
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtein acidification and hydrolysis by pepsin ensure efficient trypsin-catalyzed hydrolysis†
Andrea
Rivera del Rio
 *,
Julia K.
Keppler
,
Remko M.
Boom
and
Anja E. M.
Janssen
*,
Julia K.
Keppler
,
Remko M.
Boom
and
Anja E. M.
Janssen
Food Process Engineering, Wageningen University, P.O. Box 176700 AA, Wageningen, The Netherlands. E-mail: andrea.riveradelrio@wur.nl
First published on 24th April 2021
Abstract
Enzyme-catalysed hydrolysis is important in protein digestion. Protein hydrolysis is initiated by pepsin at low pH in the stomach. However, pepsin action and acidification happen simultaneously to gastric emptying, especially for liquid meals. Therefore, different extents of exposure to the gastric environment change the composition of the chyme that is emptied from the stomach into the small intestine over time. We assessed the susceptibility of a protein to trypsin-catalysed hydrolysis in the small intestine, depending on its pH and hydrolysis history, simulating chyme at different times after the onset of gastric emptying. Isothermal titration calorimetry was used to study the kinetics of pepsin and trypsin-catalysed hydrolysis. Bovine serum albumin (BSA) that was acidified and hydrolysed with pepsin, showed the highest extent and most efficient hydrolysis by trypsin. BSA in the chyme that would be first emptied from the stomach, virtually bypassing gastric acidity and peptic action, reduced trypsin-catalysed hydrolysis by up to 58% compared to the acidified, intact protein, and 77% less than the acidified, pepsin-hydrolysate. The least efficient substrate for trypsin-catalysed hydrolysis was the acidified, intact protein with a specificity constant (kcat/Km) nearly five times lower than that of the acidified, pepsin-hydrolysate. Our results illustrate the synergy between pepsin and trypsin hydrolysis, and indicate that gastric hydrolysis increases the efficiency of the subsequent trypsin-catalysed hydrolysis of a model protein in the small intestine.
1. Introduction
During food digestion, proteins are hydrolysed into polypeptides and ultimately down to single amino acids, prior to absorption into the bloodstream. The first protease that catalyses the hydrolysis of these intact proteins is pepsin, which is secreted in the stomach. Pepsin is active at pH ranging from 1 to 6, with a maximum activity between 1.5 to 2.5.1 However, this optimum depends on the type and denaturation state of the substrate.2,3 Meanwhile, trypsin, chymotrypsin and elastase are secreted from the pancreas into the small intestine. Of these, trypsin seems to be the endopeptidase with the most significant activity as it has been found in human duodenal aspirates, in higher proportion in mass and activity than chymotrypsin.4 Trypsin further hydrolyses proteins and polypeptides into smaller peptides that are better accessible for exopeptidases.5 Pepsin-catalysed hydrolysis happens in parallel with gastric emptying.6–8 Therefore, the composition of chyme that enters the duodenum changes over time, with varying extents of gastric acidification and hydrolysis. This causes pancreatic endopeptidases to interact with different substrates over the course of the postprandial state. As chyme enters the duodenum, its pH is slowly neutralized by the bicarbonate present in the pancreatic juice approaching the pH of optimum activity of intestinal endopeptidases.5,9 An enzyme's affinity and efficiency to catalyse the hydrolysis of intact proteins, compared to hydrolysates, can be better understood by studying its kinetics as a function of the substrate and conditions.Isothermal titration calorimetry (ITC) is a suitable method to study enzyme kinetics.10 Unlike alternative methods, calorimetry allows the direct estimation, in real time, of rates of enzyme-catalysed reactions without the need to label or modify the substrate. Other methods are based on measuring the concentration of substrate or product to derive the rate of catalysis, some requiring separation techniques such as chromatography or electrophoresis after the reaction has taken place. Spectrometric methods rely on fluorogenic or chromogenic substrates. Since virtually all reactions require or release heat, calorimetry can be used to study nearly any reaction. An isothermal titration calorimeter consists of a reference and a sample cell. Usually, the sample cell is loaded with an enzyme solution and a substrate solution is injected into it. The thermal power required to maintain isothermal conditions between the two cells is recorded, being equal to the heat absorbed or released by the system after injection. This heat rate is then proportional to the rate of the reaction.10 ITC has been used to study the kinetics of pepsin-catalysed hydrolysis in gastric conditions.11 In the current work, we aim at extending the knowledge of kinetics of digestive enzymes towards trypsin in small intestinal conditions and of the role of the gastric phase in the overall digestion process of proteins. While some studies have assessed the kinetics of trypsin-catalysed hydrolysis, they have done so with proteins or short, synthetic peptides without a varying pH history and without prior hydrolysis by other enzymes.10,12–14 We compare the extent and rate of trypsin-catalysed hydrolysis of a protein with varying pH histories and prior peptic hydrolysis. Bovine serum albumin (BSA) was used as a model protein. BSA was acidified at pH 2 and hydrolysed by pepsin action, simulating the longest residence time in the stomach. Shorter residence times were simulated by acidifying and not hydrolysing the protein, and by dissolving the intact protein directly at pH 8.
2. Materials and methods
All materials, unless otherwise stated, were purchased from Sigma-Aldrich. All buffers and solutions were prepared with Milli-Q water (resistivity 18.2 MO, Merck Millipore, France).2.1. Sample preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 U ml−1) was prepared in water. A 1 mM trypsin from porcine pancreas (1655 U N-benzoyl-L-arginine ethyl ester per mg, ≤0.1% chymotrypsin, 38560 U ml−1) stock solution was prepared in 1 mM HCl. Stock solutions were prepared daily and kept at 4 °C. The pH of the stock solutions was chosen to be well away the optimum enzyme activity pH to avoid excessive autolysis prior to the substrate hydrolysis assays.
900 U ml−1) was prepared in water. A 1 mM trypsin from porcine pancreas (1655 U N-benzoyl-L-arginine ethyl ester per mg, ≤0.1% chymotrypsin, 38560 U ml−1) stock solution was prepared in 1 mM HCl. Stock solutions were prepared daily and kept at 4 °C. The pH of the stock solutions was chosen to be well away the optimum enzyme activity pH to avoid excessive autolysis prior to the substrate hydrolysis assays.
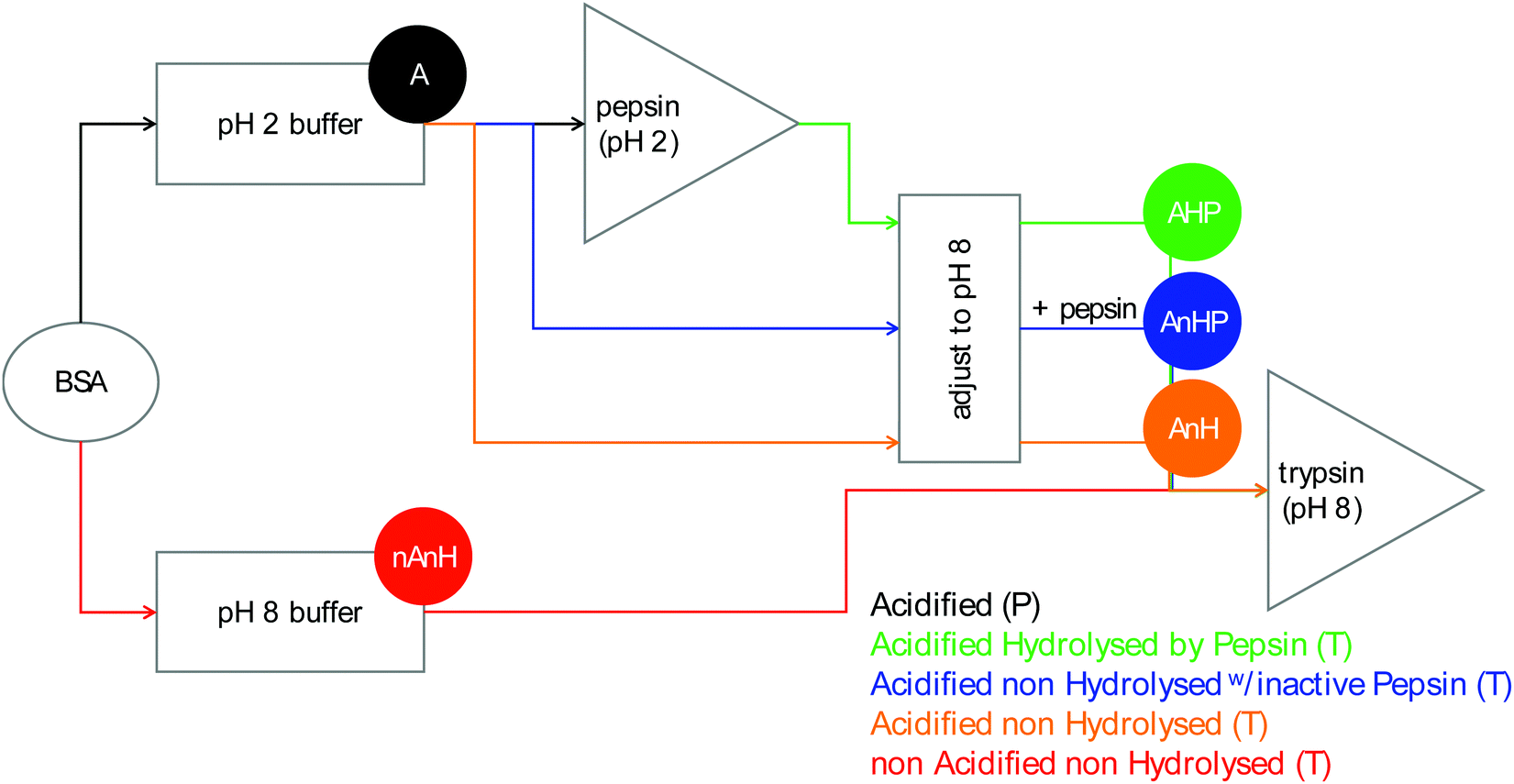 | ||
| Fig. 1 Sample preparation of the five substrates for pepsin (BSA acidified, A) and trypsin-catalysed hydrolysis (BSA acidified and hydrolysed by pepsin, AHP, BSA acidified non hydrolysed, with inactive pepsin, AnHP, BSA acidified non hydrolysed, AnH, and BSA non acidified non hydrolysed, nAnH). | ||
2.1.2.1. Pepsin-catalysed hydrolysis. Acidified BSA (A): BSA was dissolved in a sodium phosphate buffer (100 mM, I = 43 mM, pH 2) to a concentration of 300 μM. Due to the buffering capacity of the protein, pH was adjusted to 2 with phosphoric acid.
