
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enzymatic production of β-glucose 1,6-bisphosphate through manipulation of catalytic magnesium coordination†
Henry P.
Wood
 a,
Nicola J.
Baxter
ab,
F. Aaron
Cruz-Navarrete
a,
Clare R.
Trevitt
a,
Andrea M.
Hounslow
a and
Jonathan P.
Waltho
*ab
a,
Nicola J.
Baxter
ab,
F. Aaron
Cruz-Navarrete
a,
Clare R.
Trevitt
a,
Andrea M.
Hounslow
a and
Jonathan P.
Waltho
*ab
aKrebs Institute for Biomolecular Research, Department of Molecular Biology and Biotechnology, The University of Sheffield, Sheffield, S10 2TN, UK. E-mail: j.waltho@sheffield.ac.uk; Tel: +44 114 22717
bManchester Institute of Biotechnology and School of Chemistry, The University of Manchester, Manchester, M1 7DN, UK
First published on 15th January 2021
Abstract
Manipulation of enzyme behaviour represents a sustainable technology that can be harnessed to enhance the production of valuable metabolites and chemical precursors. β-Glucose 1,6-bisphosphate (βG16BP) is a native reaction intermediate in the catalytic cycle of β-phosphoglucomutase (βPGM) that has been proposed as a treatment for human congenital disorder of glycosylation involving phosphomannomutase 2. Strategies to date for the synthesis of βG16BP suffer from low yields or use chemicals and procedures with significant environmental impacts. Herein, we report the efficient enzymatic synthesis of anomer-specific βG16BP using the D170N variant of βPGM (βPGMD170N), where the aspartate to asparagine substitution at residue 170 perturbs the coordination of a catalytic magnesium ion. Through combined use of NMR spectroscopy and kinetic assays, it is shown that the weakened affinity and reactivity of βPGMD170N towards βG16BP contributes to the pronounced retardation of the second step in the two-step catalytic cycle, which causes a marked accumulation of βG16BP, especially at elevated MgCl2 concentrations. Purification, employing a simple environmentally considerate precipitation procedure requiring only a standard biochemical toolset, results in a βG16BP product with high purity and yield. Overall, this synthesis strategy illustrates how manipulation of the catalytic magnesium coordination of an enzyme can be utilised to generate large quantities of a valuable metabolite.
Enzyme engineering represents an emerging technology with the potential to deliver solutions to many sustainable development problems.1,2 Biofuel production, plastic degradation and the clean generation of industrial reagents and precursors are three examples of areas where enzymes already make a significant contribution.3–6 Research that aims to foster a deeper understanding of enzyme catalysis is therefore of great interest. Phosphoryl transfer enzymes are at the forefront of research models for investigating the origins of enzyme catalysis because they exhibit some of the largest enzymatic rate enhancements known.7,8 In addition, phosphate esters are often covalently incorporated into pharmaceutical products to improve bioavailability.9,10 β-Phosphoglucomutase (βPGM; EC 5.4.2.6) has emerged as an archetypal enzyme in the study of phosphoryl transfer, and substantial progress has been made in understanding its mechanism of catalysis.11–16 This magnesium-dependent enzyme from Lactococcus lactis (subspecies lactis IL1403) catalyses the isomerisation between β-glucose 1-phosphate (βG1P) and glucose 6-phosphate (G6P) via a β-glucose 1,6-phosphate (βG16BP) intermediate, which is released to solution before rebinding in the alternate orientation (Fig. 1).11,17 The βG1P substrate of βPGM is commercially unavailable, but appropriate quantities for research have been produced enzymatically from maltose using a simple method involving maltose phosphorylase.18 To initiate the catalytic cycle, βPGM requires priming with a phosphorylating agent to generate the active phospho-enzyme (βPGMP, phosphorylated on residue D8) and βG16BP can perform this role in vivo. Since βG16BP is also commercially unavailable, alternative phosphorylating agents such as acetyl phosphate (AcP), fructose 1,6-bisphosphate (F16BP) and α-glucose 1,6-bisphosphate (αG16BP) have been used to generate βPGMPin vitro, but these compounds are less effective and produce complicated kinetic behaviour.12,19
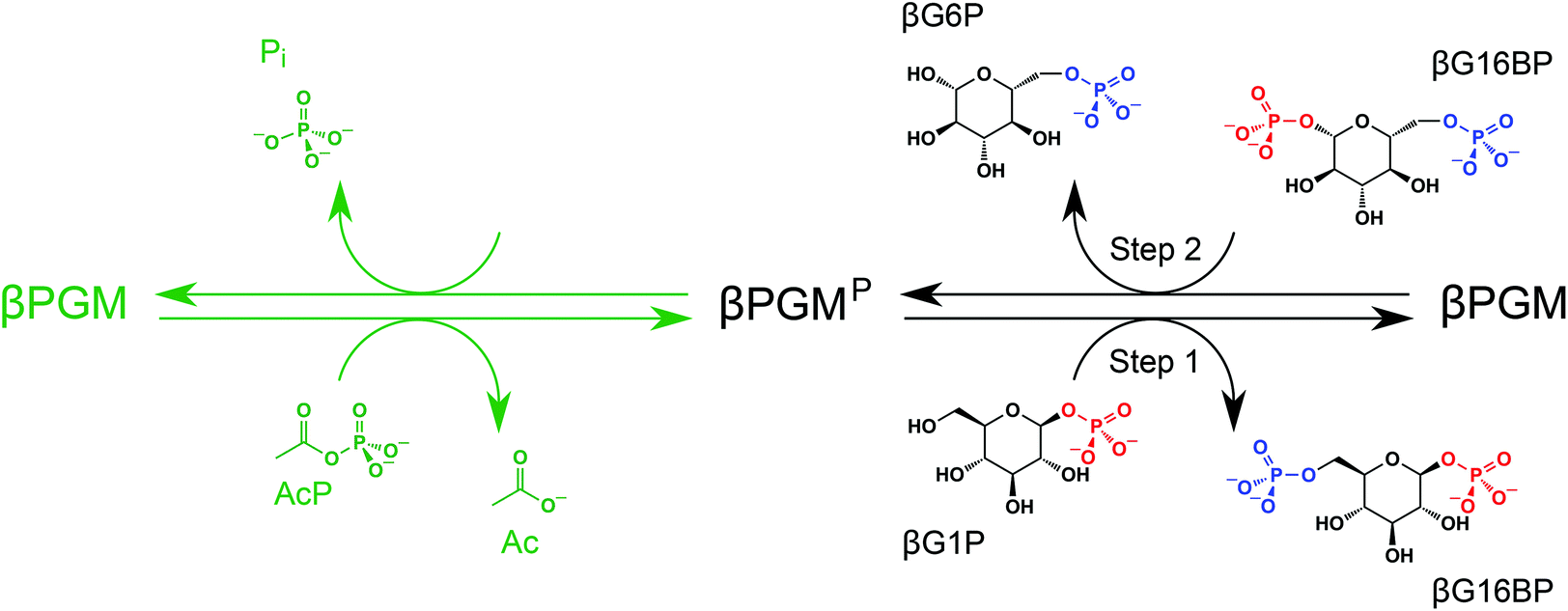 | ||
| Fig. 1 In vitro phosphorylation and catalytic cycle of βPGM. AcP phosphorylates βPGM generating βPGMP (phosphorylated on residue D8) in a Mg2+-independent reaction (green ink). In the Mg2+-dependent catalytic cycle (black ink), Step 1 involves phosphoryl transfer from βPGMP to the βG1P substrate forming the βG16BP intermediate, whereas Step 2 comprises phosphoryl transfer from βG16BP (bound in the alternate orientation) to βPGM forming the G6P product and regeneration of βPGMP. In the phosphorylated glucosaccharide structures, 1-phosphate groups are coloured red and 6-phosphate groups are coloured blue. The black arrows denote the dominant direction of the corresponding reversible reactions. In the absence of the βG1P substrate, βPGMP has a half-life of 30 s and hydrolyses readily to βPGM liberating inorganic phosphate (Pi).12 | ||
βG16BP has also been identified as a potential pharmacological chaperone for the management of a human congenital disorder of glycosylation involving phosphomannomutase 2.20 Acting as a weakly binding competitive inhibitor, βG16BP is able to rescue the compromised activity of pathological variants of phosphomannomutase 2 by stabilising the protein fold. Therefore, further investigations of phosphomannomutase 2 and of βPGM are reliant on the availability of substantial quantities of βG16BP. Three strategies have been reported previously for the synthesis of βG16BP; however, each of these methods either delivers low yields or uses chemicals and procedures with significant environmental impacts. Firstly, the chemical synthesis of βG16BP from α-glucose involves an eight-step protocol, requiring considerable time and technical expertise, together with the use of harmful and environmentally hazardous reagents.12 Low yields are obtained, since the β-anomer must be selected carefully on the basis of solubility from a racemic mixture of glucosaccharide products. Secondly, an enzymatic production method utilises a non-native reaction of phosphofructokinase to generate βG16BP from βG1P using adenosine triphosphate as the phosphoryl donor.17,20 Purification of the product, though, cannot be achieved simply using precipitation procedures, since contaminating adenosine diphosphate co-precipitates with βG16BP,21 and therefore ion-exchange HPLC purification is required. The use of HPLC columns is inherently damaging to the environment owing to the use of triethylammonium bicarbonate as a volatile buffer mobile phase, which during its production results in the release of large quantities of carbon dioxide.22 Thirdly, an extraction method involves the removal of βG16BP from a variant of βPGM that co-purifies with a stoichiometric quantity of the molecule.18 This method suffers from low yields, since it relies on very high recombinant βPGM production levels, and requires a week-long protein growth and purification procedure for each new batch of βG16BP. The limited availability of βG16BP therefore represents a significant barrier to the structural, kinetic and therapeutic investigations of phosphomutase enzymes. Herein, we describe a room-temperature, enzymatic method using the D170N variant of βPGM (βPGMD170N) for the production of 100% anomer-specific βG16BP, which requires only micromolar quantities of enzyme and a simple environmentally considerate purification procedure that can be performed easily by a non-chemist over the course of two days. Through combined use of NMR spectroscopy and kinetic assays, it is shown that the weakened affinity and reactivity of βPGMD170N towards βG16BP contributes to the pronounced retardation of the second step in the two-step catalytic cycle, which causes a marked accumulation of βG16BP, especially at elevated MgCl2 concentrations. More generally, this enzymatic synthesis strategy illustrates how manipulation of catalytic magnesium coordination can be utilised to generate large quantities of a valuable metabolite.
