derivatives bearing electron donating groups, promising yields were afforded (3g–3i), and the electron withdrawing substituents were also tolerated (3j–3l). Both polycyclic aromatic reactants (3m–3n) and heterocyclic reactants (3o–3p) underwent efficient conversion to the desired products. The alkylation of aliphatic amines with 1a was explored as well; cyclohexyl amine, octylamine and dodecylamine were used as substrates; and the corresponding products were obtained in good yields (3q–3s).

Subsequently, the scope of alcohols was examined. Both 4-methylbenzyl alcohol and 1-naphthalenemethanol took part in the reactions exhibiting high conversion and selectivity (3t–3u). Moderate yields of 3v and 3w were obtained, for which the alcohol substrate included the p-chloro and p-bromo groups, respectively. Next, reactions involving secondary alcohols were performed; 78% yield of 3x was achieved when 1-phenylethan-1-ol was used, and a higher temperature (130 °C) was required for a high yield of 3y. N-Alkylation of 2a with 1-octanol was carried out at 140 °C and gave the corresponding product 3z in 69% yield. The satisfactory yields of the heterocyclic products (3o–3p and 3aa–3cc) signify the promising application prospect of this catalytic system in pharmaceutical synthesis. Besides, biomass derived cinnamyl alcohol and vanillyl alcohol were successfully converted with acceptable results (3dd–3ee), indicating that Fe10Pd1/NC500 could play an important role in the utilization of biomass resources.
Catalyst recyclability and stability test
Recyclability is a key issue for a heterogeneous catalyst, and the recyclability of Fe10Pd1/NC500 was tested under the optimized reaction conditions (Table 1, entry 3). After the reaction, Fe10Pd1/NC500 can be easily separated using a magnet and washed with methanol, and then dried in a vacuum for reuse. As shown in Fig. 9, only a minor loss in yield was observed after eight runs, which might be a result of the slight loss of metal sites in the catalyst (Table S2,† entry 2). In addition, ICP analysis of the solutions of the first reaction cycle indicated that the leaching ratios of Fe and Pd in Fe10Pd1/NC500 were about 0.07% and 0.14% respectively, which were much less than those in Fe10Pd1/C500 (0.27% for Fe and 0.49% for Pd). This comparison result suggests that the N-doped carbon supported Fe10Pd1/NC500 has better stability than Fe10Pd1/C500. The TEM image (Fig. S5†) shows that no obvious fragmentation of the catalytic structure occurred after eight cycles. The good stability and recyclability of Fe10Pd1/NC500 were confirmed by the above results. | ||
| Fig. 9 Recyclability of Fe10Pd1/NC500. | ||
Conclusions
In summary, a novel bimetallic catalyst, Fe10Pd1/NC500, has been successfully synthesized, and the catalyst shows excellent performance for N-alkylation of amines with alcohols via a HAT methodology. The reactions proceed at 120 °C and an alcohol/amine molar ratio of 2:1 without any external base, hydrogen source or solvent. The catalytic efficiency has been significantly improved by the synergistic effects inside Fe10Pd1/NC500: (i) hydrogen-active Pd sites facilitate the dehydrogenation step while oxophilic Fe sites accelerate the intermediate reaction; (ii) the electron transfer from Fe to Pd stabilizes the Pd sites and promotes the hydrogenation process; (iii) the N dopants on the catalyst surface enhance the basic nature of Fe10Pd1/NC500 and avoid the requirement of an external base. In addition, the wide substrate scope and remarkable recyclability of Fe10Pd1/NC500 validate the advantages of this catalyst prepared from bimetallic MOF. The successful alloying of the Earth-abundant Fe with hydrogen-active Pd provides a new route for the future design and synthesis of green and multifunctional catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Key Laboratory of Biomass Energy and Material, Jiangsu Province (JSBEM201912) and the Chinese Postdoctoral Science Foundation (2015M571761 and 2016T90465) for financial support. This work was funded by the Priority Academic Program Development of Jiangsu Higher Education Institution and Instrument and Equipment Foundation of Nanjing University of Science and Technology.References
- K.-i. Shimizu, Catal. Sci. Technol., 2015, 5, 1412–1427 RSC.