2.1.2.2. Trypsin-catalysed hydrolysis. Pepsin-hydrolysate (AHP): A 13 μM pepsin solution was prepared by diluting the pepsin stock solution in pH 2 buffer and was incubated at 37 °C for 30 min in a jacketed glass vessel connected to a water bath. An aliquot of previously prepared 300 μM BSA solution in pH 2 buffer was added to the enzyme solution. The hydrolysis reaction was allowed to proceed to completion for 60 min. A 3 M NaOH solution was used to bring the pH of the pepsin-hydrolysate solution from 2 to 8, resulting in an 8 μM pepsin, 99.6 μM BSA-equivalent solution with an ionic strength of 304 mM.
Acidified, non-hydrolysed BSA with inactive pepsin (AnHP): The composition of the pepsin hydrolysate was reproduced for this solution, except the pepsin stock solution was added after the pH had been adjusted to 8, thus keeping the pepsin presence but not its activity.
Acidified, non-hydrolysed BSA (AnH): The composition was the same as the previous solution, only the stock pepsin solution was substituted with water.
Non-acidified, non-hydrolysed BSA (nAnH): BSA was dissolved in a sodium phosphate buffer (100 mM, I = 290 mM, pH 8) to a concentration of 99.6 μM. At this pH, no buffering capacity was observed.
2.2. Determination of hydrolysis ratio
Pepsin and trypsin have a preference to catalyse the cleavage of specific peptide bonds within a protein or peptide.15 This means that hydrolysis of all peptide bonds in a protein will be achieved by neither pepsin nor trypsin. Next to the enzyme's specificity and selectivity, the reaction conditions influence both the enzymatic activity and the protein conformation. It has been noted that the true substrates for proteases are peptide bonds and not proteins themselves.11,16 Hence we need to measure the real achievable hydrolysis towards the different substrates.Enzyme stock solutions were diluted in 100 mM sodium phosphate buffer, to 12.8 μM pepsin, at pH 2 and to 2.5 μM trypsin, at pH 8. 925 μl of the enzyme dilutions were incubated in Eppendorf tubes at 37 °C for 30 min, at 300 rpm in a Thermomixer (Eppendorf AG, Germany). At four time intervals of 2000 s, 20 μl of substrate solution was added. The hydrolysis ratio was determined and was defined as the molar concentration of free amino groups after hydrolysis per total protein concentration (eqn (1)).11 The hydrolysis ratio was determined for each of the four additions of all five substrate–enzyme combinations.
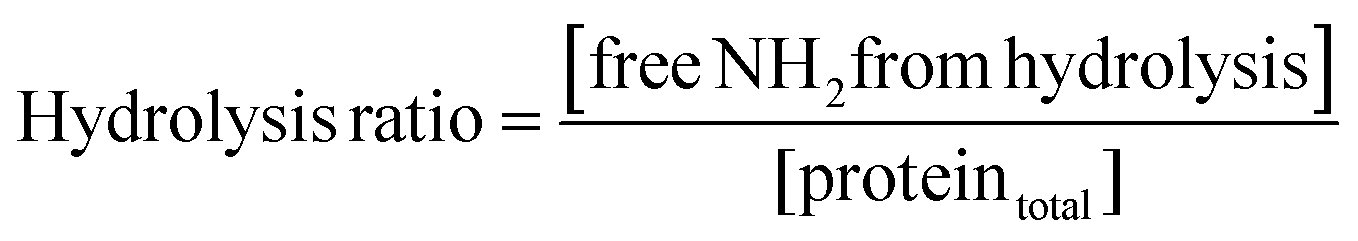 | (1) |
Free amino groups were quantified with the colorimetric reaction between o-phthalaldehyde (OPA) and free amino groups, as previously described.17 Briefly, the OPA reagent was prepared with final concentrations of 3.81% sodium tetraborate decahydrate and 0.1% SDS, 0.08% o-phthalaldehyde pre-dissolved in 2% ethanol, and 0.088% dithiothreitol. The solution was filtered through a 0.45 μm syringe filter and stored to avoid light exposure as the reagent is light sensitive. A standard curve was prepared by using L-serine (Alfa Aesar, Germany) in a concentration range of 10–200 mg L−1. The OPA assay was carried out by adding 200 μL of sample, or standard, to 1.5 mL of OPA reagent. After the reaction took place for exactly 3 minutes, the optical density was measured at 340 nm with a spectrophotometer. Measurements were conducted in duplicate for triplicate hydrolysis samples.
2.3. Tryptophan fluorescence
Samples of substrates and products of hydrolysis were diluted with the appropriate buffer to a protein concentration of 0.01 wt%. Diluted samples were transferred to a quartz cell with 10.00 mm light path. Fluorescence emission spectra were scanned between 300 and 500 nm, at an excitation wavelength of 294 nm, following the method of Heyn, et al.18 Measurements were conducted in a RF-6000 spectrofluorometer (Shimadzu Corporation, Japan). The excitation and emission slit widths were set at 5.0 nm, with a scan speed of 200 nm min−1. Spectra were measured in duplicate.2.4. Isothermal titration calorimetry
ITC assays were conducted in a power compensation, low volume isothermal titration calorimeter (Affinity ITC, TA Instruments, USA) with a 190 μl gold cell (185 μl active volume, considering the stirring paddle and needle). All ITC assays were conducted at 37 °C and a stirring speed of 75 rpm. Instrumental thermal power was recorded every 5 seconds. The reference cell was filled with degassed, deionized water for all assays.Substrate solutions were degassed in a vacuum degassing station (TA Instruments, USA) for at least 30 min. Enzyme stock solutions were degassed and appropriate dilutions were made just before the ITC assays into degassed, 100 mM sodium phosphate buffers, at pH 2 for pepsin and at pH 8 for trypsin, to avoid excessive autolysis. The sample cell was filled with enzyme solution and allowed to equilibrate until a stable baseline was measured. Baseline data was collected for 100 s. It was followed by a small injection of 0.5 μl to account for diffusion into the syringe needle. The thermal power was allowed to return to the baseline for at least 250 s. The calorimeter records the required heat for the sum of all heat effects in the reaction cell, and to maintain its temperature equal to that of the reference cell. When injecting substrate into enzyme, the reaction heat is measured as well as the dilution heat. Therefore, a blank assay of substrate solution injected into buffer is needed to account for the dilution heat. For all assays the substrate dilution heat was measured in a blank assay by injecting the substrate into buffer, with the same conditions of injection volume and interval as the reaction assay.
The total heat of the reaction was obtained by integrating the peaks of each injection with respect to time 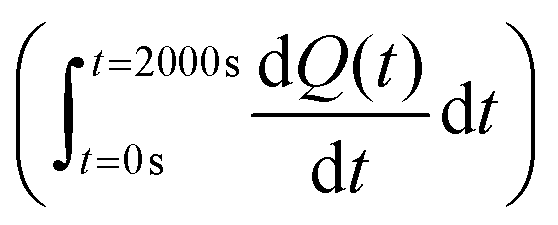 and correcting for the dilution heat from the blank assay. The apparent molar enthalpy (ΔHapp) was estimated with eqn (2), where [S]total is the concentration of substrate in the cell after each injection, and V is the volume of the cell.10
and correcting for the dilution heat from the blank assay. The apparent molar enthalpy (ΔHapp) was estimated with eqn (2), where [S]total is the concentration of substrate in the cell after each injection, and V is the volume of the cell.10
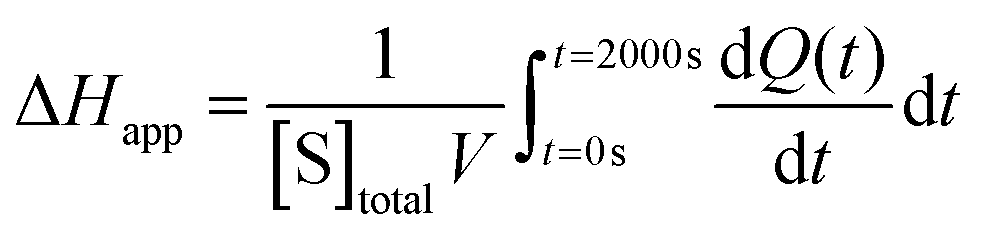 | (2) |
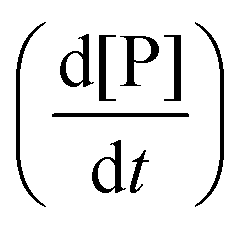 at each injection was estimated from eqn (3), where
at each injection was estimated from eqn (3), where 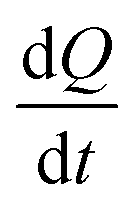 is the thermal power after each injection.10
is the thermal power after each injection.10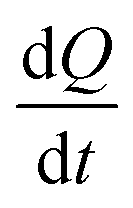 from each injection was considered as the average of the heat rate, 25 s before the following injection. ΔHapp was determined from the first injection of the continuous injection assay.