βPGM has two phosphoryl transfer steps in its catalytic cycle: Step 1 comprises phosphoryl transfer from βPGMP to the βG1P substrate forming the βG16BP reaction intermediate, whereas Step 2 involves phosphoryl transfer from βG16BP to βPGM forming the G6P product and regeneration of βPGMP (Fig. 1). When wild-type βPGM (βPGMWT) is incubated in the presence of Mg2+ ions, with 20 mM AcP as the phosphorylating agent and 10 mM βG1P as a substrate, βG16BP generated in the catalytic cycle does not accumulate to detectable levels when monitored using 31P NMR experiments.18 Instead, βG16BP rebinds the enzyme with micromolar affinity in the alternate orientation, for the Step 2 reaction. Thus, the tight binding and high reactivity of βG16BP maintain a low steady state concentration, which precludes the harvesting of this species in useful quantities. The crystal structures of substrate-free βPGMWT (PDB: 6YDL23) and of the βPGMWTP analogue complex (βPGMWT:BeF3 complex, PDB: 2WFA15) indicate that the catalytic magnesium ion (Mgcat) is coordinated through three enzyme atoms in the former and four phospho-enzyme atoms in the latter (Fig. 2). Therefore, the differential coordination of Mgcat provides an appropriate target with which to manipulate βPGM to shift the balance in the rate constants of Step 1 and Step 2 so that βG16BP will accumulate to a greater extent. Two potential strategies emerged where Step 2 could be retarded with respect to Step 1, which involved either performing the reactions of the catalytic cycle under Mg2+-free conditions or perturbing Mgcat coordination through point mutation to alter its binding properties. In either scenario, it was hypothesised that βPGM with a compromised Mgcat site could be phosphorylated efficiently by reactive phosphorylating agents such as AcP, thereby generating βPGMP and subsequent reaction with βG1P to produce βG16BP in Step 1 (Fig. 1). In contrast, phosphorylation of βPGM by βG16BP in Step 2 is less likely under these circumstances, which would lead to an accumulation of the reaction intermediate that could be harvested.
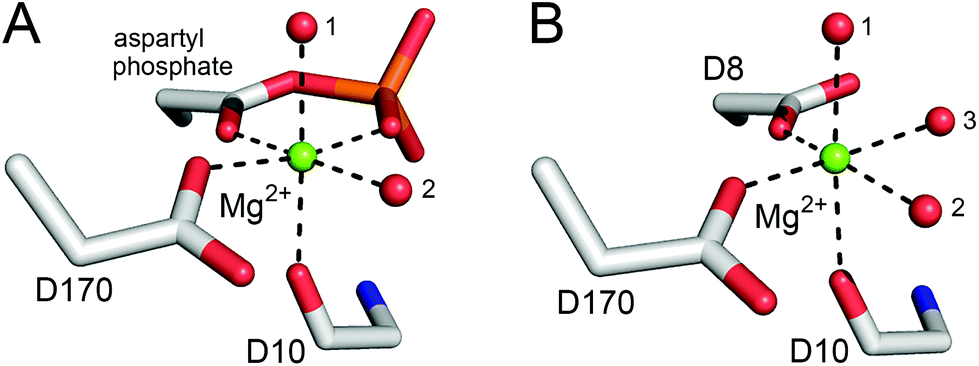 | ||
| Fig. 2 Comparison of octahedral Mgcat coordination in βPGMWTP (Step 1) and βPGMWT (Step 2). (A) A model of βPGMWTP derived from the crystal structure of the βPGMWT:BeF3 complex (PDB: 2WFA15) showing Mgcat coordination. The ligands comprise a carboxylate oxygen atom of residue D170, the carbonyl oxygen atom of residue D10 and two water molecules (indicated by numbers), together with the carboxylate oxygen atom and a phosphate oxygen atom of the D8 aspartyl phosphate moiety, creating bidentate coordination of Mgcat in a six-membered ring of atoms. (B) The crystal structure of substrate-free βPGMWT (PDB: 6YDL23) showing Mgcat coordination. The ligands involve a carboxylate oxygen atom of residue D8, a carboxylate oxygen atom of residue D170, the carbonyl oxygen atom of residue D10 and three water molecules (indicated by numbers). Mgcat is depicted as a green sphere, water molecules are illustrated as red spheres and metal ion coordination is shown as black dashes. | ||
To explore whether AcP is able to phosphorylate Mgcat-free βPGMWT, 31P NMR experiments were acquired to measure the change in AcP concentration over time in the presence and absence of 300 μM βPGMWT. The addition of βPGMWT resulted in a 25% increase in the rate of AcP hydrolysis (Fig. 3A), implying that βPGMWTP is generated and hydrolysed in the absence of Mgcat. Consequently, the Step 1 reaction between Mgcat-free βPGMWT and 10 mM βG1P in the presence of 50 mM AcP, together with the Step 2 production of G6P, was monitored using 31P NMR time-course experiments. However, there was no detectable accumulation of βG16BP (Fig. 4A–C) and the appearance of G6P product proceeded with a rate constant of 6.7 × 10−3 s−1, which is 4 orders of magnitude smaller than the rate constant observed in the presence of 5 mM MgCl2.18 Hence, the observed enzymatic activity may simply arise due to the presence of very low levels of residual Mg2+ ions associated with the reagents. Taken together, these results indicate that both Mgcat-bound βPGMWTP and Mgcat-free βPGMWTP can be generated by AcP, but both the Step 1 and Step 2 phosphoryl transfer reactions are seriously impaired by the absence of Mgcat.
