
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSirt6-mediated Nrf2/HO-1 activation alleviates angiotensin II-induced DNA DSBs and apoptosis in podocytes†
Yanqin
Fan‡
 ab,
Jing
Cheng‡
c,
Qian
Yang
ab,
Jun
Feng
ab,
Jijia
Hu
ab,
Zhilong
Ren
ab,
Hongxia
Yang
ab,
Dingping
Yang
ab and
Guohua
Ding
*ab
ab,
Jing
Cheng‡
c,
Qian
Yang
ab,
Jun
Feng
ab,
Jijia
Hu
ab,
Zhilong
Ren
ab,
Hongxia
Yang
ab,
Dingping
Yang
ab and
Guohua
Ding
*ab
aDivision of Nephrology, Renmin Hospital of Wuhan University, Wuhan, Hubei, China. E-mail: ghxding@gmail.com; Fax: +86 27 88042292; Tel: +86 27 88041919
bNephrology and Urology Research Institute of Wuhan University, Wuhan, Hubei, China
cDivision of Neurosurgery, Renmin Hospital of Wuhan University, Wuhan, Hubei, China
First published on 9th July 2021
Abstract
Recent studies suggested that DNA double-strand breaks (DSBs) were associated with the pathogenesis of chronic kidney disease (CKD). The purpose of this investigation was to determine the role of Sirtuin6 (Sirt6), a histone deacetylase related to DNA damage repair, in angiotensin (Ang) II-induced DNA DSBs and the cell injury of podocytes and explore the possible mechanism. Here we showed that an increase of DNA DSBs was accompanied by a reduction in Sirt6 expression in the glomeruli of patients with hypertensive nephropathy (HN). Similar results were found in rat kidneys infused with Ang II and in cultured podocytes stimulated with Ang II. Sirt6 overexpression inhibited Ang II-induced ROS generation and DNA DSBs, and thus served as a protection against Ang II-induced apoptosis in podocytes. Moreover, Sirt6 activation enhanced Nrf2 and HO-1 gene expressions in podocytes after Ang II treatment. Furthermore, Nrf2 knockdown could partly reverse the cytoprotective effects of Sirt6 activation. In conclusion, our observations demonstrated that the Sirt6-Nrf2-HO-1 pathway played a vital role in relieving Ang II-mediated oxidative DNA damage and podocyte injury.
Introduction
Activation of the intrarenal renin–angiotensin system (RAS) plays a crucial role in the development and progression of chronic kidney disease (CKD).1–4 As a main effector of RAS, angiotensin (Ang) II is considered to be involved in the progression of CKD.5–7 However, not all patients with RAS activation develop CKD, and an aggressive inhibition of RAS activity reduces but does not eliminate the risk of CKD, suggesting that other factors are involved in the pathogenesis.8–11 As a key component in maintaining the integrity of the glomerular filtration barrier with limited regeneration and repair ability, podocyte loss correlates with the prognosis of CKD.12–15 Numerous studies suggest that Ang II-induced podocyte injury contributes to the onset of proteinuria and the progression of chronic glomerular disease.6,7,16,17 However, it is still worth investigating whether there are other mechanisms involved in Ang II-induced podocyte injury.DNA safekeeping is regarded as a vital biological function of cells, which ensures the transfer of unchanged genetic material to the next generation and maintains proper cellular function.18 DNA damage can be induced by various stimuli, such as reactive oxygen species (ROS), mechanical stress, and UV radiation.19,20 A DNA double-strand break (DSB) is the most severe type of DNA damage because it has been found that it is intrinsically more difficult to repair this type of damage than to repair other types of DNA damage.19,21 Cell injury occurs when DNA DSB induction increases or when their repair is impaired. Previous studies demonstrated that oxidative DNA damage including DNA DSBs caused by Ang II lead to cell apoptosis, and this was thought to be involved in the pathogenesis of CKD.22–24 It is speculated that an inhibition of DNA DSBs would protect one against podocyte injury and thus mitigate the progression of CKD. Hence, an identification of the key molecules regulating DNA DSBs in podocytes may provide a potential therapeutic target for the treatment of CKD.
Sirtuin6 (Sirt6) is an important member of the sirtuin family of class III NAD+-dependent histone deacetylases, which exhibits several catalytic activities including deacetylation and ribosylation.25–28 It has been demonstrated that Sirt6 is involved in multiple cellular pathways related to DNA repair, aging, inflammation, epigenetics and cellular glucose/lipid metabolism.29–31 The role of Sirt6 as a cytoprotective protein in podocytes is now becoming clear.32 Liu et al. revealed that Sirt6 deficiency triggered a high glucose-induced inflammatory response, cytoskeletal remodeling and apoptosis in podocytes.33 Huang et al. reported that Sirt6 was important for podocyte homeostasis and the maintenance of glomerular function.51 Our recent studies suggested that Sirt6 deletion exacerbated Ang II-induced cholesterol accumulation and injury in podocytes.6 Functionally, current literature has identified that Sirt6 serves as a vital regulator of DNA damage. Nagai et al. reported that a depletion of Sirt6 caused an increase of DNA DSBs and telomere dysfunction in human chondrocytes.34 Jung et al. have elucidated that Sirt6 overexpression prevented Aβ42-induced DNA DSBs in HT22 mouse hippocampal neurons.35 Sirt6 has also been implicated in the protection of human endothelial cells from DNA damage.36 However, whether Sirt6 is involved in Ang II-induced DNA DSBs in podocytes and the related molecular mechanisms remains unknown.
In the present investigation, we detected a reduction of Sirt6 in renal biopsies in patients with hypertensive nephropathy compared to healthy subjects. Sirt6 overexpression alleviated Ang II-induced DNA damage and apoptosis in cultured podocytes, and vice versa. Mechanistically, Sirt6 regulated the Nfr2/HO-1 signaling pathway to exert cytoprotection against Ang II-induced podocyte DNA damage and cell injury. Our data revealed that Sirt6 might be a potential therapeutic target in CKD.
Materials and methods
Human renal biopsy samples
Samples from patients who had renal biopsy-proven hypertensive nephropathy (HN) and diabetic nephropathy (DN) were obtained from the Division of Nephrology, Renmin Hospital of Wuhan University, Wuhan, China. The control samples were normal kidney sections of para-carcinoma tissues from individuals who underwent tumor nephrectomies. The samples from the control subjects without DN or other renal diseases except for solitary renal cell carcinoma were obtained from the Division of Pathology, Renmin Hospital of Wuhan University, Wuhan, China. The patients signed the informed consent before this experiment was conducted. The research was conducted in accordance with the approved guidelines of Wuhan University and was approved by the Research Ethics Committee of Renmin Hospital of Wuhan University. The investigation complied with the principles of the Declaration of Helsinki.Animals
All procedures were approved by the Ethical Committee for the Experimental Use of Animals of Renmin Hospital in Wuhan, China. A total of twenty-four male specific-pathogen-free Wistar rats were provided by the Hubei Research Center of Experimental Animals. Rats were raised in an environment with an artificial light cycle with controlled temperature and humidity at the Center for Animal Experiments of Wuhan University. The animals were given free access to standard rat chow and tap water. Rats were randomly divided into the normal saline infusion group or the Ang II infusion group after being embedded with osmotic minipumps (Alzet, CA). Rats were anesthetized with 4% isoflurane containing 30% O2 and 70% N2O. Rats in the Ang II-infusion group were subjected to Ang II perfusion at 400 ng kg−1 min−1 for 14 or 28 d. The animals were sacrificed on day 28 by cervical dislocation. The kidneys were perfused with vanadate (a phosphatase inhibitor) before isolation. Some kidney sections were stored at −80 °C for biochemical analysis, some kidney sections were fixed in 4% paraformaldehyde for pathological staining, and the remaining sections were fixed in glutaraldehyde for electron microscopy observation.The glomeruli were separated by the sieving method. 100 mg of renal cortex were passed through a three-layer stainless steel screen with a diameter of 80 mesh, 120 mesh and 200 mesh successively. The glomeruli on the net were collected after they were washed with Hank's solution.
Cell culture
Conditionally immortalized human podocytes, which were kindly provided by Dr Moin A. Saleem (Academic Renal Unit, Southmead Hospital, Bristol, UK), were grown in a standard environment. Podocytes were cultured at 33 °C in RPMI 1640 medium (HyClone, USA) supplemented with 10% heat-inactivated fetal bovine serum (BI, Israel), 100 μg mL−1 streptomycin, 100 U mL−1 penicillin G, and 1× insulin–transferrin–selenium (ITS) (Invitrogen, USA) for proliferation. Podocytes were grown for 10–14 days at 37 °C without ITS for differentiation. The differentiated cells were incubated with Ang II (10−7 M) at various times. Every result was verified in at least three independent cultures of podocytes.Transfection
For the interference treatment, Sirt6 siRNA (Qiagen, Germany) were transfected into podocytes according to the manufacturer's instructions. Cells with a density of 2 × 105 were seeded into 6-well plates and transfected with a mixture containing 10 nM Sirt6 siRNA or scrambled siRNA and the HiPerFect transfection reagent (Qiagen, Germany) under normal culture conditions for 24 h.Nrf2 siRNA (Qiagen, Germany) were transfected into the podocytes according to the manufacturer's instructions. Cells with a density of 2 × 105 were seeded into 6-well plates and transfected with a mixture containing 10 nM Nrf2 siRNA or scrambled siRNA and the HiPerFect transfection reagent (Qiagen, Germany) under normal culture conditions for 24 h.
For the overexpression treatment, the Sirt6 plasmid (Addgene) was transfected into the podocytes according to the manufacturer's instructions. Cells with a density of 2 × 105 were seeded into 6-well plates and transfected with a mixture containing 2 μg Sirt6 plasmid or pcDNA3.1 and 2 μl X-tremeGENE transfection reagent (Roche) under normal culture conditions for 24 h.
Western immunoblotting
Isolated glomeruli and podocytes were homogenized in RIPA buffer (Beyotime, China) mixed with a protease inhibitor cocktail (Sigma-Aldrich, USA). The extractions were then centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min at 4 °C. Equal amounts of protein samples were subjected to separation by SDS-PAGE and then transferred to nitrocellulose membranes (GE Healthcare). After blocking in 5% milk for 1 h, the membranes were incubated with primary antibodies (Sirt6 rabbit monoclonal antibody, 1:1000, Abcam; Nrf2 rabbit polyclonal antibody, 1:1000, Abcam; HO-1 rabbit monoclonal antibody, 1:1000, Abcam; GAPDH rabbit monoclonal antibody, 1:1000, Antgene and GAPDH mouse monoclonal antibody, 1:1000, Antgene) at 4 °C overnight. An Alexa Fluor 680/790-labeled goat anti-rabbit/goat anti-mouse IgG antibody (1:10000, LI-COR Biosciences, USA) was used as the secondary antibody. The blots were detected using the LI-COR Odyssey Infrared Imaging System.