from each injection was considered as the average of the heat rate, 25 s before the following injection. ΔHapp was determined from the first injection of the continuous injection assay.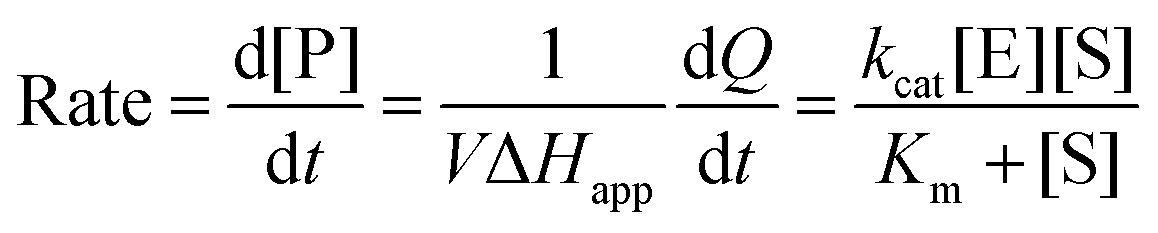 | (3) |
2.5. Data analysis
Data from the continuous injection assay were integrated with NanoAnalyze (TA Instruments, USA). Data were fit to the Michaelis Menten equation (eqn (3)) using a least-squares approach with the nonlinear fitting function ‘lsqcurvefit’ on Matlab R2019b (Mathworks, USA). Statistical analysis was performed using SPSS Statistics software (version 25, IBM, USA). One-way analysis of variance and post-hoc Tukey's honestly significant difference were used to compare significant differences between means at a confidence interval of 95% (p ≤ 0.05%).3. Results and discussion
3.1. Hydrolysis ratio
The aforementioned enzyme specificity towards certain peptide bonds15 means that we need to determine the maximum achievable hydrolysis by one combination of protease and protein, at predetermined conditions.11 This maximum achievable hydrolysis was used to convert the concentration of protein to the concentration of cleavage sites. The hydrolysis ratio was the determined for each of the four substrate additions. To obtain only the free amino groups that are formed by hydrolysis, the terminal and ε-amino groups were determined before hydrolysis (β, Table 1). The β values show that for the acidified substrates (A, AnHP and AnH), the exposure of terminal and side chain amino groups was similar (53.8 ± 0.7 to 55.5 ± 0.8). Interestingly, when the substrate was not acidified (nAnH), the exposure of the terminal or side chain amino groups was less than for the acidified substrates (45.1 ± 1.8), suggesting a more compact structure in the former. As expected, β was much higher for pepsin-hydrolysate (101.8 ± 2.0) as more terminal amino groups had been formed.| Substrate |
β
|
Hydrolysis ratio
|
|||
|---|---|---|---|---|---|
| — | Addition 1 | Addition 2 | Addition 3 | Addition 4 | |
| BSA non-hydrolysed at pH 2 (A), BSA pepsin-hydrolysate (AHP), acidified, non-hydrolysed with inactive pepsin (AnHP), acidified, non-hydrolysed (AnH) and non-acidified, non-hydrolysed (nAnH). The hydrolysis ratio is the amount of free NH2 groups after hydrolysis, minus the amount present before hydrolysis (i.e. β), normalized on the amount of protein. | |||||
| A (P) | 55.5B (0.8) | 80.9a (3.5) | 74.1b (1.7) | 71.2bc (2.7) | 66.1c (2.7) |
| AHP (T) | 101.8A (2.0) | 34.5e (3.1) | 40.9d (4.1) | 43.4d (3.9) | 44.0d (3.7) |
| AnHP (T) | 54.8B (0.8) | 14.4gh (1.2) | 23.9f (2.3) | 23.9f (1.7) | 22.3f (1.7) |
| AnH (T) | 53.8B (0.7) | 12.4hi (1.4) | 13.2hi (3.4) | 21.5f (2.0) | 21.9f (1.8) |
| nAnH (T) | 45.1C (1.8) | 8.1i (2.0) | 15.2gh (2.2) | 18.9fg (2.4) | 18.9fg (2.9) |
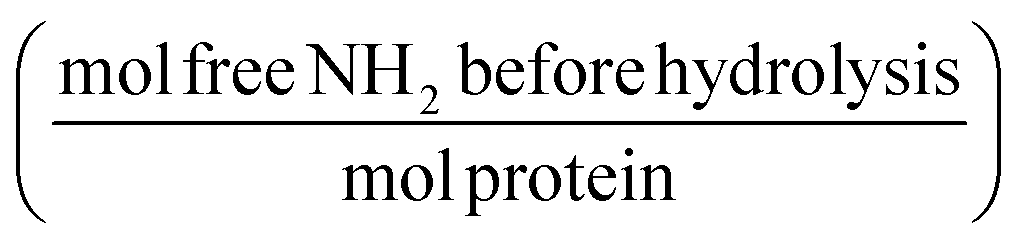
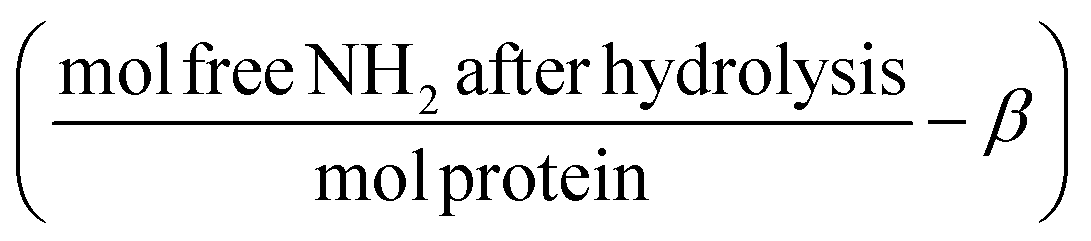
For the first time interval of the pepsin-catalysed hydrolysis of acidified BSA at pH 2, we determined a maximum hydrolysis ratio of 80.9 mol free amino groups from hydrolysis per mol non-hydrolysed protein (Table 1). The hydrolysis ratio after trypsin-catalysed hydrolysis for the pepsin-hydrolysate (AHP, 34.5 ± 3.4) was significantly larger than that of the intact protein (AnHP, AnH and nAnH, 8.1 ± 2.0 to 14.4 ± 1.2). Therefore, more peptide bonds in BSA became susceptible to trypsin-catalysed hydrolysis after the protein had been hydrolysed by pepsin, potentially increasing the overall digestibility of the protein. The pepsin hydrolysate of A and AHP, before trypsin-catalysed hydrolysis, were the same sample, only with different final pH, 2 and 8, respectively. As such, β + hydrolysis ratio1 of A would be similar to β of AHP. Approximately 34 more free amino groups were quantified for β + hydrolysis ratio1 of A than β of AHP. This suggests a pH-induced conformational change that results in a lower exposure of terminal and ε-amino groups or perhaps even aggregation of peptides in the hydrolysate at pH 8.
The pH history of the intact protein also influences the hydrolysis ratio by trypsin. For the previously acidified BSA (AnHP and AnH, 14.4 ± 1.2 and 12.4 ± 1.4), significantly more peptide bonds were cleaved than for the not-acidified protein (nAnH, 8.1 ± 2.0). This can be attributed to the more compact structure of BSA at pH 8, compared to pH 2, suggested by the low β of nAnH. A tighter protein conformation might hinder the access of trypsin to its preferred cleavage sites.19 The slightly higher hydrolysis ratio of AnHP, compared to AnH, might be explained by the trypsin-catalysed hydrolysis of the inactive pepsin in the former. This finding highlights the importance of the gastric pH reaching low enough values to alter the protein conformation for BSA to be better hydrolysed by trypsin in the following stage of food digestion.
One molecule of BSA has 582 peptide bonds. Of those, according to the ExPASy PeptideCutter tool, the maximum potential cleavage sites by pepsin (pH > 2) are 147 and by trypsin, 74 cleavage sites.15,20 This lower selectivity partly explains the significantly larger hydrolysis ratio observed for pepsin than for trypsin. Nevertheless, the hydrolysis ratio reached by pepsin is 55% of the maximum theoretical hydrolysis ratio, while for trypsin, it ranges between and 10 and 19% for the intact proteins.
The hydrolysis ratio decreased from the first to the fourth substrate addition in the pepsin-catalysed hydrolysis of acidified BSA assay (from 80.9 ± 3.5 to 66.1 ± 2.7). The opposite was seen for the trypsin-catalysed hydrolysis, where the hydrolysis ratio of subsequent substrate additions increased to an apparent maximum reached at the third addition. The reaction conditions were not the same from the first addition compared to the subsequent additions. In the first step, the substrate solution was added to an enzyme solution. For the subsequent steps, the substrate was added into a solution containing enzyme, peptides and, perhaps, intact proteins. It is uncertain whether the higher hydrolysis ratio of subsequent substrate additions were solely from products of the freshly added substrate or if some uncleaved peptide bonds from the previous additions were also targets for trypsin-catalysed hydrolysis.
The varying hydrolysis ratios for the different substrates and additions make evident that the history of the protein, in terms of pH and hydrolysis, influences the amount of peptide bonds susceptible to further hydrolysis, particularly by trypsin.
