
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Flowers of the plant genus Hypericum as versatile photoredox catalysts†
Jun-jie
Wang
 a,
Kai
Schwedtmann
a,
Kun
Liu
a,
Stephen
Schulz
a,
Jan
Haberstroh
a,
Gerrit
Schaper
a,
Anja
Wenke
b,
Julia
Naumann
b,
Torsten
Wenke
b,
Stefan
Wanke
b and
Jan J.
Weigand
*a
a,
Kai
Schwedtmann
a,
Kun
Liu
a,
Stephen
Schulz
a,
Jan
Haberstroh
a,
Gerrit
Schaper
a,
Anja
Wenke
b,
Julia
Naumann
b,
Torsten
Wenke
b,
Stefan
Wanke
b and
Jan J.
Weigand
*a
aFaculty of Chemistry and Food Chemistry, Technische Universität Dresden, Dresden, Germany. E-mail: jan.weigand@tu-dresden.de
bFaculty of Biology, Technische Universität Dresden, Dresden, Germany
First published on 24th November 2020
Abstract
Photoredox catalysis is a powerful and modern strategy for the synthesis of complex organic molecules. So far, this field has relied on the use of a limited range of metal-based chromophores or artificial organic dyes. Here, we show that the ubiquitous plant genus Hypericum can be used as an efficient photoredox catalyst. The dried flowers efficiently catalyze two typical photoredox reactions, a photoreduction and a photooxidation reaction, with a versatile substrate scope. Constitution analysis of the worldwide available plant genus indicated that naphthodianthrones, namely the compounds of the hypericin family, are crucial for the photocatalytic activity of the dried plant material. In situ UV-vis spectroelectrochemical methods provide insights into the mechanism of the photoreduction reaction where the radical dianion of hypericin (Hyp˙2−) is the catalytically active species. Our strategy provides a sustainable, efficient and an easy to handle alternative for a variety of visible light induced photocatalytic reactions.
Introduction
The trend in modern synthetic chemistry more and more endeavors to incorporate sustainable aspects which are highly desirable but also extremely challenging. Sustainable chemistry includes the design of efficient, safe and more environmentally benign chemical processes and products. In this regard, the renaissance of catalytic photochemistry, using light as a natural energy source, provides powerful solutions for various difficult and seemingly insurmountable problems in organic synthesis.1,2 Among all the categories of photocatalysts, transition metal chromophores such as ruthenium and iridium complexes stand at the forefront due to their excellent photophysical properties, which are the basis for numerous versatile applications in inorganic and organic synthesis.3–5 However, transition metal chromophores suffer from low availability, high cost and toxicity, resulting in environmental contamination. In order to overcome the disadvantages brought by transition metal complexes, synthetic organic photocatalysts, including Rhodamine B, Rhodamine 6G and Eosin Y, have been recently introduced into photoredox catalytic processes.6–8 This has resulted in many excellent works demonstrating that synthetic organic catalysts reach comparable or even better catalytic efficiency compared to transition metal chromophores.2,9 However, these artificial organic catalysts are often toxic and must be generated by laborious multi-step syntheses.10,11 Therefore, finding sustainable – renewable and environmentally friendly – and alternative photoredox catalysts still remains urgent and desirable.The plant genus Hypericum L. (Hypericaceae) comprising 460 herbal species can be found in temperate to tropical regions of the world.12Hypericum perforatum (also known as St John's Wort) is among the best known and the most frequently used medicinal herbs since the second century BC,13 covering a wide range of applications such as treatment of burns, skin injuries, neuralgia, fibrosis, sciatica and depression.13–15 Furthermore, anti-depressive,16,17 anti-viral,18 anti-oxidant,19 and anti-bacterial effects of the plant extracts have been responsible for the traditional use of Hypericum perforatum.20 Most recently, the anticancer activity of hypericin – the most active compound from the plant extracts – has attracted much interest due to the remarkable photophysical properties of hypericin making it an ideal photosensitizer for phototherapy.21 In this regard, hypericin and its homologues have long been recognized as an excellent photosensitizer to produce reactive oxygen species (ROSs).21 These ROSs generated by photoexcited hypericin are considered as the source of hypericin's phototoxicity towards bacteria and cancer cells.
The naphthodianthrones hypericin and pseudohypericin as well as their biosynthetic precursor protohypericin, and protopseudohypericin are present in fresh plant material.20,22 The unstable protoforms (protohypericin and protopseudohypericin) are efficiently converted into the stable products hypericin and pseudohypericin by the influence of light.14,20 These are the major components for Hypericum's medicinal value, although other biologically active metabolites, e.g. flavonoids and tannins, are also present.22 It was found that hypericin, pseudohypericin and protohypericin are localized and probably also synthesized in the dark glands which are dispersed over the entire above-ground plant parts (flowers, capsules, leaves, and stems) but not in the roots.23,24 The amount of hypericin produced depends on the number and size of the dark glands but not on their location.24 Nevertheless, the amounts of hypericin and pseudohypericin in flowers are higher than those in leaves.25,26
Considering the wide availability of the plant and the excellent photophysical properties of hypericin, we investigated the plant genus Hypericum as a novel and versatile photoredox catalyst. Two kinds of important model reactions, the reductive coupling reactions of aryl halides9 and the oxidative coupling reactions of N-aryl tetrahydroisoquinolines,10 were selected to investigate the photocatalytic properties of Hypericum plants (Fig. 1). The results demonstrate that the dried flowers of the plant material efficiently catalyze the model reactions under mild conditions. Furthermore, besides standard column chromatography, no additional purification procedure is needed to isolate the desired coupling products.