000 rpm for 10 min at 4 °C. Equal amounts of protein samples were subjected to separation by SDS-PAGE and then transferred to nitrocellulose membranes (GE Healthcare). After blocking in 5% milk for 1 h, the membranes were incubated with primary antibodies (Sirt6 rabbit monoclonal antibody, 1:1000, Abcam; Nrf2 rabbit polyclonal antibody, 1:1000, Abcam; HO-1 rabbit monoclonal antibody, 1:1000, Abcam; GAPDH rabbit monoclonal antibody, 1:1000, Antgene and GAPDH mouse monoclonal antibody, 1:1000, Antgene) at 4 °C overnight. An Alexa Fluor 680/790-labeled goat anti-rabbit/goat anti-mouse IgG antibody (1:10000, LI-COR Biosciences, USA) was used as the secondary antibody. The blots were detected using the LI-COR Odyssey Infrared Imaging System.
Immunofluorescence assay
Kidney paraffin sections were dewaxed and subjected to antigen repair treatment. After blocking with 5% bovine serum albumin (BSA) at room temperature for 30 min, the slides were incubated with a mixture of mouse anti-WT1 monoclonal antibody (1:100, Novus, USA) and rabbit anti-Sirt6 polyclonal antibody (1:100, Thermo Fisher Scientific, USA) or rabbit anti-γH2AX polyclonal antibody (1:100, ABclonal, China) at 4 °C overnight. The slides were incubated with a mixture of Alexa Fluor 488, Donkey anti-mouse IgG (HL) (1:200, Jackson Immuno Research Laboratories, USA) and Alexa Fluor 594, Donkey anti-Rabbit IgG (HL) (1:200, Jackson Immuno Research Laboratories, USA) or Alexa Fluor 488, Donkey anti-Rabbit IgG (HL) (1:200, Life Technologies, USA) as the fluorescent secondary antibodies at 37 °C in darkness for 60 min. The nuclei were stained with DAPI (Antgene, China) for 5 min.Podocytes grown on cover slides were fixed with 4% paraformaldehyde at 4 °C for 30 min and incubated with rabbit anti-Sirt6 polyclonal antibody (1:100, Thermo Fisher Scientific, USA) or rabbit anti-γH2AX polyclonal antibody (1:100, ABclonal, China) overnight at 4 °C. The slides were then stained with Alexa Fluor 594, Donkey anti Rabbit IgG (HL) (1:200, Jackson Immuno Research Laboratories, USA) or Alexa Fluor 488, Donkey anti-Rabbit IgG (HL) (1:200, Life Technologies, USA) at 37 °C for 60 min. The nuclei were stained with DAPI (Antgene, China) for 5 min. Images were obtained using a laser scanning confocal microscope (Olympus, Japan).
Immunohistochemistry
Sirt6 expression in the kidney tissue was evaluated by immunohistochemistry. Kidney paraffin sections were dewaxed and subjected to antigen retrieval. After blocking with 5% bovine serum albumin, the slides were incubated with Sirt6 primary antibody (1:100, Thermo Fisher Scientific, USA) overnight at 4 °C. Subsequently, the sections were incubated with polymerized horseradish peroxidase-conjugated secondary antibody for 30 min. After DAB (Dako) staining, the slides were stained with haematoxylin. Images were obtained using a microscope (Olympus, Japan).Apoptosis assay
Podocyte apoptosis in the kidney tissue was examined using a TUNEL assay kit (Roche, Germany) according to the manufacturer's instructions. Briefly, the kidney paraffin sections were dewaxed and subjected to antigen retrieval. After blocking with 5% bovine serum albumin, the slides were incubated with terminal deoxynucleotidyl transferase (TdT) and digoxigenin-11-dUTP for 1 h at room temperature. The slides were then incubated with streptavidin–biotin–peroxidase-conjugated antidigoxigenin-11-dUTP antibody for 30 min. Images were obtained using a microscope (Olympus, Japan).Podocyte apoptosis in vitro was evaluated by flow cytometry with Annexin V–FITC/PI double staining according to the manufacturer's protocol (Annexin V-FITC Apoptosis Detection Kit I, BD Pharmingen, USA). Podocytes in the upper-right and lower-right quadrants were considered as apoptotic cells.
ROS detection
The intracellular ROS generation was evaluated using the dichlorofluorescein diacetate (DCFH-DA) fluorescent probe (Beyotime, China). The cells were incubated with a final concentration of 10 μM DCFH-DA for 1 h at 37 °C, and were subsequently washed with PBS three times. Subsequently, the images were obtained using a laser scanning confocal microscope (Olympus, Japan).Statistical analyses
All values were presented as the mean ± SD and analyzed using SPSS v 17.0. Differences in the mean values were evaluated using Student's t-test or one-way ANOVA. Differences with P < 0.05 were considered statistically significant.Results
Sirt6 expression was reduced in the renal biopsies of patients with HN accompanied by an increase of DNA DSBs
To investigate the effect of Ang II on the expression of Sirt6 in the glomeruli in vivo, we first conducted immunohistochemical staining of Sirt6 in the renal biopsies from patients with HN. As shown in Fig. 1A, we observed that Sirt6 was significantly reduced in the glomeruli of patients with HN and DN relative to those from healthy subjects. We further conducted double immunofluorescence staining of Sirt6 and the podocyte marker WT1 to evaluate the Sirt6 expression in the kidney tissues. The results suggested that Sirt6 was decreased in the podocytes from individuals with HN and DN relative to the healthy group (Fig. 1B). DNA damage is one of the major triggers of cell senescence and is considered to be related to apoptosis. Only few studies have revealed that Ang II induced ROS generation and oxidative DNA damage in podocytes.23,24 To elucidate the DNA injury of podocytes in patients with HN, we performed immunofluorescence staining to evaluate the γH2AX (a marker for DNA DSBs) expression in the glomeruli of HN subjects. The results indicated that γH2AX was increased in the glomeruli from HN individuals compared with the healthy controls (Fig. 1C), which suggested the existence of severe DNA damage in hypertensive kidney. Thus, we speculated that the downregulation of Sirt6 may contribute to Ang II-induced DNA DSBs and podocyte injury.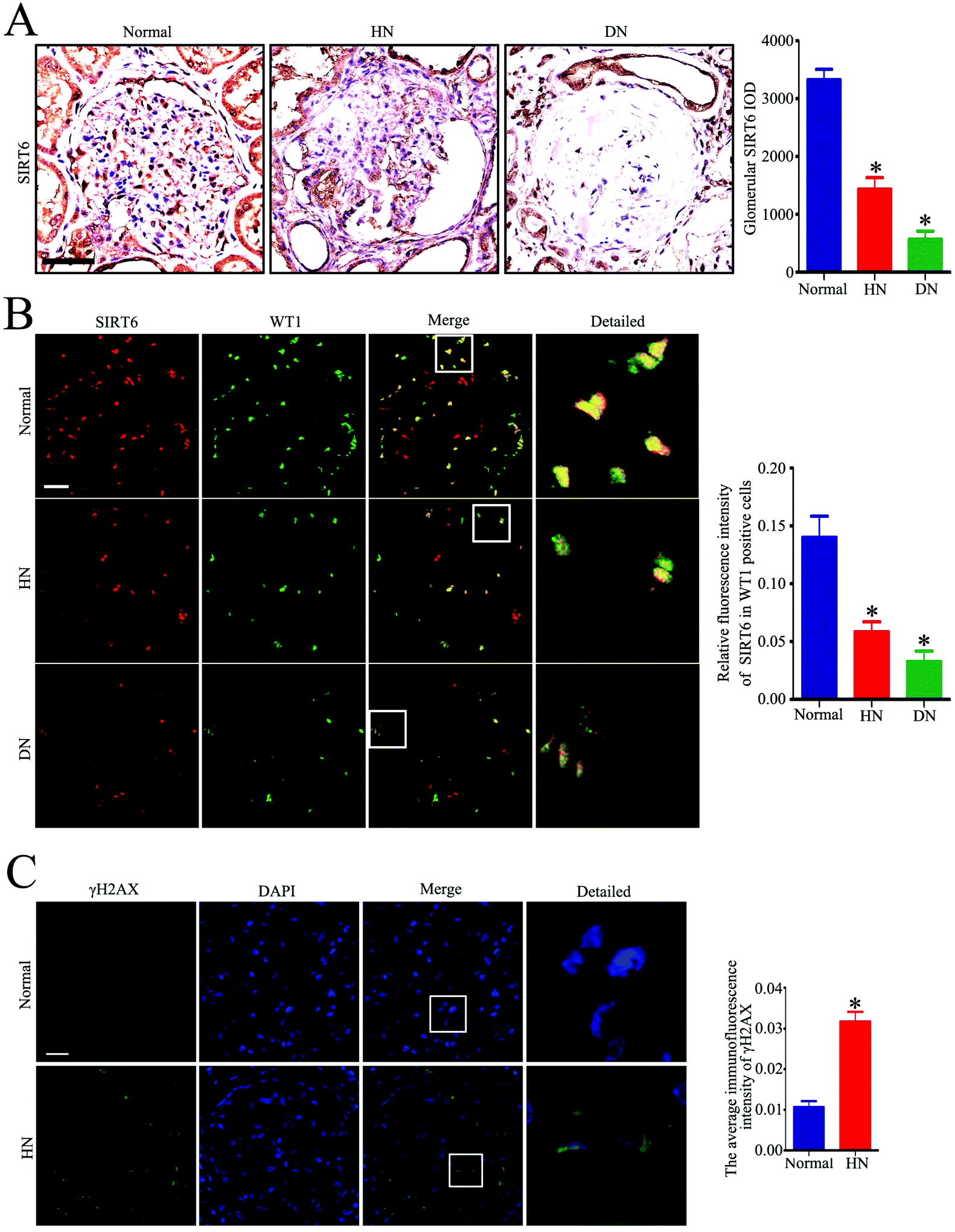 | ||
