
Recent advances in visible-light-mediated organic transformations in water†
Kai
Sun
 a,
Qi-Yan
Lv
*ab,
Xiao-Lan
Chen
*a,
Ling-Bo
Qu
a and
Bing
Yu
*a
a,
Qi-Yan
Lv
*ab,
Xiao-Lan
Chen
*a,
Ling-Bo
Qu
a and
Bing
Yu
*a
aGreen Catalysis Center, College of Chemistry, Zhengzhou University, Kexue Road No. 100, Zhengzhou 450001, China. E-mail: qiyanlv@zzu.edu.cn; chenxl@zzu.edu.cn; bingyu@zzu.edu.cn
bSchool of Life Sciences, Zhengzhou University, Kexue Road No. 100, Zhengzhou 450001, China
First published on 11th December 2020
Abstract
Water, as a green reaction medium, is safe, non-toxic, and rich in reserves on Earth, while visible light represents a renewable, clean, and abundant energy source. Organic transformations carried out under the irradiation of visible light in water are undoubtedly more attractive, and are in line with the concept of “green chemistry” to effectively minimize the environmental impact of chemical synthesis. This review focuses on the recent progress in visible-light-mediated organic transformations in water, and the related mechanisms are also discussed. Although great achievements have been made, they are just the tip of the iceberg and there is still great room for improvement. This review will facilitate the understanding of visible-light-mediated organic transformations in water and further stimulate the advancement of more novel relevant strategies.
Introduction
The increasing energy shortage and environmental pollution have prompted chemists to develop green organic transformations to reduce toxic or polluting wastes from chemical processes. Generally, the chemical pollution produced during classical organic synthesis is mainly caused by the employment of toxic and volatile organic solvents. Moreover, several potential safety issues, such as combustion and explosion, might exist in heat- or gas-releasing reactions when utilizing volatile organic solvents in organic synthesis. Compared with traditional organic solvents, water, as an important component of life, is a safe, non-toxic, and abundant natural liquid on Earth. Due to the excellent biocompatibility, non-flammability, and high specific heat capacity, the utilization of water instead of traditional volatile solvents for organic reactions is in line with the concept of “green chemistry”, and this can effectively minimize the environmental impact and potential risks of chemical synthesis.1Conventionally, successful handling of organic synthesis generally necessitates the use of organic solvents to dissolve the components and facilitate chemical reactions. Although many catalysts or reaction substrates are incompatible with or immiscible in water, water is not always considered incompatible with organic synthesis. In fact, in the childhood of organic chemistry starting from Wohler's synthesis of urea in 1828, water was the preferred choice of solvent in many named reactions developed in the 19th century, including the well-known Baeyer–Villiger oxidation, the Curtius rearrangement, the Wolff–Kishner reduction, the Hofmann degradation, the Lieben haloform reaction, the Pictet–Spengler reaction, and the Sandmeyer reaction.2 With the vigorous development of organometallic chemistry, water was supplanted by volatile organic solvents mainly because of the sensitivity to hydrolysis of the earlier developed organometallic reagents. The employment of water as a solvent was reassessed until the discovery of the greatly enhanced rate and selectivity of the Diels–Alder reactions in water compared to the same reactions in organic solvents in the early 1980s.3 Organic transformations conducted in water were further increased in the 1990s with the definition of “green chemistry” proposed by Anastas and Warner.4 Considering the potential rewards of replacing unfriendly organic solvents with water, researchers face the academic challenge of the development of novel synthetic methods that are compatible with water.
In the past two decades, researchers have been committed to exploring novel organic synthetic methods to eliminate the traditional dependence on non-renewable petrochemical feedstocks and toxic solvents. The development of organic transformations using water as a solvent has emerged recently. Organic transformations, such as the Diels–Alder reactions, pericyclic reactions, nucleophilic additions, the Mukaiyama reaction, and the Barbier–Grignard-type arylations, which were traditionally carried out in volatile organic solvents could be conducted in water, showing additional interesting features resulting from the inherent advantages of reactions performed in water over those carried out in organic solvents.5 For example, the employment of water as a solvent would effectively benefit chemical processes by omitting the use of flammable and anhydrous organic solvents, improving the overall synthesis efficiency to eliminate the tedious protection/deprotection processes for certain acidic hydrogen-containing functional groups, using water-soluble compounds such as carbohydrates directly to avoid derivatization, reducing operational costs by recycling water-soluble catalysts and other reagents via simple phase separation, allowing mild reaction conditions, and delivering unforeseen reactivity and selectivity in some typical cases.6 Therefore, the application of water as a solvent is significant and attractive.
On the other hand, visible light represents a renewable, clean, and abundant energy source. Utilizing the energy of visible light instead of burning fossil fuels to break and form chemical bonds is an attractive green prospect. Over the last decade, visible light-mediated photoredox catalysis has become a universal and powerful synthetic technique for the development of novel and valuable organic transformations.7 As a result, a series of photocatalytic reaction systems have been successfully developed and applied in modern synthesis, demonstrating the outstanding potential of photocatalysis.8 Among them, photocatalytic organic conversions in the aqueous phase are more attractive and have received dramatic attention in recent years. Undoubtedly, the combination of photocatalysis and aqueous phase reaction is of increasing interest to develop sustainable synthetic methods for organic transformations.
However, an overview to emphasize the recent advances in visible-light-mediated organic transformations in water is particularly lacking. In this review, we will focus on the recent advances in visible-light-mediated organic transformations in water, excluding those carried out in mixed solutions of organic solvents and water. In such protocols, photocatalysts such as transition metal complexes, organic dyes, solid-supported photocatalysts, and semiconductors are significantly critical. However, some photocatalytic approaches without typical photocatalysts were also reported. We sincerely hope that this review will be beneficial to scientists interested in visible-light-mediated organic transformation in water and will stimulate the advancement of more novel relevant strategies.
Transition metal complexes as photocatalysts
Transition metal complexes as photocatalysts have been extensively applied in the photocatalytic transformations due to their good stability, unique redox properties, and outstanding luminescence properties.9 For example, Ru(II)/Ir(III) polypyridyl complexes, Co complexes, and Pd catalysts have shown excellent activities in various photocatalytic reactions in the past few decades.10Ruthenium(II) polypyridyl complexes as photocatalysts
Among the well-studied ruthenium(II) polypyridyl complexes, the air- and moisture-stable commercially available [Ru(bpy)3]2+ complex demonstrated excellent performance in photoredox reactions. The reasonable maximum absorption of [Ru(bpy)3]2+ (around 450 nm) makes it suitable to be effectively excited by visible light and even sunlight, avoiding the requirement of harsh UV light irradiation and special quartz equipment. Moreover, under the irradiation of visible light, the [Ru(bpy)3]2+ complex was excited through the electron transfer from the t2g dπ orbital of the metal center Ru to the π* orbital of the ligand (2,2′-bipyridine), namely MLCT (metal to ligand charge transfer), which led to a long-lived triplet excited state (1 μs) for chemical transformations.In the presence of Ru polypyridyl complexes, aryldiazonium salts could be employed as precursors of aryl radicals and a large variety of elegant arylation reactions have been well developed.11 It is well known that N-heterocycles, especially quinolines and pyridines, are widely present in natural products, functional materials, and drugs approved by the FDA. In 2014, the visible light promoted arylation of pyridines 1a with aryldiazonium salts 1b in water was first disclosed by the Xue group (Scheme 1).12 By employing [Ru(bpy)3]Cl2·6H2O as a photocatalyst, a wide range of pyridines with electron-donating and electron-withdrawing groups were all compatible in this transformation, furnishing the desired products 1c in moderate to excellent yields with different regioselectivities. Moreover, other electron-deficient heteroarenes, including xanthenes, thiazoles, pyrazines, and pyridazines, could also react smoothly with aryldiazonium salts when using aqueous formic acid as the solvent. A trapping experiment by adding the commonly used radical scavenger 2,2,6,6-tetramethylpiperidinoxyl (TEMPO) to the model reaction was carried out to elucidate the mechanistic details. The significant suppression of the trapping experiment indicated that a radical pathway might be involved in the reaction process.
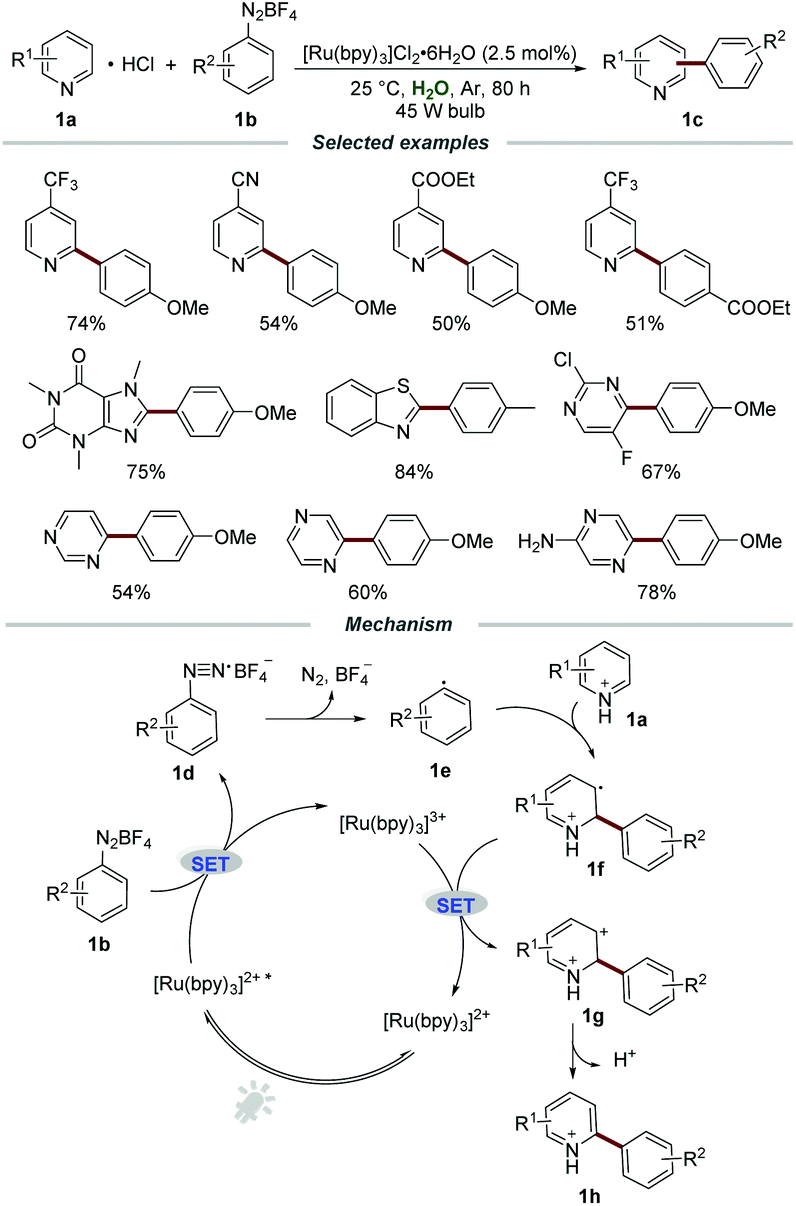 | ||
| Scheme 1 [Ru(bpy)3]Cl2·6H2O-catalyzed arylation of pyridines. | ||
The plausible mechanism showed that [Ru(bpy)3]2+* was formed under the irradiation of visible light, and it then reacted with aryldiazonium salts 1b to afford aryl radical 1evia a single-electron transfer (SET) process. Then, the aryl radical 1e added to pyridine hydrochloride 1a to produce the intermediate 1f. The radical cation 1f was further oxidized by [Ru(bpy)3]3+ to afford carbocation intermediate 1g, which was subsequently transformed into the corresponding product 1h by deprotonation.
Besides electron transfer photoredox catalysis, energy transfer photoredox catalysis utilizing the relatively high energy of the triplet state of [Ru(bpy)3]2+* could occur to activate an energy acceptor with relatively low triplet energy.13 In recent years, the development of synthetic methods for the introduction of fluorine-containing functional groups into organic molecules has become an important topic in organic chemistry due to the great significance of fluorine-containing molecules in pharmaceuticals, agrochemicals, and functional materials.14 In 2019, Wang et al. reported a straightforward and efficient synthetic methodology for the construction of trifluoromethylated dihydroisoquinolinones 2cvia the photoinduced radical cascade cyclization of N-allylbenzamides 2a with CF3SO2Na (2b) under mild reaction conditions (Scheme 2).15 They demonstrated that the reaction could proceed smoothly by employing Ru(bpy)3Cl2 as a photocatalyst and TBHP as an oxidant under the irradiation with a 3 W blue LED at room temperature. By employing the optimized reaction conditions, a wide range of substituted N-allylbenzamides 2a with different functionalities such as electron-donating groups (MeO–, EtO–, t-Bu–, Me–, Et–) or electron-withdrawing groups (–Ph, –F, –Cl, –Br, –I, –CF3, –CO2Me, –OCF3) were reacted smoothly with CF3SO2Na, affording the corresponding products 2c in moderate to good yields. Among them, an obvious electronic effect on the reactivity was observed, and the substrates 2a with electron-donating substituents exhibit higher reactivity than those with electron-withdrawing groups. It is worth noting that the reactions of substrates 2a with functional groups (–OMe, –Cl, –Br) located on the meta-position of the aromatic ring would selectively produce sole products in excellent regioselectivity with good yields.
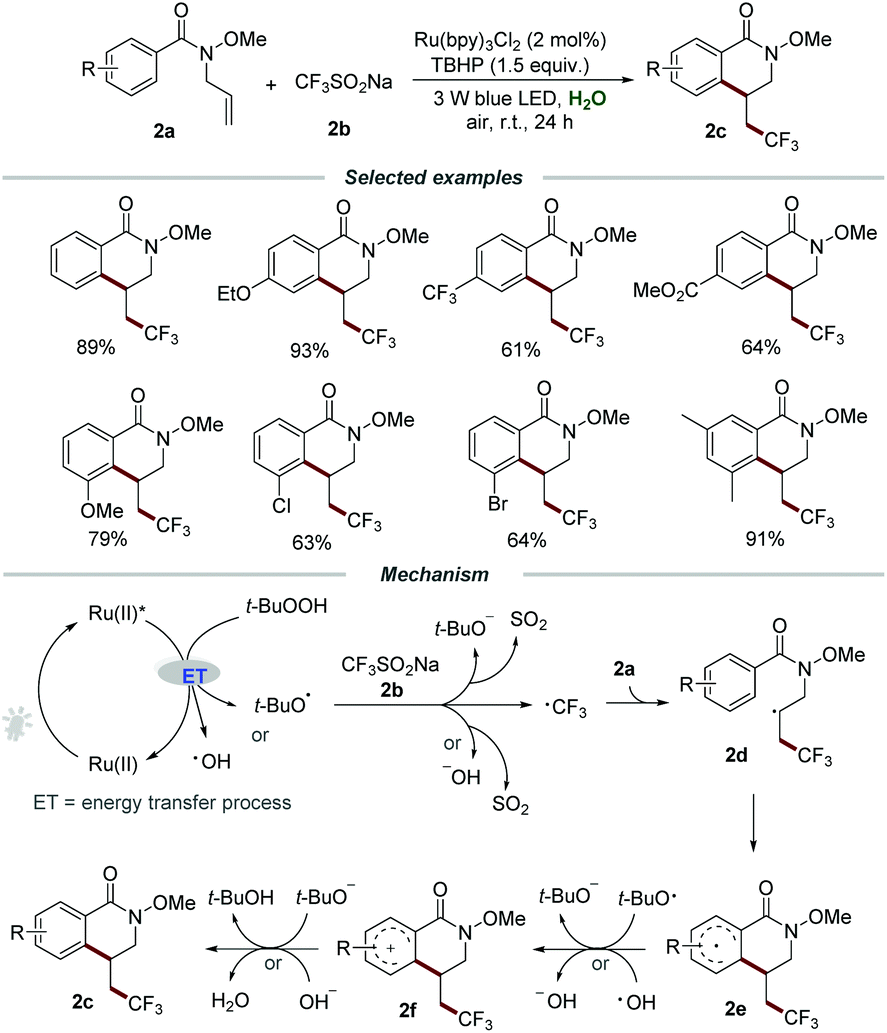 | ||
| Scheme 2 The construction of trifluoromethylated dihydroisoquinolinones. | ||
It was proposed that the energy transfer (ET) between TBHP and the excited photocatalyst Ru(II)* would result in the homolytic cleavage of the peroxide bond, leading to the formation of the hydroxyl radical and the tert-butoxy radical. Then, the reaction between CF3SO2Na and the hydroxyl radical or the tert-butoxy radical would generate ˙CF3, which subsequently added to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of 2a to afford radical intermediate 2d. The formed 2d would undergo intramolecular cyclization, followed by a SET process with the hydroxyl radical or the tert-butoxy radical to form radical cation 2f. Finally, 2f would regenerate the aromatic ring by deprotonation to produce the corresponding product 2c along with the formation of water or tert-butanol.