3.2. Tryptophan fluorescence
The pH-influenced protein conformation may be a key feature determining the extent of hydrolysis. We measured the tryptophan fluorescence of the different substrates and their hydrolysis products. Tryptophan is an intrinsic fluorophore of proteins. The maximum measured fluorescence intensity and emission wavelength provides information on the tryptophan residues’ neighbouring amino acids and accessibility to the solvent.21 For instance, a red-shifted maximum, i.e. at a higher emission wavelength, indicates greater exposure of the tryptophan residues towards a polar solvent, while the maximum intensity is a reflection of the surroundings of the residue and is quenched upon protein unfolding due to solvent effects. The structure of BSA is divided into three domains, each subdivided into two subdomains, A and B. BSA has two tryptophan residues in its structure, Trp-134 and Trp-213. The former is located in the subdomain IB near the surface of BSA, while the latter is closer to the hydrophobic core of the subdomain IIA (Fig. 2). The response from both residues in the two locations convenes in the recorded fluorescence spectrum. Nevertheless, it has been demonstrated that Trp-134 dominates the intensity over Trp-213.22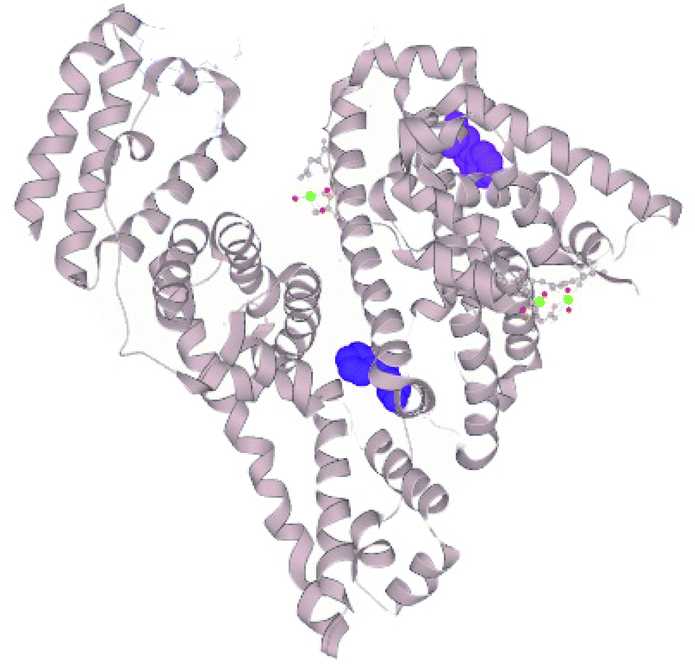 | ||
| Fig. 2 Crystal structure of BSA.23 Tryptophan residues represented as surfaces (TRP-134 in purple, TRP-213 in blue; selection made on LiteMol24). | ||
Among the five substrates, BSA dissolved in pH 2 buffer (A) showed the most blue-shifted maximum at 327 nm, i.e. lowest fluorescence emission wavelength at maximum intensity (Fig. 3, purple filled circle), indicating a more apolar environment as well as quenching of one or both Trp residues. Non-acidified and previously acidified BSA returned at pH 8 (nAnH and AnH(P)) emitted at 336 nm, at comparable maximum intensities, revealing only a slight difference in the direct environment of Trp. Meanwhile, the pepsin-hydrolysate at pH 8 showed the most red-shifted maximum implying the greatest exposure of Trp residues to the polar environment, including strong quenching effects. This substrate not only differs to the other substrates in conformation, but also in peptide length.
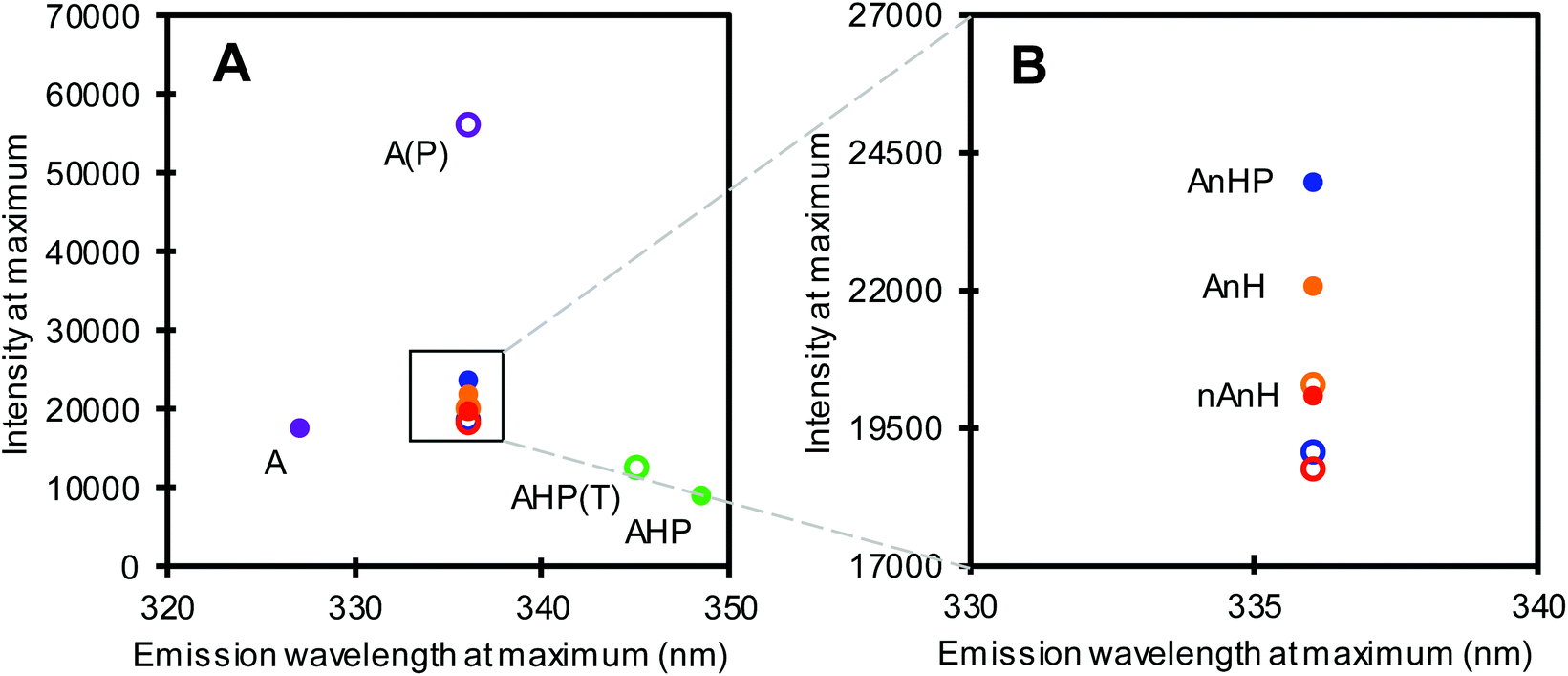 | ||
| Fig. 3 Tryptophan fluorescence intensity and emission at maximum. (A) BSA at pH 2, purple; BSA pepsin-hydrolysate at pH 2, green; acidified, non-hydrolysed with inactive pepsin, blue; acidified, non-hydrolysed, orange; non-acidified, non-hydrolysed, red. Filled circles represent samples before hydrolysis, open circles represent samples after pepsin (BSA at pH 2) and trypsin-catalysed hydrolysis (other substrates at pH 8). (B) Expanded view of samples which emitted its maximum intensity at 336 nm. Circles represent means of duplicate measurements. | ||
We also measured the fluorescence spectrum of the products of hydrolysis from the first substrate addition assay of the hydrolysis ratio determination (section 3.1). Pepsin catalysed hydrolysis (A1) resulted in peptides that were red-shifted with a markedly higher fluorescence intensity at maximum compared to the intact protein. This shows a change in the protein conformation that significantly affects the microenvironment surrounding the Trp residues, perhaps by losing the rigidity of the intact protein. Conversely, trypsin catalysed hydrolysis of the pepsin-hydrolysate (AHP1) resulted in a slight blue-shift from the substrate. Hydrolysis did not always result in a shift in fluorescence emission at maximum intensity. No change in fluorescence emission wavelength was observed for BSA trypsin-hydrolysates (AnHP1, AnH1 and nAnH1), although in all cases, fluorescence was slightly quenched after hydrolysis (Fig. 3B). Interestingly, the extent of hydrolysis was reflected in the difference in fluorescence emission and intensity at maximum, between samples before and after hydrolysis.
Our findings agree with previous research which showed a change in the tryptophan fluorescence emission of BSA under acidic and alkaline conditions, from blue to red-shifted.25,26 Protonation at low pH increases the repulsion between charged groups which releases the electrostricted water from the hydrophobic pockets.27,28 As a result, Trp remains embedded in a dry molten globule with an apolar environment.29 In contrast, the deprotonation of amino acids in alkaline conditions leads to a conformational shift resulting in increasing polarity near Trp.30 Furthermore, researchers who studied the far-UV circular dichroism spectra of BSA proposed that, at high pH, the protein has a tighter structure compared to low pH.31 This can also be related to charges and their distribution throughout the protein. At low pH, the strong repulsion between protonated groups, which are distributed uniformly, weakens the intra and inter-domain interactions and provides the largest stability in an expanded protein conformation.26,32 Meanwhile, at pH 8, the net electrical charge is nearly neutral, allowing for a folded structure.26 This is supported by the low β of nAnH BSA (Table 1).
Tryptophan residues in proteins are sensitive to changes in the local environment.33 This was observed particularly between the BSA pepsin-hydrolysate at pH 2 and at pH 8 (purple-hollow and green-filled circles in Fig. 3A). These two samples have the same protein, hydrolysed by the same enzyme at pH 2. However, the pH of the AHP substrate had been adjusted to 8 to be further hydrolysed by trypsin, avoiding differences in composition of the buffer in which the enzyme is dissolved. We therefore propose an alkalinity-induced aggregation which is supported by the aforementioned discrepancy between β + hydrolysis ratio1 of A and β of AHP (section 3.1).
Such differences were not observed in the fluorescence spectra of the intact proteins that were previously acidified (AnH(P)) and that which was directly dissolved in pH 8 buffer (nAnH, Fig. 3B). In previous research, the isomer formed at pH 3 was found to be thermodynamically less stable than the native protein at higher pH.25 Small-angle X-ray scattering showed that the refolding of BSA from the acid-induced expanded state into the alkaline, native state was reversible.34 In this study, pH increase was achieved by dialysis against a neutral solution, potentially not altering too much the ionic strength. Nevertheless, it has also been suggested that an initial acidity-induced unfolding of BSA is not rapidly reversible when titrating back to alkaline pH.35 This indicates that refolding might be influenced by the environment surrounding the protein. Helical structures in folded BSA are favoured by internal salt bridges.36 The protein in our AnH(P) sample was initially exposed not only to low pH but also to the ionic strength of the buffer. When increasing the pH to 8, we propose that BSA might have refolded less tightly because of the initial exposure to ions at pH 2, which may have blocked the formation of some salt bridges, enabling the slightly more extensive hydrolysis found for AnH(P) compared to nAnH. In sum, we suggest that pH-induced conformational isomerization, as a result of the substrates’ pH and hydrolysis history, influences the accessibility for enzymes to the preferred peptide bonds for cleavage. The tight structure reported for BSA at alkaline pH might hinder trypsin activity and thus explain the lower hydrolysis ratio compared to the substrates that had gone through an acid-denatured state.