| Fig. 1 Sirt6 is decreased in the renal biopsies in patients with HN along with increase of DNA DSBs. (A) Representative micrographs and quantification of Sirt6 staining in the glomeruli from each group (original magnification, ×100). *P < 0.05 relative to normal, n = 6. Scale bars: 50 μm. (B) Representative micrographs and quantification of Sirt6 and WT-1 double staining in the glomeruli from each group (original magnification, ×400). *P < 0.05 relative to normal, n = 6. Scale bars: 20 μm. (C) Representative micrographs and quantification of γH2AX staining in the glomeruli from each group (original magnification, ×400). *P < 0.05 relative to normal, n = 6. Scale bars: 20 μm. Normal: healthy subjects, HN: patients with hypertensive nephropathy, DN: patients with diabetic nephropathy. | ||
Sirt6 expression was decreased in the podocytes from the glomeruli of Ang II-infused rats
To assess the effect of Ang II on the Sirt6 expression in the podocytes in vivo, we established an Ang II-infused rat model. Periodic acid–Schiff (PAS) staining showed a significant increase in mesangial expansion and the accumulation of the extracellular matrix in Ang II-infused rats relative to those from the control group (Fig. 2A). Transmission electron microscopy (TEM) examination revealed exacerbated diffuse foot process fusion and chromatin agglutination (podocyte apoptosis) in Ang II-infused rats (Fig. 2B). Moreover, the Ang II-infused rats exhibited a marked increase in apoptotic podocytes in the glomeruli compared with those in the control group, as revealed by the WT1 and TUNEL double immunofluorescence staining (Fig. 2C). These findings are consistent with our previous results. We next evaluated the renal expression of Sirt6 by western blot analysis. As with the tendency of Sirt6 expression in renal biopsy specimens of patients with HN, Sirt6 protein levels were also decreased in the kidneys of Ang II-infused rats (Fig. 2D). To further explore the expression of Sirt6 in the podocytes of rats infused with Ang II, double immunofluorescence staining of Sirt6 and WT1 was performed in the kidney tissues. We found a dramatic reduction in Sirt6 in the podocytes from the Ang II-infused rats compared to that in the podocytes from the control group (Fig. 2E). These results further verified that Sirt6 expression was downregulated in the podocytes which were subjected to the Ang II challenge in vivo.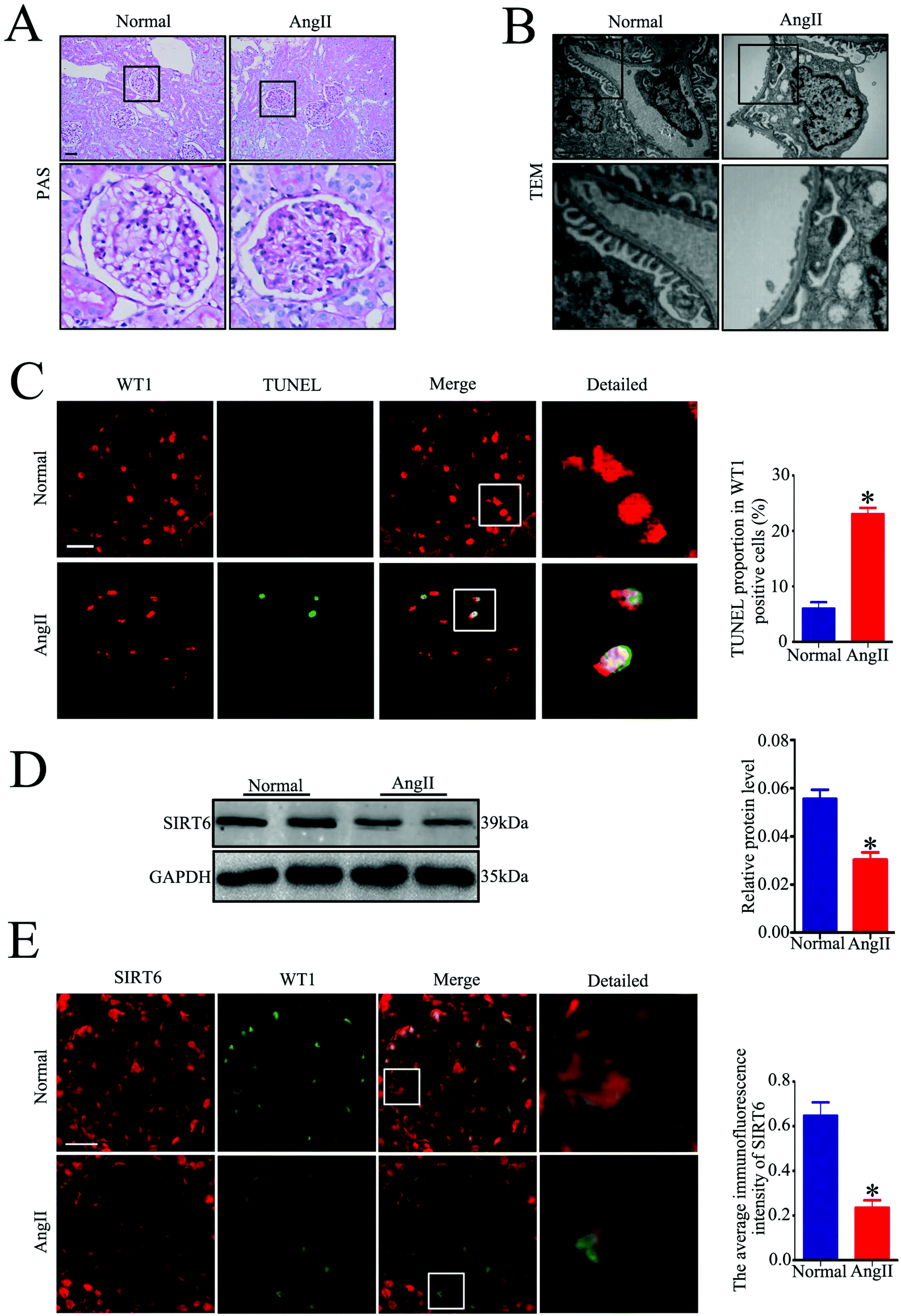 | ||
| Fig. 2 Sirt6 is decreased in the glomeruli of Ang II-infused rats. (A) Representative micrographs of PAS-stained kidney sections from each group (original magnification, ×200). n = 6. Scale bars: 50 μm. (B) Representative transmission electron micrographs of capillary loops in the glomeruli from each group (original magnification, ×10000). n = 6. Scale bars: 1900 nm. (C) Representative micrographs and quantification of WT-1 and TUNEL double staining in the glomeruli from each group (original magnification, ×400). *P < 0.05 relative to normal, n = 6. Scale bars: 20 μm. (D) Representative western blots and quantification of the Sirt6 protein level in the glomeruli from each group. *P < 0.05 relative to normal, n = 6. (E) Representative micrographs and quantification of Sirt6 and WT-1 double staining in the glomeruli from each group (original magnification, ×200). *P < 0.05 relative to normal, n = 6. Scale bars: 25 μm. Normal: normal saline-infused group, Ang II: Ang II-infused group. | ||
Sirt6 expression was downregulated in the cultured podocytes treated with Ang II along with severe DNA DSBs in vitro
We further investigated whether Ang II could induce Sirt6 reduction in the cultured podocytes in vitro. We incubated the cells with Ang II (10−7 M) at various time points (0, 1, 3, 6, 12, and 24 h). As shown in Fig. 3A, Ang II promoted a time-dependent reduction of Sirt6 fluorescence in the nuclei of podocytes, as observed via the immunofluorescence assays. In addition, the western blot analysis further revealed that Ang II-stimulated podocytes exhibited a time-dependent reduction in the Sirt6 protein levels (Fig. 3B). Ang II has been shown to induce oxidative DNA damage in podocytes in few studies. Therefore, we assessed the effects of Ang II on DNA damage in podocytes by immunofluorescence staining of activated H2AX (γH2AX, a marker for DNA damage). It suggested that Ang II markedly provoked an increase in the γH2AX expression in the podocytes compared with the cells cultured under normal conditions (Fig. 3C), which signified the dramatic DNA DSBs contributed by Ang II. Therefore, these results demonstrated that Ang II-exerted podocyte injury might be attributed to the DNA DSBs, and the suppression of Sirt6 may be involved in the regulatory mechanism.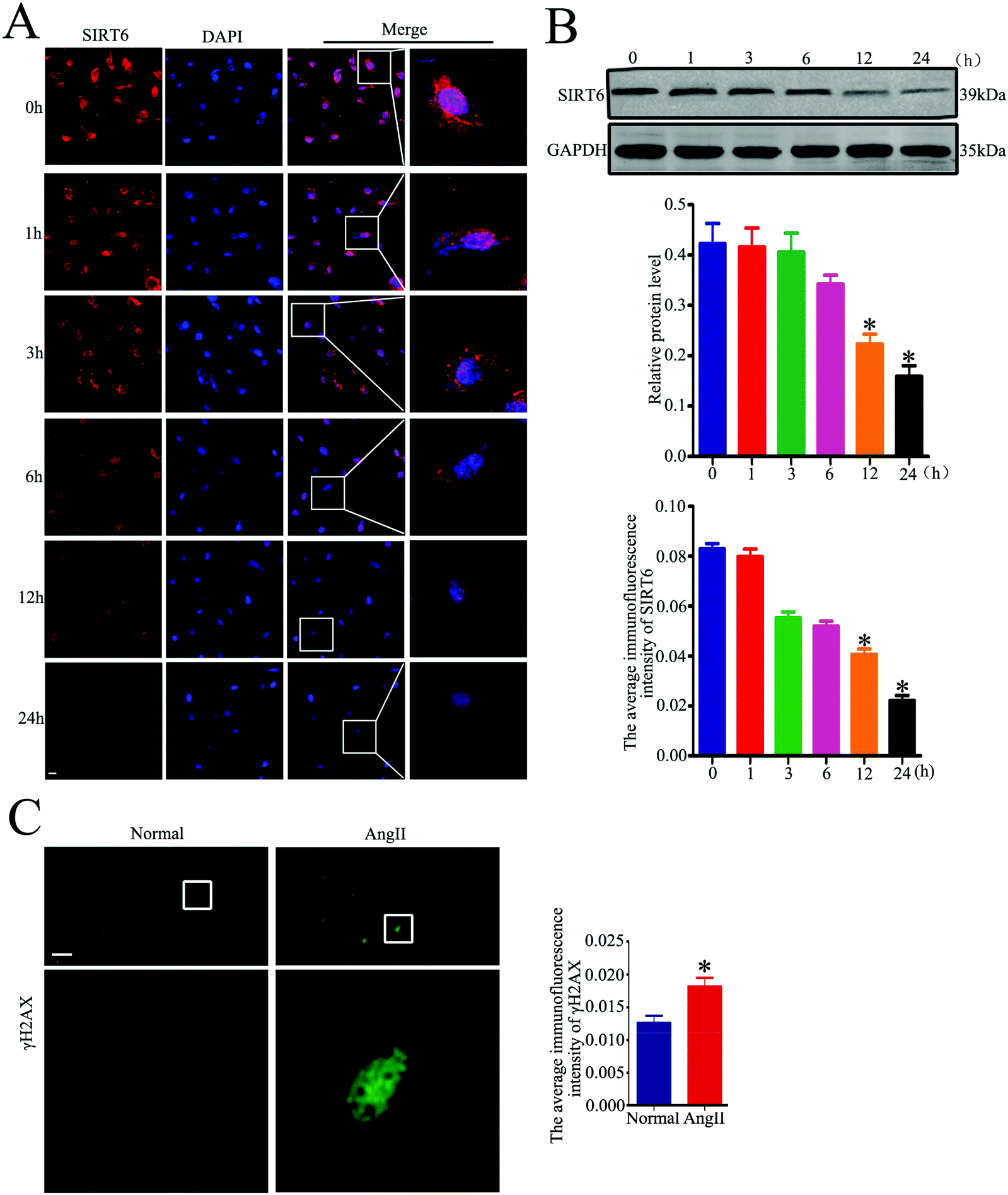 | ||
| Fig. 3 Sirt6 is reduced in Ang II-treated cultured podocytes accompanied by an increase of DNA DSBs. (A) Representative micrographs and quantification of Sirt6 staining in 10−7 M Ang II-treated podocytes at various times points (original magnification, ×400). *P < 0.05 relative to podocytes treated with Ang II for 0 h, n = 6. Scale bars: 20 μm. (B) Representative western blots and quantification of the Sirt6 protein level in 10−7 M Ang II-treated podocytes at various times points. *P < 0.05 relative to podocytes treated with Ang II for 0 h, n = 6. (C) Representative micrographs and quantification of γH2AX staining in 10−7 M Ang II-treated podocytes for 24 h (original magnification, ×400). *P < 0.05 relative to podocytes treated with Ang II for 0 h, n = 6. Scale bars: 40 μm. | ||
Sirt6 overexpression ameliorated Ang II-induced DNA DSBs and apoptosis in podocytes in vitro