C bond of 2a to afford radical intermediate 2d. The formed 2d would undergo intramolecular cyclization, followed by a SET process with the hydroxyl radical or the tert-butoxy radical to form radical cation 2f. Finally, 2f would regenerate the aromatic ring by deprotonation to produce the corresponding product 2c along with the formation of water or tert-butanol.
Iridium polypyridyl complexes as photocatalysts
The relevant isoelectronic cyclometalated iridium complexes also exhibit excellent redox ability in visible-light-induced photoredox catalysis. In 2018, Jiang and co-workers reported the methylsulfonylation/bicyclization of 1,7-enynes 3a with dimethyl sulfoxide 3b for the synthesis of sulfone-containing benzo[a]fluoren-5-ones 3c in the presence of iridium polypyridyl complexes (Scheme 3).16 The structure of the product 3c is interesting not only because of the potential pharmacophore, but also because of the challenging all-carbon quaternary centers generated in a single step. Several control experiments were conducted to gain a deep insight into the mechanism. No desired products were observed when the radical scavenger TEMPO or BHT was subjected to the reaction system. Moreover, 18O-labelling experiments revealed that the corresponding methylsulfonyl radical was in situ produced from DMSO via the cleavage of the C–S bond, and one oxygen atom in sulfone came from H2O.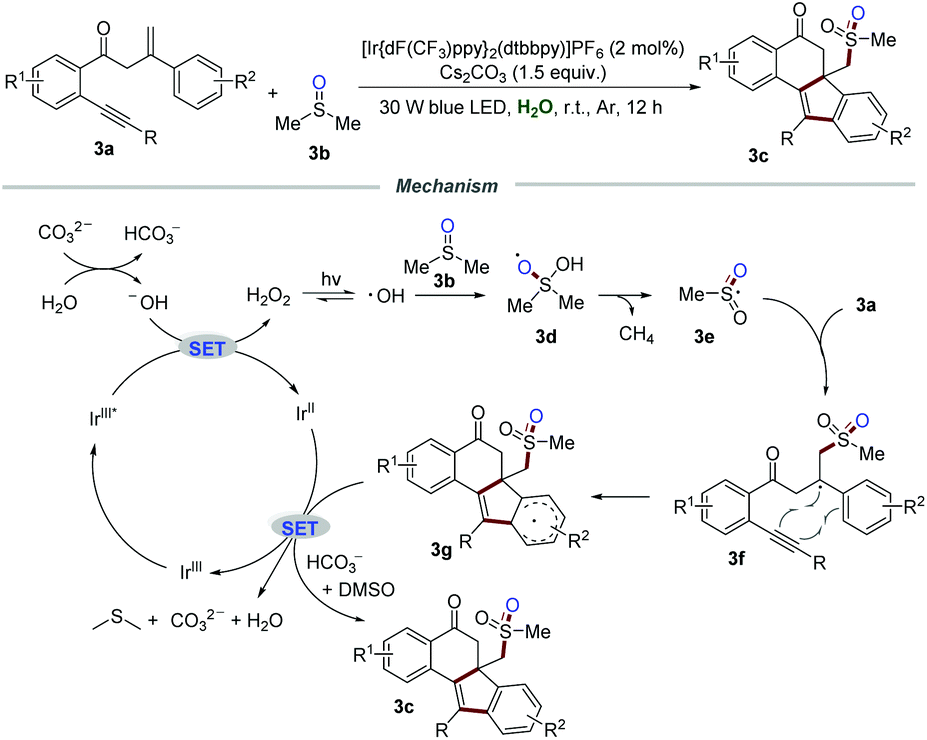 | ||
| Scheme 3 The synthesis of sulfone-containing benzo[a]fluoren-5-ones. | ||
It was postulated that H2O reacted with CO32− to produce a hydroxyl anion, which would be further oxidized by IrIII* via a SET process to form H2O2 and IrII. Under the irradiation of visible light, H2O2 would decompose into the hydroxyl radical, which subsequently reacts with DMSO to afford radical intermediate 3d. Radical intermediate 3d underwent C–S bond cleavage and H-abstraction to generate sulfonyl radical 3e. Afterward, bicyclization of radical 3e would produce radical intermediate 3g, which would further convert into the corresponding sulfone-containing benzo[a]fluoren-5-ones 3c in the presence of bicarbonate ions.
Difunctionalization of alkenes has attracted considerable attention in recent years, and is recognized as an efficient and powerful protocol to introduce diverse functional groups into a molecule in a single step.17 In the same year, Lipshutz et al. reported a photoreductive difunctionalization of alkenes by using task-specific amphiphilic iridium as a photocatalyst (Scheme 4).18 This amphiphilic photocatalyst is composed of a hydrophilic component (MPEG), a lipophilic side chain (50 carbon), and an Ir center. Without any additives and co-solvents, sulfonylation of both alkenes 4a or enol acetates 4d with sulfonyl chloride 4b proceeded smoothly to yield the desired products 4c or 4e in moderate to good yields, respectively. Notably, the aqueous medium containing the photocatalyst could be recycled by simple extraction with methyl t-butyl ether and reused for at least four runs without significant yield reduction.
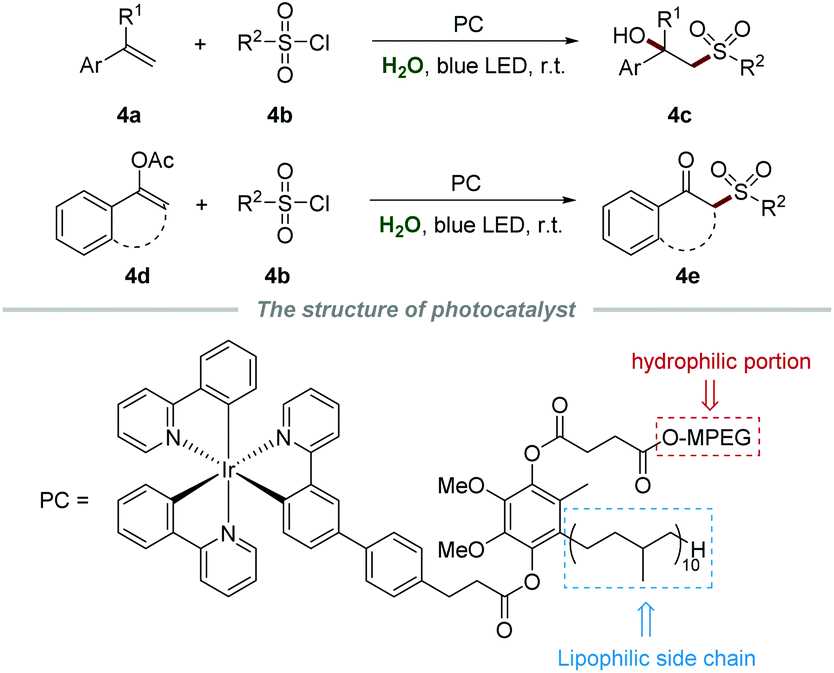 | ||
| Scheme 4 Photoreductive difunctionalization of alkenes catalyzed by an amphiphilic iridium catalyst. | ||
Other transition metal complexes as photocatalysts
Photocatalytic reactions could also be performed using other transition metal complexes such as palladium and cobalt complexes. In 2015, Wu and co-workers accomplished the cross-dehydrogenative coupling reaction of N-phenyl-1,2,3,4-tetrahydroisoquinolines 5a and indoles 5b by using the cobaloxime complex, which was generated in situ from inexpensive CoCl2 and dmgH (dimethylglyoxime) as the photocatalyst (Scheme 5).19 The indoles bearing electron-donating groups exhibit higher reactivities than those with electron-withdrawing groups, which might be due to the enhanced nucleophilicity of indoles caused by electron-donating substituents. The mechanistic studies showed that molecular oxygen played an essential role in this transformation, and the characteristic signal of O2˙− was successfully captured by using the technology of electron–spin resonance (ESR). Moreover, in situ generated CoIII(dmgH)2Cl2 from CoCl2 and dmgH was demonstrated to be a photocatalyst by UV-vis absorption spectroscopy.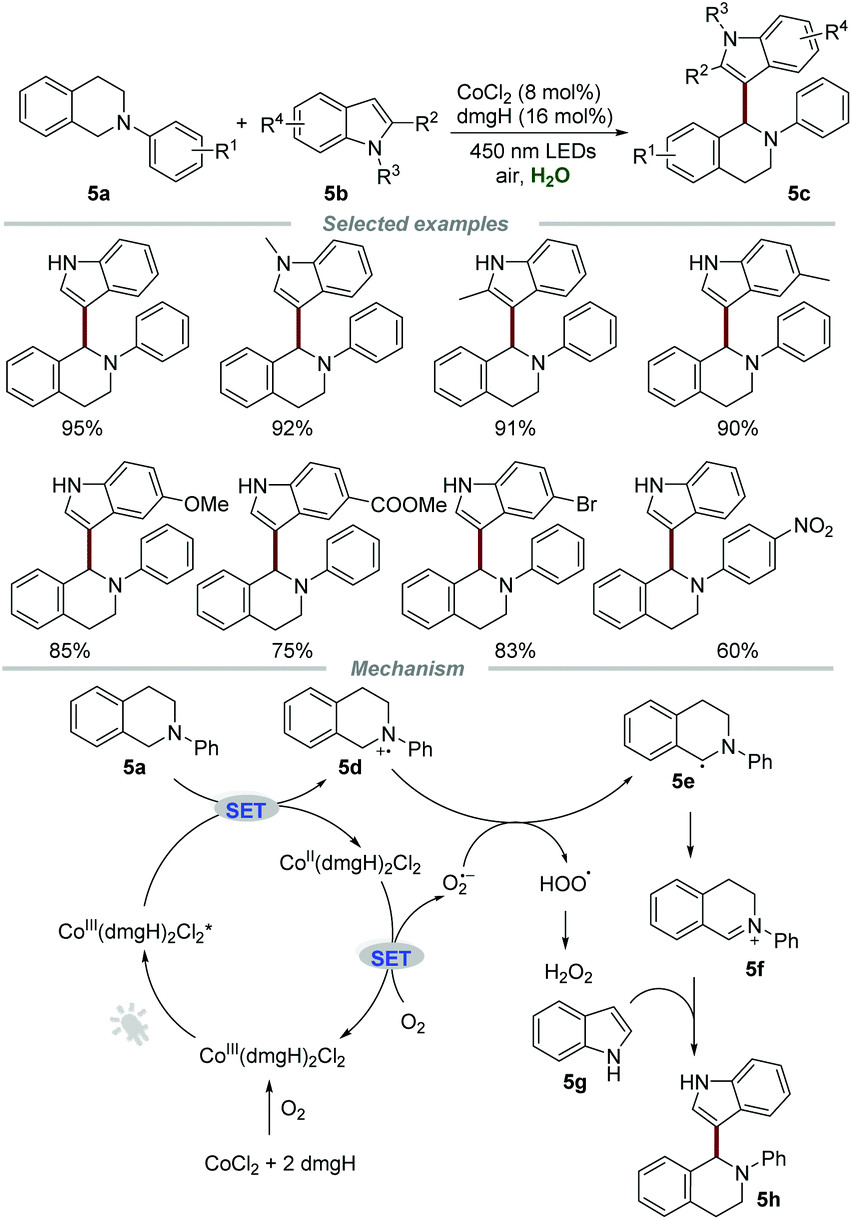 | ||
| Scheme 5 Cobalt-catalyzed cross-dehydrogenative coupling reaction. | ||
Based on the control experiments, it was reasoned that CoIII(dmgH)2Cl2* could react with 5a to generate 5d and CoII(dmgH)2Cl2via a SET process under the irradiation of visible light. Then, CoII(dmgH)2Cl2 could be oxidized by O2 to regenerate CoIII(dmgH)2Cl2 along with the formation of O2˙−, which could further react with 5d to generate 5e and HOO˙. Subsequently, 5e loses one electron to give iminium ion 5f, which further underwent nucleophilic addition with 5g to afford the desired product 5h.
In 2012, Ghosh et al. reported a PdCl2-catalyzed intermolecular Sonogashira coupling conjoined cyclization strategy for the preparation of 2-aryl/alkylbenzofurans 6cvia reactions of ortho-halophenols 6a and terminal alkynes 6b (Scheme 6).20 Both aromatic and aliphatic alkynes were suitable for this reaction, affording the corresponding 2-aryl/alkyl benzofurans in moderate to good yields. In this transformation, an excess amount of triethylamine was required to act both as a base and a co-solvent to increase the solubility of organic substrates. The proposed mechanism showed that Pd(PPh3)2Cl2 or Pd(PPh3)4 was generated when a reaction mixture of PPh3 and PdCl2 was preheated. Then, the reaction between ortho-iodophenol and Pd(0) would generate aryl radical 6d and PdIX via a SET process. Subsequently, aryl radical 6d coupled with PdIX to give aryl palladium intermediate 6e, followed by reductive elimination, leading to the formation of intermediate 6f. Finally, intermediate 6f underwent intramolecular cyclization to yield the corresponding 2-aryl/alkyl benzofurans 6c.