- A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed.
- G.-M. Zhao, H.-l. Liu, X.-r. Huang, X. Yang and Y.-p. Xie, ACS Catal., 2015, 5, 5728–5740 CrossRef CAS.
- A. Bartoszewicz, G. González Miera, R. o. Marcos, P.-O. Norrby and B. Martín-Matute, ACS Catal., 2015, 5, 3704–3716 CrossRef CAS.
- B. Guo, H.-X. Li, S.-Q. Zhang, D. J. Young and J.-P. Lang, ChemCatChem, 2018, 10, 5627–5636 CrossRef CAS.
- Y. Takahashi, R. Kondo, M. Utsunomiya, T. Suzuki, H. T. Takeshita and Y. Obora, ChemCatChem, 2019, 11, 2432–2437 CrossRef CAS.
- R. Grigg, T. R. B. Mitchell, S. Sutthivaiyakit and N. Tongpenyai, J. Chem. Soc., Chem. Commun., 1981, 611–612, 10.1039/C39810000611.
- Y. Watanabe, Y. Tsuji and Y. Ohsugi, Tetrahedron Lett., 1981, 22, 2667–2670 CrossRef CAS.
- K. Polidano, B. D. W. Allen, J. M. J. Williams and L. C. Morrill, ACS Catal., 2018, 8, 6440–6445 CrossRef CAS.
- S. P. Midya, J. Pitchaimani, V. G. Landge, V. Madhu and E. Balaraman, Catal. Sci. Technol., 2018, 8, 3469–3473 RSC.
- G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS PubMed.
- J. He, J. W. Kim, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2009, 48, 9888–9891 CrossRef CAS PubMed.
- C. Williams, J. H. Carter, N. F. Dummer, Y. K. Chow, D. J. Morgan, S. Yacob, P. Serna, D. J. Willock, R. J. Meyer, S. H. Taylor and G. J. Hutchings, ACS Catal., 2018, 8, 2567–2576 CrossRef CAS.
- J. T. Kozlowski and R. J. Davis, ACS Catal., 2013, 3, 1588–1600 CrossRef CAS.
- G. Guillena, D. J. Ramón and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS PubMed.
- R. Cano, D. J. Ramón and M. Yus, J. Org. Chem., 2011, 76, 5547–5557 CrossRef CAS PubMed.
- C. Chaudhari, S. M. A. H. Siddiki, K. Kon, A. Tomita, Y. Tai and K.-i. Shimizu, Catal. Sci. Technol., 2014, 4, 1064–1069 RSC.
- A. E. Putra, Y. Oe and T. Ohta, Eur. J. Org. Chem., 2015, 7799–7805 CrossRef CAS.
- A. S. Alshammari, K. Natte, N. V. Kalevaru, A. Bagabas and R. V. Jagadeesh, J. Catal., 2020, 382, 141–149 CrossRef CAS.
- A. Y. Li, N. Dumaresq, A. Segalla, N. Braidy and A. Moores, ChemCatChem, 2019, 11, 3959–3972 CrossRef CAS.
- Y. Wu, Y. Huang, X. Dai and F. Shi, ChemSusChem, 2019, 12, 3185–3191 CrossRef CAS PubMed.
- I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS.
- R. V. Jagadeesh, A.-E. Surkus, H. Junge, M.-M. Pohl, J. Radnik, J. Rabeah, H. Huan, V. Schünemann, A. Brückner and M. Beller, Science, 2013, 342, 1073 CrossRef CAS.
- R. Martínez, D. J. Ramón and M. Yus, Org. Biomol. Chem., 2009, 7, 2176–2181 RSC.
- M. Nallagangula, C. Sujatha, V. T. Bhat and K. Namitharan, Chem. Commun., 2019, 55, 8490–8493 RSC.