3.3. Isothermal titration calorimetry
We observed that the dilution of all substrates into pH 2 and pH 8 buffers was overall endothermic and was compensated by the ITC within approximately 100 s (dotted lines in Fig. 4 and S1†). Meanwhile, the joint signal of dilution and reaction heat for the pepsin-catalysed hydrolysis of acidified BSA at pH 2 was overall exothermic and lasted for around 1400 s (Fig. 4A). Trypsin catalysed hydrolysis was overall endothermic and its return to baseline happened sooner than for pepsin (Fig. 4B and S1†). Shorter reaction times for the trypsin-catalysed reaction were likely due the lower concentrations of both protein and enzyme, compared to the pepsin assay. The total heat of the reaction was obtained by integrating the area under the curve of each injection with respect to time. To account for the dilution heat, the integrated heat from the blank experiments was subtracted from the heat of the injection of substrate into enzyme.
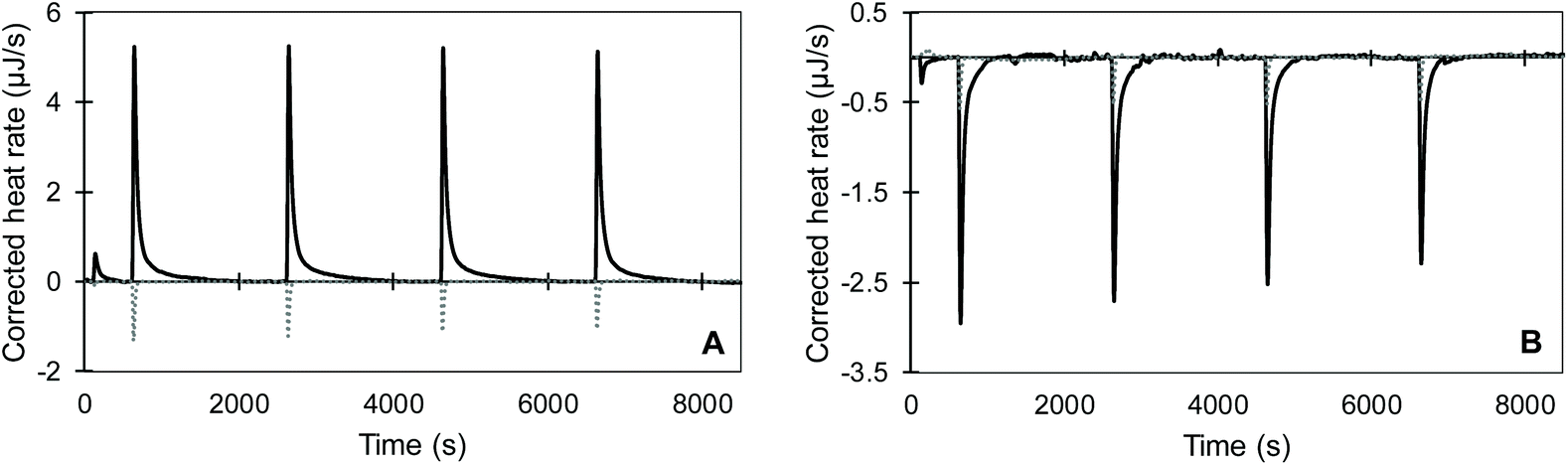 | ||
| Fig. 4 Continuous injection assay of (A) acidified BSA hydrolysed by pepsin at pH 2 and (B) BSA pepsin-hydrolysate, further hydrolysed by trypsin at pH 8. Blank injections of substrate into buffer (dotted line) and injections of substrate into enzyme solution (solid line). A small volume was injected after the initial equilibration to account for the diffusion of substrate solution from the syringe into the cell. | ||
The reaction heat rate was used to calculate the apparent molar enthalpy of the reaction (ΔHapp, eqn (2)). ΔHapp is the total heat produced or absorbed to convert one mol of substrate into product. To normalise with the number of susceptible peptide bonds of BSA in a given condition, the cleavage sites were considered as the substrate.11 The molar concentration of substrate used in eqn (2) was estimated from the hydrolysis ratio (mol free amino groups from hydrolysis per mol protein) of the first substrate addition (hydrolysis ratio1, Table 1). Both ΔHapp, expressed as kJ mol−1 BSA and as kJ mol−1 cleavage sites, are given in Table 2.
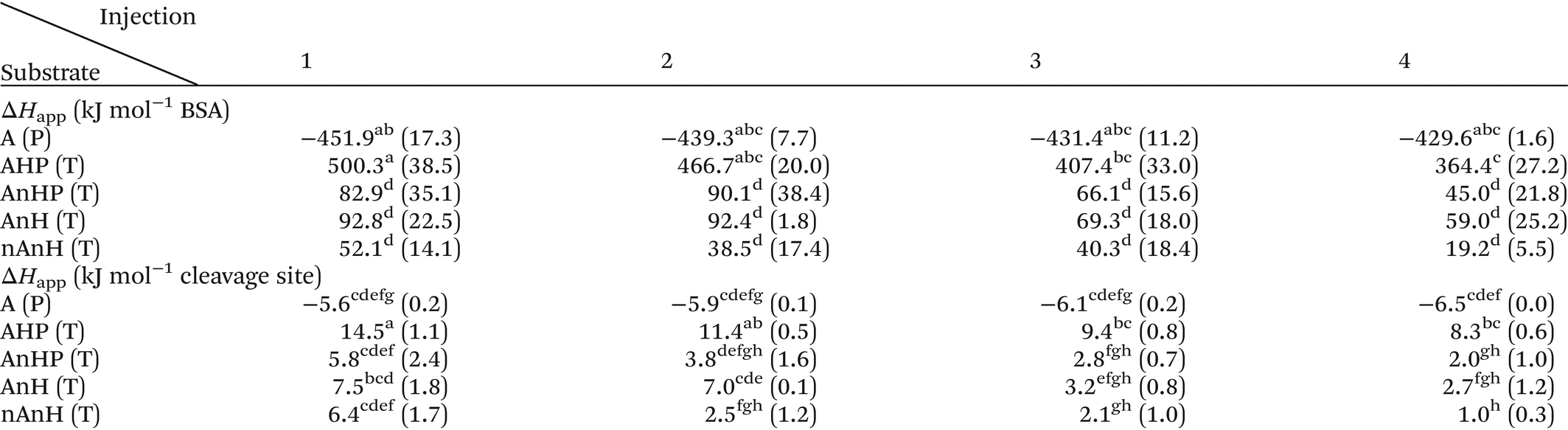
|
For the first injection, ΔHapp of the hydrolysis of peptide bonds in the intact protein did not differ significantly, regardless of the pH history (A catalysed by pepsin and, AnHP, AnH and nAnH catalysed by trypsin). Meanwhile, the enthalpy of the trypsin-catalysed hydrolysis of the pepsin-hydrolysate (AHP) was significantly higher than for intact BSA. This means that more heat is absorbed to hydrolyse a peptide bond within a shorter peptide chain than in a full protein.
The continuous injection assay was chosen over the more common single injection assay to gain insight into potential product inhibition.13,37 After the first injection, the reaction medium contains enzyme, leftover substrate and products. With each subsequent injection of substrate, products continue to accumulate. Changes in the overall magnitude and shape of the curves associated to the injections would indicate an effect on the enzyme activity due to the presence of hydrolysis products. In all assays, the magnitudes and shapes of the peaks corresponding to each of the four injections appeared similar between each other, as shown by the superimposed curves from each injection (S 2). Nevertheless, a slight decrease was observed in the total area under the curve of each subsequent injection. The decrease was not significant for the ΔHapp associated to each injection of the pepsin-catalysed hydrolysis of peptide bonds of BSA (Table 2). Conversely, for the trypsin-catalysed hydrolysis of all substrates, ΔHapp decreased from the first to the fourth injection. This can be partly explained by the lower enzyme concentration after a new injection due to the displaced volume from the sample cell. Such a decrease in the heat signal has been associated to trypsin inhibition by products of casein hydrolysis.13 Further, product inhibition has been observed on both pepsin and trypsin-catalysed hydrolysis.38,39 Contradictorily, in our study, the average conversion rate seems to increase with subsequent injections as shown by the hydrolysis ratio1,2,3 (Table 1), challenging the notion of product inhibition. This increase in the hydrolysis ratio further reduces ΔHapp (kJ mol−1 cleavage site) after each injection, implying that by increasing the ratio of substrate to enzyme, less heat is consumed to hydrolyse one peptide bond.
 was measured after each injection and corrected for the heat rate associated to the heat of dilution of the substrate. The reaction initial rate was estimated from the heat rate using eqn (3). The reaction enthalpy used for the estimation was from the first injection of the continuous injection assay as negligible product effects were observed (Table 2). The initial rates of the enzyme-catalysed hydrolysis of peptide bonds in BSA were calculated for each substrate–enzyme concentration combination (Fig. 5B and 6). The reaction rates estimated at progressive substrate concentrations were fit to the Michaelis Menten model (eqn (3)). The turnover number and Michaelis–Menten constant were obtained (kcat and Km, Table 3).
was measured after each injection and corrected for the heat rate associated to the heat of dilution of the substrate. The reaction initial rate was estimated from the heat rate using eqn (3). The reaction enthalpy used for the estimation was from the first injection of the continuous injection assay as negligible product effects were observed (Table 2). The initial rates of the enzyme-catalysed hydrolysis of peptide bonds in BSA were calculated for each substrate–enzyme concentration combination (Fig. 5B and 6). The reaction rates estimated at progressive substrate concentrations were fit to the Michaelis Menten model (eqn (3)). The turnover number and Michaelis–Menten constant were obtained (kcat and Km, Table 3).