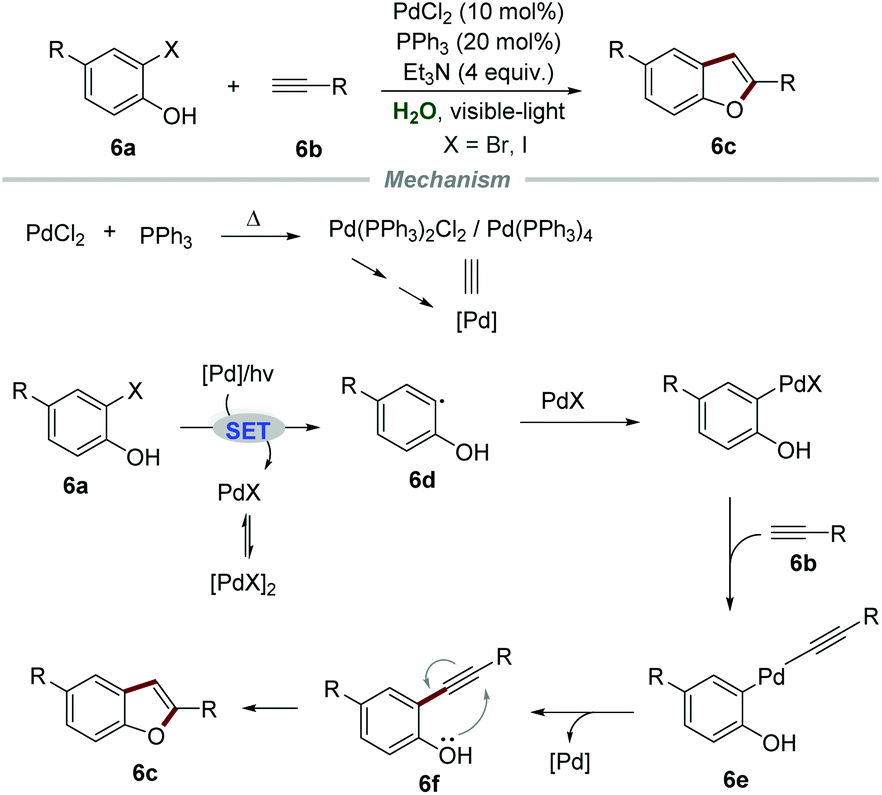 | ||
| Scheme 6 Synthesis of 2-aryl/alkyl benzofurans. | ||
Organic dyes as photocatalysts
In recent years, organic dye-based photoredox catalysts have gained increasing interest due to the significant advantages of transition-metal-free and cost-effective reactions, and hence proving to be viable alternatives to transition metal-based photoredox catalysts.21Organic dyes catalyzed visible light mediated photoredox reactions
Sulfur-containing organic molecules represent an extremely significant class of compounds which have found prominent applications in pharmaceuticals, agrochemicals, and functional materials. The development of sustainable procedures for the formation of the C–S bond to synthesize sulfur-containing compounds remains at the cutting edge of organic synthesis. In 2015, Wang and co-workers reported the direct preparation of sulfonated oxindoles 7c through the metal-free photocatalytic reactions of N-arylacrylamides 7a and sulfinic acids 7b (Scheme 7).22 Aromatic and aliphatic sulfinic acids were all compatible with this transformation to give the desired products 7c with good to high yields, demonstrating a broad substrate scope.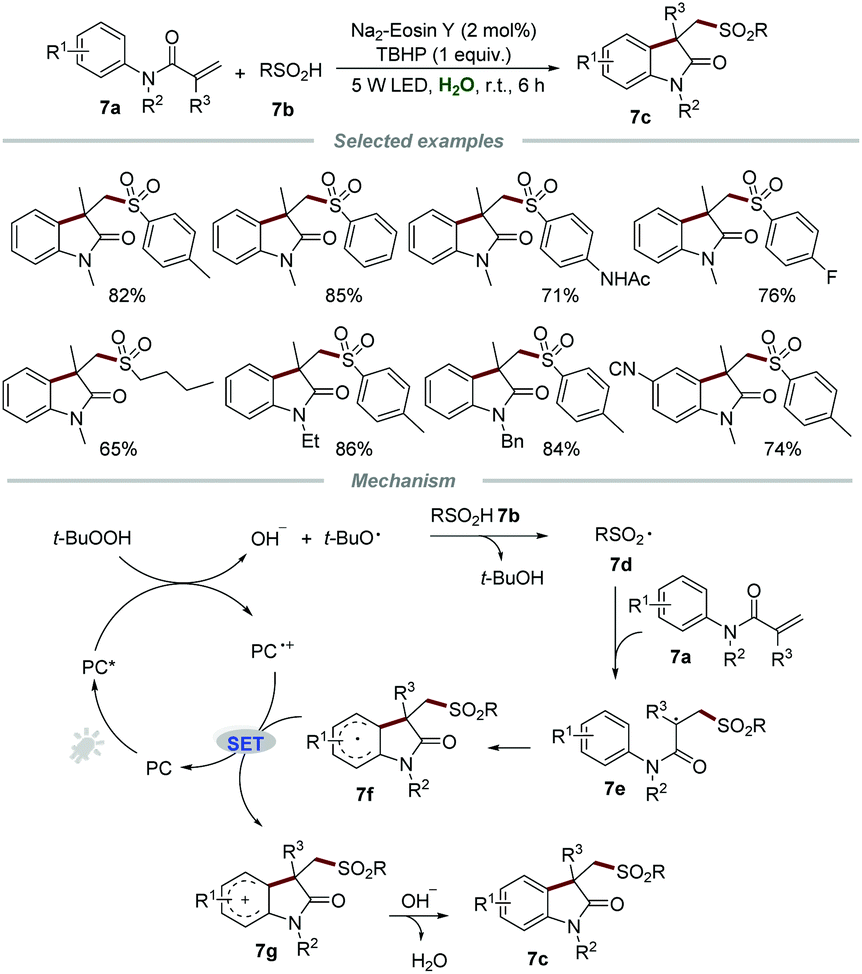 | ||
| Scheme 7 The synthesis of sulfonated oxindoles. | ||
A plausible mechanism involving a radical pathway was proposed. Sulfonyl radical 7d, generated from sulfinic acids 7b in the presence of TBHP and a photocatalyst, would add to the CC bond of 7a to produce radical intermediate 8e. Subsequently, intermediate 7e further underwent intramolecular cyclization to form the radical intermediate 7f, which would be oxidized by PC˙+ to afford carbon cation 7g along with the formation of PC. Finally, deprotonation of intermediate 7g afforded the corresponding sulfonated oxindoles 7c.
In 2018, the synthesis of unsymmetrical diaryl sulfides 8c with broad functionalities via oxidative coupling of thiols 8a and arylhydrazines 8b was conducted by employing a catalytic amount of rose bengal as a photocatalyst (Scheme 8).23 This mild and green approach allowed a wide range of thiols, including phenylthiols bearing various electron-donating and electron-withdrawing groups, benzyl mercaptan, butane-1-thiol and heteroaryl thiols, to be applicable in this transformation. Moreover, diphenyldiselenide could also be employed as a suitable reactant to produce the coupling product (4-methoxyphenyl)(phenyl)selane in good yield.
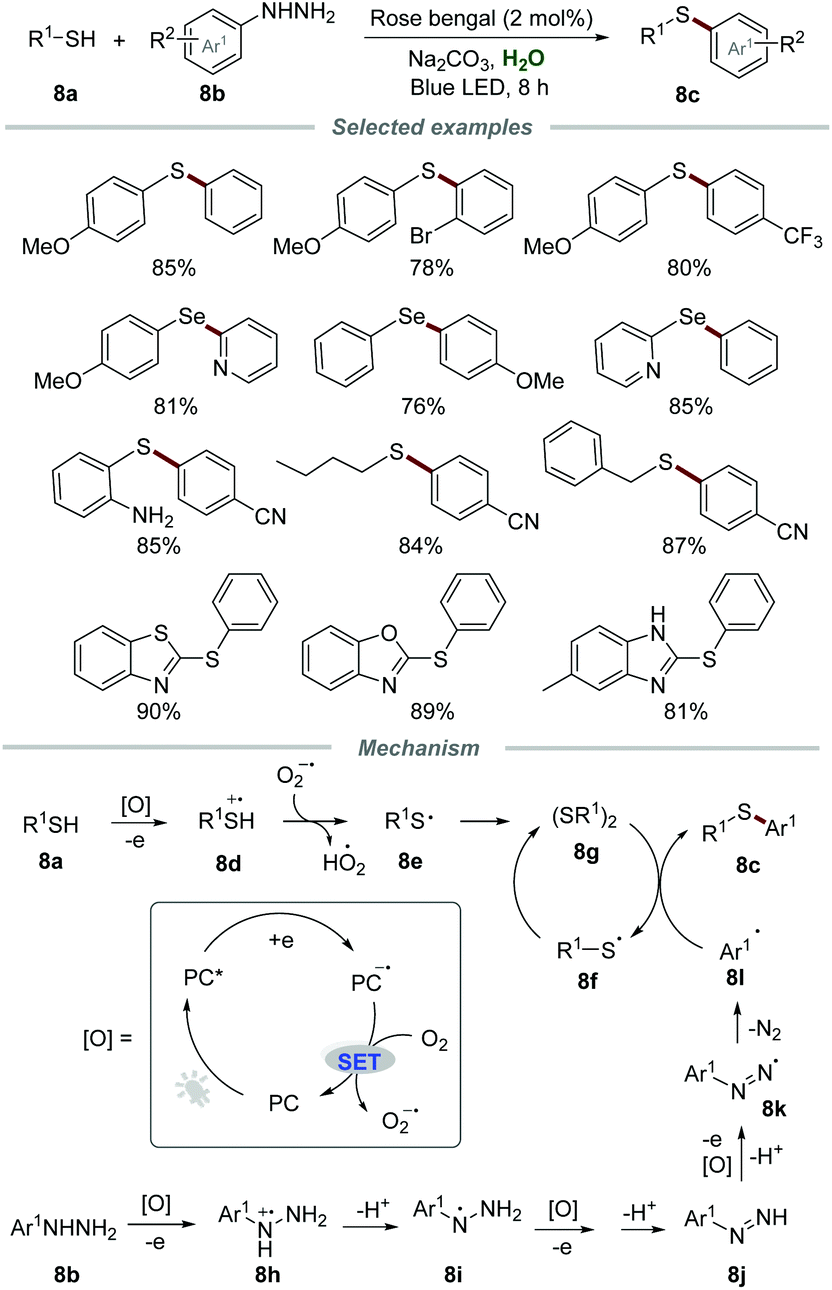 | ||
| Scheme 8 The synthesis of unsymmetrical diaryl sulfides. | ||
The proposed mechanism showed that the reaction between PC* and thiol could generate thiol radical cation 8d and PC˙−via single electron transfer (SET). Then, PC˙− could be further oxidized by O2 to regenerate PC along with the formation of O2˙−. O2˙− could abstract a proton from thiol radical cation 8d to afford thiyl radical 8e which was further transformed into diaryl disulfide 8gvia the homocoupling reaction. Furthermore, arylhydrazine 8b could also be oxidized by PC* to afford radical cation 8h, which subsequently went through deprotonation to give radical intermediate 8i. After sequential SET and deprotonation, radical intermediate 8i could transform into radical intermediate 8k, which subsequently released molecular N2, leading to the generation of aryl radical 8l. Finally, the reaction of aryl radical 8l and diaryl disulfide 8g afforded the desired product 8c and thiyl radical 8f, which again dimerizes to regenerate diaryl disulfide 8g.
In the same year, Xia and co-workers developed a visible-light-mediated nickel(II)-catalyzed approach for the preparation of pyrazole-containing compounds 9cvia C–N cross-couplings between arylamines 9a and pyrazoles 9b in water (Scheme 9). By employing H2O2 as a green oxidant and Acr+-Mes·ClO4− as a photocatalyst, a wide range of pyrazole-containing compounds 9c could be obtained in acceptable yields.24 Moreover, the pyrazole-containing compounds 9c could be hydrolyzed to the corresponding aminated products, which were further converted to the corresponding iodine-containing compounds for the Suzuki–Miyaura coupling, Heck reaction, and Sonogashira reaction. Additionally, the nickel(II) salt could be recovered by simple extraction and could be reused with eight recycles without an obvious loss of catalytic activity.
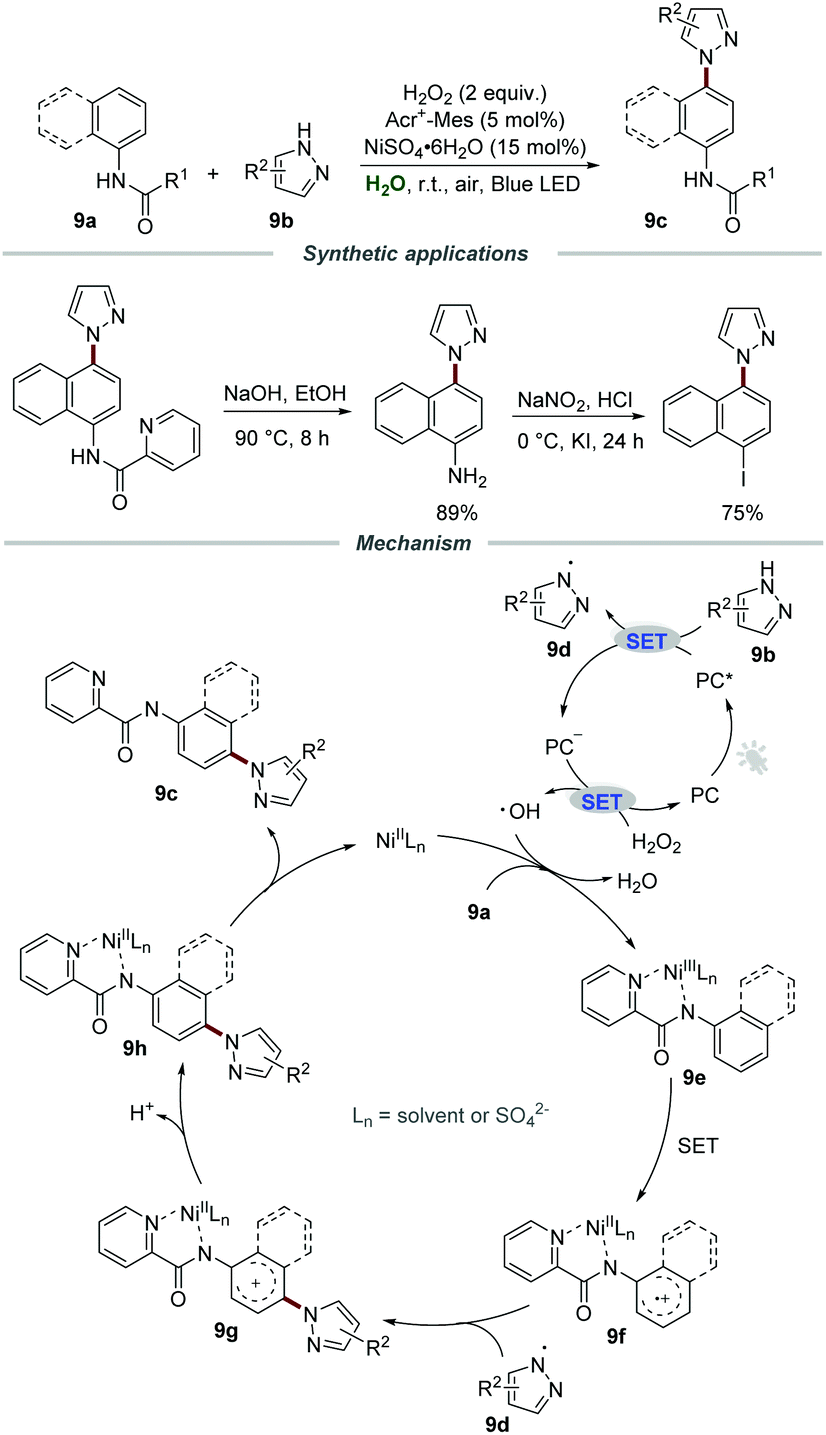 | ||
| Scheme 9 The preparation of pyrazole-containing compounds. | ||
Radical trapping experiments were conducted to gain further insight into the reaction mechanism and the results showed that a radical process might be involved in this transformation. Moreover, the low kinetic isotope effect (KIE) indicated that the cleavage of the C–H bond was not the rate-determining step. It was postulated that the reaction of pyrazole 9b and the excited photocatalyst PC* generated the pyrazole radical 9d and PC˙−. Then, PC˙− would be further oxidized by H2O2 to regenerate PC for the next cycle along with the formation of a hydroxyl radical. With the assistance of NiIILn, arylamines 9a were further oxidized by the hydroxyl radical to give aryl-NiIIILn complex 9e and H2O. After that, the aryl-NiIIILn complex underwent single electron oxidation to form radical cation intermediate 9f, which could react with pyrazole radical 9d to produce cation intermediate 9g. Subsequently, the intermediate 9g underwent deprotonation and a metal-dissociation process to yield the target product 9c.
The construction of C–P bonds is of great significance, because organophosphorus compounds are important and invaluable chemicals that have broad applications in pharmaceuticals and are also widely employed as ligands or intermediates in organic synthesis.25 Particularly, the C–P bond formation via the addition of a phosphoryl radical to alkynes for the construction of Z-alkenes is regarded as a significant value-added but very challenging conversion. Compared to lower-energy E-alkenes, the generation of higher-energy Z-alkenes is more difficult due to the thermodynamically unfavorable process. In 2018, the group of Lei reported the direct synthesis of Z-alkenylphosphine oxides 10c through the addition of phosphoryl radicals to alkynes (Scheme 10).26 Various terminal alkynes and internal alkynes were all compatible with this protocol, leading to the desired products 10c in moderate to good yields. Gratifyingly, a series of amino acid substrates were applicable to this visible-light-induced Z-selective protocol, affording the corresponding products 10c in moderate yields. Furthermore, alkynes containing active functional groups such as the hydroxyl group could be compatible with this protocol, which further demonstrates excellent functional group tolerance. Among them, the π–π stacking interaction between aromatic alkynes and phosphorus substituents could effectively improve the Z-selectivity. The control experiment showed that K2CO3 played an indispensable role in increasing the solubility of the photocatalyst in water by the acid–base reaction.