Sirt6 has been demonstrated as an independent DNA damage sensor and can regulate DNA damage repair (DDR) and gene expression. Although Sirt6 was found to be involved in the regulation of DNA damage in human chondrocytes and endothelial cells, its role in DNA damage in podocytes is unknown. To investigate whether Sirt6 activation affects Ang II-induced podocyte DNA damage and apoptosis in vitro, a plasmid of Sirt6 (pcDNA3.1-Sirt6) was transfected into the cultured podocytes to overexpress Sirt6, followed by subsequent culture under Ang II conditions. The transfection efficiency of the plasmid was evaluated by western blot analysis, and we found that Sirt6 protein levels were increased in the plasmid-transfected cells under Ang II treatment (Fig. 4A). Accumulation of excessive amounts of ROS has been confirmed to promote oxidative DNA damage and cell apoptosis. It has been reported that Ang II-treated podocytes exhibit increased ROS production. First, we analyzed the effects of Sirt6 upregulation on Ang II-induced intracellular ROS production in the podocytes using DCFH-DA as a probe. As shown in Fig. 4B, Sirt6 overexpression prevented ROS production exerted by Ang II. Second, immunofluorescence staining for γH2AX was performed to detect DNA damage. We found that Sirt6 overexpression blocked Ang II-induced podocyte DNA damage (Fig. 4C). We next evaluated cell apoptosis in the podocytes by flow cytometry analysis. As shown in Fig. 4D, upregulating Sirt6 expression attenuated Ang II-induced apoptosis in the podocytes. These results suggested that overexpression of Sirt6 inhibited ROS production and DNA DSBs in Ang II-exposed podocytes. Taken together, we speculated that the cytoprotective effect of Sirt6 on the podocytes may be related to its restriction on oxidative DNA DSBs caused by ROS generation.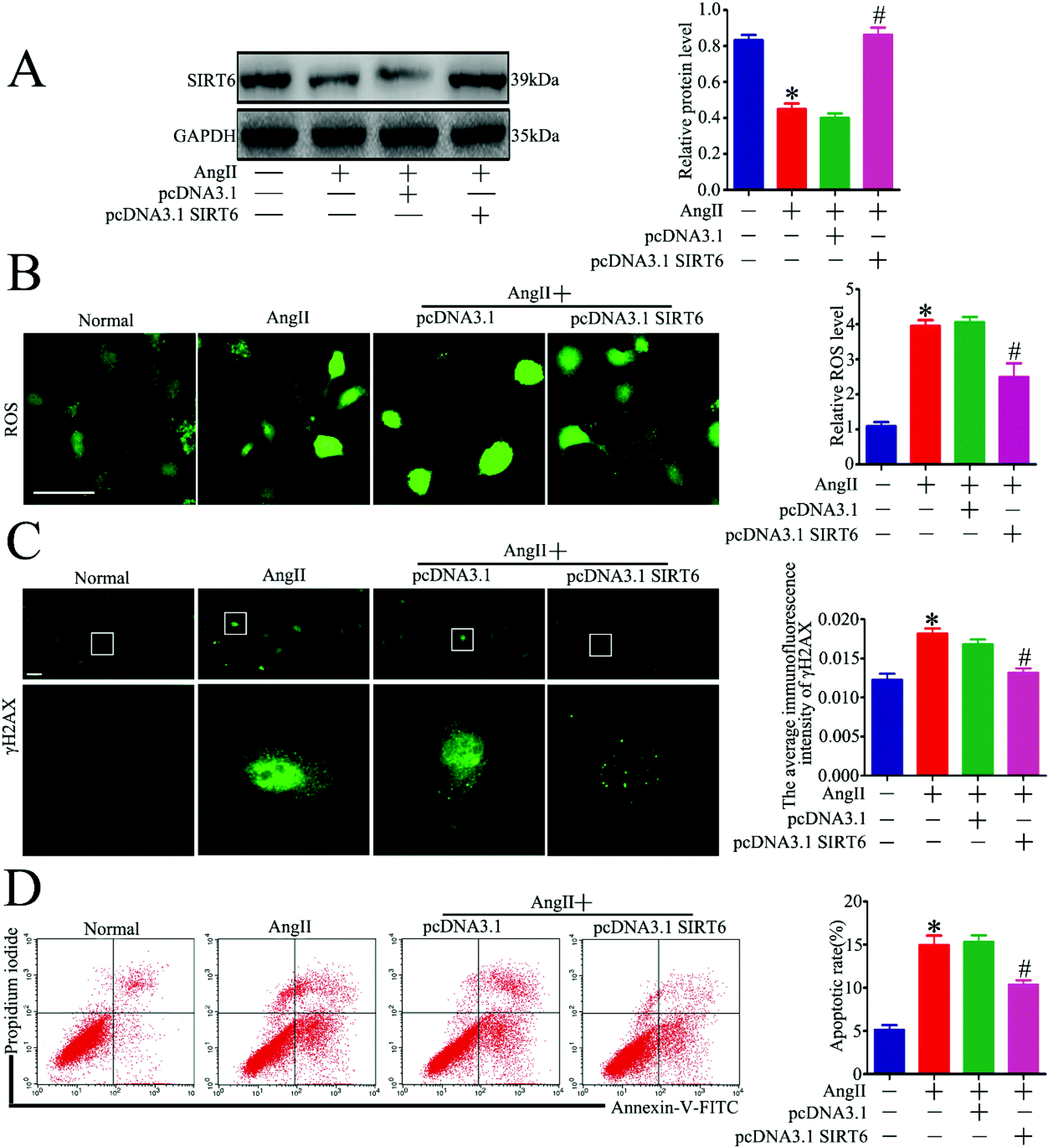 | ||
| Fig. 4 Sirt6 overexpression attenuated Ang II-induced DNA DSBs and apoptosis in podocytes in vitro. Podocytes were transfected with no plasmid, pcDNA3.1, or pcDNA3.1-SIRT6 and then treated with 10−7 M Ang II for 24 h. Untreated and untransfected podocytes were regarded as normal cells. (A) Representative western blots and quantification of Sirt6 protein levels in each group. *P < 0.05 relative to normal cells, #p < 0.05 compared with the cells stimulated with Ang II only, n = 3. (B) Representative micrographs and quantification of ROS staining in each group (original magnification, ×200). *P < 0.05 relative to normal cells, #p < 0.05 compared with the cells stimulated with Ang II only, n = 3. Scale bars: 15 μm. (C) Representative micrographs and quantification of γH2AX staining in each group (original magnification, ×400). *P < 0.05 relative to normal cells, #p < 0.05 compared with the cells stimulated with Ang II only, n = 3. Scale bars: 40 μm. (D) Flow cytometry analysis and quantification of podocyte apoptosis in each group. *P < 0.05 relative to normal cells, #p < 0.05 compared with the cells stimulated with Ang II only, n = 3. | ||
Sirt6 deletion exacerbated Ang II-induced DNA DSBs and apoptosis in podocytes in vitro
We next explored whether Sirt6 knockdown could potentiate Ang II-induced podocyte DNA damage and apoptosis in vitro. Sirt6 siRNA was transfected into the cultured podocytes to silence the Sirt6 expression before Ang II stimulation. The transfection efficiency was evaluated by western blot analysis, which showed that Sirt6 siRNA further reduced the Sirt6 expression in the podocytes incubated with Ang II (Fig. 5A). Gene silencing of Sirt6 provoked ROS production in Ang II-induced podocytes by using DCFH-DA as a probe (Fig. 5B). In addition, immunofluorescence staining for γH2AX detected that Sirt6 deficiency promoted Ang II-induced podocyte DNA damage (Fig. 5C). Moreover, Ang II-induced podocyte apoptosis was exacerbated by Sirt6 inhibition, which was evaluated by the flow cytometry analysis (Fig. 5D). These results suggested that Sirt6 upregulation inhibited ROS production and DNA DSBs in the Ang II-exposed podocytes. These results further suggested that Sirt6 exerted cytoprotection against Ang II-induced podocyte injury via preventing ROS production and DNA DSBs.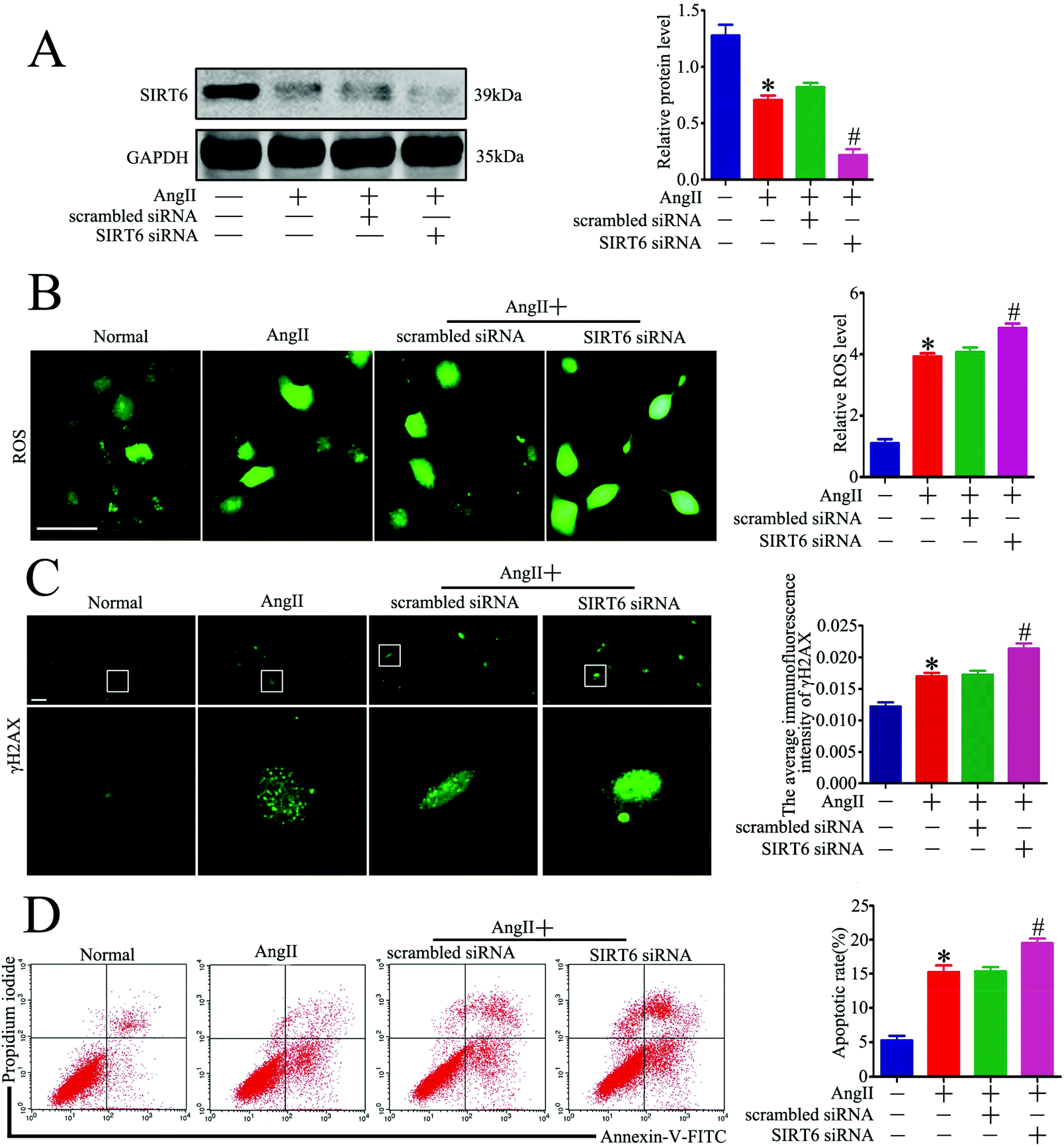 | ||
| Fig. 5 Sirt6 inhibition promoted Ang II-induced DNA DSBs and apoptosis in podocytes in vitro. Podocytes were transfected with no siRNA, scrambled siRNA, or Sirt6 siRNA and then treated with 10−7 M Ang II for 24 h. Untreated and untransfected podocytes were regarded as normal cells. (A) Representative western blots and quantification of Sirt6 protein levels in the different groups. *P < 0.05 relative to normal cells, #p < 0.05 compared with the cells treated with Ang II only, n = 3. (B) Representative micrographs and quantification of ROS staining in each group (original magnification, ×200). *P < 0.05 relative to normal cells, #p < 0.05 compared with the cells stimulated with Ang II only, n = 3. Scale bars: 15 μm. (C) Representative micrographs and quantification of γH2AX staining in the different groups (original magnification, ×400). *P < 0.05 relative to normal cells, #p < 0.05 compared with the cells treated with Ang II only, n = 3. Scale bars: 40 μm. (D) Flow cytometry analysis and quantification of podocyte apoptosis in the different groups. *P < 0.05 relative to normal cells, #p < 0.05 compared with the cells treated with Ang II only, n = 3. | ||
Sirt6 regulated Nrf2/HO-1 signaling in Ang II-treated podocytes in vitro
Oxidative stress is a critical mediator of podocyte injury caused by Ang II. And the above results have demonstrated that podocyte apoptosis injury caused by Ang II may be associated with oxidative stress induced DNA damage. The Nrf2/HO-1 signaling pathway has been suggested to be involved in restricting oxidative stress-induced DNA damage and apoptosis.47,48,50 To investigate the underlying molecular mechanism of the Sirt6-mediated neuroprotective effect in podocytes, we assessed the regulatory effect of Sirt6 on the antioxidant Nrf2/HO-1 pathway. Our results indicated that Sirt6 overexpression ameliorated Ang II-induced reduction of Nrf2 in nuclear expression. Meanwhile, Sirt6 overexpression also attenuated Ang II-induced decrease in HO-1 expression, which was a downstream target gene of Nrf2 (Fig. 6A). In contrast, gene silencing of Sirt6 had the opposite effect on Nrf2/HO-1 signaling (Fig. 6B). These data suggested that the cytoprotective effects of Sirt6 overexpression were likely mediated by activating the Nrf2/HO-1 pathway.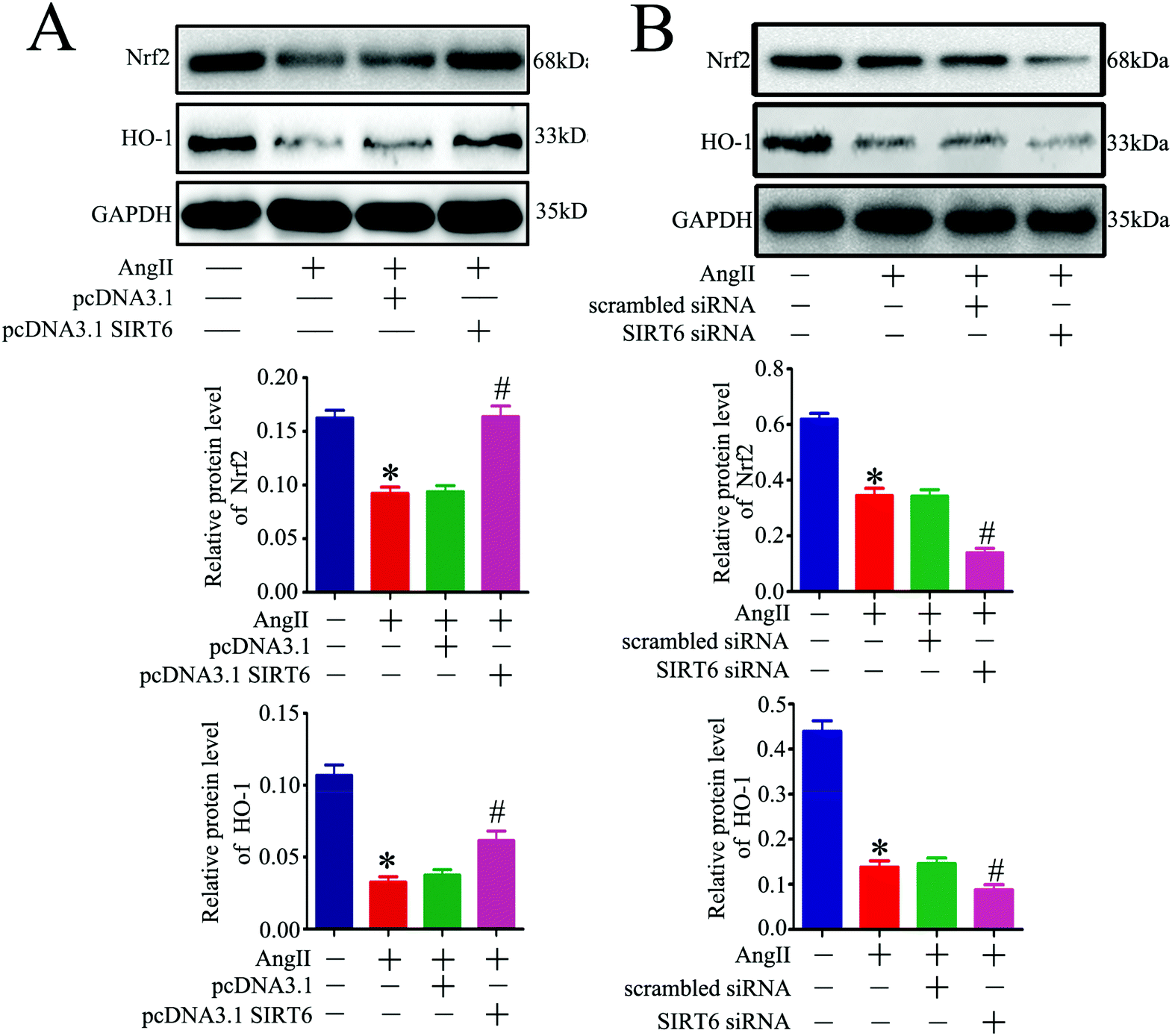 | ||
| Fig. 6 Sirt6 regulated Ang II-induced podocyte DNA damage and apoptosis through the Nrf2/HO-1 pathway. (A) Podocytes were transfected with no plasmid, pcDNA3.1, or pcDNA3.1-SIRT6 and then treated with 10−7 M Ang II for 24 h. Untreated and untransfected podocytes were regarded as normal cells. Representative western blots and quantification of the Nrf2 and HO-1 protein levels in each group. *P < 0.05 relative to normal cells, #p < 0.05 compared with the cells stimulated with Ang II only, n = 3. (B) Podocytes were transfected with no siRNA, scrambled siRNA, or Sirt6 siRNA and then treated with 10−7 M Ang II for 24 h. Untreated and untransfected podocytes were regarded as normal cells. Representative western blots and quantification of the Nrf2 and HO-1 protein levels in the different groups. *P < 0.05 relative to normal cells, #p < 0.05 compared with the cells stimulated with Ang II only, n = 3. | ||