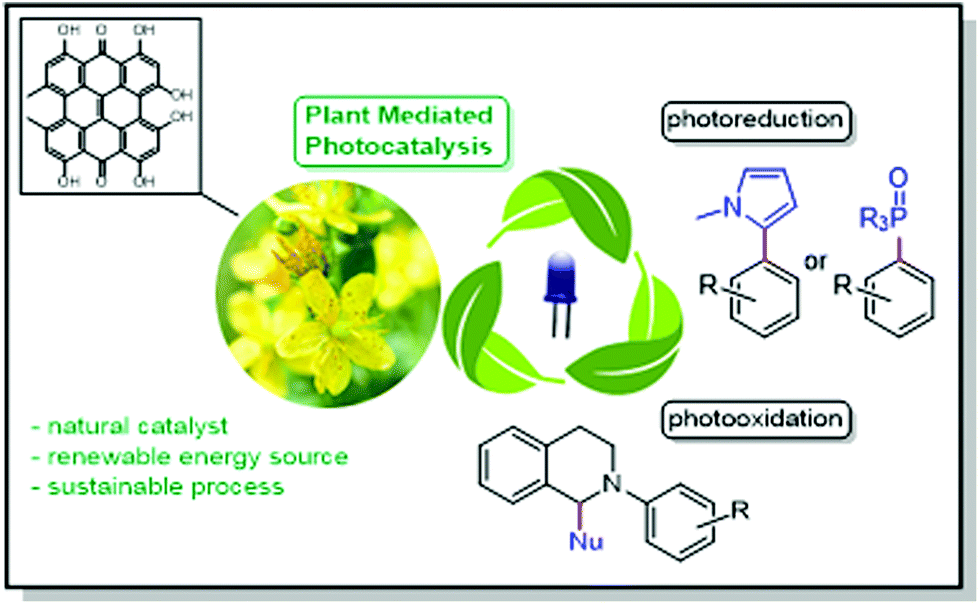 | ||
| Fig. 1 Schematic representation using flowers of plant genus Hypericum as catalysts for photooxidation and photoreduction reactions. | ||
Results and discussion
The photocatalytic reduction of aryl halides in the presence of a radical trap is an important strategy to form C–C and C–P bonds and to build up larger molecules. Using perylene diimides as catalysts, König and co-workers developed a method to activate aryl bromides or chlorides through a consecutive visible light induced electron transfer process.27 Inspired by their strategy, we started our investigation using 2-bromobenzonitrile as a substrate, N-methyl-pyrrole as a radical trap, diisopropylethylamine (DIPEA) as an electron donor and dried flowers of different Hypericum species as catalysts. In order to investigate the catalytic performance between different Hypericum species, ten species collected from Germany, Austria and Greece were tested. The plant material was dried for 24 h at 40 °C in a drying chamber under exclusion of light and the flowers were separated from the dried plant. Prior to the reaction the flowers were ground to a fine powder which was directly added to the reaction mixture without further purification (for details, see ESI 2.1†). To our delight, all Hypericum species demonstrated catalytic activity towards our benchmark reaction in yields of up to 68% (Table 1, entries 1–10). In general, with an increasing amount of hypericin analogues (hypericin, pseudohypericin, protohypericin and protopseudohypericin) the catalytic yield improved simultaneously. It is worth noting that although Hypericum spruneri and Hypericum rochelii exhibit higher contents of hypericin analogues, neither of them demonstrated superior catalytic performance compared to Hypericum vesiculosum, which suggests a different catalytic activity between these hypericin analogues. Moreover, some species in which the concentration of hypericin analogues is comparatively low, such as Hypericum cerastioides, also gave a reaction yield of more than 30% (Table 1, entry 1), indicating that further plant ingredients might also be catalytically active in the model reaction.|
|
||||
|---|---|---|---|---|
| Entry | Hypericum species | Hypericin analoguesb (w%) | Conditions | Yieldc (%) |
| a Reactions were performed using 0.1 mmol 2-bromobenzonitrile, 2.0 mmol N-methyl-pyrrole, 50 mg of plant material and 0.1 mmol DIPEA in 1.0 ml of DMSO under nitrogen with a blue LED (470 nm). b Quantification of hypericin homologues including protopseudohypericin, pseudohypericin, protohypericin and hypericin was based on external standard methods (see the ESI† for details). c Calculated from UPLC analysis by the external standard method. d 100 mg of plant material. e 25 mg of plant material. f 0.5 mmol DIPEA was used. g 1.5 mmol DIPEA was used. | ||||
| 1 | H. cerastioides | 0.016 | 470 nm, 24 h | 31 |
| 2 | H. empetrifolium | 0.018 | 470 nm, 24 h | 51 |
| 3 | H. hirsutum | 0.25 | 470 nm, 24 h | 38 |
| 4 | H. olympicum | 0.53 | 470 nm, 24 h | 40 |
| 5 | H. maculatum | 0.98 | 470 nm, 24 h | 36 |
| 6 | H. perfoliatum | 1.11 | 470 nm, 24 h | 42 |
| 7 | H. perforatum | 1.25 | 470 nm, 24 h | 37 |
| 8 | H. vesiculosum | 1.86 | 470 nm, 24 h | 68 |
| 9 | H. spruneri | 1.97 | 470 nm, 24 h | 58 |
| 10 | H. rochelii | 2.00 | 470 nm, 24 h | 63 |
| 11d | H. vesiculosum | 1.86 | 470 nm, 24 h | 70 |
| 12e | H. vesiculosum | 1.86 | 470 nm, 24 h | 53 |
| 13 | H. vesiculosum | 1.86 | 470 nm, 48 h | 67 |
| 14f | H. vesiculosum | 1.86 | 470 nm, 24 h | 48 |
| 15g | H. vesiculosum | 1.86 | 470 nm, 24 h | 56 |
| 16 | H. vesiculosum | 1.86 | 530 nm, 24 h | 29 |
| 17 | — | — | 470 nm, 24 h | 0 |
| 18 | H. vesiculosum | 1.86 | Dark, 24 h | 0 |
In order to elucidate the catalytically active component, the plant material was investigated by means of UPLC-MS. In agreement with the literature28–32 three classes of compounds have been found in the plant extracts, which were identified as flavonol glycosides (hyperoside, rutin and quercetin), naphthodianthrone (protopseudohypericin, pseudohypericin, protohypericin and hypericin) and polycyclic polyprenylated acylphloroglucinols (hyperforin, adhyperforin, hyperfirin and adhyperfirin) (Fig. 2a–d). The latter substance class can be excluded to be catalytically active as (a) a π-conjugated system is necessary for π–π* electron excitation and subsequent electron transportation within the photoredox procedure and (b) polycyclic polyprenylated acylphloroglucinols tend to decompose under light irradiation within a short period of time.33,34 The flavonol glycosides are structurally similar and differ only in the glycosyl groups linked to the hydroxy group (Fig. 2e), and therefore, quercetin was selected representatively in order to elucidate the catalytic activity of this substance class. When 2 mol% of quercetin is inserted as a catalyst in the benchmark reaction, a yield of 28% was obtained from the optimized conditions (Table 2). This explains that Hypericum species having a low amount of naphthodianthrone, such as Hypericum cerastioides, indeed show catalytic activity.