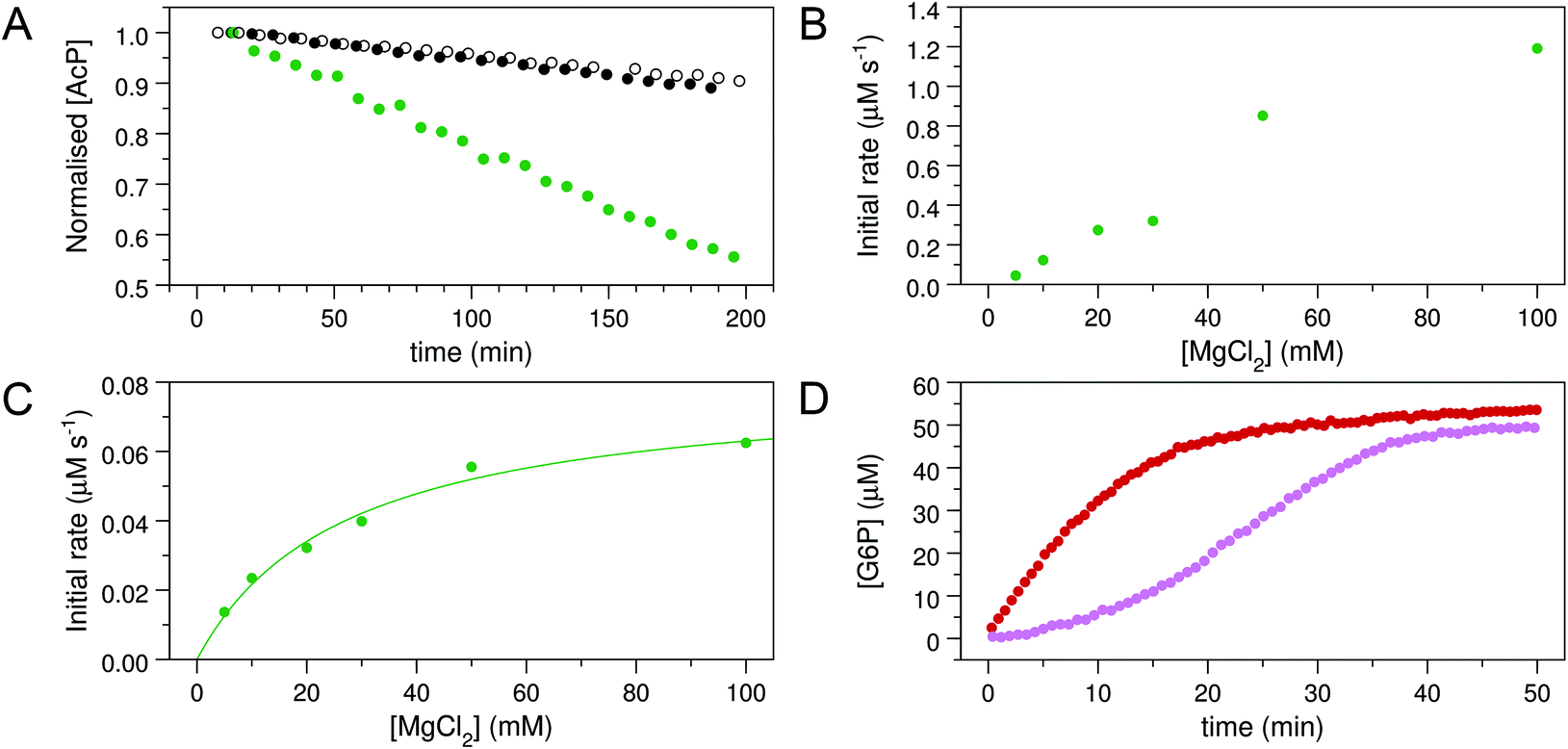 | ||
| Fig. 3 Kinetic experiments involving βPGMWT and βPGMD170N. (A) Effect of βPGMWT and βPGMD170N on the hydrolysis of AcP monitored using 31P NMR time-course experiments. AcP hydrolysis profiles were derived from normalised peak intensities obtained from reactions containing 50 mM AcP in 200 mM K+ HEPES buffer (pH 7.2) without MgCl2 (white circles) and separately in the presence of 300 μM βPGMWT (black circles) or presence of 300 μM βPGMD170N (green circles). (B and C) Activity of βPGMD170N with increasing MgCl2 concentration monitored using 31P NMR time-course experiments. Samples contained 5 μM βPGMD170N and 10 mM βG1P in 200 mM K+ HEPES buffer (pH 7.2), 10% 2H2O (v/v) and 1 mM TSP with increasing concentrations of MgCl2 (5, 10, 20, 30, 50, 100 mM). The reactions were initiated by and timed from the addition of 20 mM AcP. Initial rate measurements for (B) the Step 1 production of βG16BP and (C) the Step 2 production of G6P were obtained from linear least-squares fitting of normalised integral values of the 31P resonances of βG16BP and G6P present in the spectra. Subsequent fitting of the Step 2 initial rate values to eqn (1) using an in-house Python non-linear least squares fitting program yielded an apparent Km (Mg2+) = 27 ± 4 mM. (D) Kinetic profiles for the conversion of βG1P to G6P by βPGMWT monitored using a G6PDH coupled assay. Reactions were conducted in 200 mM K+ HEPES buffer (pH 7.2) containing 5 mM MgCl2, 1 mM NAD+, 5 U mL−1 G6PDH, 50 μM βG1P and 5 nM βPGMWT with either 1 μM of the final βG16BP product (red circles) or 8 mM AcP (pink circles) as the phosphorylating agent. For clarity, only half of the acquired data points have been included. | ||
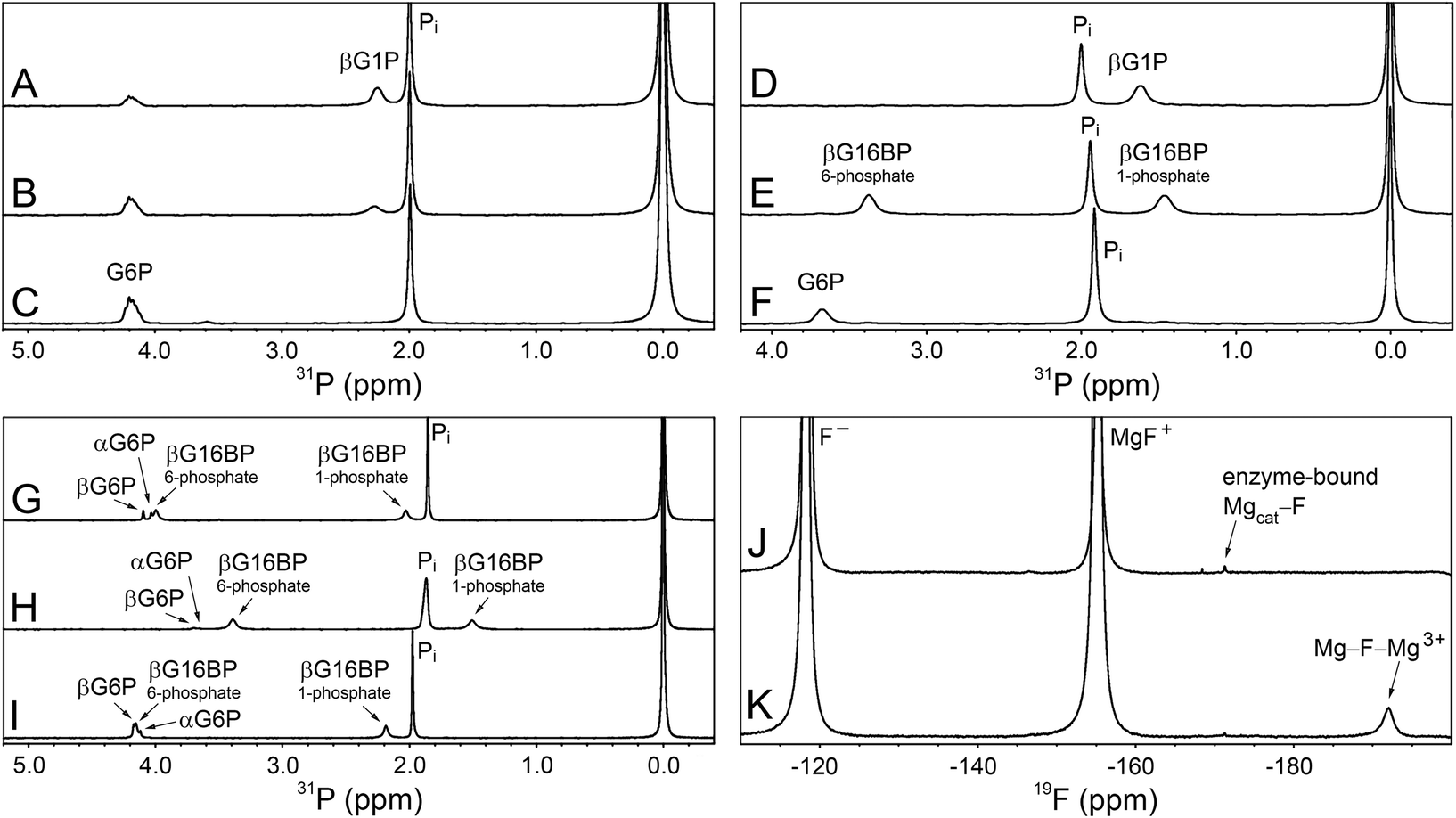 | ||
| Fig. 4 31P and 19F NMR experiments involving βPGMWT and βPGMD170N. (A–C) The βPGMWT-catalysed conversion of βG1P to G6P via βG16BP in the absence of Mg2+ ions. (A) Reaction containing 300 μM βPGMWT and 10 mM βG1P in 200 mM K+ HEPES buffer (pH 7.2) without MgCl2, acquired 23 min after the addition of 50 mM AcP. (B) Reaction after 42 min showing the presence of βG1P and G6P. (C) Reaction after 72 min showing complete conversion of βG1P to G6P. (D–F) The βPGMD170N-catalysed conversion of βG1P to G6P, together with the accumulation of βG16BP. (D) Reaction containing 20 μM βPGMD170N and 10 mM βG1P in 200 mM K+ HEPES buffer (pH 7.2) with 100 mM MgCl2, prior to the addition of 20 mM AcP. (E) Reaction after 87 min showing the generation of βG16BP. (F) Reaction after 1179 min showing complete conversion of βG1P to G6P. (G–I) The βPGMD170N-catalysed conversion of βG1P to G6P together with the accumulation of βG16BP under variable ion concentrations. (G) Reaction containing 400 μM βPGMD170N and 10 mM βG1P in 200 mM K+ HEPES buffer (pH 7.2) with 5 mM MgCl2, 35 min after the addition of 20 mM AcP. (H) Reaction containing 400 μM βPGMD170N and 10 mM βG1P in 200 mM K+ HEPES buffer (pH 7.2) with 100 mM MgCl2, 19 min after the addition of 20 mM AcP. (I) Reaction containing 400 μM βPGMD170N and 10 mM βG1P in 200 mM K+ HEPES buffer (pH 7.2) with 5 mM magnesium acetate and 200 mM NaCl, 36 min after the addition of 20 mM AcP. The peak at 1.9–2.0 ppm in panels A–I corresponds to inorganic phosphate (Pi), which is present in the stocks of both βG1P and AcP. 31P chemical shifts were referenced to external 1 M H3PO4 = 0.0 ppm, which was sealed inside a glass capillary and inserted into the sample NMR tubes. (J–K) 19F NMR experiments involving βPGMD170N acquired in 200 mM K+ HEPES buffer (pH 7.2) at 5 °C. (J) A sample containing 0.5 mM βPGMD170N with 5 mM MgCl2, 10 mM NaF and 0.2 mM deferoxamine shows an enzyme-bound 19F resonance (−171 ppm) that corresponds to a Mgcat–F moiety. (K) A sample containing 0.5 mM βPGMD170N with 100 mM MgCl2, 10 mM NaF and 0.2 mM deferoxamine reveals that an increase in Cl− ion concentration results in a decrease in the peak intensity of the Mgcat–F moiety. Three free 19F species are present in solution: Free F− (−118 ppm), free MgF+ (−155 ppm) and a free Mg–F–Mg3+ species (−192 ppm), which have been assigned based on the partitioning behaviour between discrete species as the MgCl2 concentration is increased. | ||
Given the low rate of βG16BP production in the absence of Mg2+ ions in the reaction buffer, a more subtle modification of the enzyme Mgcat site was engineered. In βPGMWT, Mgcat is coordinated octahedrally by a carboxylate oxygen atom of residue D8, a carboxylate oxygen atom of residue D170 and the carbonyl oxygen atom of residue D10, together with three water molecules. In βPGMWTP, one of the water molecules (water 3) is displaced by a phosphate oxygen atom of the D8 aspartyl phosphate moiety, creating bidentate coordination of Mgcat in a six-membered ring of atoms (Fig. 2). Point mutations involving residue D8 have been reported to result in the complete loss of measurable catalytic activity.19 Therefore, perturbation of Mgcat was achieved through the generation of the D170N variant (βPGMD170N), where the carboxamide group of residue N170 retains an oxygen atom with which to coordinate Mgcat, but the sidechain is not charged. Accordingly, the reaction of βPGMD170N with 10 mM βG1P and 20 mM AcP in the presence of 100 mM MgCl2 was monitored using 31P NMR time-course experiments and, in contrast to βPGMWT, the βG16BP intermediate was observed to accumulate to a level comparable with the initial βG1P concentration (Fig. 4D and E). The G6P product was only generated to a measurable extent once the AcP concentration had reduced significantly (Fig. 4F). Hence, perturbation of Mgcat in βPGMD170N (in the presence of excess AcP) results in a substantial retardation of phosphorylation of βPGMD170N by βG16BP (Step 2) with respect to phosphorylation of βG1P by βPGMD170NP (Step 1), therefore allowing βG16BP to accumulate.