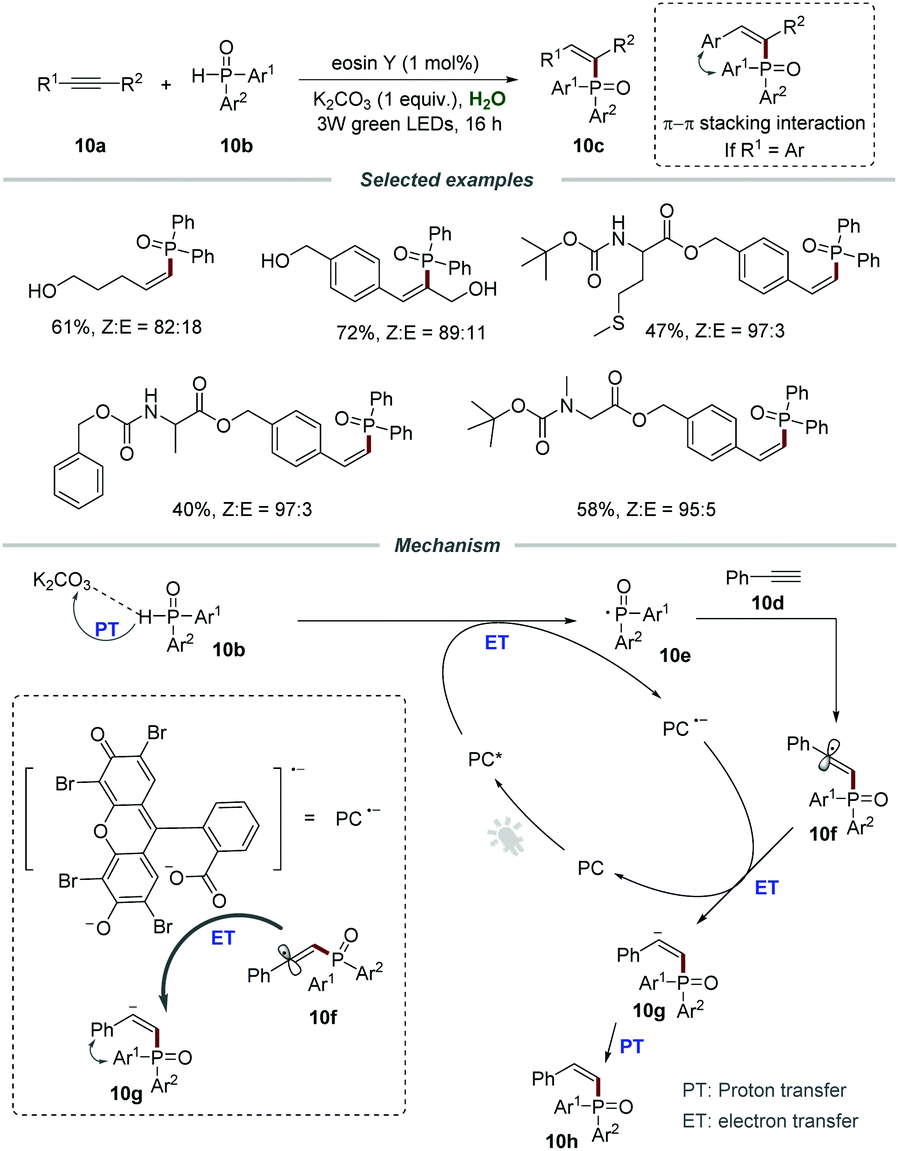 | ||
| Scheme 10 Addition of diarylphosphine oxides to alkynes. | ||
The mechanism involving a radical process is proposed in Scheme 11. The reaction between the photoexcited catalyst (PC*) and diarylphosphine oxides via the proton-coupled electron transfer (PCET) process generated phosphoryl radical 10e which would add to the C–C triple bond of phenylacetylene 10d, leading to the formation of α-alkenyl carbon radical 10f. Then, α-alkenyl carbon radical 10f reacted with PC˙− to afford Z-alkenyl anion 10g, followed by proton transfer to give the corresponding product 10h.
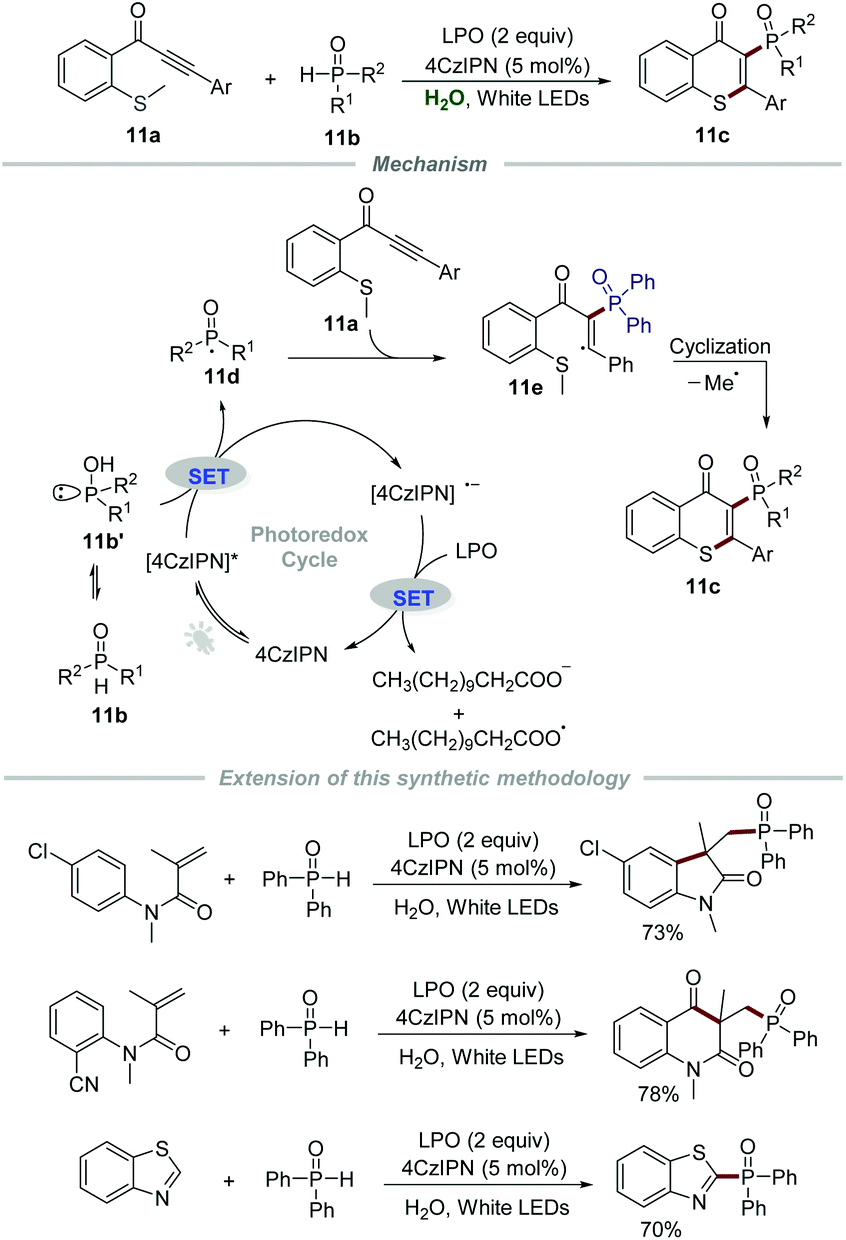 | ||
| Scheme 11 The synthesis of 3-phosphorylated thioflavones. | ||
Later in 2020, Chen and Yu's group described the synthesis of 3-phosphorylated thioflavones 11c through the reaction of methylthiolated alkynones 11a and phosphine oxides 11b by employing 4CzIPN (1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene) as a photocatalyst and lauroyl peroxide (LPO) as an oxidant (Scheme 11).27 The reaction had a broad substrate scope and was amenable to scale up, which provided efficient and green access to 3-phosphorylated thioflavones 11c in moderate to good yields. It should be mentioned that water can be reused at least 5 times after simple extraction without an obvious reduction in reactivity in this powerful protocol, which further reduced the waste. Additionally, this sustainable strategy was successfully used in the synthesis of some other phosphorylated N-heterocycles, such as phosphorylated oxindole, phosphorylated quinoline-2,4(1H,3H)-dione, and phosphorylated benzothiazole. The mechanistic studies showed that the generated phosphoryl radical 11d would react with methylthiolated alkynones 11a to afford radical intermediate 11e which would further undergo intramolecular cyclization to give the corresponding products 11c.
Afterward, the same group expanded this metal-free photocatalytic procedure for the synthesis of 3-phosphorylated benzothiophenes 12c (Scheme 12).28 Starting from easily available methylthiolated alkynes 12a and phosphine oxides 12b, a series of 3-phosphorylated benzothiophenes 12c bearing different functional groups were prepared in moderate to excellent yields. This mild phosphorylation/cyclization reaction could also be scaled up, and 1.30 g of 3-phosphorylated benzothiophene (79% yield) was obtained when the reaction of methylthiolated alkyne and diphenylphosphine oxide was performed in 4 mmol-scale under the optimized reaction conditions. Stern–Volmer fluorescence quenching experiments showed that the excited state of 4CzIPN was effectively quenched by methylthiolated alkynes 12a.
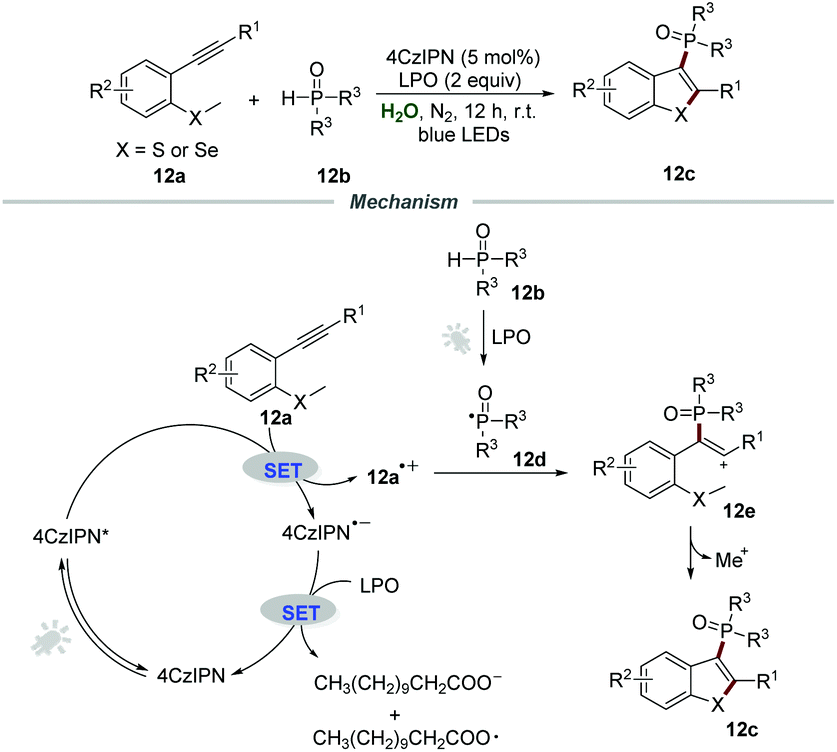 | ||
| Scheme 12 The synthesis of 3-phosphorylated benzothiophenes. | ||
Based on the control experiments, a plausible reaction pathway involving a radical pathway was proposed. With the assistance of LPO and visible light, phosphine oxides 12b would generate phosphoryl radical 12d. Then, the reaction between 4CzIPN* and methylthiolated alkynes 12a would produce intermediate 12a˙+, which would further react with phosphoryl radical 12d to yield cation intermediate 12e. Finally, cation intermediate 12e underwent an intramolecular cyclization to afford the phosphorylated benzothiophenes 12c.
Phenanthrols are widespread in naturally occurring alkaloids, agrochemicals, pharmaceuticals, and functional materials. Very recently, the construction of phenanthrols was accomplished via the intramolecular cycloaromatization reaction of alkyl 1,1′-biaryl-2-ones 13a under the irradiation of visible light, which was reported by Natarajan and co-workers (Scheme 13).29 The employment of rose bengal as a photocatalyst and K2CO3 as a base in H2O under the irradiation with a 5 W blue LED and ambient conditions gave the best result, and a wide range of 10-alkylphenanthren-9-ols 13b were prepared in good yields under the optimized reaction conditions.
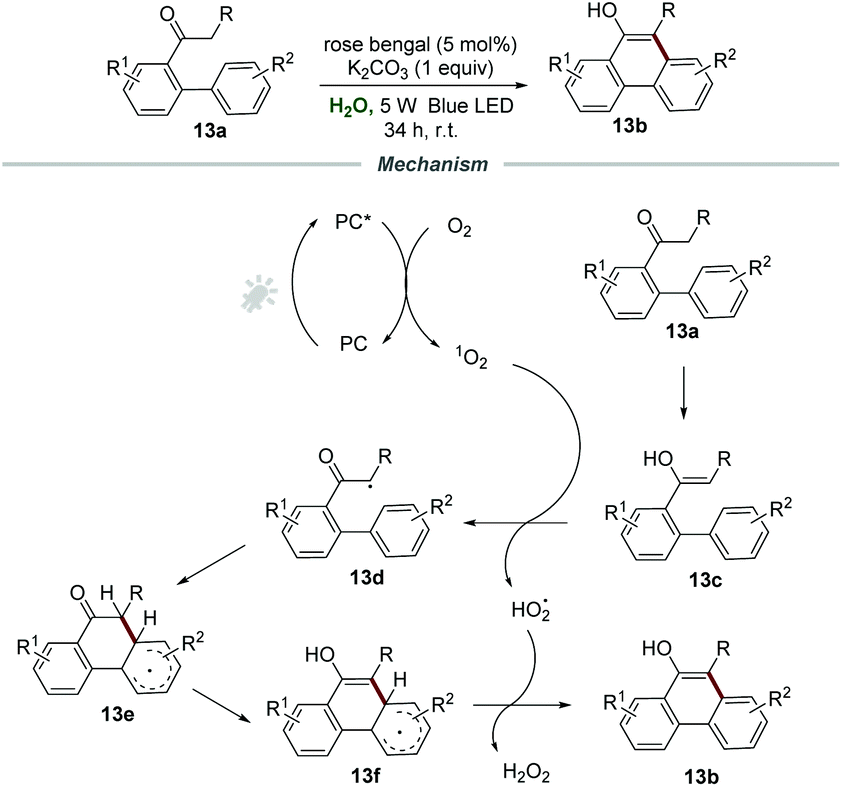 | ||
| Scheme 13 The preparation of phenanthrols. | ||
For the mechanism, it was postulated that the energy transfer between PC* and O2 produces singlet oxygen, which would react with the enol form 13c of alkyl 1,1′-biaryl-2-ones 13a to form α-carbon radical 13d and HO2˙. Afterward, the α-carbon radical 13d underwent intramolecular cyclization to afford the radical intermediate 13e, followed by the enolization of the ketone to generate radical intermediate 13f. With the assistance of HO2˙, 13f was further transformed into the desired product 13b with the formation of H2O2.
Merging visible light photoredox catalysis and micellar catalysis
Most organic substrates and organic dyes are insoluble in water, which should be a major problem for photoredox catalysis carried out in aqueous media. In order to solve this problem, a surfactant could be added to water to form micelles, allowing organic reactions to proceed in water. Combining photoredox catalysis and micellar catalysis has provided a feasible solution to carry out photoredox catalysis in aqueous media.Aromatic amines are a class of cheap, easily available, and versatile organic synthetic intermediates, which can react with t-BuONO to generate aryldiazonium salts in situ. In 2018, the arylation of heteroarenes through photocatalytic reactions of heteroarenes 14a, aromatic amines 14b, and t-BuONO was developed by using Triton X-100 as the external surfactant and eosin B as a photocatalyst, as reported by Cai's group (Scheme 14).30 In this transformation, various aromatic amines 14b bearing different functional groups, including alkyl, alkoxy, nitro, cyano, and trifluoromethyl, were all compatible. Among them, anilines 14b with electron-withdrawing and neutral substituents usually show higher yields than those with electron-donating groups. In addition, the reaction conditions can also be suitable to accomplish α-arylation of enol acetates, sulfides, and selenides, proving it to be a simple and efficient catalytic system for the arylation of heteroarenes.
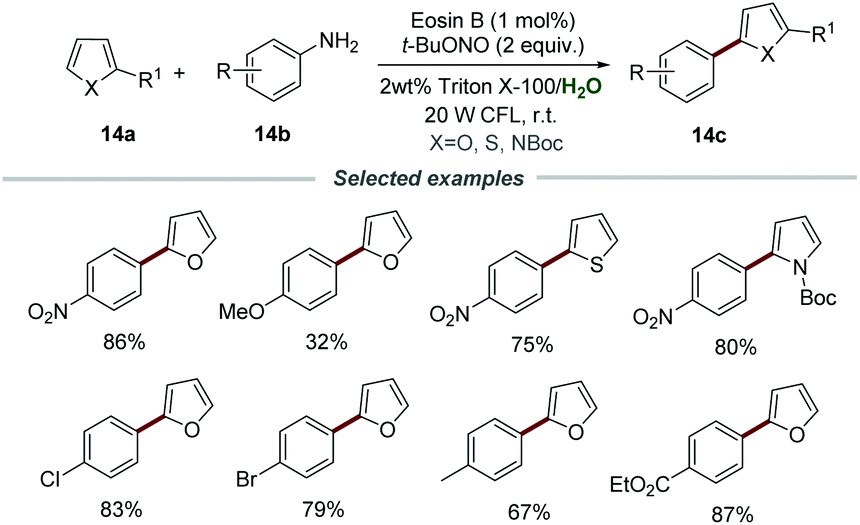 | ||
| Scheme 14 The arylation of heteroarenes. | ||
Very recently, the group of Xu reported a metal-free visible light-mediated approach for the remote 1,5-trifluomethylthio-sulfonylation of vinylcyclopropanes 15a with ArSO2SCF3 reagents 15b in the presence of eosin Y and sodium dodecyl sulfate (SDS), affording the desired products 15c in moderate to good yields with high Z/E ratios (Scheme 15).31 A large variety of vinylcyclopropanes were applicable and the aldehyde moiety could be tolerated in this transformation, demonstrating broad reaction scope and excellent tolerance of functional groups. Additionally, alkyl vinylcyclopropane was also suitable for this reaction, leading to the desired product in moderate yield with poor stereoselectivity, which might be due to the less steric hindrance effect of the benzyl group than the aryl group. Radical trapping experiments indicated that a radical pathway was involved in this transformation. Moreover, the on/off experiments indicated that continuous photocatalysis was indispensable and a radical chain pathway was not involved in this transformation.