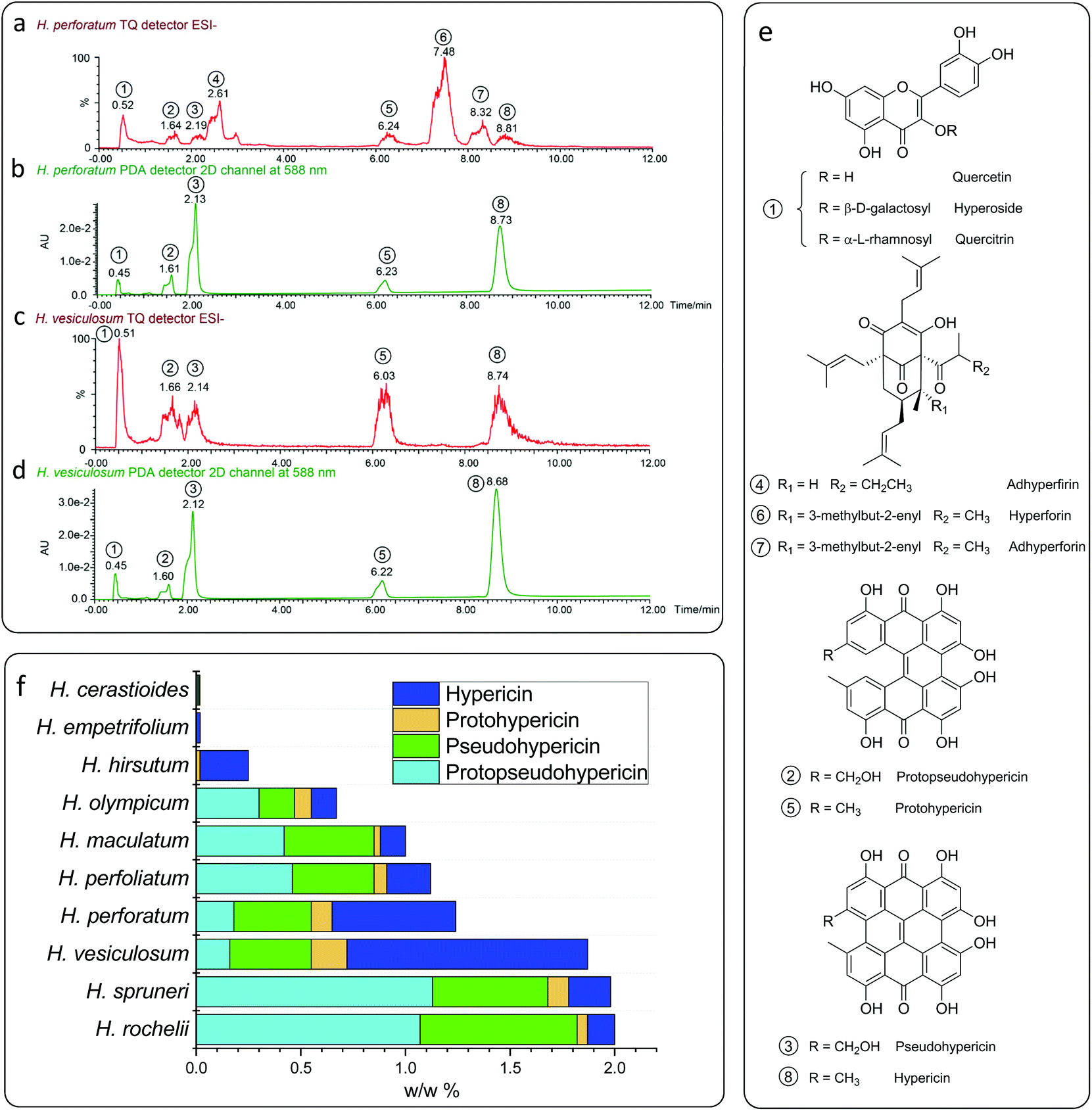 | ||
| Fig. 2 UPLC-PDA/MS analysis of the typical Hypericum plant extracts of H. perforatum (a and b) and H. vesiculosum (c and d); (a) and (c) detected using a tandem quadrupole detector in the ESI− mode; (b) and (d) detected using a photodiode array detector at 588 nm; (e) assigned compounds of the plant extracts; and (f) contents of hypericin analogues in different Hypericum species. | ||
| Entry | Hypericum species | Hypericin Analogues (mol%) | Conditions | Yieldc (%) |
|---|---|---|---|---|
| a Reactions were performed using 0.1 mmol 2-bromobenzonitrile, 2.0 mmol N-mehyl-pyrrole, 2 mol% of corresponding catalyst and 0.1 mmol DIPEA in 1.0 ml of DMSO under nitrogen with a blue LED (470 nm). b Hypericin was in situ synthesized from protohypericin. c Calculated from UPLC analysis using an external standard. | ||||
| 1 | Quercetin | 2 | 470 nm, 24 h | 28 |
| 2 | Protopseudohypericin | 2 | 470 nm, 24 h | 57 |
| 3 | Pseudohypericin | 2 | 470 nm, 24 h | 40 |
| 4 | Protohypericin | 2 | 470 nm, 24 h | 58 |
| 5 | Hypericinb | 2 | 470 nm, 24 h | 73 |
Surprisingly, when 2 mol% of naphthodianthrone are inserted as photosensitizers in the benchmark reaction, yields from 40% to 73% are obtained (Table 2), and hypericin displays the highest catalytic activity among all the naphthodianthrone (73%). The concentration of naphthodianthrone in the well investigated species H. perforatum extract has been derived from the UV-vis absorption spectra at 588 nm (Fig. 2b), revealing an amount of 1.8 mg g−1 for protopseudohypericin, 3.7 mg g−1 for pseudohypericn, 1.0 mg g−1 for protohypericin and 5.9 mg g−1 for hypericin, respectively. In contrast, the extract of H. vesiculosum shows a reduced amount of flavonol glycosides and polycyclic polyprenylated acylphloroglucinols and at the same time, more naphthodianthrone (Fig. 2c). The concentration of the latter substance class is higher in the H. vesiculosum extract (1.6 mg g−1 for protopseudohypericin, 3.9 mg g−1 for pseudohypericin, 1.7 mg g−1 for protohypericin and 11.4 mg g−1 for hypericin) (Fig. 2d), and in addition contains the highest amount of hypericin (Fig. 2f). Therefore, this plant material has been chosen as a catalyst for subsequent reactions.
Since H. vesiculosum showed the highest catalytic activity (68% yield) among all the species, we optimized the photoredox reaction conditions using H. vesiculosum as a catalyst. As expected, reducing the catalyst loading caused a lower yield of 53% (Table 1, entry 12 vs. entry 8). However, increasing the catalyst loading or extending the reaction time only led to a comparable or slightly higher yield compared to the standard conditions, respectively (Table 1, entries 11 and 13 vs. entry 8). Additionally, the concentration of the electron donor (DIPEA) has no significant influence on the reaction yield (Table 1, entries 14 and 15 vs. entry 8). No reaction is observed in the absence of plant material or visible light irradiation, indicating the involvement of a photoredox process in the reaction. In summary, the best conditions for this reaction were determined as 0.1 mmol of aryl halide, 2.0 mmol of a suitable radical trap and 50 mg of H. vesiculosum as a catalyst in DMSO under blue LED irradiation for 24 hours.
With the best conditions in hand, we expanded the reaction to different substrates and radical traps in order to elucidate the influence of functional groups. Moderate to good yields (33%–65%) were obtained with aryl bromide substrates featuring different electron withdrawing substituents including –CN, –CHO, –CF3 and –COCH3 in combination with N-methyl-pyrrole and pyrrole. In contrast, substrates bearing electron donating substituents, such as –CH3 and –OtBu, gave the corresponding products only in very low yields which is attributed to the high reduction potential (Fig. 3A).27 Notably, the reaction with bromopentafluorobenzene as a substrate gives a mixture of two isomers where the pyrrole moiety is bonded via the α C atom or the β C atom (Fig. 3A).35 Different kinds of radical traps such as 3-methyl-indole, trimethoxybenzene and trimethylphosphite were also applied to the protocol, giving yields of 23% to 59% depending on the activity of the traps. Most importantly, the product can readily be separated from the plant catalysts by simple column purification, demonstrating the ease in the application of the plant material as active catalysts.