To investigate the source of the retardation of Step 2, glucose 6-phosphate dehydrogenase (G6PDH; EC 1.1.1.49) coupled assay experiments were conducted to assess the ability of βPGMD170N to bind and convert the substrates of Step 2 (βG16BP) and Step 1 (βG1P). Initial rate measurements were recorded at increasing concentrations of βG16BP, which revealed that βPGMD170N had an apparent Km (βG16BP) = 150 ± 13 μM (Fig. S1A†). This value is 18-fold weaker than that measured for βPGMWT (Km (βG16BP) = 8.5 ± 0.3 μM (ref. 23)). Analogous experiments at increasing concentrations of βG1P indicated that βPGMD170N had an apparent Km (βG1P) = 6.9 ± 1.0 μM, which is similar in magnitude to the Km (βG1P) for βPGMWT (Km (βG1P) = 14.7 ± 0.5 μM (ref. 12)). These experiments also demonstrated that a similar level of βG1P inhibition was operating in βPGMD170N (apparent Ki (βG1P) = 1540 ± 170 μM) and βPGMWT (Ki (βG1P) = 1510 ± 100 μM (ref. 23)) (Fig. S1B†). The Km (Mg2+) value for the overall reaction of βG1P to G6P was also measured for βPGMD170N using increasing concentrations of MgCl2 (Fig. S2†). These experiments resulted in an apparent Km (Mg2+) = 690 ± 110 μM, which is only 4-fold weaker than that determined for βPGMWT using the same method (apparent Km (Mg2+) = 180 ± 40 μM). Overall, at 1 mM βG1P, 100 μM βG16BP and 5 mM MgCl2 the observed rate constant for the overall reaction of βG1P to G6P catalysed by βPGMD170N (kobs = 3.0 × 10−3 s−1) is 79![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold smaller than that for βPGMWT (kobs = 237 s−1).
000-fold smaller than that for βPGMWT (kobs = 237 s−1).
The observable accumulation of βG16BP in the 31P NMR spectra of the βPGMD170N-catalysed reaction (Fig. 4D–F) presented an opportunity to measure the effects of Mg2+ ion concentration on Step 1 and Step 2 independently within the same experiment (Fig. 1), although it should be noted that kinetic parameters obtained using 31P NMR methods and the G6PDH coupled assay differ significantly due to the effects of the different conditions employed.18,2331P NMR time-course experiments were therefore conducted at increasing concentrations of MgCl2 to measure simultaneously the initial rates of βG16BP production in Step 1 and G6P production in Step 2 (Fig. 3B and C). The initial rates of βG16BP production increased linearly with MgCl2 concentration and so could not be fitted to a Michaelis–Menten equation over the concentration range 5–100 mM (Fig. 3B), indicating that the affinity of βPGMD170N for Mgcat in Step 1 is low. Extraction of the initial rates of G6P production in Step 2 resulted in an apparent Km (Mg2+) = 27 ± 4 mM (Fig. 3C). These observations therefore reveal that βG16BP accumulation can be greatly enhanced by using elevated concentrations of Mg2+ ions. For βPGMD170N at 5 mM MgCl2, the initial rate of Step 1 (kobs = 9.0 × 10−3 s−1) is only 3-fold faster than that of Step 2 (kobs = 3.0 × 10−3 s−1), while at 100 mM MgCl2 Step 1 (kobs = 0.24 s−1) exceeds Step 2 (kobs = 1.2 × 10−2 s−1) by a factor of 20 (Fig. 3B, C and 4G, H). A control experiment employing elevated NaCl concentrations demonstrated that the increased initial rate of Step 1 at higher concentrations of MgCl2 is not caused by Cl− ions alone (Fig. 4I). Therefore, the Mg2+ ion concentration at which the production of βG16BP is performed has a strong bearing on its yield.
Taken together, this analysis demonstrates that the D170N point mutation causes a pronounced retardation of Step 2 together with a more modest change to Step 1. The reduced apparent Km value of βPGMD170N for βG16BP is in line with this behaviour. However, the substantially different apparent Km (Mg2+) values determined for Step 1 and Step 2 are not, which was surprising given the perturbation of the Mgcat binding site in βPGMD170N. One plausible explanation for these observations is that a Cl− ion binds in the active site in substrate-free βPGMD170N to mitigate the loss of the negative charge resulting from the D170N point mutation. The presence of a Cl− ion at the Mgcat binding site would rescue the binding of Mgcat but hamper the binding of βG16BP and the approach of its phosphodianion to the carboxylate group of D8 in Step 2. In contrast, AcP is able to generate βPGMD170NP in a Mgcat-independent manner (Fig. 1 and Fig. 3A) and the presence of the D8 aspartyl phosphate moiety will obviate the formation of the Mgcat–Cl moiety.
To obtain evidence for a putative Mgcat–halide moiety binding to substrate-free βPGMD170N, 10 mM NaF was added to substrate-free βPGMD170N containing 5 mM MgCl2 and 19F NMR experiments were recorded (Fig. 4J and K). A βPGMD170N-bound 19F species was observed with a chemical shift of −171 ppm, which corresponds to an analogous peak seen for substrate-free βPGMWT acquired under similar conditions.24 Elevation to 100 mM MgCl2 did not result in increased saturation of Mgcat, but instead reduced the 19F peak integral to 80% of its size at 5 mM MgCl2, suggesting that at higher concentrations, Cl− ions are displacing the F− ion bound at the Mgcat site (Fig. 4J and K). In the experiment containing 100 mM MgCl2, three free 19F species are present in solution that are separated by chemical shift differences of exactly 37 ppm. Free F− (−118 ppm) and free MgF+ (−155 ppm) have been assigned previously,24 whereas the peak resonating at −192 ppm is likely to be a free Mg–F–Mg3+species, based on the partitioning behaviour between discrete species as the MgCl2 concentration is increased. Comparison of the chemical shifts of the βPGMD170N-bound 19F species with those of the three free species suggests that it is closer in identity to MgF+. Therefore, the primary candidate for such an enzyme-bound species is a Mgcat–F moiety, which in turn provides supporting evidence for the binding of a Mgcat–Cl moiety to substrate-free βPGMD170N that would consequently play a role in the retardation of Step 2 relative to Step 1.