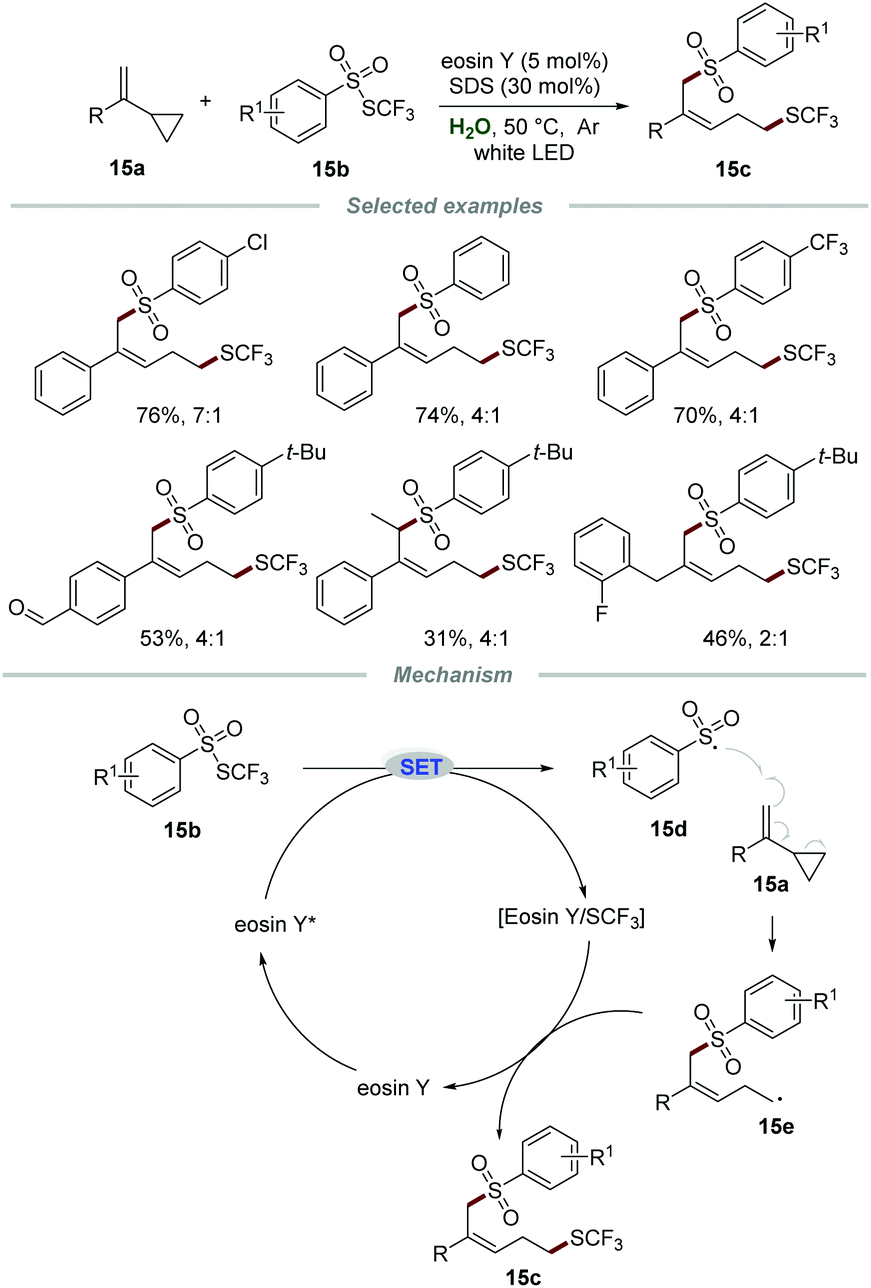 | ||
| Scheme 15 Difunctionalization of alkenes. | ||
According to the reaction mechanism, the reaction was initiated by a SET process between eosin Y* and ArSO2SCF315b to give sulfonyl radical 15d and the eosin Y/SCF3 complex. The arylsulfonyl radical 15d was added to the CC bond of substrate 15a, followed by a rearrangement of cyclopropane to afford radical intermediate 15e, which would further react with the eosin Y/SCF3 complex to furnish the corresponding product 15c along with the regeneration of eosin Y for the next cycle.
Solid-supported photocatalysts
Despite the great achievements that have been made in recent years, transition metal- and organic dye-based photocatalysts were difficult to recycle and reuse. To overcome this problem of recyclability, some elegant solid-supported photocatalysts have been developed by the immobilization of photocatalysts onto appropriate carrier materials.Compared with silica-based materials, metal–organic frameworks (MOFs), and graphene, the application of sponge as the catalyst carrier would make the photocatalysts more convenient to be separated from the reaction system without conventional filtration or centrifugation, making it more practical for industrial application. Polydimethylsiloxane (PDMS), as a sustainable and highly transparent polymer material, is a suitable sponge support for photocatalysts to let light smoothly penetrate for photocatalysis.32
In 2017, the Zhang group reported a novel and powerful protocol for the efficient coupling of tertiary amines 16a with ketones 16b in water in the presence of a sponge photocatalyst (Scheme 16).33 The sponge photocatalyst could be obtained via the modification of the polymer surface and classical solid-phase peptide synthesis. The morphology was further characterized by scanning electron microscopy (SEM) and a 3D-interconnected porous material was observed, providing strong evidence for the intact PDMS sponge which was not destroyed by surface modification. Moreover, the SEM elemental mapping images revealed the even distribution of C, N, O, Si, Cl, and I elements in this sponge photocatalyst. Additionally, the catalyst loading on the PDMS sponge was confirmed by elemental analysis to be 0.0176 mmol g−1. A hydrophobic reaction microenvironment, provided by the PDMS sponge, was the key to ensure the smooth progress of the coupling reaction. N-Phenyltetrahydroisoquinolines substituted with various functional groups, including –OMe, –Me, –Br, –Cl and –CF3, on the different positions of the N-phenyl ring were all compatible with this reaction, and the substrates bearing electron-donating groups showed slightly higher reactivities than those with electron-withdrawing groups. Moreover, diverse methyl ketones and 3-pentanone could also be suitable substrates for this transformation. Furthermore, intramolecular cross-dehydrogenative coupling (CDC) reactions could also be achieved in the presence of this sponge photocatalyst. Notably, the sponge photocatalyst could be conveniently separated and reused at least 10 times by simply removing and washing without an obvious reduction in activity.
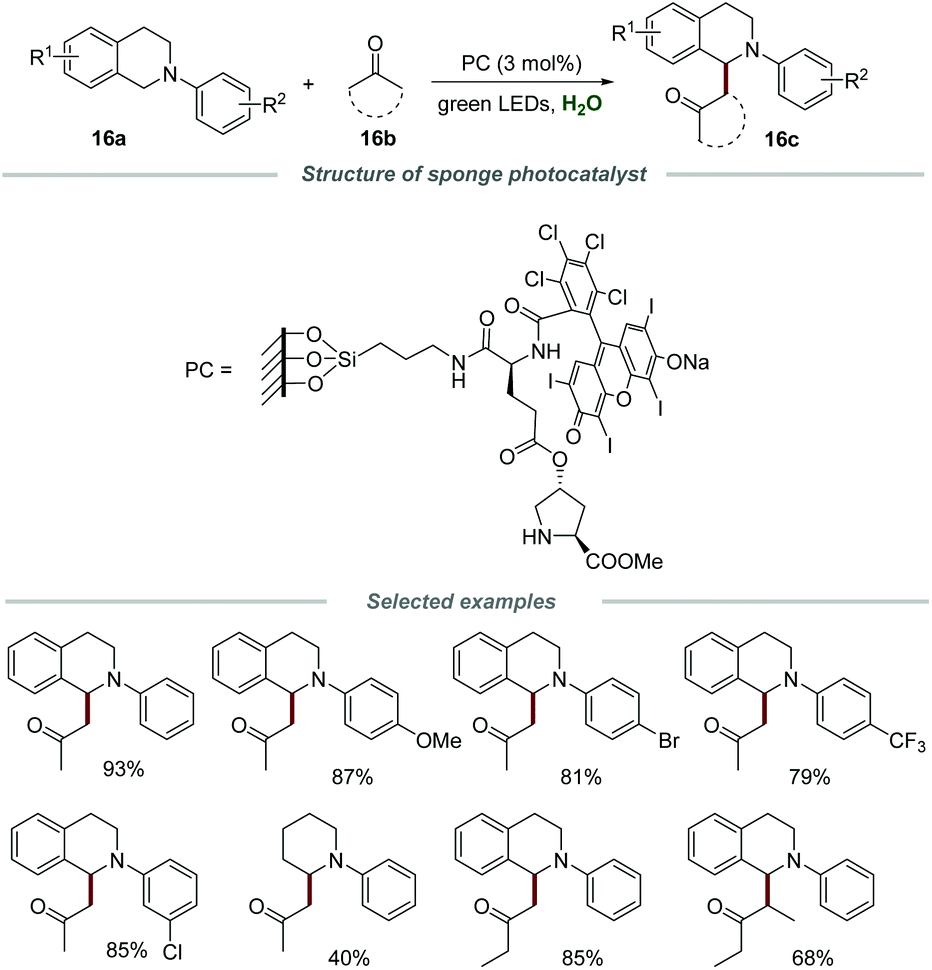 | ||
| Scheme 16 Coupling of tertiary amines with ketones. | ||
Layered double hydroxides (LDHs), as two-dimensional layered anionic clay nanomaterials, have obvious advantages such as simple preparation, low-cost, flexible composition, a high charge density of the positive layer, and even a relatively large surface area, which make them a promising candidate for immobilizing a photocatalyst. In 2018, Zhang et al. disclosed the preparation of an LDH supported organic photoredox catalyst (Fe3O4-RB/LDH), which could be easily obtained by ultrasound-assisted self-assembly among the anionic rose bengal (RB) dye, negatively charged Fe3O4 NPs, and positively charged MgAl-LDH.34 The XRD spectra of Fe3O4-RB/LDH indicated the existence of Fe3O4 and MgAl-LDH, while the red color and the solid-state UV-vis spectra indicated the presence of RB in this composite. The analysis of morphology by SEM showed a similar morphology of MgAl-LDH and Fe3O4-RB/LDH, suggesting that the structure was not destroyed after several rounds of ultrasonication. Additionally, the results of SEM mapping gave strong support for the homogeneous distribution of RB and Fe3O4 NPs in Fe3O4-RB/LDH. Further studies showed the elegant catalytic activity of Fe3O4-RB/LDH in highly efficient and selective aerobic oxidation of 17a to sulfoxides 17b, and the desired products could be obtained in moderate to good yields (Scheme 17a). Moreover, the ability of Fe3O4-RB/LDH in the cross-coupling reaction of tetrahydroquinoline derivatives 17c with nitroalkanes 17d has been elegantly demonstrated. Different functionalized tetrahydroquinoline derivatives 17c all reacted smoothly with nitroalkanes 17d to produce the corresponding products 17e in moderate to good yields, and no prominent electronic effect was observed in this transformation (Scheme 17b). Additionally, Fe3O4-RB/LDH catalyzed selective synthesis of thioethers 17h from alkenes 17f and thiols 17g has also been explored, and the desired thioethers 17h could be isolated in moderate to good yields (Scheme 17c). The recyclability of Fe3O4-RB/LDH was investigated by convenient separation using a magnet in the coupling reaction of N-phenyl tetrahydroisoquinoline and nitromethane, and it could be used at least 6 times without any significant loss of its catalytic activity.
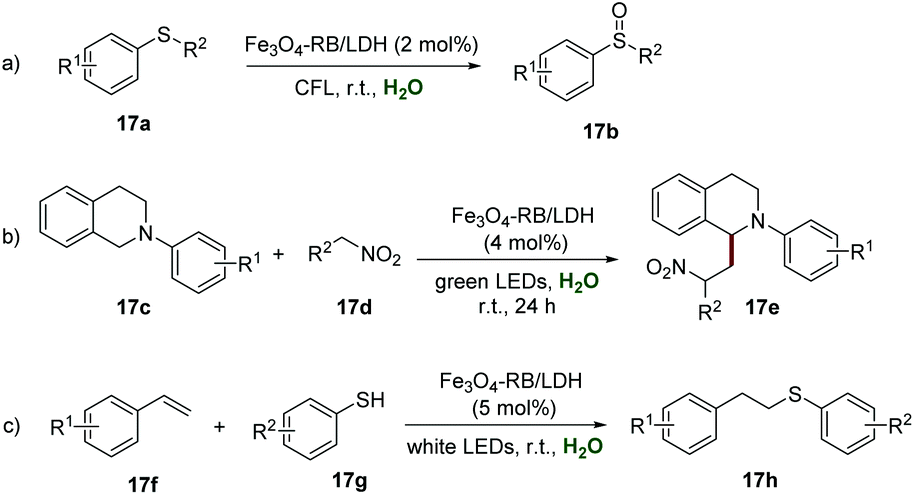 | ||
| Scheme 17 Fe3O4-RB/LDH catalyzed organic transformations. | ||
Semiconductors as photocatalysts
Semiconductor materials are generally thermally stable, photo-stable, and chemically stable. The photoexcited electron–hole pairs on the surface of semiconductors could donate or accept an electron to enable photoredox catalysis via oxidative or reductive quenching cycles. In the past few decades, the application of semiconductor materials in photochemistry mainly focused on the photolysis of water.35 However, recently the employment of semiconductors as heterogeneous photocatalysts for various organic transformations has emerged as a novel strategy in the development of a sustainable photocatalytic system.36Quantum dots as photocatalysts
Zero-dimensional (0D) nanomaterial quantum dots (QDs) possess abundant surface-binding sites and show tunable absorption spectra. Therefore, QDs should be a potential photocatalyst for organic transformations due to the ability of light harvesting and energy conversion.37Green and highly selective oxidation of alcohols to the corresponding aldehydes/ketones without over-oxidation is still enthusiastically pursued due to the important roles of aldehydes/ketones in organic transformation. In the past few years, some novel photocatalysts including rutile TiO2,38 Ru-substituted polyoxometalate39 and carbon nitride40 have been developed for the oxidation of alcohols to aldehydes/ketones in water under the irradiation of visible light. Particularly, in 2017, Wu and co-workers achieved a photoinduced selective conversion of alcohols 18a to aldehydes/ketones 18b by dehydrogenation with the assistance of 3-mercaptopropionic acid (MPA)-capped CdSe quantum dots (MPA-CdSe QDs) and NiCl2 without any external oxidant (Scheme 18).41 The general applicability of this protocol was demonstrated by employing various alcohols 18a with both electron-donating and withdrawing groups. The experimental results proved that the substrate with electron-withdrawing substituents reacted slightly slower than those with electron-donating groups. Moreover, aliphatic primary alcohols, including propanol, 3-phenylpropanol, cyclopentanol, and cyclohexanol, were all suitable for this transformation with a slower reaction rate than benzyl alcohols, which might be caused by the higher bond dissociation energies of the aliphatic C–H bonds.