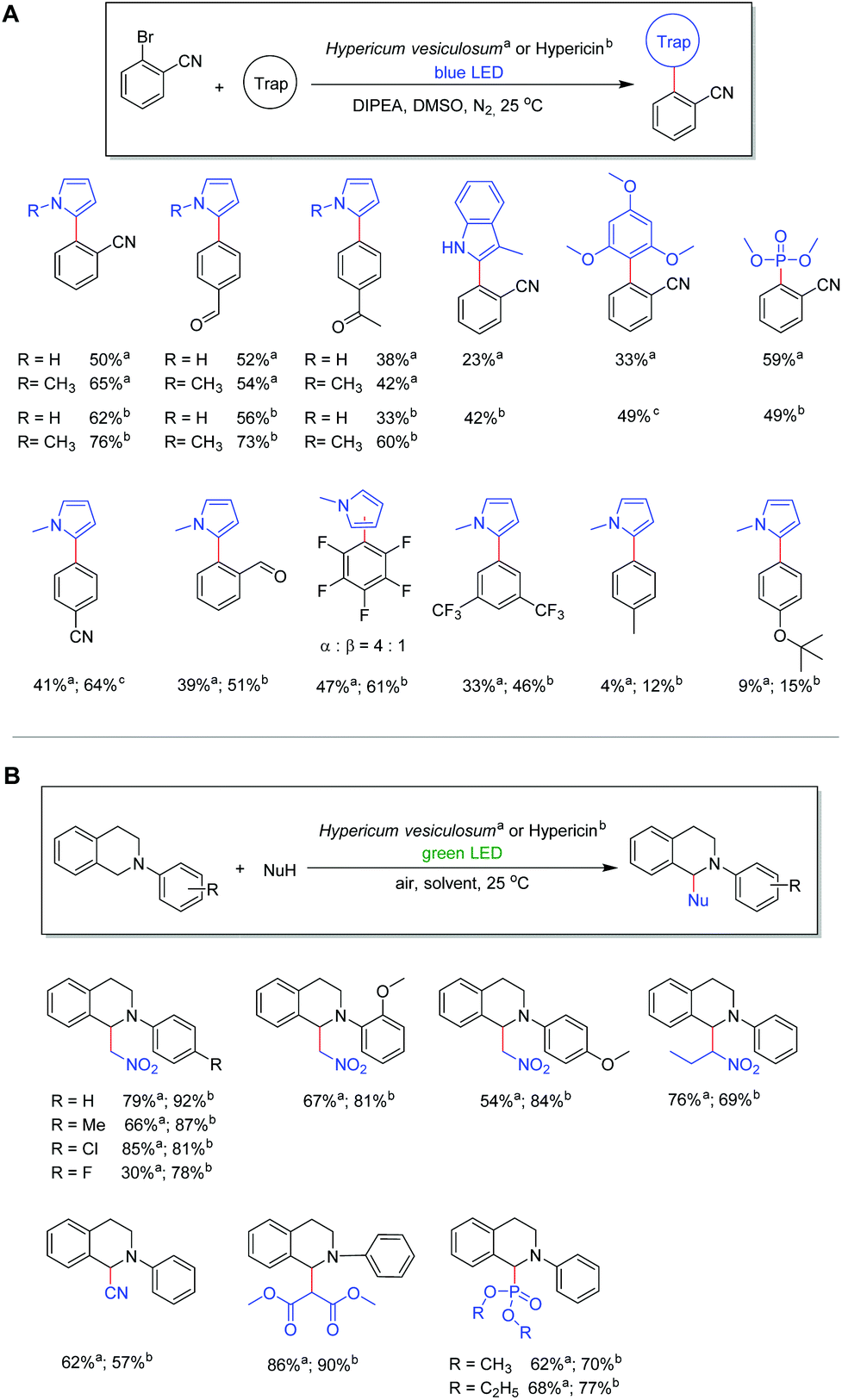 | ||
Fig. 3 (A) Photoreduction reactions using flowers of Hypericum vesiculosuma or hypericinb as catalysts. Reactions were performed using 0.2 mmol 2-bromobenzonitrile, 4.0 mmol N-methylpyrrole, and 0.2 mmol DIPEA in 1.0 mL of DMSO under nitrogen with a blue LED (470 nm). a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Using 100 mg plant material (equals 1.8 mol% naphthodianthrones) for 24 hours. bUsing 4 mmol (2 mol%) hypericin for 48 hours. cUsing 20 mmol (10 mol%) hypericin for 48 hours; (B) Hypericum vesiculosum plant or hypericin as a photoredox catalyst for photooxidative coupling reactions of N-aryl-tetra-hydroisoquinolines with nitromethane or dialkylphosphites ((OR)2P(O)H) under green light irradiation. Reactions were performed using 0.2 mmol N-aryl-tetrahydroisoquinolines and 10.0 mmol nucleophile under ambient atmosphere with a green LED (530 nm). aUsing 50 mg of Hypericum plant as a catalyst in 2.0 ml of methanol. bUsing 2 μmol (1 mol%) hypericin in 2.0 ml of DMF. Using 100 mg plant material (equals 1.8 mol% naphthodianthrones) for 24 hours. bUsing 4 mmol (2 mol%) hypericin for 48 hours. cUsing 20 mmol (10 mol%) hypericin for 48 hours; (B) Hypericum vesiculosum plant or hypericin as a photoredox catalyst for photooxidative coupling reactions of N-aryl-tetra-hydroisoquinolines with nitromethane or dialkylphosphites ((OR)2P(O)H) under green light irradiation. Reactions were performed using 0.2 mmol N-aryl-tetrahydroisoquinolines and 10.0 mmol nucleophile under ambient atmosphere with a green LED (530 nm). aUsing 50 mg of Hypericum plant as a catalyst in 2.0 ml of methanol. bUsing 2 μmol (1 mol%) hypericin in 2.0 ml of DMF. | ||
As we found that hypericin is the most efficient photosensitizer in the plant material, in situ prepared hypericin was applied as a catalyst in order to compare the catalytic activity with the dried plant material of H. vesiculosum. The in situ preparation of hypericin from protohypericin via light irradiation is necessary to increase the solubility of the active catalyst hypericin.36–38 The condition optimization (see Table S3†) demonstrates that with 2 mol% of hypericin catalyst and 1.0 equivalent of DIPEA, the best conversion can be achieved under blue light irradiation for 48 hours. Although hypericin reveals absorption maxima at 555 nm and 599 nm (Fig. S5†), blue light is necessary for further stimulation of the active species (Hyp˙2−) in this process (vide infra).27,39 Using the optimized conditions, moderate to good yields from 42% to 76% can be obtained in the reaction of aryl bromides featuring different substituents including –CN, –CHO, and –COCH3 in combination with N-methyl-pyrrole or pyrrole and hypericin as catalysts (Fig. 3A). With a few exceptions, the results reveal that hypericin provides generally higher yields compared to the plant material. However, considering the simplicity of the reactions by adding the blossoms of the plant as active catalysts, it appears as a sustainable and convenient alternative since this method is beneficial regarding time, cost and chemical waste.