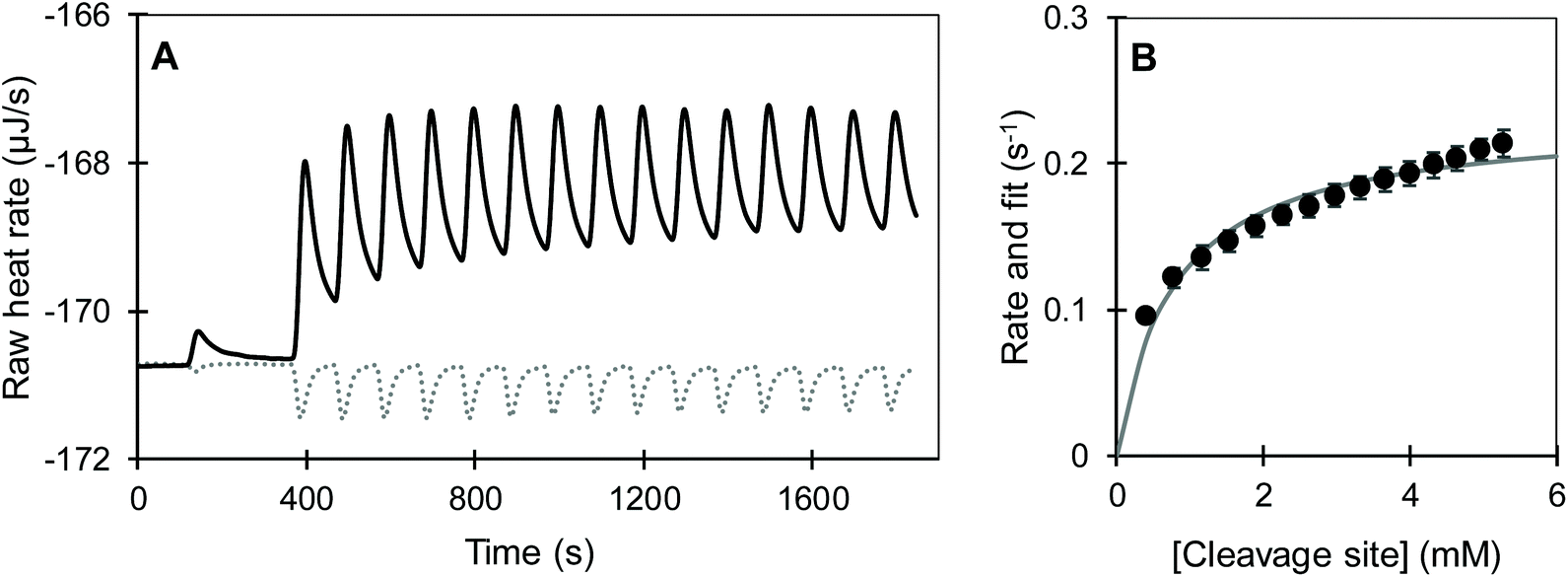 | ||
| Fig. 5 Multiple injection assay of pepsin-catalysed hydrolysis of BSA at pH 2. (A) Exemplar of calorimetric rate measurements of the blank injection of 300 μM BSA into phosphate buffer (dotted line) and reaction injection of BSA into 12.8 μM pepsin (solid line). A small volume was injected after the initial equilibration to account for the diffusion of substrate solution from the syringe into the cell. (B) Reaction rates estimated from three replicate measurements (circles), error bars represent standard deviation; line shows the fit to the Michaelis Menten model. | ||
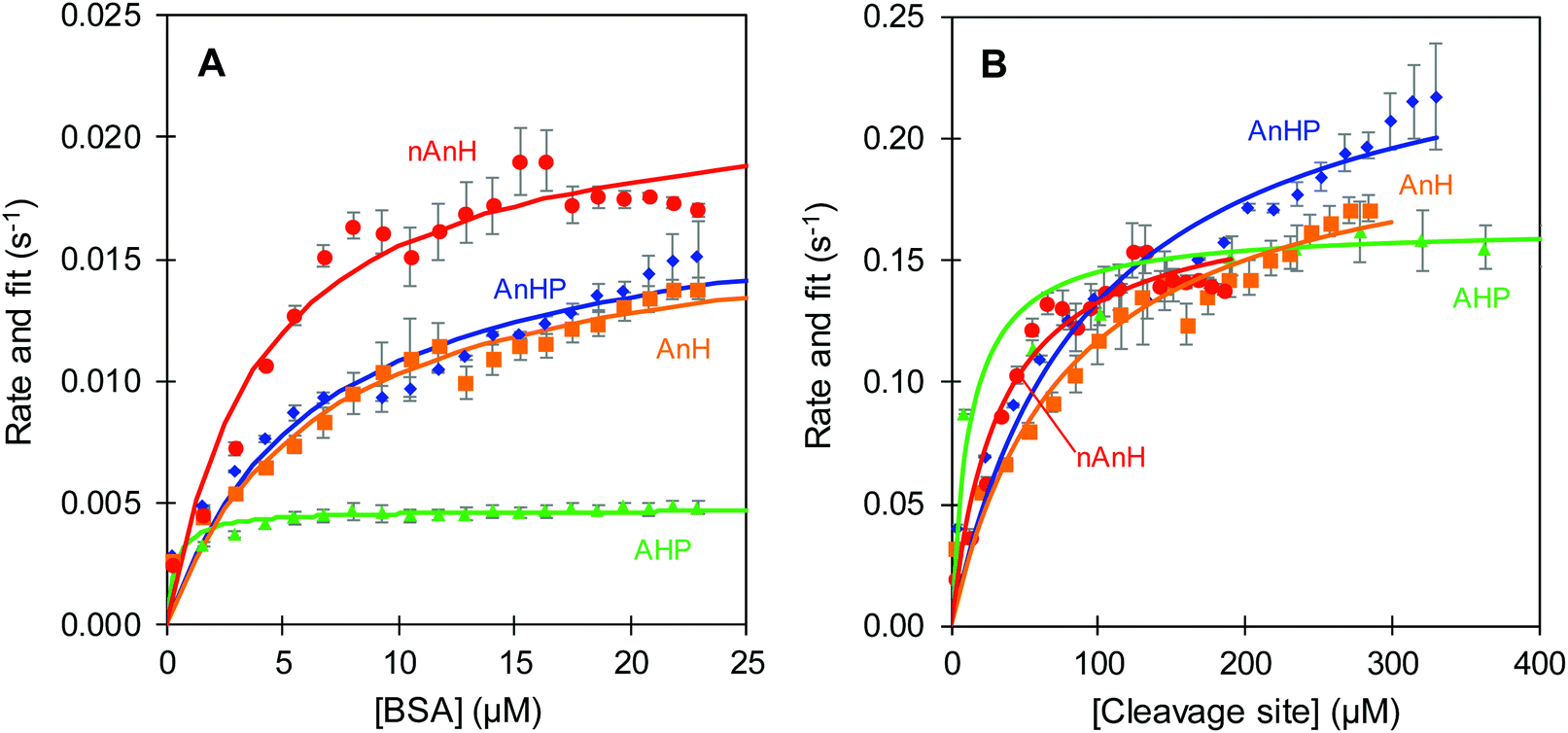 | ||
| Fig. 6 Reaction rate of trypsin-catalysed hydrolysis at pH 8 of BSA pepsin-hydrolysate (AHP, triangle), acidified, non-hydrolysed BSA with inactive pepsin (AnHP, diamond), acidified, non-hydrolysed BSA (AnH, square) and non-acidified, non-hydrolysed BSA (nAnH, circle). Error bars represent standard deviation from three replicate estimations. Fit to the Michaelis Menten model (line). Protein (A) and cleavage sites (B) considered as substrates, rates estimated from the appropriate ΔHapp (kJ mol−1 BSA or kJ mol−1 cleavage sites). The complete range of the BSA pepsin-hydrolysate reaction rate is shown in the ESI.† | ||
| Substrate (enzyme) | K m (μMcleavage sites) | k cat (s−1) | k cat/Km (1/s mM) | r 2 |
|---|---|---|---|---|
| A (P) | 776.5 (194.2) | 0.23 (0.01) | 0.3 | 0.980 |
| AHP (T) | 12.8 (4.9) | 0.16 (0.01) | 12.8 | 0.957 |
| AnHP (T) | 92.8 (35.1) | 0.26 (0.03) | 2.8 | 0.947 |
| AnH (T) | 79.1 (24.2) | 0.21 (0.02) | 2.6 | 0.966 |
| nAnH (T) | 33.2 (11.2) | 0.18 (0.02) | 5.3 | 0.960 |
A comparison was made between the initial reaction rates when protein or cleavage sites were considered as the substrate of trypsin-catalysed hydrolysis. In the former, there was a clear distinction between the hydrolysis rates for the different pH and hydrolysis history of the protein (Fig. 6A). The hydrolysis rate as a function of BSA concentration seemingly approached a rate plateau. The slowest turnover was observed for the BSA pepsin-hydrolysate at 0.004 s−1. It was followed by the previously acidified BSA, with and without inactive pepsin at 0.013 s−1. Lastly, BSA directly dissolved in pH 8 buffer was hydrolysed reaching the fastest maximum reaction rate of approximately 0.017 s−1. In the latter case, when the available cleavage sites were considered being the substrate of the reaction, the distinction between pH and hydrolysis history of BSA was far less evident and in some cases a plateau was not observed (Fig. 6B). The hydrolysis of the peptide bonds in the pepsin-hydrolysate had still the slowest turnover. Nevertheless, the previously acidified intact BSA (AnHP and AHP) and the basic BSA (nAnH) swapped positions, with AnHP and AHP seemingly having the fastest turnover. The Michaelis–Menten constant (Km) increased accordingly (Table 3). The magnitude of the rates increased by a factor of up to 30 when cleavage sites were considered as the substrate. This is not surprising as, in any case, there were more cleavage sites than moles of protein, which reduced the apparent enthalpy thus increasing the reaction rate: it can be expected that hydrolysing all the susceptible peptide bonds in a protein requires more time than a single cleavage site. Maximova and Trylska (2015) used ITC to study the trypsin-catalysed hydrolysis of casein and of a small substrate with one peptide bond that acted as a cleavage site, Nα-benzoyl-DL-arginine β-naphthylamide.13 They observed slower rates of hydrolysis for the protein compared to the single peptide bond. We make a similar comparison when considering BSA as a substrate as opposed to cleavage sites (Fig. 6).