Nrf2 knockdown attenuated the protective effect of Sirt6 against Ang II-induced podocytes
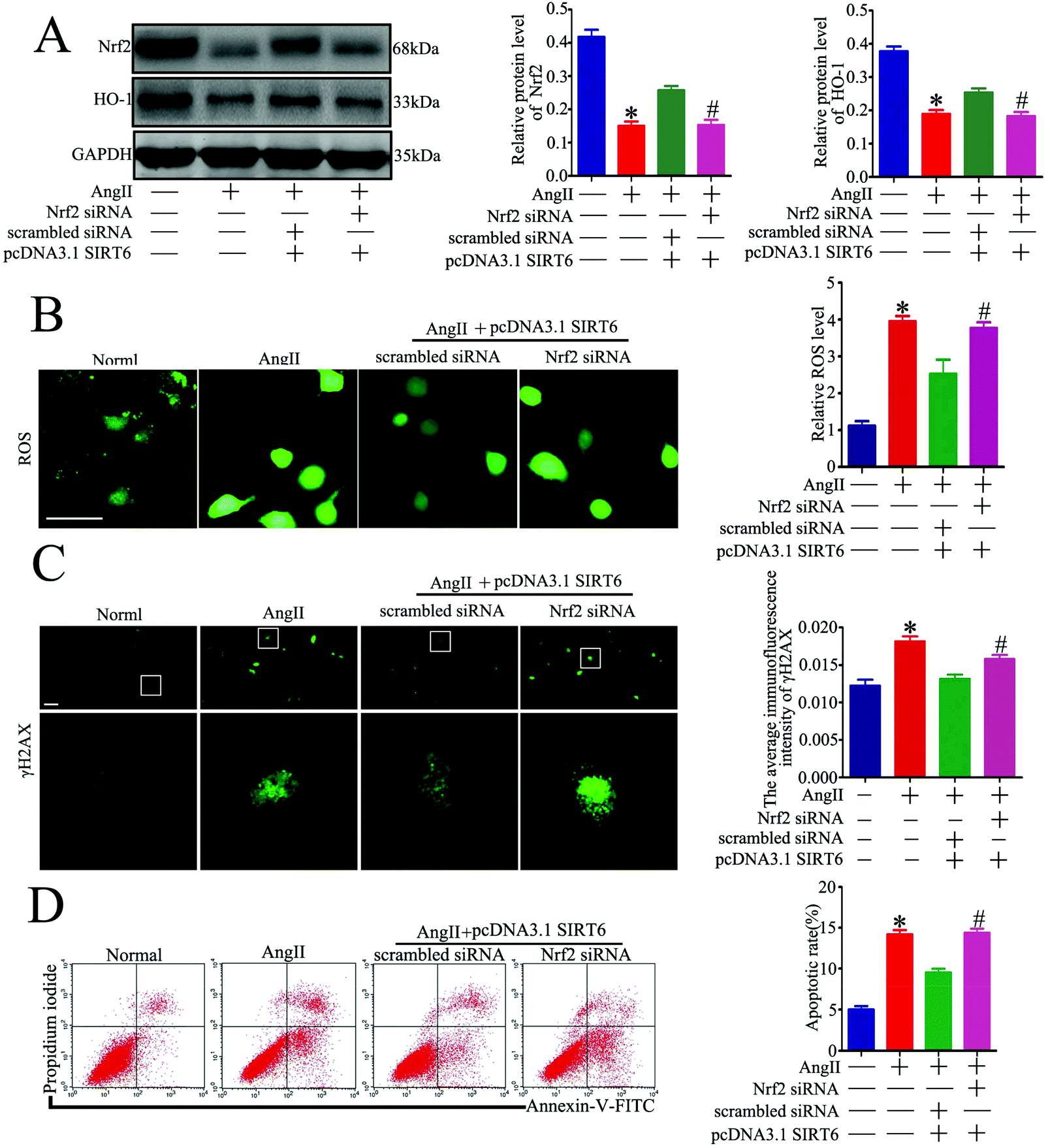 | ||
| Fig. 7 Sirt6 overexpression-induced protective effect in podocytes is blocked by Nrf2 inhibition. Podocytes were transfected with scrambled siRNA, or Nrf2 siRNA before being transfected with pcDNA3.1-SIRT6 and were then subjected to 10−7 M Ang II for 24 h. Untreated and untransfected podocytes were regarded as normal cells. (A) Representative western blots and quantification of the Nrf2 and HO-1 protein levels in the different groups. *P < 0.05 relative to normal cells, #p < 0.05 compared with the cells transfected with scrambled siRNA and pcDNA3.1-SIRT6 pretreated with Ang II, n = 3. (B) Representative micrographs and quantification of ROS staining in each group (original magnification, ×200). *P < 0.05 relative to cells treated with Ang II, #p < 0.05 compared with the cells transfected with scrambled siRNA and pcDNA3.1-SIRT6 pretreated with Ang II, n = 3. Scale bars: 15 μm. (C) Representative micrographs and quantification of γH2AX staining in the different groups (original magnification, ×400). *P < 0.05 relative to normal cells, #p < 0.05 compared with the cells transfected with scrambled siRNA and pcDNA3.1-SIRT6 pretreated with Ang II, n = 3. Scale bars: 40 μm. (D) Flow cytometry analysis and quantification of podocyte apoptosis in the different groups. *P < 0.05 relative to normal cells, #p < 0.05 compared with the cells transfected with scrambled siRNA and pcDNA3.1-SIRT6 pretreated with Ang II, n = 3. | ||
Discussion
Increased DNA damage induced by activated renin–angiotensin system and impaired DNA repair caused by decreased expression of DNA repair factors have been recently reported in kidney diseases.37 As the primary effector in chronic kidney disease (CKD) progression, angiotensin II (Ang II) can cause DNA damage including DSBs in podocytes.22–24 Accumulating evidence has revealed that DNA methylation in podocytes is involved in the pathogenesis of CKD, and impaired DNA DSB repair is recently implicated in the induction of altered DNA methylation in podocytes.19,37–41 Thus, we suppose that overproduction of DNA DSBs in podocytes contributes to glomerular disorders. In this regard, it makes sense to explore the key molecules and the potential signaling pathways involved in DNA DSB regulation in podocytes. Herein, we demonstrated that Sirt6 provided cytoprotection against Ang II-induced DNA DSBs and podocyte injury by activating the Nrf2/HO-1 pathway, which suggested that Sirt6 might be a therapeutic target for CKD.Recent research illustrated that DNA DSBs was associated with altered DNA methylation in murine models of kidney disease, and DNA DSBs and DNA methylation were involved in the pathogenesis of CKD. Hayashi et al. detected that the expression of the DNA DSB marker γH2AX in the podocytes of the glomeruli was significantly increased in patients with IgA nephropathy compared to the controls. They further observed that the glomerular γH2AX level was significantly associated with the slope of eGFR decline over one year in IgA nephropathy patients. These results suggested that renal DNA DSBs may influence renal function of human.42 However, DSBs in human kidneys have not been adequately investigated so far, and the status of DSBs in the glomeruli of patients with HN has not been reported yet. Our data showed that γH2AX-positive areas in the glomeruli were increased in patients with HN compared to the controls, which indicated that DNA DSB formation might contribute to the pathogenesis of HN. Intriguingly, very few DNA DSB sites were observed in the glomeruli of healthy subjects, suggesting that the DNA repair mechanism may be pivotal in kidneys with physiological states. In addition, these findings were further verified by corresponding in vitro experiments. Consistently, cultured podocytes treated with Ang II exhibited an increase in γH2AX foci induction, suggesting that Ang II promoted DNA DSBs in podocytes. Our findings provided additional evidence that DNA DSBs existed in podocytes under Ang II stimulation and DNA damage may be associated with podocyte injury in an Ang II environment.
Sirt6 has been considered as a DNA DSB sensor and is involved in DNA repair.18,21,43–45 It was found to participate in the regulation of DNA damage in a variety of cells. Sirt6 was suggested to play an important role in protecting chondrocytes from DNA damage, and depletion of Sirt6 led to accumulation of γH2AX foci.34 Similarly, Sirt6 overexpression prevented Aβ42-induced DNA damage in HT22 mouse hippocampal neurons.35 Cardus et al. demonstrated that the presence of Sirt6 in endothelial cells provided protection from genomic DNA damage.36 However, whether Sirt6 is involved in regulating Ang II-induced DNA damage in podocytes has not been reported yet. We demonstrated for the first time that Sirt6 expression was reduced along with increase of DNA DSBs in the glomeruli of patients with HN. Moreover, the results showed that Sirt6 expression was decreased accompanied by increase of DNA DSBs in Ang II-treated podocytes, which was consistent with the findings of previous studies. Considering this result, we speculated that Sirt6 may be involved in Ang II-induced DNA damage in podocytes. To further explore the role of Sirt6 in Ang II-induced DNA damage, podocytes transfected with the Sirt6 plasmid or Sirt6 siRNA were used to regulate the expression of Sirt6. We observed that Sirt6 overexpression inhibited Ang II-induced DNA damage and apoptosis in the cultured podocytes. Rather, Sirt6 inhibition potentiated Ang II-induced DNA damage and podocyte injury. We illustrated that Sirt6 exhibited a protective role against DNA damage. In support