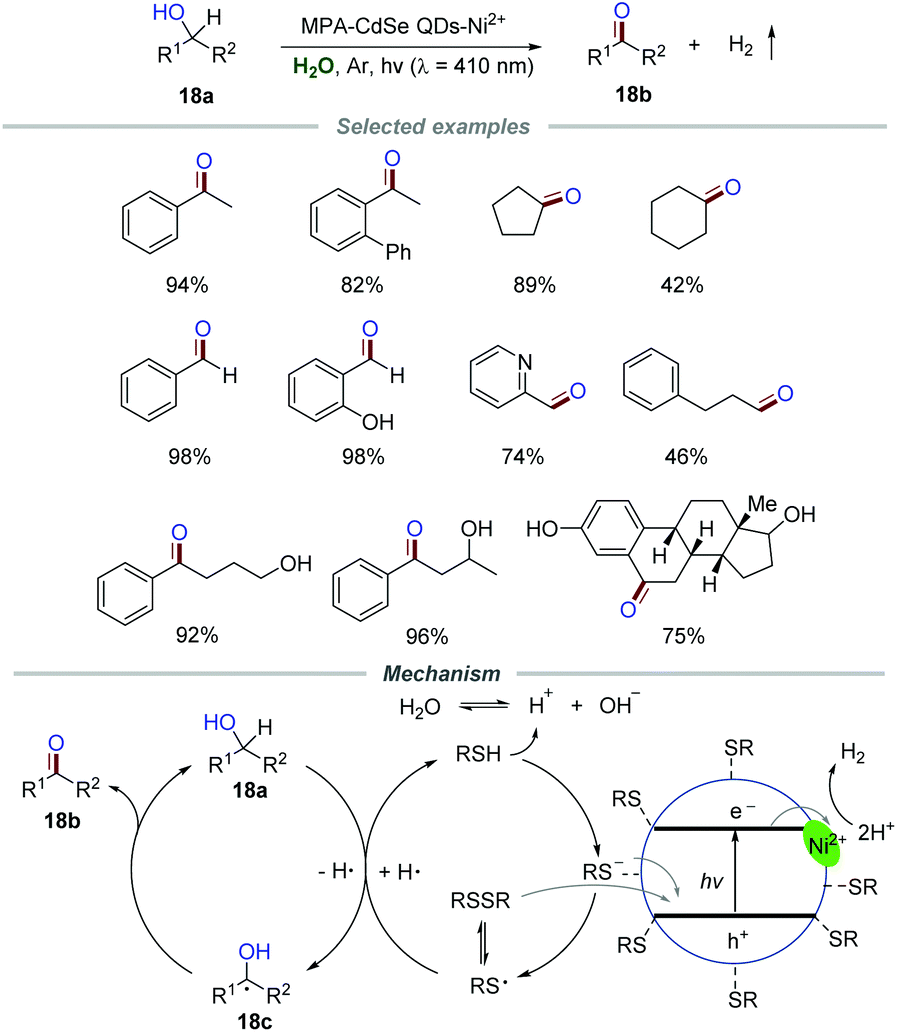 | ||
| Scheme 18 Selective oxidation of alcohols to aldehydes/ketones. | ||
The mechanistic investigation showed that the optimal pH range (8–11) for the reaction is close to the pKa value of aliphatic thiols, implying that MPA adsorbed on CdSe QDs in the form of the thiolate anion instead of neutral thiol. Moreover, the band edge emission was effectively quenched by NiCl2, suggesting that the surface interactions of QDs and NiCl2 are significant. In addition, the reaction was severely suppressed in the presence of a good electron acceptor (AgNO3). All of these results gave strong support for the electron transfer between the conduction band of CdSe QDs and the physisorbed Ni2+. Additionally, the reaction proceeded slowly when the hole quencher ammonium oxalate was added, suggesting a “hole transfer” from the valence band of CdSe QDs to the thiolate anion. Additionally, the signals corresponding to the adducts of thiyl and hydrogen radicals with the radical trapper 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) were successfully detected by using ESR. Based on these results, a mechanism was proposed. The thiyl radical generated from the adsorbed thiolate anion or disulfide via hole transfer would abstract a hydrogen atom from benzyl alcohol 18a to generate benzylic radical 18c which would further transform into the corresponding benzyl alcohol or benzaldehyde 18b. Hydrogen abstracted from benzyl alcohol by the thiyl radical would quickly equilibrate with water. The H+ from H2O was further reduced by Ni2+ adsorbed on the surface of MPA-CdSe QDs, leading to the formation of H2. The deuterium labeling experiment was in agreement with the proposed mechanism.
The formation of C–N bonds occupies an irreplaceable important position in organic synthetic chemistry and at least one C–N bond exists in about 84% of small-molecule pharmaceuticals.42 In 2017, Biswas et al. realized the formation of amide bonds via the reaction of thioacids and amines by using CdS nanoparticles (CdSNPs) as the photocatalyst under the irradiation with a household 30 W CFL in water (Scheme 19).43 This transformation had a broad substrate scope and a large number of amide products were effectively obtained in moderate to excellent yields. Impressively, not only substituted aromatic anilines with electron-donating groups or electron-withdrawing groups but also ammonia, benzylamine, and piperidine could proceed smoothly with thiobenzoic acid in this conversion.
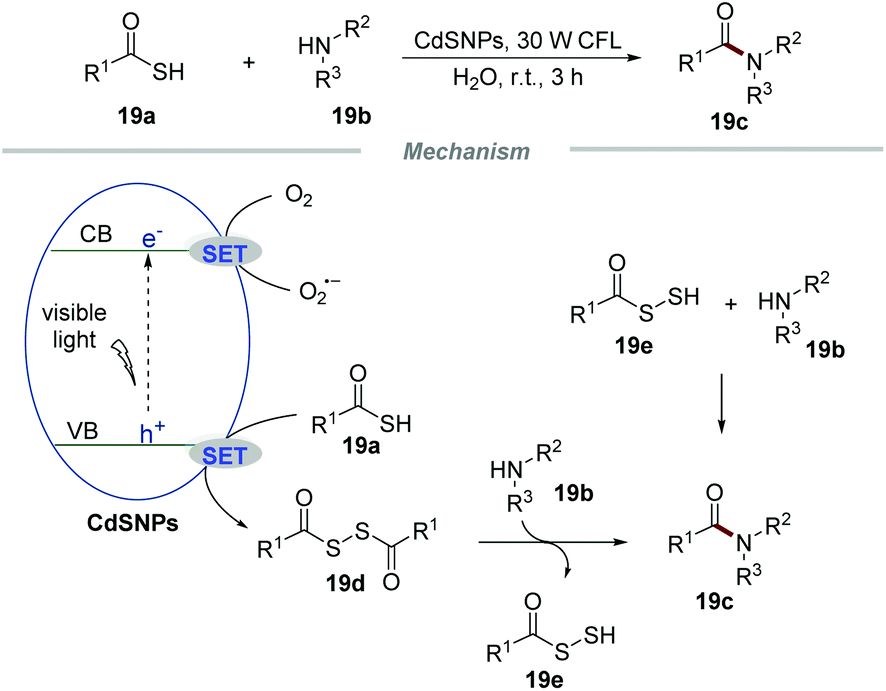 | ||
| Scheme 19 The formation of amide bonds. | ||
The mechanistic studies showed that the photocatalytic reaction of thiobenzoic acids without amines with the assistance of CdSNPs would result in the formation of the corresponding disulfides, which when freshly prepared could react with anilines effectively to afford the corresponding amide products. Based on the detailed mechanistic studies, a plausible mechanism was proposed. Initially, the electron of CdSNPs would be excited to the conduction band under the irradiation of visible light, leading to the formation of an electron (e−) and a hole (h+). Then, a SET process between thioacids 19a and holes would produce benzoyl thiyl radicals, which could readily form disulfide intermediate 19e. The intermediate 19e subsequently reacted with amines 19b to give the corresponding amides 19c along with the formation of intermediate 19e. According to the literature,44 intermediate 19e could further react with amines 19b to generate amides 19c. Additionally, oxygen can accept an electron from the conduction band, leading to the formation of O2˙− and regenerating the CdsNP catalyst.
Very recently, Wu et al. reported the borylation of diazonium salts 20a with diboronate esters 20b by employing nontoxic and easily prepared carbon dots (CDs) as photocatalysts (Scheme 20).45 The blue carbon dots were synthesized through a reaction of 2,6-diaminopyridine with acrylic acid by using ZnCl2 as a promotion agent and tetraethylene glycol as a solvent at 210 °C for 4 h. The mechanistic studies showed that the reaction was severely suppressed in the presence of the radical trapping agent 2,2,6,6-tetramethyl piperidin-1-oxyl (TEMPO) and 2-methyl-2-nitrosopropane (MNP), demonstrating that a radical pathway was involved in this transformation. Stern–Volmer fluorescence quenching studies indicated a strong interaction between diazonium salts and excited CDs. Moreover, the calculated conduction band energy was −0.94 eV, giving reasonable support for the SET process from excited CDs to diazonium salts.
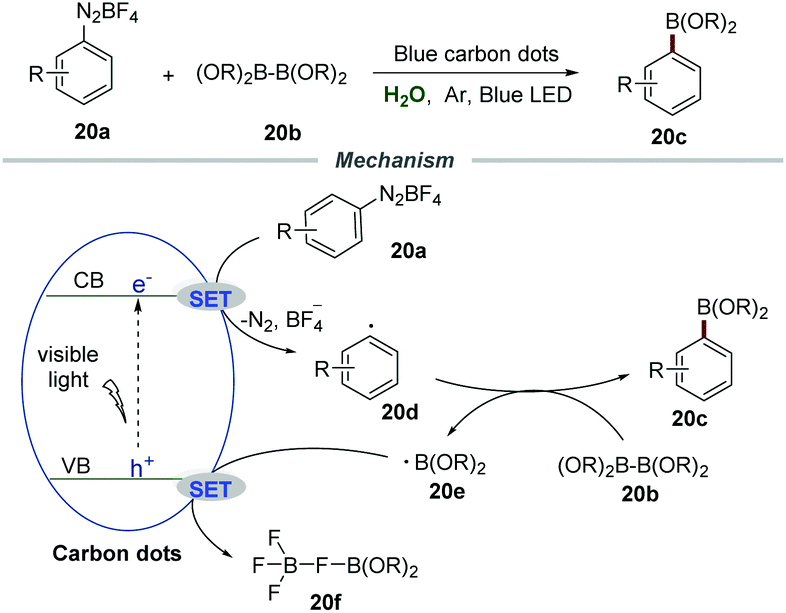 | ||
| Scheme 20 Borylation of diazonium salts. | ||
Based on the results of mechanistic studies, a plausible mechanism was proposed. The reaction between the electron on the conduction band (CB) and diazonium salts 20avia a SET process would generate aryl radical 20d, which would couple with diboronate esters 20b to furnish the corresponding arylboronate esters 20c along with a boron radical 20e. In the presence of tetrafluoroborate anions, the boron radical was converted to 20f through a SET process with the hole on the valence band (VB) of carbon dots. The high reduction potential of carbon dots was the key to promote this transformation.
C3N4 as a photocatalyst
Graphitic carbon nitrides (g-CN), with tunable optical band gaps, are a class of promising metal-free polymers for photochemical applications. They could be easily synthesized from readily available and inexpensive precursors, such as urea, cyanamide, or melamine. The employment of C3N4 as a photocatalyst has attracted increasing interest in recent years.36b,46Phenols are privileged building blocks in medical and industrial organic compounds, dyes, and important intermediates for organic synthesis.47 In 2020, Yu's group developed a copper-doped g-C3N4 (Cu@C3N4) catalyzed hydroxylation of aryl boronic acids 21a for the preparation of phenols 21b by employing environmentally benign oxygen as an oxidant under mild reaction conditions (Scheme 21).48 The catalyst Cu@C3N4 was easily obtained only by calcining melamine and copper(II) tartrate at 550 °C for 2 h under a N2 atmosphere. A number of aryl boronic acids with electron-withdrawing and electron-withdrawing groups reacted smoothly to give the desired phenols in good to excellent yields, showing the advantage of wide reactant scope. Notably, several vulnerable functional groups, such as the amino group and hydroxyl group, were well-tolerated in this procedure, exhibiting excellent functional group tolerance. The heterogeneous photocatalyst, Cu@C3N4, could be easily separated by centrifugation after the reaction and reused up to five times without any significant loss of reactivity.
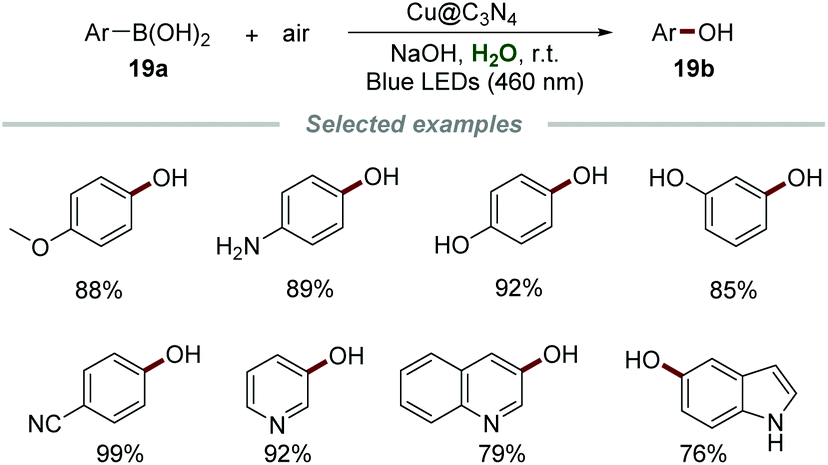 | ||
| Scheme 21 Hydroxylation of aryl boronic acids. | ||
Almost at the same time, Niu et al. successfully realized the Meerwein hydration reaction of alkenes 22b with aryldiazonium salts 22a by using mpg-C3N4 as a recyclable photocatalyst, providing various racemic alcohols 22c in good yields (Scheme 22).49 Various diazonium salts and alkenes with different functional groups all reacted smoothly under the optimized conditions. In all cases, the corresponding racemic alcohols were obtained in moderate to good yields. Among them, a lower yield was obtained when alkenes were attached with ortho-substituents, which was probably caused by steric hindrance. Moreover, this efficient and green protocol was applicable for the gram-scale synthesis of racemic alcohols in good yield driven by sunlight. More importantly, the mpg-C3N4/water system can be reused directly at least 5 times without significant loss of activity, and was recovered by simple extraction and separation after the reaction. The mechanism indicated that the reaction of aryldiazonium salts and the electron on the conduction band (CB) would generate aryl radical 22d, which added to the CC bond of alkenes 22b to give benzyl radical 22e. Then the benzyl radical 22e was oxidized by mpg-C3N4 to produce radical cation 22f which was subsequently attacked by H2O to afford the corresponding racemic alcohols 22c accompanied by deprotonation.
