
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeanut sprout rich in p-coumaric acid ameliorates obesity and lipopolysaccharide-induced inflammation and the inhibition of browning in adipocytes via mitochondrial activation†
Seok Hee
Seo‡
a,
Sang-Mi
Jo‡
a,
Tien Thi My
Truong
b,
Guiguo
Zhang
c,
Dong-Shin
Kim
d,
Myoungsook
Lee
e,
Yunkyoung
Lee
ab and
Inhae
Kang
 *ab
*ab
aDepartment of Food Science and Nutrition, Jeju National University, Jeju 63243, Korea. E-mail: inhaek@jejunu.ac.kr
bInterdisciplinary Graduate Program in Advanced Convergence Technology and Science, Jeju National University, Jeju 63243, Korea
cCollege of Animal Sciences and Technology, Shandong Provincial Key Laboratory of Animal Biotechnology and Disease Control and Prevention, Shandong Agricultural University, 61 Daizong Street, Taian City, Shandong Province 271018, China
dDepartment of Food Bioengineering, Jeju National University, Jeju 63243, Korea
eDepartment of Food and Nutrition, Sungshin Women's University, Seoul 01133, Korea
First published on 13th May 2021
Abstract
Obesity is accompanied by adipose tissue inflammation that subsequently reduces thermogenic potential in brown and beige (brown-like) adipocytes. We previously reported that peanut sprout (PS) inhibited triglyceride accumulation via fatty acid oxidation in adipocytes. However, it is unknown whether PS reverses diet-induced obesity/inflammation and protects against the inflammation-induced inhibition of browning. To investigate this, C57BL/6 male mice, as an in vivo model, were randomly assigned to three different diets and fed for 8 weeks: (i) low-fat diet (LF, 11% kcal from fat), (ii) high-fat diet (HF, 61% kcal from fat), or (iii) HF diet with PS (4% PS in diet, HF + PS). As an in vitro model, lipopolysaccharides (LPS)-induced macrophages and 3T3-L1 adipocytes in the absence (white adipocytes) or presence of dibutyryl-cAMP (Bt-cAMP, beige adipocytes) were used. The supplementation of PS improved HF-diet-mediated body weight gain, dyslipidemia, and hyperglycemia as compared to the HF group. Although there was a marginal impact on visceral hypertrophy, PS reversed the adipocyte inflammation. In parallel, LPS-mediated induction of inflammation was impeded by PS extract (PSE) in macrophages and adipocytes. PSE also protected against LPS-induced suppression of adipocyte browning in Bt-cAMP-treated adipocytes with mitochondrial activation. The phenolic acid analysis showed that among the constituent of PSE, p-coumaric acid (PCA) was identified as a polyphenol that showed a similar effect to PSE. PCA treatment was also able to maintain a higher temperature than the control group upon cold exposure. Taken together, PCA-enriched PS attenuated HF-diet-induced obesity and protected against LPS-induced inflammation and the inhibition of browning via mitochondrial activation.
Introduction
Obesity is defined as an aberrant expansion of white adipose tissue (WAT) that causes metabolic disorders, such as type II diabetes and cardiovascular diseases.1 Energy imbalances, such as a high energy intake (i.e., a high fat (HF) diet) and low energy expenditure, are the main inducements for obesity and obesity mediated metabolic complications.2 The abnormal expansion of visceral adiposity is accompanied by chronic low-grade inflammation closely associated with adipocytes’ immune cell recruitment.3Unlike white adipose tissue (WAT), brown adipose tissue (BAT) has a unique thermogenic capacity resistant to obesity.4 BAT converts energy into heat in the process of non-shivering thermogenesis.5 There is a growing body of evidence that this thermogenic activity of BAT is facilitated by numerous mitochondria that are larger and contain packed cristae with uncoupling protein 1 (UCP1) expression in the inner membrane.6,7 Beige adipocytes, also known as brown-like white adipocytes, are found under conditions such as in response to cold or other stimuli,8,9 which burn energy like BAT for thermogenesis and subsequently control energy homeostasis.8 Given that obesity is considered to be a low-grade inflammatory condition in adipose tissue, browning (white adipocyte conversion into beige adipocytes) is inhibited by an inflammatory condition.10,11 Furthermore, immune stimulants, such as lipopolysaccharide (LPS) and LPS-derived cytokines have been shown to negatively impact the function of thermogenic activity.10,12,13 Thus, immunomodulatory components targeting the white, brown, and beige adipocyte inflammation could have potential therapeutic value for treating obesity.
Peanut (Arachis hypogaea L.) is a naturally occurring nutritious food containing significant amounts of protein, dietary fiber and fat, mostly unsaturated fatty acids.14 Recent evidence demonstrated that the bioactive components such as total phenolic content and trans-resveratrol are increased during peanut germination with high antioxidant activities.15,16 Germinated peanut seeds, called peanut sprout (PS), have recently been reported to have anti-inflammatory activity17–19via inhibiting mitogen-activated protein kinase (MAPK) and induction protein kinase B (PKB, also known as Akt)/nuclear factor erythroid 2-related factor 2 (Nrf2) signaling pathways.17,20 Recently, our group has reported that PS extract (PSE) at a concentration as low as 25 μg mL−1 was able to inhibit adipogenesis and lipid accumulation in mature adipocytes through the attenuation of mitochondrial fatty acid oxidation.21 For in vivo studies, HF diet-fed Sprague-Dawley rats administered PS 30 mg kg−1 showed reduced body weight (BW) gain and lipid levels compared to the HF diet control.22 Furthermore, PSE (0.05% PSE in HF diet) supplementation significantly reduced BW gain and fat mass with the reduction of peroxisome proliferator-activated receptors γ (PPARγ) expression in adipose tissue.23 Despite accumulating evidence indicating that PS affects many signaling pathways in various models, PS's roles in obesity and the signaling pathways of inflammation in distinct adipocytes are largely unknown. Therefore, we hypothesized that PS would prevent HF-diet-induced obesity and metabolic parameters. We also postulated that PS protects from LPS-mediated inflammation and inhibition of browning. To answer this hypothesis, HF-diet-fed C57BL/6 mice, LPS-induced murine macrophages and white/beige adipocytes (3T3-L1 adipocytes treated with or without dibutyryl-cAMP (Bt-cAMP)) were used as in vitro models. The major components responsible for mimicking the lipid-lowering characteristics of PS were also identified in this study.
Materials and methods
Experimental materials and sample preparation
All cell culture dishes and flasks were purchased from SPL (Seoul, Korea) unless otherwise stated. Dulbecco's Modified Eagle's Medium (DMEM), fetal bovine serum (FBS), and penicillin/streptomycin were purchased from Gibco (Grand Island, NY, USA). Rosiglitazone (BRL49653) was purchased from Cayman Chemical (Ann Arbor, MI, USA). All other chemicals and reagents were purchased from Sigma Chemical Co. (St Louis, MO, USA) unless otherwise stated.Peanut sprout (PS) was provided by WooYoung E&T (Jeju, South Korea). Freeze-dried PS was crushed into powder (14–50 mesh) and stored in a freezer at −80 °C for animal experiments (nutritional profiles of PS in Table S2†). PS extract was prepared as previously reported for cell culture.21 Briefly, 10 g of dried PS was extracted with 100 ml of Milli-Q water by using pressurized hot water extraction methods. Extracted samples were centrifuged, filtered, and lyophilized to obtain the powdered extract. The obtained samples were dissolved in dimethyl sulfoxide at a concentration of 100 mg ml−1 for experiments. p-Coumaric acid (PCA) was purchased from Sigma Chemical Co. (St Louis, MO, USA). We freshly diluted 40 mg mL−1 PCA stock with phosphate-buffered saline (PBS) before use for animal experiments.
Animals and diets
All protocols and procedures were approved by the Institutional Animal Care and Use Committee at the Jeju National University (Approval ID # 2019-0004). All experiments were performed in compliance with national laws and all experiments followed institutional guidelines. C57BL/6J 6-week-old mice and Balb/c 7-week-old mice were purchased from the ORIENT BIO Animal Center (Seongnam-si, Korea) and housed in a dark/light cycle at Jeju National University. For the PS animal experiments, the C57BL/6 mice were randomly assigned after one week of acclimation to one of three experimental groups fed with different diets ad libitum: low-fat diet (11% kcal from fat, LF group, n = 4), high-fat diet (61% kcal from fat, HF group, n = 8), or HF plus 4% of PS powder (HF + PS group, n = 8) and fed for 8 weeks (4 animals per cage). Diet preparation was adapted from the AIN-93G diets. The AIN93G diet was used as a LF control diet, and the HF diet was modified from AIN93G and a typical HF diet (60% kcal from fat) (Table S1†).24,25 For the PCA animal experiments, 7-week-old Balb/c mice were fed with a HF diet with either 100 μL vehicle (PBS, n = 4) or PCA (50 mg per kg body weight (BW), n = 5) injection for 7 days. To measure the thermogenic potential, the mice were exposed to cold temperatures (8 °C) for 24 h based on the previous report.26 The body temperature was measured using an infrared (IR) thermometer (AD-801, Zhengzhou AiQURA Intelligent Technology Co., Henan, China). An IR camera (FLIR E5, FLIR® Systems, Wilsonville, OR, USA) was used to detect and capture the thermal release in the body surface as previously described.10 Surface heat release temperatures between 20–35 °C were displayed using the FLIR Research IR software.Serum biochemistry and glucose tolerance test
After completing the experiment, the experimental animals were fasted for 12 hours and were sacrificed by carbon dioxide narcosis. Blood was collected from a cardiac puncture, and serum samples were aliquoted. The serum total cholesterol (TC, mg dL−1) level was analyzed using an enzyme assay kit (Asan Pharmaceutical Co., Seoul, Korea) with absorbance at 550 nm. For the glucose tolerance test (GTT), mice were fasted (12 h) before intraperitoneal injection of 10% D-glucose solution (0.5 g per kg BW). The blood glucose levels were measured before the injection and at 10, 20 30, 60, and 120 min after injection using a glucose meter (Midium Blood Glucose Analyzer, Kia Ace Co., Ltd, Gyeonggi, Korea).Hematoxylin and eosin (H&E) staining and adipocyte size quantification
Upon necropsy, epididymal adipose tissue was collected from the mice and flash-fixed in 10% buffered formalin. The paraffin-embedded adipose tissues were cut into 5–7 μm sections and processed for hematoxylin and eosin (H&E) staining, as described previously.27 Bright-field images were obtained by an Invitrogen microscope (Invitrogen™ EVOS™ FL Digital Inverted Fluorescence Microscope, Invitrogen, CA, USA) under 10× magnification. H&E-stained sections of epididymal adipose tissue were quantified for measuring adipocyte size. Image J and Adiposoft from the National Institutes of Health (NIH) were used for the adipocyte size quantification.2,2-Diphenyl-1-picryl-hydrazyl-hydrate (DPPH) of PSE and PCA
The DPPH radical scavenging activity was measured by the previously described method with some modifications.28,29 Briefly, DPPH was dissolved in ethanol to a 0.2 mM concentration. In a 96-well plate, 25 μL of the samples (6 mg mL−1 of PSE or PCA), and 175 μL of DPPH solution were incubated at 37 °C for 30 min. The absorbance of the samples was measured at 517 nm. Ascorbic acid (6 mg mL−1) was used as a positive control. The free radical scavenging activity (DPPH) was calculated according to the following formula: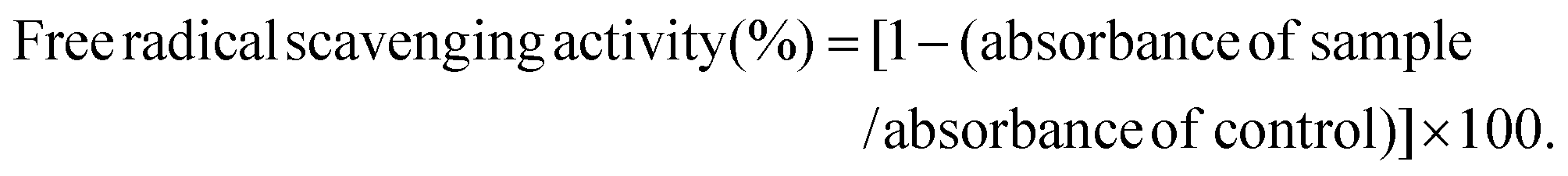
Quantification of phenolic acid components in peanut sprout extracts
Here, 100 mg PSE samples were mixed with 2 mL 1 N sodium hydroxide (NaOH) and kept for 2 h at 28 °C. A total of 500 μL 5 M hydrochloric acid was added to the sample. The samples were shaken and mixed, and then we added 2 mL ethyl acetate to the samples. The samples were centrifuged at 4000 rpm for 5 min. The ethyl acetate phase was collected in new 15 mL centrifuge tubes (repeated three times). The ethyl acetate was dried with nitrogen and dissolved in 0.5 mL methanol.Phenolic acid components were quantified using an Ultra-Performance Liquid Chromatography (UPLC) system. The analysis was performed with a Waters HSS T3 (100 × 2.1 mm, 1.8 μm) liquid chromatography column (AcQuity UPLC, Waters, USA) at a column temperature of 40 °C (mobile phase A: 0.1% formic–acetonitrile/mobile phase B: 0.1% formic–water). The sample was analyzed in single-ion detection (SIM) mode using an electrospray ion source (ESI), with simultaneous anion scanning, which can significantly improve the sensitivity. The optimized mass spectrometry analysis conditions were as follows: sheath gas 40 °C; auxiliary gas 10 °C; Ion spray voltage +3000 V; temperature 350 °C; and temperature of the ion transport tube 320 °C.