of this idea, several studies have described the mechanisms through which Sirt6 promoted DNA repair. Further studies on the underlying mechanisms revealed that Sirt6 can mono-ADP-ribosylate PARP1 to stimulate PARP1 activity and enhance DNA DSB repair under oxidative stress.46 Sirt6 was revealed to be recruited to the sites of DSBs and strongly stimulated both pathways of DSB repair. A recent study showed that Sirt6 directly recognized DNA damage through a tunnel-like structure that had high affinity for DNA DSBs.18 The transfer of Sirt6 to the damaged location is independent of the signal and known sensors. It activated downstream signaling for DNA DSB repair by triggering ATM recruitment, H2AX phosphorylation and the recruitment of proteins related to homologous recombination and non-homologous end joining pathways.18 Moreover, a comparative analysis of 18 rodent species identified the role of Sirt6-dependent DNA DSB repair as a major factor in organismal lifespan, which elucidated that Sirt6 was responsible for more efficient DNA DSB repair in long-lived species.43 Together with these previous findings, our observation indicated that the specific mechanisms by which Sirt6 affects podocyte DNA damage may be related to its influence on DNA DSB formation and repair.
The accumulation of ROS caused by DNA damage was considered to be involved in cell apoptosis. ROS acts as the second messengers in intracellular signaling cascades. Therefore, ROS overproduction plays a crucial role in the modulation of gene expression and DNA peroxidation.47 Since oxidative stress was known to be a major cause of DNA damage, we assessed the ROS content in the podocytes exposed to Ang II. The data indicated that Ang II promoted ROS production in the podocytes, which explained why Ang II caused DNA damage. We further identified that Ang II-provoked podocyte ROS generation and apoptosis were reversed by Sirt6 upregulation. Our results elucidated that Sirt6 exhibited protective effects against Ang II-induced podocyte injury and DNA damage through the suppression of intracellular ROS generation.
Regarding the specific mechanisms by which Sirt6 affects podocyte DNA DSBs, we detected that the antioxidant Nrf2/HO-1 signaling pathway was involved in Sirt6-mediated cytoprotection in podocytes. Nrf2 antioxidant signaling regulated by Sirt6 has been shown to be involved in inhibiting apoptosis and oxidative stress in Ang II-treated vascular endothelial cells.52 Nrf2 knockdown abolished the protective effect of Sirt6 against Ang II-induced podocyte DNA damage and injury. Growing evidence has shown that Nrf2 pathway activation consists of a cellular protective system that promotes DNA damage recognition and repair, and cell survival in detrimental environments.47–49 Nrf2 is considered as a master regulator of redox homeostasis to perform its anti-oxidative, anti-inflammatory and anti-apoptotic activities. Nrf2 plays a central role in protecting cells from oxidative stress-induced DNA injury and cytotoxicity by regulating the transcriptional activity of antioxidant genes. Among the Nrf2-dependent cytoprotective enzymes, HO-1 has been shown to exhibit adaptive and beneficial cellular responses to resist DNA damage and apoptosis induced by oxidative stress. Nrf2/HO-1 has been shown to participate in the regulation of oxidative DNA DSBs and apoptosis in a variety of studies.50 In the present study, we observed an inhibition of Nrf2/HO-1 in Ang II-treated podocytes and also found that Sirt6 can induce HO-1 through the transcription factor Nrf2. Although we have evidenced that HO-1 protein levels increased via Sirt6 overexpression, it is worth noting that it did not implicate HO-1 to be the only factor involved in the protective effects of Sirt6. Therefore, further studies on the relevance of various Nrf2-dependent enzymes besides HO-1 should be carried out; however, the current results demonstrated that the protective potential of Sirt6 against Ang II-induced DNA damage in podocytes was at least dependent on the activation of Nrf2/HO-1 signaling. And it will be interesting to study the in vivo effects of the Sirt6/Nfr2/HO-1 cytoprotective pathway on Ang II-induced DNA damage and tissue injury.
A lot of animal and in vitro studies recently confirmed that Sirt6 played a renal protective role in kidney disease through different signaling pathways, including DN, kidney fibrosis, kidney aging and AKI.6,32,33,51 Based on these studies and our studies, we speculate that Sirt6 may be a therapeutic target for CKD. Currently, no clinical trial has been conducted to explore the therapeutic effect of Sirt6 on renal disease. More clinical studies are needed in the future to explore whether Sirt6 can be used as a target molecule for the treatment of CKD. Nrf2 activation has been reported to ameliorate renal disease progression in many experimental studies. The Nrf2 signaling pathway serves as a protection against oxidative stress and inflammation in CKD and AKI.53–55 It exerts renoprotective effects in inhibiting long-term fibrosis after AKI and preventing fibrosis in CKD. Recent clinical trials targeting Nrf2 in renal diseases have highlighted its beneficial effects in improving renal function in patients with diabetes and stage 3 CKD. An exploratory phase 2 clinical trial showed that the Nrf2 inducer bardoxolone methyl increased GFR in patients with DN. These beneficial effects of NRF2 activation demonstrate that NRF2 acts as a vital target for kidney disease treatment.53–55
Our findings concluded that the Sirt6/Nrf2/HO-1 pathway played a critical role in the cellular defense mechanism in podocytes. Ang II-induced ROS generation and DNA DSBs were reversed by Sirt6. We also highlighted that the antioxidant effects of Sirt6 involved the regulation of the Nrf2/HO-1 defense pathway and subsequently served as a protection against podocyte apoptosis (Fig. 8). In summary, this study demonstrated that Sirt6 exerted cytoprotection against podocyte injury, at least in part through the Nfr2/HO-1 pathway. We proposed that Sirt6 may be a novel therapeutic target for CKD treatment and our observations have pathophysiological significance with translational importance.