Motivated by our successful application of the H. vesiculosum plant in a catalytic photoreductive process, the coupling reaction of N-aryl-tetrahydroisoquinoline with nitromethane was selected as another model reaction to explore the potential application in photooxidation reactions (Fig. 3B). Please note that this reaction is reported to proceed without a catalyst; however, bromotrichloromethane (BrCCl3) is necessary and the conversion using green light is comparatively low (Table S5†).40 Considering the highest UV-vis absorption of hypericin at 555 nm and 599 nm (see Fig. S5†), a green LED source (535 nm) was chosen as it may provide sufficient energy to stimulate the ground state of hypericin. To our delight, when using the Hypericum plant as a catalyst in the initial photooxidative coupling reaction of N-phenyl-tetrahydroisoquinoline with nitromethane and green light irradiation, the desired coupling product was obtained in good yield (79%; Fig. 3B). Moreover, moderate to very good yields from 30% to 85% can be observed depending on the inserted substrates under the optimized conditions (Fig. 3; for details see Table S4†). The use of dialkyl phosphite ((RO)2P(O)H; R = Me, Et) in the reaction with N-phenyl-tetrahydroisoquinoline similarly generates the corresponding dialkyl phosphonates in good yields (62%, 68%). Consistent with the aforementioned photoreduction reaction, the use of in situ prepared hypericin as a catalyst generally gives higher yields (57% to 92%) after optimization (Fig. 3B; for details see Table S5†). However, also in this case these results reveal the versatile applicability of the Hypericum plant as an efficient photocatalyst in photooxidation reactions.
Mechanistically, we propose that under the given reaction conditions, hypericin (HypH) is dissociated in DMSO solution (HypH → Hyp− + H+) as has been discussed in the literature based on NMR41,42 and electrochemical studies (Fig. 4).43 Thus, the monoanionic Hyp−, being considered as a catalyst, is stimulated under light irradiation to its excited state [Hyp−]*. In the photoreduction process, [Hyp−]* is subsequently reduced by the electron donor DIPEA to give the radical dianion Hyp˙2− along with oxidized DIPEA˙+. Under further light irradiation radical dianion Hyp˙2− is again stimulated to its excited state [Hyp˙2−]* which enables electron transfer to the inserted aryl halide causing homolytic halogen–carbon bond cleavage to give the aryl radical along with the formed halide and the inserted catalyst Hyp−. The highly reactive aryl radical subsequently reacts with the corresponding radical trap (e.g. pyrroles) to form the product radical, which is converted to the desired product upon quenching with the DIPEA˙+ radical cation. In order to justify the proposed mechanism including the intermediately formed active species of hypericin, we performed in-depth spectroscopic and in situ UV-vis spectroelectrochemical (SEC) investigations. The dissolved hyperin shows in DMSO absorption maxima of 555 nm and 599 nm consistent with the reported values of Na+Hyp−.44,45 No change of the absorption maxima is observed upon addition of an excess amount of base (e.g. DIPEA), indicating the presence of dissociated Hyp− in DMSO which is in agreement with previous reports.43
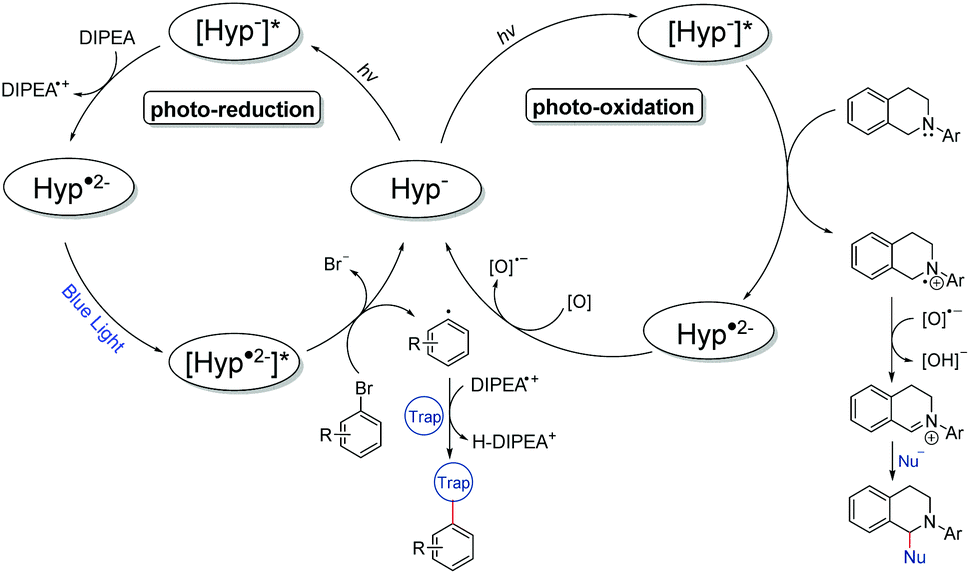 | ||
| Fig. 4 Proposed reaction mechanisms of the photoreduction (left) and photooxidation (right) processes using hypercin as a photocatalyst. | ||