Cell culture (3T3-L1, Raw264.7 cells, bone marrow-derived macrophage (BMDM) and oil-red-O staining (ORO)
The 3T3-L1 cells were purchased from American Type Culture Collection (ATCC, VA, USA) and grown as described previously.21 Briefly, cells were cultured in a basal medium—DMEM with penicillin/streptomycin and 2 mM L-glutamine (Sigma, St Louis, MO, USA)—supplemented with 10% newborn calf serum (Hyclone, USA) until confluence. Two days after the cells reached confluence (referred to as d 0), the cells were differentiated into adipocytes in a basal medium containing 10% fetal bovine serum (FBS, Gibco, Grand Island, NY, USA), 1 μM dexamethasone (Sigma, St Louis, MO, USA), 0.5 mM 3-isobutyl-1-methylxanthine (Sigma, St Louis, MO, USA), and 2 nM insulin (Sigma, St Louis, MO, USA) for 48 h. Then, fresh medium containing 2 nM of insulin was added to the cells for 48 h. The cells were subsequently maintained for up to 7–14 days with a change of media (10% FBS without insulin) every other day. To measure the lipid accumulation of PCA in adipocytes, 3T3-L1 cells were treated with or without PCA for 3–7 days (adipogenesis for 7 days, lipogenesis for 3–4 days), then stained with oil red O (ORO) as described previously.21RAW264.7 murine macrophage-like cells were cultured as previously described.30 The cells were grown in DMEM supplemented with 10% FBS and 1% (v/v) penicillin (100 U mL−1)/streptomycin (100 μg mL−1) under humidified conditions of 5% CO2 at 37 °C.
The bone marrow was collected from the femurs and tibias of the C57BL/6 mice, modified from a previous report.30 The isolated cells were suspended in DMEM containing 20% FBS, penicillin (100 U mL−1), and streptomycin (100 μg mL−1), in the presence of 30% L929-conditioned medium. BMDM were induced to differentiation using the same media as for culturing BMDM to form a monolayer for 7–10 days.
The interscapular brown adipose tissue (iBAT)-derived primary brown adipocytes were isolated and differentiated as described previously.31 Briefly, for the adipocyte differentiation, 20 nM of insulin, 1 μM of triiodothyronine, 0.5 mM isobutylmethylxanthine, 5 μM dexamethasone, 125 μM indomethacin, and 0.5 μM rosiglitazone were added to cells. Then, fresh medium containing 2 nM of insulin and 1 μM of triiodothyronine was added to the cells.
Oxygen consumption rate by Seahorse extracellular flux analyzer
The oxygen consumption rate (OCR) in the 3T3-L1 adipocytes was measured using an XF24 extracellular flux analyzer (Agilent Technologies, Santa Clara, CA, USA/Bio-Health Materials Core-Facility at Jeju National University) for measuring the mitochondrial respiration activities as previously described.21 For the PSE experiment, 3T3-L1 preadipocytes were seeded in an XFe analyzer microplate (24-well) and cultured until they reached confluency. 3T3-L1 cells were induced into differentiation with or without PSE for 7 d, followed by LPS (100 ng mL−1) treatment for 48 h and Bt-cAMP (50 mM) for 6 h. For the PCA experiment, 3T3-L1 were differentiated to become mature adipocytes until d7. Fully differentiated adipocytes (d7) were incubated in the presence or absence of PCA (200 μM) for four days. The cell medium was then exchanged for XF base medium before the experiments supplemented with 10 mM glucose, 2 mM glutamine and 1 mM of pyruvate. In OCR measurements, cells were treated with oligomycin (oligo, 2 μM) to measure the adenosine triphosphate (ATP) turnover. The maximum respiratory capacity was evaluated using carbonyl cyanide 4-trifluoromethoxy phenylhydrazone (FCCP, 0.5 μM). Then, the mitochondrial oxygen consumption was blocked by a combination of antimycin A (1 μM) and rotenone (1 μM) (A + R).Fatty acid oxidation rate by using radioactive [3H]-oleic acid (OA)
The fatty acid (FA) oxidation rate was measured using a radioactive precursor, [3H]-OA (PerkinElmer, Norwalk, CT, USA; with a final concentration of 0.5 μCi mL−1) as previously described,21,32 in mature adipocytes and human hepatoma cells. Briefly, 200 μM of PCA was used for four days on mature 3T3-L1 adipocytes. HepG2 cells were pre-incubated with PCA (200 μM) or DMSO for 48 h. The cells were provided with serum-free low glucose (1000 mg L−1D-(+)-glucose) medium overnight before the experiment. The bovine serum albumin (BSA)-sodium oleate complex (400 μM) with [3H]-OA was added to cells and incubated for 2 h. The medium was harvested and precipitated using 100% trichloroacetic acid (TCA) solution. We added 6 N NaOH to the precipitated medium to obtain an alkaline supernatant. The supernatant was run through columns filled with Dowex ion-exchange resin (Acros Organics, Geel, Belgium) to capture [3H]-H2O. The radioactivity was measured by MicroBeta Microplate counters (PerkinElmer, (Norwalk, CT, USA)).Analysis of mRNA by real-time polymerase chain reaction (real time-PCR)
After the end of the experiment, 0.2 g of the epididymal fat tissue was stored rapidly in the freezer and cells were subjected to RNA extraction using Trizol reagent (Invitrogen Co., Carlsbad, CA, USA). cDNA was synthesized by high-capacity cDNA reverse transcription kits (Applied Biosystem, USA) after measuring the extracted RNA concentration using a Nano-200 Micro-Spectrophotometer (Hangzhou City, China). Gene expression was determined by real-time PCR (CFX96™ Real-Time PCR Detection System, Bio-Rad, USA). The relative gene expression was normalized using hypoxanthine–guanine phosphoribosyltransferase (HPRT) and/or ribosomal protein lateral stalk subunit P0 (RPLP0, 36B4) (Cosmo Genetech, Table S3†).Protein Isolation and western blotting