- J.-w. Zhang, Y. Cai, G.-p. Lu and C. Cai, Green Chem., 2016, 18, 6229–6235 RSC.
- H. Zhong, M. Iguchi, M. Chatterjee, T. Ishizaka, M. Kitta, Q. Xu and H. Kawanami, ACS Catal., 2018, 8, 5355–5362 CrossRef CAS.
- H. Wang, S. Bai, Y. Pi, Q. Shao, Y. Tan and X. Huang, ACS Catal., 2019, 9, 154–159 CrossRef CAS.
- A. Dhakshinamoorthy, A. M. Asiri and H. Garcia, ACS Catal., 2019, 9, 1081–1102 CrossRef CAS.
- D. Yang and B. C. Gates, ACS Catal., 2019, 9, 1779–1798 CrossRef CAS.
- H. Konnerth, B. M. Matsagar, S. S. Chen, M. H. G. Prechtl, F.-K. Shieh and K. C. W. Wu, Coord. Chem. Rev., 2020, 416, 213319 CrossRef CAS.
- G. Li, H. Yang, H. Zhang, Z. Qi, M. Chen, W. Hu, L. Tian, R. Nie and W. Huang, ACS Catal., 2018, 8, 8396–8405 CrossRef CAS.
- K.-k. Sun, J.-l. Sun, G.-P. Lu and C. Cai, Green Chem., 2019, 21, 4334–4340 RSC.
- L. Chen, H.-F. Wang, C. Li and Q. Xu, Chem. Sci., 2020, 11, 5369–5403 RSC.
- R. N. Salvatore, C. H. Yoon and K. W. Jung, Tetrahedron, 2001, 57, 7785–7811 CrossRef CAS.
- Q. Wang, N. Tsumori, M. Kitta and Q. Xu, ACS Catal., 2018, 8, 12041–12045 CrossRef CAS.
- Ö. Metin, A. Mendoza-Garcia, D. Dalmızrak, M. S. Gültekin and S. Sun, Catal. Sci. Technol., 2016, 6, 6137–6143 RSC.
- W. Xiao, W. Lei, J. Wang, G. Gao, T. Zhao, M. A. L. Cordeiro, R. Lin, M. Gong, X. Guo, E. Stavitski, H. L. Xin, Y. Zhu and D. Wang, J. Mater. Chem. A, 2018, 6, 11346–11352 RSC.
- Z. Ye, J. A. Padilla, E. Xuriguera, E. Brillas and I. Sirés, Appl. Catal., B, 2020, 266, 118604 CrossRef CAS.
- P. Zhao, X. Hua, W. Xu, W. Luo, S. Chen and G. Cheng, Catal. Sci. Technol., 2016, 6, 6365–6371 RSC.
- T. Song, P. Ren, Y. Duan, Z. Wang, X. Chen and Y. Yang, Green Chem., 2018, 20, 4629–4637 RSC.
- L. Lin, Q. Zhu and A.-W. Xu, J. Am. Chem. Soc., 2014, 136, 11027–11033 CrossRef CAS PubMed.
- S.-H. Zhang, M.-F. Wu, T.-T. Tang, Q.-J. Xing, C.-Q. Peng, F. Li, H. Liu, X.-B. Luo, J.-P. Zou, X.-B. Min and J.-M. Luo, Chem. Eng. J., 2018, 335, 945–953 CrossRef CAS.
- J. K. Kim, J. K. Lee, K. H. Kang, J. C. Song and I. K. Song, Appl. Catal., A, 2015, 498, 142–149 CrossRef CAS.
- S. Sitthisa, W. An and D. E. Resasco, J. Catal., 2011, 284, 90–101 CrossRef CAS.
- Y. Lv, M. Han, W. Gong, D. Wang, C. Chen, G. Wang, H. Zhang and H. Zhao, Angew. Chem., Int. Ed., 2020, 59, 23521–23526 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for products. See DOI: 10.1039/d0gc03725g |
| This journal is © The Royal Society of Chemistry 2021 |