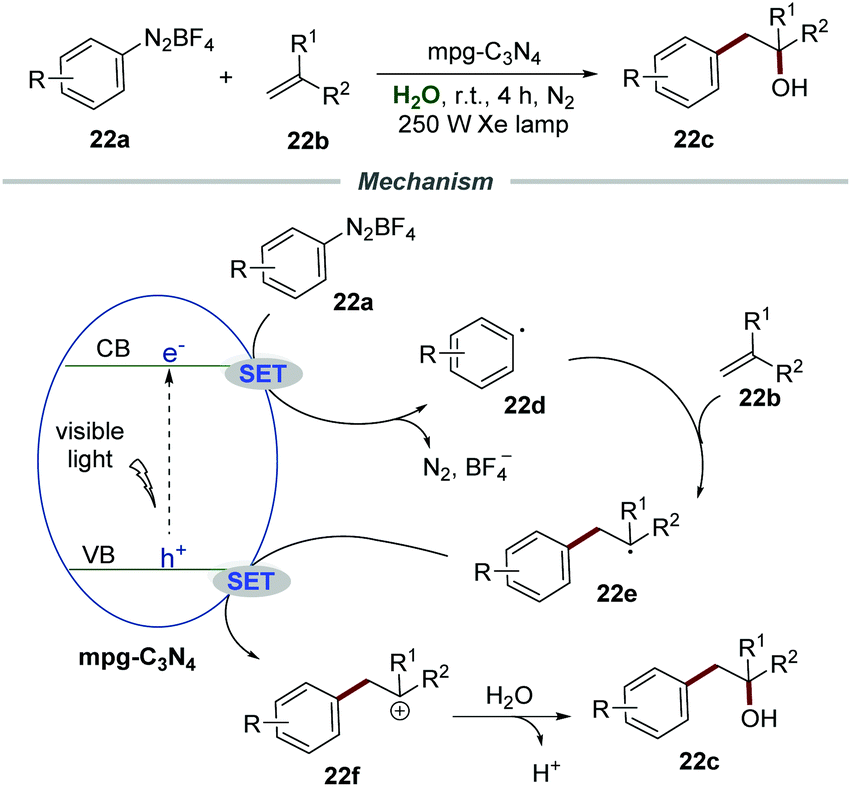 | ||
| Scheme 22 Photoinduced synthesis of racemic alcohols. | ||
Bismuth oxyhalides as photocatalysts
Bismuth oxyhalides have received increasing attention for photoredox reactions owing to their suitable band-structure, low toxicity, low cost, and well-matched band edges.50 Selective oxidative coupling of amines to imines has attracted widespread interest of chemists due to the significance of amines in the synthesis of fine chemicals and pharmaceuticals.51 Recently, Ye and coworkers developed an environmentally friendly and sustainable protocol for the selective oxidative coupling of amines 23a to imines 23b in water by using bismuth-rich Bi24O31Br10 as a photocatalyst (Scheme 23).52 This reaction was conducted at ambient temperature and pressure with wide substrate structural generality, providing an efficient and highly selective access to imines 23b in excellent yields. The control experiments showed that the selectivity decreased when the reaction was performed with K2S2O8 (a scavenger for electrons) or benzoquinone (BQ, a scavenger for O2˙−), while the conversion decreased when the reaction was conducted in the presence of ammonium oxalate (AO, a scavenger for holes) or tertiary butanol (t-BuOH, a scavenger for ˙OH). These results indicated that the photogenerated electron (e−) and hole (h+) were the driving force for this transformation and the selectivity and conversion of the reaction were dominated by O2˙− and ˙OH, respectively. Moreover, ESR was used to evaluate the amount of O2˙− and ˙OH produced in the process. Under visible light irradiation, stronger ESR signals of O2˙− and ˙OH were observed in the Bi24O31Br10-catalyzed system than those in the BiOBr-catalyzed system, providing strong evidence for the higher catalytic activity of bismuth-rich Bi24O31Br10 than BiOBr. More carriers in Bi24O31Br10 than in BiOBr was the cause of its excellent photocatalytic performance, which was confirmed from the surface photovoltage spectra, photocurrent response, and electrochemical impedance spectra. Ammonia gas was detected by on-line mass spectrometry, while NH3 and NDH2 were detected when the reaction was carried out in D2O, implying that water participates in the reaction process. In addition, in situ infrared spectroscopy proved that benzaldehydes and benzoic acids are important intermediates in the reaction.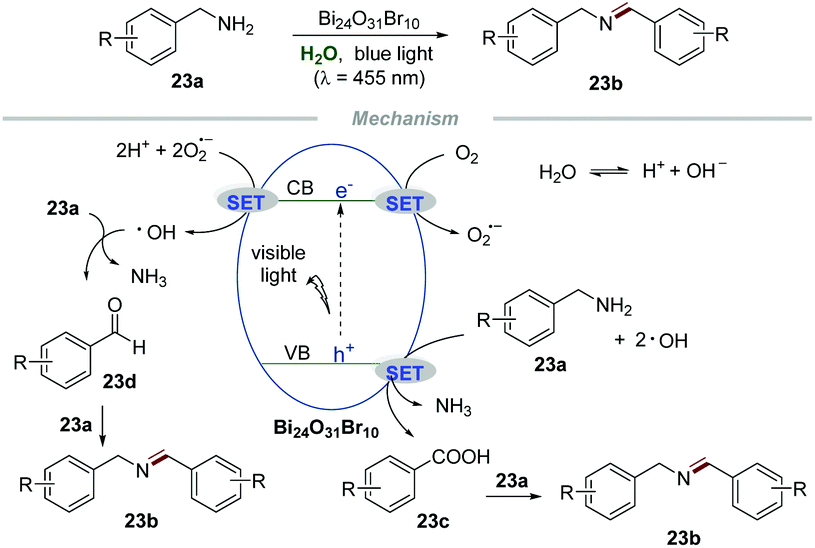 | ||
| Scheme 23 Selective oxidative coupling of amines to imines. | ||
Based on these results, a plausible mechanism was proposed. Initially, O2 would react with e− to form O2˙− which would further react with e− again in the presence of a proton, leading to the formation of ˙OH. Then, the oxidation of amines 23a would generate phenylaldehydes 23d or benzoic acids 23c. Then 23d or 23c would further react with free amines 23a to furnish the corresponding imines 23b.
Recently, Ye and co-authors achieved selective aerobic oxidation of sulfides 24a to sulfoxides 24b in water by using Bi4O5Br2 as a photocatalyst (Scheme 24).53 Sulfides bearing electron-donating and electron-withdrawing groups were all applicable to this transformation to produce the desired products in moderate to excellent yields. Among the sulfides tested, the sulfides bearing electron-donating groups are more likely to be oxidized than those bearing electron-withdrawing groups. Moreover, the catalyst Bi4O5Br2 could be recovered by simple centrifugation and reused at least three times without a significant deactivation. The control experiments carried out with various scavengers indicated that e−, h+, and O2˙− were all involved and played essential roles in this transformation.
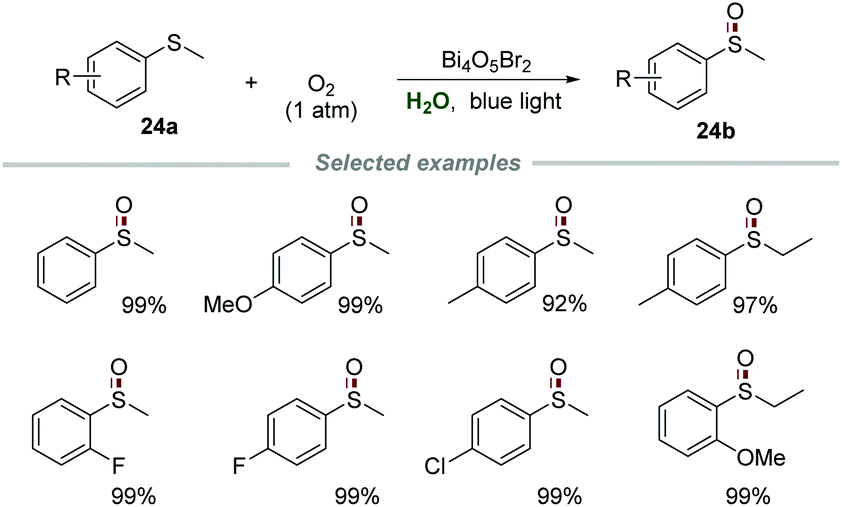 | ||
| Scheme 24 Selective aerobic oxidation of sulfides to sulfoxides. | ||
Others without a photocatalyst
Although the above-mentioned elegant catalytic systems were reported, external photocatalysts are still required. Oliveira's group successfully overcame this shortcoming in 2019, and the photo-arylation of pyridines 25a with aryldiazonium salts 25b could be realized under the irradiation with a blue LED without any additional photocatalyst (Scheme 25).54 Quinolines and quinoxalines were also suitable for this protocol. The control experiment confirmed that the employment of pyridinium salts to activate the moiety of pyridine was essential for this transformation. The proposed mechanism showed that the electron donor–acceptor (EDA) complex 25f, composed of aryldiazonium salts 25b and free pyridine 25e, could absorb blue light to generate the aryl radical 25g. Then, the addition of aryl radical 25g to the pyridinium salt 25d could yield a new radical intermediate 25h, which subsequently underwent single-electron oxidation by O2 to regenerate the aromatic ring, leading to the formation of the final products 25j.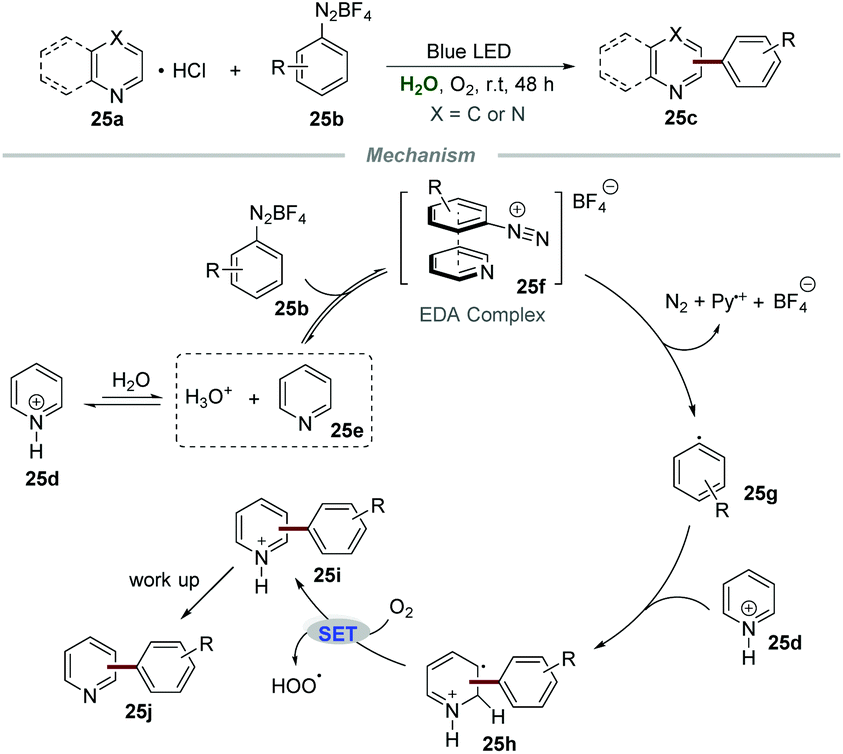 | ||
| Scheme 25 Arylation of N-heterocycles under photocatalyst free conditions. | ||
Very recently, Wang and co-workers reported the photoinduced synthesis of difluoroarylmethylated quinoxalin-2(1H)-ones 26cvia reactions of quinoxalin-2(1H)-ones 26a with potassium 2,2-difluoro-2-arylacetates 26b in water (Scheme 26).55 This efficient and eco-friendly synthetic protocol shows the great advantage of broad substituent group tolerance, and various functional groups including alkynyl, alkenyl, and carbonyl groups were all compatible with this transformation. Moreover, quinoxalines also worked smoothly in this protocol, leading to the generation of the desired products 26c in moderate to good yields. A plausible mechanism was proposed, which showed that the radical anion SO4˙− was formed initially from K2S2O8 under the irradiation of visible light. Then, the reaction of the radical anion SO4˙− and potassium 2,2-difluoro-2-phenylacetate 26d generated the radical intermediate PhCF2˙ (26e). Afterward, PhCF2˙ attacked the C3-position of quinoxalin-2(1H)-ones 26a to produce a new radical intermediate 26f, which was further oxidized by the radical anion SO4˙−, followed by deprotonation to afford the final product 26g.
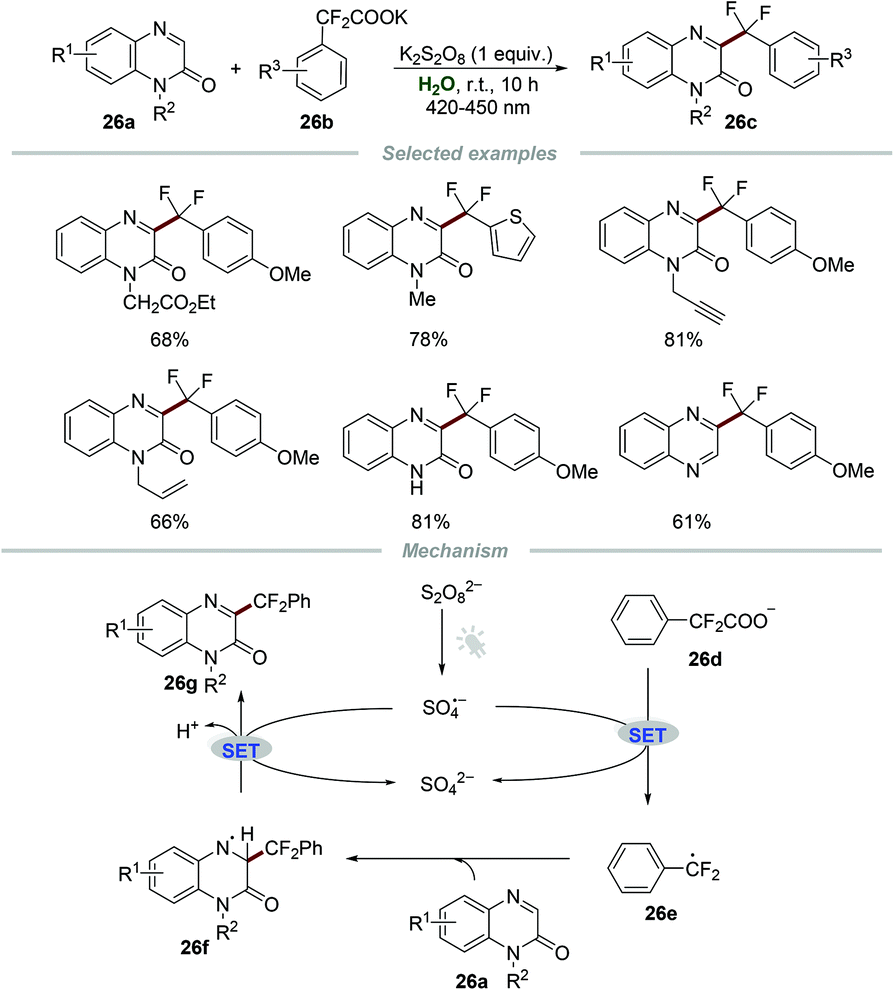 | ||
| Scheme 26 Photoinduced synthesis of difluoroarylmethylated quinoxalin-2(1H)-ones. | ||
Encouraged by the significance and importance of quinoxalin-2(1H)-ones in synthetic chemistry, materials, natural products, and pharmaceuticals, the green synthesis of different functionalized quinoxalin-2(1H)-ones is undoubtedly very meaningful and valuable.56 Recently, Xuan et al. realized the acylation of quinoxalin-2(1H)-ones 27a by employing α-oxo-carboxylic acids 27b as acylating reagents and phenyliodine(III)diacetate (PIDA) as the oxidant (Scheme 27).57 A wide range of substrates including various functionalized quinoxalin-2(1H)-ones 27a and α-oxo-carboxylic acids 27b were presented, and diverse 3-acyl quinoxalin-2(1H)-ones 27c were formed in moderate to good yields. More importantly, quinoxalin-2(1H)-ones with natural molecule fragments all performed well, demonstrating the utility of this green and simple transformation. The mechanism showed that α-oxo-carboxylic acids reacted with PIDA to generate the acyl radical, which then went through similar radical addition and single-electron oxidation to give the final product.
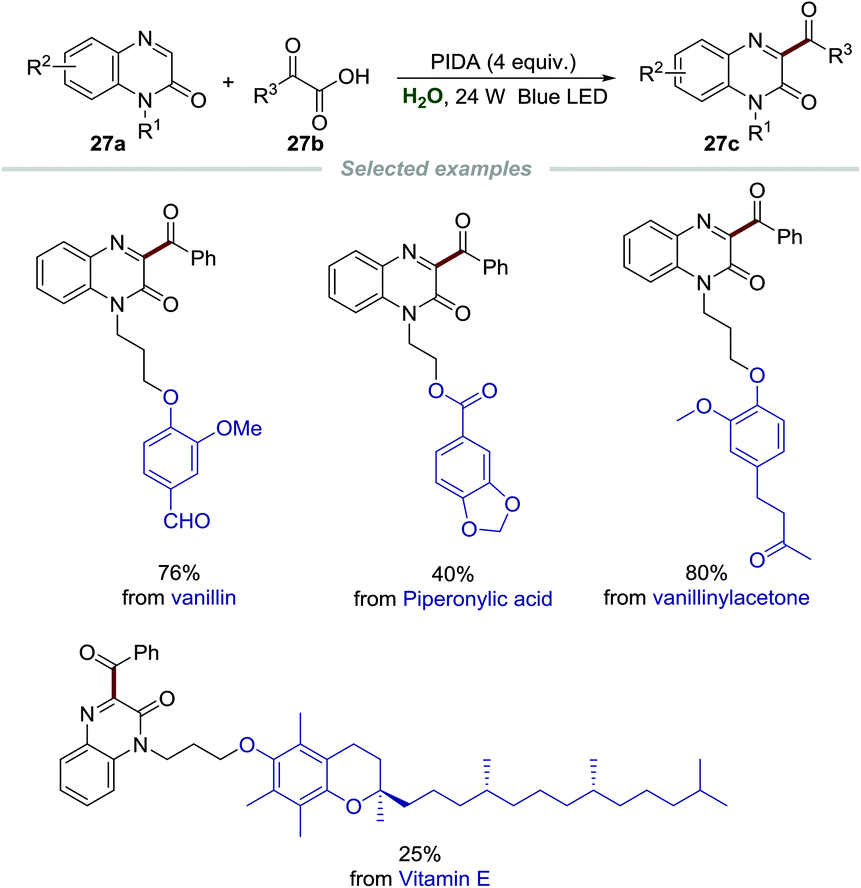 | ||
| Scheme 27 Acylation of quinoxalin-2(1H)-ones. | ||
Conclusions
Water is an abundant natural resource, which has been considered as an ideal “green solvent” with the advantages of non-toxicity and safety. Photocatalytic reactions can effectively use clean solar energy to achieve various organic conversions. The combination of photocatalysis and aqueous phase reactions is undoubtedly more attractive to develop sustainable synthetic routes, which is in line with the concept of “green chemistry” to effectively minimize the environmental impact of chemical synthesis. In this review, we have summarized the recent advances in visible-light-mediated organic transformations in water. These novel synthetic methodologies have provided a green and sustainable tool for the synthesis of various meaningful organic compounds. The feasible catalytic systems and the newly developed photocatalysts have made a great contribution to the success of the visible-light-mediated organic transformation in water. The newly developed semiconductors and carbon dots, which could be easily separated and reused through simple separation, are the most attractive photocatalysts, demonstrating promising applications to achieve novel visible-light-mediated organic conversions in water.Despite the great achievements made in recent years, photocatalytic reactions in water rather than in a mixture of water and organic solvents are still limited and the rules of these reactions have not been fully revealed. With the increasing interest in green chemistry, the pursuit of photocatalytic reactions in pure water for the construction of more structurally diverse organic compounds will never be terminated. We hope that this review will inspire more researchers interested in visible-light-mediated organic transformations in water and further stimulate the advancement of more novel relevant strategies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (21971224, 22071222), the 111 Project (D20003), the Key Research Projects of Universities in Henan Province (20A150006, 21A150053) and the Natural Science Foundation of Henan Province (202300410375).Notes and references
- T. Kitanosono, K. Masuda, P. Xu and S. Kobayashi, Chem. Rev., 2018, 118, 679 CrossRef CAS.