Harvested tissue samples were homogenized with a homogenizer in an ice-cold radioimmunoprecipitation assay (RIPA) lysis buffer (Thermo Fisher Scientific, MA, USA) with a protease and phosphatase inhibitor cocktail (Sigma, St Louis, MO, USA) and centrifuged to collect the supernatant. We separated 10–12 μg of protein using 10% SDS-PAGE and transferred the protein to a polyvinylidene difluoride (PVDF) membrane (Thermo Fisher Scientific, MA, USA) with Tris-buffered saline/Tween 20 and blocked for 1 hour at room temperature. The membranes were washed several times with Tris-buffered saline with Tween 20 (TBST) solution and reacted overnight at 4 °C with primary antibodies against fatty acid synthase (FAS), PPARγ, uncoupling protein 1 (UCP1), β-actin (Cell signaling Technology, Danvers, MA, USA), and adipocyte Protein 2 (aP2, FABP4) (Santa Cruz Biotechnology, CA, USA). The next day, the membrane was washed several times and then the secondary antibody (goat anti-rabbit, Cell Signaling Technology, Danvers, MA, USA or goat anti-mouse IgG-HRP, Santa Cruz Biotechnology, CA, USA) and reacted with the membrane for one hour. The membranes were washed and reacted with enhanced chemiluminescence (ECL) reagent (PerkinElmer, Waltham, Mass.) To identify the band, ChemiDoc MP (Bio-Rad, CA, USA) was used, and the expression level was calculated using Image Lab (Bio-Rad, CA, USA) or Image J (NIH, MD, USA).Statistical analysis
The experimental results are expressed as the mean ± standard error of the mean (SEM). Statistical calculations were performed using ANOVA (one-way analysis of variance) with Bonferroni's multiple comparison test or Student's t-test. P-Value <0.05 was considered statistically significant. All analyses were performed with GraphPad Prism 7.0 (La Jolla, California., USA.Results
Peanut sprout (PS) supplementation ameliorates HF-diet-mediated abnormal metabolic parameters and adipose tissue inflammation
We first investigated whether peanut sprout (PS) supplementation altered HF-diet-induced obesity. As seen in Fig. 1a and b, mice fed with a HF diet significantly promoted BW and BW gain in comparison to the LF diet group, which was partially normalized in mice fed with HF + PS without change in the food intake (Fig. 1d). Liver and visceral fat (epididymal fat) mass did not reach statistical significance between the HF vs. HF + PS group (Fig. 1c). Despite no significance in organ weight, the elevated levels of serum total cholesterol (TC) were normalized by PS supplementation comparable to the LF control group (Fig. 1e).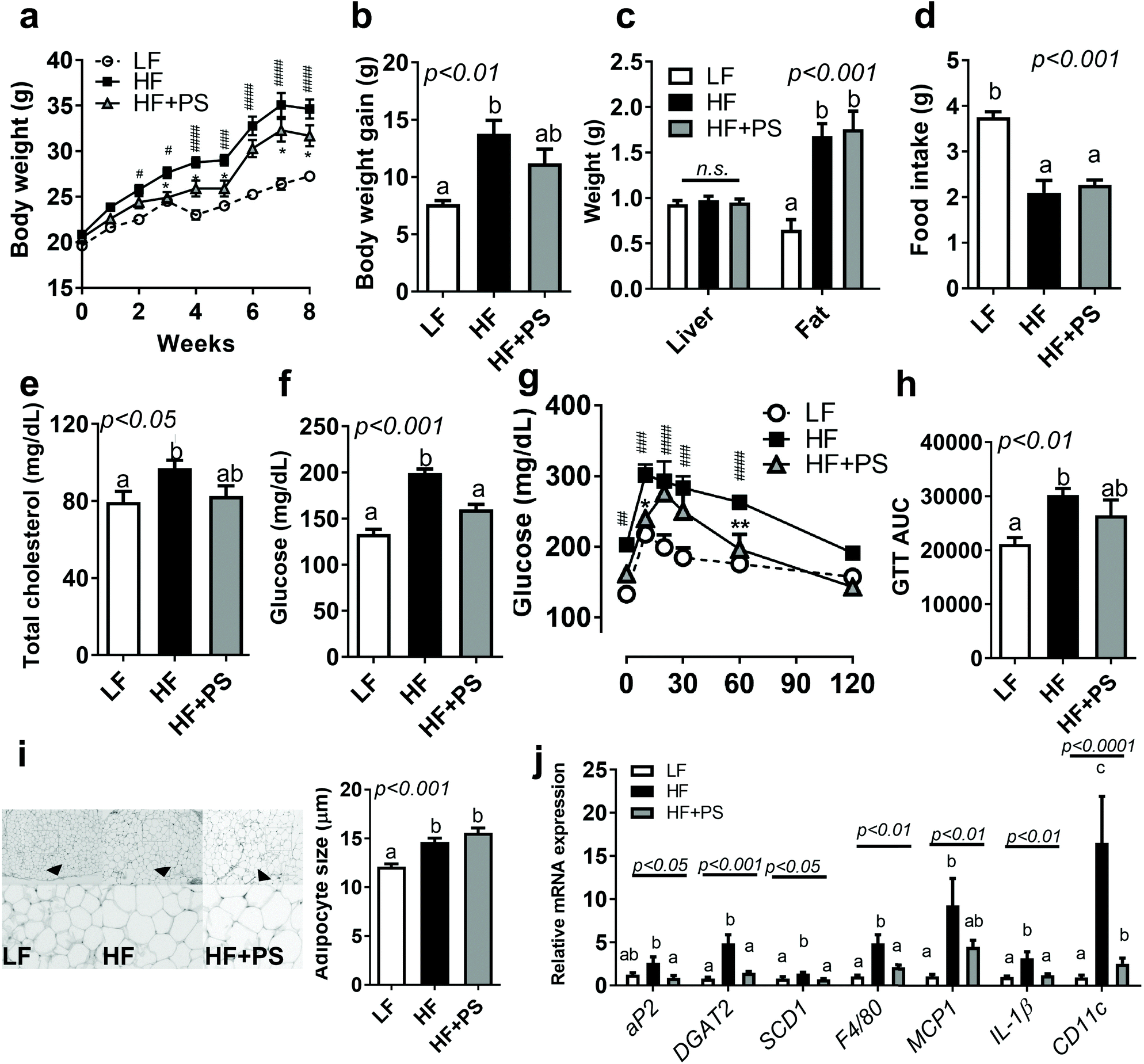 | ||
| Fig. 1 Peanut sprout (PS) supplementation ameliorated high fat (HF)-diet-mediated abnormal metabolic parameters and adipose inflammation. Seven-week-old male C57BL/6 mice were fed with low-fat (LF) (white circle, white bar), HF (black square, black bar), or HF + PS (grey triangle, grey bar) diets for 8 weeks (n = 4–8 per group). (a) Body weight (g); (b) body weight gain (g); (c) organ weight (g); (d) food intake (g); (e) fasting total cholesterol levels (mg dL−1); (f) fasting glucose (mg dL−1); (g) intraperitoneal glucose tolerance tests (GTT); (h) area under the curve (AUC) of GTT; (i) hematoxylin and eosin (H&E) staining of adipose tissue (magnified 10×) and adipocyte size (μm); (j) mRNA expression of lipogenic genes, aP2, DGAT2, and SCD1/inflammatory mRNA expression, F4/80, MCP-1, IL-β, and CD11c. Data are expressed as mean ± SEM (n = 4–8). Bars with different letters are significantly different by one-way ANOVA with Bonferroni's comparison test. n.s. represents no significance. #p < 0.05; ##p < 0.01; ###p < 0.001; ####p < 0.0001 (LF vs. HF), * p < 0.05; ** p < 0.01; and *** p < 0.001 (HF vs. HF + PS). | ||
We investigated whether PS modulated HF-mediated hyperglycemia. The HF-diet-fed mice exhibited higher fasting glucose levels than the LF-diet-fed mice, which was significantly improved by PS supplementation (Fig. 1f). Consistent with this, glucose disposal was markedly slower in the HF group than the LF group during the glucose tolerance test (GTT), which was partially attenuated by the HF + PS diet (Fig. 1g and h). The data suggested that PS supplementation caused resistance to HF-mediated obesity/dyslipidemia with blood glucose control.
Next, we examined the adipocyte morphology with lipogenic gene expression. Adipocyte hypertrophy was induced by the HF diet as compared to LF control. PS did not alter adipose expansion (Fig. 1i), which may reflect no changes in visceral adipose mass between HF vs. HF + PS (Fig. 1c). Despite no impact on adipocyte size, lipogenic gene expressions, adipocyte protein 2 (aP2, FABP4), diacylglycerol O-acyltransferase 2 (DGAT2), and stearoyl-coenzyme A desaturase-1 (SCD1) were significantly decreased with PS supplementation (Fig. 1j). Aberrant mRNA expressions of F4/80, monocyte chemoattractant protein-1 (MCP1), interleukin-1β (IL-1β) and CD11c, which were mediated by the HF diet, were completely diminished with PS supplementation (Fig. 1j).
PS extract (PSE) reduces lipopolysaccharide (LPS)-mediated inflammatory responses in macrophages and adipocytes
To gain additional insights into the underlying mechanisms of PS on inflammatory responses, we adapted murine macrophage models by using RAW264.7 cells and bone marrow-derived macrophages (BMDM). PS was extracted using a pressurized hot water extraction method as described previously21 and used to treat murine macrophage models. To determine whether PS extract (PSE) has cytotoxic effects in cells, the PSE (10–100 μg ml−1) was used to treat macrophages. The PS extract did not affect cell viability in RAW264.7 and BMDM up to a concentration of 50 μg mL−1 (Fig. 2a). Based on these results, we used 25 μg mL−1 of PSE, which we used for the previous report, for the rest of the cell-culture experiment. Upregulated levels of interleukin 6 (IL-6), tumor necrosis factor-alpha (TNFα), IL-1β, and MCP1 by lipopolysaccharide (LPS) treatment (1 μg mL−1) were attenuated by PSE treatment in RAW264.7 cells (Fig. 2b). Consistently, LPS (100 ng mL−1)-induced inflammatory gene expressions were attenuated by 25 μg mL−1 of PSE in BMDM (Fig. 2c). PSE also reduced the TNFα-gene expression in LPS-treated 3T3-L1 adipocytes (Fig. 2d). Adipocytes and macrophages create crosstalk in obesity-mediated inflammatory and insulin resistance status.33 To evaluate the association between macrophages and adipocytes, we exposed macrophage conditioned media (CM) to 3T3-L1 adipocytes with or without PSE (Fig. 2e). Macrophage conditioned media (mΦ CM) significantly induced TNFα, and IL-1β mRNA expressions. Adipocyte cultured with PSE and adipocyte exposed to PSE-pretreated CM significantly reduced TNFα, and IL-1β mRNA expression, respectively (Fig. 2f). Taken together, the data suggest that PS and/or PSE contribute to a reduction in inflammation in macrophages and adipocytes mediated by a HF diet and/or LPS.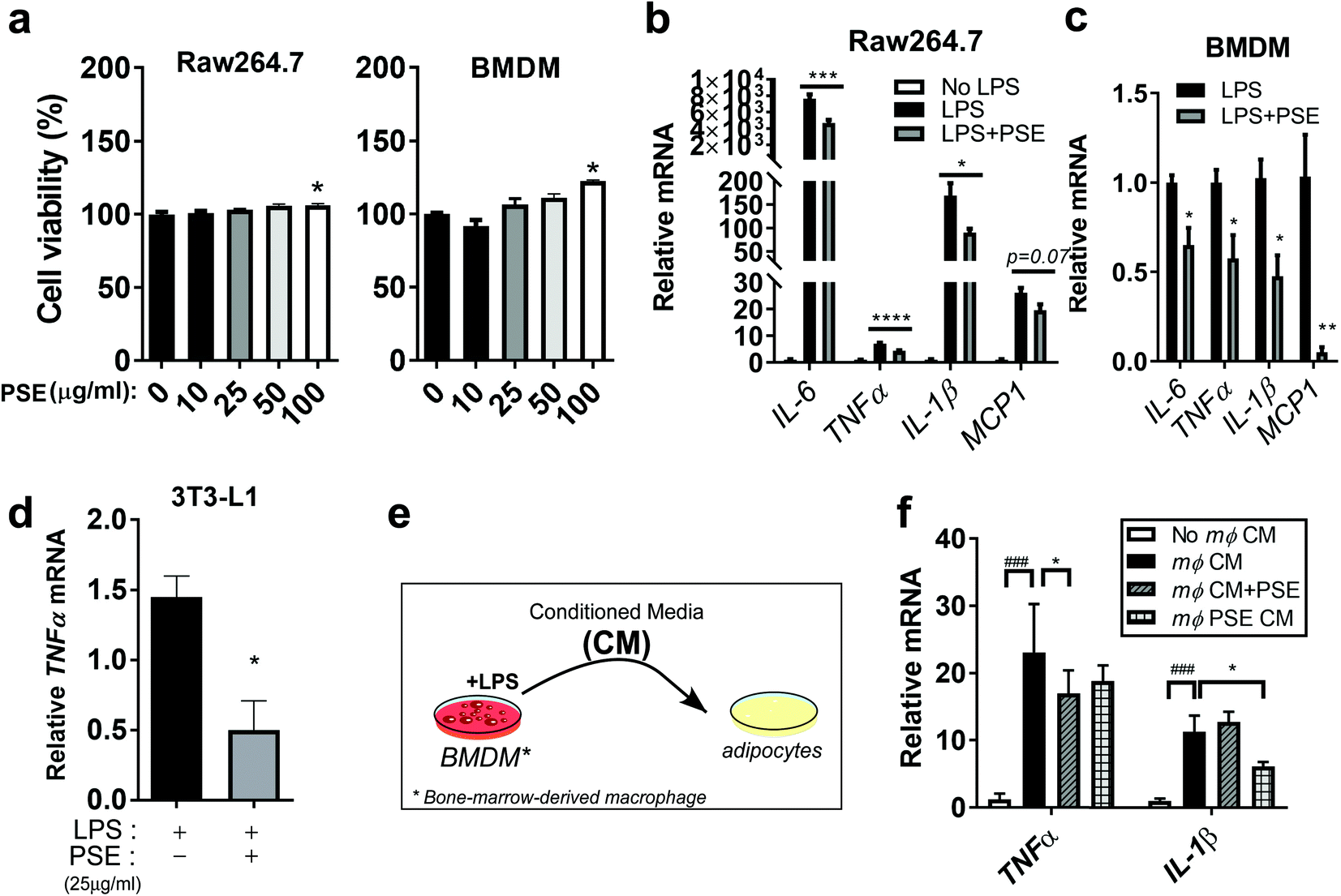 | ||