Similar to the catalysis, the hypericin for the SEC measurements was prepared in situ from protohypericin in order to increase the solubility (vide supra) in e.g. acetonitrile. The CV of hypericin supports the assumption of the dissociation into Hyp− in DMSO in comparison with acetonitrile. While the CV measurement of hypericin in DMSO shows explicitly two reversible peaks at potentials of E1/2 (1) = −1.39 V and E1/2 (2) = −1.74 V (vs. E1/2(Cp2Fe/Cp2Fe+); Fig. 5a), the CV curve of the first cycle in acetonitrile reveals a non-reversible peak at Ep = −1.30 V followed by two reversible peaks at E1/2 (1) = −1.46 V and E1/2 (2) = −1.75 V (vs. E1/2(Cp2Fe/Cp2Fe+); Fig. 5b). The non-reversible peak is attributed to the electrochemical deprotonation of HypH to Hyp− + ½H2 and the two reversible processes are assigned to the subsequent redox steps of Hyp− to Hyp˙2− and Hyp˙2− to Hyp3−, respectively.43,44
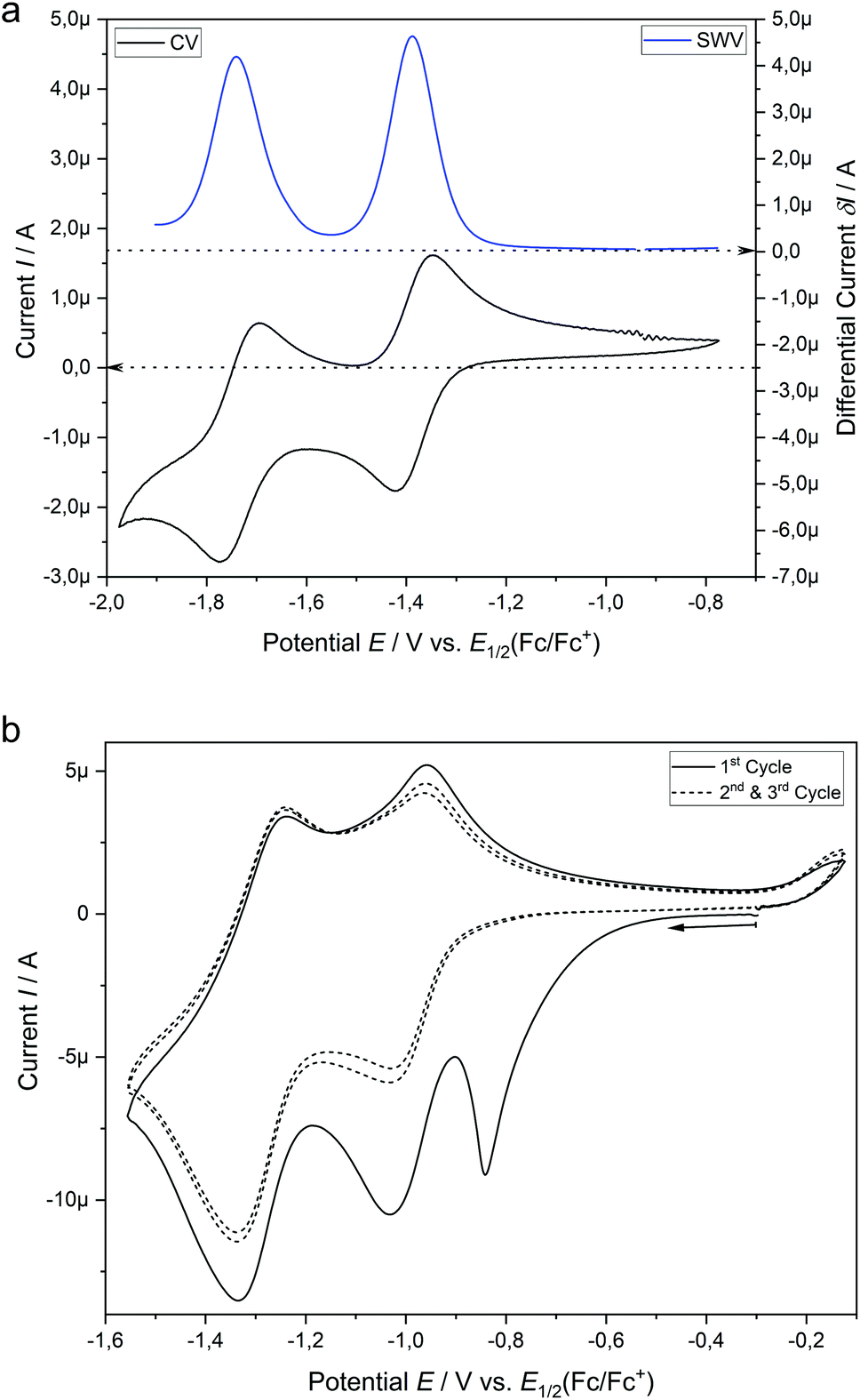 | ||
| Fig. 5 (a) Cyclic voltammetry (CV) curve and square wave voltammogram (SWV) of hypericin (1.0 mM) in DMSO with 0.1 M [nBu4N][BF4] at a 1.6 mm platinum disk electrode. (b) Three cycles of the CV measurement of hypericin (1.0 mM) in acetonitrile with 0.1 M [nBu4N][BF4] at a 1.6 mm platinum disk electrode. | ||
We continued to investigate the base induced formation of Hyp˙2− upon blue light irradiation from Hyp− in DMSO. The Stern–Volmer quenching experiments reveal that the fluorescence emission of the proposed active species [Hyp−]* gradually decreases upon consecutive addition of the electron donor DIPEA, suggesting a single electron transfer from the electron donor (DIPEA) to the excited species [Hyp−]* while the substrate 2-bromobenzonitrile has no quenching effect (see Fig. S11†). The in situ UV-vis spectroscopy of hypericin with an excess DIPEA under constant blue light (470 nm) irradiation congruently reveals a change of the absorption maxima of Hyp− (Fig. 6a, red line, 554 nm and 598 nm) while four new absorption peaks at 493 nm, 570 nm, 678 nm and 745 nm are formed (Fig. 6a, red → blue line) which are attributed to the radical dianion Hyp˙2−.
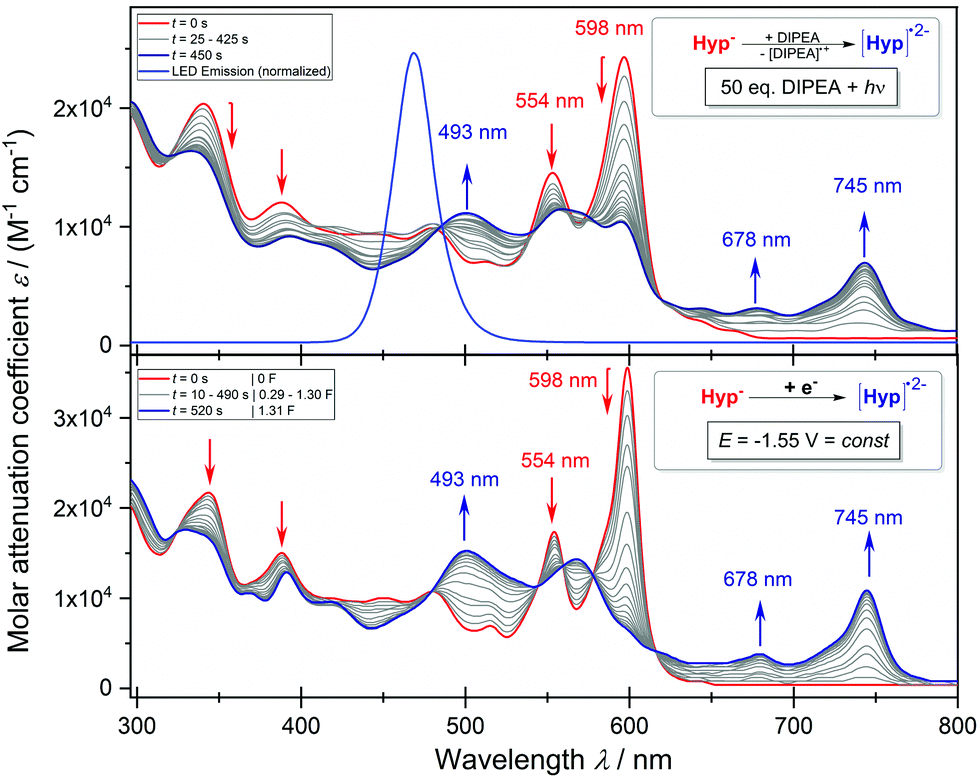 | ||
| Fig. 6 (a) In situ UV-vis spectrum of the reaction of Hyp to Hyp˙2− (0.5 mM) with an excess of DIPEA (50 equiv.) in DMSO under constant blue light irradiation (470 nm; top). (b) In situ UV-vis spectrum of the electrolysis of Hyp− to Hyp˙2− (0.5 mM) at a constant potential of −1.55 V in DMSO with 0.1 M [nBu4N][BF4] at a platinum grid electrode (bottom). | ||
In addition, the formation of the radical dianion Hyp˙2− is confirmed by the electrolysis of Hyp− to Hyp˙2− at a constant potential of −1.55 V coupled with in situ UV-vis spectroscopy (Fig. 6b, red → blue line) revealing the formation of the corresponding absorption maxima of Hyp˙2− during the reduction process. Furthermore, repetitive chronoamperometric switching between Hyp− and Hyp˙2− proves that the reduction of Hyp− to Hyp˙2− is a one-electron process (see Fig. S15 and S16†). Further in situ UV-vis SEC experiments in a double compartment cuvette cell over two cycles of CV demonstrate the reversible redox process between Hyp− and Hyp˙2− (Fig. 7). The reductive formation of Hyp˙2− is coupled with the increased absorbance at 493 nm (Fig. 7, 2D plot and blue absorption line) whereby the reoxidation to Hyp− is accompanied by a decrease of the absorbance at 493 nm and the concomitant occurrence of the absorbance at 591 nm (Fig. 7, 2D plot and red absorption line). The CV curve based on the UV-vis absorbance changes for the starting material Hyp− (Fig. 7, right, red line) and for the reduction product Hyp˙2− (Fig. 7, right, blue line) is in accordance with the in situ CV (Fig. 7, right, top).