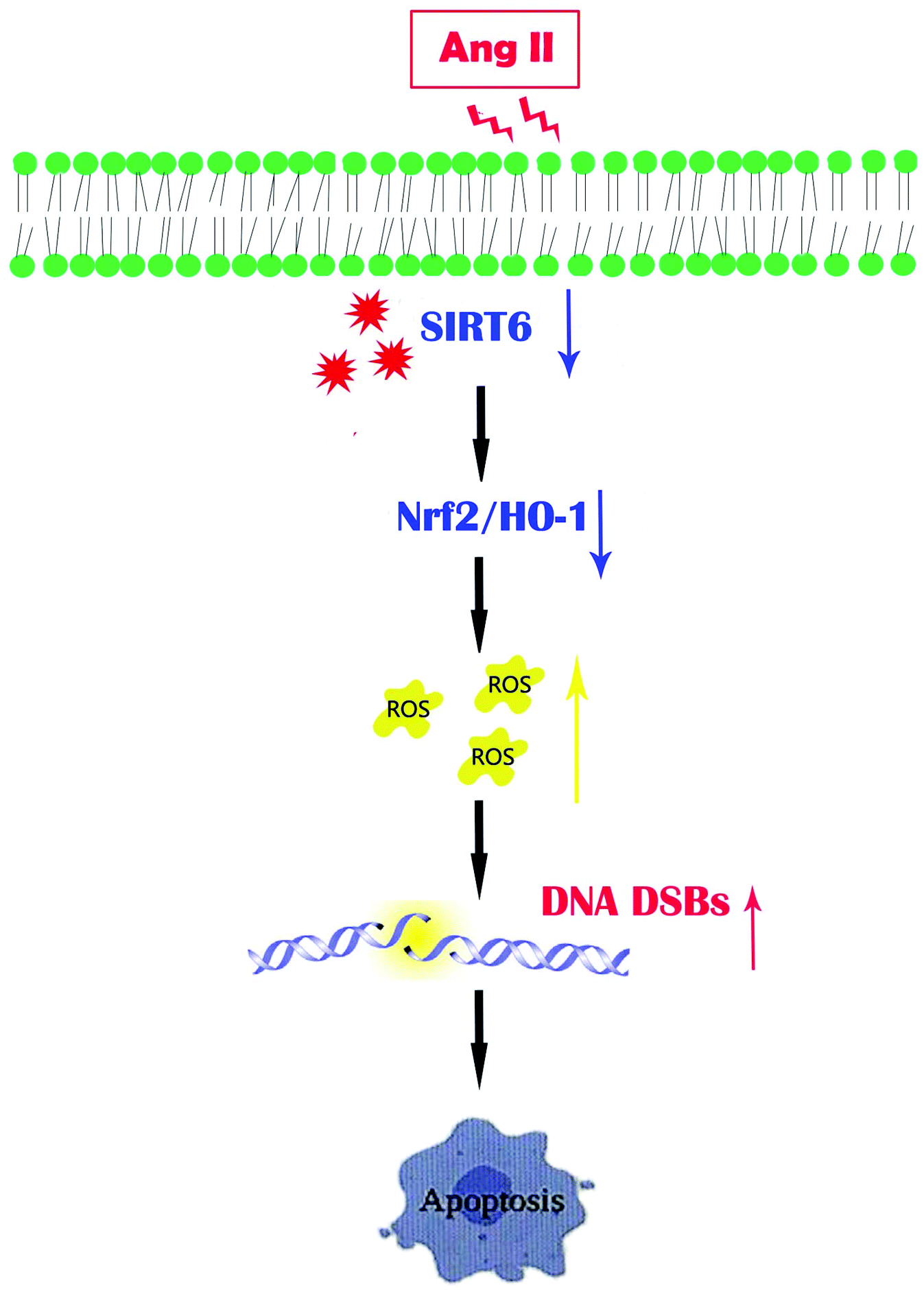 | ||
| Fig. 8 A diagram of the proposed signaling of the Sirt6-mediated Nrf2/HO-1 pathway activation in Ang II-induced podocyte injury. Ang II suppresses the Nrf2/HO-1 signaling pathway, and provokes ROS generation and DNA DSBs, which finally contribute to apoptosis in podocytes. However, Sirt6 restoration promotes Nrf2/HO-1 transduction to exert cytoprotection against Ang II-induced podocyte injury. | ||
Abbreviations
| Ang II | Angiotensin II |
| CKD | Chronic kidney disease |
| DDR | DNA damage repair |
| DSBs | DNA double-strand breaks |
| DN | Diabetic nephropathy |
| HN | Hypertensive nephropathy |
| HO-1 | Heme oxygenase 1 |
| Nrf2 | Nuclear factor erythroid 2 (NF-E2)-related factor 2 |
| PAS | Periodic acid–Schiff |
| RAS | Renin–angiotensin system |
| ROS | Reactive oxygen species |
| Sirt6 | Sirtuin6 |
| TEM | Transmission electron microscopy |
Ethics statement
Study on human kidney tissue staining was conducted in accordance with the approved guidelines of Wuhan University and was approved by the Research Ethics Committee of Renmin Hospital of Wuhan University. The investigation complied with the principles of the Declaration of Helsinki.Rats were raised in an environment with an artificial light cycle with controlled temperature and humidity at the Center for Animal Experiments of Wuhan University. All animal studies were carried out in accordance with the recommendations of “the ethical guidelines of the Animal Ethical Committee of Renmin Hospital of Wuhan University”. All procedures were approved by the Ethical Committee for the Experimental Use of Animals of Renmin Hospital in Wuhan, China.
Author contributions
G.D. designed the study and directed the manuscript. Y.F. and J.C. helped to design the study, performed most of the experiments, collected the data and wrote the manuscript. J.H. helped to perform the animal experiments. J.F. helped to culture podocytes. H.Y. help to guide pathological staining and film reading. Q.Y., Z.R. and D.Y. helped to modify the manuscript.Conflicts of interest
The authors have declared that no competing interest exists.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81570617 to G. D. and 81670631 to D. Y.) and the Fundamental Research Funds for the Central Universities (2042020kf0090 to Y. F.). All authors approved the final version of the manuscript.References
- E. Y. Chung, M. Ruospo, P. Natale, D. Bolignano, S. D. Navaneethan and S. C. Palmer, et al., Aldosterone antagonists in addition to renin angiotensin system antagonists for preventing the progression of chronic kidney disease, Cochrane Database Syst. Rev., 2020, 10, CD007004 Search PubMed.
- A. A. Saldanha da Silva, T. R. Rodrigues Prestes, A. O. Lauar, B. B. Finotti and A. C. Simoes E. Silva, Renin Angiotensin System and Cytokines in Chronic Kidney Disease: Clinical and Experimental Evidence, Protein Pept. Lett., 2017, 24(9), 799–808 CrossRef CAS PubMed.
- H. Yaribeygi, M. Maleki, T. Sathyapalan, T. Jamialahmadi and A. Sahebkar, Incretin-based therapies and renin-angiotensin system: Looking for new therapeutic potentials in the diabetic milieu, Life Sci., 2020, 256, 117916 CrossRef CAS PubMed.
- C. L. Laffer, F. Elijovich, M. Sahinoz, A. Pitzer and A. Kirabo, New Insights Into the Renin-Angiotensin System in Chronic Kidney Disease, Circ. Res., 2020, 127(5), 607–609 CrossRef CAS PubMed.
- K. Zhang, C. Fan, D. Cai, Y. Zhang, R. Zuo and L. Zhu, et al., Contribution of TGF-Beta-Mediated NLRP3-HMGB1 Activation to Tubulointerstitial Fibrosis in Rat With Angiotensin II-Induced Chronic Kidney Disease, Front. Cell Dev. Biol., 2020, 8, 1 CrossRef PubMed.
- Q. Yang, J. Hu, Y. Yang, Z. Chen, J. Feng and Z. Zhu, et al., Sirt6 deficiency aggravates angiotensin II-induced cholesterol accumulation and injury in podocytes, Theranostics, 2020, 10(16), 7465–7479 CrossRef CAS PubMed.
- Y. Ma, Q. Yang, Z. Zhong, W. Liang, L. Zhang and Y. Yang, et al., Role of c-Abl and nephrin in podocyte cytoskeletal remodeling induced by angiotensin II, Cell Death Dis., 2018, 9(2), 185 CrossRef PubMed.
- L. Koppe and D. Fouque, The Role for Protein Restriction in Addition to Renin-Angiotensin-Aldosterone System Inhibitors in the Management of CKD, Am. J. Kidney Dis., 2019, 73(2), 248–257 CrossRef CAS PubMed.
- A. C. Webster, E. V. Nagler, R. L. Morton and P. Masson, Chronic Kidney Disease, Lancet, 2017, 389(10075), 1238–1252 CrossRef.
- M. D. Breyer and K. Susztak, The next generation of therapeutics for chronic kidney disease, Nat. Rev. Drug Discovery, 2016, 15(8), 568–588 CrossRef CAS PubMed.
- S. J. Allison, Chronic kidney disease: Actin cytoskeleton alterations in podocytes: a therapeutic target for chronic kidney disease, Nat. Rev. Nephrol., 2015, 11(7), 385 CrossRef CAS PubMed.
- P. Garg, A Review of Podocyte Biology, Am. J. Nephrol., 2018, 47(Suppl 1), 3–13 CrossRef CAS PubMed.
- S. S. Hayek, K. H. Koh, M. E. Grams, C. Wei, Y. A. Ko and J. Li, et al., A tripartite complex of suPAR, APOL1 risk variants and alphavbeta3 integrin on podocytes mediates chronic kidney disease, Nat. Med., 2017, 23(8), 945–953 CrossRef CAS PubMed.
- P. Beckerman, J. Bi-Karchin, A. S. Park, C. Qiu, P. D. Dummer and I. Soomro, et al., Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice, Nat. Med., 2017, 23(4), 429–438 CrossRef CAS PubMed.
- Y. Guo, J. Pace, Z. Li, A. Ma'ayan, Z. Wang and M. P. Revelo, et al., Podocyte-Specific Induction of Kruppel-Like Factor 15 Restores Differentiation Markers and Attenuates Kidney Injury in Proteinuric Kidney Disease, J. Am. Soc. Nephrol., 2018, 29(10), 2529–2545 CrossRef CAS PubMed.
- Q. Yang, Y. Ma, Y. Liu, W. Liang, X. Chen and Z. Ren, et al., Angiotensin II down-regulates nephrin-Akt signaling and induces podocyte injury: roleof c-Abl, Mol. Biol. Cell, 2016, 27(1), 197–208 CrossRef CAS PubMed.
- L. Zhang, Z. Ren, Q. Yang and G. Ding, Csk regulates angiotensin II-induced podocyte apoptosis, Apoptosis, 2016, 21(7), 846–855 CrossRef CAS PubMed.