| Fig. 2 PS extract (PSE) reduces lipopolysaccharide (LPS)-mediated inflammatory responses in macrophages and adipocytes. RAW264.7 cells and BMDMs were treated with 10–100 μg mL−1 of PSE for 24 h. 2,3-Bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) reagent was added 3 h before OD measurement at 450 nm. (a and b) Cell viability assay in RAW264.7 cells and BMDMs; RAW264.7 cells and BMDMs were pretreated with PSE (25 μg mL−1) for 48 h in FBS-containing completed media and then starved for 12–18 h in DMEM. LPS stimulation in each cell (1 μg mL−1 for RAW264.7 cells or 100 ng mL−1 for BMDM) was conducted for 2 h in the presence or absence of PSE; (b) IL-6, TNFα, IL-1β, and MCP1 gene expression by qPCR in RAW264.7 cells; (c) IL-6, TNFα, IL-1β, and MCP1 gene expression by qPCR in BMDM cells; differentiated 3T3-L1 adipocytes in the presence or absence of PSE (25 μg mL−1) for 7 days. Cells were starved in DMEM for 12–18 h, followed by LPS (100 ng mL−1) stimulation for 2 h; (d) TNFα gene expression by qPCR. 3T3-L1 adipocytes were with or without PSE. The cells received macrophage (mΦ) CM of LPS-treated BMDMs with or without PSE; (e) experimental scheme; (f) TNFα and IL-1β gene expressions by qPCR. All values are presented as the mean ± SEM (n = 3–4 per group for each experiment). n.s. represents no significance. ### p < 0.001; compared with non-mΦ CM vs. mΦ CM. * p < 0.05; *** p < 0.001; and **** p < 0.0001 compared with the LPS-treated group or mΦ CM vs. PSE-treated group by one-way ANOVA with Bonferroni's comparison test or Student's t-test. +, treatment; −, non-treatment. | ||
PSE protects from LPS-induced adipocyte browning inhibition
Previously, our group reported that PSE was involved in white adipocyte browning.21 By using the same beige adipocyte mimicking cells, beige-specific transcript, CD137, transmembrane protein 26 (TMEM26), and short stature homeobox 2 (Shox2) were measured in Bt-cAMP-treated adipocytes. PSE was able to induce TMEM26 and Shox2, but not CD137. BAT-specific markers, PGC1α and UCP1, were re-analyzed and PSE significantly upregulated UCP1 in Bt-cAMP-treated adipocytes (Fig. 3a). We then wondered whether PSE would promote brown adipogenesis. To test this hypothesis, murine primary brown adipogenic precursor cells from brown adipose tissue were treated with 25 μg mL−1 of PSE during brown adipogenesis. Although PSE did not enhance the UCP1 protein levels, there was a significant increase in cell death-inducing DNA fragmentation factor alpha-like effector A (CIDEA, p < 0.05) mRNA expression primary brown adipogenic markers as compared with vehicle control. The UCP1 level was partially increased in the PSE group (p = 0.07) (Fig. 3b and c).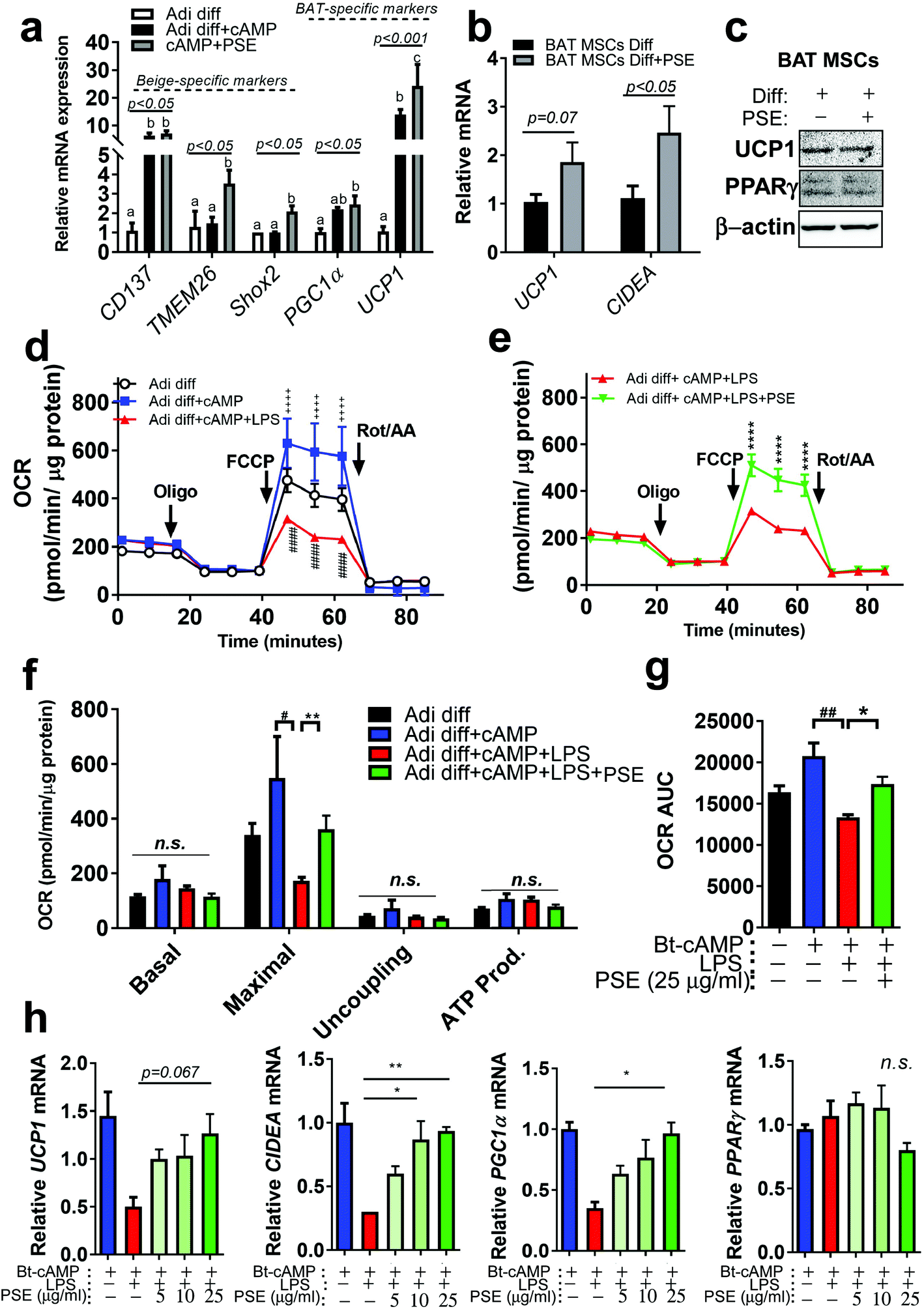 | ||
| Fig. 3 PSE protects from LPS-induced adipocyte browning inhibition. 3T3-L1 cells induced into differentiation with or without PSE for 7 d followed by Bt-cAMP (50 mM) for 6 h. 3T3-L1 adipocytes treated with Veh (Adi diff), Bt-cAMP (Adi diff + cAMP), Bt-cAMP + PSE (cAMP + PSE); (a) relative mRNA levels of CD137, TMEM26, Shox2, PGC1α, and UCP1 by real-time PCR. Brown progenitor cells were isolated from interscapular brown adipose tissue (iBAT) of C57BL/6 mice and differentiated into brown adipocytes with or without PSE (25 μg mL−1) for 10 d; (b) relative expressions of UCP1 and CIDEA by qPCR; (c) protein expressions of UCP1 and PPARγ by western blot; 3T3-L1 cells induced into differentiation with or without PSE for 7 d, followed by LPS (100 ng mL−1) treatment for 48 h and Bt-cAMP (50 mM) for 6 h. 3T3-L1 adipocytes treated with Veh (white circle), Bt-cAMP (blue square), Bt-cAMP + LPS (red triangle), or Bt-cAMP + LPS + PSE (green triangle) are shown as black, blue, red, and green bars; (d and e) the oxygen consumption rate (OCR) determined with a Seahorse extracellular analyzer. The respiratory inhibitors, oligomycin (Oligo), carbonyl cyanide 4-trifluoromethoxy phenylhydrazone (FCCP) and a combination of antimycin A and rotenone (Rot/AA) are indicated with arrows; (f and g) OCR profiles and AUC analysis from (d) and (e); (h) relative mRNA levels of UCP1, CIDEA, PGC1α, and PPARγ by real-time PCR. Dimethyl sulfoxide (DMSO) was used as a vehicle control. All values are presented as the mean ± SEM. n.s. represents no significance. ++++ p < 0.0001 compared differentiated adipocytes (Adi diff) vs. Adi diff with Bt-cAMP. # p < 0.05; ## p < 0.01; and #### p < 0.0001 compared Adi diff + Bt-cAMP vs. Adi diff + Bt-cAMP + LPS. * p < 0.05; ** p < 0.01; and **** p < 0.0001 compared Bt-cAMP + LPS vs. Bt-cAMP + LPS + PSE by Student's t-test or one-way ANOVA with Bonferroni's comparison test. Means that do not share a common superscript are significantly different as determined by one-way ANOVA with Bonferroni's comparison test. +, treatment; −, non-treatment. | ||
Pro-inflammatory signaling is regarded as a negative regulator of thermogenesis of BAT and beige adipocytes.11,34 Since PSE has anti-inflammatory activity in macrophages and adipocytes (Fig. 2) and potential for browning21/brown adipocyte activation (Fig. 3a–c), we postulated that PSE could protect against LPS-mediated inhibition of adipocyte browning. Differentiated 3T3-L1 adipocytes treated with Bt-cAMP were used for mimicking beige adipocytes (brown-like adipocytes) as previously reported.21 LPS treatment in Bt-cAMP-treated adipocytes showed a significant reduction in mitochondrial bioenergetics, evidenced by the decrease in the maximal oxygen consumption rates (OCR). PSE treatment reversed the LPS-induced down-regulation of OCR (Fig. 3d–g). Downregulated browning markers, UCP1, CIDEA, and PGC1α by LPS, were recovered by PSE treatment in a dose-dependent manner (5–25 μg mL−1) (Fig. 3h). These data suggest that PSE increased the mitochondrial bioenergetics, which consequently stimulated adipocyte browning in LPS-treated adipocytes.