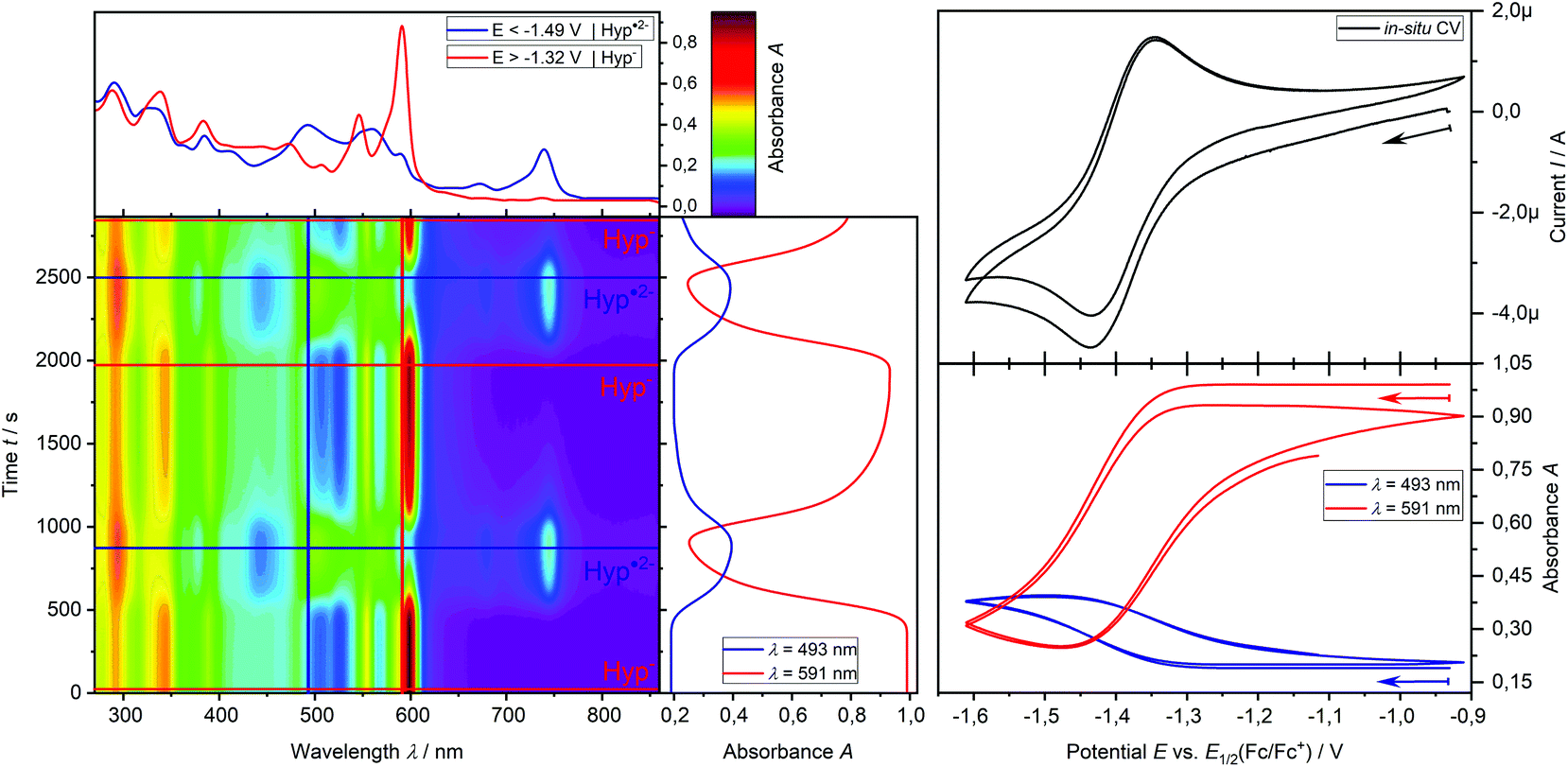 | ||
| Fig. 7 In situ UV-vis-CV SEC measurement of hypericin (0.5 mM) in DMSO with 0.1 M [nBu4N] [BF4] at a platinum grid electrode in a double compartment cuvette cell (d = 0.5 mm); left: 2D plot of UV-vis spectra during the CV measurement with the UV-vis spectra of Hyp (red) and Hyp˙2− (blue); upper right: 2 cycles of CV; lower right: potential dependent absorbance at 591 nm (red) for Hyp− and 493 nm (blue) for Hyp˙2−. | ||
Ultimately, these results confirm the formation of Hyp˙2− and thus its excited state [Hyp˙2−]* during the photoredox process under the given reaction conditions and furthermore justify the utilization of the blue LED (470 nm) as the absorption maximum of the active species (Hyp˙2−) is at 493 nm.
For the photooxidation reaction we propose a similar excitation of Hyp− to [Hyp−]* with green light as one absorption maximum of Hyp− is at 555 nm. The excited [Hyp−]* receives one electron from the inserted N-aryl-tetrahydroisoquinoline to give the corresponding aminyl radical cation along with the radical dianion Hyp˙2−. The latter is reoxidized by the adventitious oxygen to Hyp−. At the same time the aminyl radical cation is deprotonated by the oxygen radical anion to give hydroxide46 and the iminium cation which reacts with the present nucleophile to the final product. According to known reports,47 the photooxidation reaction has been proved by the control reaction under an inert (oxygen free) atmosphere or using the radical trap 2,2,6,6-tetramethylpiperidinyloxide (TEMPO) under standard conditions (Table S5†). Both reactions reveal a significant decrease of the formed product which strongly indicates an oxygen driven radical process during the reaction.48,49
Conclusions
Herein, we have successfully introduced the plant material of the genus Hypericum as efficient and renewable catalysts for photoredox organic reactions, providing a sustainable alternative to metal-based chromophores or artificial organic dyes. The investigated Hypericum species demonstrate photocatalytic activity towards two model reactions, i.e. photooxidation and photoreduction reactions. It is found that naphthodianthrone, especially hypericin, are critical for the catalytic activity of the dried plant material. In-depth mechanistic studies including in situ UV-vis spectroelectrochemical methods reveal that the radical dianion Hyp˙2− in the photoreduction process is the catalytically active species, justifying the use of blue light in this reaction. Ultimately, the results promise a versatile and facile application of plant material in photochemical reactions and contribute to the design of efficient, safe and more environmentally benign chemical processes and future developments.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank the Sächsische AufbauBank (SAB; HyperiPhen project 100315829 in TG70 Bioleben), the European Regional Development Fund, the Free State of Saxony (ERDF-InfraPro, GEPARD-100326379) and the German Federal Ministry of Education and Research (BMBF; Fenabium project 02NUK046A). We also thank Prof. Xinliang Feng for providing access to the fluorescence spectrometer.Notes and references
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS.