- U. M. Lindström, Chem. Rev., 2002, 102, 2751 CrossRef.
- D. C. Rideout and R. Breslow, J. Am. Chem. Soc., 1980, 102, 7816 CrossRef CAS.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- (a) F. Zhou and C.-J. Li, Nat. Commun., 2014, 5, 4254 CrossRef CAS; (b) W. A. Herrmann and C. W. Kohlpaintner, Angew. Chem., Int. Ed. Engl., 1993, 32, 1524 CrossRef; (c) A. Lubineau, J. Augé and Y. Queneau, Synthesis, 1994, 741 CrossRef CAS.
- T. Kitanosono and S. Kobayashi, Chem. – Eur. J., 2020, 26, 9408 CrossRef CAS.
- (a) M. M. D. Pramanik, H. Qian, W.-J. Xiao and J.-R. Chen, Org. Chem. Front., 2020, 7, 2531 RSC; (b) H. Jiang and A. Studer, CCS Chem., 2019, 1, 38 CAS; (c) L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034 CrossRef CAS; (d) T.-Y. Shang, L.-H. Lu, Z. Cao, Y. Liu, W.-M. He and B. Yu, Chem. Commun., 2019, 55, 5408 RSC; (e) H. Huang, K. Jia and Y. Chen, ACS Catal., 2016, 6, 4983 CrossRef CAS; (f) M. Melchionna and P. Fornasiero, ACS Catal., 2020, 10, 5493 CrossRef CAS; (g) C. Michelin and N. Hoffmann, ACS Catal., 2018, 8, 12046 CrossRef CAS; (h) D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355 CrossRef CAS; (i) J. A. Rossi-Ashton, A. K. Clarke, W. P. Unsworth and R. J. K. Taylor, ACS Catal., 2020, 10, 7250 CrossRef CAS; (j) G. S. Lee, J. Won, S. Choi, M.-H. Baik and S. H. Hong, Angew. Chem., 2020, 59, 16933 CrossRef CAS; (k) G. S. Lee and S. H. Hong, Chem. Sci., 2018, 9, 5810 RSC.
- (a) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Acc. Chem. Res., 2016, 49, 1911 CrossRef CAS; (b) K. Sun, Q.-Y. Lv, Y.-W. Lin, B. Yu and W.-M. He, Org. Chem. Front., 2021 10.1039/D0QO01058H; (c) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS; (d) Q. Liu and L.-Z. Wu, Natl. Sci. Rev., 2017, 4, 359 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS.
- T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562 RSC.
- (a) I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566 CrossRef CAS; (b) J. Wu, J. Li, H. Li and C. Zhu, Chin. J. Org. Chem., 2017, 37, 2203 CrossRef CAS; (c) M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261 CrossRef CAS; (d) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (e) D. P. Hari and B. König, Angew. Chem., Int. Ed., 2013, 52, 4734 CrossRef CAS.
- D. Xue, Z.-H. Jia, C.-J. Zhao, Y.-Y. Zhang, C. Wang and J. Xiao, Chem. – Eur. J., 2014, 20, 2960 CrossRef CAS.
- (a) Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586 CrossRef CAS; (b) F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190 RSC.
- (a) D.-Q. Dong, H. Yang, J.-L. Shi, W.-J. Si, Z.-L. Wang and X.-M. Xu, Org. Chem. Front., 2020, 7, 2538 RSC; (b) G. Tarantino and C. Hammond, Green Chem., 2020, 22, 5195 RSC; (c) M. Li, X.-S. Xue and J.-P. Cheng, Acc. Chem. Res., 2020, 53, 182 CrossRef CAS; (d) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef CAS; (e) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS; (f) C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765 CrossRef CAS.
- L. Zou, P. Li, B. Wang and L. Wang, Green Chem., 2019, 21, 3362 RSC.
- M.-H. Huang, C.-F. Zhu, C.-L. He, Y.-L. Zhu, W.-J. Hao, D.-C. Wang, S.-J. Tu and B. Jiang, Org. Chem. Front., 2018, 5, 1643 RSC.
- (a) C. Liu, H. Zeng, C. Zhu and H. Jiang, Chem. Commun., 2020, 56, 10442 RSC; (b) Y. Li, D. Wu, H.-G. Cheng and G. Yin, Angew. Chem., 2020, 59, 7990 CrossRef CAS; (c) Z.-L. Li, G.-C. Fang, Q.-S. Gu and X.-Y. Liu, Chem. Soc. Rev., 2020, 49, 32 RSC.
- M.-J. Bu, C. Cai, F. Gallou and B. H. Lipshutz, Green Chem., 2018, 20, 1233 RSC.
- C.-J. Wu, J.-J. Zhong, Q.-Y. Meng, T. Lei, X.-W. Gao, C.-H. Tung and L.-Z. Wu, Org. Lett., 2015, 17, 884 CrossRef CAS.
- S. Ghosh, J. Das and F. Saikh, Tetrahedron Lett., 2012, 53, 5883 CrossRef CAS.
- (a) S. Fukuzumi and K. Ohkubo, Org. Biomol. Chem., 2014, 12, 6059 RSC; (b) D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688 RSC; (c) D. Ravelli and M. Fagnoni, ChemCatChem, 2012, 4, 169 CrossRef CAS.
- D. Xia, T. Miao, P. Li and L. Wang, Chem. – Asian J., 2015, 10, 1919 CrossRef CAS.
- G. Kibriya, S. Mondal and A. Hajra, Org. Lett., 2018, 20, 7740 CrossRef CAS.
- G. You, K. Wang, X. Wang, G. Wang, J. Sun, G. Duan and C. Xia, Org. Lett., 2018, 20, 4005 CrossRef CAS.
- (a) E.-Q. Li and Y. Huang, Chem. Commun., 2020, 56, 680 RSC; (b) L. Wu, B. Yu and E.-Q. Li, Adv. Synth. Catal., 2020, 362, 4010 CrossRef CAS; (c) Y. Gao, G. Tang and Y. Zhao, Chin. J. Org. Chem., 2018, 38, 62 CrossRef CAS; (d) K. Luo, W.-C. Yang and L. Wu, Asian J. Org. Chem., 2017, 6, 350 CrossRef CAS.
- H. Wang, Y. Li, Z. Tang, S. Wang, H. Zhang, H. Cong and A. Lei, ACS Catal., 2018, 8, 10599 CrossRef CAS.
- X.-C. Liu, X.-L. Chen, Y. Liu, K. Sun, Y.-Y. Peng, L.-B. Qu and B. Yu, ChemSusChem, 2020, 13, 298 CrossRef CAS.
- X.-Y. Yuan, F.-L. Zeng, H.-L. Zhu, Y. Liu, Q.-Y. Lv, X.-L. Chen, L. Peng and B. Yu, Org. Chem. Front., 2020, 7, 1884 RSC.
- P. Natarajan, N. Kumar, R. Chaudhary and P. Venugopalan, Tetrahedron Lett., 2020, 61, 151823 CrossRef CAS.
- M.-j. Bu, G.-p. Lu, J. Jiang and C. Cai, Catal. Sci. Technol., 2018, 8, 3728 RSC.
- J. Liu, H. Yao, X. Li, H. Wu, A. Lin, H. Yao, J. Xu and S. Xu, Org. Chem. Front., 2020, 7, 1314 RSC.
- X. Li, Y. Li, Y. Huang, T. Zhang, Y. Liu, B. Yang, C. He, X. Zhou and J. Zhang, Green Chem., 2017, 19, 2925 RSC.
- T. Zhang, W. Liang, Y. Huang, X. Li, Y. Liu, B. Yang, C. He, X. Zhou and J. Zhang, Chem. Commun., 2017, 53, 12536 RSC.
- Y. Huang, Z. Xin, W. Yao, Q. Hu, Z. Li, L. Xiao, B. Yang and J. Zhang, Chem. Commun., 2018, 54, 13587 RSC.
- (a) S. Gisbertz and B. Pieber, ChemPhotoChem, 2020, 4, 456 CrossRef CAS; (b) L. Zhang, J. Ran, S.-Z. Qiao and M. Jaroniec, Chem. Soc. Rev., 2019, 48, 5184 RSC.
- (a) Y. Zheng, L. Lin, B. Wang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868 CrossRef CAS; (b) I. Ghosh, J. Khamrai, A. Savateev, N. Shlapakov, M. Antonietti and B. König, Science, 2019, 365, 360 CrossRef CAS; (c) C. Han, X. Zhu, J. S. Martin, Y. Lin, S. Spears and Y. Yan, ChemSusChem, 2020, 13, 4005 CrossRef CAS.
- (a) X.-B. Li, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2019, 58, 10804 CrossRef CAS; (b) X.-B. Li, C.-H. Tung and L.-Z. Wu, Nat. Rev. Chem., 2018, 2, 160 CrossRef CAS.
- (a) S. Yurdakal, G. Palmisano, V. Loddo, O. Alagöz, V. Augugliaro and L. Palmisano, Green Chem., 2009, 11, 510 RSC; (b) V. Augugliaro, T. Caronna, V. Loddo, G. Marcì, G. Palmisano, L. Palmisano and S. Yurdakal, Chem. – Eur. J., 2008, 14, 4640 CrossRef CAS; (c) S. Yurdakal, G. Palmisano, V. Loddo, V. Augugliaro and L. Palmisano, J. Am. Chem. Soc., 2008, 130, 1568 CrossRef CAS.
- T. Ishizuka, S. Ohkawa, H. Ochiai, M. Hashimoto, K. Ohkubo, H. Kotani, M. Sadakane, S. Fukuzumi and T. Kojima, Green Chem., 2018, 20, 1975 RSC.
- B. Long, Z. Ding and X. Wang, ChemSusChem, 2013, 6, 2074 CrossRef CAS.
- L.-M. Zhao, Q.-Y. Meng, X.-B. Fan, C. Ye, X.-B. Li, B. Chen, V. Ramamurthy, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2017, 56, 3020 CrossRef CAS.
- J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044 RSC.
- S. Das, S. Ray, A. B. Ghosh, P. K. Samanta, S. Samanta, B. Adhikary and P. Biswas, Appl. Organomet. Chem., 2018, 32, e4199 CrossRef.
- (a) R. Liu and L. E. Orgel, Nature, 1997, 389, 52 CrossRef CAS; (b) P. Wang, X. Li, J. Zhu, J. Chen, Y. Yuan, X. Wu and S. J. Danishefsky, J. Am. Chem. Soc., 2011, 133, 1597 CrossRef CAS.
- T. Lei, S.-M. Wei, K. Feng, B. Chen, C.-H. Tung and L.-Z. Wu, ChemSusChem, 2020, 13, 1715 CrossRef CAS.
- (a) Y.-F. Si, X.-L. Chen, X.-Y. Fu, K. Sun, X. Song, L.-B. Qu and B. Yu, ACS Sustainable Chem. Eng., 2020, 8, 10740 CAS; (b) B. Ni, B. Zhang, J. Han, B. Peng, Y. Shan and T. Niu, Org. Lett., 2020, 22, 670 CrossRef CAS; (c) Y. Cai, Y. Tang, L. Fan, Q. Lefebvre, H. Hou and M. Rueping, ACS Catal., 2018, 8, 9471 CrossRef CAS.
- L. Hao, G. Ding, D. A. Deming and Q. Zhang, Eur. J. Org. Chem., 2019, 7307 CrossRef CAS.
- M. H. Muhammad, X.-L. Chen, Y. Liu, T. Shi, Y. Peng, L. Qu and B. Yu, ACS Sustainable Chem. Eng., 2020, 8, 2682 CrossRef CAS.
- J. Wang, L. Xue, M. Hong, B. Ni and T. Niu, Green Chem., 2020, 22, 411 RSC.
- L. Ye, Y. Deng, L. Wang, H. Xie and F. Su, ChemSusChem, 2019, 12, 3671 CrossRef CAS.
- X. Lang, H. Ji, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2011, 50, 3934 CrossRef CAS.
- W. Zhao, C. Yang, X. Zhang, Y. Deng, C. Han, Z. Ma, L. Wang and L. Ye, ChemSusChem, 2020, 13, 116 CrossRef CAS.
- W. Zhao, C. Yang, J. Huang, X. Jin, Y. Deng, L. Wang, F. Su, H. Xie, P. K. Wong and L. Ye, Green Chem., 2020, 22, 4884 RSC.
- A. d. A. Bartolomeu, R. C. Silva, T. J. Brocksom, T. Noël and K. T. de Oliveira, J. Org. Chem., 2019, 84, 10459 CrossRef CAS.
- Y. Gao, L. Zhao, T. Xiang, P. Li and L. Wang, RSC Adv., 2020, 10, 10559 RSC.
- (a) Y.-F. Si, K. Sun, X.-L. Chen, X.-Y. Fu, Y. Liu, F.-L. Zeng, T. Shi, L.-B. Qu and B. Yu, Org. Lett., 2020, 22, 6960 CrossRef CAS; (b) L.-Y. Xie, Y.-S. Liu, H.-R. Ding, S.-F. Gong, J.-X. Tan, J.-Y. He, Z. Cao and W.-M. He, Chin. J. Catal., 2020, 41, 1168 CrossRef CAS; (c) L.-Y. Xie, J.-L. Hu, Y.-X. Song, G.-K. Jia, Y.-W. Lin, J.-Y. He, Z. Cao and W.-M. He, ACS Sustainable Chem. Eng., 2019, 7, 19993 CrossRef CAS; (d) W. Wei, L. Wang, H. Yue, P. Bao, W. Liu, C. Hu, D. Yang and H. Wang, ACS Sustainable Chem. Eng., 2018, 6, 17252 CrossRef CAS; (e) W. Wei, L. Wang, P. Bao, Y. Shao, H. Yue, D. Yang, X. Yang, X. Zhao and H. Wang, Org. Lett., 2018, 20, 7125 CrossRef CAS; (f) L. Wang, P. Bao, W. Liu, S. Liu, C. Hu, H. Yue, D. Yang and W. Wei, Chin. J. Org. Chem., 2018, 38, 3189 CrossRef CAS; (g) P. Mao, J. Zhu, J. Yuan, L. Yang, Y. Xiao and C. Zhang, Chin. J. Org. Chem., 2019, 39, 1529 CrossRef CAS.
- J. Lu, X.-K. He, X. Cheng, A.-J. Zhang, G.-Y. Xu and J. Xuan, Adv. Synth. Catal., 2020, 362, 2178 CrossRef.
Footnote |
| † Dedicated to the 100th anniversary of Chemistry at Nankai University. |
| This journal is © The Royal Society of Chemistry 2021 |