PS has natural bioactive components rich in p-coumaric acid with high antioxidant capacity
We then wondered what kind of bioactive components are responsible for PS's anti-obesity and adipocyte modulatory effects in vivo and in vitro (Fig. 1 and 2).21 We performed the content analysis for 15 phenolic acids using ultra-performance liquid chromatography (UPLC). PSE mainly contained p-coumaric acid, caffeic acid, and trans-ferulic acid with 429.90 μg per g fresh weight (F.W.), 222.52 μg per g F.W., and 56.84 μg per g F.W., respectively (Fig. 4a and b). We then tested the DPPH radical scavenging assay to determine the antioxidant capacity of PSE and PCA. The DPPH radical scavenging assay showed that PSE had two-times higher radical scavenging activities than PCA, which was around 43% of the activity as compared to ascorbic acid, a powerful antioxidant used as a positive control (Fig. 4c). PCA is the most abundant polyphenol among PSE's constituents; we regarded PCA as a major bioactive component with high anti-antioxidant properties in PSE.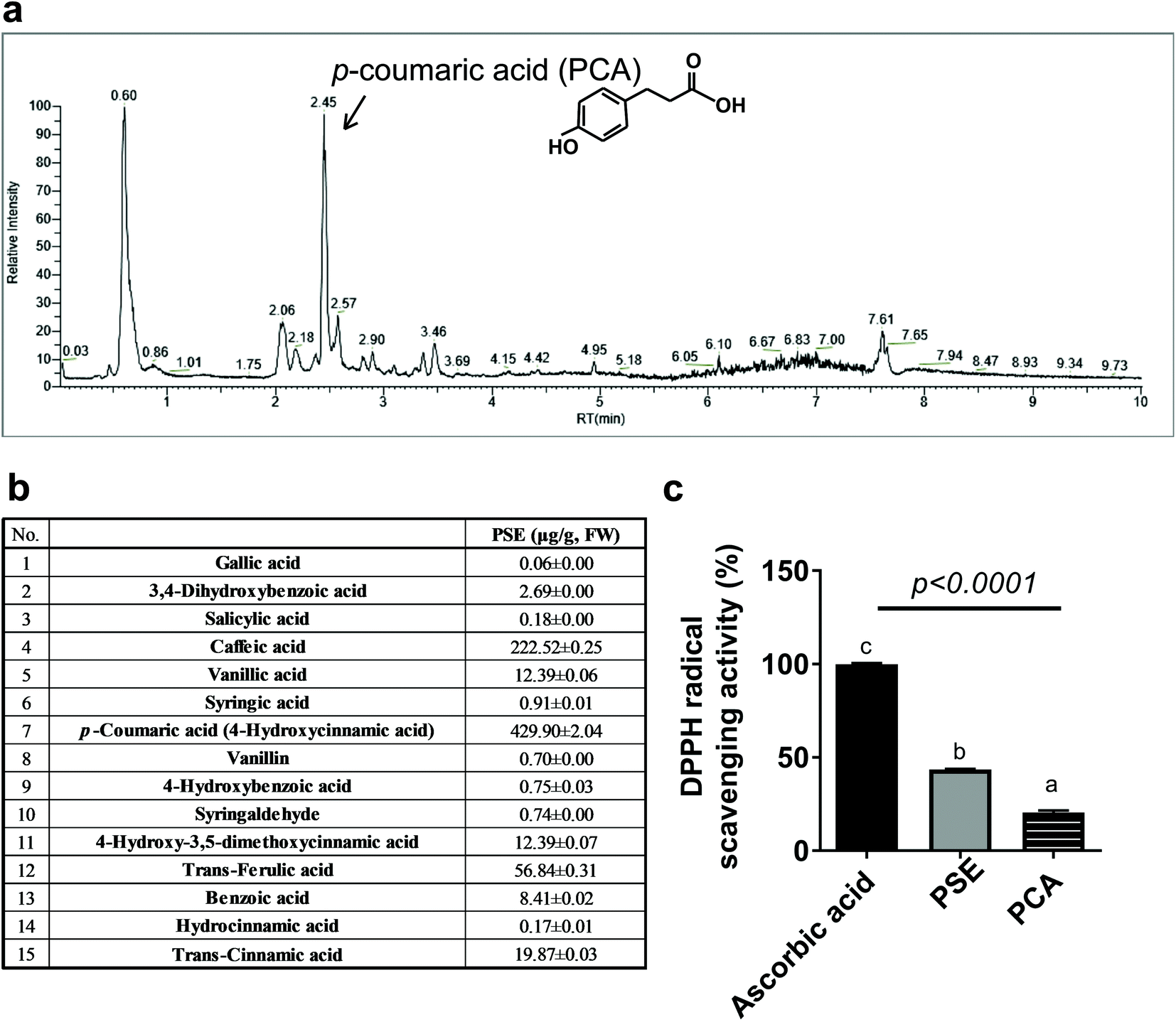 | ||
| Fig. 4 PS is a natural bioactive component rich in p-coumaric acid with high antioxidant capacity. (a and b) Identification of phenolic acid components by UPLC; (c) DPPH radical scavenging activity. All values are presented as the mean ± SEM. Means that do not share a common superscript are significantly different as determined by one-way ANOVA with Bonferroni's comparison test. | ||
PCA attenuates lipid accumulation in adipocytes by promoting fatty acid oxidation and mitochondria activities
Since PCA is the most abundant polyphenol in PSE, we wondered whether it would have similar anti-adipogenic and anti-lipogenic properties to PSE with increasing mitochondrial fatty acid oxidation, as we previously reported.21 To answer this question, we first treated 3T3-L1 cells with different PCA doses (50–400 μM) during adipogenesis (Fig. 5a upper). The presence of PCA (50–400 μM) significantly reduced the lipid accumulation measured by oil-red-O staining (ORO) (Fig. 5a lower). The reduction in TG accumulation in adipocytes was confirmed by adipogenic gene and protein expression. Although the PPARγ and aP2 levels did not reach statistical significance, the FAS and CCAAT/enhancer-binding protein alpha (C/EBPα) gene expressions were significantly reduced by 200 μM of PCA treatment (Fig. 5b). Consistently, 200 μM of PCA also suppressed adipogenic protein expression, including Fas, PPARγ, and aP2 (Fig. 5c).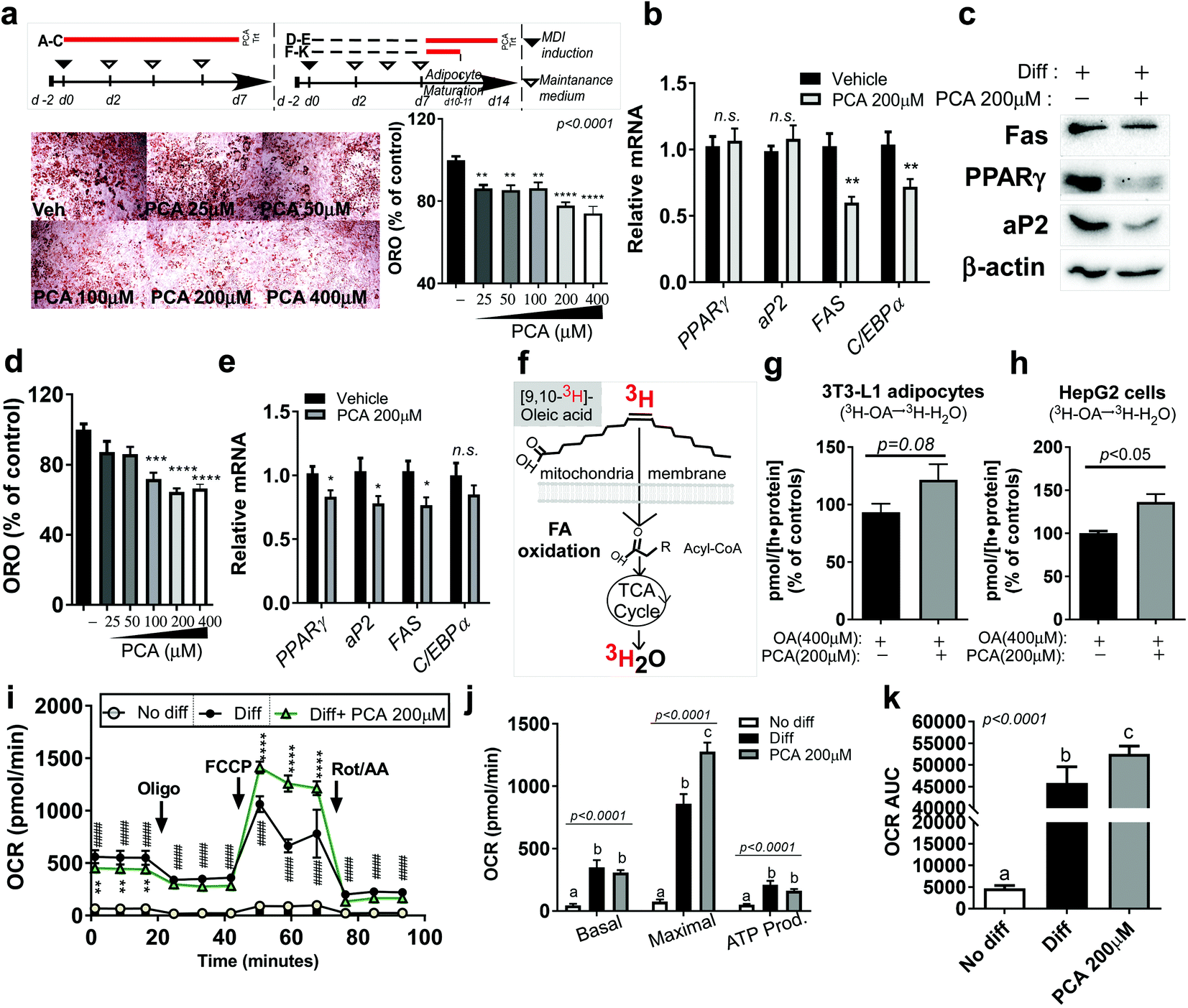 | ||
| Fig. 5 PCA attenuates lipid accumulation in adipocytes by promoting fatty acid oxidation and mitochondria activities. 3T3-L1 were seeded (d-2) and allowed to reach confluency for 2 d. Cells were induced to differentiation (d0, MDI: methyl isobutyl-xanthine, dexamethasone, and insulin). PSE treatment was conducted during differentiation until d7. DMSO was used as a vehicle control; (a) experimental scheme (upper). Visualization of lipid accumulation by ORO staining (magnified 4×); ORO was extracted and quantified (OD 500 nm, down); (b) relative PPARγ, aP2, FAS, and C/EBPα mRNA expressions; (c) adipogenic protein expressions of Fas, PPARγ, aP2, and β-actin by western blot analysis; 3T3-L1 are differentiated to become mature adipocytes until d7. Fully differentiated adipocytes (d7) were incubated with or without PSE (25 μg mL−1) for an additional seven days; (d) quantified ORO staining; (e) relative PPARγ, aP2, FAS, and C/EBPα mRNA expressions; 3T3-L1 are differentiated to become mature adipocytes until d7. Fully differentiated adipocytes (d7) were incubated in the presence or absence of PCA (200 μM) for four days; (f) the experimental scheme for measuring the fatty acid oxidation rate using radioactive precursor; (g) fatty acid oxidation rate (conversion of [3H]-OA into [3H]-H2O) in mature adipocytes; HepG2 cells were pre-incubated with PCA (200 μM) for 48 h; 0.8 mM of BSA–OA complex (or BSA control) was loaded for 2 h; (h) fatty acid oxidation rate ([3H]-OA in [3H]-H2O) in HepG2 cells; (i) oxygen consumption rate (OCR) determined using a Seahorse extracellular analyzer. The respiratory inhibitors, Oligo, FCCP and a combination of Rot/AA were indicated with arrows; (j) OCR profiles; (k) OCR AUC analysis. #### p < 0.0001 compared No diff vs. diff. * p < 0.05; ** p < 0.01; *** p < 0.001; and **** p < 0.0001 compared with the vehicle control by Student's t-test or one-way ANOVA with Bonferroni's comparison test. Means that do not share a common superscript are significantly different as determined by one-way ANOVA with Bonferroni's comparison test. +, treatment; −, non-treatment. | ||
Next, we investigated whether PCA also antagonized adipocyte hypertrophy. Fully differentiated 3T3-L1 adipocytes were exposed to PCA for 7 days (Fig. 5a upper), and we evaluated the lipid accumulation and lipogenic gene expressions. We found that PCA exposure (100–400 μM) was sufficient to reduce lipid accumulation in fully differentiated adipocytes as measured by ORO (Fig. 5d). Lipogenic genes, including PPARγ, aP2, and FAS, were reduced by PCA (200 μM). C/EBPα had a tendency to decrease gene expression by PCA but was not significantly different from the vehicle control (Fig. 5e).