- J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS.
- D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS.
- D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97–113 RSC.
- V. Srivastava and P. P. Singh, RSC Adv., 2017, 7, 31377–31392 RSC.
- D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC.
- I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566–1577 CrossRef CAS.
- M. A. Brown and S. C. De Vito, Crit. Rev. Environ. Sci. Technol., 1993, 23, 249–324 CrossRef CAS.
- T. K. M. Prashantha Kumar and S. K. Ashok Kumar, Photochem. Photobiol. Sci., 2019, 18, 148–154 RSC.
- S. L. Crockett and N. K. B. Robson, Med. Aromat. Plant Sci. Biotechnol., 2011, 5, 1–13 Search PubMed.
- J. Patočka, J. Appl. Biomed., 2003, 1, 61–70 CrossRef.
- H. Falk, Angew. Chem., Int. Ed., 1999, 38, 3116–3136 CrossRef.
- H. Du, B.-H. Bay, R. Mahendran and M. Olivo, Cancer Lett., 2006, 235, 202–208 CrossRef CAS.
- B. L. Fiebich, R. Knörle, K. Appel, T. Kammler and G. Weiss, Fitoterapia, 2011, 82, 474–480 CrossRef CAS.
- V. Butterweck, CNS Drugs, 2003, 17, 539–562 CrossRef CAS.
- D. F. Birt, M. P. Widrlechner, K. D. P. Hammer, M. L. Hillwig, J. Wei, G. A. Kraus, P. A. Murphy, J.-A. McCoy, E. S. Wurtele, J. D. Neighbors, D. F. Wiemer, W. J. Maury and J. P. Price, Pharm. Biol., 2009, 47, 774–782 CrossRef.
- B. A. Silva, F. Ferreres, J. O. Malva and A. C. P. Dias, Food Chem., 2005, 90, 157–167 CrossRef CAS.
- J. Barnes, L. A. Anderson and J. D. Phillipson, J. Pharm. Pharmacol., 2001, 53, 583–600 CrossRef CAS.
- P. Agostinis, A. Vantieghem, W. Merlevede and P. A. M. de Witte, Int. J. Biochem. Cell Biol., 2002, 34, 221–241 CrossRef CAS.
- A. Nahrstedt and V. Butterweck, Pharmacopsychiatry, 1997, 30, 129–134 CrossRef CAS.
- S. Kusari, S. Sezgin, K. Nigutova, E. Cellarova and M. Spiteller, Anal. Bioanal. Chem., 2015, 407, 4779–4791 CrossRef CAS.
- S. M. A. Zobayed, F. Afreen, E. Goto and T. Kozai, Ann. Bot., 2006, 98, 793–804 CrossRef CAS.
- C. Cirak, J. Radusiene, V. Jakstas, L. Ivanauskas, F. Yayla, F. Seyis and N. Camas, S. Afr. J. Bot., 2016, 104, 82–90 CrossRef CAS.
- C. Cirak, J. Radusiene, V. Jakstas, L. Ivanauskas, F. Seyis and F. Yayla, Pharm. Biol., 2016, 54, 2244–2253 CrossRef CAS.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS.
- A. Agapouda, A. Booker, T. Kiss, J. Hohmann, M. Heinrich and D. Csupor, J. Pharm. Pharmacol., 2019, 71, 15–37 CrossRef CAS.
- K. P. Mitsopoulou, V. P. Vidali, A. Maranti and E. A. Couladouros, Eur. J. Org. Chem., 2015, 287–290 CrossRef CAS.
- P. Mauri and P. Pietta, Rapid Commun. Mass Spectrom., 2000, 14, 95–99 CrossRef CAS.
- E. C. Tatsis, S. Boeren, V. Exarchou, A. N. Troganis, J. Vervoort and I. P. Gerothanassis, Phytochemistry, 2007, 68, 383–393 CrossRef CAS.
- A. Smelcerovic, M. Spiteller and S. Zuehlke, J. Agric. Food Chem., 2006, 54, 2750–2753 CrossRef CAS.
- P. Maisenbacher and K. A. Kovar, Planta Med., 1992, 58, 351–354 CrossRef CAS.
- F. Liu, C. Pan, P. Drumm and C. Y. W. Ang, J. Pharm. Biomed. Anal., 2005, 37, 303–312 CrossRef CAS.
- A. U. Meyer, T. Slanina, C.-J. Yao and B. König, ACS Catal., 2016, 6, 369–375 CrossRef CAS.
- G. Bánó, J. Staničová, D. Jancura, J. Marek, M. Bánó, J. Uličný, A. Strejčková and P. Miškovský, J. Phys. Chem. B, 2011, 115, 2417–2423 CrossRef.
- H. Falk and J. Meyer, Monatsh. Chem., 1994, 125, 753–762 CrossRef CAS.
- T. Yamazaki, N. Ohta, I. Yamazaki and P. S. Song, J. Phys. Chem., 1993, 97, 7870–7875 CrossRef CAS.
- J. I. Bardagi, I. Ghosh, M. Schmalzbauer, T. Ghosh and B. König, Eur. J. Org. Chem., 2018, 34–40 CrossRef CAS.
- J. F. Franz, W. B. Kraus and K. Zeitler, Chem. Commun., 2018, 51, 8280–8283 RSC.
- D. Freeman, L. Konstantinovskii and Y. Mazur, Photochem. Photobiol., 2001, 74, 206–210 CrossRef CAS.
- D. Skalkos, E. Tatsis, I. P. Gerothanassis and A. Troganis, Tetrahedron, 2002, 58, 4925–4929 CrossRef CAS.
- F. Gerson, G. Gescheidt, P. Häring, Y. Mazur, D. Freeman, H. Spreitzer and J. Daub, J. Am. Chem. Soc., 1995, 117, 11861–11866 CrossRef CAS.
- T. Sagara, H. Kawamura, K. Ezoe and N. Nakashima, J. Electroanal. Chem., 1998, 445, 171–177 CrossRef CAS.
- D. Freeman, F. Frolow, E. Kapinus, D. Lavie, G. Lavie, D. Meruelo and Y. Mazur, J. Chem. Soc., Chem. Commun., 1994, 7, 891–892 RSC.
- Please note that the formation of hydroxide in this mechanism follows the assumptions given in the subsequent references.
- K. Chen, Y. Cheng, Y. Chang, E. Li, Q.-L. Xu, C. Zhang, X. Wen and H. Sun, Tetrahedron, 2018, 74, 483–489 CrossRef CAS.
- A. G. Condie, J. C. González-Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464–1465 CrossRef CAS.
- D. P. Hari and B. König, Org. Lett., 2011, 13, 3852–3855 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc03281f |
| This journal is © The Royal Society of Chemistry 2021 |