- L. Onn, M. Portillo, S. Ilic, G. Cleitman, D. Stein and S. Kaluski, et al., SIRT6 is a DNA double-strand break sensor, eLife, 2020, 29(9), e51636 CrossRef PubMed.
- A. Hishikawa, K. Hayashi, T. Abe, M. Kaneko, H. Yokoi and T. Azegami, et al., Decreased KAT5 Expression Impairs DNA Repair and Induces Altered DNA Methylation in Kidney Podocytes, Cell Rep., 2019, 26(5), 1318–1332 CrossRef CAS PubMed.
- A. Geng, H. Tang, J. Huang, Z. Qian, N. Qin and Y. Yao, et al., The deacetylase SIRT6 promotes the repair of UV-induced DNA damage by targeting DDB2, Nucleic Acids Res., 2020, 48(16), 9181–9194 CrossRef CAS PubMed.
- T. Hou, Z. Cao, J. Zhang, M. Tang, Y. Tian and Y. Li, et al., SIRT6 coordinates with CHD4 to promote chromatin relaxation and DNA repair, Nucleic Acids Res., 2020, 48(6), 2982–3000 CrossRef CAS PubMed.
- K. Fukami, S. Yamagishi, K. Kaifu, T. Matsui, Y. Kaida and S. Ueda, et al., Telmisartan inhibits AGE-induced podocyte damage and detachment, Microvasc. Res., 2013, 88, 79–83 CrossRef CAS PubMed.
- G. J. Weber, B. Purkayastha, L. Ren, S. Pushpakumar and U. Sen, Hypertension exaggerates renovascular resistance via miR-122-associated stress response in aging, J. Hypertens., 2018, 36(11), 2226–2236 CrossRef CAS.
- P. Rai, T. Singh, R. Lederman, A. Chawla, D. Kumar and K. Cheng, et al., Hyperglycemia enhances kidney cell injury in HIVAN through down-regulation of vitamin D receptors, Cell. Signalling, 2015, 27(3), 460–469 CrossRef CAS PubMed.
- A. R. Chang, C. M. Ferrer and R. Mostoslavsky, SIRT6, a Mammalian Deacylase with Multitasking Abilities, Physiol. Rev., 2020, 100(1), 145–169 CrossRef CAS PubMed.
- M. de Ceu Teixeira, E. Sanchez-Lopez, M. Espina, M. L. Garcia, A. Durazzo and M. Lucarini, et al., Sirtuins and SIRT6 in Carcinogenesis and in Diet, Int. J. Mol. Sci., 2019, 20(19), 4945 CrossRef PubMed.
- R. I. Khan, S. S. R. Nirzhor and R. Akter, A Review of the Recent Advances Made with SIRT6 and its Implications on Aging Related Processes, Major Human Diseases, and Possible Therapeutic Targets, Biomolecules, 2018, 8(3), 44 CrossRef PubMed.
- M. Vitiello, A. Zullo, L. Servillo, F. P. Mancini, A. Borriello and A. Giovane, et al., Multiple pathways of SIRT6 at the crossroads in the control of longevity, cancer, and cardiovascular diseases, Ageing Res. Rev., 2017, 35, 301–311 CrossRef CAS PubMed.
- J. Kuang, L. Chen, Q. Tang, J. Zhang, Y. Li and J. He, The Role of Sirt6 in Obesity and Diabetes, Front Physiol., 2018, 9, 135 CrossRef PubMed.
- L. Tasselli, W. Zheng and K. F. Chua, SIRT6: Novel Mechanisms and Links to Aging and Disease, Trends Endocrinol. Metab., 2017, 28(3), 168–185 CrossRef CAS PubMed.
- M. Kitada, Y. Ogura, I. Monno and D. Koya, Sirtuins and Type 2 Diabetes: Role in Inflammation, Oxidative Stress, and Mitochondrial Function, Front. Endocrinol., 2019, 10, 187 CrossRef PubMed.
- M. Morigi, L. Perico and A. Benigni, Sirtuins in Renal Health and Disease, J. Am. Soc. Nephrol., 2018, 29(7), 1799–1809 CrossRef CAS PubMed.
- M. Liu, K. Liang, J. Zhen, M. Zhou, X. Wang and Z. Wang, et al., Sirt6 deficiency exacerbates podocyte injury and proteinuria through targeting Notch signaling, Nat. Commun., 2017, 8(1), 413 CrossRef PubMed.
- K. Nagai, T. Matsushita, T. Matsuzaki, K. Takayama, T. Matsumoto and R. Kuroda, et al., Depletion of SIRT6 causes cellular senescence, DNA damage, and telomere dysfunction in human chondrocytes, Osteoarthritis cartilage, 2015, 23(8), 1412–1420 CrossRef CAS PubMed.
- E. S. Jung, H. Choi, H. Song, Y. J. Hwang, A. Kim and H. Ryu, et al., p53-dependent SIRT6 expression protects Abeta42-induced DNA damage, Sci. Rep., 2016, 6, 25628 CrossRef CAS PubMed.
- A. Cardus, A. K. Uryga, G. Walters and J. D. Erusalimsky, SIRT6 protects human endothelial cells from DNA damage, telomere dysfunction, and senescence, Cardiovasc. Res., 2013, 97(3), 571–579 CrossRef CAS PubMed.
- K. Hayashi, A. Hishikawa and H. Itoh, DNA Damage Repair and DNA Methylation in the Kidney, Am. J. Nephrol., 2019, 50(2), 81–91 CrossRef CAS PubMed.
- H. Hamatani, T. Sakairi, H. Ikeuchi, Y. Kaneko, A. Maeshima and Y. Nojima, et al., TGF-beta1 alters DNA methylation levels in promoter and enhancer regions of the WT1 gene in human podocytes, Nephrology, 2019, 24(5), 575–584 CrossRef CAS PubMed.
- H. Li, W. Zhang, F. Zhong, G. C. Das, Y. Xie and Z. Li, et al., Epigenetic regulation of RCAN1 expression in kidney disease and its role in podocyte injury, Kidney Int., 2018, 94(6), 1160–1176 CrossRef CAS PubMed.
- L. Zhang, Q. Zhang, S. Liu, Y. Chen, R. Li and T. Lin, et al., DNA methyltransferase 1 may be a therapy target for attenuating diabetic nephropathy and podocyte injury, Kidney Int., 2017, 92(1), 140–153 CrossRef CAS PubMed.
- Z. Li, H. Chen, F. Zhong, W. Zhang, K. Lee and J. C. He, Expression of Glutamate Receptor Subtype 3 Is Epigenetically Regulated in Podocytes under Diabetic Conditions, Kidney Dis., 2019, 5(1), 34–42 CrossRef PubMed.
- K. Hayashi, A. Hishikawa, A. Hashiguchi, T. Azegami, N. Yoshimoto and R. Nakamichi, et al., Association of glomerular DNA damage and DNA methylation with one-year eGFR decline in IgA nephropathy, Sci. Rep., 2020, 10(1), 237 CrossRef CAS PubMed.
- X. Tian, D. Firsanov, Z. Zhang, Y. Cheng, L. Luo and G. Tombline, et al., SIRT6 Is Responsible for More Efficient DNA Double-Strand Break Repair in Long-Lived Species, Cell, 2019, 177(3), 622–638 CrossRef CAS PubMed.
- F. Meng, M. Qian, B. Peng, L. Peng and B. Liu, Synergy between SIRT1 and SIRT6 helps recognize DNA breaks and potentiates the DNA damage response and repair in humans and mice, eLife, 2020, 15(9), 55828 CrossRef PubMed.
- R. A. McCord, E. Michishita, T. Hong, E. Berber, L. D. Boxer and R. Kusumoto, et al., SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair, Aging, 2009, 1(1), 109–121 CrossRef CAS PubMed.
- Z. Mao, C. Hine, X. Tian, M. Van Meter, M. Au and A. Vaidya, et al., SIRT6 promotes DNA repair under stress by activating PARP1, Science, 2011, 332(6036), 1443–1446 CrossRef CAS PubMed.
- Z. Shi, Y. Niu, Q. Wang, L. Shi, H. Guo and Y. Liu, et al., Reduction of DNA damage induced by titanium dioxide nanoparticles through Nrf2 in vitro and in vivo, J. Hazard. Mater., 2015, 298, 310–319 CrossRef CAS PubMed.
- B. L. Ya, Q. Liu, H. F. Li, H. J. Cheng, T. Yu and L. Chen, et al., Uric Acid Protects against Focal Cerebral Ischemia/Reperfusion-Induced Oxidative Stress via Activating Nrf2 and Regulating Neurotrophic Factor Expression, Oxid. Med. Cell. Longevity, 2018, 2018, 6069150 Search PubMed.
- B. M. Barakat, H. I. Ahmed, H. I. Bahr and A. M. Elbahaie, Protective Effect of Boswellic Acids against Doxorubicin-Induced Hepatotoxicity: Impact on Nrf2/HO-1 Defense Pathway, Oxid. Med. Cell. Longevity, 2018, 2018, 8296451 Search PubMed.
- C. Park, H. J. Cha, S. H. Hong, G. Y. Kim, S. Kim and H. S. Kim, et al., Protective Effect of Phloroglucinol on Oxidative Stress-Induced DNA Damage and Apoptosis through Activation of the Nrf2/HO-1 Signaling Pathway in HaCaT Human Keratinocytes, Mar. Drugs, 2019, 17(4), 225 CrossRef CAS PubMed.
- W. Huang, H. Liu, S. Zhu, M. Woodson, R. Liu, R. G. Tilton, J. D. Miller and W. Zhang, Sirt6 deficiency results in progression of glomerular injury in the kidney, Aging, 2017, 9(3), 1069–1083 CrossRef CAS PubMed.
- Y. Yang, T. Tian, Y. Wang, Z. Li, K. Xing and G. Tian, SIRT6 protects vascular endothelial cells from angiotensin II-induced apoptosis and oxidative stress by promoting the activation of Nrf2/ARE signaling, Eur. J. Pharmacol., 2019, 859, 172516 CrossRef CAS PubMed.
- M. Guerrero-Hue, S. Rayego-Mateos, C. Vázquez-Carballo, A. Palomino-Antolín, C. García-Caballero, L. Opazo-Rios, J. L. Morgado-Pascual, C. Herencia, S. Mas, A. Ortiz, A. Rubio-Navarro, J. Egea, J. M. Villalba, J. Egido and J. A. Moreno, Protective Role of Nrf2 in Renal Disease, Antioxidants, 2020, 10(1), 39 CrossRef PubMed.
- M. Guerrero-Hue, V. Farre-Alins, A. Palomino-Antolin, E. Parada, A. Rubio-Navarro, J. Egido, J. Egea and J. A. Moreno, Targeting Nrf2 in Protection Against Renal Disease, Curr. Med. Chem., 2017, 24(33), 3583–3605 CrossRef CAS PubMed.
- M. Nezu, N. Suzuki and M. Yamamoto, Targeting the KEAP1-NRF2 System to Prevent Kidney Disease Progression, Am. J. Nephrol., 2017, 45(6), 473–483 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0fo03467c |
| ‡ Co-first authors. |
| This journal is © The Royal Society of Chemistry 2021 |