To test whether the TG reduction was due to the upregulation of FA oxidation, the adipocytes were incubated with the radioactive precursor, 3[H]-oleic acid. The incorporation rate of 3[H]-oleic acid into 3[H]-H2O was used for measuring the FA oxidation rate (Fig. 3f,21). The FA oxidation rate (3[H]-oleic acid → 3[H]-H2O) was significantly increased by PCA treatment in adipocytes. Although it did not reach significance, PCA treatment partially augmented the FA oxidation rate in human hepatoma HepG2 cells exposed to BSA–OA complex (p = 0.08) (Fig. 2g and h). We assumed that the induction of fatty oxidation with lipid reduction by PCA would be followed by mitochondrial activation. Strikingly, PCA treatment for 4 days in fully differentiated adipocytes significantly promoted the mitochondrial respiration rate and, in particular, the maximal OCR compared to fully differentiated adipocytes (Fig. 5i–k).
We then performed a small pilot study to demonstrate whether PCA promoted thermogenic activity. The 7-week-old Balb/c mice were treated with PCA or vehicle fed with HF diet for 7 days at ambient temperature (25 °C) and then exposed at cold-temperature (8 °C) for 24 h (Fig. 6a). Interestingly, heat release and body temperature were significantly higher in the PCA treated group than the control (Fig. 6b and c), indicating that PCA may have a potential thermoregulatory function. However, brown-specific UCP1 protein expression was insufficient to reach statistical significance in the PCA-treated group (Fig. 6d). Taken together, these data suggest that PCA mimics PSE's lipid-lowering effects with mitochondrial fatty acid oxidation in adipocytes.
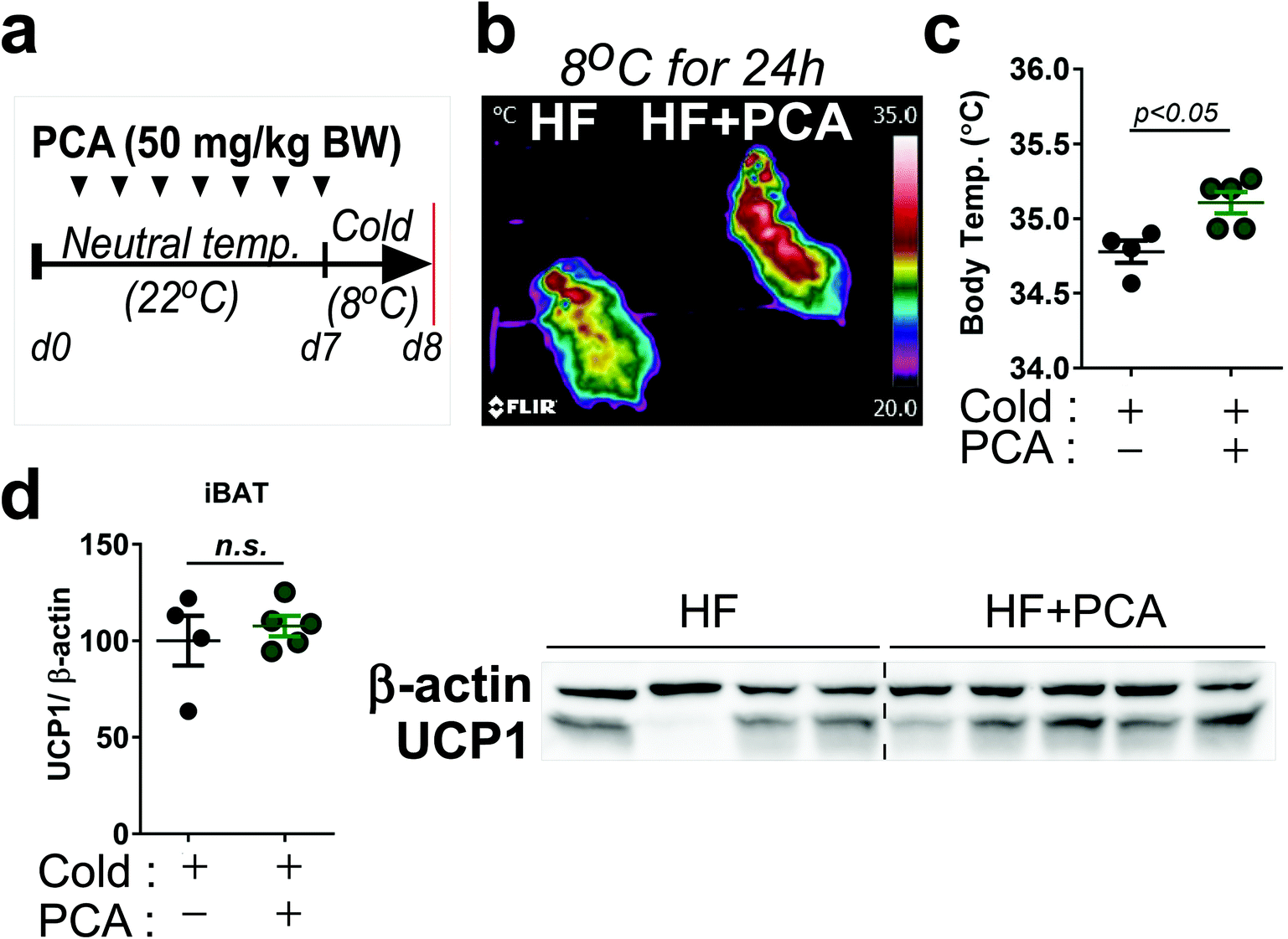 | ||
| Fig. 6 PCA can maintain higher temperature upon cold exposure. Seven-week-old Balb/c mice were intraperitoneally injected with vehicle (0.9% PBS) or PCA (50 mg per kg BW) with HF diet for 7 days (n = 4–5 per group) followed by cold-exposure (8 °C) for 24 h; (a) experimental scheme; (b) representative thermography captured by an IR camera; (c) body temperature upon cold-exposure for 24 h; (d) UCP1 protein expression of iBAT after cold acclimation. All values are presented as the mean ± SEM. n.s. represents no significance. p < 0.05; compared with the control by Student's t-test. +, treatment; −, non-treatment. | ||
Discussion
Excessive expansion of visceral adiposity is accompanied by adipose remodeling: angiogenesis, collagen accumulation, and immune cell recruitments.3 Thus, inflammatory cytokines and metabolic precursors in adipocytes damage cells, thereby, leading to metabolic dysfunctions. Low-grade chronic inflammation caused by a HF diet and/or LPS activates pro-inflammatory signaling, such as toll-like receptor 4 (TLR4) pathways, leading to reprogramming of the immune systems in WAT.35 Recent evidence showed that inflammation reduced thermogenic activities in BAT and brown-like adipocytes (beige adipocyte).10,34 Thus, the therapeutic agents and components with an immune-modulatory function have been recently studied to control energy homeostasis. We previously demonstrated that PSE contains lots of bioactive molecules that reduced the lipid accumulation in adipocytes via mitochondrial fatty acid oxidation.21 However, it is still unknown whether the lipid-lowering properties of PS are translated into animal models and the impact of PS extract on inflammation in both white and beige adipocytes.In this study, we asked two fundamental questions: (i) does PS supplementation diminish HF diet-mediated obesity and adipocyte inflammation? (ii) Does PSE preserve the inflammation-mediated inhibition of adipocyte browning and what components of PSE are most responsible for this? To answer these questions, we performed animal experiments using HF-diet-induced obese mice as an in vivo model and LPS-treated murine macrophages, adipocytes, and beige adipocytes (Bt2-cAMP-induced beige adipocytes) as an in vitro model.
Regarding the first question, we found that PS supplementation improved HF-diet-induced body weight gain (BWG), total cholesterol, glucose, and glucose disposal during GTT, despite no significant changes in the liver and fat mass between HF vs. HF + PS (Fig. 1). Consistent with our results, Ha et al. reported that PSE (30 mg of PSE kg−1 diet) significantly reduced obesity, evidenced by diminished BWG, TG, and hepatic lipid with high fecal lipid excretion compared to the HF diet control.22 They also mentioned that TG reduction in the PSE group may be involved in the sterol regulatory element-binding protein-c (SREBP-1c) pathway. A recent clinical study (double-blind randomized trial) demonstrated that high PSE (5.8 g PSE day−1) consumption for 4 weeks dramatically reduced the waist circumference, serum low-density lipoprotein, and TG levels (pre vs. post) in overweight and obese women (BMI ≧ 23 kg m−2).36 In our study, PS consumption marginally reduced (p = 0.06) the TC levels when compared with the HF diet (Fig. 1e). Cholesterol-lowering effects in systemic levels are mainly due to changes in (i) the cholesterol absorption, (ii) the de novo synthesis of cholesterol, (iii) modulation of the lipoprotein levels (low LDL and high HDL), and (iv) cholesterol excretion, etc. Although we have not tested all the pathways to reduce cholesterol, Ha et al.22 reported SREBP-1c reduction by PSE related to the de novo synthesis of lipids. Thus, it is plausible to assume that PS may reduce the new synthesis of lipids. Our unpublished data also showed that PS supplementation reduced FAS gene expression in hepatic tissue as compared to the HF diet control (data not shown), indicating that the fatty acid metabolism indirectly affects PS's lipid-lowering effects. We assume that the reduction in TC levels by PS supplementation may increase glucose homeostasis (Fig. 1f–h). However, it is still vague as to which pathways PS directly targets to improve dyslipidemia; therefore, further study is required to unravel these issues.