The large-scale generation of βG16BP by the βPGMD170N-catalysed reaction at high concentrations of MgCl2 thus presented an opportunity for harvesting significant quantities of this valuable compound and so a robust production protocol was devised. Recombinant βPGMD170N is overexpressed in high yields from Escherichia coli BL21(DE3) cells (>100 mg L−1) using routine culture techniques and is readily purified using a two-step protocol involving ion-exchange chromatography followed by a size-exclusion chromatography step. βPGMD170N can be stored at −20 °C for long periods and responds well to multiple freeze–thaw cycles, meaning that once purified, a batch of enzyme can be used for numerous βG16BP preparations. In order to characterise βPGMD170N further, 1H15N–TROSY NMR spectra were recorded using samples of 15N-βPGMD170N and 15N-βPGMWT. Comparison of the spectra revealed that βPGMD170N has a similar solution behaviour and overall protein fold to βPGMWT (Fig. S3†). The slow-exchange behaviour that arises in βPGMWT from cis–trans proline isomerisation at the K145-P146 peptide bond is also observable in βPGMD170N.23 Notably, around 15 peaks are present for βPGMD170N that are absent in the spectrum of βPGMWT. These additional peaks indicate that a backbone conformational exchange process, occurring on the millisecond timescale in βPGMWT, has been abolished in βPGMD170N.23
To assess the stability of βPGMD170N and to check for time-dependent reversion to βPGMWT by deamidation,25 a sample of βPGMD170N was incubated at 25 °C for 48 h and both 1H15N–TROSY NMR spectra and 31P NMR time-course experiments were acquired every 24 h. A comparison of 1H15N–TROSY spectra recorded for βPGMD170N preincubated at 25 °C for 0 h and 48 h shows a near-identical correspondence of peaks indicating that the incubation process has a negligible effect on the integrity of βPGMD170N (Fig. S4A†). In comparisons of βPGMD170N and βPGMWT1H15N–TROSY spectra, the absence of observable βPGMWT peaks in the βPGMD170N spectra indicates that reversion of βPGMD170N to βPGMWT through deamidation is not a process that occurs readily under these sample conditions (Fig. S4B and C†). Analysis of the 31P NMR time-course experiments for the equilibration of 10 mM βG1P with G6P catalysed by βPGMD170N (preincubated at 25 °C for 0 h, 24 h and 48 h) demonstrates a consistent behaviour of βG16BP production followed by conversion to G6P as product with no change in kobs, further confirming the stability of βPGMD170N (Fig. S5A†).
To mimic the effect of βPGMD170N reversion to βPGMWT through deamidation, a control 31P NMR time-course experiment was also conducted using a sample of βPGMD170N that had been spiked with 0.1% βPGMWT (Fig. S5B†). The kinetic profile shows an initial burst of G6P production by βPGMWT together with a decrease in the ratio of the βG16BP concentration at its maximum (βG16BPmax) to the concentration of G6P at its maximum (G6Pmax). The 31P NMR time-course experiments testing βPGMD170N stability revealed no burst of G6P production nor any change in either kobs or the βG16BPmax:G6Pmax ratio. Together, these results indicate that βPGMD170N does not undergo detectable deamidation to βPGMWT and is stable at 25 °C over a 48-hour time frame.
31P NMR time-course experiments were used to monitor the βPGMD170N-catalysed conversion of βG1P to G6P to determine the optimal point at which to harvest βG16BP. In a representative reaction (see Materials and Methods) the βG16BP concentration reached a maximum after 265 min at 25 °C. Following quenching of the reaction at this point, and removal of βPGMD170N, the solution was found to contain βG16BP alongside contaminants that included significant amounts of βG1P, G6P and inorganic phosphate (Pi), in a ratio of 1:0.07:0.2:3.9, respectively. As substrates of βPGM, these phosphorylated impurities are undesirable, therefore the solution was subjected to a barium salt precipitation and ion-exchange protocol to obtain the sodium salt of βG16BP with high purity. Barium salts of phosphate species are relatively insoluble,26 and the difference in relative solubility of the βG16BP barium salt compared with those of βG1P and G6P was exploited to enable purification.27–29
To confirm the identity and assess the purity of the final βG16BP product, a sample of the fine powder was analysed using 1H, 13C and 31P NMR experiments (Fig. 5A–D). The identity of the resulting compound was established to be βG16BP by comparison of 1H and 13C chemical shifts with previously reported values.12 Glucose and maltose contaminants were identified in the sample using 1H chemical shifts and scalar coupling constants (BMRB: bmse000015, BMRB: bmse000017). Based on integral values of the anomeric proton signals and of the phosphorus signals in quantitative 1H and 31P NMR spectra, the βG16BP concentration was determined to be 67 mM, which represented 98% of the total phosphorylated glucosaccharide components and 72% of the total glucosaccharide components present in the final sample. βG1P, G6P and glucose comprised <1%, 1% and 3%, respectively, of the total glucosaccharide content. Maltose was present at a greater concentration in the sample (24%), but as a bystander in the reactions of βPGM, and not known to bind to phosphomannomutase 2, this contamination is unlikely to be problematic for users. Pi was also present at a concentration 2.9-fold higher than that of βG16BP. The glucose, maltose and Pi components, which were carried through into the final βG16BP product, are contaminants derived from the enzymatic synthesis of βG1P and would otherwise not be present if a purer source of βG1P were used. Residual HEPES buffer and acetate were also present as minor contaminants. The final yield for the βPGMD170N-catalysed conversion of βG1P to βG16BP was 33.6% and the yield for the overall conversion of maltose to βG16BP was 7.7%. Since the equilibrium for the enzymatic conversion of maltose to βG1P lies in favour of maltose, conducting the reactions for