The differences in kcat and Km for the trypsin-catalysed hydrolysis of BSA with varying pH and hydrolysis histories might be attributed to the denatured state and resulting average size of the proteins. We speculate that the intact, previously acidified BSA (AnH(P)) was less tightly refolded when the pH was increased to 8, where the folded state prevailed again.40 Therefore, we propose that the average size of AnH(P) was slightly larger than that of the protein which was not acidified (nAnH). Naturally, the average size of the pepsin-hydrolysate (AHP) would be even smaller due to the hydrolysis. A direct relationship between polypeptide chain length and hydrolysis rate has been long proposed.41,42 Further, according to the Km estimates, the affinity of trypsin towards smaller structures seems to be higher, with the BSA pepsin-hydrolysate (AHP) having lower Km compared to the intact protein (AnH(P) and nAnH). In this case, affinity might be a reflection of the poorer accessibility of larger structures to the active site of trypsin. Previous research has related the ease of dissociation of the enzyme–product complex with the volume of the product.43 It was suggested that a product of larger volume would be more readily released due to space repulsion.
As a plateau was not observed for the intact proteins, a more reliable measure of the hydrolytic efficiency towards the different substrates is the ratio of kcat over Km (Table 3). This ratio indicates how often a productive enzyme–substrate complex is formed; effectively, telling us how often an active site-bound sequence surrounding a susceptible peptide bond is broken. The cleavage sites of the BSA pepsin-hydrolysate (AHP) showed the best hydrolytic efficiency by trypsin. These were followed by those of BSA at pH 8 (nAnH), leading to the previously acidified BSA, with and without inactive pepsin (AnH(P)), as the least efficient substrate for trypsin-catalysed hydrolysis. As previously stated, we hypothesize that trypsin has a higher affinity towards the BSA pepsin-hydrolysate because of its smaller size and thus greater flexibility which allows easier exposure of cleavage sites, resulting in the most efficient trypsin-catalysed hydrolysis compared to the intact protein. For the intact protein, the initial exposure to low pH for AnH(P) had a significant impact in the hydrolytic efficiency compared to the protein dissolved directly in pH 8 (nAnH). Regardless of being the substrate with the lowest hydrolytic efficiency, previously acidified BSA could potentially reach faster rates at maximum.
The hydrolysis ratio was used to convert the protein concentrations to cleavage site concentrations. This approach corrects for the enzyme's real potential to hydrolyse certain peptide bonds of a given protein with its own pH and hydrolysis history. Nevertheless, it assumes an equal hydrolysis rate on any cleavage site, when in actual fact it will be an average rate for the various cleavage sites. Within a protein, there are multiple cleavage sites with their unique exposure and surrounding amino acid sequences. It is known that pepsin and trypsin-catalysed hydrolysis, after different amino acid sequences, results in a wide range of kinetic parameters.2,12,44–47
It has been suggested that pepsin-catalysed hydrolysis of a peptide bond within a protein is more efficient than of short, synthetic peptides.11 Our findings do not corroborate this. We believe this is because the different injection interval used in our assays. The kinetic parameters found in the current study lie between those of synthetic peptides with one cleavage site. While the specificities estimated in this study for pepsin and trypsin-catalysed hydrolysis of peptide bonds in BSA were larger than for some of the substrates reported in literature, they were still smaller than others (S 5, S 6). This illustrates how the parameters estimated in this study are averages that likely disguise the influence of the amino acid sequence surrounding the to-be-cleaved peptide bond and subsite binding to the enzymes’ active site. Hence, pepsin or trypsin catalysed hydrolysis of peptide bonds are not necessarily more efficient when located within a protein or within a short peptide. In our following work, we will address the role of the gastric phase on the gastro-duodenal digestion of other proteins. Future research should asses to what extent limited trypsin-catalysed hydrolysis might influence exopeptidase action and ultimately absorption of small peptides and amino acids.
Our findings have interesting implications in vivo, particularly for populations for whom optimal protein utilization is essential such as athletes or aging adults. In optimal conditions, parietal and chief cells secrete adequate amounts of acid and pepsinogen to reduce the pH and catalyse the hydrolysis of proteins in the stomach. This physiological function is affected by age. A reduced output of pepsinogen has been observed in healthy aging adults.48,49 In contrast, age alone does not seem to be a determining factor of acid hyposecretion or hypochloridria. Nevertheless, hypochloridria is often observed in cases of atrophic gastritis,50 which are somewhat common among the elderly.51 In any case, gastric pH is expected to rise with increasing bolus size, requiring a longer time and higher amount of acid secretion for pH to be low enough for pepsinogen activation. Perhaps by reducing meal size and thus bolus entering the stomach, the pH can be maintained low enough for pepsinogen activation, and for protein digestion to be closer to the optimum.
4. Conclusion
After consuming a meal with protein, some of it might bypass the gastric environment by being emptied quickly from the stomach, some might only be present in the stomach long enough to experience the acid pH but not pepsin-catalysed hydrolysis, and lastly, some, with the longest gastric residence time, might be acidified and hydrolysed by pepsin action. We show that there are real differences in the hydrolysis because of this. The gastric phase, and the history of the protein in terms of pH and pepsin-catalysed hydrolysis, influences the extent and efficiency of trypsin activity in small intestinal conditions. Sequential pepsin and trypsin-catalysed hydrolysis of BSA results in the most efficient overall hydrolysis regarding both degree and rate of hydrolysis. Bypassing gastric hydrolysis reduced the catalytic efficiency of trypsin. Interestingly, just exposure to acid ensued the least efficient substrate while increasing the peptide bonds susceptible to tryptic hydrolysis. We propose that conformation and peptide length, as a consequence of pH and hydrolysis history of the protein, are among the main determinants of the course of trypsin-catalysed hydrolysis. Our findings indicate that eating a large meal (causing a sharp temporary increase in pH) may result in a slower or possibly incomplete digestion of proteins. Similarly, protein digestion by elderly, who generally secrete less enzyme and acid, may also be compromised for the same reason.Author contributions
A. Rivera del Rio, conceptualization, formal analysis, investigation, methodology, visualization, writing –original draft. J. K. Keppler, conceptualization, methodology, writing –review & editing. R. M. Boom, conceptualization, supervision, writing –review & editing. A. E. M. Janssen, conceptualization, methodology, supervision, writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Consejo Nacional de Ciencia y Tecnología, Mexico (grant number 480085).References
- D. Piper and B. H. Fenton, pH stability and activity curves of pepsin with special reference to their clinical importance, Gut, 1965, 6, 506 CrossRef CAS PubMed.
- A. J. Cornish-Bowden and J. Knowles, The pH-dependence of pepsin-catalysed reactions, Biochem. J., 1969, 113, 353–362 CrossRef CAS PubMed.
- L. K. Christensen, Concerning the pH optimum of peptic hydrolysis, Arch. Biochem. Biophys., 1955, 57, 163–173 CrossRef CAS PubMed.
- D. Goldberg and K. Wormsley, The interrelationships of pancreatic enzymes in human duodenal aspirate, Gut, 1970, 11, 859–866 CrossRef CAS PubMed.
- J. Feher, Digestion and Absorption of the Macronutrients, in Quantitative Human Physiology, ed. J. Feher, Academic Press, Boston, 2012, pp. 731–743 Search PubMed.
- D. Gentilcore, T. Hausken, M. Horowitz and K. Jones, Measurements of gastric emptying of low–and high–nutrient liquids using 3D ultrasonography and scintigraphy in healthy subjects, Neurogastroenterol. Motil., 2006, 18, 1062–1068 CrossRef CAS PubMed.
- L. Marciani, N. Hall, S. E. Pritchard, E. F. Cox, J. J. Totman, M. Lad, C. L. Hoad, T. J. Foster, P. A. Gowland and R. C. Spiller, Preventing gastric sieving by blending a solid/water meal enhances satiation in healthy humans, J. Nutr., 2012, 142, 1253–1258 CrossRef CAS PubMed.
- A. Pal, J. G. Brasseur and B. Abrahamsson, A stomach road or “Magenstrasse” for gastric emptying, J. Biomech., 2007, 40, 1202–1210 CrossRef PubMed.
- T. Sipos and J. R. Merkel, Effect of calcium ions on the activity, heat stability, and structure of trypsin, Biochemistry, 1970, 9, 2766–2775 CrossRef CAS PubMed.
- M. J. Todd and J. Gomez, Enzyme Kinetics Determined Using Calorimetry: A General Assay for Enzyme Activity?, Anal. Biochem., 2001, 296, 179–187 CrossRef CAS PubMed.
- Q. Luo, D. Chen, R. M. Boom and A. E. M. Janssen, Revisiting the enzymatic kinetics of pepsin using isothermal titration calorimetry, Food Chem., 2018, 268, 94–100 CrossRef CAS PubMed.
- A. P. Lobo, J. D. Wos, M. Y. Sherman and W. B. Lawson, Active site studies of human thrombin and bovine trypsin: peptide substrates, Arch. Biochem. Biophys., 1976, 177, 235–244 CrossRef CAS PubMed.