Improved metabolic parameters, such as BWG, TC, and glucose disposal during GTT, are followed by reduced adipose inflammation (Fig. 1j). In parallel, an in vitro study was performed to confirm PSE's anti-inflammatory properties in murine macrophages and adipocytes (Fig. 2). These results are well-aligned with the Lee et al. study where PS root extract (PSRE) reduced nitric oxide (NO) production and pro-inflammatory cytokines and cyclooxygenase-2 (COX-2) protein expression in LPS-stimulated RAW264.7 cells.20 Choi et al. also showed that PSE treatment reduced COX-2 and nerve growth factor (NGF) levels in oxazolone-induced contact dermatitis models.37 COX-2 is the enzyme mostly responsible for causing inflammation,38 and this is well aligned with PS alleviating inflammation in macrophages and adipocytes (Fig. 2), which is due to COX-2 mechanisms. We are now adopting the ‘gain-of-function’ and ‘loss-of-function’ strategies in macrophages and adipocytes to rule out anti-inflammatory effects of PS via COX-2.
The second question that we asked is regarding the protecting effects of PS on inflammation-induced adipocyte browning inhibition. Previously, we reported that PS was able to increase adipocyte browning as evidenced by the upregulation of UCP1 and PGC1α gene expression in a brown-like adipocyte cell model.21 Along with BAT markers, the beige-specific markers, TMEM26 and Shox2 were also increased by PS extract (Fig. 3a). However, the browning effect of PS is determined by Bt2-cAMP-treated 3T3-L1 adipocytes, which are a classical white adipocyte model, not a “good” beige-adipocyte model. Thus, it is necessary to have further experiments using an in vivo or primary stromal vascular fraction from inguinal adipose tissue to support the browning effect of PS. In our study, we found that brown adipose tissue activation was augmented by increasing thermogenic gene expression in BAT primary cells (Fig. 3b) although the UCP1 protein expression does not differ. Moreover, our data showed that the protection of LPS-mediated mitochondrial dysfunction by PSE preserves thermogenic activity (Fig. 3b–f). PCA, the major component in PSE (429.90 μg PCA g−1 extract) (Fig. 4), may have similar effects in adipocytes (Fig. 5). Recent evidence showed that PCA prevented obesity via upregulation of BAT function in diet-induced obese mice and leptin receptor-deficient (db/db) mice.39 Our study also found that PCA treatment for only 7 days increased heat release and body temperature when mice were exposed to cold temperatures (8 °C). Although an iBAT-marker, UCP1, was not altered with PCA; we assumed that the PCA concentration and duration were not sufficient to increase protein levels. We are currently undertaking animal experiments to demonstrate whether PCA enables the reverse of the LPS-induced inhibition of white adipocyte browning in vivo. Based on our data (Fig. 3–5) and previous studies,21 we assume that protecting the role against inflammation-induced inhibition of adipocyte browning by PS and/or PSE might be due to PCA. Resveratrol, which is also a constituent in PSE, is a well-known activator of sirtuin 1 (SIRT1) and PGC1α,40 and so it is plausible that resveratrol also contributes to the LPS-mediated inhibition of adipocyte browning. We are currently performing synergistic experiments by comparing different combinations of PSE components. Our data showed that FA oxidation, which is evidenced by the conversion of 3[H]-oleic acid into 3[H]-H2O, was significantly increased in BSA–OA-treated HepG2 cells (Fig. 5 h). This is well-aligned with the recent evidence that showed that PCA inhibited lipid accumulation in palmitic acid-treated human hepatoma HepG2 cells.41 Thus, future studies are warranted to evaluate the comprehensive characteristics of PCA, the principal constituents of PSE, and lipid modulation in nutrient-extensive conditions.
Although 4% of PS diet, 25 μg ml−1 of PSE, and/or 200 μM of PCA showed health-promoting effects in terms of obesity and its mediated metabolic complications in different models, there are some limitations. The primary reason we used 4% PS is based on the high bioavailability of PCA.42 According to Pei et al.,43 PCA has a high absorption rate. The serum concentration of free PCA peaked 10 min after the oral administration of 100 μmol kg−1 of PCA (Cmax = 165 μmol L−1). From a theoretical point of view, we fed 4% PS powder (254 μg PCA g−1 dry PS sample) to mice, which provided ∼30 μg of PCA per day based upon food intake of 3 g day−1 (average 3 g of food intake, Fig. 1d). Given that >97% PCA was estimated to be absorbed in the gastrointestinal tract,42 the total amount of PCA would be roughly 29.1 μg ml−1 PCA in serum, which is equivalent to 177.51 μmol L−1. It is plausible that for attaining ∼200 μM of PCA, 4% of PS is required for cell culture experiments. Moreover, PS powder has 4 kcal g−1 energy, which indicated high energy intake in the HF + PS group throughout the animal experiments (4.2 kcal g−1 diet in the HF group vs. 4.3 kcal g−1 diet in the HF + PS group). Although our study provided the impact of the PS anti-obesity properties against the HF diet, 4% of PS is hardly achievable. Future research on the optimal dose–period range of PS and/or PSE is warranted to obtain the physiological efficacy of PS.
In summary, the present study evaluated the anti-obesity characteristics of PS in C57BL/6 mice fed a HF-diet. The anti-inflammatory effects of PSE in LPS-stimulated mouse macrophages and adipocytes and its crosstalk were also investigated. PSE was found to protect against the inflammation-induced inhibition of adipocyte browning. PCA, the most abundant phenolic acid form in PSE, is regarded as a potent polyphenol to mimic PSE's lipid-lowering effects by the induction of maximal mitochondrial respiration. The following are some unanswered questions that remain from our research: (i) is 4% of PS in the diet physiologically possible in human physiology? (ii) Does the PSE supplementation also have similar outcomes to PS supplementation (direct comparison of PS and PSE)? (iii) If PCA is the most abundant polyphenol in PS, how much PCA is absorbed systemically and in adipose tissue? (iv) What specific mechanisms are involved in controlling the adipocyte metabolism through PS and/or PCA? Although further investigations are necessary to validate the above-mentioned limitations, we have herein presented evidence of the pivotal role of PS in obesity and inflammation via mitochondrial activation.
Conclusions
In this study, we investigated PS-attenuated HF-diet-mediated obesity and adipose inflammation. We have shown that PSE reversed inflammation and adipocyte browning in the presence of LPS with mitochondrial activation. We evaluated PSE's major components, PCA, as abundant and responsible for mimicking PS, and/or PSE's lipid-lowering effects with mitochondrial fatty acid oxidation. To our knowledge, this is the first study to report the previously unrecognized impact of PS rich in PCA in the alleviation of HF-diet-mediated metabolic dysfunctions and the protective role of PSE against LPS-induced inflammation and the suppression of adipocyte browning.Abbreviations
| aP2 | Adipocyte protein 2 |
| BAT | Brown adipose tissue |
| BMDM | Bone marrow-derived macrophages |
| Bt-cAMP | Dibutyryl-cyclic adenosine monophosphate |
| BW | Body weight |
| C/EBPα | CCAAT/enhancer-binding protein alpha |
| CIDEA | Cell death-inducing DNA fragmentation factor alpha-like effector A |
| CM | Conditioned media |
| DPPH | 2,2-Diphenyl-1-picryl-hydrazyl-hydrate |
| FA | Fatty acid |
| FAS | Fatty acid synthase |
| GTT | Glucose tolerance test |
| H&E | Hematoxylin and Eosin |
| iBAT | Interscapular brown adipose tissue |
| IL-1β | Interleukin 1β |
| IL-6 | Interleukin 6 |
| LF | Low-fat |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| MCP1 | Monocyte chemoattractant protein-1 |
| mΦ CM | Macrophage conditioned media |
| OA | Oleic acid |
| OCR | Oxygen consumption rates |
| PCA | p-Coumaric acid |
| PGC1α | PPAR gamma coactivator 1 α |
| PPARγ | Peroxisome proliferator-activated receptors γ |
| PS | Peanut sprout |
| PSE | Peanut sprout extract |
| TNFα | Tumor necrosis factor alpha |
| UCP1 | Uncoupling protein 1 |
| WAT | White adipose tissue |
Author contributions
S. S., S. J. performed experiments, collected and analyzed data. T. T. assisted in data collection of in vivo studies. D. K. and G. Z. participated in polyphenol analysis and drafted the results. M. L. and Y. L. critically reviewed the manuscript. I. K. designed the supervised work and wrote the manuscript. All the authors reviewed the final manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) 2020R1C1C1012547 and Basic Science Research Program to Research Institute for Basic Sciences (RIBS) of Jeju National University through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2019R1A6A1A10072987).References
- B. E. Wisse, The inflammatory syndrome: the role of adipose tissue cytokines in metabolic disorders linked to obesity, J. Am. Soc. Nephrol., 2004, 15, 2792–2800 CrossRef CAS PubMed.
- R. Buettner, J. Scholmerich and L. C. Bollheimer, High-fat diets: modeling the metabolic disorders of human obesity in rodents, Obesity, 2007, 15, 798–808 CrossRef CAS PubMed.
- K. J. Strissel, Z. Stancheva, H. Miyoshi, J. W. Perfield 2nd, J. DeFuria, Z. Jick, A. S. Greenberg and M. S. Obin, Adipocyte death, adipose tissue remodeling, and obesity complications, Diabetes, 2007, 56, 2910–2918 CrossRef CAS PubMed.
- W. D. van Marken Lichtenbelt, J. W. Vanhommerig, N. M. Smulders, J. M. Drossaerts, G. J. Kemerink, N. D. Bouvy, P. Schrauwen and G. J. Teule, Cold-activated brown adipose tissue in healthy men, N. Engl. J. Med., 2009, 360, 1500–1508 CrossRef CAS PubMed.
- K. A. Lo and L. Sun, Turning WAT into BAT: a review on regulators controlling the browning of white adipocytes, Biosci. Rep., 2013, 33 DOI:10.1042/BSR20130046.
- S. Cinti, Transdifferentiation properties of adipocytes in the adipose organ, Am. J. Physiol. Endocrinol. Metab., 2009, 297, E977–E986 CrossRef CAS PubMed.
- B. Cannon and J. Nedergaard, Brown adipose tissue: function and physiological significance, Physiol. Rev., 2004, 84, 277–359 CrossRef CAS PubMed.