the maltose phosphorylase synthesis of βG1P and the βPGMD170N synthesis of βG16BP in a one-pot system is likely to lead to higher βG16BP yields. The removal of βG1P by βPGMD170N would drive the maltose reaction to produce more βG1P, which in turn would result in a greater overall yield of βG16BP. This approach has been demonstrated previously for the protocol involving maltose phosphorylase and phosphofructokinase.17 To demonstrate the biochemical effectiveness of the final βG16BP product at activating βPGMWT, a kinetic experiment was conducted using the G6PDH coupled assay. βPGMWT was mixed with the βG1P substrate and activated using either 1 μM βG16BP (produced using βPGMD170N) or 8 mM AcP as the phosphorylating agent. The kinetic profile obtained was linear for the βG16BP-containing reaction, but exhibited a lag phase when AcP was used (Fig. 3D). As βG16BP is the only phosphorylating agent known to induce linear initial kinetics in βPGM,23 this experiment provided a clear demonstration of the activity of the final βG16BP product.
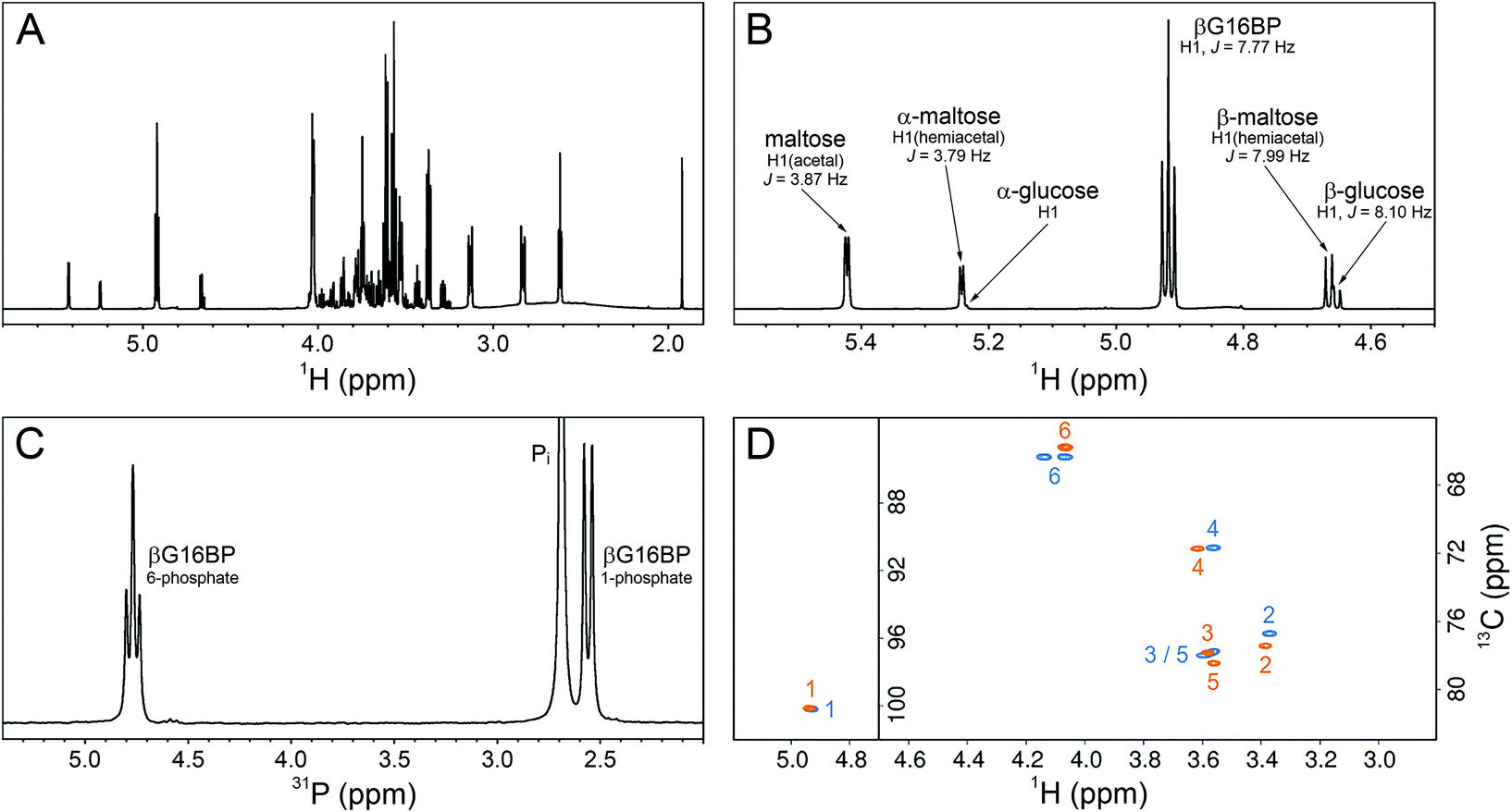 | ||
| Fig. 5 NMR experiments recorded on a sample of the final βG16BP product, purified following its production by βPGMD170N and prepared in 100% 2H2O. (A) 1H spectrum showing βG16BP and other glucosaccharide species present in the sample. (B) A region of the 1H spectrum showing the anomeric proton glucosaccharide signals, together with their assignments. (C) 31P spectrum showing the two phosphorus signals of βG16BP (6-phosphate, 4.76 ppm (triplet) and 1-phosphate, 2.55 ppm (doublet)) and the signal corresponding to inorganic phosphate (Pi, 2.70 ppm (singlet), truncated for clarity). (D) Natural abundance 1H13C–HSQC spectra comparing the final βG16BP product (orange) with chemically synthesised βG16BP (blue).12 Peaks are labelled with carbon ring atom assignments. | ||
Conclusions
The successful manipulation of βPGM behaviour to facilitate βG16BP production is a demonstration of how detailed structural and mechanistic knowledge of an enzyme can lead to novel engineering strategies. Specifically, the modification of the metal binding site of the enzyme dramatically increases the steady state concentration of its reactive intermediate. This highlights the transformative potential that enzymes have within chemical industries and vindicates the intensive study of these useful biomolecules.Materials and methods
Reagents
Unless stated otherwise, reagents were purchased from Sigma-Aldrich, Fischer Scientific, Alfa Aesar and VWR. Isotopically enriched 15NH4Cl was purchased from CortecNet.Gene sequence for βPGMD170N
CATATGTTTAAAGCAGTATTGTTTGATTTAGATGGTGTAATTACAGATACCGCAGAGTATCATTTTAGAGCTTGGAAAGCTTTGGCTGAAGAAATTGGCATTAATGGTGTTGACCGCCAATTTAATGAGCAATTAAAAGGGGTCTCACGAGAAGACTCGCTTCAGAAAATTCTAGATTTAGCTGATAAAAAAGTATCAGCTGAGGAATTTAAAGAACTTGCTAAGAGAAAAAATGATAACTATGTGAAAATGATTCAGGATGTGTCGCCAGCCGATGTCTATCCTGGAATTTTACAATTACTCAAAGATTTACGTTCAAATAAAATCAAAATTGCTTTAGCGTCGGCTTCTAAGAATGGTCCATTTTTATTAGAGAGAATGAATTTAACTGGATATTTTGATGCAATTGCTGATCCGGCTGAAGTTGCAGCATCAAAACCAGCACCAGATATTTTTATTGCAGCAGCACATGCAGTGGGTGTTGCCCCCTCTGAATCAATTGGGTTAGAGAATTCTCAAGCTGGAATTCAAGCCATCAAAGATTCAGGGGCTTTACCAATTGGTGTAGGGCGCCCAGAAGATTTGGGAGATGATATCGTCATTGTGCCTGATACTTCACACTATACATTAGAATTTTTGAAAGAAGTTTGGCTTCAAAAGCAAAAATAACTCGAG.βPGM expression and purification
The βPGMD170N gene sequence was created by modifying the pgmB gene (encoding the βPGMWT enzyme) from Lactococcus lactis (subspecies lactis IL1403) (NCBI: 1114041). The βPGMD170N gene was generated and cloned by GenScript into a pET22b(+) vector. The βPGMWT and βPGMD170N plasmids were transformed into Escherichia coli BL21(DE3) cells and grown using 15N isotopically enriched M9 minimal media.30 Cells were grown to an OD600 of 0.6 at 37 °C and overexpression was induced with 0.5 mM (final concentration) isopropyl β-D-1-thiogalactopyranoside (IPTG) before a 16-hour incubation at 25 °C and centrifugation (Sigma Model 3-15; 9000 rpm for 10 min) to harvest the cells. The βPGMWT and βPGMD170N proteins were purified using the following protocol. The cell pellet was resuspended in ice-cold standard buffer (50 mM K+ HEPES buffer (pH 7.2), 5 mM MgCl2, 2 mM NaN3, 1 mM EDTA) containing a cOmplete protease inhibitor cocktail. The cell suspension was sonicated on ice for 6 × 20 s pulses separated by 60 s intervals. The cell lysate was separated from the insoluble cell debris using centrifugation (Beckman Coulter Avanti centrifuge, Rotor: JA-25-50) at 20000 rpm for 30 min at 4 °C. The soluble fraction was loaded onto a DEAE-Sepharose anion-exchange column connected to an ÄKTA Prime purification system, which had been washed previously with 1 M NaOH and 6 M guanidinium chloride and equilibrated with 5 column volumes of standard buffer. Bound proteins were eluted using a gradient of 0 to 50% standard buffer supplemented with 1 M NaCl over 300 mL. Fractions containing βPGM were identified using SDS-PAGE and were concentrated to a 5–10 mL volume using centrifugation in a Vivaspin (10 kDa molecular weight cut off; Sartorius) at 4500 rpm and 4 °C (Thermo Scientific Heraeus Labofuge 400 R). The concentrated protein sample was loaded onto a pre-packed Hiload 26/600 Superdex 75 size-exclusion column connected to an ÄKTA Prime purification system, which had been washed previously with degassed 1 M NaOH and equilibrated with 3 column volumes of degassed standard buffer supplemented with 1 M NaCl. Following elution, the fractions containing βPGM were checked for purity and were pooled and buffer-exchanged into standard buffer for βPGMWT and standard buffer containing 50 mM MgCl2 for βPGMD170N. Mgcat-free βPGMWT and Mgcat-free βPGMD170N were prepared by buffer-exchanging into standard buffer without MgCl2. The final protein samples were concentrated using a Vivaspin (10 kDa molecular weight cut off; Sartorius) to a 1 mM concentration, as measured by Nanodrop OneC (Thermo Scientific) (βPGM molecular weight = 24.2 kDa, ε280 = 19940 M−1 cm−1), and were stored at −20 °C.
NMR spectroscopy
All NMR spectra were acquired at 298 K, unless stated otherwise. 31P NMR time-course experiments were acquired using a Bruker 500 MHz Avance II spectrometer (operating at 202.456 MHz for 31P) equipped with a 5 mm room-temperature broadband probe and running TopSpin version 3.5. One-dimensional experiments consisting of 256 transients were recorded with a recycle delay of 1 s with proton-phosphorus decoupling and took 479.4 s to acquire. Where stated, experiments were acquired without phosphorus decoupling and took 370.4 s to record. For the AcP hydrolysis experiments, no proton-phosphorus decoupling was used and samples contained 50 mM AcP in 200 mM K+ HEPES buffer (pH 7.2), 10% 2H2O (v/v) and 1 mM trimethylsilyl propionate (TSP) without MgCl2, together with either 300 μM βPGMWT or 300 μM βPGMD170N. For the reactions involving the βPGM-catalysed conversion of βG1P to G6P, samples contained: 300 μM βPGMWT, 10 mM βG1P and 50 mM AcP in 200 mM K+ HEPES buffer (pH 7.2), 10% 2H2O (v/v) and 1 mM TSP without MgCl2 and with no proton-phosphorus decoupling used; 20 μM βPGMD170N, 10 mM βG1P and 20 mM AcP in 200 mM K+ HEPES buffer (pH 7.2), 10% 2H2O (v/v) and 1 mM TSP with 100 mM MgCl2 and with no proton-phosphorus decoupling used; 400 μM βPGMD170N, 10 mM βG1P and 20 mM AcP in 200 mM K+ HEPES buffer (pH 7.2), 10% 2H2O (v/v) and 1 mM TSP with 5 mM MgCl2; 400 μM βPGMD170N, 10 mM βG1P and 20 mM AcP in 200 mM K+ HEPES buffer (pH 7.2), 10% 2H2O (v/v) and 1 mM TSP with 100 mM MgCl2; 400 μM βPGMD170N, 10 mM βG1P and 20 mM AcP in 200 mM K+ HEPES buffer (pH 7.2), 10% 2H2O (v/v) and 1 mM TSP with 5 mM magnesium acetate and 200 mM NaCl. For the βPGMD170N stability measurements, samples contained 200 μM βPGMD170N (preincubated at 25 °C either for 0 h, 24 h or 48 h), 10 mM βG1P and 20 mM AcP in 200 mM K+ HEPES buffer (pH 7.2), 10% 2H2O (v/v) and 1 mM TSP with 100 mM MgCl2. For the experiment representative of 0.1% reversion of βPGMD170N to βPGMWT through deamidation, the sample contained 200 μM βPGMD170N and 200 nM βPGMWT, 10 mM βG1P and 20 mM AcP in 200 mM K+ HEPES buffer (pH 7.2), 10% 2H2O (v/v) and 1 mM TSP with 100 mM MgCl2. To measure apparent Km (Mg2+) values for the Step 1 production of βG16BP and the Step 2 production of G6P, samples contained 5 μM βPGMD170N, 10 mM βG1P and 20 mM AcP in 200 mM K+ HEPES buffer (pH 7.2), 10% 2H2O (v/v) and 1 mM TSP with increasing concentrations of MgCl2 (5, 10, 20, 30, 50, 100 mM). 19F experiments were recorded at 278 K using a Bruker 500 MHz Avance III spectrometer (operating at 470.59 MHz for 19F) equipped with a 5 mm QCI-F cryoprobe and z-axis gradients running TopSpin version 3.5. One-dimensional experiments consisting of 16384 transients were recorded with a recycle delay of 1 s and took 352 min to acquire. Samples of 0.5 mM βPGMD170N were prepared in 200 mM K+ HEPES buffer (pH 7.2) containing 5 mM or 100 mM MgCl2, 10 mM NaF, 0.2 mM deferoxamine, 10% 2H2O (v/v) and 1 mM TSP. 1H15N–TROSY NMR spectra were recorded for βPGMWT using a Bruker 500 MHz Avance III spectrometer equipped with a 5 mm QCI-F cryoprobe and z-axis gradients running TopSpin version 3.5. The sample contained 1 mM 15N-βPGMWT in 50 mM K+ HEPES buffer (pH 7.2) with 5 mM MgCl2, 2 mM NaN3, 10% (v/v) 2H2O and 1 mM TSP. 1H15N–TROSY NMR spectra were recorded for βPGMD170N using a Bruker 800 MHz Neo spectrometer equipped with a 5 mm TCI cryoprobe and z-axis gradients running TopSpin version 4.0. The sample contained 0.5 mM 15N-βPGMD170N (preincubated at 25 °C either for 0 h, 24 h or 48 h) in 50 mM K+ HEPES buffer (pH 7.2) with 5 mM MgCl2, 2 mM NaN3, 10% (v/v) 2H2O and 1 mM TSP. 1H15N–TROSY experiments were acquired with 16 transients with 256 increments and spectral widths of 32 or 36 ppm centred at 120 ppm or 118 ppm in the indirect 15N dimension. For the final βG16BP product prepared in 100% 2H2O containing 1 mM TSP, 1H and natural abundance 1H13C–HSQC experiments were recorded using standard Bruker pulse sequences on an 800 MHz Bruker Neo spectrometer with a 5 mm TCI cryoprobe equipped with z-axis gradients and running TopSpin version 4.0. 31P experiments were also recorded for this sample, as described above. 1H and 13C chemical shifts were referenced to TSP resonating at 0.0 ppm. 31P experiments were either referenced to 1 M H3PO4 resonating at 0.0 ppm, sealed inside a glass capillary and inserted into the sample NMR tube or were referenced indirectly to TSP using the gyromagnetic ratios of the 1H and 31P nuclei. 19F experiments were referenced indirectly to TSP using the gyromagnetic ratios of the 1H and 19F nuclei. NMR data were processed with baseline correction and Lorentzian apodisation using either FELIX (Felix NMR, Inc.) or TopSpin version 4.0 (Bruker). Quantitative NMR experiments were performed using a recycle delay of 60 s. To measure apparent Km (Mg2+) values from 31P NMR time-course experiments, the initial rate for the Step 1 production of βG16BP and the Step 2 production of G6P was obtained using an in-house Python linear least-squares fitting program. The initial rates were subsequently fitted to eqn (1) using an in-house Python non-linear least-squares fitting program, which uses bootstrap error estimation.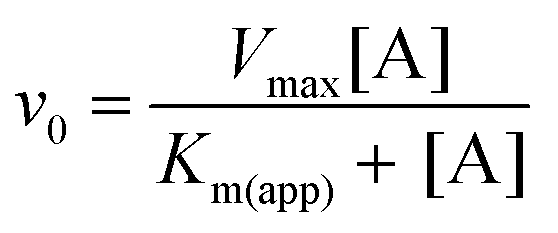 | (1) |
Kinetic assays
Kinetic assays for βPGMWT and βPGMD170N were conducted using a G6PDH coupled assay. Here, the G6P product of βPGM activity is oxidised to 6-phosphogluconolactone by G6PDH, while the concomitant reduction of NAD+ to NADH is monitored by measuring changes in absorbance at 340 nm (ε340 = 6220 M−1 cm−1). Reactions were run at 25 °C using a FLUOstar OMEGA microplate reader (BMG Labtech). To measure the MgCl2 dependence of the βPGMWT- and βPGMD170N-catalysed conversion of βG1P to G6P, reactions (160 μL) were conducted in 200 mM K+ HEPES buffer (pH 7.2) containing different concentrations of MgCl2 (0, 0.1, 0.3, 0.6, 1.0, 1.5, 2.5, 5, 10, 20, 50 and 100 mM), 1 mM NAD+, 5 U mL−1 G6PDH, 1 mM βG1P and either 1 nM βPGMWT with 100 μM βG16BP, or 10 μM βPGMD170N with 1250 μM βG16BP. Initial rates of the reactions were obtained using an in-house Python linear least-squares fitting program. Initial rates were subsequently fitted to eqn (2) using an in-house Python non-linear least-squares fitting program, which uses bootstrap error estimation.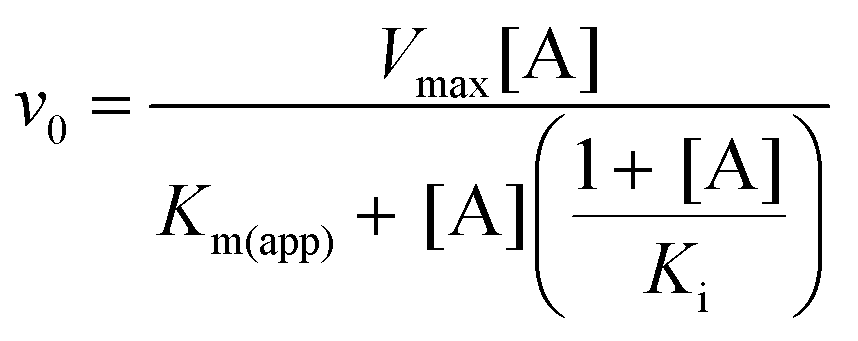 | (2) |
βG1P preparation
βG1P was prepared enzymatically from maltose using maltose phosphorylase (EC 2.4.1.8). A solution of 611 mM maltose was incubated overnight with 1.2 U mL−1 maltose phosphorylase in 0.5 M sodium phosphate buffer (pH 7.0) at 30 °C. The production of βG1P was confirmed using 31P NMR spectroscopy. Maltose phosphorylase (molecular weight = 90 kDa) was removed from the solution by centrifugation using a Vivaspin (5 kDa molecular weight cut off; Sartorius). The concentration of βG1P in the flow-through was measured to be 149 mM using quantitative 31P NMR experiments in which a known amount of G6P had been added to a sample of the βG1P product, along with 10% 2H2O (v/v) and 1 mM TSP. This concentration represents a yield of 24%. The βG1P product was contaminated with glucose, maltose and Pi (estimated concentrations were 150 mM, 850 mM and 350 mM, respectively), and was not purified further since these compounds are bystanders in the reaction catalysed by βPGM.βG16BP production and purification
A 15 mL reaction was prepared containing 20 μM βPGMD170N in 200 mM K+ HEPES buffer together with 100 mM MgCl2, 2 mM NaN3, 20 mM βG1P and was initiated with 40 mM AcP. The concentration of βG16BP reached a maximum after 265 min, whereupon the reaction was quenched by heat-denaturation of βPGMD170N at 90 °C for 10 min. Precipitated enzyme was pelleted using centrifugation (Sigma Model 3-15) and the βG16BP-rich supernatant was collected and filtered with a Vivaspin (5 kDa molecular weight cut off; Sartorius) using a benchtop centrifuge (Thermo Scientific Heraeus Labofuge 400 R). The resulting enzyme-free solution was passed through a 20 × 10 mm column packed with IR120 (H+) ion-exchange resin, which had been washed with 15 mL of milliQ water. This step acidified the solution, which was then neutralised using 0.2 M barium hydroxide solution, resulting in significant precipitation. The solution was kept on ice during neutralisation to increase the solubility of the mono-phosphorylated glucosaccharide barium salts.31 Fractions obtained along the course of the barium salt formation were analysed using 31P NMR experiments, which indicated that the βG16BP barium salt was contained mainly in the precipitate, and that the βG1P and G6P barium salts remained in solution. The precipitate was pelleted using centrifugation at 4 °C (4500 rpm, Thermo Scientific Heraeus Labofuge 400 R) and the supernatant was discarded. To convert the βG16BP barium salt to the more soluble sodium salt the pellet was resolubilised in a large volume (∼1 L) of cold milliQ water and passed through a 20 × 10 mm column packed with IR120 (Na+) ion-exchange resin. The flow-through was then frozen at −80 °C and lyophilised to leave a fine powder as the final βG16BP product.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Prof. Nicholas Williams for useful insight into βG16BP purification. This research was supported by the Biotechnology and Biological Sciences Research Council (BBSRC; H. P. W. – Grant Number X/009906-20-26, N. J. B. – Grant Number BB/M021637/1 and BB/S007965/1, C. R. T. – Grant Number BB/P007066/1) and Consejo Nacional de Ciencia y Tecnologia, Mexico (CONACYT; F. A. C. N. – Grant Number 472448).References
- P.-S. Huang, S. E. Boyken and D. Baker, The coming of age of de novo protein design, Nature, 2016, 357, 320–327 CrossRef.
- K. Chen and F. H. Arnold, Engineering new catalytic activities in enzymes, Nat. Catal., 2020, 3, 203–213 CrossRef CAS.
- D. N. Bolon and S. L. Mayo, Enzyme-like proteins by computational design, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14274–14279 CrossRef CAS.
- F. Wen, N. U. Nair and H. Zhao, Protein engineering in designing tailored enzymes and microorganisms for biofuels production, Curr. Opin. Biotechnol., 2009, 20, 412–419 CrossRef CAS.
- A. Zanghellini, De novo computational enzyme design, Curr. Opin. Biotechnol., 2014, 29, 132–138 CrossRef CAS.
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, A bacterium that degrades and assimilates poly(ethylene terephthalate), Science, 2016, 351, 1196–1199 CrossRef CAS.
- R. Wolfenden and M. J. Snider, The depth of chemical time and the power of enzymes as catalysts, Acc. Chem. Res., 2001, 34, 938–945 CrossRef CAS.
- C. Lad, N. H. Williams and R. Wolfenden, The rate of hydrolysis of phosphomonoester dianions and the exceptional catalytic proficiencies of protein and inositol phosphatases, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5607–5610 CrossRef CAS.
- S. Jones and D. Selitsianos, A simple and effective method for phosphoryl transfer using TiCl4 catalysis, Org. Lett., 2002, 4, 3671–3673 CrossRef CAS.
- S. Jones, D. Selitsianos, K. J. Thompson and S. M. Toms, An improved method for Lewis acid catalyzed phosphoryl transfer with Ti(t-BuO)4, J. Org. Chem., 2003, 68, 5211–5216 CrossRef CAS.
- J. Dai, L. Finci, C. Zhang, S. Lahiri, G. Zhang, E. Peisach, K. N. Allen and D. Dunaway-Mariano, Analysis of the structural determinants underlying discrimination between substrate and solvent in β-phosphoglucomutase catalysis, Biochemistry, 2009, 48, 1984–1995 CrossRef CAS.
- M. Goličnik, L. F. Olguin, G. Feng, N. J. Baxter, J. P. Waltho, N. H. Williams and F. Hollfelder, Kinetic analysis of β-phosphoglucomutase and its inhibition by magnesium fluoride, J. Am. Chem. Soc., 2009, 131, 1575–1588 CrossRef.
- N. J. Baxter, M. W. Bowler, T. Alizadeh, M. J. Cliff, A. M. Hounslow, B. Wu, D. B. Berkowitz, N. H. Williams, G. M. Blackburn and J. P. Waltho, Atomic details of near-transition state conformers for enzyme phosphoryl transfer revealed by MgF3− rather than by phosphoranes, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4555–4560 CrossRef CAS.
- B. Elsässer, S. Dohmeier-Fischer and G. Fels, Theoretical investigation of the enzymatic phosphoryl transfer of β-phosphoglucomutase: Revisiting both steps of the catalytic cycle, J. Mol. Model., 2012, 18, 3169–3179 CrossRef.
- J. L. Griffin, M. W. Bowler, N. J. Baxter, K. N. Leigh, H. R. W. Dannatt, A. M. Hounslow, G. M. Blackburn, C. E. Webster, M. J. Cliff and J. P. Waltho, Near attack conformers dominate β-phosphoglucomutase complexes where geometry and charge distribution reflect those of substrate, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6910–6915 CrossRef CAS.
- Y. Jin, D. Bhattasali, E. Pellegrini, S. M. Forget, N. J. Baxter, M. J. Cliff, M. W. Bowler, D. L. Jakeman, G. M. Blackburn and J. P. Waltho, α-Fluorophosphonates reveal how a phosphomutase conserves transition state conformation over hexose recognition in its two-step reaction, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12384–12389 CrossRef CAS.
- J. Dai, L. Wang, K. N. Allen, P. Rådström and D. Dunaway-Mariano, Conformational cycling in β-phosphoglucomutase catalysis: Reorientation of the β-D-Glucose 1,6-(bis)phosphate intermediate, Biochemistry, 2006, 45, 7818–7824 CrossRef CAS.
- L. Johnson, A. J. Robertson, N. J. Baxter, C. R. Trevitt, C. Bisson, Y. Jin, H. P. Wood, A. M. Hounslow, M. J. Cliff, G. M. Blackburn, M. W. Bowler and J. P. Waltho, van der Waals contact between nucleophile and transferring phosphorous is insufficient to achieve enzyme transition-state architecture, ACS Catal., 2018, 8, 8140–8153 CrossRef CAS.
- G. Zhang, J. Dai, L. Wang, D. Dunaway-Mariano, L. W. Tremblay and K. N. Allen, Catalytic cycling in β-phosphoglucomutase: A kinetic and structural analysis, Biochemistry, 2005, 44, 9404–9416 CrossRef CAS.
- M. Monticelli, L. Liguori, M. Allocca, G. Andreotti and M. V. Cubellis, β-Glucose-1,6-bisphosphate stabilizes pathological phophomannomutase2 mutants in vitro and represents a lead compound to develop pharmacological chaperones for the most common disorder of glycosylation, PMM2-CDG, Int. J. Mol. Sci., 2019, 20, 4164 CrossRef.
- P. S. Chan, C. T. Black and B. J. Williams, Separation of cyclic 3′,5′-AMP from ATPADP, and 5′-AMP by precipitation with inorganic compounds, Anal. Biochem., 1973, 55, 16–25 CrossRef CAS.
- M. Carlson, J. D. Carter and J. Rohloff, Improved preparation of 2 M triethylammonium bicarbonate, Green Chem. Lett. Rev., 2015, 8, 37–39 CrossRef.
- H. P. Wood, F. A. Cruz-Navarrete, N. J. Baxter, C. R. Trevitt, A. J. Robertson, S. R. Dix, A. M. Hounslow, M. J. Cliff and J. P. Waltho, Allomorphy as a mechanism of post-translational control of enzyme activity, Nat. Commun., 2020, 11, 1–12 Search PubMed.
- N. J. Baxter, L. F. Olguin, M. Goličnik, G. Feng, A. M. Hounslow, W. Bermel, G. M. Blackburn, F. Hollfelder, J. P. Waltho and N. H. Williams, A Trojan horse transition state analogue generated by MgF3− formation in an enzyme active site, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14732–14737 CrossRef CAS.
- F. Paradisi, J. L. Dean, K. F. Geoghegan and P. C. Engel, Spontaneous chemical reversion of an active site mutation: Deamidation of an asparagine residue replacing the catalytic aspartic acid of glutamate dehydrogenase, Biochemistry, 2005, 44, 3636–3643 CrossRef CAS.
- L. E. Holt, J. A. Pierce and C. N. Kajdi, The solubility of the phosphates of strontium, barium, and magnesium and their relation to the problem of calcification, J. Colloid Sci., 1954, 9, 409–426 CrossRef CAS.
- J. E. Seegmiller and B. L. Horecker, The synthesis of glucose-6-phosphate and 6-phosphogluconate, J. Biol. Chem., 1951, 192, 175–180 CrossRef CAS.
- D. M. Brown and D. A. Usher, Hydrolysis of hydroxyalkyl phosphate esters: The epoxide route, J. Chem. Soc., 1965, 1965, 6547–6558 RSC.
- D. M. Brown and D. A. Usher, Hydrolysis of hydroxyalkyl phosphate esters: Effect of changing ester group, J. Chem. Soc., 1965, 1965, 6558–6564 RSC.
- M. A. C. Reed, A. M. Hounslow, K. H. Sze, I. G. Barsukov, L. L. P. Hosszu, A. R. Clarke, C. J. Craven and J. P. Waltho, Effects of domain dissection on the folding and stability of the 43 kDa protein PGK probed by NMR, J. Mol. Biol., 2003, 330, 1189–1201 CrossRef CAS.
- T. Posternak, Synthesis of α- and β-glucose-1,6-diphosphate, J. Biol. Chem., 1949, 180, 1269–1278 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc03290e |
| This journal is © The Royal Society of Chemistry 2021 |