- K. Maximova and J. Trylska, Kinetics of trypsin-catalyzed hydrolysis determined by isothermal titration calorimetry, Anal. Biochem., 2015, 486, 24–34 CrossRef CAS PubMed.
- K. Olsen, J. Otte and L. H. Skibsted, Steady-state kinetics and thermodynamics of the hydrolysis of β-lactoglobulin by trypsin, J. Agric. Food Chem., 2000, 48, 3086–3089 CrossRef CAS PubMed.
- B. Keil, Specificity of proteolysis, Springer Science & Business Media, 2012 Search PubMed.
- M. Ye, Y. Pan, K. Cheng and H. Zou, Protein digestion priority is independent of protein abundances, Nat. Methods, 2014, 11, 220–222 CrossRef CAS PubMed.
- P. Nielsen, D. Petersen and C. Dambmann, Improved method for determining food protein degree of hydrolysis, J. Food Sci., 2001, 66, 642–646 CrossRef CAS.
- T. R. Heyn, V. M. Garamus, H. R. Neumann, M. J. Uttinger, T. Guckeisen, M. Heuer, C. Selhuber-Unkel, W. Peukert and J. K. Keppler, Influence of the polydispersity of pH 2 and pH 3.5 beta-lactoglobulin amyloid fibril solutions on analytical methods, Eur. Polym. J., 2019, 120, 109211 CrossRef CAS.
- A. L. Robertson, S. J. Headey, N. M. Ng, L. C. Wijeyewickrema, M. J. Scanlon, R. N. Pike and S. P. Bottomley, Protein unfolding is essential for cleavage within the α-helix of a model protein substrate by the serine protease, thrombin, Biochimie, 2016, 122, 227–234 CrossRef CAS PubMed.
- E. Gasteiger, C. Hoogland, A. Gattiker, M. R. Wilkins, R. D. Appel and A. Bairoch, Protein identification and analysis tools on the ExPASy server, in The proteomics protocols handbook, Springer, 2005, pp. 571–607 Search PubMed.
- C. A. Royer, Probing Protein Folding and Conformational Transitions with Fluorescence, Chem. Rev., 2006, 106, 1769–1784 CrossRef CAS PubMed.
- P. M. Viallet, T. Vo-Dinh, A.-C. Ribou, J. Vigo and J.-M. Salmon, Native fluorescence and mag-indo-1-protein interaction as tools for probing unfolding and refolding sequences of the bovine serum albumin subdomain in the presence of guanidine hydrochloride, J. Protein Chem., 2000, 19, 431–439 CrossRef CAS PubMed.
- K. A. Majorek, P. J. Porebski, A. Dayal, M. D. Zimmerman, K. Jablonska, A. J. Stewart, M. Chruszcz and W. Minor, Structural and immunologic characterization of bovine, horse, and rabbit serum albumins, Mol. Immunol., 2012, 52, 174–182 CrossRef CAS PubMed.
- D. Sehnal, M. Deshpande, R. S. Vařeková, S. Mir, K. Berka, A. Midlik, L. Pravda, S. Velankar and J. Koča, LiteMol suite: interactive web-based visualization of large-scale macromolecular structure data, Nat. Methods, 2017, 14, 1121–1122 CrossRef CAS PubMed.
- M. Bhattacharya, N. Jain, K. Bhasne, V. Kumari and S. Mukhopadhyay, pH-induced Conformational Isomerization of Bovine Serum Albumin Studied by Extrinsic and Intrinsic Protein Fluorescence, J. Fluoresc., 2011, 21, 1083–1090 CrossRef CAS PubMed.
- H. Lan, H. Liu, Y. Ye and Z. Yin, The Role of Surface Properties on Protein Aggregation Behavior in Aqueous Solution of Different pH Values, AAPS PharmSciTech, 2020, 21, 1–13 CrossRef PubMed.
- E. Katchalski, G. S. Benjamin and V. Gross, The availability of the disulfide bonds of human and bovine serum albumin and of bovine γ-globulin to reduction by thioglycolic acid, J. Am. Chem. Soc., 1957, 79, 4096–4099 CrossRef CAS.
- T. Peters, The albumin molecule: its structure and chemical properties, in All about albumin, 1996, pp. 9–75 Search PubMed.
- N. Acharya, P. Mishra and S. K. Jha, Evidence for dry molten globule-like domains in the pH-induced equilibrium folding intermediate of a multidomain protein, J. Phys. Chem. Lett., 2016, 7, 173–179 CrossRef CAS PubMed.
- N. El Kadi, N. Taulier, J. Le Huerou, M. Gindre, W. Urbach, I. Nwigwe, P. C. Kahn and M. Waks, Unfolding and refolding of bovine serum albumin at acid pH: ultrasound and structural studies, Biophys. J., 2006, 91, 3397–3404 CrossRef CAS PubMed.
- R. Kun, M. Szekeres and I. Dékány, Isothermal titration calorimetric studies of the pH induced conformational changes of bovine serum albumin, J. Therm. Anal. Calorim., 2009, 96, 1009–1017 CrossRef CAS.
- S. Muzammil, Y. Kumar and S. Tayyab, Molten globule–like state of human serum albumin at low pH, Eur. J. Biochem., 1999, 266, 26–32 CrossRef CAS PubMed.
- P. R. Callis and T. Liu, Quantitative prediction of fluorescence quantum yields for tryptophan in proteins, J. Phys. Chem. B, 2004, 108(14), 4248–4259 CrossRef CAS.
- Y.-Q. Yeh, K.-F. Liao, O. Shih, Y.-J. Shiu, W.-R. Wu, C.-J. Su, P.-C. Lin and U.-S. Jeng, Probing the acid-induced packing structure changes of the molten globule domains of a protein near equilibrium unfolding, J. Phys. Chem. Lett., 2017, 8, 470–477 CrossRef CAS PubMed.
- J. Steinhardt, J. Krijn and J. G. Leidy, Differences between bovine and human serum albumins. Binding isotherms, optical rotatory dispersion, viscosity, hydrogen ion titration, and fluorescence effects, Biochemistry, 1971, 10, 4005–4015 CrossRef CAS PubMed.
- S. Servagent-Noinville, M. Revault, H. Quiquampoix and M.-H. Baron, Conformational changes of bovine serum albumin induced by adsorption on different clay surfaces: FTIR analysis, J. Colloid Interface Sci., 2000, 221, 273–283 CrossRef CAS PubMed.
- K. Maximova, J. Wojtczak and J. Trylska, Enzyme kinetics in crowded solutions from isothermal titration calorimetry, Anal. Biochem., 2019, 567, 96–105 CrossRef CAS PubMed.
- T. Kitson and J. Knowles, The inhibition of pepsin-catalysed reactions by structural and stereochemical product analogues, Biochem. J., 1971, 122, 241–247 CrossRef CAS PubMed.
- Y. Deng, C. I. Butré and P. A. Wierenga, Influence of substrate concentration on the extent of protein enzymatic hydrolysis, Int. Dairy J., 2018, 86, 39–48 CrossRef CAS.
- P. J. Sadler and A. Tucker, pH–induced structural transitions of bovine serum albumin: Histidine pKa values and unfolding of the N–terminus during the N to F transition, Eur. J. Biochem., 1993, 212, 811–817 CrossRef CAS PubMed.
- M. M. Nachlas, R. E. Plapinger and A. M. Seligman, Role of some structural features of subtrates on trypsin activity, Arch. Biochem. Biophys., 1964, 108, 266–274 CrossRef CAS PubMed.
- M. Pozsgay, G. C. Szabó, P. Elödi, R. Gáspár, S. Bajusz and R. Simonsson, Investigation of the Substrate–Binding Site of Trypsin by the Aid of Tripeptidyl–p–nitroanilide Substrates, Eur. J. Biochem., 1981, 115, 497–502 CrossRef CAS PubMed.
- D. Shi, Z. He and W. Qi, Lumping kinetic study on the process of tryptic hydrolysis of bovine serum albumin, Process Biochem., 2005, 40, 1943–1949 CrossRef CAS.
- R. M. Caprioli and L. Smith, Determination of Km and Vmax for tryptic peptide hydrolysis using fast atom bombardment mass spectrometry, Anal. Chem., 1986, 58, 1080–1083 CrossRef CAS.
- A. Cornish-Bowden, P. Greenwell and J. Knowles, The rate-determining step in pepsin-catalysed reactions, and evidence against an acyl-enzyme intermediate, Biochem. J., 1969, 113, 369–375 CrossRef CAS PubMed.
- T. R. Hollands and J. S. Fruton, Kinetics of the hydrolysis of synthetic substrates by pepsin and by acetyl-pepsin, Biochemistry, 1968, 7, 2045–2053 CrossRef CAS PubMed.
- G. P. Sachdev and J. S. Fruton, Pyridyl esters of peptides as synthetic substrates of pepsin, Biochemistry, 1969, 8, 4231–4238 CrossRef CAS PubMed.
- M. Feldman, B. Cryer, K. E. McArthur, B. A. Huet and E. Lee, Effects of aging and gastritis on gastric acid and pepsin secretion in humans: a prospective study, Gastroenterology, 1996, 110, 1043–1052 CrossRef CAS PubMed.
- A. Hurwitz, D. A. Brady, S. E. Schaal, I. M. Samloff, J. Dedon and C. E. Ruhl, Gastric acidity in older adults, J. Am. Med. Assoc., 1997, 278, 659–662 CrossRef CAS PubMed.
- E. J. Kuipers and T. A. Grool, The dynamics of gastritis, Curr. Gastroenterol. Rep., 2001, 3, 509–515 CrossRef CAS PubMed.
- X. Gao, Y. Zhang and H. Brenner, Associations of Helicobacter pylori infection and chronic atrophic gastritis with accelerated epigenetic ageing in older adults, Br. J. Cancer, 2017, 117, 1211–1214 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1fo00413a |
| This journal is © The Royal Society of Chemistry 2021 |