- J. Wu, P. Bostrom, L. M. Sparks, L. Ye, J. H. Choi, A. H. Giang, M. Khandekar, K. A. Virtanen, P. Nuutila, G. Schaart, K. Huang, H. Tu, W. D. van Marken Lichtenbelt, J. Hoeks, S. Enerback, P. Schrauwen and B. M. Spiegelman, Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human, Cell, 2012, 150, 366–376 CrossRef CAS PubMed.
- N. Petrovic, T. B. Walden, I. G. Shabalina, J. A. Timmons, B. Cannon and J. Nedergaard, Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes, J. Biol. Chem., 2010, 285, 7153–7164 CrossRef CAS PubMed.
- M. Okla, W. Wang, I. Kang, A. Pashaj, T. Carr and S. Chung, Activation of Toll-like receptor 4 (TLR4) attenuates adaptive thermogenesis via endoplasmic reticulum stress, J. Biol. Chem., 2015, 290, 26476–26490 CrossRef CAS PubMed.
- M. K. Nohr, N. Bobba, B. Richelsen, S. Lund and S. B. Pedersen, Inflammation Downregulates UCP1 Expression in Brown Adipocytes Potentially via SIRT1 and DBC1 Interaction, Int. J. Mol. Sci., 2017, 18 DOI:10.3390/ijms18051006.
- L. Gan, Z. Liu, F. Feng, T. Wu, D. Luo, C. Hu and C. Sun, Foxc2 coordinates inflammation and browning of white adipose by leptin-STAT3-PRDM16 signal in mice, Int. J. Obes., 2018, 42, 252–259 CrossRef CAS PubMed.
- F. Omran and M. Christian, Inflammatory Signaling and Brown Fat Activity, Front. Endocrinol., 2020, 11, 156 CrossRef PubMed.
- O. T. Toomer, Nutritional chemistry of the peanut (Arachis hypogaea), Crit. Rev. Food Sci. Nutr., 2018, 58, 3042–3053 CrossRef CAS PubMed.
- A. E. Griel, B. Eissenstat, V. Juturu, G. Hsieh and P. M. Kris-Etherton, Improved diet quality with peanut consumption, J. Am. Coll. Nutr., 2004, 23, 660–668 CrossRef CAS PubMed.
- D. T. Khang, T. N. Dung, A. A. Elzaawely and T. D. Xuan, Phenolic Profiles and Antioxidant Activity of Germinated Legumes, Foods, 2016, 5 DOI:10.3390/foods5020027.
- C. K. Youn, E. R. Jo, J. H. Sim and S. I. Cho, Peanut sprout extract attenuates cisplatin-induced ototoxicity by induction of the Akt/Nrf2-mediated redox pathway, Int. J. Pediatr. Otorhinolaryngol., 2017, 92, 61–66 CrossRef PubMed.
- K. H. Pyo, Y. W. Lee, S. H. Lee, C. F. Xin, J. H. Shin and E. H. Shin, Preventive Effects of Resveratrol-enriched Extract of Peanut Sprout on Bacteria- and Estradiol-induced Prostatitis in Mice, Nat. Prod. Commun., 2017, 12, 73–78 CrossRef.
- J. H. Song, B. Hwang, H. J. Chung, B. Moon, J. W. Kim, K. Ko, B. W. Kim, W. R. Kim, W. J. Kim, S. C. Myung and S. K. Moon, Peanut Sprout Extracts Cultivated with Fermented Sawdust Medium Inhibits Benign Prostatic Hyperplasia In Vitro and In Vivo, World J. Men's Health, 2020, 38, 385–396 CrossRef PubMed.
- Y. M. Lee, E. Son and D. S. Kim, Treatment with Peanut Sprout Root Extract Alleviates Inflammation in a Lipopolysaccharide-Stimulated Mouse Macrophage Cell Line by Inhibiting the MAPK Signaling Pathway, Int. J. Mol. Sci., 2019, 20 DOI:10.3390/ijms20235907.
- S. H. Seo, S. M. Jo, J. Kim, M. Lee, Y. Lee and I. Kang, Peanut Sprout Extracts Attenuate Triglyceride Accumulation by Promoting Mitochondrial Fatty Acid Oxidation in Adipocytes, Int. J. Mol. Sci., 2019, 20 DOI:10.3390/ijms20051216.
- A. W. Ha, N. E. Kang and W. K. Kim, Ethanol Extract of Peanut Sprout Lowers Blood Triglyceride Levels, Possibly Through a Pathway Involving SREBP-1c in Rats Fed a High-Fat Diet, J. Med. Food, 2015, 18, 850–855 CrossRef CAS PubMed.
- N. E. Kang, A. W. Ha, H. W. Woo and W. K. Kim, Peanut sprouts extract (Arachis hypogaea L.) has anti-obesity effects by controlling the protein expressions of PPARgamma and adiponectin of adipose tissue in rats fed high-fat diet, Nutr. Res. Pract., 2014, 8, 158–164 CrossRef PubMed.
- M. K. Judge, J. Zhang, N. Tumer, C. Carter, M. J. Daniels and P. J. Scarpace, Prolonged hyperphagia with high-fat feeding contributes to exacerbated weight gain in rats with adult-onset obesity, Am. J. Physiol.: Regul., Integr. Comp. Physiol., 2008, 295, R773–R780 CrossRef CAS PubMed.
- P. G. Reeves, F. H. Nielsen and G. C. Fahey Jr., AIN-93 purified diets for laboratory rodents: final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet, J. Nutr., 1993, 123, 1939–1951 CrossRef CAS PubMed.
- A. Bartelt, O. T. Bruns, R. Reimer, H. Hohenberg, H. Ittrich, K. Peldschus, M. G. Kaul, U. I. Tromsdorf, H. Weller, C. Waurisch, A. Eychmuller, P. L. Gordts, F. Rinninger, K. Bruegelmann, B. Freund, P. Nielsen, M. Merkel and J. Heeren, Brown adipose tissue activity controls triglyceride clearance, Nat. Med., 2011, 17, 200–205 CrossRef CAS PubMed.
- I. Kang, J. C. Espin, T. P. Carr, F. A. Tomas-Barberan and S. Chung, Raspberry seed flour attenuates high-sucrose diet-mediated hepatic stress and adipose tissue inflammation, J. Nutr. Biochem., 2016, 32, 64–72 CrossRef CAS PubMed.
- D. S. Kim and S. B. Lim, Extraction of flavanones from immature Citrus unshiu pomace: process optimization and antioxidant evaluation, Sci. Rep., 2020, 10, 19950 CrossRef CAS PubMed.
- K. B. Kim, S. Lee, I. Kang and J. H. Kim, Momordica charantia Ethanol Extract Attenuates H(2)O(2)-Induced Cell Death by Its Antioxidant and Anti-Apoptotic Properties in Human Neuroblastoma SK-N-MC Cells, Nutrients, 2018, 10 DOI:10.3390/nu10101368.
- H. J. Park, S. M. Jo, S. H. Seo, M. Lee, Y. Lee and I. Kang, Anti-Inflammatory Potential of Cultured Ginseng Roots Extract in Lipopolysaccharide-Stimulated Mouse Macrophages and Adipocytes, Int. J. Environ. Res. Public Health, 2020, 17 DOI:10.3390/ijerph17134716.
- J. Kim, M. Okla, A. Erickson, T. Carr, S. K. Natarajan and S. Chung, Eicosapentaenoic Acid Potentiates Brown Thermogenesis through FFAR4-dependent Up-regulation of miR-30b and miR-378, J. Biol. Chem., 2016, 291, 20551–20562 CrossRef CAS PubMed.
- I. Kang, Y. Kim, F. A. Tomas-Barberan, J. C. Espin and S. Chung, Urolithin A, C, and D, but not iso-urolithin A and urolithin B, attenuate triglyceride accumulation in human cultures of adipocytes and hepatocytes, Mol. Nutr. Food Res., 2016, 60, 1129–1138 CrossRef CAS PubMed.
- T. Romacho, M. Elsen, D. Rohrborn and J. Eckel, Adipose tissue and its role in organ crosstalk, Acta Physiol., 2014, 210, 733–753 CrossRef CAS PubMed.
- M. Alcala, M. Calderon-Dominguez, E. Bustos, P. Ramos, N. Casals, D. Serra, M. Viana and L. Herrero, Increased inflammation, oxidative stress and mitochondrial respiration in brown adipose tissue from obese mice, Sci. Rep., 2017, 7, 16082 CrossRef PubMed.
- L. K. Heilbronn and L. V. Campbell, Adipose tissue macrophages, low grade inflammation and insulin resistance in human obesity, Curr. Pharm. Des., 2008, 14, 1225–1230 CrossRef CAS PubMed.
- A. W. Ha, W. K. Kim, J. H. Kim and N. E. Kang, The supplementation effects of peanut sprout on reduction of abdominal fat and health indices in overweight and obese women, Nutr. Res. Pract., 2015, 9, 249–255 CrossRef CAS PubMed.
- D. I. Choi, J. Y. Choi, Y. J. Kim, J. B. Lee, S. O. Kim, H. T. Shin and S. C. Lee, Ethanol Extract of Peanut Sprout Exhibits a Potent Anti-Inflammatory Activity in Both an Oxazolone-Induced Contact Dermatitis Mouse Model and Compound 48/80-Treated HaCaT Cells, Ann. Dermatol., 2015, 27, 142–151 CrossRef PubMed.
- C. S. Williams, M. Mann and R. N. DuBois, The role of cyclooxygenases in inflammation, cancer, and development, Oncogene, 1999, 18, 7908–7916 CrossRef CAS PubMed.
- X. Han, J. Guo, Y. You, J. Zhan and W. Huang, p-Coumaric acid prevents obesity via activating thermogenesis in brown adipose tissue mediated by mTORC1-RPS6, FASEB J., 2020, 34, 7810–7824 CrossRef CAS PubMed.
- M. Lagouge, C. Argmann, Z. Gerhart-Hines, H. Meziane, C. Lerin, F. Daussin, N. Messadeq, J. Milne, P. Lambert, P. Elliott, B. Geny, M. Laakso, P. Puigserver and J. Auwerx, Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha, Cell, 2006, 127, 1109–1122 CrossRef CAS PubMed.
- X. Yan, X. Chen, X. Xu, J. Liu, C. Fu, D. Zhao, W. Zhao, R. Ma and L. Sun, Mechanism Underlying p-Coumaric Acid Alleviation of Lipid Accumulation in Palmitic Acid-Treated Human Hepatoma Cells, J. Agric. Food Chem., 2020, 68, 3742–3749 CrossRef CAS PubMed.
- Y. Konishi, Y. Hitomi and E. Yoshioka, Intestinal absorption of p-coumaric and gallic acids in rats after oral administration, J. Agric. Food Chem., 2004, 52, 2527–2532 CrossRef CAS PubMed.
- K. Pei, J. Ou, J. Huang and S. Ou, p-Coumaric acid and its conjugates: dietary sources, pharmacokinetic properties and biological activities, J. Sci. Food Agric., 2016, 96, 2952–2962 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1fo00342a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |